
Abstract
Mitochondrial metabolism is crucial for hepatocellular carcinoma (HCC) to thrive. Although phospholipids modulate mitochondrial metabolism, their impact on metabolism in HCC remains unknown. Here we report that the mitochondrial phospholipidome is unaltered in HCC mitochondria, suggesting HCC maintain their mitochondrial phospholipidome to enable efficient metabolism and promote thriftiness. Consistent with this, silencing phosphatidylserine decarboxylase (PISD), the inner mitochondrial membrane protein that generates mitochondrial phosphatidylethanolamine (PE), in HEPA1-6 cells impairs mitochondrial metabolism of fatty acid and glucose-derived substrates and reduces electron transport chain I and IV abundance. Moreover, PISD deficiency increased mitochondrial superoxide generation and altered mitochondria dynamics by augmenting mitochondrial fission, mitophagy, and mitochondrial extracellular efflux. Despite compensatory increases in anaerobic glycolysis and peroxisome fat oxidation, mitochondrial PE deficiency reduced DNA synthesis and cell proliferation, effects associated with reduced mTOR signaling and peptide levels. We conclude that targeting mitochondrial PE synthesis may be a viable therapy to slow HCC progression.

Similar content being viewed by others

Introduction
The most common form of liver cancer is hepatocellular carcinoma (HCC), which accounts for ~90% of liver cancer cases [1]. Current therapies for HCC have limited effectiveness, while the development of more effective treatment options has been slow due to a lack of understanding of its molecular pathogenesis and progression. However, emerging evidence in the field has identified that most cancers, including HCC, display altered phospholipid metabolism [2,3,4]. Phospholipids are fundamental for the proper structural integrity of cell membranes [5]. Rapidly growing tumors upregulate phospholipid synthesis to match the tumor growth rate with the need for new cell membranes, while lipid synthesis also alters membrane properties to protect cancer cells from endogenous and exogenous insults [6]. Mitochondrial metabolism is crucial for HCC growth and survival [7,8,9,10] whereas mitochondrial phospholipids are fundamental for the structural integrity of mitochondrial membranes [5] and through lipid-protein interactions modulate mitochondria protein localization, stability, and function [11]. Although the whole cell phospholipidome is altered in HCC [12,13,14], it is not clear whether mitochondrial phospholipid content is altered in HCC.
Phosphatidylethanolamine (PE) is one of the most abundant phospholipids within mammalian cells and is primarily synthesized through the CDP-ethanolamine pathway (Kennedy pathway) or within mitochondria through decarboxylation of phosphatidylserine (PS) to PE via the mitochondrial enzyme phosphatidylserine decarboxylase (PISD) [15,16,17,18]. The PISD pathway generates most mitochondria PE in normal cells, but its importance in the context of HCC is undefined. A previous study demonstrated in Chinese Hamster Ovary cells that silencing PISD results in severe mitochondrial defects and reduces the cell growth rate [19], suggesting that PISD may be a promising target for slowing cancer cell progression. However, another study reported that reducing PISD in skeletal muscle did not alter mitochondrial respiration but increased DNA synthesis [20], a finding that might promote tumor progression. In breast cancer, overexpression of PISD reduced tumor initiating potential and metastasis [21, 22]. We next searched The Human Protein Atlas [23] (www.proteinatlas.org) which revealed PISD was a potential prognostic marker in melanoma and pancreatic cancer, but not HCC. In melanoma, low expression of PISD was more favorable for survival, while in pancreatic cancer high expression of PISD was more favorable for survival, suggesting that PISD may have a different role depending on the cancer type. Taken together, PISD deficiency may alter mitochondrial bioenergetics and cell proliferation differentially depending on cancer type, while the role of mitochondrial PE in HCC metabolism remains unknown.
The purpose of this study was to characterize the phospholipidome of mitochondria isolated from a mouse model of chemical induced HCC and to examine how silencing PISD and reducing mitochondria PE in liver cancer cells alters metabolism, mitochondrial function, and cell proliferation. Our data indicates that the phospholipidome of liver tumor mitochondria is unaltered compared to mitochondria from adjacent non-tumor liver tissue, indicating that the mitochondrial phospholipidome remains intact in HCC and is likely essential to maintain mitochondria function. Consistent with this, silencing PISD impaired mitochondria bioenergetics, an effect associated with reduced DNA synthesis rates and cell proliferation.
Results
Mitochondrial lipidomics in a mouse model of HCC
To determine whether the mitochondrial phospholipidome is altered in HCC, we induced HCC in wildtype C57BL/6J male and female mice using a modification of a previously published protocol involving the carcinogens diethylnitrosamine (DEN) and thioacetamide (TAA) in addition to a high sucrose/high fat Western diet (WD) [24] (Supplementary Fig. 1A). Our protocol was effective at inducing large visible liver tumors and HCC by 34 weeks of age in both male and female mice (Fig. 1A). Hematoxylin and eosin staining revealed HCC with clusters of dense nuclei indicating a high rate of cell proliferation. Serum alanine transaminase (ALT) and aspartate aminotransferase (AST), markers of liver damage that are elevated in HCC, were increased in the WD + DEN/TAA treated groups (Fig. 1B, C). Moreover, the fluorescent intensity of the proliferation marker Ki-67 was elevated in liver sections of both male and female mice in the WD + DEN/TAA group (Fig. 1D, E), further confirming the development of HCC in our model.

A Images of representative liver dissected from 34-week-old mice (top) with a corresponding hematoxylin and eosin (H&E) stain (bottom). The inset H&E image is illustrating normal blood vessels (circular or oval areas that do not stain with H&E) in liver sections from LFD mice, or areas of HCC in WD + DEN/TAA treated mice, which also do not stain with H&E but are larger, irregularly shaped, and surrounded by nuclei (blue staining) (n = 6 mice per group). B Serum ALT activity was elevated in WD + DEN/TAA groups. The male and female LFD groups and female WD + DEN/TAA group had an n = 3. The male WD + DEN/TAA group had an n = 4. C Serum AST activity was elevated in WD + DEN/TAA groups (n = 3 per group). D Nuclei (Hoechst) and Ki-67 (proliferation marker, upregulated in HCC) fluorescent images of liver sections. Ki-67 staining was higher in WD + DEN/TAA treated liver sections (n = 6 per group). E Relative fluorescent intensity of Ki-67 staining from images shown in D (n = 6 per group). F PCA plot of mitochondrial lipids between non-tumor and tumor mitochondria (n = 6 per group). G Heatmap of select lipids that were differentially altered between non-tumor and tumor mitochondria (n = 6 per group). H The abundance of mitochondrial phospholipids was not different between non-tumor and tumor mitochondria (n = 6 per group). The data are presented as means ± S.D.
Mitochondria from non-tumor liver tissue and liver tumor tissue were isolated and rapidly frozen in liquid nitrogen until being prepped for quantitative lipidomic analysis (Supplementary Fig. 1B). For the lipidomic analysis, samples from both male and female mice were used, including three males and three females, and each mouse had an adjacent non-tumor mitochondria and tumor mitochondria fraction analyzed. A total of 1 631 mitochondria lipid species were detected (Supplementary Table 1). A PCA plot did not reveal significant separation in lipid species between non-tumor and tumor mitochondria (Fig. 1F), indicating that the mitochondria from liver tumors are largely unaltered compared to non-tumor mitochondria. However, nine total lipids were significantly different between non-tumor and tumor mitochondria (Fig. 1G). This included an increased tumor mitochondria abundance of carnitine C6-2OH, diacylglycerol 16:0/16:0, free fatty acids 21:2 and 25:0, and PC O-18:2/20:4, while the lipid species lyso-phosphatidylcholine O-24:0, PI 20:2/20:4 and 20:2/20:3, and TG 15:0/15:0/15:1 were significantly reduced in tumor mitochondria. The total abundance of the phospholipid species PE, phosphatidylcholine (PC), phosphatidylinositol (PI), phosphatidylserine (PS), phosphatidylglycerol (PG), phosphatidic acid (PA), and sphingomyelin (SM) was not different between non-tumor and tumor mitochondria (Fig. 1H, Supplementary Fig. 1C–E), suggesting that the phospholipidome of tumor mitochondria is intact and required to maintain proper mitochondrial function and support rapid cell proliferation.
Silencing PISD reduces mitochondrial PE content and alters mitochondrial bioenergetics
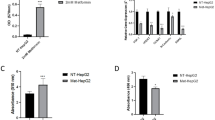
Given that the mitochondrial phospholipid content was similar between tumor and non-tumor mitochondria indicates that maintenance of mitochondrial phospholipid content may be important in HCC to promote optimal mitochondrial metabolism. To test this hypothesis, we sought to determine if mitochondrial phospholipid synthesis is a metabolic vulnerability that could be targeted to disrupt mitochondrial metabolism and impair HCC progression. We focused on PE synthesis, which in mitochondria is generated through a decarboxylation reaction of phosphatidylserine via the enzyme PISD (Fig. 2A), as a previous study has shown PISD deficiency reduces cell viability [19]. To determine how inhibiting mitochondrial PE synthesis impacts HCC metabolism, a 2nd generation lentivirus system was used to knockdown PISD (Fig. 2B). A control scrambled (shSCR) lentivirus or shPISD lentivirus were made using HEK293T cells, and subsequently the lentiviruses produced were used to infect HEPA1-6 cells. The shPISD lentivirus was effective as PISD mRNA abundance was significantly reduced (Fig. 2C). We also determined if shPISD treated cells had altered levels of other genes involved in phospholipid metabolism, and the mRNA abundance of choline/ethanolamine- phosphotransferase-1 (CEPT1) was increased in shPISD treated cells, while the mRNA abundance of choline kinase alpha (CHKA) and tafazzin (TAZ) displayed modest, but significant, reductions. In addition to reduced PISD mRNA abundance, PISD activity in isolated mitochondria was reduced (Fig. 2D). We next isolated mitochondria and measured the phospholipid content of the major mitochondrial phospholipids including PE, cardiolipin (CL), PI, PC, and PS using thin-layer chromatography (TLC). Mitochondrial PE was significantly reduced in shPISD treated cells, while no significant differences between shSCR and shPISD treated cells were observed in other mitochondrial phospholipids (Fig. 2E, F).

