
Abstract
The year 2024 marks the 60th anniversary of the discovery of the Epstein-Barr virus (EBV), the first virus confirmed to cause human cancer. Viral infections significantly contribute to the global cancer burden, with seven known Group 1 oncogenic viruses, including hepatitis B virus (HBV), human papillomavirus (HPV), EBV, Kaposi sarcoma-associated herpesvirus (KSHV), hepatitis C virus (HCV), human T-cell leukemia virus type 1 (HTLV-1), and human immunodeficiency virus (HIV). These oncogenic viruses induce cellular transformation and cancer development by altering various biological processes within host cells, particularly under immunosuppression or co-carcinogenic exposures. These viruses are primarily associated with hepatocellular carcinoma, gastric cancer, cervical cancer, nasopharyngeal carcinoma, Kaposi sarcoma, lymphoma, and adult T-cell leukemia/lymphoma. Understanding the mechanisms of viral oncogenesis is crucial for identifying and characterizing the early biological processes of virus-related cancers, providing new targets and strategies for treatment or prevention. This review first outlines the global epidemiology of virus-related tumors, milestone events in research, and the process by which oncogenic viruses infect target cells. It then focuses on the molecular mechanisms by which these viruses induce tumors directly or indirectly, including the regulation of oncogenes or tumor suppressor genes, induction of genomic instability, disruption of regular life cycle of cells, immune suppression, chronic inflammation, and inducing angiogenesis. Finally, current therapeutic strategies for virus-related tumors and recent advances in preclinical and clinical research are discussed.
Similar content being viewed by others
Introduction
Cancer is a major public health and economic concern of the 21st century. Globally, almost 1.4 million new cancer cases are ascribed to viral infections, accounting for 8% of all cancer cases. While this attributable estimate is large, it is likely to be significantly underestimated, as developing countries are particularly severely affected by virus-related cancers, but often lack cancer registries. The International Agency for Research on Cancer (IARC) has identified seven viruses that are classified as Group 1 carcinogens (Carcinogenic to humans), including human papillomavirus (HPV), hepatitis B virus (HBV), Hepatitis C virus (HCV), Epstein–Barr virus (EBV), Kaposi sarcoma-associated herpesvirus (KSHV), human T cell lymphotropic virus type-1 (HTLV-1), and human immunodeficiency virus (HIV). Each group of viruses can cause various cancers.1
Most people will contract at least one known human oncogenic virus during their lifetime.2 The majority of viral infections are self-limiting and resolve within a few months without any oncogenic potential. However, infected cells can acquire oncogenesis features, especially when immunosuppressed or exposed to co-oncogenic stimuli.2,3 In fact, there is a long latency period between primary infection and the occurrence of cancer. This means that the onset of cancer is not part of the viral replication cycle, but the combination of multiple factors is necessary for the virus to manifest its oncogenic potential in a susceptible host.4 Viral oncogenesis is mainly achieved through multifaceted intricate mechanisms, including insertion of viral genes, regulation of host cell genomes and signaling pathways mediated by viral proteins, chronic inflammation resulting from persistent infection, and disorder of tumor suppressor genes and host oncogenes involved in cell growth.5,6 The oncogenic mechanisms of viruses are complex and have not been fully elucidated.
Understanding the mechanisms targeting viral oncogenesis has provided vital clues for developing new therapeutic strategies. Current treatment strategies primarily involve antiviral medications, inhibition of viral-encoded proteins to disrupt viral oncogenic mechanisms, or developing vaccines to control viral infections and prevent virus-related cancers.7,8 Nevertheless, current treatment outcomes remain challenging. Consequently, it is essential to have a thorough knowledge of the mechanisms by which oncogenic viruses function and cutting-edge treatment approaches. This article aims to comprehensively summarize the characteristics of oncogenic viruses, the mechanisms by which they induce tumors, and the therapeutic strategies for targeting virus-induced oncogenesis. This review will first provide an overview of the worldwide prevalence of oncogenic-virus-related tumors, then review the milestone events in the research history of oncogenic viruses. Subsequently, it will describe in detail the mechanisms of initial infection of target cells by oncogenic viruses and the molecular mechanisms that mediate tumorigenesis during the long-term chronic infection process. Finally, current targeted therapeutic strategies for virus-induced oncogenesis, as well as the progress in pre-clinical and clinical studies, will be introduced. This knowledge is expected to significantly contribute to the future development of basic research and clinical translation in virus-induced tumorigenesis.
The prevalence of virus-related cancer globally
More than 1.4 million cancer cases are caused by viral infections every year, which brings a significant health burden to the whole world.9,10 Data from 2018 indicated that among virus-related cancers, 49% were induced by HPV, 26% were triggered by HBV, 11% were related to HCV, 11% were caused by EBV, and 3% were associated with other viruses (KSHV and HTLV-1).11 The age-standardized incidence rates (ASIR) of virus-related cancers show significant regional differences in global distribution (Table 1), and are affected to some extent by gender and national income.
In 2018, HBV was responsible for 360,000 new cancer cases (ASIR 4.1 per 100,000 person-years); HCV led to 160,000 new cancer cases (ASIR 1.7 per 100,000 person-years), mainly hepatocellular carcinoma (HCC) (90%) and non-Hodgkin lymphoma (NHL) (10%).11 15%–40% of untreated chronic HBV patients progress to cirrhosis and HCC, with higher male risk.12 Owing to the inadequacy of early prevention, screening, and antiviral treatment, less developed regions, such as Asia and Africa, have become the main endemic areas for HBV and HCV.13,14 The promotion of HBV vaccination has reduced the prevalence of HBsAg (hepatitis B surface antigen) in children under 5 years old from over 5% to 0.9%.12,15 Although no preventive HCV vaccine is available currently, direct-acting antivirals (DAAs) treatment has enabled over 98% of HCV patients to achieve viral clearance.16 From 2010–2019, ASIR of HBV-related and HCV-related HCC declined except in Americas, due to vaccination and antiviral therapy success.17 However, the number of HCV-related HCC cases and disability-adjusted life years (DALYs) are increasing because of large early infection base.18,19
The ASIR of HPV-attributable cancer is five or more per 100,000 person-years in most countries and regions except the Middle East.11 HPV is predominantly prevalent in Asia, Africa, and the Americas. The number of infection cases in these regions accounts for 54%, 19%, and 16% of the global total, respectively.20 HPV types 16 and 18 account for approximately 72% of all HPV-induced cancer cases, while HPV types 31, 33, 45, 52, and 58 account for an additional 17%.11,21 Infection with HPV is responsible for nearly all cases of cervical cancer (CC) and a significant proportion of oropharyngeal (31%), anal cancers (88%).22 The ASIR of CC shows a significant negative correlation with income levels, a trend primarily influenced by the accessibility of CC screening.11,23 Over the past few decades, the incidence and mortality of CC have steadily declined, particularly in many high-income countries in North America and Europe. However, the incidence of oropharyngeal and anal cancers has been rising in most countries, with a significantly higher burden in more developed countries compared to less developed ones.20,22 70%–90% of HPV-attributable cancer cases can be prevented by widespread high-coverage HPV vaccination.24,25 Universal vaccination and early screening are the keys to preventing the majority of HPV-attributable cancer cases.26
EBV is a highly prevalent and persistent human viral infection, with about 95% of the global population having asymptomatic EBV infections lifelong.27,28 EBV-associated cancers account for around 1.3–1.9% of the global cancer burden, encompassing nasopharyngeal carcinoma (NPC), Burkitt lymphoma (BL), Hodgkin lymphoma (HL), and gastric cancer (GC).29 Among the four EBV-attributable cancers, males have a higher proportion than females.30 95% of NPC cases are attributable to EBV, with a predominant occurrence in East Asia, Southeast Asia, and the Middle East.31 The estimated number of new EBV-attributable BL cases in 2018 was 6,600, mainly in children aged 5–10 years.32 EBV-attributable HL makes up about 40% of global HL cases. Its incidence is positively correlated with a country’s social demographic index (SDI), but the mortality burden is mainly in low and low middle SDI countries, indicating insufficient HL treatment resources in low-income areas.33 The EBV prevalence in GC is about 8%, and EBV infection increases the risk of GC by more than 18 times.34,35 Given the current lack of effective anti-EBV drugs or vaccines, EBV-attributable cancers pose a challenging aspect of the global disease burden.36
The prevalence of HTLV-1 varies from less than one case per 10,000 individuals to over 10%, with the highest prevalence observed in Japan, South America, the Caribbean, Central Australia, and Western, Central, and Southern Africa.37 Adult T-cell leukemia/lymphoma (ATLL), a rare tumor induced by HTLV-1, had 3,600 new cases in 2018.11 Another rare virus-related cancer is Kaposi sarcoma (KS), which is closely associated with KSHV infection and had 42,000 new cases in 2018.11 HIV, a Group 1 carcinogen by IARC, has indirect carcinogenicity, complicating the classification of HIV-related cancers.38,39 In general, NHL, HL, and oropharyngeal cancer are more prevalent in men living with HIV, while CC and NHL are more incident in women living with HIV.40,41,42
The research history and milestone events of viral oncogenesis in cancers
The year 2024 commemorates the 60th anniversary of the human discovery of EBV, the first virus proven to cause cancer in humans. The history of the finding of viruses that cause cancer in humans dates back to the early 1900s. In 1908, when Ellerman and Bang tested whether cancer could be an infectious disease, they found that leukemia could be transmitted from one bird to another by injection.43 In addition, they obtained avian leukosis virus (ALV), the first known tumor virus.44 For a variety of reasons, their work was not given due attention at the time. In 1910, Rous successfully transplanted sarcoma from a barred Plymouth Rock hen to other chickens.45 By 1911, his research had demonstrated that cancer could be transmitted through one virus, which he named Rous sarcoma virus (RSV).46 As before, this work was not generally accepted as evidence that a virus could cause cancer. Rous’s contributions to the field were not widely acknowledged until 1966, when he was awarded the Nobel Prize in Physiology or Medicine. In 1933, Shope and Hurst found rabbit papillomavirus (CRPV) in wild cottontail rabbits and those infected rabbits, which exhibited weird protrusions on their heads and necks.47 In 1951, Gross discovered that murine leukemia virus (MLV) could cause spontaneous leukemia in C3H mice.48 In 1962, Eddy et al. found that simian virus 40 (SV40) was shown to induce tumors in rhesus monkeys.49 These studies further strengthened the association between viruses and cancer, thus triggering the first strong interest in tumor virology.
In 1964, Epstein and Barr et al. first found EBV in BL patients.50 EBV was later shown to cause mononucleosis and nasopharyngeal carcinoma.51 In 1984, Baer et al. sequenced and analyzed the expression of EBV genome.52 In 1965, Blumberg identified a novel antigen in the blood of an Aboriginal Australian, which was subsequently designated HBsAg.53 This groundbreaking achievement earned him the 1976 Nobel Prize. In 1981, a study in Taiwan found that HBV infection was associated with HCC.54 And in the same year, the first human anti-tumor vaccine, the HBV vaccine, was approved. In 1970, Temin and Baltimore proposed independent but related hypotheses for reverse transcriptase. Temin proposed a role for reverse transcriptase in the replication of RSV viruses.55 Baltimore independently proposed a similar idea and revealed that the RNA oncolytic virus has an enzyme in its viral particles that synthesizes DNA from RNA templates.56 In 1975, together with their mentor Dulbecco, they received a Nobel Prize for discovering transcriptase. Combined with the fact that Rous won the Nobel Prize in 1966 for the discovery of RSV, an event that prompted Peter Duesburg (a retroviralist) to jest, “One sick chicken, two Nobel Prizes.” But that’s not the end of the story.
The discovery of proto-oncogenes by Bishop and Varmus in 1976, during their research on RSV, initiated the perception that viral infections cause cancer cell growth.57 After their research was published, one oncogene after another was discovered. They were awarded the 1989 Nobel Prize for this work. In 1976, Stehelin et al. identified the transforming gene, SRC, in avian sarcoma viruses (ASV) as a key oncogene in poultry.57 This study pioneered the discovery of the relationship between viral oncogenes and the host genome. In 1953, Rowe et al. discovered adenoviruses.58 In 1977, Roberts and Sharp discovered “RNA Splicing” in adenoviruses and were awarded the Nobel Prize in 1993. In 1979, Linzer et al. found that the tumor antigen p53 was associated with the replication and transformation of the SV40 virus.59 Lane et al. found that the SV40 virus T-antigen interacted with many proteins in the host cell, which may affect key biological processes.60 These two studies suggest the complexity of virus-host cell interactions and contribute to understanding the mechanisms of viral cancers. In 1980, Poiesz et al. identified HTLV-1 from T cell lymphoblastoid cell lines and cutaneous T cell lymphoma patients.61 In 1981, Hinuma et al. found an association between HTLV-1 and ATLL.62 In 1976, Hausen proposed the hypothesis that HPV was the leading cause of cervical cancer.63 In 1983, two high-risk viruses, HPV16 and HPV18, were identified in cervical cancer, thus laying a solid foundation for the development of the cervical cancer vaccine.64 In 2006, the FDA approved the HPV vaccine. Hausen, known as the “father of the HPV vaccine” was awarded the Nobel Prize in 2008. In 1983, Barré-Sinoussi et al. first reported the discovery of HIV.65 In 2012, the IARC identified HIV as an oncogenic virus, (IARC data, https://monographs.iarc.who.int/agents-classified-by-the-iarc/) associated explicitly with KS and Non-Hodgkin’s lymphoma. In 1989, Alter et al. cloned a cDNA sequence from a hepatitis patient, marking the discovery of HCV.66 In 1991, Houghton et al. successfully discerned the HCV genome.67 The 2020 Nobel Prize was awarded to Alter, Houghton, and Rice for their contributions to the discovery of HCV. Chang and Moore discovered KSHV in AIDS patients in 1994,68 and isolated KSHV from primary effusion lymphoma (PEL) cells in 1995.69 Subsequently, they discovered Merkel Cell Polyomavirus (MCV) in 2008.70 On the whole, viruses have contributed to our understanding of the molecular basis of oncogenesis (Fig. 1).

History of research and milestones in viral oncogenesis. The exploration embarked in the early 20th century. Initially, the discovery of the ALK laid the groundwork for oncovirus research. Subsequently, the RSV came to the fore, and in due sequence, the EBV and HBV emerged. A cascade of milestone accomplishments followed. For example, the identification of oncogenes revolutionized the field; the proposition that HPV induces cancer spurred the development of the cervical cancer vaccine. Moreover, viruses like HCV, KSHV, and MCV were unearthed. Collectively, these events have vividly traced the trajectory of the unceasing deepening of scientific research and the ceaseless efforts to battle cancer. This figure was created with BioRender.com
The characteristics and infection mechanisms of oncogenic viruses
Human oncogenic viruses exhibit a range of different characteristics.71 Some viruses have large double-stranded DNA genomes, such as EBV and KSHV. Others have small double-stranded DNA genomes, such as HPV and HBV. In addition, some viruses have positive-sense single-stranded RNA genomes, such as HCV, while HTLV-1 and HIV belong to the retrovirus family.72 Viral diversity is associated with different pathogenesis of DNA and RNA viruses, and this diversity contributes to rapid intrahost evolution.73 Most human oncogenic viruses infect their hosts with chronic monoclonal infections that persist for years after initial infection, which suggests that infection is only one factor in oncogenesis under a multifactorial environment and complex process (Fig. 2 and Table 2).74

Types of virus-related cancer and mechanisms of viral infection. Major steps in the process of viral infection include Attachment, Entry, Uncoating, Replication and Transcription, Protein Synthesis, Assembly, and Release. Newly released viral particles can infect neighboring cells, continuing the infection process. Persistent viral infection increases the risk of virus-related cancer, including hepatocellular carcinoma, gastric cancer, cervical cancer, nasopharyngeal carcinoma, Kaposi sarcoma, lymphoma, and adult T-cell leukemia/lymphoma. Text in host cell color: intracellular components; Text in virus color: viral infection process; Black text: viral components. This figure was created with BioRender.com
DNA virus
HBV
HBV is a partially double-stranded, hepatophilic DNA virus that primarily infects human hepatocytes, causing liver diseases.75 The HBV envelope encapsulates a nucleocapsid that contains partially double-stranded, relaxed circular DNA (rcDNA).76 The virus binds to the cell surface via pre-S glycoprotein and interacts with the primary receptor, the hepatic bile acid transporter sodium taurocholate cotransporting polypeptide (NTCP).77 Upon entry into hepatocytes, the rcDNA genome is translocated into the nucleus and converted into covalently closed circular DNA (cccDNA).78 Subsequently, cccDNA serves as a transcriptional template to form pregnenomic RNA (pgRNA) and messenger RNA (mRNA) in the presence of the regulatory HBV X protein (HBx).79 The pgRNA is selectively packaged into the nucleocapsid and serves as a template for the reverse transcription of HBV DNA in the cytoplasm of the liver, generating the core proteins and the polymerase.80 It also serves as a template for reverse transcription to produce new rcDNA. Viral envelope assembly takes place in multivesicular bodies (MVBs) to produce and secrete viral particles, which can also be recirculated into the nucleus to participate in cccDNA amplification and maintenance, so that cccDNA libraries can be sustained without requiring the entry of new HBV.81 Most acute HBV infections achieve viral clearance in a non-cytolytic manner (without directly killing infected hepatocytes), mainly involving cytokine-mediated inhibition of HBV replication via Interferon-gamma (IFN-γ) and tumor necrosis factor (TNF) secreted by virus-specific CD8+ T cells.76 In contrast, patients with chronic HBV infection usually pursue a lifelong course.82
HPV
HPV is a non-enveloped, double-stranded, circular DNA virus of the papillomavirus family comprising three regions: the long control region (LCR), the early region encoding virions (E1-E7), and the late region encoding virions (L1 and L2).83 More than 200 HPV genotypes have been identified, and these include mainly high-risk types (HPV16, HPV18), which cause cancer, and low-risk types (HPV6, HPV11), which do not cause cancer.84 Despite discrepancies in the size and number of open reading frames (ORFs) of different HPV molecules, all HPVs contain conserved core genes that are involved in viral replication (E1 and E2) and packaging (L1 and L2); the rest of the genes (E4, E5, E6, and E7) function in driving the cell cycle, immune escape, and viral release.22 HPV predominantly infects basal epithelial cells, binding to the extracellular matrix (ECM) or cell surface via L1.3,85 This process has been shown to involve multiple receptors/co-receptors, including heparan sulfate (HS), α6β4 integrin, growth factor receptors and annexin-A2.86,87 Upon internalization, the capsid is disassembled in endosomes, and the multifunctional L2 protein directs viral DNA to the host cell nucleus.88,89 Once infection is established in the cell, the viral life cycle is regulated to maintain host-viral protein homeostasis.90 Most HPV infections are self-limiting, while persistent high-risk HPV infections can increase the risk of cervical intraepithelial neoplasia (CIN) and CC.91
EBV
EBV is a γ-herpesvirus whose structure consists mainly of a core and an envelope. The core contains a linear double-stranded DNA involved in viral replication and transcription. The envelope consists of a variety of glycoproteins (gp350/gp220 and gp42) and membrane proteins (BLLF1, glycoprotein B (gB), and glycoprotein H/glycoprotein L (gH/gL)), which mediate the viral entry into the host cell.92 The target cells of EBV are B lymphocytes and epithelial cells, and its mechanism for entering host cells is similar to that of other members of the herpesvirus family.93 The infection of B cells by EBV is initiated by the binding of the EBV envelope protein gp350 to B cell surface receptor CD21. Upon binding to CD21, EBV gp42 interacts with the host cell surface MHC-II, resulting in its binding to the heterodimerization protein gH/gL. EBV gH/gL then activates the EBV fusion protein gB, which directly mediates the fusion of the virus with the host cell membrane. At this point, the viral glycoprotein gB and the heterodimeric gH/gL form the core of the EBV fusion machinery, which ultimately allows the virus to be translocated into the nucleus.94,95 Upon EBV infection of epithelial cells, EBV BMRF2 binds to integrins. Subsequently, gH/gL binds to integrins and ephrin receptor A2, triggering activation of gB and fusion of the viral envelope to the plasma membrane of the epithelial cell.96 EBV infection is categorized into latent and lytic infections. During latent infection, four modes are classified as latency III, latency II, latency I, and latency 0 based on how these genes are expressed in different combinations in EBV-infected cells, which is characterized by a gradual restriction of the viral gene expression pattern to evade immune surveillance.97 Eventually, EBV establishes a persistent residency in memory B cells characterized by a lack of viral antigen expression (latency 0), thus evading T cell recognition and acting as a viral reservoir.98 EBV can periodically transit to the lytic cycle, leading to viral replication, shedding, and subsequent dissemination, which is involved in B cell transformation and oncogenesis.95,99 EBV exhibits four distinct latency phases, each contributing uniquely to carcinogenesis.100 In latency III, expressing all 6 latent genes, it is implicated in severely immunosuppressed diffuse large B-cell lymphoma.101 Latency I, with only EBNA1 expressed, aids in maintaining the EBV genome and is associated with cancers like Burkitt lymphoma.102 Latency II, expressing EBNA1 and LMPs, correlates with cancers like nasopharyngeal carcinoma, where LMP1 impacts cell growth and apoptosis.102 Even latency 0, despite having no antigen expression, might play a part in cancer development under specific circumstances.103 Besides latency, EBV’s lytic replication also plays a role in carcinogenesis. Some lytic proteins, like BZLF1 and BRLF1, can activate genes involved in DNA amplification and virus production, potentially disrupting normal cell functions and fueling cancer development.104
KSHV
KSHV, also known as human herpesvirus 8 (HHV-8), belongs to the γ-herpesvirus family.105 KSHV is a linear double-stranded DNA virus with a central coding region surrounded by two sides of non-coding terminal repeat units with high GC content on both sides.106 KSHV is a broadly cytophilic virus that can infect B cells, fibroblasts, epithelial cells, and endothelial cells.51,107 The glycoprotein gB on the viral envelope mediates virus-cell binding and entry by interacting with HS and entry receptors on the surface of host target cells.108 After the virus enters the host cell, the capsid of the virus particle detaches and releases its genome into the cytoplasm, followed by the import of the KSHV genome into the nucleus.109 KSHV is often found in the host in both latent and lytic states, and the latency-associated nuclear antigen (LANA) is the essential protein that maintains its latency. The latent KSHV can attach its genome to the host chromosome and be retained as a free body mediated by LANA.110 However, various stimuli can cause the virus to withdraw from latency. The regulator of transcription activator (RTA) is a nuclear DNA-binding protein that regulates the transition of the virus from latency to lytic replication. The expression of RTA initiates the KSHV gene expression in a lytic cascade. Subsequently, the replication of the genome and the production of new viruses are facilitated.111 Chronic infection with KSHV is one of the etiologic causes of KS, B cell lymphoma and PEL.112
RNA virus
HCV
HCV is a single-stranded, positive-sense hepatotropic RNA virus, consisting of an envelope and a nucleocapsid. The main components of the envelope are glycoproteins E1 and E2, which mediate the internalization of virus particles.113,114 E1 acts as a fusion protein during internalization, while E2 is responsible for host cell receptor binding.113,115,116 Once internalized, the viral RNA is released and translated into a variety of viral proteins, including three structural proteins (Core, E1, and E2) and seven non-structural proteins (p7, NS2, NS3, NS4A, NS4B, NS5A, and NS5).117 The structural proteins are used for the assembly of the zygotic virosome.118 In contrast, non-structural proteins can bind to lipid droplets, forming highly lipidated lipoviral particles that enable viral replication and transmission.119 Various errors are observed during the replication process, mainly attributable to the uncorrected function of RNA polymerase and the absence of an effective mechanism for error correction.120 This ultimately contributes to the high degree of heterogeneity observed in HCV.121 Additionally, the envelope protein E2/NS1 region, which stimulates the production of neutralizing antibodies, exhibits the highest degree of variability, resulting in a robust capacity of the virus to evade adaptive immunity.122 In the acute phase of infection, the clearance of viral particles is predominantly contingent upon intrinsic immunity. The lysis and clearance of most HCV-infected cells is mediated by perforin and granzyme B, which are released by cytotoxic T-lymphocytes (CTLs) and natural killers (NKs), respectively.123 A minority of HCV-infected cells are cleared non-lytically by IFN-γ secreted by CTLs and NK cells.124 The majority of individuals infected with HCV will eventually progress to chronic infection. The proliferation of HCV-specific CTLs during chronic infection is impaired, accompanied by a diminished ability to produce IFN-γ and an increased susceptibility to exhaustion. This increases the probability that the patients will develop HCC.125,126
HIV
HIV is a single-stranded RNA virus that predominantly infects CD4+ T cells, macrophages, dendritic cells, and others.127,128 HIV comprises two distinct components: the Gag polyprotein and the envelope. Gag polyproteins are composed of a capsid protein (CA),129,130 which contains two identical viral single-stranded positive-stranded RNAs; a nucleocapsid protein (NC); and a collection of enzymes essential for viral replication, including reverse transcriptase (RT), integrase (IN), and protease (PR).131 The outermost layer of the virus is the envelope, which is embedded with the outer membrane glycoprotein gp120 and the transmembrane glycoprotein gp41. Beneath the envelope structure are matrix proteins (MA) that form the viral inner capsid.132 Following the binding of the viral outer membrane protein gp120 to the CC-chemokine receptor/CXC chemokine receptor 4 (CCR5/CXCR4) on the membrane surface of the target cell, the virus initiates its invasion of the target cell. Upon entering the target cell, the virus releases its NC.133,134 With the effect of RT, HIV RNA is reverse-transcribed in the host cytoplasm to form DNA, which further migrates into the host cell nucleus.135 However, as RT cannot correct errors, the HIV genome is susceptible to mutation during the reverse transcription process.136 Typically, most of the viral genome is integrated into silent and non-replicating genomic regions that serve as long-term HIV reservoirs without being destroyed by immune surveillance. Nevertheless, minor quantities of viral DNA may integrate into more active genome regions.137 Finally, Gag drives the assembly and release of nascent HIV particles and causes damage and exhaustion of target cells.131,138 The course of HIV infection can be categorized as acute, asymptomatic, or AIDS phases. With the use of antiretroviral therapy (ART), there has been a notable reduction in people living with HIV (PLWH) progressing to the AIDS Phase.139,140 However, the persistent damage to the immune system caused by HIV infection still increases the risk of cancer in PLWH.141
HTLV-1
HTLV-1 is a delta retrovirus that infects peripheral blood mononuclear cells.142,143 Like most retroviruses, HTLV-1 is composed of an envelope and a capsid. The capsid contains enzymes essential for viral replication and a nucleocapsid with the viral genome.144 HTLV-1 is mainly transmitted through cell-to-cell contact. Virological synapse, a key structure in this process, promotes the binding of HTLV-1 viral envelope glycoproteins to specific receptors on target cells and mediates the endocytosis of viral particles into cells.142 Neuropilin 1 (NRP1), glucose transporter 1 (GLUT-1) and heparan sulfate proteoglycans (HSPGs) are HTLV-1-specific receptors.145,146 With the fusion of the viral envelope with the cytoplasmic membrane, HTLV-1 RNA is reverse transcribed into HTLV-1 DNA and integrated into the host DNA, which is transcribed and translated along with the host DNA.147 HTLV-1 DNA contains multiple coding regions and long terminal repeats (LTRs) located at the 5’ and 3’ ends of the genome. The 5’ LTR cis-sequence contains all viral structural genes and most regulatory genes; the 3’ LTR is a reverse transcription of the original viral gene that produces the essential zipper protein (HBZ).148 HBZ, a highly conserved protein, is expressed in all HTLV-1-infected cells and is vital for maintaining the malignant phenotype of ATLL.149,150 HTLV-1 replication is active only during the initial stages of infection, after which it transitions to a latent state.151 The vast majority of HTLV-1-infected individuals remain carriers for long periods or for life; only a minority develop ATLL or lymphoma.152
Oncogenic mechanisms in specific cancer types
Oncoviruses induce cellular transformation by hijacking host cell homeostasis.6 They mainly exert their biological functions by encoding oncogenic proteins of viral origin, and the specific mechanisms include: (1) Activation of oncogenes or inactivation of tumor suppressor genes: Oncoviruses hijack host cells and induce tumorigenesis by activating oncogenes or inactivating tumor suppressor genes.1 (2) Inducing genomic instability: Oncoviruses trigger genomic instability in host cells, including point mutations, insertions/deletions, structural variations, and DNA damage.153 (3) Interference with normal cell life processes: Oncoviruses disrupt critical signaling pathways, such as cell proliferation, cell cycle regulation, and apoptosis, leading to abnormal host cell growth and proliferation.5 For instance, EBV latency proteins LMP1, LMP2a, EBNA2, EBNA3A, EBNA3B, EBNA3C, and EBNA LP intersect and manipulate several cellular signaling pathways, which ultimately contribute to EBV-mediated B cell proliferation.154,155 (4) Evolving immune evasion strategies: Oncoviruses evolve multiple immune evasion strategies, including inhibition of antigen presentation, upregulation of immune checkpoint molecules like PD-L1, and activation of regulatory T cells, thereby suppressing the immune system’s ability to effectively recognize and eliminate tumor cells.156,157 (5) Persistent inflammation and oxidative stress: Oncoviruses promote the release of inflammatory factors and chemokines, leading to a disrupted cellular microenvironment, and induce abnormal oxidative stress, resulting in oxidative damage to DNA, proteins, and lipids, further driving the carcinogenic process.158,159 (6) Remodeling the ECM: Oncoviruses alter the composition and structure of the ECM, affecting the surrounding microenvironment and promoting tumor growth and invasion.160,161,162 (7) Regulating angiogenesis: Oncoviruses regulate angiogenesis-related signaling pathways. For instance, the oncoproteins E6 and E7 of HPV can induce the expression of vascular endothelial growth factor (VEGF), which promotes the formation of new blood vessels.163,164,165 (8) Oncoviruses induce metabolic changes, including enhancing glycolysis and lipid metabolism, to support the rapid growth and proliferation of transformed cells.166,167 For example, HPV early-phase proteins E1, E2, E5, E6 and E7 can activate plethora of metabolic pathways and directly influence enzymes of the glycolysis pathway to promote the Warburg effect by increasing glucose uptake, activating glycolysis and pentose phosphate pathway.168 Generally, the complex and unique features that drive tumor formation and metastatic spread are summarized into ten hallmarks of cancer, including self-sufficiency in growth signals, insensitivity to anti-growth signals, resisting cell death, limitless replicative potential, sustained angiogenesis, tissue invasion and metastasis, avoiding immune destruction, tumor-promoting inflammation, deregulating cellular energetics, and genome instability and mutation (Fig. 3).169,170

An overview of viral oncogenic mechanisms. Oncoviruses induce cellular transformation by hijacking the homeostasis of host cells. The specific mechanisms include: oncogene activation and tumor suppressor gene inhibition; induction of genomic instability; interfering with cell proliferation, cell cycle and apoptosis; immunosuppression and immune evasion; chronic inflammation stimulation; extracellular matrix remodeling; inducing angiogenesis; metabolic reprogramming. These mechanisms work together to drive tumor formation and metastatic spread. Red text: direct oncogenesis; Blue text: indirect oncogenesis. This figure was created with BioRender.com
Hepatocellular carcinoma (HCC)
HBV-related HCC
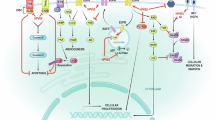
HBV is the most diverse DNA virus and its genome consists of four open reading frames (ORFs): the presurface antigen/surface antigen gene (preS/S), the precore/core gene (preC/C), the polymerase gene (P), and the X gene (X).171 HBV infection leads to liver fibrosis and cirrhosis, and ultimately to HCC.172 Integration of viral DNA into the host genome is thought to be the initiating event for HBV-induced carcinogenesis, with integration preferentially targeting promoters and coding regions.173 In HCC, the most common recurrent HBV DNA integrations occur at the telomerase reverse transcriptase (TERT), mixed-lineage leukemia 4 (MLL4), cyclin E1 (CCNE1), and fibronectin (FN1) loci.174 Integration is amplified when integrating cells undergo mitosis. This allows HBV DNA integration to persist and become a major source of HBV antigen in the later stages of chronic infection.175 Although HBV DNA integration is often unable to transcribe pre-core mRNAs and pgRNAs due to loss of the upstream basic core promoter, the promoters of the S and X ORFs are intact, allowing synthesis of functional HBsAg and C-terminally truncated HBx proteins.176 Since HBx is mainly localized in the cytoplasm, it regulates multiple signaling pathways, such as mitogen-activated protein kinase (MAPK), NF-κB, rat sarcoma virus (Ras), rapidly accelerated fibrosarcoma (Raf) and JAK-STAT.177,178,179,180 HBx is also involved in cell cycle regulation, DNA repair, apoptosis, and immunosuppression, and plays an important role in HCC development (Fig. 4).181

Oncogenic mechanisms of HBV and HCV in hepatocellular carcinoma. a HBV infection causes chromosomal abnormalities, mitotic defects, and increased genetic mutations. b HBx activates glycolysis via the NF-κB/HK2 pathway, and excess lactic acid enhances HCC proliferation through the PI3K/AKT pathway. c HBV evades T cell responses by enhancing the expression of PD-L1 in HCC cells, and its related factors construct an inflammatory microenvironment while the infection leads to ROS accumulation and triggers oxidative stress. d HBV promotes angiogenesis in HCC by increasing HIF-1α, HMGB1, and VEGF expression, and activates MAPK pathways to facilitate metastasis. e HBx protein upregulates p65 and HK2, downregulates TXNIP to enhance host cell glycolysis. f HCV promotes HCC proliferation by inhibiting EGFR degradation, activating Wnt/β-catenin signaling, inducing c-Myc and cyclin D1 expression, blocking K+ channels, and enhancing the expression of abnormal DNA polymerase. g HCV suppresses IFN signaling, promotes TGF-β1 secretion, and increases inhibitory receptor expression on immune cells, thereby dampening the immune response. h HCV infection enriches inflammatory factors in the TME, contributing to liver fibrosis. i HCV can enhance glycolysis, increase the accumulation of long-chain fatty acids, and interfere with insulin signaling. Blue part: HBV carcinogenic mechanism; Red part: HCV carcinogenic mechanism; Text in virus color: viral components; Black text: host cell components; Direct oncogenesis: a, b (HBV), f (HCV); Indirect oncogenesis: c, d (HBV), g–i (HCV). This figure was created with BioRender.com
Direct oncogenesis: leading to the emergence of virus-related tumors
Oncogene activation and tumor suppressor gene inhibition: Proto-oncogenes regulate the cell cycle, control cell growth, and division, and their abnormal activation can lead to uncontrolled cell proliferation, ultimately causing cellular transformation into cancer.182 HBx protein can trans-activate multiple proto-oncogenes, including c-Myc and c-Jun, thereby contributing to HCC development.178,183,184,185,186 HBV DNA integration can also transform proto-oncogenes into oncogenes or cause mutations in tumor suppressor genes, leading to the loss of their normal biological functions, disrupting normal hepatocyte proliferation, and causing HCC development.187 Tumorigenesis is closely associated with oncogene activation and the functional inhibition of tumor suppressor genes.188 The p53 protein is a well-known tumor suppressor that plays a crucial role in promoting apoptosis, repairing DNA damage, and other biological functions. The downregulation or inactivation of p53 is often linked to tumor formation.189 Research has shown that the HBx protein can bind to p53 in the cytoplasm, forming a complex that hinders p53’s translocation to the nucleus, thereby reducing its nuclear concentration and impairing its ability to induce apoptosis.190 The C-terminal domain of p53, which is responsible for binding to damaged DNA, is competitively bound by HBx, obstructing p53’s DNA repair function.191 Additionally, HBx binding to the C-terminal of p53 leads to the inactivation of the promoter of another tumor suppressor gene, pRb, resulting in cell cycle alterations that signal the onset of carcinogenesis. Collectively, HBx protein inhibits the activity of tumor suppressor genes p53 and pRb through multiple pathways, promoting the development of HCC (Fig. 4a).
Genome instability: Random integration of HBV DNA into the human genome contributes to host genomic instability, including chromosomal deletions, amplifications, breaks, inversions, and translocations.192 During the early stages of clonal tumor expansion, HBV integrates into the host genome at sites of double-stranded DNA breaks, leading to cis-acting interference, chromosomal instability, and the expression of mutated HBV genes, which are characteristic of HCC cells.193 HBx affects centrosome replication and cell division by regulating the hepatitis B X-interacting protein (HBXIP), leading to mitotic abnormalities and loss of genetic stability, thereby promoting HCC development.194 HBx also interacts with TAX1-binding protein 2 (TAX1BP2), disrupting centrosome-microtubule dynamics and leading to chromosomal instability.195 For proper chromosomal duplication and segregation, the higher-order chromosome structure must be organized by structural maintenance of chromosomes (SMC) complexes, which include cohesin, condensin, and SMC5/6.196 HBx binds to the cellular E3 ligase complex containing DNA damage-binding protein 1 (DDB1), leading to the degradation of SMC 5/6 proteins and promoting host cell genetic instability.197,198 Additionally, HBx interacts with the epigenetic regulator WD repeat domain 5 protein (WDR5), affecting H3K4me3 modification and further influencing HBV transcription and HCC progression (Fig. 4a).199
Interfering cell proliferation, cell cycle and apoptosis: HBx interferes with cell proliferation, cell cycle, and apoptosis through various mechanisms during the occurrence of HCC. First, HBx can activate multiple molecular pathways and critical proteins involved in cell proliferation, including (MAPK)/Ras/Raf/c-Jun, NF-κB, JAK-STAT, protein kinase C, SMAD4, DEAD-box RNA helicase 17 (DDX17)/ZW10 interacting protein (ZWINT), Src, decorin, decorin-derived peptides, survivin, and PI3K cascades.200,201,202,203 Additionally, HBx can shift hepatocytic TGF-β signaling from the tumor-suppressive pSmad3 pathway to the oncogenic pSmad3 pathway during the early stages of carcinogenesis.204,205 Other studies have found that HBx promotes hepatocyte division by upregulating cyclooxygenase-2 (COX-2), 5-lipoxygenase (5-LOX), and phosphorylated extracellular signal-regulated protein kinases 1/2 (p-ERK1/2).206 Additionally, HBx can regulate various miRNAs and lncRNAs, leading to pathway dysregulation, including RB1-TP53, PI3K-MAPK, WNT-β/catenin, and JAK/STAT, which are involved in cell proliferation, angiogenesis, and metastasis.190,191.
