
Abstract
Hypoplastic Left Heart Syndrome (HLHS) is a life-threatening congenital heart disease characterized by underdevelopment of the left heart and aorta. Animal models of HLHS are used to study mechanisms of disease onset and progression and generally fall into two in vivo categories: genetic and mechanical. Genetic models primarily employ zebrafish and mice, whereas mechanically induced models are developed in chick embryos, fetal lambs, and rodents. Together, genetic and mechanical models provide insight into developmental and hemodynamic mechanisms of HLHS but differ in their ability to reproduce key anatomical and physiological features. Genetic models have identified genes and pathways involved in structural abnormalities and disrupted cell lineage. Mechanical models commonly restrict left-heart inflow using surgical or catheter-based techniques to induce hypoplasia of the left ventricle, valves, and aorta. Findings across chick embryos, fetal lambs, and mouse models support the “no flow, no grow” theory. This review synthesizes current HLHS animal models, evaluates their advantages and limitations, and considers their translational relevance from genetic and hemodynamic perspectives, while emphasizing species-specific limitations.
Impact
-
Systematically evaluate genetically and mechanically induced HLHS models across zebrafish, mice, rats, chick embryos, and fetal lambs.
-
Discuss how these models elucidate developmental and hemodynamic mechanisms of HLHS, highlight innovations such as CRISPR-based gene editing and staged in utero flow restriction, and assess their translational relevance, particularly for fetal intervention research.
-
Provide practical criteria for model selection and interpretation, weighing strengths, limitations, and fidelity to human HLHS for mechanistic and translational aims.
Similar content being viewed by others

Introduction
Hypoplastic Left Heart Syndrome (HLHS) is one of the most severe types of congenital heart disease (CHD), characterized by an underdeveloped left side of the heart and aorta at birth.1 In the United States, HLHS affects more than 2000 infants a year, and its prevalence is 2.3 per 10,000 births.2,3
Because the underdeveloped left ventricle (LV) cannot adequately support systemic perfusion, the circulation becomes functionally univentricular and solely reliant on the right ventricle (RV) to provide systemic output. Therefore, HLHS typically necessitates a series of staged open-heart surgeries, including the Norwood procedure, which is considered one of the most challenging procedures in pediatric cardiac surgery. The Norwood procedure is usually performed within the first few days to 2 weeks of life and consists of reconstruction of the hypoplastic aortic arch and placement of an artificial shunt to the pulmonary artery to balance pulmonary and systemic circulations. Subsequently, patients undergo bidirectional Glenn and Fontan procedures in early childhood as part of the standard single-ventricle surgical pathway.
Although surgical techniques and patient outcomes have improved, palliation remains the only available treatment option, and many of these patients still progress to heart failure.4 According to a recent study,5 even among newborns with HLHS who survived to undergo the Norwood or hybrid procedure, transplant-free survival was only 31.0% at 35 years. Survival improved over time until the early 2000s but has plateaued over the past 20 years, despite improvements in perioperative morbidity and mortality for the initial palliative procedure.5 In this context, prenatal diagnosis of HLHS and interventions to promote LV growth have received increasing attention.
The characteristic anatomical features of HLHS include (1) hypoplasia of the LV, (2) hypoplasia of the aorta, (3) atretic or stenotic aortic (AA or AS) and mitral valves (MA or MS), (4) a small or hypoplastic left atrium (LA), (5) a dilated right ventricle (RV), (6) dilatation of the main pulmonary artery, and (7) LV endocardial fibroelastosis in the presence of AA or AS.6 A marked discrepancy in chamber size between the hypoplastic LV and RV is a key clue suggesting HLHS during fetal cardiac screening. Absence or severe reduction of anterograde flow across the aortic valve, togheter with retrograde flow in the aortic arch, can be detected on color Doppler imaging in the fetal three-vessel and aortic arch views and are considered hallmark signs of HLHS.6
The etiology of HLHS is thought to involve genetic factors that influence multiple aspects of cardiac development, as suggested by the high incidence of heart defects among first-degree relatives of HLHS patients.7 Although the cellular mechanisms underlying HLHS remain poorly characterized, recent genetic studies have identified several genes implicated in HLHS, such as NOTCH1,8 SAP130/Pcdha9,9 Rbfox2,10 and LRP2,11 many of which are involved in cardiac cell proliferation and differentiation.
In addition to genetic factors, alterations in embryonic cardiac blood flow are widely recognized as playing a critical role in the pathogenesis of HLHS. The etiology of HLHS has been conceptualized by the “flow theory”, which proposes that cardiac malformations, including HLHS, arise when the coupling between normal blood flow patterns through the developing heart and the growth of cardiac valves and myocardium is disturbed in utero.12 Thus, both genetic and hemodynamic aspects are thought to be important contributors to the development of HLHS. Nevertheless, our understanding of HLHS development remains limited. To address this gap, robust animal models are essential for understanding the mechanisms underlying the development of HLHS, including its onset and progression. These models are also crucial for identifying novel and effective therapeutic targets that may lead to curative strategies.
In this review, we evaluate the current options for animal models of HLHS, including zebrafish, rodents, chick embryos, and fetal lambs, and discuss their potential utility, advantages, and limitations in improving our understanding of HLHS pathology from both genetic and anatomical perspectives, as well as their implications for advancing therapeutic strategies and outcomes for patients with HLHS.
Genetically induced HLHS models
While the genetic basis of HLHS is still mostly undetermined and no transgenic animal models have succeeded in recapitulating a human HLHS phenotype, studies that showed a link between human HLHS and certain chromosomes13,14 support the involvement of genetic factors in HLHS development. Advanced imaging techniques in zebrafish embryos and transgenic or knockout mice provide advantages for exploring the etiology and pathology of HLHS. Representative genes and their functions in animal models and humans associated with HLHS phenotypes are summarized in Table 1.