A Schematic created using BioRender illustrating PISD enzyme reaction with phosphatidylserine (PS) to form CO2 and phosphatidylethanolamine (PE). B Schematic created using BioRender showing generation of shScrambled (shSCR) and shPISD lentivirus in 293T cells and subsequent infection and gene silencing in HEPA1-6 cells. C The shPISD lentivirus reduced PISD mRNA abundance (n = 2 per group) and altered the abundance of various genes involved in phospholipid metabolism (n = 5 per group). D PISD activity was reduced in shPISD treated cells (n = 3 per group). E, F Thin layer chromatography images indicating shPISD treated cells had reduced mitochondrial PE content (E) and densitometry quantification of thin layer chromatography images presented in E (n = 3 per group) (F). G A Seahorse mitochondrial stress test reveals shPISD treated cells have reduced FCCP stimulated respiration rates (n = 10 per group). H shPISD treated cells have elevated proton leak, calculated by subtracting oxygen consumption rates (OCR) during basal conditions from OCR during oligomycin (oligo) treated conditions (n = 10 per group). I, J Western blot image indicating shPISD treated cells have reduced complex I and IV protein abundance (I) and densitometry quantification of Western blot images shown in I (n = 6 per group) (J). K 1-14C oleate oxidation is reduced in shPISD treated cells (n = 6 per group). L 1-14C lignoceric acid oxidation is increased in shPISD treated cells (n = 6 per group). M PCA plot of untargeted metabolomics data illustrating that shPISD treated cells have an altered metabolome compared to shSCR treated cells (n = 3 per group). N Volcano plot of untargeted metabolomics data illustrating differences in metabolites between shSCR and shPISD treated cells (n = 3 per group). O Heatmap of select lipid metabolism related metabolites that were differentially expressed between shSCR and shPISD treated cells (n = 3 per group). The data are presented as means ± S.D.
To determine how loss of mitochondrial PE impacted mitochondrial bioenergetics and metabolism, we performed a series of cell phenotyping experiments. A Seahorse mitochondrial stress test revealed that shPISD treated cells had reduced FCCP-stimulated maximal aerobic capacity, although basal respiration rates were not altered (Fig. 2G). Mitochondrial proton leak increased in shPISD treated cells (Fig. 2H) suggesting that the membranes of PE deficient mitochondria leak more protons into the mitochondrial matrix independently of ATP synthase (i.e. ATP production). The reduced capacity for oxidative metabolism was associated with reduced abundance of Complex I and IV of the electron transport chain, located on the inner mitochondrial membrane (Fig. 2I, J). These data suggest that mitochondrial PE may play a crucial role in maintaining the stability of these complexes. The protein abundance of the voltage-dependent anion channel (VDAC), which is located on the outer mitochondrial membrane and functions to transport mitochondrial metabolites, was not altered in shPISD treated cells suggesting that PE synthesis mainly impacts inner mitochondrial membrane proteins. The peroxisome marker PEX5 protein abundance was not altered, indicating that peroxisome abundance was not altered. Complete oxidation of 1-14C oleate into 14CO2 was reduced in shPISD treated cells, indicating a reduction in mitochondrial fatty acid oxidation (Fig. 2K). This coincided with an increase in the complete oxidation of 1-14C lignoceric acid into 14CO2, which is a very long chain fatty acid that is initially metabolized in peroxisomes, suggesting that peroxisomal fatty acid metabolism increases to compensate for mitochondrial dysfunction and reductions in fatty acid metabolism in mitochondria (Fig. 2L). This increase in peroxisomal lignoceric acid oxidation was due to increased flux of very long chain fatty acids through peroxisomal fatty acid catabolism, not increased peroxisomal abundance, and greater capacity for very long chain fatty acid metabolism.
To better gauge alterations in global cell metabolism induced by mitochondrial PE deficiency, a metabolomics analysis on whole cell samples was performed (Supplemental Table 2). A PCA plot revealed a clear separation between shSCR and shPISD treated cells (Fig. 2M). A volcano plot revealed a total of 1489 metabolites that were identified (Fig. 2N), with a differential expression analysis revealing that 247 of these metabolites were differentially expressed between shSCR and shPISD treated cells, with 222 metabolites lower in abundance and 25 higher in abundance in shPISD treated cells. A fatty acid metabolite that was more abundant in shPISD treated cells included carnitine C18:1, which is a metabolite required for the transfer of long chain fatty acids across the inner mitochondrial membrane for oxidation. These data suggest that carnitine C18:1 might accumulate due to reduced flux through fatty acid oxidation. Fatty acid species that were less abundant in shPISD treated cells included 16-hydroxyhexadecanoic acid, palmitic amide, cis-3-hexenyl acetate, traumatic acid, oleoyl oxazolopyridine, hexadecanedioic acid, carnitine C5:1, 2,3-dinor thromboxane B1, Cis-8,11,14,17-eicosatetraenoic acid, FFA(16:1), and floionolic acid (Fig. 2O).
PISD deficiency promotes anaerobic glycolysis
Given that mitochondrial PE deficiency reduced mitochondrial fatty acid oxidation, we next tested if glycolysis was altered as a compensatory mechanism. A glycolysis stress test revealed that the extracellular acidification rate (ECAR), a measure of glycolysis, was not different between control and shPISD treated cells when glucose was absent from the media, indicating no change in endogenous glycogen metabolism (Fig. 3A). When glucose was subsequently added to the media, ECAR increased in both control and shPISD treated cells, but this increase was greater in shPISD treated cells. The addition of oligomycin did not alter the differences in ECAR, while inhibiting glycolysis with 2-deoxy-d-glucose (2-DG) reduced ECAR to comparable levels in shSCR and shPISD treated cells. The shPISD treated cells also had elevated rates of lactate appearance in the media (Fig. 3B), indicating increased anaerobic glycolysis. A limitation of the glycolysis stress test is that the pH of the extracellular media is used, which is not only impacted by lactate but also by CO2. To specifically measure glucose oxidation and aerobic glycolysis, 1-14C glucose and 6-14C glucose were used. The 1 labeled carbon on glucose can enter either glycolysis or the pentose phosphate pathway, while the 6 labeled carbon on glucose only enters glycolysis [25]. Both 1-14C and 6-14C glucose oxidation were significantly reduced in shPISD treated cells (Fig. 3C, D), indicating both reduced aerobic glycolysis and flux through the oxidative branch of the pentose phosphate pathway. Glucose uptake, as assessed by the % decrease in glucose from the media over 24 h (Fig. 3E), 1-14C 2-deoxy-glucose uptake (Fig. 3F), or uptake of a fluorescent glucose analog (Fig. 3G, H) all revealed a modest increase in glucose uptake in shPISD treated cells, which helps support increased anaerobic glycolysis. Consistent with increased glucose uptake in shPISD treated cells, GLUT 1 protein abundance increased (Fig. 3I, J). Neither the protein abundance of the pentose phosphate pathway enzyme transketolase (TKT) or enzymes involved in glycolysis including phosphoenolpyruvate carboxykinase 1 (PCK1), enolase (ENO1), aldolase A (ALDOA), glyceraldehyde-3-phosphate dehydrogenase (GAPDH), or phosphoglycerate mutase (PGAM) were altered, suggesting that increased anaerobic glycolysis with mitochondrial PE deficiency occurs due to increased glycolytic flux rather than alterations in protein machinery. Metabolomics also revealed alterations in several carbohydrate metabolites including increased abundance of aldehyde-d-galactose, sorbitol, and xylose with reduced abundance of 3’-fucosyllactose, N-Acetyl-D-glucosamine, xylitol, linustatin, arabinose-5-phosphate, and erythrulose in shPISD treated cells (Fig. 3K). Ribose-1-phosphate is a secondary metabolite in the pentose phosphate pathway that can be converted to ribose-5-phosphate, which is used for nucleotide synthesis. Thus, its accumulation in shPISD treated cells may indicate a reduced flux through the pentose phosphate pathway, which is consistent with the reduction in 1-14C glucose oxidation.