HBx can extend the S phase of the cell cycle by binding to DDB1, inducing mitotic abnormalities and genomic instability in hepatocytes, which promotes HCC development.207,208 Additionally, HBx can directly interact with cyclin-dependent kinase 2 (CDK2), promoting HCC development. This interaction induces phosphorylation of ubiquitin-like with PHD and ring finger domains 2 (UHRF2) at serine 643, inhibiting UHRF2 ubiquitination.209 CDK2 is a cyclin-dependent kinase, and UHRF2 is a nuclear protein involved in cell cycle regulation, both playing critical roles in the development of HCC.210,211 Furthermore, HBx can upregulate arrestin beta 1 (ARRB1) protein, which drives HBV-related HCC by regulating autophagy and the CDKN1B-CDK2-CCNE1-E2F1 axis.212
Research indicates that HBx can inhibit hepatocyte apoptosis and increase HBV replication by activating the NF-κB signaling pathway, leading to the upregulation of TNF-related apoptosis-inducing ligand receptor 3 (TRAIL-R3), thereby providing favorable conditions for HCC development.213 HBx also interacts with apoptosis-inducing factor (AIF) and the homologous AIF-homolog mitochondrion-associated inducer of death (AMID), inhibiting the translocation of AIF from the mitochondria to the nucleus, resulting in a significant downregulation of caspase-independent apoptosis pathways.214 The apoptosis-inducing effects of HBx can vary, partly due to HBx mutations, such as C-terminal truncation (trHBx).215 Additionally, HBx can influence cell proliferation, migration, apoptosis, and the cell cycle by regulating centromere protein M (CENPM), thereby promoting HCC development.216 Known HBx mutants can also increase the risk of HCC development. For instance, the HBx mutation F30V, localized in the HBx N terminus, enhances the anti-apoptotic activity of HBx in vitro, promoting the survival of infected hepatocytes and thereby initiating HBV-driven HCC (Fig. 4b).217
Indirect oncogenesis: promoting the progression of virus-related tumors
Immunosuppression: HBx primarily exerts its immunosuppressive effects by increasing HBV replication, stabilizing cccDNA, and inhibiting IFN production in the host.218 HBx can enhance PD-L1 expression by modulating the PTEN/β-catenin/c-Myc signaling pathway, inhibiting T-cell responses and promoting HBV immune evasion.219 HBV-specific CD8+ T cells exhibit T cell exhaustion in patients with chronic infection, impairing their ability to clear the virus effectively, thus failing to control viral replication.220,221,222 HBx has also been reported to interact with Spindlin1, shifting chromatin from an H3K9me3-repressed state to an H3K4me3-active state, thereby promoting the transcription of the HBV chromatin genome.81 Additionally, the HBx protein can bind to cccDNA microchromosomes, playing a crucial role in initiating and maintaining cccDNA transcription, with accumulated cccDNA contributing to HCC development.81,223 HBx downregulates IFN-γ production by targeting critical signaling components, including RIG I, TRAF3, Cardif, TRIF, Nemo, TBK1, IKKε, interferon regulatory factor 3 (IRF3), and cGAS.224,225,226 This interference by HBx helps create a favorable environment for HBV replication and survival within the host. Furthermore, HBx has been shown to directly bind to and impair the function of IFN-γ, counteracting the antiviral activity of MHC II transactivator (CIITA). This may represent a novel immune evasion mechanism employed by HBx, further illustrating its multifaceted approach to hindering host immune responses.227
The host’s innate and adaptive immune systems play a crucial role in clearing HBV after infection. However, HBV has evolved and developed effective strategies to evade the host’s immune surveillance, resulting in persistent infection.228 Most HBV infections occur in neonates with immune deficiencies, characterized by lower quality and quantity of HBV-specific T cells and B cells. In addition, maternal HBeAg can induce PD-L1 upregulation in Kupffer cells (KCs) in offspring, thereby inhibiting the HBV-specific CD8+ T cell response and thus supporting the persistence of HBV after birth.229 In chronic viral hepatitis, the upregulation of immune checkpoint, including PD-1/PD-L1, CTLA-4 and TIM-3, plays an important role in immunosuppression by inhibiting the T cell response.230 HBV infection induces immunosuppression, and then peripheral immune tolerance occurs as chronic infection progresses; finally, due to impaired immune surveillance, it mediates the occurrence of HCC (Fig. 4c).231
Chronic inflammation stimulation: Accumulation of ROS and excessive oxidative stress are detected in almost all cancers and are considered to induce tumor initiation and progression.232 ROS production induced by the C-terminal region of HBx leads to mitochondrial DNA damage in hepatocytes, which may contribute to HCC development.233 During HBV-induced inflammatory responses, the HBx protein can upregulate the transcription level of interleukin-6 (IL-6) by activating the NF-κB pathway.234 Activated IL-6 can trigger the JAK/STAT signaling pathway, leading to the upregulation of VEGF expression, thereby promoting angiogenesis.235 Additionally, the activated STAT pathway can induce the overproduction of IL-6 and other oncogenic inflammatory cytokines or chemokines, creating a vicious cycle that ultimately leads to HCC.236 Several other inflammatory factors, such as granulocyte colony-stimulating factor (G-CSF), granulocyte-macrophage colony-stimulating factor (GM-CSF), VEGF, monocyte chemoattractant protein-1 (MCP-1), and interleukins, contribute to the inflammatory microenvironment essential for HCC development.237 These inflammatory factors promote other cancer hallmarks by delivering bioactive molecules to the microenvironment, such as anti-apoptotic survival factors, growth factors, ECM-modifying enzymes driving angiogenesis, pro-angiogenic factors, and signals that facilitate cellular transformation, thereby promoting tumorigenesis.170,238
Non-resolving inflammation is driven by external factors (e.g., pathogen-associated molecular patterns (PAMPs) released by gut microbiota) and internal factors (e.g., damage-associated molecular patterns (DAMPs) released by apoptotic hepatocytes). These DAMPs include nuclear and cytoplasmic proteins, such as histones, IL-1, high-mobility group box 1 (HMGB1), and heat shock proteins (HSPs). Intrinsic pathways (including those where the tumor itself attracts inflammatory cells) also contribute to HCC development through chronic inflammation stimulation and oxidative stress induced by HBV.239,240 In summary, HBV-induced chronic inflammation and oxidative stress increase the risk of HCC. Persistent inflammatory states lead to the continuous release of inflammatory factors, promoting abnormal cell proliferation. Oxidative stress causes DNA damage, further exacerbating the carcinogenic process in hepatocytes (Fig. 4c). Since the liver is directly connected to the intestines through the hepatic portal vein circulation, the pathogenesis of HCC is associated with alterations in the gut microbiota.241 Compared with non-viral-related HCC patients, those with HBV-related HCC exhibit significantly greater richness in gut microbiota species.242 In patients with HBV-related HCC, there is a notable shift in the bacterial composition of the gut. Specifically, there are more potentially anti-inflammatory bacteria, such as Prevotella and Faecalibacterium, and fewer pro-inflammatory bacteria, like Escherichia-Shigella and Enterococcus. This divergence strongly implies that the gut microbiota plays a crucial role in the progression of HBV-related HCC.242
Inducing angiogenesis: HBx promotes angiogenesis through various pathways, a critical process in the growth and metastasis of HCC.243 Studies indicate that HBx enhances the expression of HMGB1, promoting epithelial-mesenchymal transition (EMT) and angiogenesis in HBV-related HCC.244 Furthermore, HBx stabilizes and activates hypoxia-inducible factor 1-alpha (HIF-1α) and the MAPK pathway, leading to increased VEGF expression and angiogenesis.245 HBx also cooperates with lysine-deficient protein kinase-1 (WNK1) to promote tumor-induced angiogenesis. Specifically, WNK1 activates downstream effectors oxidative stress-response kinase-1 (OSR1) to stimulate the migration of endothelial cells and HCC cells246 These findings highlight the complex molecular regulatory mechanisms through which HBx, including HMGB1, HIF-1α, WNK1, and their downstream effectors, plays a crucial role in promoting angiogenesis (Fig. 4d).
Metabolic reprogramming: Viral infections often alter the energy metabolism of host cells to support their replication and survival.247 This metabolic reprogramming may involve enhanced glycolysis, altered lipid metabolism, impact on calcium channels, and regulation of mitochondrial function, providing essential nutrients and energy for the virus while promoting its replication and pathogenicity.248 Studies have found that C-terminal truncated HBx (Ct-HBx) initiates HCC by downregulating thioredoxin-interacting protein (TXNIP), a key regulator of glucose sensing and the redox system, and reprogramming glucose metabolism.249 TXNIP is considered a tumor suppressor, and its expression is low or redundant in HCC cells.250 Additionally, HBx induces lactate fermentation metabolism by activating the NF-κBp65/hexokinase 2 (HK2) signaling pathway, with excessive lactate significantly promoting HBx-induced spontaneous HCC via the PI3K/AKT pathway.178 HBx also enhances insulin signaling sensitivity by promoting the expression of HER2, thereby facilitating HCC cell growth and migration.251 Moreover, HBx stabilizes glucose-regulated protein 78 (GRP78) through TRIM25, promoting the expression of mannosidase alpha class 1B member 1 (MAN1B1), a process critical in HBV-induced tumorigenesis. Additionally, HBx amplifies TRPM7-mediated calcium influx through hsa-miR-128-3p/SPG21, thereby activating the c-Jun N-terminal kinase (JNK) pathway and contributing to liver oncogenesis (Fig. 4e).252
HCV-related HCC
Chronic HCV infection can also cause progressive liver fibrosis, cirrhosis, and HCC.253 Unlike other oncogenic viruses, HCV does not integrate into the host genome or stably exists as an episome. Instead, it maintains its presence by forming complexes with low-density lipoproteins and other molecules.254 During natural infection, HCV typically exhibits transient replication within individual cells but requires continuous infection of new cells to establish persistent infection.255 HCV produces five proteins associated with oncogenesis: the core protein and four nonstructural proteins (NS3, NS4B, NS5A, and NS5B). Specifically, the core protein can regulate intracellular pathways (e.g., NF-κB) and upregulate various cellular proteins (e.g., IL-6, STAT-3), whose dysregulation may induce hepatocyte transformation.256 The nonstructural proteins of HCV also have oncogenic effects by inhibiting hepatocyte apoptosis and increasing cellular proliferation.257 HCV-related HCC is typically the result of chronic inflammation and fibrosis caused by long-term infection, a process that may take decades.258
Direct oncogenesis: leading to the emergence of virus-related tumors
Oncogene activation and tumor suppressor gene inhibition: The oncogene epidermal growth factor receptor (EGFR) assists HCV in entering host cells. EGFR signaling is active during ligand-induced receptor dimerization and internalization and is regulated by phosphatases and endosomal recycling/degradation.259 HCV has evolved mechanisms to exploit EGFR signaling to enhance its survival and replication. HCV infection induces EGFR signaling and alters the expression of other erythroblastic oncogene B (ERBB) receptors in early endosomes via NS5A, sustaining EGFR signaling and leading to the accumulation of more EGFR in infected cells.260 HCV-NS5A promotes viral replication by binding to Raf-1 kinase, indicating that HCV infection leads to direct virus-host dependencies and pathway-related transcriptional changes.261 In summary, oncogene activation is a crucial strategy for HCV to hijack host cell signaling pathways and drive malignant transformation.
Like other oncogenic viruses, one of the functions of the HCV core protein is to inactivate tumor suppressors, such as p53 and pRb, to enhance genomic instability and induce uncontrolled cell proliferation.262 The core protein can bind to tumor suppressor proteins p53, p73, and pRb,263 while NS3 and NS5B bind to pRb, interfering with its suppressive function.264,265 NS5A physically interacts with p53 in vitro and in vivo, sequestering p53 in the perinuclear membrane. The subsequent reduction in nuclear p53 may lead to the downregulation of p53-mediated gene expression necessary for normal cell growth.266 NS5B interacts with RB in the perinuclear region, forming a stable complex and degrading it before its transport to the nucleus. Conversely, the HCV core protein appears to lower pRb levels by downregulating RB mRNA.267 Collectively, the HCV core protein and nonstructural proteins enhance genomic instability and promote cell proliferation by binding to and interfering with tumor suppressor proteins (Fig. 4f).
Genome instability: HCV can induce genomic instability in at least three ways: (i) by directly interfering with mitotic regulatory proteins, (ii) by integrating the viral genome into multiple sites in the host genome, and (iii) by causing persistent inflammation in the liver.268 The mutational profile of HCV-related HCC generally lacks specificity, but drive mutations in the telomerase reverse transcriptase (TERT) promoter and CTNNB1 (β-catenin) are ubiquitous.269,270 Perez et al. found an enrichment of C- > T transcriptional mutations in HCV-related HCC, suggesting that this contributes to the increased susceptibility to HCC in chronic HCV-infected individuals.271 The overexpressed HCV viral protein NS5A can interfere with the mitotic process and cause genomic instability. Interfering with these mitotic regulatory proteins can lead to chromosomal instability.268 HCV promotes genomic instability by activating activation-induced cytidine deaminase (AICDA), leading to DNA repair defects and an increased mutational phenotype in cellular genes, such as tumor suppressors or oncogenes, although no specific viral oncogenes have been identified.271 Peng et al. also found a strong association between methylation of the Ras association domain family protein 1 isoform A (RASSF1A) gene promoter and increased susceptibility to HCV-related HCC, highlighting RASSF1A methylation as a critical indicator of HCC tumorigenesis.272 Chronic HCV infection induces aberrant AICDA expression, which has been proposed to cause tumors by introducing translocations and somatic mutations into proto-oncogenes (Fig. 4f).273
Interference cell proliferation, cell cycle, and apoptosis: During HCV infection, viral proteins stimulate the expression of oncogenes, disrupt the cell cycle, and inactivate tumor suppressor genes, leading to the proliferation of quiescent hepatocytes.257 The HCV core protein and NS4B promote HCC development by directly activating the Wnt/β-catenin signaling pathway.274 Additionally, NS5A interacts with intracellular signaling pathways by inhibiting protein kinase R (PKR) activity and activating the PI3K/AKT pathway.275 HCV activates β-catenin, inducing the expression of oncogenes like c-Myc and cyclin D1, thereby accelerating the cell cycle and triggering a cascade of pro-carcinogenic events.276,277 The HCV core protein also modulates the expression of the cell cycle inhibitor p21, controlling unnecessary cell cycle progression and tumor formation.278 Furthermore, NS5A interacts with growth factor receptor-bound protein 2 (GRB2), p53, p21, and cyclins, inhibiting hepatocyte apoptosis.279 NS5A also activates PPARα and inhibits voltage-gated K+ channel Kv2.1 through ROS, further preventing hepatocyte apoptosis.280 Together, these mechanisms enable HCV to promote cell proliferation and transformation, ultimately leading to HCC development (Fig. 4f).
Indirect oncogenesis: promoting the progression of virus-related tumors
Immunosuppression: Innate immune sensors within hepatocytes, such as retinoic acid-inducible gene I protein (RIG-I), recognize HCV PAMP RNA and induce IFN-mediated local antiviral responses.259 However, HCV suppresses downstream IFN signaling and limits the expression of interferon-stimulated genes (ISGs).281 This persistent yet ineffective antiviral response within the liver microenvironment allows HCV to persist, leading to tissue damage and the development of HCC.282 Initially, HCV infection activates macrophages (Kupffer cells) in the liver and triggers the release of antiviral cytokines, such as IL-1β and IFN-γ.283 However, HCV core proteins and other inhibitory factors, such as PD-L1, suppress Kupffer cell function. During chronic HCV infection, the function and phenotype of NK cells are significantly altered, characterized by the downregulation of natural killer group 2 member D (NKG2D) expression.284 Additionally, specific HCV proteins can induce dysregulation of cellular surveillance, stem cell-like cell induction, and alterations in apoptotic signaling, becoming potential drivers of HCC.285 HCV proteins, including NS3A/4A, NS4B, and NS5A, activate TGF-β1 secretion through ROS and calcium-dependent mechanisms, thereby inhibiting immune responses.286 NS5A induces TGF-β1 secretion through interaction with TLR4, reducing NK cell cytotoxicity. NS5A also upregulates COX-2 expression, leading to the production of prostaglandins via the ERK/JNK signaling pathway and blocking IFN via the JAK/STAT pathway.287 Similarly, mucosal-associated invariant T cells (MAIT cells) are affected during HCV infection, exhibiting reduced numbers but an activated phenotype with impaired responsiveness to antigen stimulation.288 Additionally, the sustained stimulation of T cells by HCV antigens leads to T cell exhaustion, characterized by the upregulation of inhibitory receptors, such as PD-1, CTLA4, and TIM-3, resulting in the loss of T cell effector functions and reduced proliferative capacity.289 A large number of research have stated that HCV actively suppresses the immune response by altering the differentiation of innate immune cells, resulting in the impairment of the subsequent robust antiviral adaptive response.290 HCV infection can generate direct mechanisms to produce Tregs to suppress T cell responses. Patients with chronic infections also accumulate a large number of circulating Treg cells, which express CD45RO, high levels of intracellular CTLA-4 and PD-1, and have the ability to inhibit the proliferation of CD4+ and CD8+ T cells as well as the production of T helper 1 (Th1) cytokines.291 At the cytokine level, HCV can promote the production of a large number of immunosuppressive cytokines in the liver microenvironment, such as IL-10 and TGF-β. IL-10 can inhibit the activation and proliferation of T cells. By inhibiting the expression of co-stimulatory molecules on the surface of antigen-presenting cells (APCs), it reduces the activation signals received by T cells, thereby weakening the immune response of T cells.292 In addition, HCV infection is associated with the activation of inflammasomes, such as the release of NLRP3, ASC, caspase-1 and IL-1β secretion.293 Many of these cytokines and chemokines are important for the survival and differentiation of CD4+ T cells.294 The impairment of antiviral CD8+ and CD4+ Th1 T cell responses is associated with the persistence of HCV infection. These dysregulations and functional impairments of immune cells not only promote chronic liver damage and fibrosis but also ultimately contribute to the development of HCC (Fig. 4g).295 During HCV infection, the abundance of Clostridiales decreases, while the Lactobacillus and Streptococcus genera increase.242 Moreover, overgrowth and enrichment of Streptococcus salivarius have been observed. This bacterium is known to downregulate the immune response, especially in the context of advanced liver cirrhosis and HCC induced by HCV, indicating that it plays a role in the development and progression of HCC.296
Chronic inflammation stimulation: Chronic inflammation is a major driver of HCV-induced HCC. The immune system recognizes viral replication products, known as pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs), which trigger the release of pro-inflammatory cytokines, such as IL-1α, IL-1β, and TNF-α. This leads to chronic liver inflammation and fibrosis.160,297 The HCV NS5A protein is an activator of the NF-κB pathway, which encodes several cytokines, including IL-6, TNF-α, and IL-1β.298 Additionally, HCV-infected cells secrete TGF-β1 following ER stress and ROS generation, which activates hepatic stellate cells and promotes fibrosis.299 While TGF-β1 initially inhibits tumor formation, it later promotes proliferation, angiogenesis and EMT, accelerating tumor progression. Oxidative damage exacerbates the inherent risk of mutations due to faulty DNA replication. This is primarily caused by ROS secreted by immune cells or released by mitochondria in infected hepatocytes.300 Furthermore, HCV induces ROS production as part of its replication strategy.301 The carcinogenesis resulting from HCV-induced chronic liver injury is a complex process, which is influenced by multiple factors.262 After HCV infects the liver, it will trigger a persistent inflammatory response. Inflammatory cells, such as macrophages and neutrophils, are recruited to the liver tissue and release a large number of inflammatory factors.302 Meanwhile, ROS generated in the chronic inflammatory environment will directly damage the DNA of hepatocytes, causing gene mutations or chromosomal abnormalities.303 Long-term HCV infection will also lead to liver fibrosis. Hepatic stellate cells are activated and proliferate excessively, secreting a large amount of extracellular matrix and destroying the normal structure and microenvironment of the liver.304 In the fibrotic liver, the intercellular signal transduction becomes disordered, and the interaction between hepatocytes and surrounding cells as well as the extracellular matrix is out of balance, further promoting the malignant transformation of cells (Fig. 4h).305
Metabolic reprogramming: HCV influences oncogenesis through the metabolic reprogramming of host cells. This reprogramming includes the regulation of energy metabolism pathways, lipid synthesis and metabolism, and alterations in cellular nutrient requirements. HCV promotes viral replication and survival through these mechanisms while driving tumor development in host hepatocytes.200 It has been reported that HCV induces the activation of inflammatory pathways like STAT3, which impairs peroxisome function by downregulating peroxisome proliferator-activated receptor α (PPAR-α), leading to the accumulation of long-chain fatty acids and potentially providing a replication advantage for the virus.306 The HCV protein NS5A interacts with glucokinase via its D2 domain (NS5A-D2), reprogramming central carbon metabolism to align with the energy and glycolytic phenotype required for HCV replication. Simultaneously, NS5A-D2 suppresses the interferon response triggered by HCV activation.307 HCV infection significantly impacts glucose metabolism, inducing aerobic glycolysis to stimulate anabolic pathways necessary for viral replication.308 Furthermore, HCV directly or indirectly interferes with insulin signaling, leading to systemic insulin resistance.308 Combined with lipid metabolism disorders and steatosis, the resulting hyperinsulinemia creates an anti-apoptotic, pro-proliferative environment in the liver that fosters tumor development (Fig. 4i).309
Gastric cancer (GC)
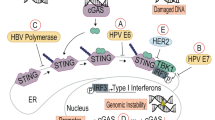
EBV-associated gastric cancer (EBVaGC) accounts for 10% of all GC and is more common in males and younger individuals.310 Gastric cancer is a multi-step process that develops from chronic gastritis, atrophy, gastric intestinal metaplasia, and dysplasia, ultimately leading to gastric cancer. More than 95% of GC patients have a history of Helicobacter pylori (H. pylori) infection, which is a significant risk factor for non-EBV gastric cancer, although no correlation between H. pylori and EBVaGC has been found.311 EBV enters gastric epithelial cells via B lymphocytes, kills epithelial cells, and remains latent to promote cancer.312 In addition, elevated protein titers associated with EBV reactivation have been shown to precede the development of precancerous and malignant gastric lesions or to be associated with EBVaGC.313 EBV is often dormant in host cells but occasionally switches to the lysis cycle when stimulated; immunosuppressed state and other infections may trigger reactivation of EBV.314 The latent mode of EBVaGC corresponds to a unique latency I/II phase, characterized by the expression of EBV nuclear antigen 1 (EBNA1), non-coding RNAs (EBER1, EBER2), and EBV BamHI-A rightward transcripts (EBV-BART miRNA), typical of latency type I. Additionally, latent membrane protein 2A (LMP2A) is a feature of latency type II, expressed in approximately 50% of cases, while LMP-1 expression is rare.315 EBV latent gene products and EBV-encoded proteins may cooperate with the host genome to promote malignant transformation by regulating multiple signaling pathways, including cell cycle control, apoptosis, and tumor suppression (Fig. 5).102,316

Oncogenic mechanisms of EBV in gastric cancer. a EBV induces genomic instability by promoting DNA methylation and inhibiting P53-mediated DNA repair. b EBV-encoded proteins disrupt the TGF-β1/Smad pathway, activate NF-κB and STAT3 signaling pathways, upregulate oncogene expression, and promote the degradation of tumor suppressor proteins, resulting in malignant cell proliferation. c EBV promotes immune evasion by impairing antigen presentation, increasing PD-L1 expression, and disrupting the function of CTLs and NK cells while increasing Tregs and TAMs in the TME. d EBV facilitates angiogenesis via KHSRP/VHL/HIF-1α/VEGFA and PI3K/AKT/mTOR/HIF-1α pathways and enhances extracellular matrix remodeling, aiding metastasis. Red text: EBV components; Black text: host cell components; Direct oncogenesis: a, b; Indirect oncogenesis: c, d. This figure was created with BioRender.com
Direct oncogenesis: leading to the emergence of virus-related tumors
Tumor suppressor gene inhibition
The p53 pathway is critical in the oncogenic process driven by EBV. Compared to EBV-negative GC, EBVaGC is characterized by a significant reduction in p53 expression, though mutations in p53 are rare.315 EBNA1 and EBNA3C suppress p53 transcription and promote its degradation. EBNA1 reduces p53 expression by targeting ubiquitin-specific protease 7 (USP7).317 The EBV-encoded immediate early protein—BamHI Z fragment leftward open reading frame 1 (BZLF1) also suppresses p53-dependent transcription by acting as a component of the ECS ubiquitin ligase complex (Elongin B/C-Cul2/5-SOCS-box proteins),318,319 facilitating p53 degradation.320 This direct inhibition of p53 by EBV may explain the low frequency of p53 gene mutations in EBVaGC.321
EBV is also the first human virus identified to encode multiple microRNAs.322 EBV-encoded EBV-miR-BART3-3p and EBV-miR-BART5-3p can bind to p53, reducing cellular senescence in EBVaGC, accelerating cell cycle progression, and preventing apoptosis.323 This suggests that EBV-encoded products also inhibit p53 function. It has been suggested that normal or elevated levels of p53 may confer a survival advantage to virus-infected cells, as these cells are less susceptible to apoptosis induced by JNK and rapamycin.324 Li et al. found that LMP1 induces hypermethylation of the H19 promoter, suppressing H19 and hsa-miR-675-5p expression, leading to overexpression of p53 protein in EBVaGC cells, which favors EBV latency.325 EBV-miR-BART9-5p maintains EBV latency by reducing the expression of the oncogene MUS81, promoting the progression of EBVaGC.326 Additionally, EBV-encoded EBV-miR-BART5-3p promotes p53 protein degradation, marking it as the first identified EBV-microRNA to inhibit p53 expression, highlighting EBV’s multiple strategies to maintain latency and promote EBV-related cancer development.327 Overall, p53 mutations do not play a major role in the early stages of carcinogenesis, while p53 reduction may promote tumor development as an independent event (Fig. 5a).
Genome instability
Upon entering gastric epithelial cells and establishing latency, EBV undergoes whole-genome methylation.328 Normal gastric tissue shows a promoter CpG island methylation level of 1-2%, while EBVaGC exhibits a CpG island methylation frequency of 19%.329 EBVaGC is typically characterized by somatic mutations in PIK3CA, loss of p16 (CDKN2A) expression, extensive DNA hypermethylation, and localized amplification of the 9p24.1 region, including JAK2, PD-L1, and PD-L2.330,331 In EBVaGC, abnormal DNA methylation of multiple genes or loci, including THBS1, APC, and p16, is widely observed.332 Other methylated genes include those involved in cell cycle regulation (p14ARF, p15, and p73), DNA repair (hMLH1, MGMT, and GSTP1), cell adhesion and metastasis (E-cadherin, TIMP1, and TIMP3), apoptosis (DAPK and Bcl-2), and signal transduction (APC, PTEN, and RASSF1A) (Fig. 5a).333,334,335
Interfering cell proliferation, cell cycle and apoptosis
EBV interferes with cell proliferation, cell cycle, and apoptosis in various ways during the occurrence and development of EBVaGC. EBV promotes the growth and survival of EBVaGC cells through its encoded proteins and small RNAs, such as EBNA1, LMP2A, and EBER.336 EBNA1 upregulates DNA methyltransferase 3a (DNMT3a) expression by activating the E2F1 transcription factor, thereby promoting gastric cancer cell proliferation, inducing cell cycle progression, inhibiting apoptosis, and accelerating migration in vitro.337 EBNA1 also causes the loss of promyelocytic leukemia (PML) nuclear bodies (NBs), weakening the DNA damage response and promoting gastric cancer cell survival.338 LMP2A activates DNMT1 transcription by phosphorylating STAT3, promoting EBVaGC cell growth.339,340 It also inhibits mTORC1 activation by inactivating the TGF-β1/Smad pathway and downregulating GCNT3 expression, thereby promoting cell proliferation and migration while preventing G0/G1 phase arrest.341 EBER induces insulin-like growth factor 1 (IGF-I) as an autocrine growth factor in EBVaGC, accelerating gastric cancer cell growth. EBER1 reduces the expression of the m6A “writer” Wilms’ tumor 1-associating protein (WTAP) by activating the NF-κB signaling pathway, further promoting the migration of EBVaGC cells.342
EBV-encoded miRNAs are divided into two main clusters, BamHI fragment H rightward open reading frame 1 (BHRF1) and BART miRNAs.333,343 In EBVaGC, the 44 mature miRNAs encoded by BART miRNA may interfere with genes related to cell death and cell cycle regulation.333,344,345 Specifically, EBV-miR-BART5-5p targets protein inhibitor of activated STAT 3 (PIAS3) and enhances PD-L1 expression, promoting gastric cancer cell proliferation, anti-apoptosis, invasion, and migration.322 EBV-miR-BART1-3p targets DAB2, promoting EBVaGC cell migration and reducing apoptosis.346 EBV-miR-BART6-3p regulates the expression of cell cycle-related proteins through the LOC553103-STMN1 axis, thereby inhibiting the proliferation of EBV-related tumor cells.347 EBV-miR-BART10-3p may promote EBVaGC proliferation and migration by targeting Dickkopf 1 (DKK1).336 EBV-miR-BART20-5p regulates cell proliferation and apoptosis by targeting Bcl-2-associated death promoter (BAD), promoting the development of EBVaGC.348 In short, EBV-encoded miRNAs play a significant regulatory role in the development of EBVaGC.
Like other cancers, EBV disrupts multiple cell cycle checkpoints, such as p16 and p21. p16 (CDKN2A) is a tumor suppressor gene that inhibits the cyclin D1/CDK4 complex, and it is commonly hypermethylated and silenced in EBVaGC.349 LMP1 also inactivates p16 by inducing the sequestration of E2F4 and E2F5 (E2F4/5) and the E26 transformation-specific transcription factor (Ets2) in the cytoplasm, leading to p16 dysfunction in these cells.350,351
EBNA1 promotes tumorigenesis by ubiquitinating p53, inhibiting TGF-β signaling, and enhancing the transcription of the anti-apoptotic protein survivin.352 Conversely, LMP2A can activate the PI3K/AKT proliferative pathway, increasing the survival of infected cells by upregulating survivin gene expression, inhibiting TGF-β1-induced apoptosis, and promoting cell migration through the Notch signaling pathway.353,354 EBV is known to encode over 40 different miRNAs. Among them, EBV-miR-BART1-3p directly targets disabled homolog 2 (DAB2), increasing the migration of EBVaGC cells and reducing apoptosis.346 EBV-miR-BART4-5p exerts anti-apoptotic effects in EBVaGC by modulating the expression of the BH3-interacting domain death agonist (Bid).343 EBV-miR-BART5 inhibits the expression of the pro-apoptotic Bcl-2 family member Puma, preventing Puma-mediated p53-independent apoptosis in EBVaGC cells.355 BART miRNAs also downregulate pro-apoptotic and anti-apoptotic mediators, such as caspase 3.356 In summary, EBV promotes the development and progression of EBVaGC by interfering with cell apoptosis through multiple mechanisms, including inhibiting p53 function, enhancing survivin expression, activating the PI3K/AKT pathway, and targeting apoptosis-related genes. These mechanisms interact with each other and jointly promote the occurrence and development of EBVaGC by interfering with cell proliferation, cell cycle, and apoptosis (Fig. 5b).
Indirect oncogenesis: promoting the progression of virus-related tumors
Immunosuppression and chronic inflammatory stimulation
EBV can persist and evade immune clearance, inducing an immunosuppressive tumor microenvironment (TME).357,358 EBVaGC is characterized by active immune infiltration, including CD8+ T cells, NK cells, macrophages, and Treg cells,323,359 along with elevated levels of IL-1β and IL-10, indicating a pro-inflammatory environment.360 The microenvironment also contains unique immune cell subsets, including highly proliferative T and B cells, B cells expressing T cell markers, and proliferative T cell clusters with follicular T helper cell markers.361 Antigen-specific ISG-15+CD8+ T cell populations are highly enriched in EBVaGC patients and exhibit a transitory exhaustion state.362 Due to this pro-inflammatory state, tumors show high PD-1, PD-L1, and LAG-3 expression.359,363 Additionally, elevated IFN-γ levels deplete tryptophan, inhibiting tryptophan-sensitive CTL and NK cells. Other molecules contributing to immunosuppression include CCL22, which increases Treg recruitment, and indoleamine 2,3-dioxygenase 1 (IDO1).364 Although the exact mechanism of PD-L1 overexpression in EBVaGC has not yet been clarified, it has been reported that EBV-miR-BART5-5p directly targets PIAS3 and controls and enhances PD-L1 through the EBV-miR-BART5/PIAS3/pSTAT3/PD-L1 axis, which contributes to anti-apoptosis, tumor cell proliferation, invasion, migration and immune escape.322 EBV-miR-BART11 and EBV-miR-BART17-3p inhibit FOXP1 and PBRM1 respectively, and enhance the transcription of PD-L1, facilitating tumor immune escape.365 Additionally, elevated IFN-γ levels deplete tryptophan, inhibiting tryptophan-sensitive CTL and NK cells.364 Other molecules that cause immune suppression include CCL22, which can increase Treg recruitment, and indoleamine 2,3-dioxygenase 1 (IDO1). The EBNA1 repeat sequence and the early lysis gene BNLF2A also inhibit antigen processing in EBVaGC, leading to immune evasion (Fig. 5c).364,366
The gut microbiota encompasses various pathogens, among which H. pylori is a notable example. This pathogen has the potential to induce epigenetic alterations and mutagenesis within the host genome. A significant majority of the bacteria implicated in the development of GC belong to the category of Gram-negative bacteria. Moreover, the co-infection scenario involving Gram-negative bacteria and viruses like EBV has been identified as a risk factor for GC.367 Fusobacteria are another kind of Gram-negative bacteria that are regarded as a risk factor for GC. GC, pancreatic, and colorectal cancers are often more abundant with Fusobacterium species, primarily F. nucleatum, than non-cancerous tissues.368
Extracellular matrix remodeling
LMP1-containing extracellular vesicles (EVs) enhance the adhesion, proliferation, migration, and invasion of recipient cells and remodel the TME by increasing the expression of ECM remodeling interaction proteins, such as matrix metalloproteinase 9 (MMP9), MMP2, and integrin-α5.369 LMP2A, BARF0, EBER, and EBNA1 affect cell behavior by downregulating key cell adhesion molecules, such as E-cadherin.333,370 Additionally, the absence of EBV-encoded EBV-miR-BART9 leads to elevated E-cadherin expression, reducing the proliferation and invasion potential of EBVaGC.371 Molecules important for cell invasion, such as THBS1, E-cadherin (CDH1), and TIMP2, also have their expression suppressed by promoter methylation, which may be related to the carcinogenesis process.203,372 In summary, EBV influences the invasion and growth potential of EBVaGC cells through multiple mechanisms, including ECM remodeling, downregulation of crucial adhesion molecules, and the expression of miRNAs (Fig. 5d).
Inducing angiogenesis
Angiogenesis is a key factor in tumor development and metastasis because the formation of new blood vessels enhances the delivery of nutrients and oxygen to rapidly proliferating cells.373 In EBVaGC, hypoxia-induced EBV-encoded circular RNA LMP2A (EBV-circLMP2A) promotes angiogenesis through the KHSRP/VHL/HIF-1α/VEGFA pathway.373 EBV-miRNA-BART12 inhibits tubulin polymerization-promoting protein 1 (TPPP1), suppressing α-tubulin acetylation, promoting microtubule dynamic assembly and acetylation, and activating EMT. These processes collectively enhance the invasion and metastasis of gastric cancer cells.374 Additionally, EBVaGC upregulates IHH (a gene that increases metastatic potential through angiogenesis) and Snail protein expression while reducing E-cadherin and tight junctions, further enhancing the invasive and metastatic capabilities of gastric cancer cells.310,312 EBV infection also induces the loss of PTEN expression through CpG island methylation of gene promoters, activating the PI3K-AKT signaling pathway.340 The loss of PTEN activates the PI3K/AKT pathway, leading to increased angiogenesis, cell migration, and loss of cell cycle adhesion.375 Moreover, EBV promotes the formation of vasculogenic mimicry through the PI3K/AKT/mTOR/HIF-1α pathway.376 In summary, EBV promotes gastric cancer cell growth, invasion, and metastasis by enhancing angiogenesis, thereby increasing the blood supply and metastatic potential of tumors (Fig. 5d).