Zebrafish model
Zebrafish have a two-chambered heart consisting of a single atrium and ventricle, accompanied by a bulbus arteriosus, and therefore have a single circulatory loop; in contrast, humans possess a four-chambered heart with dual pulmonary and systemic circulations.15 Additionally, the zebrafish myocardium is dominated by a trabecular layer with only a thin compact layer. Their valve architecture comprises a simple atrioventricular valve and a ventriculo-bulbar valve rather than the four distinct valves seen in humans.16 Given these fundamental architectural differences, zebrafish are frequently used as a model to investigate cardiac developmental mechanisms relevant to HLHS. Several intrinsic features make them particularly advantageous for cardiovascular research: their embryos are optically transparent and highly susceptible to chemical exposure,17 and they can survive through oxygen diffusion in the absence of a functional cardiovascular system,18 which greatly facilitates in vivo experimentation and observation. Additionally, their developing hearts are prone to looping defects that can be caused by alterations in myocardial cell polarity, cell number, or blood flow.19 With these features, zebrafish serve as a valuable platform for understanding cardiac development, identifying disease-associated genes, and establishing links between genetic mutations and CHD.20
To date, only one genetically induced HLHS model in zebrafish has been reported to reproduce the full spectrum of HLHS phenotypes, including defects in the ventricle, valves, and aorta, as described by Huang et al. in 2022.21 They demonstrated that zebrafish lacking two orthologs of the RNA-binding protein Rbfox2 develop HLHS-like malformations (Fig. 1). Rbfox2 is a highly conserved protein known to be involved in splicing and has been implicated in HLHS-related abnormalities in humans.22,23

a Brightfield images of 72hpf CTRL and DKO embryos. Black arrow indicates pericardial edema and open arrow indicated pooled blood. b Although ventricular and atrial areas are similar between cohorts during systole, a 32% and 38% reduction was observed in DKO during diastole, respectively. Additionally, %FAC approached zero in both DKO chambers. Each dot represents one embryo in all graphs. Data are presented as mean values ± one SD. Statistical significance was determined by an unpaired, two-tailed Student’s t-test assuming equal variances. P values are shown. c Single optical sections of hearts from live 72 hpf CTRL and DKO embryos carrying the Tg (kdrl:mCherry) transgene. The endothelial lining of the aorta appears partially or fully obstructed. CTRL control, DKO double knock out, hpf hours postfertilization, FAC fractional area change, AVC atrioventricular cushion, a atrium, v ventricle. These images were adapted/reproduced from ref. 21
According to Gallagher et al., deletion of this gene is known to cause severe morphological and functional cardiac defects in zebrafish as well.24 To create zebrafish lacking Rbfox1-like and Rbfox2, Huang et al. employed the CRISPR-Cas9 genome editing technique, which is a widely used method in mouse studies,25 and observed that inducing a single-gene mutation in zebrafish at post-embryonic day 3 led to the full spectrum of HLHS-like malformations. Additionally, Rbfox1-/-/Rbfox2-/- double knockout embryos exhibited paralysis, pericardial edema, poor circulation, and 100% mortality by post-embryonic days 4–5, whereas single mutants failed to undergo embryonic cardiogenesis. The study also showed that these structural cardiac defects were caused by impaired pump function, and the phenotypes were rescued when Rbfox1 was specifically expressed in the myocardium. Rbfox1 proteins are essential for sarcomere assembly and mitochondrial metabolism, and defects in these processes contribute to the observed functional deficiencies.21
Although not specifically aiming to create HLHS phenotypes, several reports have addressed genetically induced left ventricular outflow tract obstruction (LVOTO) or left ventricular hypoplasia. Liu et al.9 reported in 2017 that a CRISPR-Cas9-targeted Sap130a loss-of-function allele resulted in a small ventricle phenotype at 72 h post-fertilization, with 36% penetrance in maternal-zygotic (MZ) zebrafish. This phenotype was associated with impaired cardiomyocyte proliferation and differentiation, increased cell death, and a reduction in ventricular cardiomyocytes. Similarly, DeMoya et al.26 recently found that MZ Sap130a mutants displayed small ventricles in 48% of the population by 48 h post-fertilization, impacting zebrafish cardiac sarcomere regulation. Moreover, they observed that the combination of Sap130a and hdac1, both of which are required for ventricular formation in zebrafish,27,28 exacerbated the small ventricle phenotype.
Another zebrafish study by Edwards et al.20 in 2020 demonstrated that WAVE2 complex genes (brk1, nckap1, and wasf2), along with regulators of small GTPase signaling (cul3a and racgap1), are critical for cardiac development in zebrafish. Knockdown of brk1, cul3a, nckap1, and wasf2 resulted in reversed cardiac looping, while racgap1 knockdown led to a hypoplastic ventricle with poorly contractile and dilated atria in 40% of the population. Around the same time, Theis et al.11 conducted a study using whole-genome and iPSC RNA sequencing from an HLHS family trio. They injected morpholino and sgRNA/CRISPR directed against LRP2 (an LDL receptor-related protein) into zebrafish embryos, which led to a hypoplastic phenotype with decreased cardiomyocyte numbers at 72 h post-fertilization. These results suggest that LRP2 plays a crucial role in heart development, particularly in regulating cardiomyocyte generation in the ventricular chamber. As such, zebrafish also serve as a complementary platform to validate HLHS-related genes.
Mouse model
The mouse heart has a four-chambered structure, dual circulation, and basic valve anatomy, similar to those of the human heart.29 However, the left ventricle is dominated by a prominent trabecular myocardium with a relatively thin compact layer,30 a configuration that influences how ventricular growth and wall stress respond to developmental perturbations. Additionally, the coronary artery system is markedly simpler, with limited secondary branching and a less complex collateral network.31 These anatomical and developmental features suggest that mice may not spontaneously manifest the full spectrum of human HLHS morphology and that targeted genetic manipulation is required to reproduce the phenotype. In this context, several genetically induced HLHS mouse models have been developed, beginning with the first report in 2017.9
The first genetically induced HLHS mouse model was published in 2017,9 and has since been commonly employed in this field as the Ohia mutant line32 (Fig. 2). Through a mutagenesis screen of 3000 mice, mutations in Sap130 and Pcdha9 (which encodes the cell-adhesion protein protocadherin A9) were identified as genetic contributors to the HLHS phenotype between embryonic day 13.5 and 18.5, with 26% penetrance. Both genes are expressed in the embryonic heart, and these findings were further validated using CRISPR-Cas9 genome editing.25 HLHS was also observed through the interaction between Sap130 and Pcdha9 mutations. Homozygous mutants exhibited embryonic lethality before or at mid-gestation or, in rare cases, survived with severe cardiac defects. Furthermore, neither single mutants nor double heterozygous mutants displayed CHD.9,33 The hypoplastic left ventricle (LV) was observed only in Sap130 mutants, accompanied by disrupted cardiomyocyte proliferation and differentiation. In contrast, Pcdha9 mutants showed a normal-sized LV but developed isolated aortic hypoplasia, aortic stenosis, and a bicuspid aortic valve.9

a Representative images of newborn (P0) or E16.5 hearts from wild-type and HLHS mutants. Hypoplastic aorta and LV are visible in the HLHS mutant. Scale bars: 1.0 mm. b Compared with controls, the HLHS mutant exhibited hypoplastic aorta and aortic valve atresia, hypertrophied LV with no lumen, and MV stenosis and patent FO (arrowhead). Scale bars: 0.5 mm. AO aorta, PA pulmonary artery, RV right ventricle, LV left ventricle, HLHS hypoplastic left heart syndrome, FO fossa ovalis, LA left atrium, RA right atrium. These images were adapted/reproduced from ref. 32
In a similar context, Teekakirikul et al.34 used Pcdha9 mutant mice generated by CRISPR gene editing and demonstrated that homozygous mutations in Pcdha9 can lead to bicuspid aortic valve and LVOTO phenotypes from embryonic day 14.5, with a penetrance of over 25%, which decreased to 20% at postnatal day 1 and further dropped to 10% in adulthood. Although this does not fully meet the definition of HLHS,1 it should be considered a subtype of HLHS because LVOTO includes obstructive anomalies of the LV and aorta.35
Previously, no cardiovascular defects were observed in Rbfox2 knockout mice, despite its identification as a high-risk gene for CHD.23,36,37 Verna et al.10 however, found several molecular and phenotypic features of HLHS in Rbfox2 knockout mice. Rbfox2 is essential for embryonic survival and for the proper formation of the cardiac chambers, LVOT, and yolk sac vasculature between embryonic days 9.5 and 10.5. By day 10.5, 100% of Rbfox2 knockout mice showed pericardial edema, similar to that seen in congestive heart failure, and their growth was severely impaired, leading to embryonic lethality. Early deletion of Rbfox2 leads to embryonic lethality by post-embryonic day 11.5 due to defects in heart development that resemble HLHS. Additionally, RNA sequencing performed on Rbfox2 mutants revealed that Rbfox2-regulated alternative splicing (AS) networks impact Rho GTPase cycling and signaling, which is crucial for cell organization, extracellular matrix composition, and cytoskeletal dynamics in the embryonic heart.10,38 The identification of Rbfox2-regulated AS networks may not only accelerate HLHS research further but also highlight the lethality of the genetically induced HLHS-like model.