A Extracellular acidification rates (ECAR) during a Seahorse mitochondrial stress test were elevated in shPISD treated cells after the injection of glucose and oligomycin into the media (n = 10 per group). B Percentage change in media lactate over a 24-h period revealed shPISD treated cells generate more lactate (n = 6 per group). C 1-14C glucose oxidation was reduced in shPISD treated cells (n = 12 per group). D 6-14C glucose oxidation was reduced in shPISD treated cells (n = 12 per group). E Percentage decrease in media glucose concentration was greater in shPISD treated cells, indicating an increase in glucose uptake (n = 6 per group). F 1-14C 2-deoxy-d-glucose uptake was greater in shPISD treated cells (n = 12 for shSCR, n = 11 for shPISD). G, H Representative fluorescent images illustrating greater glucose uptake in shPISD treated cells (G) and quantification of fluorescent intensity of images presented in G (n = 25 per group) (H). I, J Western blot images illustrating shPISD treated cells have increased GLUT 1 protein abundance, while other markers of glucose metabolism were unaltered (I) and densitometry quantification of Western blot images presented in I (n = 6 per group) (J). K Heatmap of carbohydrate related metabolites that were significantly different between shSCR and shPISD treated cells (n = 3 per group). L Number of metabolites in each KEGG classification that were significantly different between shSCR and shPISD treated cells. M Percentage of significantly different metabolites within each class. N Heatmap of significantly different glycerophospholipids between shSCR and shPISD treated cells (n = 3 per group). The data are presented as means ± S.D.
Consistent with our mitochondrial and glycolysis functional experiments, a Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis of the significantly impacted cell metabolites revealed that metabolic pathways were most impacted in shPISD treated cells (Fig. 3L). Additionally, the KEGG pathway analysis and a class count ring (Fig. 3M) indicated that glycerophospholipids were significantly impacted, with shPISD treated cells having lower abundance of the lysophosphatidylcholine (LPC) species LPC(22:6/0:0), LPC(18:0/0:0), LPC(0:0/20:4), LPC(0:0/22:5), and LPC(0:0/20:3) as well as the lysophosphatidylethanolamine (LPE) species LPE(18:2/0:0), LPE(18:1/0:0), and LPE(0:0/18:0) (Fig. 3N).
PISD deficiency increases mitochondrial superoxide production and alters mitochondrial dynamics
A cellular process that occurs during normal cellular metabolism is the generation of reactive oxygen species (ROS). Mitochondria generate the ROS species superoxide during electron transfer. Given that mitochondria are an important source of ROS, it was next tested whether mitochondrial PE deficiency altered mitochondria ROS production. The mitochondria specific superoxide indicator MitoSOX TM (Thermo Fisher Scientific, Plaquemine, LA, USA) was used to assess mitochondrial superoxide production and revealed that shPISD treated cells had greater mitochondrial superoxide production (Fig. 4A, B). Cells have antioxidant defense enzymes in place to combat ROS. The primary antioxidant within cells is glutathione (GSH), and metabolomics revealed that PISD deficiency did not alter either GSH or its reduced form, GSSG, indicating that loss of GSH is not the cause of increased ROS production (Fig. 4C). GSH is localized to the cytosol, while superoxide dismutase 2 (SOD2) is localized to the mitochondrial matrix where it neutralizes superoxide originating from the respiratory chain. Therefore, we next measured the protein abundance of SOD2, which revealed shPISD treated cells had reduced protein abundance of SOD2 (Fig. 4D, E). This suggests that PE deficiency disrupts the capacity of mitochondria to neutralize superoxide by reducing SOD2 protein abundance.

A Representative images of MitoSOX staining illustrating greater mitochondrial superoxide content in shPISD treated cells. B Quantification of MitoSOX fluorescent intensity from images presented in A (n = 30 per group). C Relative abundance (measured using metabolomics) of reduced glutathione (GSH) and glutathione disulfide (GSSG) in shSCR and shPISD treated cells (n = 3 per group). D The relative protein abundance of superoxide dismutase 2 (SOD2) was reduced in shPISD treated cells (n = 6 per group). E Western blot image of SOD2 protein abundance. F Western blot image showing protein abundances of markers of mitochondrial fission (DRP, MFF), fusion (OPA1, MFN1, MFN2), or mitophagy (Parkin) (n = 3 per group). G Densitometry quantification of images shown in F (n = 3 per group). H, I Representative fluorescent images of MitoTracker and LysoTracker colocalization. The white arrows are pointing to extracellular mitochondria stained green with MitoTracker. J Greater Manders colocalization coefficient of MitoTracker and LysoTracker indicates more mitophagy in shPISD treated cells (n = 5 per group). K Number of extracellular mitochondria puncta was greater around shPISD treated cells (n = 3 per group). The data are presented as means ± S.D.
Previous studies have demonstrated that loss of PISD results in altered mitochondrial morphology which may impact mitochondria dynamics and the ability of mitochondria to undergo fusion, fission, and mitophagy. Furthermore, the excessive generation of ROS by mitochondria serves as a signal that targets mitochondria for mitophagy. Therefore, it was next determined if mitochondrial PE deficiency would alter mitochondrial dynamics. The protein abundance of dynamin-related protein 1 (DRP1), a marker of mitochondrial fission, was elevated in isolated mitochondria from shPISD treated cells (Fig. 4F, G) suggesting mitochondrial PE deficiency increases fission, although mitochondrial fission factor (MFF) was not altered in shPISD treated cells. The protein abundance of markers of mitochondrial fusion including optic atrophy protein 1 (OPA1), mitofusin 1 (MFN1), and mitofusin 2 (MFN2) were reduced in shPISD treated cells, which is consistent with an increase in mitochondrial fission and suggests that mitochondrial from shPISD treated cells undergo fission into smaller mitochondrial and oppose mitochondrial fusion into larger mitochondria. Further, the protein abundance of the mitophagy marker Parkin was elevated in isolated mitochondria from shPISD treated cells, indicating that loss of mitochondrial PE promotes mitophagy. To further assess mitophagy, the co-localization of mitotracker and lysotracker were used (Fig. 4H, I). shPISD treated cells had a significantly higher Manders colocalization coefficient suggesting greater mitophagy (Fig. 4J), which is consistent with an increase in the mitophagy marker Parkin. Unexpectedly, we also observed a rise in extracellular mitochondria around shPISD treated cells (Fig. 4K), suggesting that in addition to mitophagy, shPISD treated cells may get rid of dysfunctional mitochondria by releasing them extracellularly.
PISD deficiency alters cell proliferation and protein homeostasis
Energetic stress and excessive ROS can slow cellular growth. Therefore, we next tested whether the alterations in metabolism in shPISD treated cells would alter DNA synthesis and cell proliferation. The fluorescent thymidine analog EdU was used to measure DNA synthesis during a one-hour period in full growth media (10% FBS). Fluorescence microscopy revealed that shPISD treated cells had a significantly lower number of cells with EdU fluorescence (Fig. 5A, B). Cell proliferation was also measured using the tetrazolium salt, WST-8, at baseline and 72 h later, and the absorbance produced at 72 h was significantly reduced in shPISD treated cells, suggesting reduced cell proliferation (Fig. 5C). To better understand mechanistically why shPISD cells reduced DNA synthesis and cell proliferation rates, the protein abundance of the known tumor suppressor liver kinase B1 (LKB1) and its downstream target and known energy sensor adenosine monophosphate-activated protein kinase (AMPK) were measured. The shPISD treated cells had normal protein abundance of liver kinase B1 (LKB1) as well as the protein abundance of phosphorylated LKB1 at Serine 428 (p-LKB1S428) (Fig. 5D, E). Similarly, shPISD treated cells had a similar protein abundance of total and phosphorylated adenosine monophosphate activated protein kinase (AMPK) at Threonine 172 (p-AMPKT172), which is a downstream target of LKB1. Although the protein abundance of mechanistic target of rapamycin (mTOR) or its phosphorylation at serine 2448 was not different, the phosphorylation of mTOR’s downstream target eukaryotic translation initiations factor 4E-binding protein 1 (4E-BP1) at serine 65 was reduced. The 4E-BP1 protein binds to eukaryotic translation initiation factor 4E (eIF4E) to inhibit cap dependent transcription. Its affinity to bind to eIF4E depends on its phosphorylation status, with phosphorylation at serine 65 reducing its ability to bind eIF4E and inhibit cap dependent transcription. Therefore, reduced phosphorylation of 4E-BP1 in shPISD treated cells indicates reduced cap dependent translation, which is consistent with reduced cell growth.

A Representative fluorescent images of EdU staining illustrate reduced EdU incorporation and DNA synthesis in shPISD treated cells. B Quantification of EdU fluorescent images presented in A (n = 3 per group). C Absorbance values of WST8 compound at baseline and 72 h in shSCR and shPISD treated cells reveals slower cell proliferation in shPISD treated cells (n = 30 per group). Western blot images of regulators of cellular energy homeostasis (LKB1 and AMPK) as well as protein homeostasis (mTOR and 4E-BP1) (D) and densitometry quantification of images presented in D (n = 6 per group) (E). F Heatmap of significantly affected amino acids and peptides in shSCR and shPISD cells illustrates overall protein homeostasis is disturbed in shPISD treated cells (n = 3 per group). The data are presented as means ± S.D.
Amino acids and peptides alter mTOR signaling. Our metabolomics analysis revealed that amino acids and related metabolites represented the largest class of affected metabolites accounting for 17.41% of the total (Fig. 5F). Valine was the lone amino acid in higher abundance in shPISD treated cells. Meanwhile, numerous other amino acid related metabolites including various tripeptides, tetrapeptides, and pentapeptides were all lower in abundance in shPISD treated cells, which may reduce mTOR signaling.