Cervical cancer (CC)
HPV causes various malignancies in the anogenital and oropharyngeal regions.377 Most of HPV-associated cancers in women are cervical cancers (CC), whereas in men mostly HPV-associated cancer is head and neck squamous cell carcinoma (HNCC) related to HPV infection, which includes 30-60% of oropharyngeal carcinoma, 12% of pharyngeal cancer and 3% of oral cancer.378 It should be noted 90% of HPV infections get eliminated within a two-year period. But if infection endures may causes abnormal cells.22 The transformation into CC only takes place when these cervical cells traverse the basal membrane and infiltrate the tissues beneath (Fig. 6a).22

Oncogenic mechanisms of HPV in cervical cancer. a Progression of CC caused by HPV. b HPV infection disrupts DNA damage response and induces R-loop formation, leading to instability in DNA repair genes. c HPV promotes telomerase reverse transcriptase (TERT) accumulation, resulting in telomere elongation. d HPV-encoded proteins drive malignancy by activating EGFR signaling, degrading cell cycle inhibitors, and pro-apoptotic proteins. e HPV facilitates immune escape by inhibiting the cGAS-STING pathway, reducing antigen presentation, and inducing cytokine release. f HPV increases ROS and COX-2 production, causing chronic inflammation and oxidative stress. g HPV-positive cells can release extracellular vesicles (EVs) that promote angiogenesis and malignant transformation of epithelial cells. h HPV proteins drive cervical cancer aerobic glycolysis via upregulating HIF-1α/MYC transcription and activating PKM. Red text: Components of HPV; Black text: Components and biological processes of the host cell; Direct oncogenesis: b–d; Indirect oncogenesis: e–h. This figure was created with BioRender.com
The occurrence of HPV-associated cancers is mainly caused by HPV viral proteins, such as E5, E6, E7. E5 primarily regulates cell growth and signaling pathways in HPV-infected cells;379 E6 promotes the ubiquitin-dependent degradation of p53, allowing the infected cells to evade apoptosis;380 and E7 disrupts cell cycle regulation, leading to cervical dysplasia.381 Notably, E6 and E7 are the principal oncogenic proteins expressed by both high-risk (HR) and low-risk (LR) HPV types. But only HR-HPV types encode the E5 protein, as LR-HPV lacks a defined homologous E5 ORF or the translation initiation codon for this protein.382 HPV16 and HPV18 are HR-HPV, but with distinctions. HPV16 is more prevalent and often associated with squamous cell carcinomas. It has a high replicative activity and is commonly found in a wide range of cervical cancers.383 In contrast, HPV18 is more likely to integrate into the host genome and is associated with adenocarcinomas.384
Direct oncogenesis: leading to the emergence of virus-related tumors
Oncogene activation and tumor suppressor gene inhibition
Both the HPV integration process and the E6 and E7 proteins can increase the risk of oncogene activation.385 The Apolipoprotein B mRNA editing enzyme catalytic polypeptide like (APOBEC) gene family, which belongs to the cytosine deaminase superfamily, is an oncogene that increases genomic instability when overexpressed.386 Study has shown that APOBEC3 mutations occur following the integration of high-risk HPV into the host cells, indicating that APOBEC is a molecular factor driving CC development by HPV.387 Additionally, HR-HPV E6 protein has been found to activate super-enhancers of oncogenes, such as EGFR and c-MET by reducing the stability of the histone demethylase KDM5C, thereby promoting oncogene activation.388
HPV-mediated cervical carcinogenesis hinges on the inactivation of two critical tumor suppressor genes: p53 and pRb.389 HPV E6 binds to p53 and recruits various ubiquitin ligases to facilitate p53 degradation.390 Research has found that the E6 protein directly binds to the short leucine consensus sequence of the ubiquitin ligase E6-associated protein (E6AP) to form an E6/E6AP heterodimer that ubiquitinates and degrades p53.391 HPV also promotes the nuclear export of human heterochromatin protein 1γ, which recruits UBE2L3 to target p53 for polyubiquitination and degradation.392 The LXCXE motif in the CR2 domain of HPV E7 binds to pRB and recruits cullin 2-containing E3 ubiquitin ligases to degrade pRB.393 On the whole, HPV E6 and E7 work together to inhibit the expression of tumor suppressor genes in host cells, leading to the oncogenic transformation of HPV-infected cells (Fig. 6b).
Genome instability and telomere maintenance
Whole-genome sequencing of HPV-associated cancer has revealed that HPV integration often leads to genome structural alterations, including duplications, deletions, translocations, and inversions.394 It has been found that after HPV integrates into host DNA may decrease the gene stability of p53,395 elevating the cancer risk. Moreover, HPV integration can hijack the DNA damage response (DDR) and utilize it to promote the malignant proliferation of host cells.396 Previous studies have indicated that E6 primarily induces DDR by promoting p53 degradation.397 R-loops are trimeric RNA consisting of an RNA-DNA hybrid and a displaced DNA strand.398 Recent studies have shown that HPV16 E6 induces abnormal R-loop accumulation by inhibiting the transcriptional activity of p53, leading to the instability of DNA repair genes, such as Fanconi anemia D2 protein (FANCD2) and ATR.399 E7 also induces DDR through multiple pathways, including binding to and inactivating pRb, interacting with the pRb-like protein CBP/p107,397 and directly interacting with DDR proteins to increase their activity,400 ultimately promoting DDR gene translation induced by E2F1 expression.401 In cervical epithelial cells co-infected with HPV and HIV, the frequency of catastrophic dysregulated alternative splicing is significantly higher than in cells infected with HPV or HIV alone, increasing the risk of malignancy (Fig. 6b).402
Telomeres are specialized DNA sequences at the ends of chromosomes that maintain chromosomal stability and naturally shorten with cell division, and are generally not or lowly expressed in keratinocytes.403 Abnormal accumulation or activation of TERT, leading to telomere elongation.404 Research also found that HPV-associated CC has higher TERT expression than HPV-infected NPE cells, suggesting that HPV interference with telomerase function is a potential mechanism of carcinogenesis.405 E6 typically extends telomeres through mechanisms including methylation of the TERT transcription start site,406 downregulation of the TERT transcription inhibitor NFX-1,407 and upregulation of the TERT transcription activator c-Myc.408 However, the effect of E7 on telomeres is complex, capable of inducing either telomere shortening or elongation depending on the context. In cell lines that over-express E7, TERT expression is low and telomeres are excessively shortened, suggesting that E7 can impair telomere length.409 Conversely, some studies have shown that E7 upregulates FANCD2, activating an ALT-dependent pathway to elongate telomeres (Fig. 6c).410
Interfering cell proliferation, cell cycle, and apoptosis
The oncogenic activity of E5 is mainly through hyperactivation of the EGFR.379,411 EGFR is a known stimulator of cell growth and contributes to tumor progression.412 E5 interacts with the 16-kDa subunit of vacuolar ATPase, disrupting endosomal acidification, which promotes EGFR recycling at the cell membrane and enhances its mitogenic activity.413 Moreover, EGFR is degraded by the E3 ubiquitin ligase c-Cbl protein, but such activity is inhibited by HPV16 E5, reducing EGFR degradation.414 Downregulation of pRB, p107, and p130 by the viral E7 protein contributes to the uncontrolled proliferation and malignant transformation of HPV-infected cervical epithelial cells.415 TGF-β significantly suppresses epithelial cell proliferation. Studies have shown that HPV-encoded viral proteins bind to TGF-β receptors and signaling sensors, leading to their degradation and promoting the proliferation of infected cells.416 Similarly, the hedgehog pathway activated by HPV16 E6 and E7 may transform esophageal basal cells and promote esophageal squamous cell carcinoma.417
The DREAM repressor complex inhibits the transcription of genes associated with cell division.418 HPV E7 primarily interferes with the cell cycle by modulating the DREAM complex.419 On one hand, HPV E7 directly interacts with the B-Myb-MuvB complex to promote the transcriptional activation of genes associated with the S and M phases of the cell cycle.420 On the other hand, HPV E7 activates inhibitory proteins of DREAM, such as cyclin-dependent kinase inhibitors p21 and p27, thereby reducing the activity of the DREAM complex.421 Additionally, HPV E7 can upregulate the degradation of DREAM activators p107 and p130.422 Moreover, by modulating CDK inhibitors, such as p21CIP1 and p27KIP1, E7 disrupts the G1/S checkpoint.423 Unlike HPV E7, HPV E6 primarily overcomes cell cycle checkpoints by inactivating p53, thereby loosening cell cycle regulation.424 The E6-AP-E6-p53 complex downregulates hsa-miR-34a expression, leading to upregulation of p18Ink4c and driving cells into the S phase.425 In addition, E6 interacts with Aurora kinase A to regulate cyclin E and phosphorylated histone H3 expression, accelerating G1/S transition and mitosis.426
HPV proteins E5 and E6 evade apoptosis through various mechanisms. For example, E5 downregulates Fas expression,427 activates the Ras-Raf-MAP kinase pathway and the PI3K-Akt pathway,428 and degrades the pro-apoptotic protein Bax to escape apoptotic signals.429 In contrast, E6 reduces Bak expressionand degrades FADD and procaspase-8 to inhibit apoptosis.430 By contrast, HPV E2 and E7 are primarily involved in inducing host cell apoptosis,431 suggesting that HPV induction of apoptosis escape in host cells is a complex, dynamic process influenced by multiple factors. HIV Tat protein are capable of upregulating the expression of the HPV E6 protein. This process inhibits the expression of p53, thereby conferring the potential for immortality upon the infected cells (Fig. 6d).432
Indirect oncogenesis: promoting the progression of virus-related tumors
Immunosuppression
In CC, HPV E6 and E7 produce a sustained immunosuppressive state by inhibiting the cGAS-STING signaling pathway, reducing HPV-associated antigen presentation, decreasing immune cell recruitment, and suppressing cytokine release.433 HPV18 and 16 E7 proteins inhibit STING through the LCXCE sequence and NLRX1, respectively.434 Additionally, HPV16 E6 can inhibit IFN regulatory transcription factor IRF3, reducing the activation of the cGAS-STING signaling pathway.435 Large-scale genome-wide association studies (GWAS) have found that the HPV genome frequently integrates near the MHC-I polypeptide-related sequence A, leading to an increase in MHC-I gene mutations, which is a potential mechanism of HPV immune evasion.436 Furthermore, the disruption of chemokine regulation caused by HPV E6 and E7,437 such as the upregulation of CCL-2 and CCL-5 and the downregulation of IFN-γ,438 reduces immune cell recruitment and promotes an immunosuppressive microenvironment at the site of infection.439
In CC, HPV E5 protein directly interferes with the translation, transport, and degradation of MHC I, preventing the activation of adaptive immunity.440 In addition to affecting MHC-I expression, E5 also directly reduces the expression of MHC-II complexes,441 calnexin, and CD1 receptors that activate immune cells, establishing a sustained immunosuppressive state.87,442
The normal vaginal microbiota (VMB) is dominated by Lactobacillus, which can maintain an acidic environment and produce antibacterial proteins to resist pathogens.377 Many studies have shown that the imbalance of vaginal microbiota is closely related to the development of cervical intraepithelial lesions and CC.443 In relevant studies, L. crispatus, E. eligen, G. vaginalis, U. urealyticum, P. stutzeri and A. vaginae have been identified as high-risk factors for HPV-CC, while high abundances of D. invisus, F. magna, G. vaginalis, P. buccalis, P. timonensis and L. johnsonii significantly reduce the risk of HPV-CC.444 Specifically, high abundances of Prevotella can affect the occurrence of cervical lesions related to persistent HR-HPV infection through the host NF-kB and C-myc signals,445 and M. microronuciformis mainly induces or maintains the production of viral oncoproteins E6 and E7 to promote cancer.446 In addition, studies have also shown that the imbalance of VMB is closely related to the expression levels of immune checkpoint molecules in HPV-CC. PD-L1 and LAG-3 are negatively correlated with Lactobacillus species and positively correlated with dysbiotic bacteria, whereas TLR2 is negatively correlated with dysbiotic bacteria and positively correlated with lactobacilli.447
Overall, in cervical cancer, HPV E6 and E7 mainly influence the cGAS-STING signal, and E5 interferes with the expression of MHC. However, the impact of HPV on the immune microenvironment of head and neck cancers differs from that in CC. Firstly, in HNCC, it is E5, rather than E6 and E7, that suppresses immunity by inhibiting the immunoproteasome and STING pathway.413,448 Secondly, E6 and E7 no longer play an immunosuppressive role but function as potent immunostimulants instead. They can effectively trigger specific T-cell responses and enhance the anti-tumor activity within the microenvironment of HNCC.449 This unique mechanism results in differences in clinical prognosis. It has been reported that compared with non-HPV-infected CC, the prognosis of HPV-infected CC is poorer. Nevertheless, the prognosis of HPV-infected OPSCC is better than that of non-HPV-infected OPSCC (Fig. 6e).450
Chronic inflammation stimulation
The persistent presence of chronic inflammation increases the risk of malignant cellular transformation.451 HPV proteins E6, E7, and their miRNAs can promote the continuous release of inflammatory mediators directly through signaling pathways, such as STAT3, NF-κB, and EGFR, including COX-2 and its metabolites like PGE2.452 HPV proteins E5, E6, and E7 all activate the EGFR-Ras-MAPK-AP-1 pathway, increasing COX-2 transcription and maintaining the body in a state of chronic inflammation.453 This process is a positive feedback loop where COX-2 is continuously expressed.453 Following HPV infection, the host and microenvironment’s immune cells experience miRNA dysregulation, such as the silencing of hsa-miR-21 or the overexpression of hsa-miR-155, which contributes to the establishment of a chronic inflammatory state.454 The chronic inflammation induced by HPV infection also leads to the production and release of ROS and reactive nitrogen species in HPV-infected cells.455 Compared to non-HPV-infected cervical epithelial cells, HPV-induced intraepithelial neoplasia and CC exhibit significantly increased oxidative stress products. These oxidative stresses are considered direct consequences of HPV proteins. For example, co-expression of E1 and E2 increases ROS levels and γH2AX deposition, and ROS levels also increase in HPV-negative cervical epithelial cells transfected with E6 alone.456 In short, HPV-induced chronic inflammation and oxidative stress accelerate the malignant transformation of epithelial cells. HIV infected T cells contain hsa-miR-155-5p, which can be internalized by cervical epithelial cells and activate the ERCC5-NF-κB signaling pathway, leading to the secretion of pro-inflammatory cytokines IL-6 and IL-8. This enhances the invasive capacity of cervical epithelial cells (Fig. 6f).457
Inducing angiogenesis
EVs are mediators of communication between cancer cells and the TME and are potential factors in HPV-induced carcinogenesis.458 HPV16 and 18 E6 upregulate Wnt7b mRNA expression in host cell lines and host-derived EVs. These Wnt7b mRNA-rich EVs subsequently activate β-catenin in HUVE cells, ultimately promoting angiogenesis and malignant transformation of epithelial cells.459 Additionally, HPV-induced circRNA abnormalities contribute to the progression of CC by MAPK, and PI3K/AKT/mTOR signaling pathways.460 HIV also leads to an upregulation of neurotrophic factors BDNF and NGF, increasing local nerve infiltration, vascular invasion, and lymph node metastasis, ultimately inducing the occurrence of CC (Fig. 6g).461
Metabolic reprogramming
HPV can also promote the metabolic reprogramming of host cells.462 In HPV-infected CC, HPV E6 can increase the expression and stability of HIF-1α and upregulate the expression of HIF-1α target genes, such as GLUT1, LDHA, CAIX, MCT4 and CD147, thus promoting the Warburg effect.463 Meanwhile, the HR-HPV E7 protein can activate the glycolytic enzyme pyruvate kinase M2 (PKM2) to induce the Warburg effect.464 In addition, HPV E6 and E7 proteins can activate IGF2BP2 to stabilize m6A-MYC, which can promote MYC mRNA transcription and then drive aerobic glycolysis in cervical cancer.465 Similarly, in HPV-infected HNSCC, HR-HPV E6 proteins activate the HIF-1α and Akt/mTOR signaling pathways, and HR-HPV E6 activates PKM2 and COX, ultimately promoting glycolysis (Fig. 6h).466
Nasopharyngeal carcinoma (NPC)
The establishment of type II latency is the initial step in EBV-driven epithelial malignancies. In nasopharyngeal epithelial (NPE) cells, EBV typically exists in a lytic state, while in precancerous lesions and nasopharyngeal carcinoma (NPC), EBV is in type II latency. This suggests that type II latent EBV is closely associated with NPC development.467 Type II latent EBV mainly expresses non-coding RNAs (EBER), Epstein-Barr nuclear antigen 1 (EBNA1), latent membrane proteins (LMPs), and BamHI A transcripts (EBV-miR-BARTs).310 Constitutively expressed latent proteins and EBV-miR-BARTs, which contribute to the evasion of NPE cell death, suppression of host immune responses, and genomic instability.468 Meanwhile, heterogeneously expressed LMPs drive tumor progression in invasive epithelial cancers through metabolic reprogramming and inducing NPE cell dedifferentiation.469 Notably, although type II latency EBV induces some oncogenic features and abnormal signaling pathways, it still requires some additional acquired genetic alterations for the malignant transformation (Fig. 7a).470

Oncogenic mechanisms of EBV in nasopharyngeal carcinoma. a Progression of NPC caused by EBV. b EBV induces genetic instability through inhibition of DNA repair, increased DNA methylation, chromosomal breaks at 11q23, and telomere elongation. c EBV promotes cell proliferation by activating NF-κB, Hedgehog, mTOR signaling pathways. d EBV accelerates G1/S progression in host cells by downregulating GSK3β expression, promoting degradation of p18, and activating the JAK2/STAT3 signaling pathway. e EBV infection promotes anti-apoptotic protein expression, inactivates pro-apoptotic proteins, reduces fumarate hydratase (FH) activity, and inhibits ferroptosis through the p62-Keap1-NRF2 pathway. f EBV evades immune surveillance by modulating PD-L1, MIC-A/B, and B7-H3 expression, and induces lactate, creatinine, and cytokine release, which recruits Treg cells and expands myeloid-derived suppressor cells (MDSCs), creating an immunosuppressive microenvironment. g EBV proteins promote cytokine/chemokine release, inhibit monocyte recruitment, induce TAM, and enhance mitochondrial activity via ROS suppression. h EBV-encoded proteins activate ANXA3-HIF-1α-VEGF and PI3K/AKT/mTOR/HIF-1α pathways to promote angiogenesis. i EBV stimulates glycolysis, increases glutamine uptake, and inhibits lipid degradation via the mTORC1/NF-κB and HIF-1α pathways. Red text: EBV components; Black text: host cell components; Direct oncogenesis: b–e; Indirect oncogenesis: f–i. This figure was created with BioRender.com
Direct oncogenesis: leading to the emergence of virus-related tumors
Genome instability
EBV can both induce and counteract host genome instability. The genetic vulnerability caused by EBV integration mainly depends on the activation of fibroblast growth factor (FGF) and NF-κB signaling pathways.471 LMP1 protein inhibits the activity of DNA-dependent protein kinase, reducing the repair of double-strand breaks (DSBs).472 EBV infection often leads to hypermethylation of promoter regions of genes related to DNA repair, significantly downregulating their transcription, and in vitro experiments have confirmed that DNA repair capacity is diminished in EBV infected NPE cells.473 Additionally, the sequence-specific DNA-binding domain of EBNA1 can bind to EBV like palindromic sequences on human chromosome 11q23, which may trigger chromosome breakage.474 However, some reports suggest that EBV infection can enhance DNA repair capacity, particularly after radiotherapy and chemotherapy. The EBNA1 protein promotes DNA damage in NPE cells by inducing ROS and upregulating oxidative stress response proteins, such as SOD1 and Prx1.475 TCAB1 is involved in telomere elongation and ribonucleoprotein biogenesis, thus plays a role in activating telomerase and repairing DNA damage.476 While numerous studies have revealed various DNA repair mechanisms in NPC, the relationship between these DNA repair processes and EBV remains unclear and requires further investigation (Fig. 7b).477
Interfering cell proliferation, cell cycle, and apoptosis
EBV promotes malignant cell proliferation primarily through the activation of the NF-κB complex.478 The activation of the NF-κB signaling pathway by the LMP1 protein is a major cause of malignant proliferation in NPE cells.479 Mechanistically, LMP1 induces increased activity of Yes-associated protein (YAP) and transcriptional coactivator with PDZ-binding motif TAZ, upregulates the transcription levels of EGFR and c-Met, and downregulates cell cycle inhibitors p16 and p21, thereby activating the NF-κB pathway and promoting malignant proliferation in NPE cells.480 Additionally, EBV maintains NPE cell stemness by promoting Hedgehog autocrine signaling.481 Furthermore, EBV-miR-BART13 interacts with NF-κB inhibitor-interacting Ras-like 2 (NKIRAS2) to inhibit the activation of NF-κB signaling, inducing NPE cell proliferation.482 EBV-miR-18a can upregulate SMG1 and activate the mTOR signaling pathway, further enhancing the proliferative capacity of NPE cells (Fig. 7c).483
The regulatory effect of EBV on the cell cycle of NPE cells depends on whether the NPE cells have acquired additional abnormal genetic changes, such as p16 deletion. After the NPE cells acquire abnormal genetic changes, the products encoded by EBV accelerate the G1/S progression by regulating cell cycle checkpoints and related regulatory factors, further promoting the progression of NPC.484 EBV-encoded vIL-10 can activate the JAK2/STAT3 signaling pathway, increasing IL-6 expression and promoting G1/S phase progression.485 EBV-miR-BART22 upregulates hsa-miR-4721 transcription, leading to decreased GSK3β expression, thereby increasing nuclear accumulation of β-catenin and activating CCND1 and c-Myc.486 EBV infection can also downregulate the deubiquitinating enzyme cylindromatosis, leading to increased degradation of the cell cycle negative regulator p18, accelerating the G1/S phase transition.487 However, some EBV-derived products inhibit NPE cell proliferation. For instance, EBV-miR-BART6-3p negatively regulates the LOC553103/STMN1 axis, downregulating cyclin proteins (like CCNE1, CCND1, and CDK4) and causing cell cycle arrest in EBV-associated tumor cells (Fig. 7d).347
A large number of miR-BARTs are present in epithelial cells infected by EBV. These abundantly expressed miR-BARTs promote the occurrence of NPC by inhibiting apoptosis.488 Beyond EBV-miR-BARTs, LMP1 also demonstrates anti-apoptotic activity by promoting the expression of anti-apoptotic proteins (like survivin and Mcl-1) and inactivating pro-apoptotic proteins (like Bad and Foxo3a).469,489 LMP1 can decrease fumarate hydratase activity, protecting NPC cells from TNF-α-induced necroptosis in the microenvironment.490 EBV also reduces the sensitivity of NPC cells to ferroptosis by activating the p62-Keap1-NRF2 signaling pathway and upregulating SLC7A11 and GPX4, thereby ensuring NPC survival under high oxidative stress and promoting the development of NPC.491 Furthermore, EBV can upregulate transferrin receptor through unknown pathways, which inhibits apoptosis via activation of the PI3K/AKT/mTOR signaling axis (Fig. 7e).492
Indirect oncogenesis: promoting the progression of virus-related tumors
Immunosuppression
EBV contributes to immune evasion in NPC cells by upregulating inhibitory receptors and downregulating MHC class I and II antigens. EBV primarily promotes immune evasion in NPC by upregulating PD-L1. EBER1,493 LMP1,494 and circBART2.2 can activate the RIG-I pathway to upregulate PD-L1 transcription.495 EBV-miR-BART11 and EBV-miR-BART17-3p inhibit FOXP1 and PBRM1, respectively, enhancing PD-L1 transcription.365 Additionally, EBV increases DNA methylation, which constitutively activates NF-κB signaling and promotes the activation of genes associated with T cell exhaustion, such as CD74.496 EBV induces the expression of inhibitory receptors on NK cells, such as TIGIT, LAG3, and ZNF683.497 Furthermore, LMP1’s activation of the PI3K/AKT/mTOR signaling pathway increases B7-H3 expression, inhibiting NK cell cytotoxicity.498 Research also shows that EBV upregulates F3, leading to the accumulation of activated platelets, forming a physical barrier that prevents NK cells from entering the TME (Fig. 7f).357
Chronic inflammation stimulation
EBV integration downregulates the expression of inflammation-related genes, such as TNFAIP3, PARK2, and CDK15, activating the TNF-α-induced apoptosis and NF-κB pathway, which is closely associated with NPC development.499 The EBV-encoded transcriptional activator ZTA upregulates inflammatory cytokines GM-CSF, IL-8, and GRO-α, which suppresses monocyte chemotaxis and induces tumor-associated macrophage phenotypes.500 Additionally, EBV activates the ATR and Rad-3-related pathway, promoting PPAR-δ and inhibiting c-Jun and p-JNK expression. This leads to increased expression of inflammatory factors VEGF and CCL22, polarizing TAMs toward an M2 phenotype and accelerating malignant transformation in NPE cells.501 EBV-encoded BHRF1 protein localizes to mitochondria, causing mitochondrial membrane permeabilization transition (MMPT), increasing ROS production, and enhancing mitochondrial activation, thereby promoting tumorigenesis (Fig. 7g).502
Vascular and extracellular matrix remodeling
EBV supports NPC cell progression through the enrichment of vascular networks.316 LMP2A protein activates the PI3K/AKT/mTOR/HIF-1α signaling promoting the growth of tumor vascular networks.373 Moreover, EBV stimulates vasculogenic mimicry (VM) through various pathways,376,503 such as activation of the PI3K/AKT/mTOR/HIF-1α pathway.376,504 Interaction between LMP1 and TNF-α activates the EGFR/Src/ERK/cortactin and Cdc42/N-WASP signaling axis, mobilizing invadopodia to overcome matrix barriers.505 Additionally, exosomes from EBV-infected cells are rich in HMGA2, which upregulates Snail expression, promoting endothelial-to-mesenchymal transition (EndMT) in endothelial cells, thereby enhancing tumor metastasis (Fig. 7h).506
Metabolic reprogramming
EBV is responsible for the promotion of NPC through the increase in glycolysis. Initially, the LMP1 protein can upregulate HIF-1α expression,507 which in turn activates the mTORC1/NF-κB signaling pathway. This then leads to the upregulation of GLUT1 transcription, which enhances glucose uptake in epithelial cells.508 Additionally, LMP1 enhances HK2 activity and upregulates the expression of glycolytic enzymes, such as pyruvate dehydrogenase kinase 1 (PDK1) and pyruvate kinase M2 (PKM2).509 Besides LMP1, EBV-miR-BART1-5P also activates mTOR and HIF-1α, further increasing glycolysis in host cells.510 Given that the energy generated from glycolysis is insufficient to meet the demands of rapidly dividing cancer cells, EBV also increases glutamine uptake to sustain growth and metabolic needs,511 upregulating mitochondrial glutamate dehydrogenase enzymes GLUD1 and GLUD2 to catalyze the conversion of glutamine into α-KG.512 EBV miRNAs and proteins stimulate increased lipase activity in adipocytes and enhance the AMPK metabolic pathway, promoting lipolysis to provide energy (Fig. 7i).513
Kaposi sarcoma (KS)
Kaposi sarcoma (KS) is characterized by abnormal angiogenesis, inflammation, and proliferation, with Kaposi sarcoma-associated herpesvirus (KSHV) being the primary cause of KS development.514 In host cells, KSHV typically persists as a latent infection, encoding latent-associated nuclear antigen (LANA), vCyclin, vFLIP, and KS miRNAs during latency. These transcripts stimulate cell growth and proliferation, promote angiogenesis, and activate inflammatory signals.515 Additionally, KSHV can transition to lytic replication upon activation by various biological signals, producing lytic replication products, such as K1, viral interferon regulatory factors (VIRFs), vIL-6, vCCLs, vGPCR, and K15, which regulate host cell signaling pathways and accelerate KS progression (Fig. 8a).

Oncogenic mechanisms of KSHV in Kaposi sarcoma. a Progression of KS caused by KSHV. b KSHV infection induces intracellular DNA damage, mitotic disruption, and chromosomal structural abnormalities. c KSHV maintains telomere length in host cells by reducing TRF1 degradation. d KSHV enhances cell proliferation via NF-κB and Notch signaling, while vBcl-2 and vPK inhibit apoptosis. e KSHV facilitates immune evasion by inhibiting cGAS activity, suppressing IFN-I transcription, downregulating Nectin-2, CD155, and I-selectin ligands, and fostering a Treg and TAM-rich immunosuppressive microenvironment. f KSHV induces chronic inflammation and oxidative stress via promoting inflammatory factor release, inhibiting GR signals, and upregulating HO-1 transcription. g KSHV promotes angiogenesis and metastasis by increasing angiogenic factors and matrix metalloproteinases (MMPs). h KSHV infection augments aerobic glycolysis, pyrimidine synthesis, amino acid metabolism, and lipid generation in host cells. Blue text: Components of KSHV; Black text: Components of host cell; Direct oncogenesis: b–d; Indirect oncogenesis: e–h. This figure was created with BioRender.com
Direct oncogenesis: leading to the emergence of virus-related tumors
Genome instability and telomere maintenance
KSHV-encoded viral proteins induce the DDR and chromosomal instability, leading to genomic instability.516 The Np9 protein directly causes DDR or indirectly induces DDR by upregulating LANA and c-Myc.517 LANA protein interferes with the localization of Sgo1 to the centromere, leading to the degradation of Securin and Cyclin B1, which significantly increases chromosomal instability.518 vFLIP activates the NF-κB pathway, enhancing L1 retrotransposition, with ICAM-1 involved in regulating this process, potentially accelerating the development of KS (Fig. 8b).516
To maintain unlimited proliferative capacity, cancer cells must activate telomere maintenance mechanisms to prevent telomere shortening.403,519 KSHV-transformed cells utilize two mechanisms to maintain telomere length. Recent studies have also found that KSHV induces alternative lengthening of telomeres activity (ATL) in host cells, enhancing cell proliferation, but the specific mechanism is not yet clear.109,520 Collectively, these processes highlight KSHV's dual role in promoting genomic instability and sustaining telomere integrity to drive Kaposi sarcoma pathogenesis (Fig. 8c).
Interfering cell proliferation and apoptosis
KSHV-miR-K4-5p and KSHV-miR-K1-5p activate the mTORC1 pathway by downregulating cytosolic arginine sensor for mTORC1 subunits 1 and 2, thereby promoting cell proliferation and malignant transformation.521 Moreover, KSHV miRNAs and vFLIP upregulate forkhead box protein O1 (FoxO1) by activating the NF-κB pathway, further promoting cell proliferation.522 KSHV infection also increases the expression of Notch signaling pathway proteins, including Notch1, NICD, RBP-Jĸ, and Hes1, further promoting cell proliferation.523 Additionally, vIRF1 upregulates SPAG9, enhancing the interaction between MKK4 and JNK1/2, leading to increased VEGFA expression and cell proliferation.524 KSHV regulates the apoptosis signaling pathways through various viral-encoded proteins to enhance the survival of infected cells. Specifically, ORF 16 encodes vBcl-2, a viral homolog of the human proto-oncogene Bcl-2, with similar anti-apoptotic functions.525 K15 interacts with the anti-apoptotic protein HAX1 and induces the expression of multiple anti-apoptotic genes, providing a survival advantage.526 Additionally, KSHV ORF36 encodes viral protein kinase (vPK), which binds to AKT1 through its PH domain, upregulating AKT1 activity and inhibiting apoptosis, thus conferring a survival advantage to endothelial cells (Fig. 8d).523
Indirect oncogenesis: promoting the progression of virus-related tumors
Immunosuppression
KSHV suppresses the innate immune responses through multiple mechanisms. The KicGAS protein, produced by ORF52, directly inhibits the enzymatic activity of cGAS, interfering with DNA-induced phase separation and thereby suppressing the host’s antimicrobial and antitumor responses.527 Additionally, KSHV encodes ubiquitin ligases K3 and K5, which reduce the expression of antigen-presenting molecules and NK cell recognition ligands in host cells,528 including the downregulation of Nectin-2, CD155, and I-selectin.529 The evasion of adaptive immunity by KSHV principally hinges on the aberrant secretion of cytokines,530 such as those that induce a hyperinflammatory state and increase the infiltration of tumor-associated macrophages.531 In addition, the majority of KS patients are co-infected with HIV and KSHV. The immunodeficiency resulting from HIV constitutes a significant backdrop for the occurrence of KS (Fig. 8e).532
Chronic inflammation stimulation
Hyperinflammation is a hallmark of KS.533 KSHV infection drives the abnormal activation of various pro-inflammatory pathways. LANA can increase the activation of TLR4 and its adapters, activating the STAT3 pathway, which is considered a key viral protein responsible for the sustained release of pro-inflammatory factors IL-6, IL-1β, and IL-18.534 Additionally, elevated vIL-6 enhances the recruitment of BRD4 and the persistent binding of NF-κB p65, leading to the increased release of inflammatory factors.535 KSHV also encodes ORF45, which dissociates the Linker1-UPA complex, releasing the C-terminal domain of hNLRP1, thereby activating the NLRP1 inflammasome and promoting the maturation and release of IL-1β and IL-18, exacerbating inflammation.536,537 Studies have shown that KSHV miRNAs and vFLIP can activate the NF-κB pathway and upregulate FoxO1, accelerating the clearance of ROS within cells.522 Additionally, KSHV-miR-K12-11 targets BACH1 mRNA, reducing its translation, thereby increasing the transcription of the antioxidant enzyme heme oxygenase-1 and activating the NRF2-dependent antioxidant stress pathway (Fig. 8f).538
Inducing angiogenesis
KS is a highly angiogenic endothelial cell tumor, and KSHV-induced angiogenesis plays a crucial role in KS development.539 KSHV primarily promotes angiogenesis by upregulating HIF, VEGF, and their receptors, thereby activating angiogenesis-related pathways.540 KSHV encoded LANA,541 vIRF3,524,542 vIL-6,535,543, and vGPCR increase angiogenic factors,544 including VEGF, PDGF, TGF-α, TGF-β, ANGPT-2, and ANGPTL-4, in an mTOR-dependent manner. KSHV-encoded ORF57 can upregulate HIF-2α, stabilize the eIF4E2 translation initiation complex, and activate PDGFRA and secrete VEGF in a non-mTOR-dependent manner.544 Abnormal angiogenesis is often accompanied by ECM remodeling, with both processes complementing each other. The KSHV genome encodes 25 mature miRNAs to varying degrees, which have been shown to increase the expression of MMP1, MMP13, VEGFA, and VEGFR2 transcripts.541,545 In summary, KSHV primarily promotes KS growth and metastasis by enhancing angiogenic factors and MMP-mediated ECM remodeling.
KSHV has a neurotropic tendency. It can infect neuronal cells and promote the proliferation of neuronal cells through the Notch and Hippo signaling pathways.546,547 These infected neurons can spontaneously switch between the latent and lytic phases of infection and play an important role in regulating host genes related to tumorigenesis.547 NGS and bioinformatics analysis also found that KSHV infection leads to the expression of multiple neuronal and neuroendocrine genes in epithelial cells, thus providing survival advantages and immune evasion pathways for host cells.548 However, the relationship between these abnormal neuronal cells and neuroendocrine factors in the development of KS still requires further investigation.