Mechanically induced HLHS models
Mechanically induced hypoplasia of the left-sided heart structures by restricting blood flow has been a well-established approach for modeling HLHS, with early studies in chick embryos and later expanding to fetal lambs and, more recently, rodents. These interventions, which alter intracardiac hemodynamics through surgical manipulation, are grounded in the concept that both normal and abnormal cardiac morphogenesis are influenced by flow patterns within the embryonic circulatory system.39 As early as the 1960s and 1970s, local surgical modifications of blood flow were shown to induce specific pathological changes in heart development.40,41,42 In the following sections, we summarize and discuss the advantages and limitations of key mechanical interventions in chick, fetal lamb, and rodent models of HLHS.
Chick embryo model
Among the various model organisms used to study heart development, the chick embryo (fertile White Leghorn eggs) is preferred because of its ease of culture and a mature four-chambered heart structure.43,44 Chick embryonic hearts have atrial and ventricular septation and distinct atrioventricular and outflow valves, closely resembling the basic anatomical layout of the human heart. Although avian hearts exhibit thinner ventricular walls, simplified coronary arterial architecture, and differences in outflow tract alignment, the planar orientation of the chick embryo on top of the yolk allows easy access to surgical procedures and imaging modalities, which in turn enables detailed spatiotemporal analysis and supports precise microsurgical interventions.45 Additional advantages of the chick embryo model include its relatively longer avian cardiogenic period compared to fish, frog, or mouse, and fewer ethical concerns.
As described above, investigations into the relationship between the pattern of blood flow through the chick embryonic heart and cardiogenesis began as early as the 1960s.40,41 The first attempt to create HLHS-like aortic valvular abnormalities was conducted by Harah et al. in 1973.46 Since then, multiple experimental approaches have been developed, including mitral valve obstruction, left atrial clipping, CTB (banding of the heart tube’s outflow tract), right vitelline artery ligation (VAL) (inflow tract banding of the heart tube), and left atrial ligation (LAL).
Early experimental approaches associated with HLHS-like anatomical abnormalities are summarized in Table 2. Most of these studies did not specifically aim to induce HLHS pathology in chick embryos, but they have played an important role in understanding the mechanisms of cardiac morphogenesis, at least partially related to HLHS-associated structures. Harah et al. created mitral valve occlusion by placing a nylon device at the left atrioventricular canal at incubation days of 4.5–5 (Hamburger-Hamilton (HH) stages 23–25) and successfully demonstrated a small LA, hypoplastic LV, and hypoplastic ascending aorta in 67% of the surviving experimental models.46 They were inspired by the conotruncal banding (CTB) method performed by Gessner et al.40,41 and the clipping method by Rycheter et al.,47 who applied fine silver clips to the 3rd, 4th, and 6th aortic arches in various combinations, resulting in ventricular septal defects with a malformed aortic arch.
CTB, although originally used to investigate aortic dextroposition,40,41 was later employed by Celik et al.48 to induce hypoplastic aortic arches and investigate their development in chick embryos. They observed distinct matrix remodeling mechanisms during vascular development in the CTB group compared to the control group, triggered by altered wall shear stress, which contributed to a chronic increase in vessel lumen area. However, with this method, the heart is forced to work against increased resistance (afterload), and thus, left heart hypoplasia cannot be induced, resulting only in hypoplastic development of the aorta.
In contrast to CTB, which targets the outflow tract of the heart tube, another approach involved occlusion of the inflow tract of the heart tube through right vitelline vein ligation to induce HLHS-like cardiac morphogenesis in chick embryos. Hogers et al.49 developed a chick model in which intracardiac flow was altered by ligating a vitelline vein with a microclip, effectively rerouting venous inflow. This intervention redirected blood flow toward the heart via collateral pathways and caused long-term structural defects such as VSD, semilunar valve anomalies, atrioventricular anomalies, and arch artery malformations. Using the same technique, Ursem et al.50 reported impaired diastolic ventricular filling, which further led to changes in cardiac function and morphology.
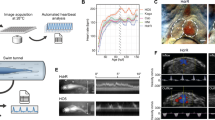
Currently, the most widely accepted approach for specifically inducing HLHS pathology is LAL (Table 3), which redirects blood flow from the left to the right heart, resulting in underdevelopment of the left heart structures.51,52,53,54 This model may also lead to compensatory overdevelopment of the right ventricle,51 which is also a fundamental feature of HLHS. In LAL, a 10-0 nylon suture loop is placed around the LA and tied to restrict the left atrioventricular orifice and reduce the volume of the LA by 50–75% (Fig. 3). Typically, this procedure has been performed at HH stages 21–23 (incubation days of 3–4),55 during the looping stage before ventricular septation. The resulting chronic alterations in flow patterns disrupt normal looping, septation, and valve formation. Chronic preload reduction following LAL has been shown to modify myocardial architecture prior to the onset of cardiac defects, including changes in ventricular wall stiffness, compact myocardium thickness, and ventricular dimensions.51,56 In addition, computational approaches such as in silico modeling44 or computational fluid dynamics analysis45 have been used to visualize blood flow alterations in the atrioventricular canal.