Discussion
Although Otto Warburg initially hypothesized that the increase in glycolysis in cancer cells was due to reduced mitochondrial function (i.e. Warburg effect), subsequent studies have shown that proper functioning mitochondria are required for some cancer cells to thrive [26, 27]. Mitochondria programming is heterogeneous in cancer and is highly adaptable to meet energy supply and demand within the ever-changing tumor microenvironment [28]. The data herein further supports the notion that properly functioning mitochondria are essential for tumor cells to thrive. We find that isolated mitochondria from mouse HCC tumors maintain their phospholipidome compared to mitochondria isolated from adjacent, non-tumor mitochondria, indicating that normal mitochondrial phospholipid content might be required for HCC cells to thrive. Indeed, this idea was supported by experiments in HEPA1-6 cells that showed that reducing the expression of mitochondria PISD reduces mitochondria PE content, mitochondria respiration, and aerobic glycolysis. Furthermore, mitochondrial PE deficiency increased mitochondrial superoxide abundance, fission, mitophagy, and mitochondrial extracellular efflux, effects that were associated with reduced cell proliferation rates and mTOR signaling. Together, these data suggest that mitochondrial PE is required for the proper function of mitochondria in HCC cells and may be a vulnerability that could be targeted to slow HCC progression.
The transmembrane electron transport chain enzymes are embedded within the inner mitochondrial membrane and are tightly associated with membrane phospholipids. This tight association allows the coordinated assembly of specifically configured supercomplexes which allow electron transfer to function correctly. Our data suggests that PE is not only essential for optimal mitochondrial electron transport chain respiration but also the stability of electron transport chain complexes I and IV, but not complexes II, III, or V, in liver cancer. Consistent with our findings, other work has shown that mitochondrial PE is bound to bovine heart mitochondrial complex I and the catalytic activity of the enzyme is determined, in part, by the amount of PE present [29]. In the porcine heart, PE interacts with complex I, particularly with various segments of the NDUFA11 subunit [30]. In the bovine heart, mitochondrial PE is tightly bound to Complex IV [31]. In Chinese hamster ovary cells, PISD deficiency led to reduced abundance of supercomplexes, which impaired electron transfer and mitochondrial respiration [19]. Consistent with this, the impairments we observed in electron transport chain abundance were associated with reduced respiration rates and the complete oxidation of fatty acids and glucose, which mitochondria are required for. Thus, mitochondrial PE deficiency renders HCC cells less metabolically flexible, which may translate into being less robust and more vulnerable to treatment.
Mitochondria are the primary site of ROS production in cells [32]. A total of 11 sites between Complex I – III of the electron transport chain within mitochondria produce ROS (superoxide or hydrogen peroxide) from substrate oxidation [32]. A major mitochondrial antioxidant is SOD2 which catalyzes the conversion of superoxide radicals into oxygen and hydrogen peroxide [33]. ROS production is normal and acts as an important second messenger in various intracellular signaling pathways [34]. Excessive ROS can be detrimental to cells as they can interact and damage cell components and may play a role in the initiation of HCC [35, 36]. However, ROS may be a double-edged sword, as after HCC is initiated excessive ROS in cancer cells can halt progression and trigger apoptosis in some contexts [36]. Consistent with this, the slower cell proliferation observed herein with mitochondrial PE deficiency was associated with increased mitochondrial superoxide content and reduced SOD2 protein abundance. This cellular ROS stress may in turn play a role in slowing cell proliferation.
Mitochondria are highly dynamic organelles that are constantly undergoing fission, fusion, mitophagy, or transport cycles [37]. These processes help maintain optimal mitochondrial and cellular function while also dictating mitochondrial morphology, number, quality, and cellular location. Previous reports have indicated that mitochondrial dynamics are altered in HCC. For instance, HCC mitochondria were reported to be shorter compared to mitochondria from adjacent, non-tumor tissue while increased mitochondrial fission promoted HCC cell survival [38]. Consistent with this, MFN2 overexpression, which promoted mitochondrial fusion, reduced HCC cell proliferation [39]. Mitophagy is also required for HCC cells to thrive, while mitophagy inhibition reduces HCC cell growth and promotes apoptosis [40, 41]. Thus, increased mitochondrial fission and mitophagy in our model were likely an adaptation to promote HCC cell survival in the setting of mitochondrial PE deficiency, which reduced mitochondrial substrate metabolism and increased mitochondrial ROS generation. Unexpectedly, we also observed increased extracellular mitochondria around cells with mitochondrial PE deficiency, suggesting an increase in the extracellular release of dysfunctional mitochondrial to help maintain the quality of the mitochondrial pool. Cells can release mitochondria or parts of mitochondria in two fashions including 1) a non-membrane bound free form via secretory vesicles, similar to how hormones and neurotransmitters are released, or 2) through membrane-enclosed extracellular vesicles or EV’s, which can be referred to as migrasomes and in a process referred to as mitocytosis where mitochondria are positioned at the cell periphery or tip of protrusions, where they are then pinched off from the cell [42,43,44,45,46]. To our knowledge, this is the first study to show that HCC cells efflux dysfunctional mitochondria. However, it is important to note limitations of this data including that only fluorescently labeled mitochondria were used to measure extracellular mitochondria, while additional techniques that could better visualize extracellular mitochondria, such as electron microscopy, were not used.
mTOR signaling regulates multiple cellular processes including cell growth, proliferation, metabolism, and survival. Mitochondria dysfunction induced by PISD deficiency was associated with reduced mTOR signaling, cap-dependent translation, and cell proliferation, indicating that it may be an effective target to slow HCC progression through its modulation of the mTOR signaling pathway. How PISD deficiency modified mTOR is not clear. Metabolomics revealed that the abundance of most cellular amino acids, except for valine, were in similar abundance between shSCR and shPISD treated cells. However, PISD treated cells had reduced abundance of various tripeptides, tetrapeptides, and pentapeptides, suggesting that sensing of these molecules by the mTOR pathway may be responsible for the reduction in cell proliferation.
In conclusion, this work provides evidence that targeting mitochondrial PE synthesis is sufficient to disrupt mitochondrial metabolism and promote oxidative stress, effects that are associated with reduced DNA synthesis and cell proliferation. Thus, targeting mitochondrial PE synthesis may be a metabolic vulnerability that can be targeted to slow HCC progression.
Material and methods
Animal experiments
This study was approved by the Pennington Biomedical Research Center IACUC. No method of randomization was used to determine which animals were placed in each group. The investigators were not blind to the group allocation. Starting at two weeks of age, male (n = 6) and female (n = 6) C57BL/6J mice (Strain 000664, Jackson Labs, Bar Harbor, ME, USA) were intraperitoneal injected with the carcinogen diethylnitrosamine (DEN, D0516, TCI America, Montgomeryville, PA, USA) once a week for eight weeks (20 mg DEN per kg of bodyweight week one, 30 mg DEN per kg of bodyweight week two, and 50 mg DEN per kg of bodyweight weeks three through eight). All mice were weaned from their mothers at three weeks of age and received a Western diet (TD.88137, Inotiv, West Lafayette, IN, USA, 15.2% protein, 42.7% carbohydrate (with 34% sucrose), and 42% fat, by calories) which is known to accelerate liver cancer progression [47, 48]. After injecting DEN once per week for eight weeks, there was a one-week washout, and then to further promote HCC the mice received thioacetamide (TAA, 172502, Millipore Sigma, Saint Louis, MO, USA) for four weeks in their drinking water (300 mg per liter of water). Control male (n = 6) and female (n = 6) mice received a purified low-fat diet (LFD, TD.05230, Inotiv, 18.7% protein, 68.7% carbohydrate, and 12.6% fat, by calories) starting when they were weaned. All the animals were group housed at 21–23 °C with a 12-h light and 12-h dark cycle for the entire study. Mice were euthanized after a 4 h fast. The mice received a cocktail of ketamine (10004027, Zoetis Inc, Kalamazoo, MI, USA, 120 mg of ketamine per kg of bodyweight), xylazine (061035, Covetrus North America, Dublin, OH, USA, 9 mg of xylazine per kg of bodyweight), and acepromazine (003845, Covetrus North America, 2 mg of acepromazine per kg of bodyweight) to induce anesthesia, after which a cardiac stick was performed to collect blood. Following the blood collection, the mice were cervically dislocated, and tissues were harvested. Blood samples were stored on ice until spinning at 5000 × g for 10 min at 4 °C to separate cells from serum. The serum was aliquoted into a new tube and stored at −80 °C until analysis.
Tissue fixation and hematoxylin and eosin (H & E) staining
Liver sections were fixed overnight in 10% formalin (HT501128, Millipore Sigma) and then transferred to 70% ethanol the following morning. The samples were stored in 70% ethanol until embedding, sectioning, and staining. Liver slides were then stained for H & E in the Molecular Mechanisms Core at Pennington Biomedical Research Center.