HIV infection can also facilitate the angiogenesis induced by KSHV. HIV Tat can directly degrade IκBα or inhibit IκBα activity by upregulating hsa-miR-891a-5p, thereby activating the NF-κB/VEGF pathway, which synergizes with KSHV K1-induced angiogenesis.549 Similarly, the HIV-Nef protein can directly downregulate PTEN or inhibit PTEN activity, thereby activating the PI3K/AKT/mTOR pathway, which synergizes with KSHV K1 protein to promote angiogenesis (Fig. 8g).549
Metabolic reprogramming
The metabolic reprogramming of host cells driven by KSHV has been demonstrated to play a role as a catalyst in the malignant transformation process.550 KSHV encodes vGPCR and KSHV miRNAs that prevents the degradation of HIF-1α and activates HIF-1α.551 Recent studies have shown that KSHV vCyclin binds and hijacks cyclin-dependent kinase CDK6, phosphorylating Ser-1900 on CAD, thereby activating pyrimidine synthesis and glycolytic reprogramming.552 KSHV infection also increases non-essential amino acid metabolites, with in vitro 3D culture further confirming that this change is driven by the activation of PYCR enzyme.553 This increase in amino acid catabolism is a crucial pathway for energy replenishment under glucose metabolism restriction. In addition to enhancing aerobic glycolysis and amino acid catabolism, KSHV also enhances lipogenesis and peroxisome-mediated lipid β-oxidation, though the specific mechanisms require further research.554 Studies have found that LPS of Escherichiacoli can promote KSHV-induced cell transformation and tumorigenesis in a KS-like mouse model.546 Oral involvement is one of the common clinical manifestations of this KS. A cross-sectional study found that compared with individuals without oral KS, the α-diversity and richness of the oral microbiota in HIV/KSHV co-infected individuals with oral KS were significantly reduced, specifically manifested as an increase in the Firmicutes and Actinobacteria phyla and a decrease in the Proteobacteria phylum.555 Among them, Firmicutes and its metabolites may promote KS tumorigenesis by inducing an inflammatory response and reactivating KSHV.555 Another study has shown that Staphylococcus aureus in the oral cavity can promote KSHV lytic reactivation by upregulating ROS and inhibiting the cyclinD1-Dicer-viral microRNA axis, leading to KS tumorigenesis.556 These studies explain why HIV/KSHV-infected individuals who suffer from opportunistic bacterial infections and metabolic complications are prone to developing KS (Fig. 8h).557
Lymphoma
EBV is implicated in the pathogenesis of several lymphomas, including extranodal natural killer/T-cell lymphoma (NKTCL), Burkitt lymphoma (BL), and Hodgkin lymphoma (HL).558 (1) NKTCL frequently occurs in extranodal sites, particularly in the nasopharyngeal region, skin, and gastrointestinal tract.559 EBV can be detected in nearly all NKTCL cells, with most cases exhibiting Latency II, characterized by the expression of EBNA1, BART, LMP1, LMP2A, and LMP2B.560,561 (2) BL has three subtypes: endemic (eBL), sporadic (sBL), and immunodeficiency-related form (idBL).562 The association with EBV varies among these subtypes; almost all eBL tumors are EBV-positive, while about 15-30% of sBL and 25-40% of idBL cases harbor EBV.32,563,564 BL is associated with EBV Latency I and II.41 (3) HL is divided into classical HL (cHL) and nodular lymphocyte-predominant HL (NLPHL).565 In cHL, tumor cells are known as Hodgkin/Reed Sternberg (HRS) cells, whereas in NLPHL, they are referred to as lymphocyte-predominant (LP) cells.566,567,568 In Western countries, approximately 30% of cHL cases show latent EBV infection in HRS cells. The three EBV-encoded proteins expressed in infected HRS cells—EBNA1, LMP1, and LMP2A—as well as viral miRNAs, are implicated in the pathogenesis of cHL.569,570 Based on the viral gene expression patterns, EBV establishes one of the three latency patterns. In latency type I, EBNA-1 and two small non-coding EBERs are expressed, and it is generally considered to be associated with BL.571 In latency type II, the virus expresses EBNA-1, EBER, and latent membrane proteins (LMP), namely LMP-1, LMP-2A, and LMP-2B.571 This latency pattern is related to Hodgkin lymphoma. Finally, latency type III leads to the expression of the entire EBV repertoire, including EBNA, EBER, and LMP, and is associated with post-transplant lymphoproliferative disorders (PTLD) and other immunocompromised states.571,572 EBV-positive DLBCL is associated with latency type II and type III patterns.573 Lytic EBV replication occurs after plasma cells differentiate from this persistent pool and is associated with PEL.574 HBV and HCV infections can not only cause liver complications, such as cirrhosis and HCC, but also extrahepatic complications including B-NHL.575 And lymphoma is the most common malignant tumor in PLWH.576,577 The risk of developing NHL in PLWH is 23 times higher than in the general population, and approximately 7% of NHL cases can be attributed to HIV infection (Fig. 9a).577

Oncogenic mechanisms of EBV in lymphoma. a Progression of lymphoma caused by EBV. b EBV infection induces high CpG island methylation, frequent gene mutations, and increased DNA damage. c LMP1 induces telomere instability through the downregulation of TRF2. d EBV activates NF-κB, AKT/PI3K, and JNK signaling pathways to enhance cell proliferation. e EBV inhibits cell apoptosis by increasing IL-10 secretion, activating NF-κB and JAK-STAT pathways, and suppressing IFN-related apoptotic signaling pathways. f EBV promotes immune evasion by reducing antigen presentation and upregulating PD-L1, contributing to an immunosuppressive microenvironment through Treg cell recruitment, M2 macrophage infiltration, and enhanced inhibitory receptor expression on CD8+ T cells, alongside increased oxidative stress. g LMP1 inhibits ROS-induced DNA damage by activating the PGC1β/HKDC1/OGG1 signaling pathway. h EBV induces metabolic reprogramming by upregulating intracellular MCT1/4 expression and promoting lipid synthesis. Red text: Components of EBV; Black text: Components and biological processes of the host cell; Direct oncogenesis: b–e; Indirect oncogenesis: g, h. This figure was created with BioRender.com
Direct oncogenesis: leading to the emergence of virus-related tumors
Oncogene activation and tumor suppressor gene inhibition
EBV plays multiple roles in the development of B-cell lymphomas. EBNA2 promotes B-cell immortalization and acts as a transcriptional activator that directly upregulates the oncogene c-Myc.578 Almost all Burkitt lymphomas carry chromosomal translocations related to the c-Myc oncogene.579 Recent studies have found that latent EBV infection, in synergy with c-Myc overexpression, can induce BL-like human B-cell lymphoma in mouse models.580 Additionally, Ikeda et al. observed that BL expressing LMP1 exhibits a lower frequency of p53 mutations, suggesting that LMP1 may reduce the incidence of p53 mutations in human BL, possibly indicating a novel pathogenic role.581 Furthermore, EBV promotes B-cell lymphoma by downregulating a novel p53-responsive lncRNA, IGFBP7-AS1.582
Genome instability
The interaction between EBV and the host significantly influences the development of B-cell lymphomas, particularly through the role of epigenetic markers in gene regulation.583,584 In EBV-related BL, the host genome typically exhibits global hypomethylation, although specific sites, particularly within CpG islands, may be hypermethylated.585 LMP1 can induce the aberrant activation of multiple signaling pathways in HRS cells, such as NF-κB and JAK/STAT, which are also activated in specific cell mutations.586,587 Additionally, in EBV-related cHL, negative regulators of the NF-κB pathway, such as TNFAIP3 and NFKBIA, are frequently mutated.588,589 In NKTCL, EBNA3B exhibits a high density of nonsynonymous mutations, potentially affecting immune escape mechanisms related to HLA-restricted epitopes, thereby promoting NKTCL development and progression.589,590 LMP1 induces telomere instability by downregulating telomere repeat binding factor 2 (TRF2), promoting the formation of HRS cells (Fig. 9b, c).586
Interfering cell proliferation and cell cycle
EBV is critical in developing B-cell lymphomas through its oncoproteins and ncRNAs. The proliferative activity of LMP1, LMP2A, and EBNA2, along with inhibiting terminal differentiation by EBNA3A, drives the progression of EBV-mediated B-cell lymphomas.591 EBNA2 disrupts c-Myc expression, increasing cell proliferation.580 Cells co-expressing LMP1 and EBNA2 can develop lymphomas in NOG mice, demonstrating the importance of EBV oncoproteins in B-cell transformation.592 Additionally, LMP1 can activate the oncogenic JNK signaling pathway through IkB kinase 2 (IKK2) and its cofactor NEMO, with p62 playing a pivotal role in EBV LMP1-mediated signaling; p62 deficiency enhances apoptosis and inhibits the proliferation of lymphoblastoid cell lines (LCLs).593 LMP2A can synergize with c-Myc and mutant Cyclin D3 to promote B-cell proliferation and survival, suppressing c-Myc-induced apoptosis and advancing the cell cycle by inhibiting RB1 function, thus facilitating B-cell malignant transformation.594 EBNA1 is crucial for the persistence of the viral genome during latency, preventing the clearance of infected cells by the immune system, promoting cell proliferation and growth, and protecting infected cells from apoptosis.595 LMP1 can transform B cells in vitro, significantly enhancing malignant traits by activating the NF-κB, PI3K/AKT/mTOR, and RAS/MAPK signaling pathways.470 The small EBV-encoded RNA EBER is implicated in the malignant phenotype of BL and is associated with resistance to apoptosis in BL cells, as well as increased autocrine expression of the growth factor IL-10.596 EBV synergizes with c-Myc to induce BL-like lymphomas, where c-Myc promotes the development of BL by inhibiting the expression of LMP1.580 Additionally, EBV-encoded proteins can prevent the differentiation of activated B cells, contributing to mutations that drive tumor transformation. LMP1, for example, rescues germinal center B cells lacking B-cell receptors (BCR) from apoptosis and plays a crucial role in the formation of HRS cells, potentially inducing B-cell survival and proliferation signaling through NF-κB, JAK-STAT, c-Myc, and Cyclin A pathways.597
Furthermore, EBV ncRNAs also significantly impact the development of B-cell lymphomas. Several EBV miRNAs are highly expressed in BL, particularly EBV-miR-BART7, EBV-miR-BART10, EBV-miR-BART11-3p, EBV-miR-BART6-3p, and EBV-miR-BART17-5p.598,599 EBER2 promotes the expansion of latently infected B cells by inhibiting their terminal differentiation.600 Additionally, EBER2 accelerates B-cell proliferation by enhancing the expression of the deubiquitinase UCHL1 and increasing the levels of Aurora kinase and Cyclin B1, an effect also observed in BL.601 Various non-coding RNA transcripts in infected cells help activate proliferation-promoting signaling pathways (e.g., BPLF1), inhibit apoptosis (e.g., BART), and regulate mechanisms that shield infected cells from immune system attacks (e.g., BORF2, BNLF2A, BDLF3, vIL-10, BZLF2).470,602 In EBV-positive BL, the inhibition of apoptosis is at least partially mediated by viral EBV-BART miRNAs, which reduce the expression of apoptosis-inducing proteins like caspase 3 (Fig. 9d, e).603,604
Indirect oncogenesis: promoting the progression of virus-related tumors
Immunosuppression
In EBV-positive NHL, there is a higher infiltration of CD8+ T cells, along with increased expression of Tregs, M2 macrophages, and immunosuppressive molecules, creating a more tolerant immune environment compared to EBV-negative cases.605 In EBV-positive cHL, the TME is characterized by a high density of CD8+ T cells expressing inhibitory receptors like PD-1 and TIGIT, along with specific macrophage subsets and an in situ inflammatory molecular profile associated with TCR and BCR signaling pathways.606 Li et al. used single-cell analysis to demonstrate the significance of LMP1+ malignant NK cells in NKTCL, showing that these cells secrete dipeptidyl peptidase-4 (DPP4) and regulate chemokines, affecting immune cell chemotaxis and exhibiting immunosuppressive effects on T cells, thereby promoting tumor development and progression.607 EBV latent genes also contribute to the unique TME of cHL. LMP1 recruits TME cells by inducing the expression of various chemokines and cytokines in infected HRS cells.608 EBNA1 has been found to increase the expression of the T-cell chemokine CCL20, facilitating the recruitment of Treg cells.609 EBV-encoded BART miRNAs promote lymphoma formation by regulating viral and cellular gene expression and promote lymphoma formation by regulating viral and cellular gene expression and impact innate and adaptive immunity.610 Specifically, EBV-miR-BART1 and EBV-miR-BART2 reduce antigen presentation, while EBV-miR-BART17 targets Transporter associated with Antigen Processing 2 (TAP2), which is involved in antigen processing and presentation.610,611,612
EBV infection induces PD-L1 expression in cHL, a process mediated by LMP1 and primarily involving the JAK/STAT and AP-1 signaling pathways.613,614 Blocking the PD-1/PD-L1 interaction can successfully inhibit the growth of EBV-induced lymphomas in mouse models, suggesting a possible interaction between EBV and the PD-1/PD-L1 pathway in tumor immunology.615 Furthermore, EBV infection in NKTCL may induce immune tolerance by upregulating PD-L1 expression through LMP1 (Fig. 9f).616,617 Although HIV does not directly cause NHL, due to the impaired immune function of PLWH, they are more susceptible to coinfection with other oncogenic viruses, such as EBV.618,619,620 In addition, HIV can directly participate in viral activity through immune suppression, immune dysregulation, and chronic antigen stimulation, playing a role in carcinogenesis and lymphoma621 H. pylori, is also thought to trigger lymphoma, which continuously stimulates antigen presentation, thereby inducing the expansion of B cells. In humans, this phenomenon is manifested as the overexpression of specific V genes.622 Although lymphocytes and microorganisms are typically separated by the epithelial barrier, bacteria, antigens, or metabolites can traverse the mucosal barrier via dendritic cells or M cells, which constantly sample the lumen.623
Chronic inflammation stimulation and oxidative stress
LMP1 is a potential driver of EBV-mediated lymphoma, functioning through the activation of the PGC1β/HKDC1/OGG1 signaling pathway. Additionally, LMP1 activates the SREBP1-mediated hexokinase domain component 1 (HKDC1) signaling pathway, leading to mitochondrial dysfunction, and forming a positive feedback loop of ROS bursts. This mechanism inhibits EBV replication and suppresses the growth of NKTCL via the LMP1/PGC1β signaling pathway (Fig. 9g).624
Metabolic reprogramming
EBV plays a crucial role in regulating B-cell metabolism and cell cycle progression through the interaction between EBNA2 and EBF1 during EBV infection.625 LMP1, a direct target gene of EBNA2, acts as a significant metabolic regulator in many EBV-related tumor cells by enhancing aerobic glycolysis.626 Additionally, EBV-infected B cells induce the expression of monocarboxylate transporter 1 (MCT1) and monocarboxylate transporter 4 (MCT4) through EBNA2 and LMP1, respectively, which are crucial for B-cell proliferation. Dual inhibition of MCT1/4 results in growth arrest of LCL cells and lactate accumulation, impacting oxygen consumption and glutathione levels, further revealing the metabolic characteristics of EBV-infected cells.627 LMP1 also drives metabolic reprogramming of B cells by inducing fatty acid synthase (FASN) and lipid droplet formation. Inhibiting lipogenesis effectively kills LMP1+ B cells and prevents EBV-induced B-cell immortalization, suggesting that targeting LMP1-induced lipogenesis could be an effective strategy for treating EBV-related tumors (Fig. 9h).628
Adult T-cell leukemia/lymphoma (ATLL)
HTLV-1 induces carcinogenesis through insertional mutagenesis and epigenetic modifications (Fig. 10).629 Therefore, high HTLV-1 proviral load (PVL) is a risk factor for HTLV-1-associated inflammation and ATLL.630 Besides, high PVL often closely relates to poor treatment efficacy and prognosis of ATLL.631

Oncogenic mechanisms of HTLV-1 in adult T-cell leukemia/lymphoma. a Progression of ATLL caused by HTLV-1. b HTLV-1 encoded proteins upregulate oncogenes and silence tumor suppressor genes via NF-κB-dependent and NF-κB-independent pathways, recruiting transcription factors to gene promoters. c The viral Tax protein increases genomic instability by promoting DNA damage and inhibiting DNA double-strand break repair. d HTLV-1 infection boosts cell proliferation through persistent activation of AKT/NF-κB and TGF-β/Smad pathways, and promotes YAP nuclear translocation. e HTLV-1 encoded proteins inhibit apoptosis by suppressing caspase signaling, activating the NF-κB pathway, and preventing FoxO3a nuclear translocation. f HTLV-1-infected host cells achieve immune escape by reducing Tax protein expression, generating Tax mutants, and suppressing Th1 cytokine production. g HTLV-1 mediates immune suppression by inducing inhibitory receptor expression on T cells and recruiting Treg cells. h HTLV-1 induces chronic inflammation by causing host cells to overproduce IL-10. i HTLV-1 infection enhances intracellular glycolysis, pyrimidine biosynthesis, and lipid synthesis. Fuchsia text: Components of HTLV-1; Black text: Components of host cell; Direct oncogenesis: b–e; Indirect oncogenesis: f–i. This figure was created with BioRender.com
The oncogenic effects of HTLV-1 primarily depend on two viral proteins: Tax and HBZ. After HTLV-1 proviral integration, HBZ is continuously expressed at a low level,632,633 while Tax is expressed intermittently in a burst manner. In the early stage of infection, Tax, as a transcriptional activator, can promote viral replication and the proliferation of infected cells, which is conducive to viral reactivation and de novo infection.634 Due to the high immunogenicity of Tax, CTLs exert selective pressure on Tax-positive cells. Moreover, the continuously expressed HBZ will inhibit Tax expression to evade immune surveillance, and eventually prompt the virus to enter the latent period.635,636 The long latent period provides ample time for the virus to interact with host cells (Fig. 10a).
Direct oncogenesis: leading to the emergence of virus-related tumors
Oncogene activation and tumor suppressor gene inhibition
HTLV-1 provirus integrates near proto-oncogenes, influencing their expression through cis-perturbation.637 Mechanistically, HTLV-1 recruits host transcription factors SRF and ELK-1 to nucleosome-free regions within the provirus DNA, promoting the expression of oncogenes near HTLV-1 integration sites.638
HTLV-1 integration can lead to mutations or functional inactivation of tumor suppressor genes, such as p53 and FBXW7, and the silencing of these tumor suppressor genes is one of HTLV-1’s key carcinogenic mechanisms.639,640 Moreover, Tax protein activation of the NF-κB signaling pathway also inhibits the expression of p53 and FBXW7.142,635,641,642 HBZ protein cooperates with CARD11, a protein required for T and B cell activation,643 and upregulates oncogenes like IRF4, BATF3, MYC, and E2F via non-canonical NF-κB pathways, inducing ATLL.644,645 In summary, HTLV-1 disrupts the balance between oncogene activation and tumor suppressor gene inhibition, leading to uncontrolled cell growth (Fig. 10b).
Genome instability
HTLV-1 infection causes oncogenic mutations, chromosomal rearrangements, and DNA damage, compromising the genomic stability of host cells. Studies have shown that HTLV-1 infection increases cell mutation rates and copy number variations.646 Additionally, Tax protein also downregulates RNase H1, causing excessive R-loop accumulation and inducing DSBs.647 Simultaneously, Tax triggers the activation of RNF8, thereby giving rise to Tax-speckle structures (TSS). These TSSs sequester DDR factors, disrupting DSBs repair and exacerbating genomic instability in host cells.648 The latest research further reveals that Tax coordinates transcriptional and alternative splicing events during NF-κB activation, upregulates intra-Topologically associating domain inter-contact remodeling of 3D chromatin structure in host cells, which lays a poor genetic foundation for the occurrence and development of HTLV-1-related diseases (Fig. 10c).649,650
Interfering cell proliferation and apoptosis
Tax and HBZ proteins play crucial roles in the proliferation and survival of T cells. Tax upregulates p65, which stabilizes YAP and promotes its nuclear translocation, leading to the expression of proliferation-related genes.634 Recent studies have also found that Tax induced hsa-miR-31 loss,651 resulting in sustained activation of the AKT/NF-κB oncogenic signaling pathway.652 In contrast, the regulatory effect of HBZ protein on T-cell proliferation exhibits a dual nature. HBZ can inhibit Tax-activated NK-κB signaling and suppress T-cell proliferation in vitro.653,654 However, in vivo experiments suggest that HBZ promotes malignant T-cell proliferation. For instance, HBZ binds to human APOBEC3G, activates the TGF-β/Smad signaling pathway, upregulates BATF3/IRF4 and MYC transcription, and promotes T-cell proliferation (Fig. 10d).655
Tax protein activates the NF-κB pathway, enhancing the expression of anti-apoptotic proteins such as, survivin, and BCL family molecules.656,657 Similarly, HBZ protein inhibits apoptosis by reducing HAX-1 protein ubiquitination and inhibiting caspase-9 activation.658 Additionally, HBZ interferes with the localization and function of transcriptional activator FoxO3a, reducing the expression of apoptotic genes Bim and its ligand gene FasL, further inhibiting apoptosis (Fig. 10e).659
Indirect oncogenesis: promoting the progression of virus-related tumors
Immunosuppression
HTLV-1 employs multiple strategies to downregulate the immune surveillance mechanism, thereby evading the attacks of the host immune system. Tax, as a viral protein with strong immunogenicity, can activate the classical NF-κB signaling pathway in CTLs, and then mediate cellular immunity.641 However, Tax is usually expressed in the early stage of infection or intermittently expressed in the late stage of infection, and this expression pattern reduces the specific recognition of it by CTLs.636 In addition, studies have shown that after HTLV-1 infection, host cells are more prone to 5'-LTR deletion,660 hypermethylation of viral promoter DNA,661 and Tax mutants that are not easily recognized by CTLs will be generated.655 These mechanisms work together to promote the immune evasion of HTLV-1. Different from Tax, HBZ not only selectively inhibits the classical NF-κB signaling pathway to achieve immune evasion,662 but also reduce the expression of Th1 cytokines, such as IFN-γ, IL-2, and TNF-α, thereby mediating the establishment of an inhibitory immune microenvironment (Fig. 10f).663
The transformation of T cells into ATLL cells induced by HTLV-1 is accompanied by an increase in immunosuppressive characteristics, such as the upregulation of CTLA4, LAG3, and MHCII.151 The HBZ protein can directly induce the expression of immunosuppressive receptors, such as Foxp3, CCR4, and TIGIT.664 Additionally, the Tax protein upregulates the secretion of the chemokine CCL22, which attracts and maintains a high-frequency circulation of CD4+ Foxp3+ CCR4+ Treg cells (Fig. 10g).665
Chronic inflammation stimulation
IL-10 is a key factor in conferring malignant proliferative abilities to HTLV-1-infected T cells. Studies have shown that the HBZ protein upregulates IL-10 levels by inducing the overexpression of TIGIT, Foxp3, and CCR4.666 Additionally, the persistent activation of interferons due to chronic HTLV-1 infection is another cause of IL-10 over-secretion.667,668 High levels of IL-10 promote STAT3 phosphorylation, increasing the expression of BIRC5, MYC, and IRF4, which contributes to the spontaneous growth and survival of T cells.669 The immunosuppressive microenvironment of HTLV-1 is also influenced by IL-10. Cells infected with HTLV-1 secrete IL-10, which can suppress host immune responses through paracrine signaling while supporting the proliferation of ATLL cells through autocrine signaling, achieving a dual effect.149,669 These studies suggest that the excessive secretion of IL-10 helps HTLV-1 infected cells evade host immune surveillance, thereby exacerbating the progression of ATLL (Fig. 10h).
Metabolic reprogramming
HTLV-1 hijacks glucose, lipid, and pyrimidine nucleotide metabolism, leading to the malignant transformation of T cells. The HBZ protein reduces EZH2 occupancy at the TAp73 promoter by binding to EZH2, while HBZ RNA activates TAp73 transcription. As TAp73 is upregulated, the transcription of lactate transporters MCT1 and MCT4 increases, enhancing glycolytic flux.150 In terms of pyrimidine nucleotide metabolism, UCK2 is a key regulator closely associated with cell proliferative capacity.670 The activation of the TCR-NF-κB signaling pathway by HTLV-1 leads to the accumulation of UCK2, thereby increasing pyrimidine biosynthesis and supporting the proliferation of transformed T cells (Fig. 10i).671
Treatment strategies in specific cancer types
Given that viral infection is a primary pathogenic factor in the development of virus-related malignancies, preventive measures such as vaccination are crucial. For example, the HBV vaccine can reduce the risk of HCC,672 while the HPV vaccine is effective in preventing CC.673 For individuals already infected with these viruses, rigorous antiviral therapy is crucial for reducing cancer risk and is associated with improved long-term survival in those with virus-related malignancies.674 Some anti-tumor agents may lead to viral reactivation, which can have severe repercussions, making vigilant monitoring for viral reactivation necessary.675 Despite the detrimental role of viruses in tumor development, they also present opportunities for targeting and destroying virus-infected cancer cells. Approaches such as therapeutic vaccines, T cell receptor-engineered T (TCR-T) cells, and chimeric antigen receptor T (CAR-T) cells, which target viral antigens, have shown promise in treating virus-related tumors.676,677 Additionally, intervening in virus-related cancer signaling pathways can effectively inhibit tumor growth and spread. Changes in tumor immunity due to viral infections offer various treatment strategies, including immune checkpoint inhibitors (ICIs), bifunctional fusion proteins, and the combination of ICIs and targeted therapy.678 For the treatment of virus-related malignancies, viral therapy is crucial. It is a double-edged sword, showing many advantages as well as facing numerous challenges. The main advantages are as follows: First, it has a preventive effect. Viral preventive vaccines can induce the production of neutralizing antibodies, reducing the risk of cancer occurrence from the source.679 Second, precise gene-targeted therapy holds the promise of precisely attacking the viral oncogenes in tumor cells, minimizing damage to normal cells.680 Third, some new immune therapy targets against viruses have been discovered, which can provide new directions for treatment.362 Fourth, combination therapy has obvious advantages. It can be combined with chemotherapy, targeted therapy, immunotherapy, etc. For example, when combined with CAR-T cell therapy, it can activate and enhance the killing activity of immune cells against tumors.681 However, the disadvantages cannot be ignored. There are differences in the immune responses of individuals to vaccines. Some people may not produce sufficient antibodies after receiving preventive vaccines, and therapeutic vaccines are still not mature.682 Gene-editing technology has potential risks. The combination therapy is complex, and the optimal dosage and sequence of drugs need to be further studied and determined.
HCC
HBV-related HCC
Chronic HBV infection is considered the main risk factor for the occurrence and development of HCC, with the lifetime risk of HCC in HBV carriers ranging from 10% to 25%.679,683 The treatment of HCC is characterized by multidisciplinary participation and the coexistence of various treatment methods, including surgical resection, liver transplantation, local ablation, targeted therapy (tyrosine kinase receptor inhibitors, multi-kinase inhibitors, VEGF monoclonal antibodies), immunotherapy (PD-1, PD-L1, CTLA4 monoclonal antibodies), vascular interventional therapy, radiotherapy, systemic therapies, and traditional Chinese medicine treatment.672,684 Clinically, most HCC patients are diagnosed at an advanced stage, having missed the opportunity for curative surgical treatment.685
Given the causes of HCC, early vaccination against HBV is crucial for preventing the occurrence of HCC.686 For patients with viral hepatitis, appropriate antiviral therapy can effectively inhibit viral replication, enhance virus-specific T-cell function, improve liver inflammation levels, and thus reduce the risk of HCC.687 Studies have shown that continuous antiviral treatment can reduce the risk of HCC by more than 50% in patients with chronic hepatitis B (CHB).688 In the current treatment strategies for HBV-related HCC, aside from routine vaccination for primary prevention and the utilization of antiviral drugs to inhibit viral replication and optimize the liver microenvironment, a rational combination of emerging therapies, such as targeted therapy, immunotherapy, and cell therapy, can intervene in the processes of tumor initiation, development, and metastasis via multiple biological pathways. This significantly enhances the comprehensive treatment efficacy of HBV-related HCC.
Virus vaccines and antiviral treatment
HBV vaccines are mainly divided into preventive vaccines and therapeutic vaccines. Common HBV vaccines are preventive and only prevent HBV infection in individuals who have not yet been infected.689 Therefore, a novel type of vaccine, the therapeutic HBV vaccine, has shown unique application value for HBV-infected individuals.690 A multicenter phase 2 clinical study showed that the DNA vaccine GS-4774 could enhance the activity of HBV-specific CD8+ T cells but failed to significantly reduce HBsAg levels. This suggests that GS-4774 alone is limited in efficacy and might be more effective when combined with other antiviral drugs to enhance antiviral immune responses.691 A lipid nanoparticle lipopeptide vaccine, εPA-44, demonstrated good safety in phase 2 clinical trials and continuously reduced HBsAg levels.692 The phase 2 clinical study results of the NASVAC vaccine suggest that it can induce anti-HBs and reduce HBsAg, with some patients achieving functional cure.693 Additionally, a therapeutic vaccine based on HBx,694 a dual-targeted HBV preS1 nanoparticle vaccine based on ferritin nanoparticles,695 and an mRNA vaccine encapsulated in lipid nanoparticles (LNP) have shown lasting potential in clearing HBV in mice in preclinical studies.696,697
Currently, antiviral drugs used to treat CHB include pegylated interferon (PEG-IFN) and oral Nucleos(t)ide analogs (NAs).698,699 Third-generation NAs, such as entecavir and tenofovir-based therapies (e.g., tenofovir disoproxil fumarate and tenofovir alafenamide) are recommended as the main treatment for CHB due to their high efficacy and low resistance.700 Studies have shown that, whether used alone or in combination, entecavir improves OS and PFS in patients compared to lamivudine.701,702 There is debate regarding whether tenofovir reduces the risk of HCC development in a secondary prevention setting compared to entecavir. Some studies indicate that patients receiving tenofovir after liver resection have a lower risk of HCC recurrence compared to those treated with entecavir.703 Recently, Chung et al. revealed that patients with HBV-related HCC who were treated with tenofovir therapy experienced a more favorable prognosis compared to those who received entecavir treatment.704 Despite long-term NAs therapy for HBV, less than 5% of patients have HBsAg loss after 12 months of treatment, highlighting the need for therapies capable of achieving functional cure.705 It has been reported that PD-1/PD-L1 can be used not only for cancer treatment but also for treating CHB. In HCC patients receiving ICI treatment, functional cure of HBV was observed, with a higher cumulative incidence of HBsAg loss compared to the control group.706 Additionally, GSK836/GSK3228836, an investigational antisense oligonucleotide drug, showed in its phase IIb clinical trial (B-Clear) that HBsAg clearance and HBV DNA negativity could be achieved following treatment, marking a significant advancement in the management of CHB.707 Lowering HBsAg levels can improve OS rates and reduce recurrence rates in HCC patients.708,709 Therefore, during HCC treatment, it is recommended to regularly monitor HBsAg levels and HBV DNA and carry out antiviral prevention to prevent fatal complications related to viral reactivation and enhance antitumor effects. In addition, HBsAg-specific antibodies can suppress plasma HBsAg and viral DNA loads in patients, but they are cumbersome and expensive to produce, and the virus will rebound once antibody treatment is stopped. A single-chain variable fragment (scFv) with high affinity for the HBV envelope protein antigenic loop (AGL) can be endocytosed by hepatocytes to inhibit viral particle secretion.710,711 Chimeric antigen receptor (CAR) forms and their Fc fusion forms (G12-scFv-Fc, G12-CAR-Fc) developed on this basis can more strongly inhibit serum HBsAg levels in HBV mouse models.712 Therefore, Fc-linked G12-scFv and G12-CAR may become new methods for reducing circulating HBsAg levels.
Targeting tumor cell signaling
HBV-associated proteins induce various intracellular signaling changes in tumor cells, and multi-targeted combination therapy is an important direction for the treatment of viral HCC. Sorafenib, a multi-kinase inhibitor, was the first targeted drug approved for HCC. Two large, randomized, controlled, international multicenter phase III clinical trials have demonstrated that sorafenib can delay tumor progression and extend the survival of patients with advanced HCC.713,714 Compared to patients with HBV-related HCC, patients with HCV-related HCC benefit more from sorafenib treatment.715 Another multi-kinase inhibitor, regorafenib, may be more beneficial for HBV-related HCC patients.716 It has been confirmed that TGF-β variants in liver diseases are closely related to viral hepatitis and HCC. Galunisertib, a specific inhibitor of TGF-β, achieves its antitumor effects by specifically inhibiting HCC growth and migration.717 Some studies suggest that Galunisertib does not show synergistic effects when combined with ramucirumab, an anti-VEGF receptor 2 antibody, for treating HCC,717 but combining it with sorafenib shows prolonged OS.718 Additionally, GPC3 is an important diagnostic marker for HCC, and its expression level in virus-related HCC is higher than in non-viral HCC,719 indicating the potential application of GPC3-targeted therapy in virus-related HCC. A phase I clinical trial demonstrated the safety and immunogenicity of a DNA vaccine encoding GPC3 in HCC.720
Immunotherapy
ICIs play a crucial role in the treatment of HCC. Common ICIs include PD-1 monoclonal antibodies (Nivolumab, Pembrolizumab, Sintilimab, Camrelizumab, Tislelizumab, Toripalimab, Penpulimab); PD-L1 monoclonal antibodies (Tezolizumab, Durvalumab, Envafolimab); and CTLA-4 monoclonal antibodies (Ipilimumab, Tremelimumab). It is worth noting that the strategy of combining immunotherapy with HCC treatment may provide better survival benefits in the HBV-related population,721,722 possibly due to the dual antiviral and antitumor effects of ICIs.723 However, studies have reported that caution is needed regarding viral reactivation during chemotherapy and ICIs treatment of virus-related HCC. Viral reactivation may lead to liver function deterioration. In a study, HBV reactivation occurred in HBsAg-positive HCC patients receiving anti-PD-1 or anti-PD-L1 immunotherapy,724 while no HBV reactivation or HBV-related liver damage occurred in patients who continued antiviral treatment.725 Moreover, combining IFN-α with anti-PD-1 treatment can improve the response rate in patients with unresectable HCC,726 highlighting the importance of combining immunotherapy with antiviral treatment.
Cell therapy
HBV-specific CAR-T and TCR-T cells targeting the HBV surface protein are two promising therapeutic approaches for virus-related HCC. These approaches involve genetically engineered T cells expressing classical T cell receptors or chimeric antigen receptors that specifically target HBV antigen.727 Preclinical studies have shown that CAR-T cells targeting HBV surface proteins exhibit potential anti-HCC activity, particularly CD39+ HBV-CAR-T cells.728 However, there are two major challenges in the clinical application of HBV-CAR-T cells. First, the high concentration of HBsAg in the serum of HBV-positive patients can directly bind to HBV-CAR-T cells, interfering with their ability to target cells. Additionally, HBV-CAR-T cells cannot recognize HCC cells with integrated fragments of HBV DNA.676 Therefore, there is still a long way to go before virus-specific CAR-T therapy can be effectively used in HCC treatment.
Wang and colleagues conducted a phase I clinical trial on HBV-TCR-T therapy targeting HBsAg in patients with advanced HBV-related HCC who had relapsed or failed previous first-line systemic treatments. The results demonstrated that the adoptive transfer of HBV-TCR-T cells is generally safe and well-tolerated, with specific targeting effects.729 SCG101 is an autologous, HBV-specific T-cell product expressing a TCR that recognizes an envelope-derived peptide (S20-28) on HLA-A2. Recent studies have shown that SCG101 exhibits significant antiviral and antitumor dual activity in treating patients with advanced HBV-related HCC, significantly extending patient survival.730 Due to the limited effectiveness of TCR-T in solid tumors, it is essential to develop next-generation HBV-TCR-T products, such as TCR-T cells that express IL-21R to improve their activity and survival.731 Additionally, ongoing investigations are exploring the combination of HBV-TCR-T cells with kinase inhibitors and/or PD-1 inhibitors for treating patients with advanced HCC (Table 3). This could provide more effective treatment options for HCC patients. Continued research efforts aim to identify more effective immune targets and optimize TCR-T transfection methods. TCR-T therapy promises to be a key player in the treatment of cancer, infectious diseases, and autoimmune disorders.
HCV-related HCC
Chronic HCV infection is a well-established risk factor for HCC, increasing the risk by 10–20 times.679 Unlike HBV infection, HCV can be cured.732 Following HCV clearance, overall mortality and the risk of developing HCC decrease significantly.733 Currently, no effective HCV vaccine is available.734 Besides vaccination and antiviral therapy, targeted therapy, immunotherapy, and CAR-T cell therapy are actively being explored for HCV-related HCC.
Virus vaccines and antiviral treatment
Currently, HCV vaccines are still under clinical investigation, and vector vaccines targeting T cells and heterodimeric E1-E2 vaccines targeting B cells have not shown promising results.735,736,737,738 Han et al. developed a DNA vaccine, GL-6150, and preliminary results showed that GRS-6150 could reduce the frequency of Treg cells and enhance HCV-specific T cell responses,124 potentially preventing reinfection in patients cured by DAAs.
The main strategy for reducing HCC incidence is achieving SVR (sustained virologic response) through antiviral therapy.739 In recent years, HCV treatment has entered the DAAs era, with interferon-free regimens being the preferred choice for HCV antiviral therapy. A systematic review shows that DAA treatment significantly reduces the risk of developing HCC compared to untreated individuals.740 Among chronic HCV patients who have never experienced HCC, the incidence of HCC after achieving SVR through treatment is 1.3%. However, among patients with a history of HCC, the recurrence rate remains as high as 29.6% after achieving SVR with DAAs.741 Recently, a 15-year follow-up study of Korean HCV patients further demonstrated that the use of DAAs is significantly associated with a reduced risk of liver fibrosis and HCC.742 Therefore, HCV-infected patients, regardless of their history of HCC, should receive DAA treatment early on to achieve SVR as soon as possible. Bourlière et al. reported in an RCT that sofosbuvir/ velpatasvir/voxilaprevir (SOF/VEL/VOX) achieved an SVR rate of 98% in patients who previously failed DAAs therapy.743 Thus, SOF/VEL/VOX monotherapy is the preferred retreatment strategy for those who experienced a previous DAA treatment failure.16
Targeting tumor cell signaling
For HCV-related HCC patients, sorafenib treatment slows tumor growth, alleviates liver function deterioration, and thus extends survival.744 An exploratory subgroup analysis of the SHARP trial indicated that HBV-negative and HCV-positive patients had increased OS with sorafenib treatment compared to placebo.745 This was confirmed by a pooled analysis of two randomized trials, suggesting that sorafenib may be more effective in HCV-related HCC patients.746 Additionally, an unsupervised transcriptome analysis conducted by Boyault et al. identified HCC subgroups with distinct genetic and molecular characteristics.747 Specifically, RAF-1,8 upregulation induced by HCV can be inhibited by sorafenib, which may contribute to the delay in HCV-related HCC progression mediated by sorafenib. Moreover, preclinical data indicate that sorafenib directly inhibits HCV viral replication,748 further supporting its efficacy in HCV-related HCC.
Immunotherapy
Research has shown that persistent HCV viremia leads to sustained upregulation of PD-1 and CTLA-4.749 To our knowledge, many previous studies on PD-1 inhibitors excluded patients with HCV infection. In 2013, a phase I clinical trial evaluated the efficacy of the CTLA-4 checkpoint inhibitor tremelimumab in a small cohort of advanced HCC patients with HCV infection. The trial reported manageable safety and preliminary evidence of antitumor and antiviral activity.750 However, evidence supporting the use of ICIs in treating advanced HCC patients with concurrent viral infections is still limited. El-Khoueiry et al. evaluated the safety and clinical benefits of nivolumab in the CheckMate 040 trial across multiple HCC etiologies, including HCV-infected patients. The results showed that disease control was achieved in 66% of HCV-related HCC patients, with a 6-month OS of 85%. However, nivolumab’s antiviral activity was limited, with some HCV-infected patients showing temporary reductions in HCV RNA,751 indicating that antiviral treatment might need to be combined. The controlled safety of nivolumab in this study lays the foundation for further research into PD-1 monoclonal antibodies as a treatment option for advanced HCV-related HCC patients.
Cell therapy
The first two CARs targeting HCV were designed based on broadly cross-reactive and cross-neutralizing human monoclonal antibodies targeting conserved epitopes on the HCV E2 glycoprotein (HCV/E2). Anti-HCV CAR-T cells have demonstrated good antiviral activity and the ability to lyse HCV/E2-transfected and HCV-infected target cells.752 This study suggests that the CAR-T cell may also apply to treating HCV. Additionally, HCV/E2 is the primary target of the host immune response and is prone to mutations.753 Therefore, identifying other conserved and essential antigens is crucial.