Top row: Embryos are cultures within their shells and opened at embryonic day (ED) 3 (a). The egg opening creates an easy access for surgery (b) and echocardiographic imaging (c), respectively. Bottom row: a Schematic representation of LAL steps. b Representative pictures of LAL steps. Step 1, Opening chorionic and allantoic membranes and exposing the animal. Step 2, Flipping the animal vertically to expose its left side shown in Step 3. Step 4, Opening the pericardium and exposing the left atrium. Step 5, Placing the pre-prepared surgical knot on the top of the atrium. Step 6, Tightening the knot around the atrium. Step 7, Cutting the extra ends of the suture and finally flipping back the animal to its original orientation. These images were adapted/reproduced from ref. 45
Sedmera et al.51 combined LAL with other methods to evaluate CTB as an increased afterload model and LAL as a mixed model of elevated right ventricular preload and reduced LV preload. They concluded that increasing pressure load is the main stimulus for myocardial growth in embryogenesis, while increased volume load is compensated primarily by dilatation of the heart. In a separate study, Lucitti et al.57 compared the manipulated arterial flow induced by LAL with the altered load from right VAL, revealing that maintaining arterial pressure and circulatory energy efficiency is more critical than flow volume during early embryogenesis. However, the long-term consequences of this mechanism have not been fully elucidated.
In-utero fetal lamb model
The sheep heart closely resembles the human heart with respect to its overall four-chamber configuration, atrioventricular and semilunar valve structure, and coronary artery dominance, making it highly suitable for translational cardiac research. Compared with the human heart, the sheep heart is more vertically oriented within the thorax and has subtle differences in papillary muscle geometry and coronary branching patterns.
The fetal sheep is the most frequently used model for human congenital heart disease, since it tolerates chronic instrumentation and surgical interventions and has a size at birth similar to that of human neonates.58 Notably, cardiomyocyte numbers in humans and sheep plateau at similar levels, and cardiomyocyte cell cycle activity declines similarly during the perinatal periods.59,60
The first HLHS model using a large animal was reported using a fetal lamb model by Fishman et al.61 in 1978. They selected fetal lambs because of the similarity between fetal lambs and humans in body weight, blood pressure, oxygen tension, ventricular stroke volume, and internal distribution of blood flows at corresponding gestational stages.61,62,63 Partial LV inflow obstruction was induced by inflating a balloon catheter in the LA of the fetuses, at 90–120 gestational days (0.6–0.8 gestation). Within seven days, LV output decreased to 30% of the control (p < 0.01), and LV/RV chamber volume was reduced to less than half of the control (p < 0.0001). However, the survival time was only 2–7 days (mean 4 days), and the fetuses in this model were unable to adapt to the abrupt hemodynamic alterations.
After more than four decades, novel attempts to establish an HLHS model in fetal lambs have recently emerged. One such approach involves in-utero occlusion of the fetal foramen ovale (FO) during mid-gestation, performed via fetal cardiac catheterization using transhepatic access.64 In this procedure, a stent is positioned across the FO, and an occluder is subsequently anchored within the stent (Fig. 4a), leading to the collapse of the fetal LV. The intervention was performed between 104 and 117 gestational days, and flow restriction and euthanasia were planned 21 days after occluder implantation. Among a total of 26 fetuses that underwent this procedure, 16 fetuses experienced successful implantation of the FO occluder. By the time of the planned euthanasia 3 weeks later, two fetuses died 1–2 days after the procedure from intra-abdominal hemorrhage, and one fetus died 1 day before euthanasia due to a dislodged occluder. At euthanasia, seven animals were found to have incomplete FO occlusion, and thus the remaining six animals with successful FO occlusion were evaluated. For these six fetuses, a smaller mitral valve annular diameter and reduced LV width compared to the control group (n = 9) were observed on echocardiography, and significantly smaller aortic valve diameter, lower mitral/tricuspid valve ratio, and lower LV/RV ratio were confirmed at termination.

a Occlusion of foramen ovale, b coiling occlusion of the left atrium, and c restriction of blood flow through the mitral valve by an inflated balloon inside the left atrium. IVC inferior vena cava, LA left atrium, LV left ventricle, RA right atrium, RV right ventricle, FO foramen ovale, BCT brachiocephalic trunk, PDA patent ductus arteriosus. These images were adapted/reproduced from refs. 64,65,66
Another attempt involved implanting coils in the LA via transcatheter delivery, as reported by Reuter et al.65 in 2023. The concept of occluding LV inflow remained the same, but the method differed in that platinum coils (0.018” IDC, Boston Scientific) were deployed above the mitral valve (Fig. 4b). An underfilled LV and either decreased or retrograde ascending aortic flow was confirmed under continuous ultrasound guidance. This intervention was performed at 76 days’ gestation (0.52 gestation), followed by echocardiographic evaluation, which was conducted 45–50 days after the intervention (0.84 gestation), and the animals were subsequently terminated at 147 days’ gestation. Of the 34 fetuses that underwent LA coil implantation, 15 fetuses (44%) survived until the echocardiographic evaluation, while the remaining 19 fetuses (56%) died due to acute circulatory changes. One of the 15 surviving fetuses was not included in further analyses due to severe coil-induced mitral regurgitation. Among the 14 fetuses, nine showed retrograde flow in the ascending aorta and subsequently developed severe (n = 4) or intermediate (n = 5) left heart hypoplasia. For the other five fetuses without retrograde flow, normal left heart growth was observed. Anatomically, the size of the left heart in the nine fetuses with retrograde ascending aortic flow was compared to that of the control group (n = 12), but due to large variations in fetal body weight (ranging from 2 to 5 kg), the reported left heart measurements were significantly smaller when normalized to right heart structures or total heart weight. They also performed transcriptomic analyses, which revealed an increase in fibroblasts, a decrease in cardiomyocytes, and an upregulation of extracellular matrix components.
The most recent development in the HLHS fetal lamb model is a modified LA balloon method,66 which utilizes a balloon catheter device inflated in the fetal LA (Fig. 4c). To address the previously observed high mortality caused by abrupt hemodynamic changes from LA balloon use, this method employed gradual balloon inflation after implantation to achieve staged LV unloading. The implantation procedure was performed at 120 days’ gestation (0.8 gestation), and balloon inflation was started 2 days later to achieve restriction of mitral inflow to the LV between 125- and 127-days’ gestation. The fetuses were terminated at around 135 days’ gestation after 8 days of flow restriction. Although the overall fetal mortality rate was 42%, it decreased to 17% in the latter half of the cases. Evaluations using transesophageal echocardiography were performed before termination, showing a significantly lower LV/RV cross-sectional area ratio and decreased LV end-diastolic and end-systolic volumes compared to the control group. On the right heart, RV end-diastolic and end-systolic volumes did not differ between experimental and control fetuses. Morphologically, the LV/RV mass ratio was significantly smaller, and RV mass was greater in the experimental group than in the control group. These findings suggest asymmetric cardiac development, with suppressed left ventricular growth and compensatory remodeling of the right ventricle in response to volume overload.