Serum alanine aminotransferase (ALT) and aspartate aminotransferase (AST) activity
Serum ALT activity was measured using a commercially available assay kit (E-BC-K235-M, MSE Supplies LLC, Tucson, AZ, USA). Briefly, standards or serum samples were added to a clear plastic 96 well plate. Next, substrate solution was added to each well and the plate was incubated at 37 °C for 30 min. Following this incubation, chromogenic agent was added to the wells, and the plate was incubated at 37 °C for an additional 20 min. Lastly, the plate was left standing for 15 min at room temperature before measuring the OD (505 nm) with a microplate reader. Serum AST activity was measured using a commercially available fluorometric assay kit (E-BC-F043, MSE Supplies LLC). Briefly, serum samples and standards were loaded into a black 96-well microplate, and then the substrate solution was added to each well. After 60 min of incubation at room temperature, the fluorescence intensity was measured with an excitation of 535 nm and an emission of 587 nm.
Ki-67 staining
Immunofluorescence staining of liver sections was performed in the Molecular Mechanisms Core at Pennington Biomedical Research Center. Briefly, slides were baked for 30 min at 60 °C before being run on the Leica Bond RXm using a Research Detection Kit. After standard bake and dewax on the Bond, slides were blocked for 15 min with casein blocking solution (37583, ThermoFisher Scientific) before incubating for 1 h at 37 °C with the primary antibody at a concentration of 1:300 (ab15580, abcam). After washing, the slides were incubated with a goat anti-rabbit Alexa Fluor 647 secondary antibody A32733, Invitrogen, Waltham, MA, USA) at a 1:300 dilution for 120 min. The slides were counterstained with Hoechst, coverslipped with Vector Vectashield Vibrance and scanned using a Zeiss Axioscan 7 slide scanner.
Mitochondrial isolation
Non-tumor liver tissue and liver tumors were dissected from the same mouse and immediately placed in separate ice-cold homogenization tubes containing mitochondrial isolation media (300 mM sucrose, 10 mM HEPES, 1 mM EGTA, and 1 mg/ml BSA). The tissues were gently homogenized using a Teflon pestle, and the homogenized tissue was spun at 800 × g for 10 min at 4 °C. The supernatant containing mitochondria was transferred to a new tube and spun at 12,000 × g for 10 min at 4 °C to pellet mitochondria. After removing the supernatant, the mitochondria were resuspended in mitochondrial isolation media and then spun again at 12,000 × g for 10 min at 4 °C to pellet mitochondria. The supernatant was removed, and the mitochondrial pellet was immediately frozen in liquid nitrogen.
Quantitative lipidomics
The quantitative lipidomics analysis of non-tumor and tumor mitochondria was performed by MetwareBio (Woburn, MA, USA) using ultra-performance liquid chromatography (LC) (Nexera LC-40, Shimadzu, Kyoto, Japan) tandem mass spectrometry (MS) (SCIEX, Triple Quad 6500+, Danaher Corporation, Framingham, MA, USA). Lipids were extracted by adding extraction solvent (Methyl tert-butyl ether: Methanol, 3:1, v/v) containing internal standards to each sample. After mixing samples for 15 min, ultrapure water was added, and the sample was vortexed for 1 min prior to undergoing centrifugation for 10 min at 12 000 rpm. After centrifugation, the upper organic layer was collected and evaporated using a vacuum concentrator. The dry extract was dissolved (Acetonitrile:Isopropyl Alcohol, 1:1, v/v) prior to LC-MS/MS analysis.
Cell lines
HEPA1-6 cells (CRL-1830, ATCC, Manassas, VA, USA) were maintained in high-glucose Dulbecco’s modified Eagle’s medium (10313021, Thermo Fisher Scientific) with the addition of 10% fetal bovine serum (FBS, A5256701, Thermo Fisher Scientific) and 1% penicillin/streptomycin (15140122, Thermo Fisher Scientific). The human embryonic kidney cell line HEK293T/17 cell line (ACS-4500, ATCC) was maintained in high-glucose Dulbecco’s modified Eagle’s medium (11995065, Thermo Fisher Scientific) with addition of 10% FBS and 1% penicillin/streptomycin.
Lentivirus production
PISD expression was inhibited using a 2nd generation lentivirus system, as previously described by our lab [49]. Briefly, 3 μg of lentiviral packing plasmids psPAX2 (plasmid #12260, Addgene, Watertown, MA, USA), 1 μg of envelope expressing plasmid pMD2.G (plasmid #12259, Addgene), and 3 µg of either scrambled shRNA (plasmid #1864, Addgene) or shPISD (TRCN0000115415, Millipore Sigma) were transfected into HEK293T/17 cells to produce the lentivirus over 48 h. After 48 h the media containing the virus was collected and used to infect HEPA1-6 cells. Polybrene was added to improve transfection efficiency, and after 48 h in viral media the HEPA1-6 cells underwent puromycin selection for 72 h prior to experiments.
RNA isolation and quantitative PCR
RNA isolation and quantitative PCR were performed as previously described by our lab [49,50,51,52]. The primer sequences used included PISD/F 5’- TCTGGGGACCTTACAGAAATTGC -3’, PISD/R 5’- GGCACAGATTTATACAGGGACAC -3’, CEPT1/F 5’- ATGAGTGGGCATCGGTCAAC -3’, CEPT1/R 5’- GTGGTGTCGGTAACTGAAACAA -3’, CHKA/F 5’- GGGTGGTCTCAGTAACATGCT -3’, CHKA/R 5’- GAACCCTGGACTCACCATCTT -3’, PTDSS1/F 5’- GCAGGACTCTGAGCAAGGATG -3’, PTDSS1/R 5’- GGCGAAGTACATGAGGCTGAT – 3’, PCYT2/F 5’- CGATGGCTGCTATGACATGGT -3’, PCYT2/R 5’- GCCCCTTATGCTTGGCAATCT -3’, TAZ/F 5’- CCCCCGCTTTGGACAGAAAAT -3’, TAZ/R 5’- AGGCTGGAAATGATTGTGGAG -3’.
PISD activity assay
PISD activity in cell homogenates was measured using a fluorescence assay as previously described [53]. Briefly, homogenized sample from shSCR or shPISD cells (25 µg of protein) were added to microcentrifuge tubes. Next, a reaction buffer containing 0.5 mM PS, 3.1 mM Triton X-100, 50 mM NaCl, and 10 mM potassium phosphate in pH 7.4 was added to the tubes. The PISD reaction was allowed to perform while shaking at 100 rpm at 37 °C for 1 hour and immediately terminated with the addition of 100 mM sodium tetraborate buffer, pH 9.85. The sample was moved into a black (clear bottom) 96 well plate and subsequently a fluorescence detection buffer containing 1,2-DAB, β-ME, sodium tetraborate buffer, pH = 9.85, potassium phosphate in pH 7.4, and Triton X-100 was added to each well and the plate was incubated for 30 min at room temperature while shaking at 100 rpm. Fluorescence intensity (Excitation: 364, Emission: 425) of the sample was measured twice including immediately after adding the fluorescence detection buffer (baseline measurement) as well as 30 min after adding the fluorescence detection buffer, and the difference in fluorescence between 30 min and baseline was used as a measure of PISD activity.
Lipid extraction and thin layer chromatography
Lipids were extracted by mixing samples with a 70:30 (v:v) chloroform:methanol solution with 0.05% BHT added. Next, samples were vortexed for ~45–60 s, 2.3 ml of 0.88% KCl was added, and the mix was centrifuged at 800 × g for 10 min at room temperature. The bottom layer of the sample was aspirated into a new tube and then dried using nitrogen gas. The dried lipids were suspended in chloroform and dried using nitrogen gas. This step was repeated twice. After drying for a second time, the dried lipids were suspended in chloroform before being loaded on a TLC plate (10 × 10 or 20 × 10 cm, silica gel) and allowed to dry. PE, CL, PC, PI, and PS standards were added on each plate so that the lipids in each sample could be accurately identified. The plates were developed using either a chloroform:glacial acetic acid:methanol:water (65:35:5:2) mobile phase or a chloroform:glacial acetic acid:methanol:water (85:25:5:2) mobile phase so that all phospholipids could be better visualized. The developed plates were dried prior to being sprayed with charring solution (4% phosphoric acid/5% copper sulfate) and then allowed to dry further for ~2 min. The plates were then heated at 190 °C for ~15 min and an image of the charred lipid spots was taken using an Odyssey Fc Imager. The intensity of the spots was quantified using ImageJ software.
Seahorse assays
Oxygen consumption rates (OCR) and extracellular acidification rates (ECAR) were measured with a Seahorse Flux Analyzer XFe96 (Seahorse Bioscience, Billerica, MA, USA) using mitochondrial or glycolysis stress test kits, respectively, as previously described [49]. After the lentiviral protocol (described above), ~20,000 cells were plated in the Seahorse XFe96 plate. Approximately 24 h after plating the cells the media was removed and either the mitochondrial stress test (Assay Medium: 5 mM glucose, 2 mM pyruvate and 2 mM glutamine) or glycolysis stress test (XF Assay Medium Modified DMEM (pH = 7.4) was added to the plate for ~1 h prior to performing each test. After each test, the protein concentration of each well was measured using a BCA protein kit and the data was normalized to the total protein amount of the well.