Given that viral infection is a primary pathogenic factor in the development of virus-related malignancies, preventive measures such as vaccination are crucial. For example, the HBV vaccine can reduce the risk of HCC,672 while the HPV vaccine is effective in preventing CC.673 For individuals already infected with these viruses, rigorous antiviral therapy is crucial for reducing cancer risk and is associated with improved long-term survival in those with virus-related malignancies.674 Some anti-tumor agents may lead to viral reactivation, which can have severe repercussions, making vigilant monitoring for viral reactivation necessary.675 Despite the detrimental role of viruses in tumor development, they also present opportunities for targeting and destroying virus-infected cancer cells. Approaches like therapeutic vaccines, T cell receptor-engineered T (TCR-T) cells, and chimeric antigen receptor T (CAR-T) cells, which target viral antigens, have shown promise in treating virus-related tumors.676,677
Gastric cancer
EBVaGC is a specific subtype of gastric cancer, making up about 10% of all cases. Current gastric cancer treatment guidelines do not distinguish between EBV-positive and EBV-negative cases.754 There is no specific treatment for EBVaGC. Most patients undergo surgical resection, similar to other gastric cancers, but postoperative chemotherapy lacks targeted drugs.755 Early-stage EBVaGC typically shows a lower rate of lymph node metastasis and forms well-defined nodular lesions in the submucosa. It has less fibrosis than non-EBVaGC, which may allow for smaller surgical margins and the possibility of endoscopic electrocoagulation or dissection.756 For advanced EBVaGC, new treatment options have emerged alongside traditional surgical methods, including vaccines, targeted therapies, immunotherapies, and cellular therapies.757
Since EBVaGC is primarily caused by EBV infection, preventing and treating EBV is essential. One of the most promising approaches is developing an EBV vaccine.758 Research on EBV vaccines focuses on both prophylactic and therapeutic aspects, and developing an effective EBV vaccine is crucial for preventing and treating EBVaGC. Clinically, EBV infections are typically treated with supportive care and specific disease treatments. Routine antiviral therapy is not recommended.759 EBVaGC is characterized by significant lymphocytic infiltration and the presence of key immune molecules and potential therapeutic targets in the TME. Clinically available PD-1/PD-L1 ICIs and immunotherapies are expected to be applied in EBVaGC.760,761 Enhancing the anti-tumor capability of immune cells by targeting tumor cell signaling to improve therapeutic efficacy has been a research focus in recent years.
Virus vaccines
To date, no prophylactic vaccine for EBV has been marketed worldwide. EBV needs several membrane proteins to enter the cytoplasm of epithelial cells, B cells, and NK/T cells. EBV infection of B cells requires five membrane proteins (gp350, gH, gL, gB, and gp42), while EBV infection of epithelial cells requires four membrane proteins (BMFR2, gH, gL, and gB). These proteins on EBV could be effective targets for preventive vaccines against EBV.762,763 Two clinical trials with recombinant gp350 vaccines have shown good efficacy in preventing infectious mononucleosis caused by EBV infection, with an average efficacy of 78.0%. However, they did not affect asymptomatic EBV infections.764,765 This may be because antibodies from gp350 do not protect epithelial cells from EBV infection. Further research has found that EBV glycoproteins gH/gL and gB together mediate EBV fusion to B cells or epithelial cells. Vaccination with these proteins can induce antibodies that broadly prevent EBV infection.766 Two reports indicate that serum-neutralizing antibody levels from the gp350 monomer are lower than those from three dimers and trimers of gH/gL, trimeric gB, or four gp350 polymers.767,768 This may be due to the critical role of gH/gL and gB in EBV fusion and entry into B cells or epithelial cells. Thus, EBV gH/gL and gB may be more effective targets for a preventive EBV vaccine than gp350 alone. Additionally, combining gp350 with gH/gL and gB could create a next-generation candidate vaccine with clinical potential.758
Tumor treatment vaccines represent a promising new approach to cancer therapy. These vaccines improve the presentation of tumor antigens and stimulate the body’s immune surveillance by supplying external tumor antigens.769 An ideal tumor vaccine should deliver high-quality specific tumor antigens to dendritic cells (DCs), which then activate cytotoxic T lymphocytes (CTLs) to exert anti-tumor effects. Currently, EBVaGC-related vaccines are still under research, with no commercial vaccines available for EBV. The first approved EBV-related therapeutic mRNA vaccine, “WGc-043 Injection,” developed by WisTGen Biopharma, has recently received dual approvals in China and the United States. It is also the first therapeutic mRNA vaccine approved for clinical trials worldwide. The EBV mRNA vaccine shows significant advantages in terms of immunogenicity, safety, and efficacy, bringing new hope and options for the treatment of EBV-related tumors. Additionally, related clinical trials are underway to investigate the efficacy of the mRNA vaccine in GC (NCT05714748) (Table 4), and the results are eagerly anticipated.
Targeting tumor cell signaling
Key targets for gastric cancer treatment include human epidermal growth factor receptor-2 (HER2), vascular endothelial growth factor receptor (VEGFR), Claudin 18.2, and microsatellite instability or mismatch repair deficiency (MSI/dMMR).770 For HER2-positive gastric cancer patients, adding trastuzumab to first-line or second-line chemotherapy improves survival rates, regardless of EBV infection status. Additionally, two novel antibody-drug conjugates (ADCs) targeting HER2, trastuzumab Deruxtecan and trastuzumab emtansine, are available for third-line and subsequent treatments of HER2-positive advanced gastric cancer. Furthermore, VEGF-targeting drugs like ramucirumab and apatinib are recommended for the second-line and third-line treatment of advanced or metastatic gastric cancer. Other targeted drugs for gastric cancer include the Claudin 18.2-targeting antibody Zolbetuximab, which has shown positive results in the GLOW and SPOTLIGHT studies, significantly prolonging PFS and OS. Currently, ongoing studies on EBV-related targets are providing new avenues for gastric cancer treatment.
Immunotherapy
Patients with HER-2-positive gastric cancer respond better to HER-2 targeted therapy and have more favorable prognoses than those with HER-2-negative gastric cancer.771 However, the majority of gastric cancers are HER-2 negative. Therefore, combining immunotherapy approaches is essential for improving outcomes in patients with advanced gastric cancer. The primary types of immunotherapy currently available are ICIs and cellular immunotherapy. Among these therapies, ICIs are the most widely used, particularly PD-1/PD-L1 monoclonal antibodies, which have garnered significant attention.772,773 Cellular immunotherapy is an emerging approach, including CAR-T therapy and NK cell therapy.
Around 80% of patients have PD-L1 expression in their tumor cells, while only 1.3% and 11.4% express PD-1 in tumor cells and infiltrating immune cells, respectively.774 These markers serve as significant therapeutic targets in cancer immunotherapy. EBVaGC exhibits substantial lymphocytic infiltration in the tumor immune microenvironment and responds well to immunotherapy,331 with high expression of PD-L1 and Ki67.775 In HER2-positive EBVaGC patients who are resistant to trastuzumab, clinical benefits can be achieved with PD-1 inhibitors used as monotherapy.776,777 Another study reported a 100% overall response rate (ORR) in EBVaGC patients treated with PD-1 inhibitors.778 Emerging data indicates that the ORR to PD-1 inhibitors in EBVaGC is approximately 25% higher compared to EBV-negative gastric cancer.779,780,781 Presently, PD-1 antibodies are widely used in the clinical treatment of gastric cancer. For first-line treatment of HER2-negative gastric cancer, pembrolizumab and nivolumab have shown survival benefits in patients with PD-L1 CPS ≥ 1.772,773,782,783 For HER2-positive gastric cancer, pembrolizumab has become the first anti-PD-1 drug approved for use in combination with trastuzumab and chemotherapy for first-line treatment of advanced patients.784,785,786,787 In 2020, Kim et al. validated the efficacy of immunotherapy in EBV-positive gastric cancer in a cohort of 300 gastric cancer patients, where 59.3% of the gastric cancer patients expressed PD-L1, associated with MSI and EBV-positive status.788 Moreover, pembrolizumab and nivolumab are recommended for second-line and beyond treatment in gastric cancer patients.785,789,790,791 While PD-1/PD-L1 antibodies have made remarkable advancements in gastric cancer treatment, their clinical efficacy as monotherapy is only observed in approximately 10% of patients. Thus, there is an urgent need for biomarkers that can predict clinical responses. Recently, Qiu et al. discovered high expression of the inhibitory immune checkpoint LAG3 in infiltrating CD8+ T cells in immune-resistant tumors. Subsequently, two patients with refractory EBV-positive gastric cancer participated in a clinical trial of LAG3 inhibitors and found that the LAG3 antibody effectively inhibited tumor growth and reduced peripheral blood EBV-DNA copy numbers. Targeting LAG3 could be a new therapeutic direction for EBVaGC patients.362
Cell therapy
CAR-T cell therapy is also being considered for the treatment of EBVaGC, with a variety of antigens including HER2, CEA, EpCAM, Claudin 18.2, and NKG2D potentially serving as targets for CAR T cell therapy.310,792 It has been reported that CAR T cell therapy can provide an additional 5 quality-adjusted life years, compared to non-drug therapies (surgery, radiotherapy, stem cell transplantation) which can provide 4.6 years, with similar levels of cost-effectiveness.793 Approximately 48% of gastric cancer patients have CLDN18.2 in their tumor tissues, which is linked to a poorer prognosis.794 In response to this, the only FDA-approved CAR-T cell therapy targeting CLDN18.2 is CT041. Recent results from a phase I trial revealed an ORR of 38.8% and a disease control rate (DCR) of 91.8%. The median PFS was 4.4 months, while the median OS was 8.8 months.727,795
As an alternative to CAR-T cell therapy, CAR-NK cell therapy has shown promising results against human GC cell lines in mouse models.681 Small phase I clinical trials indicate that NK cell therapy is safe and may have antitumor effects for advanced gastric cancer,796,797 which led to the FDA granting orphan drug designation to the NK cell therapy DF1001 in 2021.
Cervical cancer
The development of CC is closely associated with HPV infection.798,799 In clinical practice, early-stage CC patients often undergo surgery or radiation therapy, with a 5-year OS rate of 80%–90%. Late-stage CC cases are primarily managed with systemic chemotherapy, but the survival outcomes remain poor.800,801 Most CC diagnostic and prognostic biomarkers are related to the molecular mechanisms of the HPV virus. Common ones include HPV DNA, E6/E7 mRNA, p53, Rb, and p16INK4a.802 HPV DNA in cervical epithelial cells is the most widely and deeply applied biomarker.803 With the development of liquid biopsy, circulating cell-free HPV DNA (cf HPV DNA) detection has emerged as a highly promising non-invasive method. It has a reported positive predictive value of 87.5% and a negative predictive value of 89.3% for disease progression.804 CircRNAs are also significant molecular markers for HPV-related cancers, useful for early diagnosis and prognosis prediction.805 Moreover, HPV E6/E7 mRNA can detect HPV infection and reflect the E6/E7 gene’s expression and activity, which helps predict the progression of cervical lesions and the clinical outcome of CIN2 p16-positive patients. Similar to HBV,806 vaccination is highly effective in preventing HPV infections.807 Beyond vaccination, emerging strategies in CC treatment include targeting tumor cell signaling pathways, immune checkpoint blockade, and boosting the cytotoxic T-cell response against HPV oncogenes.808 PLWH have a higher risk of HPV infection compared to HIV-negative individuals.809 Consequently, HPV-related CC is more prevalent among PLWH, and it not only progresses more rapidly but also results in a poorer prognosis.810,811 Currently, the treatment of HIV+ HPV-related CC mainly involves combining ART with antitumor therapy, with a focus on restoring immune function as part of individualized treatment.812
Virus vaccines
The oncogenic potential of HPV primarily depends on the continuous expression of viral proteins E6 and E7. Direct inhibition of E6 and E7 expression or their inactivation can help prevent HPV-related CC progression.84 Although no therapeutic vaccine is currently available for CC, research is advancing. The HPV nanoparticle vaccine cPANHPVAX, based on the E7 oncogene, shows promise in preclinical studies and is moving toward clinical trials.813 Phase I/IIa clinical trial confirmed the safety and efficacy of the therapeutic DNA vaccines VB10.16 and MEDI0457 (INO-3112) in HPV-related CC.682,814 Other vaccines, like Vvax001 and GX-188E, have demonstrated effectiveness in early clinical trials.815,816 Additionally, the HPV 16/18/31/33/45/52/58 vaccine has shown great efficacy, providing long-lasting protection.817 Moreover, combining vaccines with immunotherapy has shown potential by increasing CD4/CD8 T cell infiltration and preliminary antitumor activity.818,819
Targeting tumor cell signaling
Currently, the CRISPR/Cas9 system offers a targeted approach for treating HPV-related CC by specifically cutting the HPV E7 oncogene, thereby reducing E7 expression and inhibiting CC cell proliferation.680 Other CRISPR/Cas9 systems targeting E6 and E7 have also been proven to inhibit HPV-related CC tumor growth.820,821,822 Clinical trials exploring these CRISPR-based approaches are currently underway, potentially paving the way for novel therapeutic strategies. In addition, cervical cancer cells often upregulate PARP-1 and PARP-2, which participate in double-strand DNA break repair and promote tumor cell growth.823,824,825 The genomic instability of cells caused by HPV infection makes cancer cells more reliant on the DNA repair mechanism mediated by PARP. And PARP-1 has been found to be associated with HPV infection and the development of CC, especially significantly correlated with HPV-positive status in high-grade squamous intraepithelial lesions.825 Clinical studies have shown that the PARP inhibitor veliparib, when used in combination with paclitaxel and cisplatin, is effective in managing R/R CC.826
HIV promotes angiogenesis and ECM remodeling through various pathways, which contributes to the progression of CIN to cervical cancer. HIV protease inhibitors (HIV-PI) inhibit the MMP-9/VEGF angiogenesis axis by upregulating TIMP-3, making them potential drugs for preventing the progression and metastasis of CIN and cervical cancer.827 HIV protease inhibitors, such as nelfinavir, saquinavir, and lopinavir, have also been shown to reduce HPV16 E6 and E7 protein levels, restore wild-type p53 expression, and effectively induce apoptosis in HPV-CC.828 Tat, a key molecule in HIV’s acceleration of HPV-related CC, is a promising target for controlling the progression of HPV-related CC.829 Various approaches targeting Tat, such as Tat-conjugated H3 peptides,830 HIV TAT peptide-coated gold nanoparticles (GNPs)-ROR1 siRNA,831 TAT-chitosan-SPION nanoparticles,831 and anticancer agent P-bi-TAT,832 have demonstrated strong anticancer activity in vitro and in vivo, suggesting their potential as therapeutic candidates for HIV-related CC.
Immunotherapy
Immunostimulants and ICIs have shown potential in enhancing immune cell efficacy and improving treatment outcomes of HPV-related CC.833 Currently, the main therapeutic agents include PD-1 antibodies, PD-L1 antibodies, CTLA-4 antibodies, bispecific antibodies, and 4-1BB agonists.
HIV-related HPV+ CC shows increased PD-L1 expression.834 PD-1/PD-L1 inhibitors like pembrolizumab not only show therapeutic effects in HPV-related CC but also directly inhibit the positive transcription elongation factor b (P-TEFb) signaling pathway, activating HIV transcription.835 Reversing HIV latency could help clear HIV reservoirs.836,837 Similarly, a CTLA-4 inhibitor targets HIV-related cancers and reverses HIV latency, which helps eliminate cells with replication-competent HIV.838 Additionally, a Phase I clinical trial (GOG-9929) found that the CTLA-4 inhibitor ipilimumab exhibited immunomodulatory activity in patients with locally advanced CC, suggesting its potential as a promising treatment option.839 Research into dual-target immunotherapy is also advancing. Cadonilimab, a bispecific antibody targeting PD-1 and CTLA-4, showed an ORR of 66.7% in R/R CC patients when combined with chemotherapy in a Phase II clinical study.840
Preclinical investigations have revealed that immunostimulants can inhibit the proliferation of CC cells. Specifically, the immunostimulant imiquimod enhances local antiviral and antitumor immune responses by stimulating Toll-like receptor 7 (TLR7).841 A Phase I/II clinical study demonstrated that PF-04518600 (OX40 agonist) had an ORR of 11% in R/R CC patients, with no dose-limiting toxicity or grade 3-5 immune-related adverse events, suggesting it is safe and effective.842 In another Phase II clinical study, imiquimod was found to significantly increase the population of CD4+/CD8+ T cells in cases of high-grade CIN, leading to regression to grade 1 or lower CIN.819 Additionally, the combination of ipilimumab with chemotherapy displayed the capacity to elevate E6/E7-specific CTL responses, thus enhancing the treatment outcomes for CC patients.839
Cell therapy
Engineered TCR-T cells are currently being developed as a therapeutic approach for CC.843,844,845 Preclinical studies have shown that E7 TCR-T cells can specifically recognize and destroy HPV-positive tumor cell lines by targeting the E7 11-19 epitope complexed with HLA-A*02:01. In a Phase I clinical trial, E7 TCR-T cells demonstrated excellent safety and efficacy, achieving PR in 50% of patients.846 Building on these findings, Mansour Poorebrahim et al. combined E7-TCR with a CAR targeting TROP2 to develop E7-TCR/TROP2-CAR NK-92 cells, which demonstrated enhanced cytotoxic activity against HPV-positive CC cells.847 Similarly, Isaac Quiros-Fernandez et al. developed E7-TCR/L1CAM-CAR NK 92 cells with enhanced the cytotoxic capability.848 Numerous other HPV-specific engineered TCR-T cells are also undergoing clinical trials (Table 5).
EBV-related NPC
Over 90% of undifferentiated NPC cases are associated with EBV infection.357,849,850,851 There is currently no curative treatment for NPC.852 For stage I NPC, definitive radiotherapy alone is often effective.853 In stage II NPC, there is debate over adding concurrent chemotherapy to radiotherapy, while T2N1 NPC often requires radiotherapy combined with cisplatin-based chemotherapy due to a higher risk of distant metastasis.854 For locally advanced (stage III-IVa) NPC, concurrent platinum-based chemoradiotherapy is required.855 Depending on the stage and individual patient circumstances, chemotherapy intensity may be further increased by adding induction or adjuvant chemotherapy to concurrent chemoradiotherapy.762 Challenges, such as drug resistance and adverse reactions limit chemotherapy’s effectiveness in NPC treatment.856,857,858 EBV infection is the primary cause of NPC, leading to the malignant proliferation of nasopharyngeal mucosal cells and eventually resulting in malignancy.859 Due to the pervasive nature of EBV and its transmission challenges, vaccination is considered the most effective preventive measure against EBV infection and the most cost-effective method for treating EBV-related diseases.95,860 Advances in understanding NPC’s molecular mechanisms have highlighted the potential of preventive and therapeutic vaccines, along with other strategies like tumor cell signaling targeting, immune checkpoint blockade, and cellular immunotherapy.861,862
Virus vaccines
Despite numerous attempts to develop an EBV vaccine, none have been approved to date. Preventive peptide vaccines targeting EBV, such as gp350, EBV gH/gL and gB, have been discussed in EBVaGC. This section will focus on therapeutic vaccines for NPC. Virus-like particles (VLPs) represent promising vaccine candidates. VLP-based vaccines for HBV and HPV have been widely applied in clinical practice, suggesting that a similar approach might be effective for an EBV vaccine.863,864,865 The first complete EBV VLP was developed by knocking out EBV’s terminal oncogenes and repeat sequences, such as LMP1 and BZLF1. These EBV VLPs elicited specific responses in mice.95 LMP1, LMP2, EBNA1, and EBNA3, either alone or in combination, could serve as target antigens for an anti-EBV vaccine due to their crucial roles in EBV infection and the subsequent malignant transformation of infected cells.36 Clinical trial results have shown that immunization with EBV peptide-pulsed DC cells can induce a reduction in serum EBV DNA levels and lead to tumor regression of EBV-related NPC.866,867 Additionally, EBV-related NPC aberrantly expresses CD137, which is induced by LMP.868 CD137L-DCs exhibit superior T-cell stimulation capabilities in vitro compared to conventional monocyte-derived DCs, indicating potential application in NPC.869 Besides DC vaccines, recombinant viral vector vaccines, such as those based on modified vaccinia Ankara (MVA), have also shown promise in NPC.870,871 One study demonstrated that MVA fusion proteins could effectively reactivate CD4+ memory T-cell responses in vitro, with MVA fusion proteins containing the C-terminal of EBNA1 and full-length LMP2 (MVA-EL).872 Two clinical trials involving the MVA-EL vaccine showed enhanced CD4+ and CD8+ T-cell responses to antigens in NPC patients.873 Additionally, the results of two clinical trials related to this vaccine, NCT01800071 and NCT01094405, remain undisclosed. We anticipate the release of the relevant findings (Table 6).
Targeting tumor cell signaling
Currently, there is no specific antiviral treatment for EBV-related NPC. After anti-tumor treatment, EBV levels can drop to normal.857 Molecular targeted therapy aims to inhibit tumor cell growth by specifically binding antibodies or ligands to target molecules on tumor cells, thereby blocking downstream signaling pathways crucial for tumor progression.854,874 This approach is primarily used for locally recurrent or metastatic NPC (RM-NPC) and includes therapies, such as EGFR monoclonal antibodies and anti-angiogenic agents.875,876
In most studies, EBNA1 and LMP1 have been identified as key targets for expanding EBV-specific CTLs (EBV-CTL).877 A clinical trial using EBV-CTL to treat NPC patients showed that EBV DNA levels in the plasma of all participants decreased to undetectable levels.878 Additionally, EBV-CTL treatment for NPC can stimulate LMP2-specific immune responses, which are beneficial in managing disease progression in stage IV NPC that is resistant to conventional therapies.879,880 Chia et al. conducted a phase II clinical trial revealing that the combination of chemotherapy and EBV-CTLs achieved a 71.4% effectiveness rate, with five patients not requiring further chemotherapy within 34 months of starting CTL treatment. This trial achieved the best outcomes among similar studies in advanced NPC patients, suggesting that chemotherapy combined with EBV-CTL is a highly promising treatment method.881
Immunotherapy
Immunotherapy has had a significant impact on the treatment of RM-NPC, particularly in cases induced by EBV. In EBV-induced NPC, high levels of PD-L1 and extensive lymphocyte infiltration make PD-1 blockade immunotherapy a potentially beneficial approach.882 Preclinical studies have shown that EBV proteins, such as LMP1 and EBNA1/2 can regulate PD-L1 levels, thus influencing the extent of immune evasion.883 ICIs have garnered considerable attention in the treatment of NPC. These inhibitors can disrupt immune defenses and reinvigorate the body’s natural anti-tumor immune response. The primary targets for ICIs include PD-1/PD-L1 and cytotoxic T-lymphocyte-associated protein 4 (CTLA-4).
Clinical trials targeting RM-NPC patients with anti-PD-1 therapy have reported ORRs ranging from 20.5% to 34.1%.884,885,886 To date, several ICIs have received approval from the FDA. Pembrolizumab has shown good anti-tumor activity and safety in previously treated RM-NPC patients.887,888,889 In one case report, an NPC patient with high PD-L1 expression achieved complete tumor remission after receiving treatment with the PD-L1 inhibitor nivolumab, with no recurrence after 22 months of treatment. This successful outcome paved the way for further trials involving a larger number of NPC patients.890 A phase III clinical trial also demonstrated significantly prolonged PFS in RM-NPC patients treated with chemotherapy combined with camrelizumab.891 While single ICIs therapy may have limited activity in EBV-related NPC, dual ICIs therapy has shown increased efficacy in solid tumors. In a phase II trial, patients with EBV-positive RM-NPC who had previously failed chemotherapy were treated with a combination of nivolumab and ipilimumab, resulting in a best overall response (BOR) rate of 38%, a median PFS of 5.3 months, and a median OS of 19.5 months, with good tolerance. Biomarker analysis indicated that these results were independent of PD-L1 expression or tumor mutational burden. Although the BOR did not reach the predetermined estimate, patients with low plasma EBV-DNA titers (<7800 IU/ml) tended to have better responses and PFS.892 Recently, two registered phase I trials found that advanced nasopharyngeal carcinoma patients treated with anti-PD-1 had better survival and treatment responses if they had a rapid decrease in EBV DNA and high levels of certain EBV genes in their blood and tumors.893
Cell therapy
Currently, CAR-T cell therapy has not been approved for the treatment of solid tumors. Nevertheless, eight registered clinical trials are investigating its efficacy for NPC (Table 5). A phase I trial demonstrated the feasibility and tolerability of epithelial cell adhesion molecule (EpCAM)-CAR-T therapy in patients with NPC or breast cancer.894 In vitro studies have shown that LMP1-specific CAR-T cells killed 70% of NPC cells overexpressing LMP1. Although the effectiveness of LMP1-specific CAR-T cells against NPC cells with lower LMP1 expression remains uncertain, these findings are promising.895 Clinical trials of CAR-T cells targeting LMP1 for EBV-related malignancies are ongoing (NCT02980315).
TCR-T cell therapy has quietly entered the field of cancer treatment and has shown unprecedented potential for solid tumors, due to its ability to target multiple tumor antigens. Research into TCR-T cell therapy for NPC is expanding, with the LMP2 antigen emerging as a promising target. LMP2-specific TCR-T cells have been shown to inhibit tumor progression in NPC cell lines expressing LMP2/LMP1 in mouse models.896 Currently, four clinical trials are evaluating TCR-T therapy for NPC (Table 6). The most recent trial, initiated in October 2022, aims to compare the efficacy of CAR-T and TCR-T cells in treating NPC (NCT05587543). TCR-T therapy represents an exciting and rapidly advancing field.
KSHV-related KS
KS is a malignancy strongly associated with KSHV infection. It is frequently observed in patients with HIV/AIDS and those undergoing chronic immunosuppression.897,898 Currently, there is no definitive cure for KS. Chemotherapy is commonly employed for advanced cases, with most drugs demonstrating some level of efficacy. However, treatment options are limited for patients who do not respond to chemotherapy, particularly those who develop resistance to multiple chemotherapeutic agents.899 Given that KSHV infection is the primary cause of KS, preventing and managing KSHV infection is crucial,541,900 and exploring viral vaccines presents a promising treatment option.533 For HIV-positive patients, ART should be initiated alongside anti-tumor therapy.41 Systemic treatments for KS primarily include immunomodulation, anti-inflammatory therapies, and targeting VEGF receptors.901 Immunotherapeutic options mainly include IFN, PD-1 inhibitors, pomalidomide, among others.902
Virus vaccines and antiviral treatment
KSHV vaccines are developed to target specific KSHV viral proteins (like gB, gH, gL, K8.1, ORF27, ORF28, ORF68) and have been shown to alleviate disease in KS patients.903 Additionally, VGC, an anti-KSHV drug, modulates CD4+ T cell subsets and the TNF/TNFR axis, enhancing the regulation of inflammatory states and serving as an effective treatment for AIDS-related KS.904,905 Primaquine (PQ) also shows positive effects in treating KS.906
For AIDS-related KS patients, combined ART also exhibits anti-cancer effects.41,105,907,908,909 Regardless of CD4+ T cell levels, it is essential to initiate anti-HIV therapy immediately, after excluding conditions, such as tuberculous and cryptococcal meningitis.910 Lifelong treatment is required, and efforts should be made to avoid interruptions due to anti-tumor treatments.911 Paclitaxel combined with ART has been shown to improve OS in AIDS-related KS patients.912 Additionally, early administration of etoposide alongside ART can reduce mortality in AIDS-related KS patient.913
Targeting tumor cell signaling
Inhibitors targeting the signaling pathways of KS tumor cells include cytokine Inhibitors, VEGF Inhibitors and proteasome Inhibitors. Tacrolimus, which inhibits IL-2 release and suppresses T lymphocyte activity, has shown a 100% ORR in the treatment of superficial KS with topical application.914 VEGF, a key factor in KS endothelial cell proliferation, makes VEGFR inhibitors ideal targets for KS treatment.531,915,916 Liposomal doxorubicin combined with the VEGF inhibitor bevacizumab has shown efficacy in treating advanced KS.917 Proteasome inhibitors have direct anti-tumor activity, induces lytic activation of KSHV, and inhibits HIV infectivity, thereby improving control of KS and HIV. Bortezomib has demonstrated good tolerability and effectiveness in treating AIDS-related KS.918
Immunotherapy
Immune dysfunction has been closely associated with the development of KS.908 Given that immunogenic viral neoantigens can be harnessed, immune checkpoint inhibitors (ICIs) have emerged as a primary treatment modality for virus-related tumors, including KS. Increased PD-L1 expression in KS-infected monocytes and its association with an inflammatory environment suggest that lytic virus replication may trigger PD-1/PD-L1 activation in KS.919 Pembrolizumab have achieved a 71% ORR in KS with promising efficacy and acceptable safety.920 Nivolumab and Ipilimumab has demonstrated an 87% ORR in KS, though 22% experienced grade 3-4 adverse events.921 Despite the potential of ICIs, the rarity of KS and the absence of randomized clinical trials mean that ICIs treatment for KS still lacks robust clinical evidence. Another challenge is the lack of evidence regarding the optimal systemic treatment sequence for KS. For example, interferon treatment has been shown to induce PD-1 expression in certain malignancies (e.g., ovarian cancer and melanoma).922,923 Before using ICIs in KS patients, the therapeutic benefits of drugs like interferon should be considered. Overall, ICIs are an effective treatment option for KS patients. However, further research is needed to determine the efficacy of ICIs and their optimal treatment sequence to maximize therapeutic outcomes. Ongoing clinical trials, such as NCT03469804, NCT02595866, NCT03316274, and NCT03367754, aim to address these uncertainties and refine ICIs treatment strategies for KS (Table 7).
Lymphoma
The increased risk of lymphoma is associated with various viral infections, including EBV, HIV and HCV. Among them, EBV is the most extensively studied virus associated with lymphoma, especially in ENKTCL, BL and cHL. ENKTCL is a type of T-cell lymphoma that is closely linked to EBV infection. Almost all patients with ENKTCL test positive for EBV. BL and cHL are additional types of B-cell lymphomas associated with EBV. BL is highly invasive and sensitive to chemotherapy. cHL is the most common subtype of HL, with the majority (75%-80%) of cHL patients achieving good tumor remission following first-line standard chemotherapy.924 However, the prognosis for patients with relapsed/refractory lymphoma remains poor.925 Currently, new treatment strategies under development, such as EBV-CTL, pathway inhibitors, ICIs, and CAR-T therapies, hold promise for these patients.926,927 The two most common subtypes of HIV-related non-Hodgkin lymphoma are HIV-related DLBCL and HIV-related BL.928 The immune functional defects in HIV-infected individuals exhibit distinct characteristics when comparing HIV-related lymphoma to non-HIV infections. Therefore, anti-lymphoma therapy must consider these immune deficiencies, making antiviral treatment a key component of the overall strategy.
Antiviral treatment
As elaborated in the previous sections regarding EBVaGC and NPC, the vaccines targeting EBV have been thoroughly discussed. In the context of EBV-related lymphomas, routine antiviral therapeutic interventions are not recommended at present. Virus clearance plays a significant role in lymphomas associated with HCV or HIV. In a study conducted in Italy, 250 NHL patients related to HCV infection were included in the research, with DLBCL and Marginal Zone Lymphoma being the most common types. The majority of patients had tested positive for HCV before the diagnosis of NHL, with a median time of 11 years. Antiviral therapy was effective for indolent NHL patients, achieving an overall response rate of 90%, and significantly prolonging the overall survival after a 7-year follow-up.929 And a study on patients with HCV-RNA positive diffuse large B-cell lymphoma showed that, compared to those who did not receive antiviral therapy, undergoing antiviral therapy after achieving remission from chemotherapy could improve their 2-year OS and PFS, leading to a better prognosis.930 Additionally, after confirming an HIV infection, it is important to start ART immediately, regardless of CD4 + T lymphocyte levels, as long as tuberculous and cryptococcal meningitis have been ruled out.910 According to the AMC-034 study, patients with HIV-related lymphoma who received combined ART experienced accelerated immune recovery while undergoing the EPOCH regimen.931
Targeting tumor cell signaling
Potential inhibitors of signaling pathways for EBV-related lymphomas include CD30 inhibitors, HDAC inhibitors, XPO1 inhibitors, and the NF-κB signaling pathway, among others. Around 70% of patients with NKTCL show CD30 overexpression.932 Brentuximabvedotin (BV) is the first antibody-drug conjugate that targets CD30. It has demonstrated sustained clinical activity and manageable toxicity in patients with relapsed/refractory CD30+ and EBV+ lymphomas.933,934 In patients with R/R ENKTCL, the potent HDAC inhibitor panobinostat, when combined with bortezomib, can provide clinical benefits and reduce EBV PCR titers.935 However, HDAC inhibition can trigger EBV reactivation; romidepsin (an HDAC inhibitor) has been reported to cause EBV reactivation.936,937 Therefore, the risk of EBV reactivation must be carefully considered when using HDAC inhibitors. While evidence for signaling pathway inhibitors is currently limited, using them in combination with ICIs or other therapies may help strengthen patient outcomes.
Additionally, adding Rituximab to the CODOX-M/IVAC regimen for HIV-positive BL can enhance survival rates without increasing treatment-related toxicity.938,939,940 Based on the high frequency of TP53 mutations in patients with HIV-related DLBCL, researchers are currently exploring the combination of R-EPOCH with Selinexor. A prospective, single-arm, open-label clinical study in China assessed the efficacy and safety of the XR-EPOCH regimen, which combines Selinexor with R-EPOCH, in newly diagnosed patients with HIV-DLBCL. As of May 2024, 10 patients had been enrolled in the study, and all demonstrated sustained treatment responses with manageable safety profiles.941
Immunotherapy
Approximately 40% of tumor cells in lymphoma patients express LMP1 and LMP2 during latent phase II, making them ideal targets for immunotherapy.942,943 EBV-targeted cell therapies include autologous EBV-specific T cell therapy ballaleucel-T, adoptive transfer of EBV LMP1/2-specific CTLs, and autologous LMP-specific CTLs. Among these therapies, autologous EBV-specific T cell therapy ballaleucel-T can induce remission in patients with R/R ENKTCL. The adoptive transfer of EBV LMP1-specific CTLs has been confirmed as a treatment option for EBV-positive cHL.944 Furthermore, the adoptive transfer of EBV LMP1/2-specific CTLs has demonstrated efficacy in maintaining sustained clinical responses in ENKTCL following remission.945 Additionally, autologous LMP-specific CTLs can induce sustained remission in patients with relapsed or refractory EBV-related lymphoma without obvious toxicity.946 However, the clinical application of these therapies may be limited by long manufacturing cycles and high manufacturing failure rates, making them inaccessible to most patients. Prockop et al. utilized an off-the-shelf donor-derived EBV-CTL bank, which can produce durable CR without noticeable toxicity in EBV-related lymphoma patients resistant to rituximab after transplantation.947 One of the most vulnerable groups affected by EBV is hematopoietic stem cell transplant (HSCT) recipients. Primary or reactivated EBV in immunocompromised transplant patients may be associated with post-transplant lymphoproliferative disorders (PTLD).948 Once a diagnosis of possible or confirmed EBV-PTLD is made, rituximab and reduction of immunosuppression are recommended as first-line treatments.949 Second-line options include adoptive cell therapy (like EBV-CTL) and chemotherapy with or without rituximab.950 Tabelecleucel is the first allogeneic, off-the-shelf EBV-specific T-cell immunotherapy approved for the treatment of relapsed or refractory EBV-positive post-transplant lymphoproliferative disorders.951,952 An ongoing global, multicenter, open-label phase 3 trial (NCT03394365) and expanded access study (NCT02822495) have demonstrated clinical benefits for patients with relapsed or refractory EBV-positive post-transplant lymphoproliferative disorders who have no other approved treatment options.951,952 EBV-targeted cell therapy is a favorable treatment strategy for EBV-related lymphoma patients. The availability of an off-the-shelf donor-derived EBV-CTL bank could provide an attractive and innovative treatment option that reduces the risk of graft-versus-host disease.947,953
Many ENKTCL patients express PD-L1, with estimates ranging from 39% to 100%.954 This suggests that the PD-1/PD-L1 axis is a promising target for immunotherapy.616,955 Research indicates that high serum PD-L1 levels correlate with a poor prognosis in ENKTCL patients.956 Several monoclonal antibodies targeting PD-1 and PD-L1, including the dual-targeting antibody IBI318, have demonstrated improved clinical outcomes in patients with R/R ENKTCL.957,958,959,960 Numerous ongoing clinical trials are investigating PD-1/PD-L1 inhibitors in combination with other therapies (Table 8). Furthermore, research on HIV-related DLBCL is currently scarce, with ongoing clinical trials for gene therapy, BTK inhibitors and CAR-T cell therapy (Table 8).
CAR-T therapy
Recent preclinical studies indicate that four CAR-T cell lines (CD38-CAR, LMP1-CAR, and their tandem variant CAR 2) exhibited significant cytotoxicity against NKTCL cells in both in vitro and in vivo settings.961 CD38-CAR-T cells exhibited a remarkable inhibitory effect on NKTCL in vitro and mouse xenograft models.962 Currently, a phase I trial is underway to evaluate the safety and feasibility of using fully human anti-CD30 CAR cells for treating advanced CD30-expressing lymphomas. CD30 CAR-T cells have demonstrated antitumor efficacy and low toxicity in R/R HL. However, the use of off-the-shelf allogeneic T-cell therapies is limited due to the risk of graft-versus-host disease.963 To address this issue, promising results from a study presented at the 17th International Conference on Malignant Lymphoma (ICML) showed that modifying EBV-specific T cells (EBVST) with a CD30-CAR (CD30CAR EBVST) could provide a safe and effective treatment for R/R HL.964 A single-center study conducted in China revealed that CD19/CD22 CAR-T therapy is effective and has manageable toxicity in adults with R/R BL.965 For children R/R BL, patients who have reached CR after infusion of CD19 CAR-T continue to infuse the CD22 CAR-T, which can produce lasting effects. And the patient with the suffering of the central nervous system has benefited obviously.966 Although clinical trial data on CAR-T therapy for EBV-related lymphomas are limited, the outlook remains optimistic.