In-utero rodent model
Mice and humans have homologous heart structures with highly conserved stages of heart development, and the short gestational period, as well as non-seasonal breeding of mice, offer significant advantages over large animal models. However, due to their small size, mice have been mainly used for investigating genetic aspects of HLHS, and available surgical manipulation remains very limited. The first and currently only attempt to utilize mice to induce HLHS by in-utero surgical manipulation was reported by Rahman et al. in 2021.67
In their murine study, an embolizing agent, a shear-thinning biomaterial (STB), was delivered to the fetal LA, combined with ultrasound-guided microinjection techniques. The LA was embolized at embryonic day 14.5 to block blood flow into the left side of the fetal heart structure, inducing hypoplasia of the LV, ascending aorta, and aortic valve by gestational term at 18.5 days. Survival rates at gestational term were 56% for embolized fetuses (76% for sham fetuses), and among the surviving fetuses, 48% (12 out of 25) showed successful LA embolization, as confirmed by magnetic resonance imaging. These mice with confirmed LA embolization demonstrated retrograde aortic arch flow at gestational term, and they had non-apex-forming LV and hypoplastic ascending aorta. This study established the first mouse model of isolated HLHS with a fully penetrant cardiac phenotype and survival to term.
As a unique attempt utilizing rats, Watson et al.68 created heart hypoplasia disorders by inducing congenital diaphragmatic hernia (CDH) in rats. Nitrofen is an herbicide that is known to induce CDH in rodents. Back in the 1990s, the relationship between morphologic changes in several organs and CDH was reported,69,70 and follow-up studies were performed in 2022.68 Nitrofen was given to pregnant rats at gestational day E10, and the hearts from the fetuses positive for CDH at E21 (80-85% of the fetuses treated with Nitrofen) were investigated. These hearts were smaller and showed decreased maturity, probably because normal stretching of the heart wall was diminished due to mechanical compression by visceral organs. Although it was unclear whether the induced heart hypoplasia was left-side specific, the findings suggest that reduced blood flow (mechanical force) could contribute to hypoplastic cardiac development in rats.
Rescue and reversal attempt in HLHS animal models
Recently, several centers have offered fetal aortic valvuloplasty (FAV) as an in utero intervention for mid-gestation fetuses with critical aortic stenosis. Critical aortic stenosis causes major biomechanical and functional aberrations71 and is associated with a high probability of progression to HLHS.72 FAV may prevent progression of aortic stenosis to HLHS in mid-gestation, with reported technical success rates of 81–87%; among live-born infants, 33-59% achieve a biventricular circulation according to recent reports.73,74,75 However, in the context of establishing HLHS animal models, no attempts have yet been made to apply such fetal valvuloplasty approaches. Only Sedmera et al.76 have performed a reverse intervention in their HLHS chick embryo model, using right atrial clipping following left atrial clipping, which resulted in increased myocyte proliferation and greater myocardial volume in both ventricles.
Discussion
Current in vivo models of HLHS are broadly categorized into two types: genetically induced and mechanically induced models. Zebrafish and mice are commonly used in genetic models to study molecular and developmental pathways, whereas chick embryos, fetal lambs, and rodents are employed in mechanical models to investigate the impact of altered hemodynamics. Both approaches have contributed significantly to understanding the physiological and biomechanical mechanisms underlying HLHS. The ability of these models to replicate HLHS-like features varies depending on the developmental stage targeted and the specific experimental design.
Zebrafish and mice are widely used as models for evaluating human cardiac function in developmental and functional genomics, as well as in studies of cardiac structure, due to their physiological and morphological similarities to the human heart.77,78 They are particularly valuable for understanding the function of each gene in development and disease, owing to their ease of genetic manipulation.17,25 Additionally, their short lifespans and rapidly developing embryos make them ideal models for studying cardiac development.79,80,81 These models are especially useful because testing multiple genes in mammalian in vivo models can be challenging due to cost and throughput limitations.11
Historically, HLHS was considered a valve disease82 and was thought to result from reduced blood flow caused by LVOTO, typically due to aortic valve atresia secondary to LV growth deficiency.21 Therefore, only one study has reported a genetically induced zebrafish model that mimics the full spectrum of HLHS, including deficiencies in the ventricle, valve, and aorta.21 However, employing zebrafish as a model for HLHS in human conditions or for investigating the chronic phase of the disease remains challenging, due to their two-chambered heart79 and the high morbidity associated with HLHS-like phenotypes.9,20,21
Humans and mice share a high degree of genetic similarity, with ~90% of their genomes consisting of conserved gene order regions.83 This makes mice valuable for studying cardiac development and disease, particularly through genetic modification.25,78 Despite multiple attempts, early efforts to create HLHS mouse models were largely unsuccessful, possibly due to the short gestation period and the presumed hemodynamic etiology of HLHS.81 In this context, the development of the Ohia mutant line, which exhibits mutations in Sap130 and Pcdha9, represented a significant advancement.9 This model demonstrated hypoplastic left ventricles and aortic atresia, reflecting key features of HLHS. However, it also suffered from low disease penetrance and high postnatal lethality, limiting its reproducibility and translational value.
One contributing factor to the limitations of the Ohia model is the variability in the presence and combination of mutations among individual mice, which complicates phenotype consistency and interpretation.9,32,33 This genetic heterogeneity highlights the ongoing difficulty in defining the precise genetic architecture of HLHS. Even so, the Ohia model remains a valuable foundation for the development of improved genetic systems that can more precisely dissect gene-specific roles in cardiac morphogenesis. While each model has its limitations, genetic models of HLHS are beginning to shed light on significant genetic contributors to left heart hypoplasia, which may help close knowledge gaps regarding structural defects and cell-specific contributors.82
Regarding mechanically induced HLHS models, non-genetic factors, especially abnormalities in hemodynamics and/or biomechanical loading, are believed to play a larger role in HLHS etiology, and thus more experience and knowledge have been accumulated compared to genetically induced models. The approach to such mechanical factors is based on the ‘no flow, no grow’ theory,84,85 which suggests that an abnormal cardiac valve could restrict blood flow, leading to an underdeveloped (hypoplastic) ventricle during cardiogenesis. This type of mechanical approach is a straightforward yet effective method for recapitulating HLHS physiology and hemodynamics, because the degree of induced abnormality can be surgically controlled and adjusted, and the effects are easily visualized as the severity of heart defects. Additionally, therapeutic interventions can be made targeting these mechanical and hemodynamic aspects, enabling their rapid transition to clinical use. Indeed, fetal heart intervention on HLHS fetuses has already been used in clinical settings.73,86
Looking back at the historical development of chick embryo models, two main approaches were used to create cardiac defects: (1) occlusion of the outflow tract of the heart loop (e.g., CTB and VAL) to create a hypoplastic arch, and (2) restriction or occlusion of inflow (e.g., LA clip, LAL, vitelline vein ligation, LA coil, and LA balloon) to induce underdevelopment of the ventricle and heart valves. The former attempt succeeded in achieving the development of a hypoplastic arch, but the systemic ventricle usually became dilated and hypertrophic and did not become hypoplastic. On the other hand, in the latter attempt, although each methodology varied, the results were generally convincing as HLHS models. These results are well explained by the ‘no flow, no grow’ theory, and currently, inducing obstruction to the left heart inflow by various means is a common method for creating HLHS animal models.