Western blotting
Samples were lysed using RIPA buffer (89900, Thermo Fisher Scientific) containing phosphatase and protease inhibitors. Total protein was measured using a PierceTM BCA Protein Assay Kit (23225, Thermo Fisher Scientific). Between 12 and 20 μg of protein was resolved by SDS-PAGE (Bio-Rad Laboratories, Hercules, CA, USA), transferred onto nitrocellulose membrane or methanol-activated polyvinylidene fluoride (PVDF), normalized using Ponceau S staining solution (40000279, Thermo Fisher Scientific), cut to appropriate size, and blocked in 5% non-fat milk for 75 min. Blots were incubated overnight at 4 °C with total OXPHOS rodent WB antibody cocktail (ab110413, Abcam), voltage-dependent anion channel (VDAC, 4866, Cell Signaling Technology, Danvers, MA, USA), peroxin-5 (PEX5, 83020, Cell Signaling Technology), glucose transporter 1 (Glut 1, 12939, Cell Signaling Technology), transketolase (TKT, 64414, Cell Signaling Technology), phosphoenolpyruvate carboxykinase 1 (PCK1, 12940, Cell Signaling Technology), enolase 1 (ENO1, 3810, Cell Signaling Technology), aldolase a (ALDOA, 8060, Cell Signaling Technology), glyceraldehyde-3-phosphate dehydrogenase (GAPDH, 2118S, Cell Signaling Technology), phosphoglycerate mutase (PGAM1, Cell Signaling Technology), superoxide dismutase 2 (SOD2, 13194, Cell Signaling Technology), dynamin-related protein 1 (DRP1, 8570, Cell Signaling Technology), mitochondrial fission factor (MFF, 84580, Cell Signaling Technology), optic atrophy 1 (OPA1, 80471, Cell Signaling Technology), mitofusin 1 (MFN1, 14739, Cell Signaling Technology), mitofusin 2 (MFN2, 9482, Cell Signaling Technology), Parkin (4211, Cell Signaling Technology), liver kinase B1 (LKB1, 3047, Cell Signaling Technology), phosphorylated LKB1S428 (3482, Cell Signaling Technology), adenosine monophosphate activated protein kinase (AMPK, 2532, Cell Signaling Technology), phosphorylated AMPK (2535, Cell Signaling Technology), mammalian target of rapamycin (mTOR, 2983, Cell Signaling Technology), phosphorylated mTORS2448 (2971, Cell Signaling Technology), eukaryotic translation initiation factor 4E binding protein 1 (4E-BP1, 9452, Cell Signaling Technology), or phosphorylated 4E-BP1S65 (9451, Cell Signaling Technology). All primary antibodies were used at a 1:1000 dilution. Blots were washed and incubated in the appropriate secondary antibody for 1 h. Blots were imaged with Odyssey Fc Imager (LI-COR Biosciences, Lincoln, NE, USA) and the image analysis and densitometry quantification were performed using Li-Cor Image Studio.
Oleate and lignoceric acid oxidation
1-14C oleate was purchased from PerkinElmer (NEC317050UC, Shelton, CT, USA). Fatty acid oxidation was measured using a protocol previously described [54]. Briefly, cells were exposed to 0.2 µCi/ml 1-14C oleate for 3 h. The media was high-glucose Dulbecco’s modified Eagle’s medium (10313021, Thermo Fisher Scientific) with 10% fetal bovine serum (FBS, A5256701, Thermo Fisher Scientific) and 1% penicillin/streptomycin (15140122, Thermo Fisher Scientific). After the incubation, the media was collected and perchloric acid was added to release 14CO2 which was subsequently trapped in filter paper containing NaOH. The radioactivity of the filter paper was measured using a scintillation counter as a measure of complete fatty acid oxidation. The fatty acid oxidation data was then normalized to the total protein amount of each well.
1-14C Lignoceric Acid was purchased from American Radiolabeled Chemicals (ARC-0805-50, Saint Louis, MO, USA). Lignoceric acid oxidation was measured using a protocol adapted from elsewhere [55,56,57]. Briefly, 1-14C lignoceric acid was dissolved in alpha-cyclodextrin prior to being added to high-glucose Dulbecco’s modified Eagle’s medium (10313021, Thermo Fisher Scientific) with 10% fetal bovine serum (FBS, A5256701, Thermo Fisher Scientific) and 1% penicillin/streptomycin (15140122, Thermo Fisher Scientific). The cells were then incubated for 5 h with 0.625 µCi/ml of 1-14C lignoceric acid. After the incubation, the media was collected and perchloric acid was added to release 14CO2 which was subsequently trapped in filter paper containing NaOH. The radioactivity of the filter paper was measured using a scintillation counter as a measure of complete lignoceric acid oxidation. The oxidation data was then normalized to the total protein amount of each well.
Untargeted metabolomics
The untargeted metabolomics analysis was performed by MetwareBio using ultraperformance liquid chromatography-tandem mass spectrometry (LC-MS). Briefly, cell pellets were thawed on ice and a 500 µl solution (methanol:water = 4:1, V/V) containing internal standard was mixed with the cell sample and vortexed for 3 min. The sample was placed in liquid nitrogen for 5 min, on the dry ice for 5 min, and then thawed on ice and vortexed for 2 min. This freeze-thaw cycle was repeated three times in total. The sample was centrifuged at 12,000 × g for 10 min at 4 °C. Next, 300 µl of the supernatant was collected and placed in −20 °C for 30 min. The sample was centrifuged again at 12,000 × g for 3 min at 4 °C. A 200 µl aliquot of the supernatant was used for LC-MS analysis. The metabolite abundance was normalized to total protein for each sample.
Media glucose and lactate concentrations
The rate of glucose disappearance or lactate appearance in the media was measured over a 24-h period. The media used was high-glucose Dulbecco’s modified Eagle’s medium (10313021, Thermo Fisher Scientific) with 10% fetal bovine serum (FBS, A5256701, Thermo Fisher Scientific) and 1% penicillin/streptomycin (15140122, Thermo Fisher Scientific). Briefly, the media of cells was refreshed, and an aliquot of fresh media was stored at −80 °C. After 24 h, an aliquot of the used media incubating with the cells was collected and stored at −80 °C. The glucose concentration of the fresh and used media was measured using a commercially available colorimetric kit (937-03001, FUJIFILM Healthcare Americas Corporation, Lexington, MA, USA). The lactate concentration of the fresh and used media was measured using a commercially available colorimetric kit (NBP3-25788, Novus Biologicals LLC, Centennial, CO, USA). The difference in glucose or lactate concentration between the fresh and used media was calculated to determine the change in concentration over the 24-h incubation period. The glucose or lactate concentration was normalized to total protein content of the well to correct for any differences in cell number.
Glucose uptake and oxidation
1-14C Glucose (NEC043X050UC), 6-14C glucose (NEC045X050UC), and 1-14C 2-deoxyglucose (NEC495A050UC) were purchased from PerkinElmer.
For experiments involving 1-14C or 6-14C glucose, the cells were serum starved for 3 h before experiments. The serum starve media was high-glucose Dulbecco’s modified Eagle’s medium (10313021, Thermo Fisher Scientific) with 1% penicillin/streptomycin (15140122, Thermo Fisher Scientific). After serum starvation, cells were exposed to 0.2 µCi/ml of 1-14C or 6-14C glucose for 3 h in serum free media. After the incubation, the media was collected and perchloric acid was added to release 14CO2 which was subsequently trapped in filter paper containing NaOH. The radioactivity of the filter paper was measured using a scintillation counter. The oxidation data was normalized to the total protein content of each well.
For experiments involving 1-14C 2-deoxy-glucose the cells were serum starved for 3 h prior to experiments. After serum starvation, 0.5 µCi/ml of tracer was added to cells for 20 min. Following the incubation, the cells were washed twice with ice cold phosphate-buffered saline (10010023, Thermo Fisher Scientific) and then RIPA buffer (89900, Thermo Fisher Scientific) was added to lyse the cells, and the radioactivity of the cell lysate was measured using a scintillation counter.
Fluorescent imaging of glucose uptake
A commercially available kit and glucose probe (UP03-10, Dojindo Molecular Technologies, Inc, Rockville, MD, USA) were used to measure glucose uptake with an ECHO Revolve fluorescent microscope (ECHO: A BICO Company, San Diego, CA, USA). Nucblue (R37605, Thermo Fisher Scientific) was used to stain nuclei during these glucose uptake experiments. Briefly, the glucose probe and NucBlue were added to glucose and fetal bovine serum free Dulbecco’s Modified Eagle Medium (A1443001, ThermoFisher Scientific). Next, the media on the cells was suctioned off and the cells were washed once with the glucose and fetal bovine serum free media. Next, the media containing the glucose probe and NucBlue were added to the cells for 10 min at 37°C. After 10 min, the cells were washed twice with PBS, glucose and serum free media without the glucose probe and NucBlue was added back to the wells, and the plates were imaged.
Mitochondrial reactive oxygen species (ROS) determination
The MitoSOXTM (M36008, Thermo Fisher Scientific) red mitochondrial dye was used to measure mitochondria superoxide production. Cells were treated with 500 nM MitoSOXTM red reagent, which was added to high-glucose Dulbecco’s modified Eagle’s medium (10313021, Thermo Fisher Scientific) with 1% penicillin/streptomycin (15140122, Thermo Fisher Scientific) for 20 min at 37 °C. Cells were washed three times in PBS, stained in NucBlue for 10 min at 37 °C, washed three times in PBS, and then imaged in phenol free media with an ECHO Revolve fluorescence microscope.