Currently, CAR-T therapy for HIV-related lymphomas (HAL) is supported only by case reports, as key trials have not included HIV-positive patients. In six reported cases, three patients achieved CR, one achieved PR, and the treatment was considered safe.967 As research continues, increased data will enable us to better evaluate the long-term effects and safety of CAR-T therapy for virus-associated lymphoma.
Adult T-cell leukemia/lymphoma (ATLL)
After the Human T-cell leukemia virus type 1 (HTLV-1) invades the human body, the incubation period can last for decades, with about 3% of HTLV-1 infected individuals eventually developing ATLL.968 Currently, there is no curative treatment for HTLV-1 and ATLL. Many ATLL patients experience infections, requiring aggressive infection control and enhancement of organ function. When the disease progresses, prompt and aggressive treatment measures, such as chemotherapy, targeted therapy, and biological therapy are necessary.969 Although some patients experience alleviation of clinical symptoms during the treatment process, these therapeutic methods still have certain limitations. For instance, patients who undergo chemotherapy suffer from a severe weakening of their immunity, while stem cell transplantation is prone to lead to a high recurrence of the disease.635 As HTLV-1 infection is the primary cause of ATLL, it is crucial to prioritize the prevention and control of HTLV-1 infection. Currently, there are no available antiviral drugs or vaccines for HTLV-1, making research into antiviral drugs and vaccines a potential therapeutic direction.970
Virus vaccines and antiviral treatment
Patients with ATLL typically opt for a combination therapy of Interferon-alpha (IFN-α) and Zidovudine (AZT) as the first-line treatment plan.971 Compared to monotherapy with first-line chemotherapy or chemotherapy followed by antiviral maintenance therapy, first-line antiviral treatment primarily benefits patients with acute, chronic, and smoldering types of ATLL.972 It has been reported that the five-year survival rate for patients with chronic and smoldering ATLL who received combined IFN-α and AZT therapy is 100%, significantly improving the long-term prognosis of patients.973 Although patients treated with the combination of IFN-α and AZT show a clear survival advantage, and there is theoretical research supporting the synergistic antiviral effect of IFN-α and AZT, the potential threat of cytotoxicity cannot be ruled out due to the often ineffective low doses of IFN-α and AZT. As a long-term management strategy, whether this approach can lead to a complete cure for patients remains to be further studied, and it is necessary to conduct clinical trials to evaluate new targeted therapies.974 Epigenetic abnormalities contribute to the pathogenesis of ATLL, and a phase 2 clinical study evaluated the efficacy and safety of Valemetostat, an EZH2 inhibitor, in patients with R/R ATLL. The study found an ORR of 45.8%, and the median DOR has not yet been reached. These results suggest that Valemetostat demonstrates promising efficacy and tolerability in patients with R/R ATLL.975
Therapeutic vaccines represent a potential therapeutic strategy against HTLV-1 infection, aiming to control ATLL by activating the body’s immune response. A novel immunogenic chimeric peptide vaccine that includes epitopes from Tax, gp21, gp46, and gag (p19) encompasses multiple antigenic epitopes related to HTLV-1. The vaccine is immunized with MPLA and ISCOMATRIX adjuvants, and experimental results indicate that this chimeric peptide vaccine can induce systemic and mucosal immune responses, especially when MPLA and IMX adjuvants are used in combination.976 This suggests that the vaccine has potential in eliciting cellular and mucosal immune responses, offering new insights for combating HTLV-1 infection. Additionally, a multi-epitope vaccine delivered via PLGA nanoparticles can significantly enhance systemic and mucosal immune responses.977 The application of multi-epitope vaccines and immunostimulants can improve immune responses, providing a new avenue for the development of therapeutic vaccines for HTLV-1.
Targeting tumor cell signaling
In the process of developing anti-HTLV-1 treatment methods, the greatest obstacle is the difficulty in controlling the viral permanence and the reactivated provirus. However, the emergence of a new treatment method, gene editing technology, has brought hope for treating this difficult problem.978,979 As early as 2013, zinc finger nucleases (ZFNs) specifically recognizing the HTLV-1 LTR were confirmed to be able to disrupt the LTR promoter function of HTLV-1 and inhibit the proliferation of HTLV-1-positive cell lines.980 Subsequently, the gDNA developed based on the CRISPR/Cas9 technology and targeting the HTLV-1 oncogenes HBZ and Tax was also confirmed to be able to inhibit the proliferation of ATLL cells to varying degrees.981 The latest research also reported that using substrate-linked directed evolution (SLiDE) to engineer Cre-derived site-specific recombinases, named RecHTLV recombinase, and recombinase can accurately target and effectively excise the HTLV-1 provirus in HTLV-1-infected cells, potentially preventing the development of HTLV-1-associated diseases.982 In conclusion, gene editing technology has broad application prospects in the treatment of anti-HTLV-1.
Conclusions and perspectives
Since the identification of the first human oncogenic virus, EBV, in the lymphocytes of Burkitt lymphoma patients in 1964, the number of recognized oncogenic viruses has increased with advancements in high-throughput technologies, deepening our understanding of these viruses. IARC has classified seven viruses as Group 1 carcinogens, including four DNA viruses and three RNA viruses. The distribution of virus-induced tumors varies globally, indicating that multiple factors influence the oncogenic risks and incidence rates of these viruses in different regions, necessitating further research to enhance our understanding.
The role of oncogenic viruses in cancer development is complex and multifaceted. On the molecular level, this review discusses how these viruses infect target cells and induce carcinogenesis. Different oncoviruses vary in genomic structure, ways of invasion, replication, and assembly; and these differences directly influence their subsequent carcinogenic mechanisms. First, various oncoviruses enter cells by binding to specific receptors on the surface of host cells through different structural proteins. DNA viruses (HBV, HPV, EBV, KSHV, MCV) can directly integrate into the genome of target cells, while retroviruses (HIV, HTLV-1) need to be reverse-transcribed into DNA first and then integrate into the host genome. After the oncovirus integrates into the host genome, it may activate oncogenes or inactivate tumor-suppressor genes, triggering carcinogenesis. Although HCV does not integrate, the chronic inflammation caused by persistent infection can increase the risk of cell mutation and thus lead to cancer. Meanwhile, the cell tropism of the virus determines the type of cancer it causes. For instance, EBV mainly infects B lymphocytes and epithelial cells and is associated with NPC and B-cell lymphoma; HPV infects basal epithelial cells, and persistent infection with high-risk types increases the risk of CC.
Oncogenic viruses affect host cells in various ways, including directly inducing genomic instability, regulating the expression of oncogenes and tumor suppressor genes, disrupting cell cycles, causing immune suppression, triggering metabolic reprogramming, and promoting chronic inflammation. These mechanisms provide critical insights into how oncogenic viruses lead to cancer. Although these viruses share many direct or indirect carcinogenic mechanisms, there are differences between DNA and RNA viruses. DNA viruses tend to cause cellular transformation by encoding viral oncoproteins, such as the HBx protein of HBV and E6/E7 proteins of HPV, which directly alter host cell signaling pathways and cell cycles. In contrast, RNA viruses often promote oncogenesis through chronic infection, leading to sustained inflammation or immune suppression, facilitating mutations and cancer. This tendency is most evident in the case of HIV, which, despite being classified as an oncogenic virus, is widely believed not to cause cancer directly but rather to promote certain cancers indirectly by compromising the immune system. Some viruses have also been found to invade the nervous system, and their presence might disrupt normal neural-regulatory mechanisms, which could influence the microenvironment conducive to cancer initiation or progression. Meanwhile, a dysbiotic microbiome might either enhance or suppress the replication and oncogenic potential of viruses. Some bacteria in the microbiome could produce metabolites that directly interfere with virus-host interactions or indirectly modulate the host’s immune system. These mechanisms not only promote cancer development but also complicate and challenge cancer treatment.
Targeted virus therapy for virus-related tumors offers significant advantages. Virus vaccines can trigger a highly specific immune response against relevant antigens, helping to maintain long-term immune surveillance. Precision gene-targeted therapy modifies viruses to target tumor-causing genes without harming normal cells. ICIs can be combined with chemotherapy to enhance anti-tumor immunity. Meanwhile, the treatment of virus-related cancers encounters numerous clinical obstacles. Oncogenic viruses, such as HPV, exhibit high variability with multiple subtypes and genetic mutations, which not only complicates the determination of stable therapeutic targets but also render existing drugs ineffective when mutations occur. Additionally, the latent infection state, as seen in EBV, poses a significant challenge. During latency, the virus is scarcely detectable by the immune system and resistant to antiviral drugs. Moreover, cancers linked to these viruses often possess intricate immune-escape mechanisms, like HBV-induced HCC downregulating MHC molecules to evade immune surveillance. These treatment challenges are closely associated with the carcinogenic mechanisms in which viruses integrate into the host genome. Thus, the key points of treatment are to block virus integration to impede cancer development and to mitigate virus-induced inflammation to lower the risk of cell mutation.
While significant progress has been made in understanding viral oncogenesis and developing therapeutic strategies, many challenges and unknowns remain. Developing therapeutic vaccines will be a crucial breakthrough in the future, helping to enhance immune protection in virus-infected populations and reduce the incidence of virus-related cancers. In addition to vaccines, antiviral therapies and emerging anticancer treatments are also areas of interest. For example, antiviral therapy for chronic hepatitis has been proven to reduce the risk of liver cancer significantly. Therefore, antiviral therapy should be maintained throughout the course of curative or systemic treatment. In terms of targeted and immunotherapies, although current therapies offer limited survival benefits, further understanding of the immune system may improve treatment outcomes through checkpoint blockade strategies and engineered T-cell immunotherapy. Lastly, strengthening public health education, promoting vaccination, and expanding antiviral treatment access will provide a solid foundation for reducing the incidence of virus-related cancers. In the future, in-depth research into carcinogenic mechanisms, accurate identification of key targets, and the development of novel therapies capable of breaking virus latency, overcoming immune escape, and blocking virus integration will be essential for surmounting these treatment challenges.
References
Contreras, A., Sanchez, S. A., Rodriguez-Medina, C. & Botero, J. E. The role and impact of viruses on cancer development. Periodontology 2000. https://doi.org/10.1111/prd.12566 (2024).
Gaglia, M. M. & Munger, K. More than just oncogenes: mechanisms of tumorigenesis by human viruses. Curr. Opin. Virol. 32, 48–59 (2018).
Tashiro, H. & Brenner, M. K. Immunotherapy against cancer-related viruses. Cell Res. 27, 59–73 (2017).
Chen, Y. et al. Viral carcinogenesis: factors inducing DNA damage and virus integration. Cancers 6, 2155–2186 (2014).
Ahmed, K. & Jha, S. Oncoviruses: How do they hijack their host and current treatment regimes. Biochim. Biophys. Acta Rev. Cancer 1878, 188960 (2023).
Krump, N. A. & You, J. Molecular mechanisms of viral oncogenesis in humans. Nat. Rev. Microbiol. 16, 684–698 (2018).
Lavanchy, D. Hepatitis B virus epidemiology, disease burden, treatment, and current and emerging prevention and control measures. J. Viral Hepat. 11, 97–107 (2004).
Goldie, S. J. et al. Projected clinical benefits and cost-effectiveness of a human papillomavirus 16/18 vaccine. J. Natl. Cancer Inst. 96, 604–615 (2004).
Schiller, J. T. & Lowy, D. R. An introduction to virus infections and human cancer. Recent Results Cancer Res. 217, 1–11 (2021).
Plummer, M. et al. Global burden of cancers attributable to infections in 2012: a synthetic analysis. Lancet Glob. Health 4, e609–e616 (2016).
de Martel, C. et al. Global burden of cancer attributable to infections in 2018: a worldwide incidence analysis. Lancet Glob. Health 8, e180–e190 (2020).
Hsu, Y. C., Huang, D. Q. & Nguyen, M. H. Global burden of hepatitis B virus: current status, missed opportunities and a call for action. Nat. Rev. Gastroenterol. Hepatol. 20, 524–537 (2023).
Alberts, C. J. et al. Worldwide prevalence of hepatitis B virus and hepatitis C virus among patients with cirrhosis at country, region, and global levels: a systematic review. Lancet Gastroenterol. Hepatol. 7, 724–735 (2022).
Paik, J. M. et al. Recent trends in the global burden of Hepatitis B virus: 2007–2017. Gastroenterology 160, 1845–1846.e1843 (2021).
Li, C. & He, W. Q. The impact of universal hepatitis B vaccine on the trend of liver cancer from the global burden of disease study 2017. Liver Int. 41, 1762–1774 (2021).
Manns, M. P. & Maasoumy, B. Breakthroughs in hepatitis C research: from discovery to cure. Nat. Rev. Gastroenterol. Hepatol. 19, 533–550 (2022).
Huang, D. Q. et al. Changing global epidemiology of liver cancer from 2010 to 2019: NASH is the fastest-growing cause of liver cancer. Cell Metab. 34, 969–977.e962 (2022).
GBD 2019 Europe Hepatitis B & C Collaborators Hepatitis B and C in Europe: an update from the global burden of disease study 2019. Lancet Public Health. 8, e701–e716 (2023).
Lin, L. et al. The burden and trends of primary liver cancer caused by specific etiologies from 1990 to 2017 at the global, regional, national, age, and sex level results from the global burden of disease study 2017. Liver Cancer 9, 563–582 (2020).
de Martel, C., Plummer, M., Vignat, J. & Franceschi, S. Worldwide burden of cancer attributable to HPV by site, country and HPV type. Int. J. Cancer 141, 664–670 (2017).
Crow, J. M. HPV: The global burden. Nature 488, S2–S3 (2012).
Malagón, T., Franco, E. L., Tejada, R. & Vaccarella, S. Epidemiology of HPV-associated cancers past, present and future: towards prevention and elimination. Nat. Rev. Clin. Oncol. 21, 522–538 (2024).
Maza, M. & Gage, J. C. Considerations for HPV primary screening in lower-middle income countries. Prev. Med. 98, 39–41 (2017).
Bonjour, M. et al. Global estimates of expected and preventable cervical cancers among girls born between 2005 and 2014: a birth cohort analysis. Lancet Public Health 6, e510–e521 (2021).
Luttjeboer, J., Wondimu, A., Van der Schans, J. & Postma, M. J. Maximising the potential of HPV vaccines. Lancet Glob. Health 8, e460–e461 (2020).
He, W. Q. & Li, C. Recent global burden of cervical cancer incidence and mortality, predictors, and temporal trends. Gynecol. Oncol. 163, 583–592 (2021).
Young, L. S., Yap, L. F. & Murray, P. G. Epstein-Barr virus: more than 50 years old and still providing surprises. Nat. Rev. Cancer. 16, 789–802 (2016).
Coghill, A. E. & Hildesheim, A. Epstein-Barr virus antibodies and the risk of associated malignancies: review of the literature. Am. J. Epidemiol. 180, 687–695 (2014).
Wong, Y. et al. Estimating the global burden of Epstein-Barr virus-related cancers. J Cancer Res. Clin. Oncol. 148, 31–46 (2022).
Khan, G. & Hashim, M. J. Global burden of deaths from Epstein-Barr virus attributable malignancies 1990–2010. Infect. Agent. Cancer. 9, 38 (2014).
Khan, G., Fitzmaurice, C., Naghavi, M. & Ahmed, L. A. Global and regional incidence, mortality and disability-adjusted life-years for Epstein-Barr virus-attributable malignancies, 1990–2017. BMJ Open 10, e037505 (2020).
Mawson, A. R. & Majumdar, S. Malaria, Epstein-Barr virus infection and the pathogenesis of Burkitt’s lymphoma. Int. J. Cancer. 141, 1849–1855 (2017).
Shannon-Lowe, C. & Rickinson, A. The global landscape of EBV-associated tumors. Front. Oncol. 9, 713 (2019).
Hirabayashi, M., Georges, D., Clifford, G. M. & de Martel, C. Estimating the global burden of Epstein-Barr virus-associated gastric cancer: a systematic review and meta-analysis. Clin. Gastroenterol. Hepatol. 21, 922–930 e921 (2023).
Tavakoli, A. et al. Association between Epstein-Barr virus infection and gastric cancer: a systematic review and meta-analysis. BMC Cancer 20, 493 (2020).
Sun, C., Chen, X. C., Kang, Y. F. & Zeng, M. S. The status and prospects of epstein-barr virus prophylactic vaccine development. Front. Immunol. 12, 677027 (2021).
Bangham, C. R., Araujo, A., Yamano, Y. & Taylor, G. P. HTLV-1-associated myelopathy/tropical spastic paraparesis. Nat. Rev. Dis. Primers 1, 15012 (2015).
Ruffieux, Y. et al. Age and cancer incidence in 5.2 million people with human immunodeficiency virus (HIV): the South African HIV cancer match study. Clin. Infect. Dis. 76, 1440–1448 (2023).
Wan, Z. et al. Global and regional estimates of cervical cancer burden associated with human immunodeficiency virus infection from 1990 to 2019. J. Med. Virol. 95, e28891 (2023).
Stelzle, D. et al. Estimates of the global burden of cervical cancer associated with HIV. Lancet Glob Health 9, e161–e169 (2021).
Carbone, A., Vaccher, E. & Gloghini, A. Hematologic cancers in individuals infected by HIV. Blood 139, 995–1012 (2022).
Park, B. et al. Cancer incidence among adults with HIV in a population-based cohort in Korea. Jama Netw. Open 5, e2224897 (2022).
Moore, P. S. & Chang, Y. Why do viruses cause cancer? Highlights of the first century of human tumour virology. Nat. Rev. Cancer 10, 878–889 (2010).
Rous, P. Transmission of a malignant new growth by means of a cell-free filtrate. Conn. Med. 37, 526 (1973).
Rous, P. A transmissible avian neoplasm. (sarcoma of the common fowl.). J. Exp. Med. 12, 696–705 (1910).
Rous, P. A sarcoma of the fowl transmissible by an agent separable from the tumor cells. J. Exp. Med. 13, 397–411 (1911).
Shope, R. E. & Hurst, E. W. Infectious papillomatosis of rabbits: with a note on the histopathology. J. Exp. Med. 58, 607–624 (1933).
Gross, L. “Spontaneous” leukemia developing in C3H mice following inoculation in infancy, with AK-leukemic extracts, or AK-embrvos. Proc. Soc. Exp. Biol. Med. 76, 27–32 (1951).
Eddy, B. E., Borman, G. S., Grubbs, G. E. & Young, R. D. Identification of the oncogenic substance in rhesus monkey kidney cell culture as simian virus 40. Virology 17, 65–75 (1962).
Epstein, M. A., Achong, B. G. & Barr, Y. M. Virus particles in cultured lymphoblasts from Burkitt’s lymphoma. Lancet 1, 702–703 (1964).
Chen, J. et al. Ephrin receptor A2 is a functional entry receptor for Epstein-Barr virus. Nat. Microbiol. 3, 172–180 (2018).
Baer, R. et al. DNA sequence and expression of the B95-8 Epstein-Barr virus genome. Nature 310, 207–211 (1984).
Blumberg, B. S., Alter, H. J. & Visnich, S. A “New” antigen in leukemia sera. JAMA 191, 541–546 (1965).
Beasley, R. P., Hwang, L. Y., Lin, C. C. & Chien, C. S. Hepatocellular carcinoma and hepatitis B virus. a prospective study of 22,707 men in Taiwan. Lancet 2, 1129–1133 (1981).
Temin, H. M. & Mizutani, S. RNA-dependent DNA polymerase in virions of Rous sarcoma virus. Nature 226, 1211–1213 (1970).
Baltimore, D. RNA-dependent DNA polymerase in virions of RNA tumour viruses. Nature 226, 1209–1211 (1970).
Stehelin, D., Varmus, H. E., Bishop, J. M. & Vogt, P. K. DNA related to the transforming gene(s) of avian sarcoma viruses is present in normal avian DNA. Nature 260, 170–173 (1976).
Rowe, W. P. et al. Isolation of a cytopathogenic agent from human adenoids undergoing spontaneous degeneration in tissue culture. Proc. Soc. Exp. Biol. Med. 84, 570–573 (1953).
Linzer, D. I. & Levine, A. J. Characterization of a 54K Dalton cellular SV40 tumor antigen present in SV40-transformed cells and uninfected embryonal carcinoma cells. Cell 17, 43–52 (1979).
Lane, D. P. & Crawford, L. V. T antigen is bound to a host protein in SV40-transformed cells. Nature 278, 261–263 (1979).
Poiesz, B. J. et al. Detection and isolation of type C retrovirus particles from fresh and cultured lymphocytes of a patient with cutaneous T-cell lymphoma. Proc. Natl. Acad. Sci/USA 77, 7415–7419 (1980).
Hinuma, Y. et al. Adult T-cell leukemia: antigen in an ATL cell line and detection of antibodies to the antigen in human sera. Proc. Natl. Acad. Sci. USA 78, 6476–6480 (1981).
zur Hausen, H. Condylomata acuminata and human genital cancer. Cancer Res. 36, 794 (1976).
Durst, M., Gissmann, L., Ikenberg, H. & zur Hausen, H. A papillomavirus DNA from a cervical carcinoma and its prevalence in cancer biopsy samples from different geographic regions. Proc. Natl. Acad. Sci. USA 80, 3812–3815 (1983).
Barre-Sinoussi, F. et al. Isolation of a T-lymphotropic retrovirus from a patient at risk for acquired immune deficiency syndrome (AIDS). Science 220, 868–871 (1983).
Alter, H. J. et al. Detection of antibody to hepatitis C virus in prospectively followed transfusion recipients with acute and chronic non-A, non-B hepatitis. N. Engl. J. Med. 321, 1494–1500 (1989).
Houghton, M. et al. Molecular biology of the hepatitis C viruses: implications for diagnosis, development and control of viral disease. Hepatology 14, 381–388 (1991).
Chang, Y. et al. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi’s sarcoma. Science 266, 1865–1869 (1994).
Cesarman, E. et al. Kaposi’s sarcoma-associated herpesvirus-like DNA sequences in AIDS-related body-cavity-based lymphomas. N. Engl. J. Med. 332, 1186–1191 (1995).
Feng, H., Shuda, M., Chang, Y. & Moore, P. S. Clonal integration of a polyomavirus in human Merkel cell carcinoma. Science 319, 1096–1100 (2008).
Taheri, F., Goudarzi, H. & Faghihloo, E. Aneuploidy and oncoviruses. Rev. Med. Virol. 29, e2076 (2019).
Liu, S., Han, Y., Li, W. X. & Ding, S. W. Infection Defects of RNA and DNA Viruses Induced by Antiviral RNA Interference. Microbiol. Mol. Biol. Rev. 87, e0003522 (2023).
Fan, Y. M., Zhang, Y. L., Luo, H. & Mohamud, Y. Crosstalk between RNA viruses and DNA sensors: role of the cGAS-STING signalling pathway. Rev. Med. Virol. 32, e2343 (2022).
Cesarman, E. et al. Kaposi sarcoma. Nat. Rev. Dis. Primers 5, 9 (2019).
Nguyen, M. H. et al. Hepatitis B virus: advances in prevention, diagnosis, and therapy. Clin. Microbiol. Rev. 33, (2020).
Yuen, M. F. et al. Hepatitis B virus infection. Nat. Rev. Dis. Primers 4, 18035 (2018).
Lin, C. L. & Kao, J. H. Review article: the prevention of hepatitis B-related hepatocellular carcinoma. Aliment. Pharmacol. Ther. 48, 5–14 (2018).
Schreiner, S. & Nassal, M. A role for the host DNA damage response in hepatitis B virus cccDNA formation-and beyond? Viruses 9, 125 (2017).
Song, H. et al. TRIM25 inhibits HBV replication by promoting HBx degradation and the RIG-I-mediated pgRNA recognition. Chin. Med. J. 136, 799–806 (2023).
Zhang, T. et al. RNA binding protein TIAR modulates HBV replication by tipping the balance of pgRNA translation. Signal Transduct. Target. Ther. 8, 346 (2023).
Liu, W. et al. Molecular insights into Spindlin1-HBx interplay and its impact on HBV transcription from cccDNA minichromosome. Nat. Commun. 14, 4663 (2023).
Chen, B. F. Hepatitis B virus pre-S/S variants in liver diseases. World J. Gastroenterol. 24, 1507–1520 (2018).
Assarzadegan, N., Brooks, E. & Voltaggio, L. HPV-driven anal neoplasia: review and recent developments. Pathology 54, 184–194 (2022).
Wolf, J. et al. Human papillomavirus infection: Epidemiology, biology, host interactions, cancer development, prevention, and therapeutics. Rev. Med. Virol. 34, e2537 (2024).
Mlynarczyk-Bonikowska, B. & Rudnicka, L. HPV infections-classification, pathogenesis, and potential new therapies. Int. J. Mol. Sci. 25, 7616 (2024).
Speckhart, K., Choi, J., DiMaio, D. & Tsai, B. The BICD2 dynein cargo adaptor binds to the HPV16 L2 capsid protein and promotes HPV infection. PLoS Pathog. 20, e1012289 (2024).
Jain, M. et al. Epidemiology, molecular pathogenesis, immuno-pathogenesis, immune escape mechanisms and vaccine evaluation for HPV-associated carcinogenesis. Pathogens 12, 1380 (2023).
DiGiuseppe, S., Bienkowska-Haba, M., Guion, L. G. & Sapp, M. Cruising the cellular highways: How human papillomavirus travels from the surface to the nucleus. Virus Res. 231, 1–9 (2017).
Campos, S. K. Subcellular trafficking of the papillomavirus genome during initial infection: the remarkable abilities of minor capsid protein L2. Viruses 9, 370 (2017).
Murakami, I. et al. Roles for E1-independent replication and E6-mediated p53 degradation during low-risk and high-risk human papillomavirus genome maintenance. PLoS Pathog. 15, e1007755 (2019).
Sand, F. L. et al. Risk of CIN3 or worse with the persistence of 13 individual oncogenic HPV types. Int. J. Cancer. 144, 1975–1982 (2019).
Torne, A. S. & Robertson, E. S. Epigenetic mechanisms in latent Epstein-Barr virus infection and associated cancers. Cancers 16, 991 (2024).
Jambunathan, N. et al. Two sides to every story: herpes simplex type-1 viral glycoproteins gB, gD, gH/gL, gK, and cellular receptors function as key players in membrane fusion. Viruses 13, 1849 (2021).
Neuhierl, B., Feederle, R., Hammerschmidt, W. & Delecluse, H. J. Glycoprotein gp110 of Epstein-Barr virus determines viral tropism and efficiency of infection. Proc. Natl. Acad. Sci. USA 99, 15036–15041 (2002).
Cui, X. & Snapper, C. M. Epstein Barr virus: development of vaccines and immune cell therapy for EBV-associated diseases. Front. Immunol. 12, 734471 (2021).
Yang, Y. et al. Interferon-induced transmembrane protein-1 competitively blocks Ephrin receptor A2-mediated Epstein-Barr virus entry into epithelial cells. Nat. Microbiol. 9, 1256–1270 (2024).
Westhoff Smith, D. et al. The Epstein-Barr virus oncogene EBNA1 suppresses natural killer cell responses and apoptosis early after infection of peripheral B cells. mBio 12, e0224321 (2021).
Hayman, I. R. et al. New insight into Epstein-Barr virus infection using models of stratified epithelium. PLoS Pathog. 19, e1011040 (2023).
Connolly, S. A., Jardetzky, T. S. & Longnecker, R. The structural basis of herpesvirus entry. Nat. Rev. Microbiol. 19, 110–121 (2021).
Huang, W., Bai, L. & Tang, H. Epstein-Barr virus infection: the micro and macro worlds. Virol. J. 20, 220 (2023).
Volaric, A. K. et al. Epstein-Barr virus latency patterns in polymorphic lymphoproliferative disorders and lymphomas in immunodeficiency settings: diagnostic implications. Ann. Diagn. Pathol. 70, 152286 (2024).
Munz, C. Latency and lytic replication in Epstein-Barr virus-associated oncogenesis. Nat. Rev. Microbiol. 17, 691–700 (2019).
Murata, T. et al. Molecular basis of Epstein-Barr virus latency establishment and lytic reactivation. Viruses. 13, 2344 (2021).
Yang, J. et al. Epstein-Barr virus BZLF1 protein impairs accumulation of host DNA damage proteins at damage sites in response to DNA damage. Lab. Investig. 95, 937–950 (2015).
Cesarman, E., Chadburn, A. & Rubinstein, P. G. KSHV/HHV8-mediated hematologic diseases. Blood 139, 1013–1025 (2022).
Lu, J. et al. The RBP-Jkappa binding sites within the RTA promoter regulate KSHV latent infection and cell proliferation. PLoS Pathog. 8, e1002479 (2012).
Kumar, B. & Chandran, B. KSHV entry and trafficking in target cells-hijacking of cell signal pathways, actin and membrane dynamics. Viruses 8, 305 (2016).
Aalam, F., Nabiee, R., Castano, J. R. & Totonchy, J. Analysis of KSHV B lymphocyte lineage tropism in human tonsil reveals efficient infection of CD138+ plasma cells. PLoS Pathog. 16, e1008968 (2020).
Blumenthal, M. J. et al. Evidence for altered host genetic factors in KSHV infection and KSHV-related disease development. Rev. Med. Virol. 31, e2160 (2021).
Vladimirova, O. et al. Phase separation and DAXX redistribution contribute to LANA nuclear body and KSHV genome dynamics during latency and reactivation. PLoS Pathog. 17, e1009231 (2021).
Han, C. et al. KSHV RTA antagonizes SMC5/6 complex-induced viral chromatin compaction by hijacking the ubiquitin-proteasome system. PLoS Pathog. 18, e1010744 (2022).
Zhao, Y. et al. RIG-I like receptor sensing of host RNAs facilitates the cell-intrinsic immune response to KSHV infection. Nat. Commun. 9, 4841 (2018).
Torrents de la Pena, A. et al. Structure of the hepatitis C virus E1E2 glycoprotein complex. Science 378, 263–269 (2022).
Wrighton, K. H. Structure of the HCV glycoprotein. Nat. Struct. Mol. Biol. 29, 1147 (2022).
Dearborn, A. D. & Marcotrigiano, J. Hepatitis C virus structure: defined by what it is not. Cold Spring Harb. Perspect. Med. 10, a036822 (2020).
Kumar, A. et al. Structural insights into hepatitis C virus receptor binding and entry. Nature 598, 521–525 (2021).
Laugel, E. et al. Full-length genome sequencing of RNA viruses-How the approach can enlighten us on hepatitis C and hepatitis E viruses. Rev. Med. Virol. 31, e2197 (2021).
Niepmann, M. & Gerresheim, G. K. Hepatitis C virus translation regulation. Int. J. Mol. Sci. 21, 2328 (2020).
Shimotohno, K. HCV assembly and egress via modifications in host lipid metabolic systems. Cold Spring Harb. Perspect. Med. 11, a036814 (2021).
Zhou, Z. et al. Small molecule NS5B RdRp non-nucleoside inhibitors for the treatment of HCV infection: a medicinal chemistry perspective. Eur. J. Med. Chem. 240, 114595 (2022).
Sherwood, A. V. et al. Hepatitis C virus RNA is 5’-capped with flavin adenine dinucleotide. Nature 619, 811–818 (2023).
Frumento, N. et al. Neutralizing antibodies evolve to exploit vulnerable sites in the HCV envelope glycoprotein E2 and mediate spontaneous clearance of infection. Immunity 57, 40–51.e45 (2024).
Rosen, H. R. & Golden-Mason, L. Control of HCV infection by natural killer cells and macrophages. Cold Spring Harb. Perspect. Med. 10, a037101 (2020).
Han, J. W. et al. IFNL3-adjuvanted HCV DNA vaccine reduces regulatory T cell frequency and increases virus-specific T cell responses. J. Hepatol. 73, 72–83 (2020).
Hensel, N. et al. Memory-like HCV-specific CD8(+) T cells retain a molecular scar after cure of chronic HCV infection. Nat. Immunol. 22, 229–239 (2021).
Tonnerre, P. et al. Differentiation of exhausted CD8(+) T cells after termination of chronic antigen stimulation stops short of achieving functional T cell memory. Nat. Immunol. 22, 1030–1041 (2021).
Clark, I. C. et al. HIV silencing and cell survival signatures in infected T cell reservoirs. Nature 614, 318–325 (2023).
Veenhuis, R. T. et al. HIV replication and latency in monocytes and macrophages. Semin. Immunol. 51, 101472 (2021).
Summers, B. J. et al. Modular HIV-1 capsid assemblies reveal diverse host-capsid recognition mechanisms. Cell Host Microbe 26, 203–216.e206 (2019).
Xue, G. et al. The HIV-1 capsid core is an opportunistic nuclear import receptor. Nat. Commun. 14, 3782 (2023).
Qu, K. et al. Maturation of the matrix and viral membrane of HIV-1. Science 373, 700–704 (2021).
Maertens, G. N., Engelman, A. N. & Cherepanov, P. Structure and function of retroviral integrase. Nat. Rev. Microbiol. 20, 20–34 (2022).
Jang, S. & Engelman, A. N. Capsid-host interactions for HIV-1 ingress. Microbiol. Mol. Biol. Rev. 87, e0004822 (2023).
Ha, B. et al. High-resolution view of HIV-1 reverse transcriptase initiation complexes and inhibition by NNRTI drugs. Nat. Commun. 12, 2500 (2021).
Muller, T. G., Zila, V., Muller, B. & Krausslich, H. G. Nuclear capsid uncoating and reverse transcription of HIV-1. Annu. Rev. Virol. 9, 261–284 (2022).
McLaren, P. J. & Fellay, J. HIV-1 and human genetic variation. Nat. Rev. Genet. 22, 645–657 (2021).
Jiang, C. et al. Distinct viral reservoirs in individuals with spontaneous control of HIV-1. Nature 585, 261–267 (2020).
Bou-Nader, C. et al. HIV-1 matrix-tRNA complex structure reveals basis for host control of Gag localization. Cell Host Microbe 29, 1421–1436.e1427 (2021).
Ullah Nayan, M. et al. Advances in long-acting slow effective release antiretroviral therapies for treatment and prevention of HIV infection. Adv. Drug Deliv. Rev. 200, 115009 (2023).
Cohn, L. B., Chomont, N. & Deeks, S. G. The 33gy of the HIV-1 latent reservoir and implications for cure strategies. Cell Host Microbe 27, 519–530 (2020).
Duggan, N. N., Dragic, T., Chanda, S. K. & Pache, L. Breaking the silence: regulation of HIV transcription and latency on the road to a cure. Viruses 15, 2435 (2023).
Ahmadi Ghezeldasht, S. et al. HTLV-1 oncovirus-host interactions: from entry to the manifestation of associated diseases. Rev. Med. Virol. 31, e2235 (2021).
Bangham, C. R. M. HTLV-1 persistence and the oncogenesis of adult T-cell leukemia/lymphoma. Blood 141, 2299–2306 (2023).
Hirons, A., Khoury, G. & Purcell, D. F. J. Human T-cell lymphotropic virus type-1: a lifelong persistent infection, yet never truly silent. Lancet Infect. Dis. 21, e2–e10 (2021).
Lu, Z. Z. et al. Neuropilin 1 is an entry receptor for KSHV infection of mesenchymal stem cell through TGFBR1/2-mediated macropinocytosis. Sci. Adv. 9, eadg1778 (2023).
Acchioni, C., Sandini, S., Acchioni, M. & Sgarbanti, M. Co-infections and superinfections between HIV-1 and other human viruses at the cellular level. Pathogens 13, 349 (2024).
Cook, L., Melamed, A., Yaguchi, H. & Bangham, C. R. The impact of HTLV-1 on the cellular genome. Curr. Opin. Virol. 26, 125–131 (2017).
Nakano, K. & Watanabe, T. Tuning Rex rules HTLV-1 pathogenesis. Front. Immunol. 13, 959962 (2022).
Higuchi, Y. et al. HTLV-1 induces T cell malignancy and inflammation by viral antisense factor-mediated modulation of the cytokine signaling. Proc. Natl. Acad. Sci. USA 117, 13740–13749 (2020).
Toyoda, K. et al. HTLV-1 bZIP factor-induced reprogramming of lactate metabolism and epigenetic status promote leukemic cell expansion. Blood Cancer Discov. 4, 374–393 (2023).
Tan, B. J. et al. HTLV-1 infection promotes excessive T cell activation and transformation into adult T cell leukemia/lymphoma. J. Clin. Investig. 131, e150472 (2021).
Legrand, N. et al. Clinical and public health implications of human T-lymphotropic virus type 1 infection. Clin. Microbiol. Rev. 35, e0007821 (2022).
Zapatka, M. et al. The landscape of viral associations in human cancers. Nat. Genet. 52, 320–330 (2020).
Ayee, R., Ofori, M. E. O., Wright, E. & Quaye, O. Epstein Barr virus-associated lymphomas and epithelia cancers in humans. J. Cancer. 11, 1737–1750 (2020).
Koganti, S., de la Paz, A., Freeman, A. F. & Bhaduri-McIntosh, S. B lymphocytes from patients with a hypomorphic mutation in STAT3 resist Epstein-Barr virus-driven cell proliferation. J. Virol. 88, 516–524 (2014).
Swanton, C. et al. Embracing cancer complexity: hallmarks of systemic disease. Cell 187, 1589–1616 (2024).
Sharma, P. & Allison, J. P. Immune checkpoint targeting in cancer therapy: toward combination strategies with curative potential. Cell 161, 205–214 (2015).
Reuter, S., Gupta, S. C., Chaturvedi, M. M. & Aggarwal, B. B. Oxidative stress, inflammation, and cancer: how are they linked? Free Radic. Biol. Med. 49, 1603–1616 (2010).