In-utero fetal lamb models have utilized several strategies to restrict left heart inflow, namely, occlusion of the FO, LA obstruction using coils, and mitral inflow restriction via balloon placement. The timing and survival rates for these three methods were as follows: FO occlusion was performed at 0.7–0.75 of gestation with 50% survival; LA coiling at 0.5 of gestation with 44% survival; and balloon placement at 0.8 of gestation with 58% survival. Among these, balloon-mediated mitral inflow obstruction demonstrated the highest survival rate, possibly due to the gradual inflation process, which may have allowed compensatory hemodynamic adaptation. The later gestational timing of balloon placement may have also contributed to the improved outcomes. Interestingly, Reuter et al.65 observed that younger fetuses showed greater tolerance to LA coil implantation and better survival, indicating that fetal age may interact with other procedural variables. Despite the relatively short duration and later onset of flow obstruction, the balloon group achieved comparable left heart hypoplasia to that seen in the FO occlusion model. In contrast, early and prolonged LA coiling resulted in the most pronounced hypoplasia. These findings suggest that both the timing and duration of flow restriction are key determinants of the severity of left ventricular underdevelopment.
Compared to the chick embryo models, fetal lamb models are advantageous due to their larger size, which enables several surgical procedures with detailed adjustment. The experiences with fetal lamb models, however, were still generally very limited, and more data are required on variations in induction timing and methods. Regarding the mouse as a mechanically induced HLHS model, they also shared the same concept of restricting blood inflow, utilizing STB as an embolizing agent inside the LA, and demonstrated promising data. Although only one published experience exists so far, efforts with mice should be encouraged, since insights into surgical techniques obtained from a large number of mouse models may be applicable to lamb fetuses at earlier gestational stages. In addition, a mechanically induced rat model, in which CDH is created using nitrofen, has been reported, suggesting that mechanical compression may also contribute to cardiac hypoplasia.
As a common limitation across these animal models, there are species-specific differences and incomplete phenotypic presentation of HLHS, limiting the potential for translation to humans or to other animal species. Additionally, there has been no report successfully demonstrating HLHS phenotypes after delivery. While currently available models of HLHS have offered valuable insight into its etiology, a clear need remains for more sophisticated approaches to better understand and improve survival in HLHS.
All animal models introduced in this review continue to evolve, and the future direction of HLHS modeling are likely to involve both refinement of existing fetal hemodynamic approaches and expansion into complementary genetic systems. Small-animal models will remain indispensable for dissecting developmental mechanisms and genetic pathways, but their limited body size, short gestation, and high perinatal mortality constrain their usefulness in the development of new surgical or catheter-based therapies. Consequently, more refined large-animal models, particularly fetal lambs and potentially pigs, are expected to play an increasingly central role. Recent efforts in the fetal lamb model have focused on shifting surgical interventions to earlier gestational ages (~50–70 days) to improve procedural tolerance, survival, and more consistent of LV growth suppression, and similar strategies could be extended to establish a fetal porcine HLHS models, which is not yet available. In these large-animal systems, introducing and subsequently releasing controlled left heart inflow or outflow obstruction at defined developmental stages may clarify whether and when left heart growth can be rescued, thereby delineating the critical time window for effective fetal therapy. Ultimately, it is plausible that future models will integrate defined genetic susceptibility with controlled hemodynamic perturbations across multiple species, enabling a more faithful reconstruction of the clinical HLHS phenotype and providing a robust platform for testing emerging fetal and perinatal interventions.
Conclusion
Robust animal models are essential for elucidating the genetic and hemodynamic mechanisms underlying the pathogenesis of HLHS. Genetically induced models, such as zebrafish and mice, and mechanically induced models, including chick embryos, fetal lambs, and rodents, each provide unique insights into different aspects of disease development. Although the extent to which these models replicate HLHS morphology varies, both approaches have contributed significantly to our understanding of disease onset and progression. Continued refinement of these models will be critical for identifying novel therapeutic targets and improving clinical translation. Nevertheless, species-specific limitations and the absence of postnatal HLHS phenotypes highlight the need for further innovation in model development.
References
Grossfeld, P., Nie, S., Lin, L., Wang, L. & Anderson, R. H. Hypoplastic left heart syndrome: a new paradigm for an old disease? J. Cardiovasc. Dev. Dis. 6, 10 (2019).
Tsao, C. W. et al. Heart disease and stroke statistics-2023 update: a report from the American Heart Association. Circulation 147, e93–e621 (2023).
Reller, M. D., Strickland, M. J., Riehle-Colarusso, T., Mahle, W. T. & Correa, A. Prevalence of congenital heart defects in metropolitan Atlanta, 1998-2005. J. Pediatr. 153, 807–813 (2008).
Lubert, A. M. et al. Considerations for advanced heart failure consultation in individuals with fontan circulation: recommendations from action. Circ. Heart Fail 16, e010123 (2023).
Gaynor, J. W. et al. Long-term survival and patient-reported outcomes after staged reconstructive surgery for hypoplastic left heart syndrome. J. Am. Coll. Cardiol. 85, 2386–2398 (2025).
Bravo-Valenzuela, N. J. & Araujo Júnior, E. Prenatal diagnosis of hypoplastic left heart syndrome: Current knowledge. Radio. Bras. 56, 282–286 (2023).
Loffredo, C. A. et al. Prevalence of congenital cardiovascular malformations among relatives of infants with hypoplastic left heart, coarctation of the aorta, and d-transposition of the great arteries. Am. J. Med Genet A 124a, 225–230 (2004).
Theis, J. L. et al. Compound heterozygous notch1 mutations underlie impaired cardiogenesis in a patient with hypoplastic left heart syndrome. Hum. Genet 134, 1003–1011 (2015).
Liu, X. et al. The complex genetics of hypoplastic left heart syndrome. Nat. Genet 49, 1152–1159 (2017).
Verma, S. K. et al. Rbfox2 is required for establishing RNA regulatory networks essential for heart development. Nucleic Acids Res 50, 2270–2286 (2022).
Theis, J. L. et al. Patient-specific genomics and cross-species functional analysis implicate LRP2 in hypoplastic left heart syndrome. Elife 9, e59554 (2020).
Hattam, A. T. A potentially curative fetal intervention for hypoplastic left heart syndrome. Med. Hypotheses 110, 132–137 (2018).
Hinton, R. B. Jr et al. Hypoplastic left heart syndrome is heritable. J. Am. Coll. Cardiol. 50, 1590–1595 (2007).
Hinton, R. B. et al. Hypoplastic left heart syndrome links to chromosomes 10q and 6q and is genetically related to bicuspid aortic valve. J. Am. Coll. Cardiol. 53, 1065–1071 (2009).
Genge, C. E. et al. The zebrafish heart as a model of mammalian cardiac function. Rev. Physiol. Biochem. Pharmacol. 171, 99–136 (2016).
Kaiser, A. D. et al. A fluid-structure interaction model of the zebrafish aortic valve. J. Biomech. 190, 112794 (2025).