Mitophagy assay and extracellular mitochondria
Mitophagy was assessed by measuring the colocalization of MitoTracker Green (M7514, ThermoFisher Scientific) with LysoTracker Red (L7528, ThermoFisher Scientific) using an ECHO Revolve fluorescent microscope. ImageJ software was used to quantify the Manders Colocalization Coefficient. Briefly, the dyes were added to high-glucose Dulbecco’s modified Eagle’s medium (10313021, Thermo Fisher Scientific) with 1% penicillin/streptomycin (15140122, Thermo Fisher Scientific). Next, the media containing dyes was added to the cells for 30 min at 37 °C. After 30 min, the cells were washed twice with PBS and then media without dyes was added back to each well, and the cells were imaged. Extracellular mitochondria were also assessed with MitoTracker Green in these same images.
Cell proliferation
DNA synthesis (S phase of cell cycle) was measured using a Click-iT® EdU Alexa Fluor® 488 Imaging Kit (C10337, Thermo Fisher Scientific). Cells were grown in DMEM and plated on an 8 chamber ibidi slide (ibidi USA, Inc., Fitchburg, WI, USA). After 24 h the cells were labeled with EdU (5-ethynyl-2′-deoxyuridine) by incubating them with 10 μM of EdU at 37 °C for one hour. After the incubation, the cells were washed and then subsequently fixed in formaldehyde-PBS solution for 15 min and then permeabilized in Triton X-100 PBS solution for 20 min, both of which were performed at room temperature. The Click-iT reaction cocktail was then added to the cells, after which a Hoechst-PBS solution was used to stain Nuclei prior to imaging on an ECHO Revolve fluorescence microscope. Images were analyzed using CellProfiler cell image analysis software.
A Cell Counting Kit (WST-8/CCK8) (ab228554, abcam, Boston, MA, USA) was used to measure cell proliferation. Briefly, 10,000 shSCR or shPISD treated cells were plated in a 96-well clear flat bottom plate and incubated for 24 h in a 37 °C, 5% CO2 incubator. We followed the manufacturer’s instructions using a total volume of 100 µl/well of medium with cells and two blank wells (medium without cells). The first measure (baseline) was made 24 h after seeding the cells on the 96-well plate. We changed the medium to a fresh one containing 10 µl/well of WST-8 solution, followed by one hour of incubation. The colored product was quantitatively measured at 460 nm using a microplate reader (Bio-Rad Laboratories). After measuring the baseline absorbance, the media was refreshed every 24 h until a second measure was made 72 h later, using the same procedure described above. Cell proliferation is directly proportional to the absorbance measured in the media.
Statistical analysis
To estimate the appropriate sample size to achieve adequate power we used G*Power 3 software. GraphPad Prism version 10.0.2 was used to analyze the data and generate the figures. The data are presented as means ± S.D. Where appropriate either two-sided t-tests or two-way ANOVAs were used for statistical analysis. For two-way ANOVA’s, follow-up Bonferroni multiple comparison tests were used to identify specific differences. The BioRender app (www.biorender.com) was used to generate some images. ImageJ was used to determine the fluorescent intensity of the images. Bioinformatics analysis of the lipidomics and metabolomics data was performed by MetwareBio.
Data availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Baecker A, Liu X, La Vecchia C, Zhang ZF. Worldwide incidence of hepatocellular carcinoma cases attributable to major risk factors. Eur J Cancer Prev. 2018;27:205–12.
Cheng M, Bhujwalla ZM, Glunde K. Targeting phospholipid metabolism in cancer. Front Oncol. 2016;6:266.
VanSant-Webb C, Low HK, Kuramoto J, Stanley CE, Qiang H, Su AY, et al. Phospholipid isotope tracing suggests beta-catenin-driven suppression of phosphatidylcholine metabolism in hepatocellular carcinoma. Biochim Biophys Acta Mol Cell Biol Lipids. 2024;1869:159514.
Liu Q, Zhang X, Qi J, Tian X, Dovjak E, Zhang J, et al. Comprehensive profiling of lipid metabolic reprogramming expands precision medicine for HCC. Hepatology. 2024;81:1164–80.
Dai Y, Tang H, Pang S. The crucial roles of phospholipids in aging and lifespan regulation. Front Physiol. 2021;12:775648.
Rysman E, Brusselmans K, Scheys K, Timmermans L, Derua R, Munck S, et al. De novo lipogenesis protects cancer cells from free radicals and chemotherapeutics by promoting membrane lipid saturation. Cancer Res. 2010;70:8117–26.
Yang Q, Wang L, Liu J, Cao W, Pan Q, Li M. Targeting the complex I and III of mitochondrial electron transport chain as a potentially viable option in liver cancer management. Cell Death Discov. 2021;7:293.
Wheaton WW, Weinberg SE, Hamanaka RB, Soberanes S, Sullivan LB, Anso E, et al. Metformin inhibits mitochondrial complex I of cancer cells to reduce tumorigenesis. Elife. 2014;3:e02242.
Hsu CC, Wu LC, Hsia CY, Yin PH, Chi CW, Yeh TS, et al. Energy metabolism determines the sensitivity of human hepatocellular carcinoma cells to mitochondrial inhibitors and biguanide drugs. Oncol Rep. 2015;34:1620–8.
Veiga SR, Ge X, Mercer CA, Hernandez-Alvarez MI, Thomas HE, Hernandez-Losa J, et al. Phenformin-induced mitochondrial dysfunction sensitizes hepatocellular carcinoma for dual inhibition of mTOR. Clin Cancer Res. 2018;24:3767–80.
Levental I, Lyman E. Regulation of membrane protein structure and function by their lipid nano-environment. Nat Rev Mol Cell Biol. 2023;24:107–22.
Liu Z, Zhang Z, Mei H, Mao J, Zhou X. Distribution and clinical relevance of phospholipids in hepatocellular carcinoma. Hepatol Int. 2020;14:544–55.
Krautbauer S, Meier EM, Rein-Fischboeck L, Pohl R, Weiss TS, Sigruener A, et al. Ceramide and polyunsaturated phospholipids are strongly reduced in human hepatocellular carcinoma. Biochim Biophys Acta. 2016;1861:1767–74.
Muir K, Hazim A, He Y, Peyressatre M, Kim DY, Song X, et al. Proteomic and lipidomic signatures of lipid metabolism in NASH-associated hepatocellular carcinoma. Cancer Res. 2013;73:4722–31.
van der Veen JN, Kennelly JP, Wan S, Vance JE, Vance DE, Jacobs RL. The critical role of phosphatidylcholine and phosphatidylethanolamine metabolism in health and disease. Biochim Biophys Acta Biomembr. 2017;1859:1558–72.
Bleijerveld OB, Brouwers J, Vaandrager AB, Helms JB, Houweling M. The CDP-ethanolamine pathway and phosphatidylserine decarboxylation generate different phosphatidylethanolamine molecular species. J Biol Chem. 2007;282:28362–72.
Hsu P, Shi Y. Regulation of autophagy by mitochondrial phospholipids in health and diseases. Biochim Biophys Acta Mol Cell Biol Lipids. 2017;1862:114–29.
Sam PN, Calzada E, Acoba MG, Zhao T, Watanabe Y, Nejatfard A, et al. Impaired phosphatidylethanolamine metabolism activates a reversible stress response that detects and resolves mutant mitochondrial precursors. iScience. 2021;24:102196.
Tasseva G, Bai HD, Davidescu M, Haromy A, Michelakis E, Vance JE. Phosphatidylethanolamine deficiency in Mammalian mitochondria impairs oxidative phosphorylation and alters mitochondrial morphology. J Biol Chem. 2013;288:4158–73.
Selathurai A, Kowalski GM, Mason SA, Callahan DL, Foletta VC, Della Gatta PA, et al. Phosphatidylserine decarboxylase is critical for the maintenance of skeletal muscle mitochondrial integrity and muscle mass. Mol Metab. 2019;27:33–46.
Chen YC, Humphries B, Brien R, Gibbons AE, Chen YT, Qyli T, et al. Functional isolation of tumor-initiating cells using microfluidic-based migration identifies phosphatidylserine decarboxylase as a key regulator. Sci Rep. 2018;8:244.
Humphries BA, Cutter AC, Buschhaus JM, Chen YC, Qyli T, Palagama DSW, et al. Enhanced mitochondrial fission suppresses signaling and metastasis in triple-negative breast cancer. Breast Cancer Res. 2020;22:60.
Thul PJ, Lindskog C. The human protein atlas: a spatial map of the human proteome. Protein Sci. 2018;27:233–44.
Memon A, Pyao Y, Jung Y, Lee JI, Lee WK. A modified protocol of diethylnitrosamine administration in mice to model hepatocellular carcinoma. Int J Mol Sci. 2020;21:5461.
Rodriguez-Rodriguez P, Fernandez E, Bolanos JP. Underestimation of the pentose-phosphate pathway in intact primary neurons as revealed by metabolic flux analysis. J Cereb Blood Flow Metab. 2013;33:1843–5.
Weinberg F, Hamanaka R, Wheaton WW, Weinberg S, Joseph J, Lopez M, et al. Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc Natl Acad Sci USA. 2010;107:8788–93.