Mantovani, A., Allavena, P., Sica, A. & Balkwill, F. Cancer-related inflammation. Nature 454, 436–444 (2008).
Digifico, E., Balinzo, S. & Belgiovine, C. The dark side of the force: when the immune system is the fuel of tumor onset. Int. J. Mol. Sci. 22, 1224 (2021).
Eble, J. A. & Niland, S. The extracellular matrix in tumor progression and metastasis. Clin. Exp. Metastasis 36, 171–198 (2019).
Lu, P., Weaver, V. M. & Werb, Z. The extracellular matrix: a dynamic niche in cancer progression. J. Cell Biol. 196, 395–406 (2012).
Carmeliet, P. & Jain, R. K. Molecular mechanisms and clinical applications of angiogenesis. Nature 473, 298–307 (2011).
Folkman, J. Angiogenesis: an organizing principle for drug discovery? Nat. Rev. Drug Discov. 6, 273–286 (2007).
Uzun, S. et al. Comprehensive analysis of VEGFR2 expression in HPV-Positive and -negative OPSCC reveals differing VEGFR2 expression patterns. Cancers 13, 5221 (2021).
Pavlova, N. N. & Thompson, C. B. The emerging hallmarks of cancer metabolism. Cell Metab. 23, 27–47 (2016).
Vander Heiden, M. G. & DeBerardinis, R. J. Understanding the intersections between metabolism and cancer biology. Cell 168, 657–669 (2017).
Sitarz, K., Czamara, K., Szostek, S. & Kaczor, A. The impact of HPV infection on human glycogen and lipid metabolism—a review. Biochim. Biophys. Acta Rev. Cancer 1877, 188646 (2022).
Hanahan, D. & Weinberg, R. A. The hallmarks of cancer. Cell 100, 57–70 (2000).
Hanahan, D. & Weinberg, R. A. Hallmarks of cancer: the next generation. Cell 144, 646–674 (2011).
Kumar, R. Review on hepatitis B virus precore/core promoter mutations and their correlation with genotypes and liver disease severity. World J. Hepatol. 14, 708–718 (2022).
Wang, Q. et al. Evaluation of polymer nanoformulations in hepatoma therapy by established rodent models. Theranostics 9, 1426–1452 (2019).
Mason, W. S. et al. HBV DNA integration and clonal hepatocyte expansion in chronic hepatitis B patients considered immune tolerant. Gastroenterology 151, 986–998.e984 (2016).
Ruan, P. et al. Different types of viral‑host junction found in HBV integration breakpoints in HBV‑infected patients. Mol. Med. Rep. 19, 1410–1416 (2019).
Wooddell, C. I. et al. RNAi-based treatment of chronically infected patients and chimpanzees reveals that integrated hepatitis B virus DNA is a source of HBsAg. Sci. Transl. Med. 9, eaan0241 (2017).
Peng, Z. et al. Integration of the hepatitis B virus X fragment in hepatocellular carcinoma and its effects on the expression of multiple molecules: a key to the cell cycle and apoptosis. Int. J. Oncol. 26, 467–473 (2005).
Ye, H. et al. Synergistic function of Kras mutation and HBx in initiation and progression of hepatocellular carcinoma in mice. Oncogene 33, 5133–5138 (2014).
Chen, L. et al. Aerobic glycolysis enhances HBx-initiated hepatocellular carcinogenesis via NF-kappaBp65/HK2 signalling. J. Exp. Clin. Cancer Res. 41, 329 (2022).
Arbuthnot, P., Capovilla, A. & Kew, M. Putative role of hepatitis B virus X protein in hepatocarcinogenesis: effects on apoptosis, DNA repair, mitogen-activated protein kinase and JAK/STAT pathways. J. Gastroenterol. Hepatol. 15, 357–368 (2000).
McBrearty, N. et al. Short chain fatty acids delay the development of hepatocellular carcinoma in HBx transgenic mice. Neoplasia 23, 529–538 (2021).
Ali, A. et al. Hepatitis B virus, HBx mutants and their role in hepatocellular carcinoma. World J. Gastroenterol. 20, 10238–10248 (2014).
Welcker, M. & Clurman, B. E. FBW7 ubiquitin ligase: a tumour suppressor at the crossroads of cell division, growth and differentiation. Nat. Rev. Cancer 8, 83–93 (2008).
Tsuchiya, H. et al. HBx and c-MYC cooperate to induce URI1 expression in HBV-related hepatocellular carcinoma. Int. J. Mol. Sci. 20, 5714 (2019).
Fu, Y. et al. Compartmentalisation of Hepatitis B virus X gene evolution in hepatocellular carcinoma microenvironment and the genotype-phenotype correlation of tumorigenicity in HBV-related patients with hepatocellular carcinoma. Emerg. Microbes Infect. 11, 2486–2501 (2022).
Dong, K. S. et al. TGF-beta1 accelerates the hepatitis B virus X-induced malignant transformation of hepatic progenitor cells by upregulating miR-199a-3p. Oncogene 39, 1807–1820 (2020).
Yuan, S. et al. HBV X protein induces degradation of UBXN7, a novel negative regulator of NF-kappaB signaling, to promote HBV replication. Cell. Mol. Gastroenterol. Hepatol. 15, 179–195 (2023).
Zhu, G. Z. et al. Comprehensive investigation of p53, p21, nm23, and VEGF expression in hepatitis B virus-related hepatocellular carcinoma overall survival after hepatectomy. J. Cancer. 11, 906–918 (2020).
Joerger, A. C., Stiewe, T. & Soussi, T. TP53: the unluckiest of genes? Cell Death Differ. 32, 219–224 (2024).
Boutelle, A. M. & Attardi, L. D. p53 and tumor suppression: it takes a network. Trends Cell Biol. 31, 298–310 (2021).
Yang, M. & Wei, W. SNHG16: A novel long-non coding RNA in human cancers. Onco. Targets Ther. 12, 11679–11690 (2019).
Liu, N. et al. Hepatitis B virus-upregulated LNC-HUR1 promotes cell proliferation and tumorigenesis by blocking p53 activity. Hepatology 68, 2130–2144 (2018).
Torrecilla, S. et al. Trunk mutational events present minimal intra- and inter-tumoral heterogeneity in hepatocellular carcinoma. J. Hepatol. 67, 1222–1231 (2017).
Tu, T., Zhang, H. & Urban, S. Hepatitis B virus DNA integration: in vitro models for investigating 2831 viral pathogenesis and persistence. Viruses 13, 180 (2021).
Fujii, R. et al. HBXIP, cellular target of hepatitis B virus oncoprotein, is a regulator of centrosome dynamics and cytokinesis. Cancer Res. 66, 9099–9107 (2006).
Li, S. K. et al. Centrosomal protein TAX1BP2 inhibits centrosome-microtubules aberrations induced by hepatitis B virus X oncoprotein. Cancer Lett. 492, 147–161 (2020).
Alt, A. et al. Specialized interfaces of Smc5/6 control hinge stability and DNA association. Nat. Commun. 8, 14011 (2017).
Murphy, C. M. et al. Hepatitis B virus X protein promotes degradation of SMC5/6 to enhance HBV replication. Cell Rep. 16, 2846–2854 (2016).
Sekiba, K. et al. HBx-induced degradation of Smc5/6 complex impairs homologous recombination-mediated repair of damaged DNA. J. Hepatol. 76, 53–62 (2022).
Gao, W. et al. HBx protein contributes to liver carcinogenesis by H3K4me3 modification through stabilizing Wd repeat domain 5 protein. Hepatology 71, 1678–1695 (2020).
D’Souza, S., Lau, K. C., Coffin, C. S. & Patel, T. R. Molecular mechanisms of viral hepatitis induced hepatocellular carcinoma. World J. Gastroenterol. 26, 5759–5783 (2020).
Qiao, L. et al. Hepatitis B virus X protein increases expression of p21(Cip-1/WAF1/MDA6) and p27(Kip-1) in primary mouse hepatocytes, leading to reduced cell cycle progression. Hepatology 34, 906–917 (2001).
Li, Y. et al. HBx downregulated decorin and decorin-derived peptides inhibit the proliferation and tumorigenicity of hepatocellular carcinoma cells. FASEB J. 37, e22871 (2023).
Chaomin, W. et al. Spatiotemporal modulation of SMAD4 by HBx is required for cellular proliferation in hepatitis B-related liver cancer. Cell Oncol. 45, 573–589 (2022).
Murata, M. et al. Hepatitis B virus X protein shifts human hepatic transforming growth factor (TGF)-beta signaling from tumor suppression to oncogenesis in early chronic hepatitis B. Hepatology 49, 1203–1217 (2009).
Rawal, P. et al. Targeted HBx gene editing by CRISPR/Cas9 system effectively reduces epithelial to mesenchymal transition and HBV replication in hepatoma cells. Liver Int. 44, 614–624 (2024).
Shan, C. et al. Hepatitis B virus X protein promotes liver cell proliferation via a positive cascade loop involving arachidonic acid metabolism and p-ERK1/2. Cell Res. 20, 563–575 (2010).
Martin-Lluesma, S. et al. Hepatitis B virus X protein affects S phase progression leading to chromosome segregation defects by binding to damaged DNA binding protein 1. Hepatology 48, 1467–1476 (2008).
Zeng, Q. et al. The nuclear matrix protein HNRNPU restricts hepatitis B virus transcription by promoting OAS3-based activation of host innate immunity. J. Med. Virol. 96, e29805 (2024).
Cheng, F. et al. HBx promotes hepatocarcinogenesis by enhancing phosphorylation and blocking ubiquitinylation of UHRF2. Hepatol. Int. 15, 707–719 (2021).
Wang, X., Lu, H., Sprangers, G. & Hallstrom, T. C. UHRF2 accumulates in early G(1)-phase after serum stimulation or mitotic exit to extend G(1) and total cell cycle length. Cell Cycle 23, 613–627 (2024).
Tong, D., Tang, Y. & Zhong, P. The emerging roles of histone demethylases in cancers. Cancer Metastasis Rev. 43, 795–821 (2024).
Lei, Y. et al. HBx induces hepatocellular carcinogenesis through ARRB1-mediated autophagy to drive the G(1)/S cycle. Autophagy 17, 4423–4441 (2021).
Suehiro, Y. et al. Hepatitis B virus (HBV) upregulates TRAIL-R3 expression in hepatocytes resulting in escape from both cell apoptosis and suppression of HBV replication by TRAIL. J. Infect. Dis. 227, 686–695 (2023).
Abe, R., Etori, K., Kato, T. & Sato, K. Experience with transurethral uretero-nephrolithotripsy using flexible nephro-ureteroscope. Nihon Hinyokika Gakkai Zasshi 81, 1667–1674 (1990).
Niller, H. H. et al. Wild type HBx and truncated HBx: pleiotropic regulators driving sequential genetic and epigenetic steps of hepatocarcinogenesis and progression of HBV-associated neoplasms. Rev. Med. Virol. 26, 57–73 (2016).
Xiao, Y. et al. Upregulation of CENPM promotes hepatocarcinogenesis through mutiple mechanisms. J. Exp. Clin. Cancer Res. 38, 458 (2019).
Salpini, R. et al. The novel HBx mutation F30V correlates with hepatocellular carcinoma in vivo, reduces hepatitis B virus replicative efficiency and enhances anti-apoptotic activity of HBx N terminus in vitro. Clin. Microbiol. Infect. 25, 906.e901–906.e907 (2019).
Yin, Y. et al. Immunosuppressive tumor microenvironment in the progression, metastasis, and therapy of hepatocellular carcinoma: from bench to bedside. Exp. Hematol. Oncol. 13, 72 (2024).
Sun, Y. et al. Hepatitis B virus-triggered PTEN/beta-catenin/c-Myc signaling enhances PD-L1 expression to promote immune evasion. Am. J. Physiol. Gastrointest. Liver Physiol. 318, G162–G173 (2020).
Montali, I. et al. Deregulated intracellular pathways define novel molecular targets for HBV-specific CD8 T cell reconstitution in chronic hepatitis B. J. Hepatol. 79, 50–60 (2023).
Bosch, M. et al. A liver immune rheostat regulates CD8 T cell immunity in chronic HBV infection. Nature 631, 867–875 (2024).
Andreata, F. et al. Therapeutic potential of co-signaling receptor modulation in hepatitis B. Cell 187, 4078–4094.e4021 (2024).
Xu, F. et al. Type III interferon-induced CBFbeta inhibits HBV replication by hijacking HBx. Cell. Mol. Immunol. 16, 357–366 (2019).
Ren, J. et al. Deubiquitylating enzymes in cancer and immunity. Adv. Sci. 10, e2303807 (2023).
Kumari, P., Narayanan, S. & Kumar, H. Herpesviruses: interfering innate immunity by targeting viral sensing and interferon pathways. Rev. Med. Virol. 25, 187–201 (2015).
Ding, S. et al. Epigenetic addition of m(5)C to HBV transcripts promotes viral replication and evasion of innate antiviral responses. Cell Death Dis. 15, 39 (2024).
Dezhbord, M. et al. Novel role of MHC class II transactivator in hepatitis B virus replication and viral counteraction. Clin. Mol. Hepatol 30, 539–560 (2024).
Fan, H. X. & Tang, H. Complex interactions between microRNAs and hepatitis B/C viruses. World J. Gastroenterol. 20, 13477–13492 (2014).
Tian, Y., Kuo, C. F., Akbari, O. & Ou, J. H. Maternal-derived hepatitis B virus e antigen alters macrophage function in offspring to drive viral persistence after vertical transmission. Immunity 44, 1204–1214 (2016).
Makarova-Rusher, O. V., Medina-Echeverz, J., Duffy, A. G. & Greten, T. F. The yin and yang of evasion and immune activation in HCC. J. Hepatol. 62, 1420–1429 (2015).
Vandeven, N. & Nghiem, P. Pathogen-driven cancers and emerging immune therapeutic strategies. Cancer Immunol. Res. 2, 9–14 (2014).
Glorieux, C., Liu, S., Trachootham, D. & Huang, P. Targeting ROS in cancer: rationale and strategies. Nat. Rev. Drug Discov. 23, 583–606 (2024).
Schollmeier, A., Glitscher, M. & Hildt, E. Relevance of HBx for hepatitis B virus-associated pathogenesis. Int. J. Mol. Sci. 24, 4964 (2023).
He, T. et al. GPR43 regulates HBV X protein (HBx)-induced inflammatory response in human LO(2) hepatocytes. Biomed. Pharmacother. 123, 109737 (2020).
Hu, Z. G. et al. DTNA promotes HBV-induced hepatocellular carcinoma progression by activating STAT3 and regulating TGFbeta1 and P53 signaling. Life Sci. 258, 118029 (2020).
Quetier, I. et al. C-terminal-truncated hepatitis B virus X protein enhances the development of diethylnitrosamine-induced hepatocellular carcinogenesis. J. Gen. Virol. 96, 614–625 (2015).
Peneau, C. et al. Hepatitis B virus integrations promote local and distant oncogenic driver alterations in hepatocellular carcinoma. Gut 71, 616–626 (2022).
Kanagal-Shamanna, R., Beck, D. B. & Calvo, K. R. Clonal hematopoiesis, inflammation, and hematologic malignancy. Annu. Rev. Pathol. 19, 479–506 (2024).
Zhou, S. L. et al. Tumor-associated neutrophils recruit macrophages and t-regulatory cells to promote progression of hepatocellular carcinoma and resistance to sorafenib. Gastroenterology 150, 1646–1658.e1617 (2016).
Yu, L. X., Ling, Y. & Wang, H. Y. Role of nonresolving inflammation in hepatocellular carcinoma development and progression. NPJ Precis. Oncol. 2, 6 (2018).
Gupta, H., Youn, G. S., Shin, M. J. & Suk, K. T. Role of gut microbiota in hepatocarcinogenesis. Microorganisms 7, (2019).
Liu, Q. et al. Alteration in gut microbiota associated with hepatitis B and non-hepatitis virus-related hepatocellular carcinoma. Gut Pathog. 11, 1 (2019).
Shigeta, K. et al. Dual programmed death receptor-1 and vascular endothelial growth factor receptor-2 blockade promotes vascular normalization and enhances antitumor immune responses in hepatocellular carcinoma. Hepatology 71, 1247–1261 (2020).
Zhang, Y. et al. Elevated HMGB1 expression induced by hepatitis B virus X protein promotes epithelial-mesenchymal transition and angiogenesis through STAT3/miR-34a/NF-kappaB in primary liver cancer. Am. J. Cancer Res. 11, 479–494 (2021).
Luo, Z. et al. Hypoxia signaling in human health and diseases: implications and prospects for therapeutics. Signal Transduct. Target Ther. 7, 218 (2022).
Hou, C. Y. et al. WNK1-OSR1 signaling regulates angiogenesis-mediated metastasis towards developing a combinatorial anti-cancer strategy. Int. J. Mol. Sci. 23, 12100 (2022).
Anderson, K. G., Stromnes, I. M. & Greenberg, P. D. Obstacles posed by the tumor microenvironment to T cell activity: a case for synergistic therapies. Cancer Cell 31, 311–325 (2017).
Phan, L. M., Yeung, S. C. & Lee, M. H. Cancer metabolic reprogramming: importance, main features, and potentials for precise targeted anti-cancer therapies. Cancer Biol. Med. 11, 1–19 (2014).
Zhang, Y. et al. C-terminal truncated HBx initiates hepatocarcinogenesis by downregulating TXNIP and reprogramming glucose metabolism. Oncogene 40, 1147–1161 (2021).
Chen, Y. et al. Research progress of TXNIP as a tumor suppressor gene participating in the metabolic reprogramming and oxidative stress of cancer cells in various cancers. Front. Oncol. 10, 568574 (2020).
You, H. et al. Hepatitis B virus X protein increases LASP1 SUMOylation to stabilize HER2 and facilitate hepatocarcinogenesis. Int. J. Biol. Macromol. 226, 996–1009 (2023).
Zhou, P. et al. SPG21, a potential oncogene targeted by miR-128-3p, amplifies HBx-induced carcinogenesis and chemoresistance via activation of TRPM7-mediated JNK pathway in hepatocellular carcinoma. Cell. Oncol. https://doi.org/10.1007/s13402-024-00955-5 (2024).
Zhang, T. T., Ye, S. S., Liang, J. & Bai, L. Prognostic value of non-invasive fibrosis indices post-curative resection in hepatitis-B-associated hepatocellular carcinoma patients. Exp. Biol. Med. 245, 703–710 (2020).
Nguyen, T. K. & Van Le, D. Identification of NS5B resistance-associated mutations in hepatitis c virus circulating in treatment naive Vietnamese patients. Infect. Drug Resist. 15, 1547–1554 (2022).
Reuther, P. et al. Persistent RNA virus infection is short-lived at the single-cell level but leaves transcriptomic footprints. J. Exp. Med. 218, e20210408 (2021).
Bunz, M. et al. CD81 suppresses NF-kappaB signaling and is downregulated in hepatitis C virus-expressing cells. Front. Cell Infect. Microbiol. 14, 1338606 (2024).
Shirvani-Dastgerdi, E., Schwartz, R. E. & Ploss, A. Hepatocarcinogenesis associated with hepatitis B, delta and C viruses. Curr. Opin. Virol. 20, 1–10 (2016).
Murata, M., Yoshida, K., Yamaguchi, T. & Matsuzaki, K. Linker phosphorylation of Smad3 promotes fibro-carcinogenesis in chronic viral hepatitis of hepatocellular carcinoma. World J. Gastroenterol. 20, 15018–15027 (2014).
Pan, Q. et al. EGFR core fucosylation, induced by hepatitis C virus, promotes TRIM40-mediated-RIG-I ubiquitination and suppresses interferon-I antiviral defenses. Nat. Commun. 15, 652 (2024).
Matsui, C. et al. Hepatitis C virus NS5A protein promotes the lysosomal degradation of hepatocyte nuclear factor 1alpha via chaperone-mediated autophagy. J. Virol. 92, e00639–00618 (2018).
Michalopoulos, G. K. & Bhushan, B. Liver regeneration: biological and pathological mechanisms and implications. Nat. Rev. Gastroenterol. Hepatol. 18, 40–55 (2021).
Capasso, M. et al. The role of hepatitis viruses as drivers of hepatocarcinogenesis. Cancers 16, 1505 (2024).
El-Shamy, A. et al. A cell culture system for distinguishing hepatitis C viruses with and without liver cancer-related mutations in the viral core gene. J. Hepatol. 63, 1323–1333 (2015).
Pascut, D. et al. HCV proteins modulate the host cell miRNA expression contributing to hepatitis C pathogenesis and hepatocellular carcinoma development. Cancers 13, 2485 (2021).
Kitab, B. & Tsukiyama-Kohara, K. Regulatory role of ribonucleotide reductase subunit M2 in hepatocyte growth and pathogenesis of hepatitis C virus. Int. J. Mol. Sci. 24, 2619 (2023).
Barik, S. Suppression of innate immunity by the hepatitis C virus (HCV): revisiting the specificity of host-virus interactive pathways. Int. J. Mol. Sci. 24, 16100 (2023).
Kwak, J., Shim, J. H., Tiwari, I. & Jang, K. L. Hepatitis C virus core protein inhibits E6AP expression via DNA methylation to escape from ubiquitin-dependent proteasomal degradation. Cancer Lett. 380, 59–68 (2016).
Baek, K. H. et al. Overexpression of hepatitis C virus NS5A protein induces chromosome instability via mitotic cell cycle dysregulation. J. Mol. Biol. 359, 22–34 (2006).
Cancer genome atlas research network. electronic address: wheeler@bcm.edu; cancer genome atlas research network. Comprehensive and integrative genomic characterization of hepatocellular carcinoma. Cell. 169, 1327–1341.e1323 (2017).
Fujita, M. et al. Proteo-genomic characterization of virus-associated liver cancers reveals potential subtypes and therapeutic targets. Nat. Commun. 13, 6481 (2022).
Perez, S. et al. High-resolution genomic profiling of liver cancer links etiology with mutation and epigenetic signatures. Cell. Mol. Gastroenterol. Hepatol. 16, 63–81 (2023).
Peng, J. L. et al. Association of RASSF1A hypermethylation with risk of HBV/HCV-induced hepatocellular carcinoma: a meta-analysis. Pathol. Res. Pract. 216, 153099 (2020).
Endo, Y. et al. Expression of activation-induced cytidine deaminase in human hepatocytes via NF-kappaB signaling. Oncogene 26, 5587–5595 (2007).
Jiang, X. H. et al. Effects of hepatitis C virus core protein and nonstructural protein 4B on the Wnt/beta-catenin pathway. BMC Microbiol. 17, 124 (2017).
McGivern, D. R. & Lemon, S. M. Virus-specific mechanisms of carcinogenesis in hepatitis C virus-associated liver cancer. Oncogene 30, 1969–1983 (2011).
Higgs, M. R., Lerat, H. & Pawlotsky, J. M. Hepatitis C virus-induced activation of beta-catenin promotes c-Myc expression and a cascade of pro-carcinogenetic events. Oncogene 32, 4683–4693 (2013).
Meyers, N. L. et al. Hepatitis C virus infects and perturbs liver stem cells. mBio 14, e0131823 (2023).
Sohn, W. et al. Isolated hepatitis B core antibody positivity and long-term liver-related mortality in Korea: a cohort study. Am. J. Gastroenterol. 118, 95–104 (2023).
Puigvehi, M., Moctezuma-Velazquez, C., Villanueva, A. & Llovet, J. M. The oncogenic role of hepatitis delta virus in hepatocellular carcinoma. JHEP Rep. 1, 120–130 (2019).
Tanaka, N. et al. PPARalpha activation is essential for HCV core protein-induced hepatic steatosis and hepatocellular carcinoma in mice. J. Clin. Invest. 118, 683–694 (2008).
Kayesh, M. E. H., Kohara, M. & Tsukiyama-Kohara, K. Toll-like receptor response to hepatitis C virus infection: a recent overview. Int. J. Mol. Sci. 23, 5475 (2022).
Westbrook, R. H. & Dusheiko, G. Natural history of hepatitis C. J. Hepatol. 61, S58–S68 (2014).
Lidofsky, A. et al. Macrophage activation marker soluble CD163 is a dynamic marker of liver fibrogenesis in human immunodeficiency virus/hepatitis C virus coinfection. J. Infect. Dis. 218, 1394–1403 (2018).
Strunz, B. et al. Chronic hepatitis C virus infection irreversibly impacts human natural killer cell repertoire diversity. Nat. Commun. 9, 2275 (2018).
Song, G. et al. Global immune characterization of HBV/HCV-related hepatocellular carcinoma identifies macrophage and T-cell subsets associated with disease progression. Cell Discov. 6, 90 (2020).
Deng, Z. et al. TGF-beta signaling in health, disease, and therapeutics. Signal Transduct. Target Ther. 9, 61 (2024).
Harhaj, E. W. & Shembade, N. Lymphotropic viruses: chronic inflammation and induction of cancers. Biology 9, 390 (2020).
Hengst, J. et al. Nonreversible MAIT cell-dysfunction in chronic hepatitis C virus infection despite successful interferon-free therapy. Eur. J. Immunol. 46, 2204–2210 (2016).
Blank, C. U. et al. Defining ‘T cell exhaustion. Nat. Rev. Immunol. 19, 665–674 (2019).
Szilveszter, R. M., Muntean, M. & Florea, A. Molecular mechanisms in tumorigenesis of hepatocellular carcinoma and in target treatments-an overview. Biomolecules. 14, 656 (2024).
Cabrera, R. et al. An immunomodulatory role for CD4(+)CD25(+) regulatory T lymphocytes in hepatitis C virus infection. Hepatology 40, 1062–1071 (2004).
Hartling, H. J. et al. Immune regulation in chronic hepatitis C virus infection. Scand. J. Gastroenterol. 51, 1387–1397 (2016).
Chen, W. et al. HCV genomic RNA activates the NLRP3 inflammasome in human myeloid cells. PLoS One 9, e84953 (2014).
Appay, V. & Rowland-Jones, S. L. RANTES: a versatile and controversial chemokine. Trends Immunol. 22, 83–87 (2001).
De Battista, D. et al. Molecular signature and immune landscape of HCV-associated hepatocellular carcinoma (HCC): differences and similarities with HBV-HCC. J. Hepatocell Carcinoma 8, 1399–1413 (2021).
El-Mowafy, M. et al. Changes of gut-microbiota-liver axis in hepatitis C virus infection. Biology 10, 55 (2021).
Wu, M. Y. et al. Molecular targets in hepatocarcinogenesis and implications for therapy. J. Clin. Med. 7, 213 (2018).
Castello, G. et al. HCV-related hepatocellular carcinoma: From chronic inflammation to cancer. Clin. Immunol. 134, 237–250 (2010).
Reungoat, E., Grigorov, B., Zoulim, F. & Pecheur, E. I. Molecular crosstalk between the hepatitis C virus and the extracellular matrix in liver fibrogenesis and early carcinogenesis. Cancers 13, 2270 (2021).
Yu, W. et al. Reactive oxygen species bridge the gap between chronic inflammation and tumor development. Oxid. Med. Cell. Longev. 2022, 2606928 (2022).
Kalantari, L. et al. A state-of-the-art review on the NRF2 in hepatitis virus-associated liver cancer. Cell Commun. Signal 21, 318 (2023).
Abouelasrar Salama, S. et al. Cytokines and serum amyloid A in the pathogenesis of hepatitis C virus infection. Cytokine Growth Factor Rev. 50, 29–42 (2019).
Dios-Barbeito, S. et al. Impact of nitric oxide in liver cancer microenvironment. Nitric Oxide 128, 1–11 (2022).
Sepulveda-Crespo, D., Resino, S. & Martinez, I. Strategies targeting the innate immune response for the treatment of hepatitis C virus-associated liver fibrosis. Drugs 81, 419–443 (2021).
Ismail, M. et al. Dynamic role of exosomal long non-coding RNA in liver diseases: pathogenesis and diagnostic aspects. Hepatol. Int., 18, 1715–1730 (2024).
Lupberger, J. et al. Combined analysis of metabolomes, proteomes, and transcriptomes of hepatitis C virus-infected cells and liver to identify pathways associated with disease development. Gastroenterology 157, 537–551 (2019).
Hiet, M. S. et al. Control of temporal activation of hepatitis C virus-induced interferon response by domain 2 of nonstructural protein 5A. J. Hepatol. 63, 829–837 (2015).
Diaz, O. et al. What role for cellular metabolism in the control of hepatitis viruses? Front. Immunol. 13, 1033314 (2022).
Leslie, J. et al. Metabolic dysfunction and cancer in HCV: shared pathways and mutual interactions. J. Hepatol. 77, 219–236 (2022).
Salnikov, M. Y., MacNeil, K. M. & Mymryk, J. S. The viral etiology of EBV-associated gastric cancers contributes to their unique pathology, clinical outcomes, treatment responses and immune landscape. Front. Immunol. 15, 1358511 (2024).
Giammanco, A. et al. Helicobacter pylori and Epstein-Barr virus co-infection in gastric disease: what is the correlation with p53 mutation, genes methylation and microsatellite instability in a cohort of sicilian population? Int. J. Mol. Sci. 24, 8104 (2023).
Yau, T. O., Tang, C. M. & Yu, J. Epigenetic dysregulation in Epstein-Barr virus-associated gastric carcinoma: disease and treatments. World J. Gastroenterol. 20, 6448–6456 (2014).
Camargo, M. C. et al. Case-case comparison of smoking and alcohol risk associations with Epstein-Barr virus-positive gastric cancer. Int. J. Cancer. 134, 948–953 (2014).
Kim, J. Y. et al. Ceramide promotes lytic reactivation of Epstein-Barr virus in gastric carcinoma. J. Virol. 98, e0177623 (2024).
Ribeiro, J. et al. P53 deregulation in Epstein-Barr virus-associated gastric cancer. Cancer Lett 404, 37–43 (2017).
Elgui de Oliveira, D., Muller-Coan, B. G. & Pagano, J. S. Viral carcinogenesis beyond malignant transformation: EBV in the progression of human cancers. Trends Microbiol 24, 649–664 (2016).
Nishikawa, J. et al. The role of epigenetic regulation in Epstein-Barr virus-associated gastric cancer. Int. J. Mol. Sci. 18, 1606 (2017).
Liu, M. T. et al. Epstein-Barr virus latent membrane protein 1 represses p53-mediated DNA repair and transcriptional activity. Oncogene 24, 2635–2646 (2005).
Kraus, R. J. et al. Reactivation of Epstein-Barr virus by HIF-1alpha requires p53. J. Virol. 94, e00722–00720 (2020).
Sato, Y. et al. Degradation of phosphorylated p53 by viral protein-ECS E3 ligase complex. PLoS Pathog. 5, e1000530 (2009).
Felismino, T. C. & Coimbra, F. J. F. Lessons in gastric adenocarcinoma from TCGA. Nat. Rev. Cancer. 23, 655 (2023).
Yoon, C. J. et al. Epstein-Barr virus-encoded miR-BART5-5p upregulates PD-L1 through PIAS3/pSTAT3 modulation, worsening clinical outcomes of PD-L1-positive gastric carcinomas. Gastric Cancer 23, 780–795 (2020).
Wang, J. et al. Epstein-Barr virus miR-BART3-3p promotes tumorigenesis by regulating the senescence pathway in gastric cancer. J. Biol. Chem. 294, 4854–4866 (2019).
Huang, S. et al. Sustained activation of the JNK cascade and rapamycin-induced apoptosis are suppressed by p53/p21(Cip1). Mol. Cell. 11, 1491–1501 (2003).
Li, J. et al. LMP1 induces p53 protein expression via the H19/miR-675-5p axis. Microbiol. Spectr. 10, e0000622 (2022).
Zhang, Y. et al. Downregulation of MUS81 expression inhibits cell migration and maintains EBV latent infection through miR-BART9-5p in EBV-associated gastric cancer. J. Med. Virol. 95, e28725 (2023).
Zheng, X. et al. Epstein-Barr virus microRNA miR-BART5-3p inhibits p53 expression. J. Virol. 92, e01022–01018 (2018).
Naseem, M. et al. Outlooks on Epstein-Barr virus associated gastric cancer. Cancer Treat. Rev. 66, 15–22 (2018).
Chen, H. Y. et al. High CpG island methylator phenotype is associated with lymph node metastasis and prognosis in gastric cancer. Cancer Sci. 103, 73–79 (2012).
Toh, J. W. T. & Wilson, R. B. Pathways of gastric carcinogenesis, helicobacter pylori virulence and interactions with antioxidant systems, vitamin C and phytochemicals. Int. J. Mol. Sci. 21, 6451 (2020).
Cancer Genome Atlas Research, N. Comprehensive molecular characterization of gastric adenocarcinoma. Nature 513, 202–209 (2014).
Elizazu, J. et al. Identification of a novel gene signature related to prognosis and metastasis in gastric cancer. Cell. Oncol. (Dordr.). 47, 1355–1373 (2024).
Shinozaki-Ushiku, A., Kunita, A. & Fukayama, M. Update on Epstein-Barr virus and gastric cancer (review). Int. J. Oncol. 46, 1421–1434 (2015).
Shi, D. et al. Research advances in the molecular classification of gastric cancer. Cell. Oncol. https://doi.org/10.1007/s13402-024-00951-9 (2024).
Choi, S. J. et al. DNA hypermethylation induced by Epstein-Barr virus in the development of Epstein-Barr virus-associated gastric carcinoma. Arch. Pharm. Res. 40, 894–905 (2017).
Min, K. & Lee, S. K. EBV miR-BART10-3p promotes cell proliferation and migration by targeting DKK1. Int. J. Biol. Sci. 15, 657–667 (2019).
Song, H. et al. Activation of DNA methyltransferase 3a by Epstein-Barr nuclear antigen 1 in gastric carcinoma. Dig. Liver Dis. 54, 973–983 (2022).
Kang, M. S. & Kieff, E. Epstein-Barr virus latent genes. Exp. Mol. Med. 47, e131 (2015).
Li, W. et al. Activation of EHF via STAT3 phosphorylation by LMP2A in Epstein-Barr virus-positive gastric cancer. Cancer Sci. 112, 3349–3362 (2021).
Hino, R. et al. Activation of DNA methyltransferase 1 by EBV latent membrane protein 2A leads to promoter hypermethylation of PTEN gene in gastric carcinoma. Cancer Res. 69, 2766–2774 (2009).
Liu, J. et al. Epstein-Barr Virus-encoded latent membrane protein 2A downregulates GCNT3 via the TGF-beta1/Smad-mTORC1 signaling axis. J. Virol. 95, e02481–02420 (2021).
Xiao, H. et al. EBV downregulates the m(6)A “writer” WTAP in EBV-associated gastric carcinoma. Virus Res. 304, 198510 (2021).
Shinozaki-Ushiku, A. et al. Profiling of virus-encoded microRNAs in Epstein-Barr virus-associated gastric carcinoma and their roles in gastric carcinogenesis. J. Virol. 89, 5581–5591 (2015).
Kase, K. et al. ARID1A deficiency in EBV-positive gastric cancer is partially regulated by EBV-encoded miRNAs, but not by DNA promotor hypermethylation. Carcinogenesis 42, 21–30 (2021).
Saito, M. & Kono, K. Landscape of EBV-positive gastric cancer. Gastric Cancer 24, 983–989 (2021).
Min, K., Kim, J. Y. & Lee, S. K. Epstein-Barr virus miR-BART1-3p suppresses apoptosis and promotes migration of gastric carcinoma cells by targeting DAB2. Int. J. Biol. Sci. 16, 694–707 (2020).
Wang, D. et al. Epstein-Barr virus-encoded miR-BART6-3p inhibits cancer cell proliferation through the LOC553103-STMN1 axis. FASEB J. 34, 8012–8027 (2020).
Kim, H., Choi, H. & Lee, S. K. Epstein-Barr virus miR-BART20-5p regulates cell proliferation and apoptosis by targeting BAD. Cancer Lett. 356, 733–742 (2015).
Leong, M. M. L. & Lung, M. L. The impact of epstein-barr virus infection on epigenetic regulation of host cell gene expression in epithelial and lymphocytic malignancies. Front. Oncol. 11, 629780 (2021).
Ohtani, N. et al. Epstein-Barr virus LMP1 blocks p16INK4a-RB pathway by promoting nuclear export of E2F4/5. J. Cell Biol. 162, 173–183 (2003).
Dong, W. et al. Virus-induced host genomic remodeling dysregulates gene expression, triggering tumorigenesis. Front. Cell Infect. Microbiol. 14, 1359766 (2024).
Frappier, L. Contributions of Epstein-Barr nuclear antigen 1 (EBNA1) to cell immortalization and survival. Viruses 4, 1537–1547 (2012).
Fukuda, M. & Longnecker, R. Latent membrane protein 2A inhibits transforming growth factor-beta 1-induced apoptosis through the phosphatidylinositol 3-kinase/Akt pathway. J. Virol. 78, 1697–1705 (2004).
Hino, R. et al. Survival advantage of EBV-associated gastric carcinoma: survivin up-regulation by viral latent membrane protein 2A. Cancer Res. 68, 1427–1435 (2008).
Choy, E. Y. et al. An Epstein-Barr virus-encoded microRNA targets PUMA to promote host cell survival. J. Exp. Med. 205, 2551–2560 (2008).
Lin, X. et al. The Epstein-Barr virus BART miRNA cluster of the M81 strain modulates multiple functions in primary B cells. PLoS Pathog. 11, e1005344 (2015).
Duan, X. et al. EBV infection in epithelial malignancies induces resistance to antitumor natural killer cells via F3-mediated platelet aggregation. Cancer Res. 82, 1070–1083 (2022).