Wilkinson, R. N., Jopling, C. & Van Eeden, F. J. Zebrafish as a model of cardiac disease. Prog. Mol. Biol. Transl. Sci. 124, 65–91 (2014).
Burggren, W. W. & Pinder, A. W. Ontogeny of cardiovascular and respiratory physiology in lower vertebrates. Annu. Rev. Physiol. 53, 107–135 (1991).
Miura, G. I. & Yelon, D. A guide to analysis of cardiac phenotypes in the zebrafish embryo. Methods Cell Biol. 101, 161–180 (2011).
Edwards, J. J. et al. Systems analysis implicates wave2 complex in the pathogenesis of developmental left-sided obstructive heart defects. JACC Basic Transl. Sci. 5, 376–386 (2020).
Huang, M. et al. Intrinsic myocardial defects underlie an Rbfox-deficient zebrafish model of hypoplastic left heart syndrome. Nat. Commun. 13, 5877 (2022).
Jin, S. C. et al. Contribution of rare inherited and de novo variants in 2,871 congenital heart disease probands. Nat. Genet 49, 1593–1601 (2017).
Homsy, J. et al. De novo mutations in congenital heart disease with neurodevelopmental and other congenital anomalies. Science 350, 1262–1266 (2015).
Gallagher, T. L. et al. Rbfox-regulated alternative splicing is critical for zebrafish cardiac and skeletal muscle functions. Dev. Biol. 359, 251–261 (2011).
Wang, H. et al. One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell 153, 910–918 (2013).
Demoya, R. A. et al. Sin3a associated protein 130 kDa, sap130, plays an evolutionary conserved role in zebrafish heart development. Front Cell Dev. Biol. 11, 1197109 (2023).
Song, Y. C., Dohn, T. E., Rydeen, A. B., Nechiporuk, A. V. & Waxman, J. S. Hdac1-mediated repression of the retinoic acid-responsive gene ripply3 promotes second heart field development. PLoS Genet. 15, e1008165 (2019).
Bühler, A. et al. Histone deacetylase 1 controls cardiomyocyte proliferation during embryonic heart development and cardiac regeneration in zebrafish. PLoS Genet. 17, e1009890 (2021).
Krishnan, A. et al. A detailed comparison of mouse and human cardiac development. Pediatr. Res. 76, 500–507 (2014).
Jensen, B. et al. The changing morphology of the ventricular walls of mouse and human with increasing gestation. J. Anat. 244, 1040–1053 (2024).
Kolesová, H., Bartoš, M., Hsieh, W. C., Olejníčková, V. & Sedmera, D. Novel approaches to study coronary vasculature development in mice. Dev. Dyn. 247, 1018–1027 (2018).
Yagi, H. et al. The genetic landscape of hypoplastic left heart syndrome. Pediatr. Cardiol. 39, 1069–1081 (2018).
Wilson, R. L., Troja, W., Courtney, J., Williams, A. & Jones, H. N. Placental and fetal characteristics of the ohia mouse line recapitulate outcomes in human hypoplastic left heart syndrome. Placenta 117, 131–138 (2022).
Teekakirikul, P. et al. Common deletion variants causing protocadherin-α deficiency contribute to the complex genetics of BAV and left-sided congenital heart disease. HGG Adv. 2, 100037 (2021).
Wessels, M. W. et al. Autosomal dominant inheritance of left ventricular outflow tract obstruction. Am. J. Med. Genet. A 134a, 171–179 (2005).
Nutter, C. A. et al. Dysregulation of rbfox2 is an early event in cardiac pathogenesis of diabetes. Cell Rep. 15, 2200–2213 (2016).
Wei, C. et al. Repression of the central splicing regulator rbfox2 is functionally linked to pressure overload-induced heart failure. Cell Rep. 10, 1521–1533 (2015).
Kalsotra, A. et al. A postnatal switch of celf and mbnl proteins reprograms alternative splicing in the developing heart. Proc. Natl. Acad. Sci. USA 105, 20333–20338 (2008).
Congdon, E. D. & Wang, H. W. The mechanical processes concerned in the formation of the differing types of aortic arches of the chick and the pig and in the divergent early development of their pulmonary arches. Am. J. Anat. 37, 499–520 (1926).
Gessner, I. H. Spectrum of congenital cardiac anomalies produced in chick embryos by mechanical interference with cardiogenesis. Circ. Res 18, 625–633 (1966).
Gessner, I. H. & Van Mierop, L. H. S. Experimental production of cardiac defects: the spectrum of dextroposition of the aorta. Am. J. Cardiol. 25, 272–278 (1970).
Rychter, Z., Rychterová, V. & Lemez, L. Formation of the heart loop and proliferation structure of its wall as a base for ventricular septation. Herz 4, 86–90 (1979).
Sevgin, B., Coban, M. N., Karatas, F. & Pekkan, K. Left atrial ligation in the avian embryo as a model for altered hemodynamic loading during early vascular development. J. Vis. Exp. https://doi.org/10.3791/65330 (2023).
Kowalski, W. J. et al. Left atrial ligation alters intracardiac flow patterns and the biomechanical landscape in the chick embryo. Dev. Dyn. 243, 652–662 (2014).
Salman, H. E. et al. Effect of left atrial ligation-driven altered inflow hemodynamics on embryonic heart development: Clues for prenatal progression of hypoplastic left heart syndrome. Biomech. Model. Mechanobiol. 20, 733–750 (2021).
Harh, J. Y., Paul, M. H., Gallen, W. J., Friedberg, D. Z. & Kaplan, S. Experimental production of hypoplastic left heart syndrome in the chick embryo. Am. J. Cardiol. 31, 51–56 (1973).
Rychter, Z. in Advances in Morphogenesis Vol. 2 (eds Abercrombie, M., Brachet, J.) 333–371 (Elsevier, 1962).
Celik, M. et al. Microstructure of early embryonic aortic arch and its reversibility following mechanically altered hemodynamic load release. Am. J. Physiol. Heart Circ. Physiol. 318, H1208–h1218 (2020).
Hogers, B., Deruiter, M. C., Gittenberger-De Groot, A. C. & Poelmann, R. E. Unilateral vitelline vein ligation alters intracardiac blood flow patterns and morphogenesis in the chick embryo. Circ. Res 80, 473–481 (1997).
Ursem, N. T. et al. Ventricular diastolic filling characteristics in stage-24 chick embryos after extra-embryonic venous obstruction. J. Exp. Biol. 207, 1487–1490 (2004).
Sedmera, D., Pexieder, T., Rychterova, V., Hu, N. & Clark, E. B. Remodeling of chick embryonic ventricular myoarchitecture under experimentally changed loading conditions. Anat. Rec. 254, 238–252 (1999).
Hu, N. et al. Dependence of aortic arch morphogenesis on intracardiac blood flow in the left atrial ligated chick embryo. Anat. Rec. 292, 652–660 (2009).
Pesevski, Z. et al. Endocardial fibroelastosis is secondary to hemodynamic alterations in the chick embryonic model of hypoplastic left heart syndrome. Dev. Dyn. 247, 509–520 (2018).