Martinez-Reyes I, Cardona LR, Kong H, Vasan K, McElroy GS, Werner M, et al. Mitochondrial ubiquinol oxidation is necessary for tumour growth. Nature. 2020;585:288–92.
Abbassi-Daloii T, El Abdellaoui S, Kan HE, van den Akker E, T Hoen PAC, Raz V, et al. Quantitative analysis of myofiber type composition in human and mouse skeletal muscles. STAR Protoc. 2023;4:102075.
Sharpley MS, Shannon RJ, Draghi F, Hirst J. Interactions between phospholipids and NADH:ubiquinone oxidoreductase (complex I) from bovine mitochondria. Biochemistry. 2006;45:241–8.
Wu M, Gu J, Guo R, Huang Y, Yang M. Structure of Mammalian Respiratory Supercomplex I(1)III(2)IV(1). Cell. 2016;167:1598–609.e10.
Shinzawa-Itoh K, Aoyama H, Muramoto K, Terada H, Kurauchi T, Tadehara Y, et al. Structures and physiological roles of 13 integral lipids of bovine heart cytochrome c oxidase. EMBO J. 2007;26:1713–25.
Goncalves RL, Quinlan CL, Perevoshchikova IV, Hey-Mogensen M, Brand MD. Sites of superoxide and hydrogen peroxide production by muscle mitochondria assessed ex vivo under conditions mimicking rest and exercise. J Biol Chem. 2015;290:209–27.
Li Y, Huang TT, Carlson EJ, Melov S, Ursell PC, Olson JL, et al. Dilated cardiomyopathy and neonatal lethality in mutant mice lacking manganese superoxide dismutase. Nat Genet. 1995;11:376–81.
Tell G, Vascotto C, Tiribelli C. Alterations in the redox state and liver damage: hints from the EASL Basic School of Hepatology. J Hepatol. 2013;58:365–74.
Zhang Z, Zhao Q, Wang Z, Xu F, Liu Y, Guo Y, et al. Hepatocellular carcinoma cells downregulate NADH:Ubiquinone Oxidoreductase Subunit B3 to maintain reactive oxygen species homeostasis. Hepatol Commun. 2024;8:e0395.
Xing L, Tang Y, Li L, Tao X. ROS in hepatocellular carcinoma: What we know. Arch Biochem Biophys. 2023;744:109699.
Chen W, Zhao H, Li Y. Mitochondrial dynamics in health and disease: mechanisms and potential targets. Sig Transduct Target Ther. 2023;8:333.
Huang Q, Zhan L, Cao H, Li J, Lyu Y, Guo X, et al. Increased mitochondrial fission promotes autophagy and hepatocellular carcinoma cell survival through the ROS-modulated coordinated regulation of the NFKB and TP53 pathways. Autophagy. 2016;12:999–1014.
Wang W, Lu J, Zhu F, Wei J, Jia C, Zhang Y, et al. Pro-apoptotic and anti-proliferative effects of mitofusin-2 via Bax signaling in hepatocellular carcinoma cells. Med Oncol. 2012;29:70–6.
Ma M, Lin XH, Liu HH, Zhang R, Chen RX. Suppression of DRP1‑mediated mitophagy increases the apoptosis of hepatocellular carcinoma cells in the setting of chemotherapy. Oncol Rep. 2020;43:1010–8.
Feng N, Zhang R, Wen X, Wang W, Zhang N, Zheng J, et al. RABIF promotes hepatocellular carcinoma progression through regulation of mitophagy and glycolysis. Commun Biol. 2024;7:1333.
Pan KH, Chang H, Yang WY. Extracellular release in the quality control of the mammalian mitochondria. J Biomed Sci. 2023;30:85.
Ma L, Li Y, Peng J, Wu D, Zhao X, Cui Y, et al. Discovery of the migrasome, an organelle mediating release of cytoplasmic contents during cell migration. Cell Res. 2015;25:24–38.
Jiao H, Jiang D, Hu X, Du W, Ji L, Yang Y, et al. Mitocytosis, a migrasome-mediated mitochondrial quality-control process. Cell. 2021;184:2896–910.e13.
Lyamzaev KG, Nepryakhina OK, Saprunova VB, Bakeeva LE, Pletjushkina OY, Chernyak BV, et al. Novel mechanism of elimination of malfunctioning mitochondria (mitoptosis): formation of mitoptotic bodies and extrusion of mitochondrial material from the cell. Biochim Biophys Acta. 2008;1777:817–25.
Nakajima A, Kurihara H, Yagita H, Okumura K, Nakano H. Mitochondrial Extrusion through the cytoplasmic vacuoles during cell death. J Biol Chem. 2008;283:24128–35.
Healy ME, Chow JD, Byrne FL, Breen DS, Leitinger N, Li C, et al. Dietary effects on liver tumor burden in mice treated with the hepatocellular carcinogen diethylnitrosamine. J Hepatol. 2015;62:599–606.
Healy ME, Lahiri S, Hargett SR, Chow JD, Byrne FL, Breen DS, et al. Dietary sugar intake increases liver tumor incidence in female mice. Sci Rep. 2016;6:22292.
Heden TD, Chow LS, Hughey CC, Mashek DG. Regulation and role of glycophagy in skeletal muscle energy metabolism. Autophagy. 2022;18:1078–89.
McCall CP, Mancini MC, Staszkiewicz J, Mashek DG, Heden TD. Heterozygous GAA knockout is nonconsequential on metabolism and the spatial liver transcriptome in high-fat diet-induced obese and prediabetic mice. Physiol Rep. 2025;13:e70276.
Heden TD, Franklin MP, Dailey C, Mashek MT, Chen C, Mashek DG. ACOT1 deficiency attenuates high-fat diet induced fat mass gain by increasing energy expenditure. JCI Insight. 2023;8:e160987.
Heden TD, Chen C, Leland G, Mashek MM, Najt CP, Shang L, et al. Isolated and combined impact of dietary olive oil and exercise on markers of health and energy metabolism in female mice. J Nutr Biochem. 2022;107:109040.
Choi JY, Black R 3rd, Lee H, Di Giovanni J, Murphy RC, Ben Mamoun C, et al. An improved and highly selective fluorescence assay for measuring phosphatidylserine decarboxylase activity. J Biol Chem. 2020;295:9211–22.
Huynh FK, Green MF, Koves TR, Hirschey MD. Measurement of fatty acid oxidation rates in animal tissues and cell lines. Methods Enzymol. 2014;542:391–405.
Huang TY, Zheng D, Houmard JA, Brault JJ, Hickner RC, Cortright RN. Overexpression of PGC-1alpha increases peroxisomal activity and mitochondrial fatty acid oxidation in human primary myotubes. Am J Physiol Endocrinol Metab. 2017;312:E253–E63.
Huang TY, Linden MA, Fuller SE, Goldsmith FR, Simon J, Batdorf HM, et al. Combined effects of a ketogenic diet and exercise training alter mitochondrial and peroxisomal substrate oxidative capacity in skeletal muscle. Am J Physiol Endocrinol Metab. 2021;320:E1053–E67.
Huang TY, Goldsmith FR, Fuller SE, Simon J, Batdorf HM, Scott MC, et al. Response of liver metabolic pathways to ketogenic diet and exercise are not additive. Med Sci Sports Exerc. 2020;52:37–48.
Acknowledgements
We are grateful to David Burk, Shirley Ennis, and Stephanie Wong for help with tissue embedding, sectioning, staining, and imaging. We are also grateful for MetwareBio for performing the lipidomics and metabolomics experiment and bioinformatics analysis. This study was financially supported by National Institute of Health grants GM154665, DK109556, DK125258, and GM135002 (subprojects 8000, 8438, and 5359) awarded to TDH. A National Institute of Health LAUNCHED pilot project grant U24DK132740 awarded to MCM supported this study (Grant PI’s were Robert L. Newton and Peter T. Katzmarzyk). This project used facilities within the Cell Biology and Bioimaging Core at PBRC that are supported in part by NIH center awards P20GM135002 (Grant PI is Jacqueline M. Stephens) and P30DK072476 (Grant PI is Leanne M. Redman). BioRender was used to create the graphical abstract and other schematic figures used in the manuscript.
Author information
Authors and Affiliations
Contributions
MCM: design of experiments, data generation and interpretation, data visualization, figure preparation, manuscript writing. CPM: data generation and interpretation, figure preparation, manuscript editing. RCN: supplied 1-14C lignoceric acid, design of experiments, data interpretation, manuscript editing. WSD: data generation and interpretation, manuscript editing. TDH: design of experiments, data generation and interpretation, data visualization, figure preparation, manuscript writing.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
All methods were performed in accordance with relevant guidelines and regulations. The animal study was approved by the Pennington Biomedical Research Center Institutional Animal Care and Use Committee. The use of chemical carcinogens was approved by the Pennington Biomedical Research Center Institutional Biosafety Committee.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Mancini, M.C., McCall, C.P., Noland, R.C. et al. Targeting mitochondrial phosphatidylethanolamine alters mitochondrial metabolism and proliferation in hepatocellular carcinoma. Oncogenesis 15, 3 (2026). https://doi.org/10.1038/s41389-025-00593-y
Received:
Revised:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41389-025-00593-y