Ho, S., Chan, W. X. & Yap, C. H. Fluid mechanics of the left atrial ligation chick embryonic model of hypoplastic left heart syndrome. Biomech. Model. Mechanobiol. 20, 1337–1351 (2021).
Hamburger, V. & Hamilton, H. L. A series of normal stages in the development of the chick embryo. 1951. Dev. Dyn. 195, 231–272 (1992).
Tobita, K. & Keller, B. B. Right and left ventricular wall deformation patterns in normal and left heart hypoplasia chick embryos. Am. J. Physiol. Heart Circ. Physiol. 279, H959–H969 (2000).
Lucitti, J. L., Tobita, K. & Keller, B. B. Arterial hemodynamics and mechanical properties after circulatory intervention in the chick embryo. J. Exp. Biol. 208, 1877–1885 (2005).
Jonker, S. S. et al. Myocyte enlargement, differentiation, and proliferation kinetics in the fetal sheep heart. J. Appl Physiol. 102, 1130–1142 (2007).
Jonker, S. S. & Louey, S. Endocrine and other physiologic modulators of perinatal cardiomyocyte endowment. J. Endocrinol. 228, R1–R18 (2016).
Jonker, S. S., Louey, S., Giraud, G. D., Thornburg, K. L. & Faber, J. J. Timing of cardiomyocyte growth, maturation, and attrition in perinatal sheep. FASEB J. 29, 4346–4357 (2015).
Fishman, N. H., Hof, R. B., Rudolph, A. M. & Heymann, M. A. Models of congenital heart disease in fetal lambs. Circulation 58, 354–364 (1978).
Rudolph, A. M., Heymann, M. A., Teramo, K. A. W., Barrett, C. T. & Räihä, N. C. R. Studies on the circulation of the previable human fetus. Pediatr. Res. 5, 452–465 (1971).
Cohn, H. E., Sacks, E. J., Heymann, M. A. & Rudolph, A. M. Cardiovascular responses to hypoxemia and acidemia in fetal lambs. Am. J. Obstet. Gynecol. 120, 817–824 (1974).
Wong, F. Y. et al. Induction of left ventricular hypoplasia by occluding the foramen ovale in the fetal lamb. Sci. Rep. 10, 880 (2020).
Reuter, M. S. et al. Decreased left heart flow in fetal lambs causes left heart hypoplasia and pro-fibrotic tissue remodeling. Commun. Biol. 6, 770 (2023).
Onohara, D. et al. Chronic in utero mitral inflow obstruction unloads left ventricular volume in a novel late gestation fetal lamb model. JTCVS Open 16, 698–707 (2023).
Rahman, A. et al. A mouse model of hypoplastic left heart syndrome demonstrating left heart hypoplasia and retrograde aortic arch flow. Dis. Model. Mech. 14, dmm049077 (2021).
Watson, M. C. et al. Extracellular matrix and cyclic stretch alter fetal cardiomyocyte proliferation and maturation in a rodent model of heart hypoplasia. Front. Cardiovasc. Med. 9, 993310 (2022).
Wickman, D. S., Siebert, J. R. & Benjamin, D. R. Nitrofen-induced congenital diaphragmatic defects in cd1 mice. Teratology 47, 119–125 (1993).
Correia-Pinto, J. et al. Heart-related indices in experimental diaphragmatic hernia. J. Pediatr. Surg. 35, 1449–1452 (2000).
Green, L. et al. Pre-intervention myocardial stress is a good predictor of aortic valvoluplasty outcome for fetal critical aortic stenosis and evolving HLHS. J. Physiol. 602, 663–681 (2024).
Marshall, A. C. et al. Aortic valvuloplasty in the fetus: technical characteristics of successful balloon dilation. J. Pediatr. 147, 535–539 (2005).
Friedman, K. G. et al. Improved technical success, postnatal outcome and refined predictors of outcome for fetal aortic valvuloplasty. Ultrasound Obstet. Gynecol. 52, 212–220 (2018).
Tulzer, A. et al. Valvuloplasty in 103 fetuses with critical aortic stenosis: outcome and new predictors for postnatal circulation. Ultrasound Obstet. Gynecol. 59, 633–641 (2022).
Gardiner, H. M. et al. Natural history of 107 cases of fetal aortic stenosis from a European multicenter retrospective study. Ultrasound Obstet. Gynecol. 48, 373–381 (2016).
Sedmera, D. HLHS: power of the chick model. J. Cardiovasc. Dev. Dis. 9, 113 (2022).
Genge, C. E. et al. The zebrafish heart as a model of mammalian cardiac function. Rev. Physiol. Biochem. Pharm. 171, 99–136 (2016).
Rao, K. S., Kameswaran, V. & Bruneau, B. G. Modeling congenital heart disease: lessons from mice, hpsc-based models, and organoids. Genes Dev. 36, 652–663 (2022).
Chico, T. J., Ingham, P. W. & Crossman, D. C. Modeling cardiovascular disease in the zebrafish. Trends Cardiovasc. Med. 18, 150–155 (2008).
Breschi, A., Gingeras, T. R. & Guigó, R. Comparative transcriptomics in human and mouse. Nat. Rev. Genet. 18, 425–440 (2017).
Gabriel, G. C., Yagi, H., Xu, X. & Lo, C. W. Novel insights into the etiology, genetics, and embryology of hypoplastic left heart syndrome. World J. Pediatr. Congenit. Heart Surg. 13, 565–570 (2022).
Alonzo, M., Contreras, J., Bering, J. & Zhao, M. T. In vivo and in vitro approaches to modeling hypoplastic left heart syndrome. Curr. Cardiol. Rep. 26, 1221–1229 (2024).
Waterston, R. H. et al. Initial sequencing and comparative analysis of the mouse genome. Nature 420, 520–562 (2002).
Miao, Y. et al. Intrinsic endocardial defects contribute to hypoplastic left heart syndrome. Cell Stem Cell 27, 574–589.e578 (2020).
Boselli, F., Freund, J. B. & Vermot, J. Blood flow mechanics in cardiovascular development. Cell Mol. Life Sci. 72, 2545–2559 (2015).
Galindo, A. et al. Fetal aortic valvuloplasty: experience and results of two tertiary centers in spain. Fetal Diagn. Ther. 42, 262–270 (2017).
Funding
This work was supported by Additional Ventures Single Ventricle Research Fund #1284695 (PI: Onohara).
Author information
Authors and Affiliations
Contributions
D.O. created the concept of the review, and C.M. and K.N. drafted the manuscript as co-first authors. M.D. added some inputs to the In-utero rodent model section. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Miyagi, C., Nakamae, K., Davis, M.E. et al. Animal models of hypoplastic left heart syndrome: genetic and anatomical approaches. Pediatr Res (2026). https://doi.org/10.1038/s41390-026-04815-w
Received:
Revised:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41390-026-04815-w