Fiches, G. N. et al. Profiling of immune related genes silenced in EBV-positive gastric carcinoma identified novel restriction factors of human gammaherpesviruses. PLoS Pathog. 16, e1008778 (2020).
Bos, J. et al. The tumor immune composition of mismatch repair deficient and Epstein-Barr virus-positive gastric cancer: a systematic review. Cancer Treat. Rev. 127, 102737 (2024).
Gong, L. P. et al. The implication of tumor-infiltrating lymphocytes in Epstein-Barr virus-associated gastric carcinoma. Hum. Pathol. 85, 82–91 (2019).
Salnikov, M. Y., Fonseca, G. J. & Mymryk, J. S. Differences in the tumor microenvironment of EBV-associated gastric cancers revealed using single-cell transcriptome analysis. Cancers 15, 3178 (2023).
Qiu, M. Z. et al. Dynamic single-cell mapping unveils Epstein‒Barr virus-imprinted T-cell exhaustion and on-treatment response. Signal Transduct. Target Ther. 8, 370 (2023).
Saito, R. et al. Overexpression and gene amplification of PD-L1 in cancer cells and PD-L1(+) immune cells in Epstein-Barr virus-associated gastric cancer: the prognostic implications. Mod. Pathol. 30, 427–439 (2017).
Strong, M. J. et al. Differences in gastric carcinoma microenvironment stratify according to EBV infection intensity: implications for possible immune adjuvant therapy. PLoS Pathog. 9, e1003341 (2013).
Wang, J. et al. EBV miRNAs BART11 and BART17-3p promote immune escape through the enhancer-mediated transcription of PD-L1. Nat. Commun. 13, 866 (2022).
Horst, D. et al. Specific targeting of the EBV lytic phase protein BNLF2a to the transporter associated with antigen processing results in impairment of HLA class I-restricted antigen presentation. J. Immunol. 182, 2313–2324 (2009).
Mathebela, P. et al. Influence of the microbiome metagenomics and epigenomics on gastric cancer. Int. J. Mol. Sci. 23, 13750 (2022).
Mima, K. et al. Fusobacterium nucleatum in colorectal carcinoma tissue and patient prognosis. Gut 65, 1973–1980 (2016).
Nkosi, D., Sun, L., Duke, L. C. & Meckes, D. G. Jr. Epstein-Barr virus LMP1 manipulates the content and functions of extracellular vesicles to enhance metastatic potential of recipient cells. PLoS Pathog. 16, e1009023 (2020).
Shinozaki, A. et al. Downregulation of microRNA-200 in EBV-associated gastric carcinoma. Cancer Res. 70, 4719–4727 (2010).
Tsai, C. Y. et al. Comprehensive profiling of virus microRNAs of Epstein-Barr virus-associated gastric carcinoma: highlighting the interactions of ebv-Bart9 and host tumor cells. J. Gastroenterol. Hepatol. 32, 82–91 (2017).
Che, L. et al. Intracellular antibody targeting HBx suppresses invasion and metastasis in hepatitis B virus-related hepatocarcinogenesis via protein phosphatase 2A-B56gamma-mediated dephosphorylation of protein kinase B. Cell Prolif. 55, e13304 (2022).
Du, Y. et al. Hypoxia-induced EBV-circLMP2A promotes angiogenesis in EBV-associated gastric carcinoma through the KHSRP/VHL/HIF1alpha/VEGFA pathway. Cancer Lett. 526, 259–272 (2022).
Wu, Y. et al. EBV-miR-BART12 accelerates migration and invasion in EBV-associated cancer cells by targeting tubulin polymerization-promoting protein 1. FASEB J. 34, 16205–16223 (2020).
Sunakawa, Y. & Lenz, H. J. Molecular classification of gastric adenocarcinoma: translating new insights from the cancer genome atlas research network. Curr. Treat. Options Oncol. 16, 17 (2015).
Xiang, T. et al. Vasculogenic mimicry formation in EBV-associated epithelial malignancies. Nat. Commun. 9, 5009 (2018).
Akbari, E., Milani, A., Seyedinkhorasani, M. & Bolhassani, A. HPV co-infections with other pathogens in cancer development: a comprehensive review. J. Med. Virol. 95, e29236 (2023).
Szymonowicz, K. A. & Chen, J. Biological and clinical aspects of HPV-related cancers. Cancer Biol. Med. 17, 864–878 (2020).
Gutierrez-Xicotencatl, L. et al. Cellular Functions of HPV16 E5 oncoprotein during oncogenic transformation. Mol. Cancer Res. 19, 167–179 (2021).
Kumar, A., Rathi, E., Hariharapura, R. C. & Kini, S. G. Is viral E6 oncoprotein a viable target? A critical analysis in the context of cervical cancer. Med. Res. Rev. 40, 2019–2048 (2020).
Vats, A., Trejo-Cerro, O., Thomas, M. & Banks, L. Human papillomavirus E6 and E7: what remains? Tumour Virus Res. 11, 200213 (2021).
Ren, S. et al. HPV E2, E4, E5 drive alternative carcinogenic pathways in HPV positive cancers. Oncogene 39, 6327–6339 (2020).
Wendel, S. O. et al. HPV 16 E7 alters translesion synthesis signaling. Virol. J. 19, 165 (2022).
Lagstrom, S. et al. HPV16 and HPV18 type-specific APOBEC3 and integration profiles in different diagnostic categories of cervical samples. Tumour Virus Res. 12, 200221 (2021).
Gagliardi, A. et al. Analysis of Ugandan cervical carcinomas identifies human papillomavirus clade-specific epigenome and transcriptome landscapes. Nat. Genet. 52, 800–810 (2020).
Butler, K. & Banday, A. R. APOBEC3-mediated mutagenesis in cancer: causes, clinical significance and therapeutic potential. J. Hematol. Oncol. 16, 31 (2023).
Revathidevi, S. et al. APOBEC: a molecular driver in cervical cancer pathogenesis. Cancer Lett. 496, 104–116 (2021).
Chen, X. et al. E6 protein expressed by high-risk HPV activates super-enhancers of the EGFR and c-MET oncogenes by destabilizing the histone demethylase KDM5C. Cancer Res. 78, 1418–1430 (2018).
Khan, A. et al. Insights into the role of complement regulatory proteins in HPV-mediated cervical carcinogenesis. Semin. Cancer Biol. 86, 583–589 (2022).
Wang, J. C. K. et al. Structure of the p53 degradation complex from HPV16. Nat. Commun. 15, 1842 (2024).
Drews, C. M., Brimer, N. & Vande Pol, S. B. Multiple regions of E6AP (UBE3A) contribute to interaction with papillomavirus E6 proteins and the activation of ubiquitin ligase activity. PLoS Pathog. 16, e1008295 (2020).
Yi, S. A. et al. HPV-mediated nuclear export of HP1gamma drives cervical tumorigenesis by downregulation of p53. Cell Death Differ. 27, 2537–2551 (2020).
Lau, L., Gray, E. E., Brunette, R. L. & Stetson, D. B. DNA tumor virus oncogenes antagonize the cGAS-STING DNA-sensing pathway. Science 350, 568–571 (2015).
Puram, S. V. et al. Cellular states are coupled to genomic and viral heterogeneity in HPV-related oropharyngeal carcinoma. Nat. Genet. 55, 640–650 (2023).
Pifer, P. M. et al. FAK drives resistance to therapy in HPV-negative head and neck cancer in a p53-dependent manner. Clin. Cancer Res. 30, 187–197 (2024).
Bruyere, D. et al. Human papillomavirus E6/E7 oncoproteins promote radiotherapy-mediated tumor suppression by globally hijacking host DNA damage repair. Theranostics 13, 1130–1149 (2023).
Squarzanti, D. F. et al. Human papillomavirus type 16 E6 and E7 oncoproteins interact with the nuclear p53-binding protein 1 in an in vitro reconstructed 3D epithelium: new insights for the virus-induced DNA damage response. Virol. J. 15, 176 (2018).
Niehrs, C. & Luke, B. Regulatory R-loops as facilitators of gene expression and genome stability. Nat. Rev. Mol. Cell Biol. 21, 167–178 (2020).
Templeton, C. W. & Laimins, L. A. p53-dependent R-loop formation and HPV pathogenesis. Proc. Natl. Acad. Sci. USA 120, e2305907120 (2023).
Porter, V. L. & Marra, M. A. The drivers, mechanisms, and consequences of genome instability in HPV-driven cancers. Cancers 14, 4623 (2022).
Gao, S. et al. E2F1 mediates the downregulation of POLD1 in replicative senescence. Cell Mol. Life Sci. 76, 2833–2850 (2019).
Marima, R. et al. The catastrophic HPV/HIV dual viral oncogenomics in concert with dysregulated alternative splicing in cervical cancer. Int. J. Mol. Sci. 22, 10115 (2021).
Gao, J. & Pickett, H. A. Targeting telomeres: advances in telomere maintenance mechanism-specific cancer therapies. Nat. Rev. Cancer. 22, 515–532 (2022).
Kumar, N. & Sethi, G. Telomerase and hallmarks of cancer: An intricate interplay governing cancer cell evolution. Cancer Lett. 578, 216459 (2023).
Wang, L. N. et al. The association of telomere maintenance and TERT expression with susceptibility to human papillomavirus infection in cervical epithelium. Cell Mol. Life Sci. 79, 110 (2022).
Yang, R. et al. Regulation and clinical potential of telomerase reverse transcriptase (TERT/hTERT) in breast cancer. Cell Commun. Signal 21, 218 (2023).
Vliet-Gregg, P. A. et al. NFX1-123 is highly expressed in cervical cancer and increases growth and telomerase activity in HPV 16E6 expressing cells. Cancer Lett. 449, 106–113 (2019).
Tornesello, M. L. et al. The molecular interplay between human oncoviruses and telomerase in cancer development. Cancers 14, 5257 (2022).
Plug-DeMaggio, A. W. et al. Telomere erosion and chromosomal instability in cells expressing the HPV oncogene 16E6. Oncogene 23, 3561–3571 (2004).
Studstill, C. J. & Moody, C. A. For better or worse: modulation of the host DNA damage response by human papillomavirus. Annu. Rev. Virol. 10, 325–345 (2023).
Skelin, J., Sabol, I. & Tomaic, V. Do or Die: HPV E5, E6 and E7 in cell death evasion. Pathogens 11, 1027 (2022).
Chen, B., Zhao, L., Yang, R. & Xu, T. Advances in molecular mechanism of HPV16 E5 oncoprotein carcinogenesis. Arch. Biochem. Biophys. 745, 109716 (2023).
Miyauchi, S. et al. Human papillomavirus E5 suppresses immunity via inhibition of the immunoproteasome and STING pathway. Cell Rep. 42, 112508 (2023).
Ilahi, N. E. & Bhatti, A. Impact of HPV E5 on viral life cycle via EGFR signaling. Microb. Pathog. 139, 103923 (2020).
Pang, C. L. & Thierry, F. Human papillomavirus proteins as prospective therapeutic targets. Microb. Pathog. 58, 55–65 (2013).
Perez-Plasencia, C. et al. Genome wide expression analysis in HPV16 cervical cancer: identification of altered metabolic pathways. Infect. Agent. Cancer. 2, 16 (2007).
Zhao, H. et al. HPV16 infection promotes the malignant transformation of the esophagus and progression of esophageal squamous cell carcinoma. J. Med. Virol. 95, e29132 (2023).
Uxa, S. et al. DREAM and RB cooperate to induce gene repression and cell-cycle arrest in response to p53 activation. Nucleic Acids Res. 47, 9087–9103 (2019).
James, C. D. et al. Restoring the DREAM complex inhibits the proliferation of high-risk HPV-positive human cells. Cancers 13, 489 (2021).
Pang, C. L. et al. A functional interaction of E7 with B-Myb-MuvB complex promotes acute cooperative transcriptional activation of both S- and M-phase genes. (129 c). Oncogene 33, 4039–4049 (2014).
Fischer, M. et al. Human papillomavirus E7 oncoprotein abrogates the p53-p21-DREAM pathway. Sci. Rep. 7, 2603 (2017).
Perez-Gonzalez, A., Cachay, E., Ocampo, A. & Poveda, E. Update on the epidemiological features and clinical implications of human papillomavirus infection (HPV) and human immunodeficiency virus (HIV) coinfection. Microorganisms 10, 1047 (2022).
Tavakolian, S., Goudarzi, H. & Faghihloo, E. Cyclin-dependent kinases and CDK inhibitors in virus-associated cancers. Infect. Agent. Cancer. 15, 27 (2020).
Engeland, K. Cell cycle regulation: p53-p21-RB signaling. Cell Death Differ. 29, 946–960 (2022).
Kwon, A. Y. et al. miR-22-3p and miR-30e-5p are associated with prognosis in cervical squamous cell carcinoma. Int. J. Mol. Sci. 23, 5623 (2022).
Li, S. et al. E6-encoded by cancer-causing human papillomavirus interacts with aurora kinase a to promote HPV-mediated carcinogenesis. J. Virol. 97, e0187222 (2023).
Khalil, M. I. et al. HPV upregulates MARCHF8 ubiquitin ligase and inhibits apoptosis by degrading the death receptors in head and neck cancer. PLoS Pathog. 19, e1011171 (2023).
Cararo-Lopes, E. et al. Autophagy buffers Ras-induced genotoxic stress enabling malignant transformation in keratinocytes primed by human papillomavirus. Cell Death Dis 12, 194 (2021).
Dan, V. M. et al. Streptomyces sp metabolite(s) promotes Bax mediated intrinsic apoptosis and autophagy involving inhibition of mTOR pathway in cervical cancer cell lines. Sci. Rep. 8, 2810 (2018).
Iyer, S. et al. Robust autoactivation for apoptosis by BAK but not BAX highlights BAK as an important therapeutic target. Cell Death Dis. 11, 268 (2020).
Antonioli, M. et al. HPV sensitizes OPSCC cells to cisplatin-induced apoptosis by inhibiting autophagy through E7-mediated degradation of AMBRA1. Autophagy 17, 2842–2855 (2021).
Zhitkevich, A. et al. HIV-1 reverse transcriptase expression in HPV16-infected epidermoid carcinoma cells alters E6 expression and cellular metabolism, and induces a hybrid epithelial/mesenchymal cell phenotype. Viruses 16, 193 (2024).
Ding, C. et al. Small molecules targeting the innate immune cGAS‒STING‒TBK1 signaling pathway. Acta Pharm. Sin B 10, 2272–2298 (2020).
Luo, X. et al. HPV16 drives cancer immune escape via NLRX1-mediated degradation of STING. J. Clin. Invest. 130, 1635–1652 (2020).
Poirson, J. et al. High-risk mucosal human papillomavirus 16 (HPV16) E6 protein and cutaneous HPV5 and HPV8 E6 proteins employ distinct strategies to interfere with interferon regulatory factor 3-mediated beta interferon expression. J. Virol. 96, e0187521 (2022).
Shamseddine, A. A. et al. Tumor immunity and immunotherapy for HPV-related cancers. Cancer Discov. 11, 1896–1912 (2021).
Israr, M., DeVoti, J. A., Papayannakos, C. J. & Bonagura, V. R. Role of chemokines in HPV-induced cancers. Semin. Cancer Biol. 87, 170–183 (2022).
Wang, W. et al. Stress keratin 17 enhances papillomavirus infection-induced disease by downregulating T cell recruitment. PLoS Pathog. 16, e1008206 (2020).
Mempel, T. R., Lill, J. K. & Altenburger, L. M. How chemokines organize the tumour microenvironment. Nat. Rev. Cancer 24, 28–50 (2024).
Graham, S. V. Keratinocyte differentiation-dependent human papillomavirus gene regulation. Viruses 9, 245 (2017).
Luo, X. et al. Immune escape of head and neck cancer mediated by the impaired MHC-I antigen presentation pathway. Oncogene 43, 388–394 (2024).
Rosendo-Chalma, P. et al. The hallmarks of cervical cancer: molecular mechanisms induced by human papillomavirus. Biology 13, 77 (2024).
Kyrgiou, M. & Moscicki, A. B. Vaginal microbiome and cervical cancer. Semin Cancer Biol. 86, 189–198 (2022).
Logel, M. et al. A narrative review of the putative etiologic role and diagnostic utility of the cervicovaginal microbiome in human papillomavirus-associated cervical carcinogenesis. J. Med. Virol. 96, e70027 (2024).
Dong, B. et al. Prevotella as the hub of the cervicovaginal microbiota affects the occurrence of persistent human papillomavirus infection and cervical lesions in women of childbearing age via host NF-κB/C-myc. J. Med. Virol. 94, 5519–5534 (2022).
Nicolò, S. et al. Bacterial species from vaginal microbiota differently affect the production of the E6 and E7 oncoproteins and of p53 and p-Rb oncosuppressors in HPV16-infected cells. Int. J. Mol. Sci. 24, (2023).
Łaniewski, P. et al. Vaginal microbiota, genital inflammation, and neoplasia impact immune checkpoint protein profiles in the cervicovaginal microenvironment. NPJ. Precis. Oncol. 4, 22 (2020).
Miyauchi, S. et al. HPV16 E5 mediates resistance to PD-L1 blockade and can be targeted with rimantadine in head and neck cancer. Cancer Res. 80, 732–746 (2020).
Santegoets, S. J. et al. CD163(+) cytokine-producing cDC2 stimulate intratumoral type 1 T cell responses in HPV16-induced oropharyngeal cancer. J. Immunother Cancer 8, (2020).
Santegoets, S. J., Stolk, A., Welters, M. J. P. & van der Burg, S. H. The combined HPV16-E2/E6/E7 T cell response in oropharyngeal cancer predicts superior survival. Cell Rep. Med. 4, 101262 (2023).
Karin, M. & Shalapour, S. Regulation of antitumor immunity by inflammation-induced epigenetic alterations. Cell. Mol. Immunol. 19, 59–66 (2022).
Ju, Z. et al. Recent development on COX-2 inhibitors as promising anti-inflammatory agents: the past 10 years. Acta Pharm Sin B 12, 2790–2807 (2022).
Garcia-Quiroz, J. et al. The interaction of human papillomavirus infection and prostaglandin E(2) signaling in carcinogenesis: a focus on cervical cancer therapeutics. Cells 11, 2528 (2022).
Mangino, G. et al. Inflammatory microenvironment and human papillomavirus-induced carcinogenesis. Cytokine Growth Factor Rev. 30, 103–111 (2016).
Cabeca, T. K. et al. HPV-mediated resistance to TNF and TRAIL is characterized by global alterations in apoptosis regulatory factors, dysregulation of death receptors, and induction of ROS/RNS. Int. J. Mol. Sci. 20, 198 (2019).
Cruz-Gregorio, A. et al. Human papillomavirus types 16 and 18 early-expressed proteins differentially modulate the cellular redox state and DNA damage. Int. J. Biol. Sci. 14, 21–35 (2018).
Li, H. et al. HIV-1-infected cell-derived exosomes promote the growth and progression of cervical cancer. Int. J. Biol. Sci. 15, 2438–2447 (2019).
Han, L., Lam, E. W. & Sun, Y. Extracellular vesicles in the tumor microenvironment: old stories, but new tales. Mol. Cancer 18, 59 (2019).
Qiu, J. J. et al. Extracellular vesicular Wnt7b mediates HPV E6-induced cervical cancer angiogenesis by activating the beta-catenin signaling pathway. J. Exp. Clin. Cancer Res. 39, 260 (2020).
Heydarnia, E. et al. Circular RNAs and cervical cancer: friends or foes? A landscape on circRNA-mediated regulation of key signaling pathways involved in the onset and progression of HPV-related cervical neoplasms. Cell Commun. Signal 22, 107 (2024).
Sirico, A. et al. BDNF and NGF expression in preneoplastic cervical disease according to HIV Status. Int. J. Mol. Sci. 24, 10729 (2023).
Tian, Y., Zhang, S. & Ni, F. Targeting glucose metabolism for HPV-associated cervical cancer: a sweet poison. Biomed. Pharmacother. 180, 117519 (2024).
Priego-Hernandez, V. D. et al. Expression of HIF-1alpha and genes involved in glucose metabolism is increased in cervical cancer and HPV-16-positive cell lines. Pathogens. 12, 33 (2022).
Zwerschke, W. et al. Modulation of type M2 pyruvate kinase activity by the human papillomavirus type 16 E7 oncoprotein. Proc. Natl. Acad. Sci. USA 96, 1291–1296 (1999).
Hu, C. et al. HPV E6/E7 promotes aerobic glycolysis in cervical cancer by regulating IGF2BP2 to stabilize m(6)A-MYC expression. Int. J. Biol. Sci. 18, 507–521 (2022).
Chandel, V. et al. Metabolic regulation in HPV-associated head and neck squamous cell carcinoma. Life Sci. 258, 118236 (2020).
Yoshizaki, T. et al. Recent advances in assessing the clinical implications of Epstein-Barr virus infection and their application to the diagnosis and treatment of nasopharyngeal carcinoma. Microorganisms 12, 14 (2023).
Low, Y. H. et al. Pathogenesis and therapeutic implications of EBV-associated epithelial cancers. Front. Oncol. 13, 1202117 (2023).
Dawson, C. W., Port, R. J. & Young, L. S. The role of the EBV-encoded latent membrane proteins LMP1 and LMP2 in the pathogenesis of nasopharyngeal carcinoma (NPC). Semin. Cancer Biol. 22, 144–153 (2012).
Damania, B., Kenney, S. C. & Raab-Traub, N. Epstein-Barr virus: biology and clinical disease. Cell 185, 3652–3670 (2022).
Tay, J. K. et al. The microdissected gene expression landscape of nasopharyngeal cancer reveals vulnerabilities in FGF and noncanonical NF-kappaB signaling. Sci. Adv. 8, eabh2445 (2022).
Lu, J. et al. EBV-LMP1 suppresses the DNA damage response through DNA-PK/AMPK signaling to promote radioresistance in nasopharyngeal carcinoma. Cancer Lett. 380, 191–200 (2016).
Leong, M. M. L. et al. EBV infection is associated with histone bivalent switch modifications in squamous epithelial cells. Proc. Natl. Acad. Sci. USA 116, 14144–14153 (2019).
Li, J. S. Z. et al. Chromosomal fragile site breakage by EBV-encoded EBNA1 at clustered repeats. Nature 616, 504–509 (2023).
Cao, J. Y., Mansouri, S. & Frappier, L. Changes in the nasopharyngeal carcinoma nuclear proteome induced by the EBNA1 protein of Epstein-Barr virus reveal potential roles for EBNA1 in metastasis and oxidative stress responses. J. Virol. 86, 382–394 (2012).
Wang, K. et al. Epstein-Barr virus-induced up-regulation of TCAB1 is involved in the DNA damage response in nasopharyngeal carcinoma. Sci. Rep. 7, 3218 (2017).
Chen, S. H. et al. O6-methylguanine-DNA methyltransferase modulates cisplatin-induced DNA double-strand breaks by targeting the homologous recombination pathway in nasopharyngeal carcinoma. J. Biomed. Sci. 28, 2 (2021).
Chung, G. T. et al. Constitutive activation of distinct NF-kappaB signals in EBV-associated nasopharyngeal carcinoma. J. Pathol. 231, 311–322 (2013).
Bruce, J. P. et al. Whole-genome profiling of nasopharyngeal carcinoma reveals viral-host co-operation in inflammatory NF-kappaB activation and immune escape. Nat. Commun. 12, 4193 (2021).
Kung, C. P. & Raab-Traub, N. Epstein-Barr virus latent membrane protein 1 induces expression of the epidermal growth factor receptor through effects on Bcl-3 and STAT3. J. Virol. 82, 5486–5493 (2008).
Port, R. J. et al. Epstein-Barr virus induction of the Hedgehog signalling pathway imposes a stem cell phenotype on human epithelial cells. J. Pathol. 231, 367–377 (2013).
Xu, Y. J. et al. Epstein-Barr virus-coded miR-BART13 promotes nasopharyngeal carcinoma cell growth and metastasis via targeting of the NKIRAS2/NF-kappaB pathway. Cancer Lett. 447, 33–40 (2019).
Mai, S. et al. MicroRNA-18a promotes cancer progression through SMG1 suppression and mTOR pathway activation in nasopharyngeal carcinoma. Cell Death Dis. 10, 819 (2019).
Yin, H., Qu, J., Peng, Q. & Gan, R. Molecular mechanisms of EBV-driven cell cycle progression and oncogenesis. Med. Microbiol. Immunol. 208, 573–583 (2019).
Ren, Y. et al. Viral IL-10 promotes cell proliferation and cell cycle progression via JAK2/STAT3 signaling pathway in nasopharyngeal carcinoma cells. Biotechnol. Appl. Biochem. 67, 929–938 (2020).
Tang, Z. et al. miR-4721, induced by EBV-miR-BART22, targets GSK3beta to enhance the tumorigenic capacity of NPC through the WNT/beta-catenin pathway. Mol. Ther. Nucleic Acids 22, 557–571 (2020).
Li, Y. et al. Stabilization of p18 by deubiquitylase CYLD is pivotal for cell cycle progression and viral replication. NPJ Precis. Oncol. 5, 14 (2021).
Marquitz, A. R. et al. Expression profile of microRNAs in Epstein-Barr virus-infected AGS gastric carcinoma cells. J. Virol. 88, 1389–1393 (2014).
Tsao, S. W. et al. The significance of LMP1 expression in nasopharyngeal carcinoma. Semin. Cancer Biol. 12, 473–487 (2002).
Shi, F. et al. EBV(LMP1)-induced metabolic reprogramming inhibits necroptosis through the hypermethylation of the RIP3 promoter. Theranostics 9, 2424–2438 (2019).
Yuan, L. et al. EBV infection-induced GPX4 promotes chemoresistance and tumor progression in nasopharyngeal carcinoma. Cell Death Differ. 29, 1513–1527 (2022).
Feng, G. et al. Knockdown of TFRC suppressed the progression of nasopharyngeal carcinoma by downregulating the PI3K/Akt/mTOR pathway. Cancer Cell Int. 23, 185 (2023).
Cheng, S. et al. Epstein-Barr virus noncoding RNAs from the extracellular vesicles of nasopharyngeal carcinoma (NPC) cells promote angiogenesis via TLR3/RIG-I-mediated VCAM-1 expression. Biochim. Biophys. Acta Mol. Basis Dis. 1865, 1201–1213 (2019).
Liao, C. et al. Anoikis resistance and immune escape mediated by Epstein-Barr virus-encoded latent membrane protein 1-induced stabilization of PGC-1alpha promotes invasion and metastasis of nasopharyngeal carcinoma. J. Exp. Clin. Cancer Res. 42, 261 (2023).
Ge, J. et al. Epstein-Barr Virus-Encoded Circular RNA CircBART2.2 Promotes Immune Escape of Nasopharyngeal Carcinoma by Regulating PD-L1. Cancer Res. 81, 5074–5088 (2021).
Ka-Yue Chow, L. et al. Epigenomic landscape study reveals molecular subtypes and EBV-associated regulatory epigenome reprogramming in nasopharyngeal carcinoma. EBioMedicine 86, 104357 (2022).
Chen, C. et al. Comprehensive single-cell transcriptomic and proteomic analysis reveals NK cell exhaustion and unique tumor cell evolutionary trajectory in non-keratinizing nasopharyngeal carcinoma. J. Transl. Med. 21, 278 (2023).
Chen, H. et al. EBV-upregulated B7-H3 inhibits NK cell-mediated antitumor function and contributes to nasopharyngeal carcinoma progression. Cancer Immunol. Res. 11, 830–846 (2023).
Xu, M. et al. Genome-wide profiling of Epstein-Barr virus integration by targeted sequencing in Epstein-Barr virus-associated malignancies. Theranostics 9, 1115–1124 (2019).
Xu, X. et al. EBV abortive lytic cycle promotes nasopharyngeal carcinoma progression through recruiting monocytes and regulating their directed differentiation. PLoS Pathog. 20, e1011934 (2024).
Zhang, B. et al. EB virus-induced ATR activation accelerates nasopharyngeal carcinoma growth via M2-type macrophages polarization. Cell Death Dis. 11, 742 (2020).
Song, S. et al. BHRF1 Enhances EBV-mediated nasopharyngeal carcinoma tumorigenesis through modulating mitophagy associated with mitochondrial membrane permeabilization transition. Cells 9, 1158 (2020).
Wei, X. et al. Mechanisms of vasculogenic mimicry in hypoxic tumor microenvironments. Mol. Cancer 20, 7 (2021).
Zhu, N. et al. EBV latent membrane proteins promote hybrid epithelial-mesenchymal and extreme mesenchymal states of nasopharyngeal carcinoma cells for tumorigenicity. PLoS Pathog. 17, e1009873 (2021).
Tang, W. C. et al. Latent membrane protein 1 and macrophage-derived TNFalpha synergistically activate and mobilize invadopodia to drive invasion of nasopharyngeal carcinoma. J. Pathol. 259, 163–179 (2023).
Li, D. K. et al. Exosomal HMGA2 protein from EBV-positive NPC cells destroys vascular endothelial barriers and induces endothelial-to-mesenchymal transition to promote metastasis. Cancer Gene. Ther. 29, 1439–1451 (2022).
Sung, W. W., Chen, P. R., Liao, M. H. & Lee, J. W. Enhanced aerobic glycolysis of nasopharyngeal carcinoma cells by Epstein-Barr virus latent membrane protein 1. Exp. Cell Res. 359, 94–100 (2017).
Zhang, J. et al. Epstein-Barr virus-encoded latent membrane protein 1 upregulates glucose transporter 1 transcription via the mTORC1/NF-kappaB signaling pathways. J. Virol. 91, e02168–02116 (2017).
Xiao, L. et al. Targeting Epstein-Barr virus oncoprotein LMP1-mediated glycolysis sensitizes nasopharyngeal carcinoma to radiation therapy. Oncogene 33, 4568–4578 (2014).
Lyu, X. et al. EBV-miR-BART1-5P activates AMPK/mTOR/HIF1 pathway via a PTEN-independent manner to promote glycolysis and angiogenesis in nasopharyngeal carcinoma. PLoS Pathog. 14, e1007484 (2018).
Lo, A. K. et al. Activation of the FGFR1 signalling pathway by the Epstein-Barr virus-encoded LMP1 promotes aerobic glycolysis and transformation of human nasopharyngeal epithelial cells. J. Pathol. 237, 238–248 (2015).
Krishna, G., Soman Pillai, V. & Valiya Veettil, M. Upregulation of GLS1 isoforms KGA and GAC facilitates mitochondrial metabolism and cell proliferation in Epstein-Barr virus-infected cells. Viruses 12, 811 (2020).
Liu, S. C. et al. Epstein-Barr virus induces adipocyte dedifferentiation to modulate the tumor microenvironment. Cancer Res. 81, 3283–3294 (2021).
Naipauer, J. & Mesri, E. A. The Kaposi’s sarcoma progenitor enigma: KSHV-induced MEndT-EndMT axis. Trends Mol. Med. 29, 188–200 (2023).
Yan, L., Majerciak, V., Zheng, Z. M. & Lan, K. Towards better understanding of KSHV life cycle: from transcription and posttranscriptional regulations to pathogenesis. Virol. Sin. 34, 135–161 (2019).
Nakayama, R., Ueno, Y., Ueda, K. & Honda, T. Latent infection with Kaposi’s sarcoma-associated herpesvirus enhances retrotransposition of long interspersed element-1. Oncogene 38, 4340–4351 (2019).
Chen, J. et al. Human endogenous retrovirus type K encoded Np9 oncoprotein induces DNA damage response. J. Med. Virol. 96, e29534 (2024).
Frohlich, J. & Grundhoff, A. Epigenetic control in Kaposi sarcoma-associated herpesvirus infection and associated disease. Semin. Immunopathol. 42, 143–157 (2020).
Roake, C. M. & Artandi, S. E. Regulation of human telomerase in homeostasis and disease. Nat. Rev. Mol. Cell Biol. 21, 384–397 (2020).
Lippert, T. P. et al. Oncogenic herpesvirus KSHV triggers hallmarks of alternative lengthening of telomeres. Nat. Commun. 12, 512 (2021).
Li, T., Ju, E. & Gao, S. J. Kaposi sarcoma-associated herpesvirus miRNAs suppress CASTOR1-mediated mTORC1 inhibition to promote tumorigenesis. J. Clin. Invest. 129, 3310–3323 (2019).
Li, T. & Gao, S. J. KSHV hijacks FoxO1 to promote cell proliferation and cellular transformation by antagonizing oxidative stress. J. Med. Virol. 95, e28676 (2023).
Wu, X. J. et al. Kaposi’s sarcoma-associated herpesvirus viral protein kinase augments cell survival. Cell Death Dis. 14, 688 (2023).
Li, W. et al. Sperm associated antigen 9 promotes oncogenic KSHV-encoded interferon regulatory factor-induced cellular transformation and angiogenesis by activating the JNK/VEGFA pathway. PLoS Pathog. 16, e1008730 (2020).
Moore, L. N. et al. Bcl-xL is required to protect endothelial cells latently infected with KSHV from virus-induced intrinsic apoptosis. PLoS Pathog. 19, e1011385 (2023).
Abere, B. et al. The Kaposi’s sarcoma-associated herpesvirus (KSHV) non-structural membrane protein K15 is required for viral lytic replication and may represent a therapeutic target. PLoS Pathog. 13, e1006639 (2017).
Bhowmik, D. et al. Cooperative DNA binding mediated by KicGAS/ORF52 oligomerization allows inhibition of DNA-induced phase separation and activation of cGAS. Nucleic Acids Res. 49, 9389–9403 (2021).
Munz, C. Natural killer cell responses to human oncogenic gamma-herpesvirus infections. Semin. Immunol. 60, 101652 (2022).
Gabaev, I. et al. Quantitative proteomics analysis of lytic KSHV infection in human endothelial cells reveals targets of viral immune modulation. Cell Rep. 33, 108249 (2020).
Nalwoga, A. & Whitby, D. Adaptive immune responses to Kaposi’s sarcoma-associated herpesvirus. Curr. Opin. Immunol. 77, 102230 (2022).
Gilardini Montani, M. S. et al. KSHV infection skews macrophage polarisation towards M2-like/TAM and activates Ire1 alpha-XBP1 axis up-regulating pro-tumorigenic cytokine release and PD-L1 expression. Br. J. Cancer 123, 298–306 (2020).
Lurain, K. A. et al. HIV-associated cancers and lymphoproliferative disorders caused by Kaposi sarcoma herpesvirus and Epstein-Barr virus. Clin. Microbiol. Rev. 37, e0002223 (2024).
Lucena Lage, S. et al. Inflammasome activation in patients with Kaposi sarcoma herpesvirus (KSHV)-associated disorders. Blood. 144, 1496–1507 (2024).
Gruffaz, M. et al. TLR4-mediated inflammation promotes KSHV-induced cellular transformation and tumorigenesis by activating the STAT3 pathway. Cancer Res. 77, 7094–7108 (2017).
Inagaki, T. et al. KSHV vIL-6 enhances inflammatory responses by epigenetic reprogramming. PLoS Pathog. 19, e1011771 (2023).
Yang, X. et al. KSHV-encoded ORF45 activates human NLRP1 inflammasome. Nat. Immunol. 23, 916–926 (2022).
Ramaswami, R. et al. Transcriptional landscape of Kaposi sarcoma tumors identifies unique immunologic signatures and key determinants of angiogenesis. J. Transl. Med. 21, 653 (2023).
Bayo Jimenez, M. T. et al. Protective actions of nuclear factor erythroid 2-related factor 2 (NRF2) and downstream pathways against environmental stressors. Free Radic. Biol. Med. 187, 72–91 (2022).
Tavakolian, S. et al. Role of the VEGF in virus-associated cancers. Rev. Med. Virol. 34, e2493 (2024).
Davis, D. A., Shrestha, P. & Yarchoan, R. Hypoxia and hypoxia-inducible factors in Kaposi sarcoma-associated herpesvirus infection and disease pathogenesis. J. Med. Virol. 95, e29071 (2023).
Sin, S. H. et al. The complete Kaposi sarcoma-associated herpesvirus genome induces early-onset, metastatic angiosarcoma in transgenic mice. Cell Host Microbe 32, 755–767.e754 (2024).
Li, W. et al. An oncogenic viral interferon regulatory factor upregulates CUB domain-containing protein 1 to promote angiogenesis by hijacking transcription factor lymphoid enhancer-binding factor 1 and metastasis suppressor CD82. Cell Death Differ. 27, 3289–3306 (2020).
Li, W. et al. Viral interleukin-6 encoded by an oncogenic virus promotes angiogenesis and cellular transformation by enhancing STAT3-mediated epigenetic silencing of caveolin 1. Oncogene 39, 4603–4618 (2020).
Medina, M. V. et al. KSHV G-protein coupled receptor vGPCR oncogenic signaling upregulation of Cyclooxygenase-2 expression mediates angiogenesis and tumorigenesis in Kaposi’s sarcoma. PLoS Pathog. 16, e1009006 (2020).
Kamali, M. J. et al. Hijacking and rewiring of host CircRNA/miRNA/mRNA competitive endogenous RNA (ceRNA) regulatory networks by oncoviruses during development of viral cancers. Rev. Med. Virol. 34, e2530 (2024).
Markazi, A., Bracci, P. M., McGrath, M. & Gao, S. J. Pseudomonas aeruginosa stimulates inflammation and enhances Kaposi’s sarcoma herpesvirus-induced cell proliferation and cellular transformation through both lipopolysaccharide and flagellin. mBio. 11, e02843 (2020).
Kong, X. et al. Bone marrow-derived SH-SY5Y neuroblastoma cells infected with Kaposi’s sarcoma-associated herpesvirus display unique infection phenotypes and growth properties. J. Virol. 95, e0000321 (2021).
Valiya Veettil, M. et al. Kaposi’s sarcoma-associated herpesvirus infection induces the expression of neuroendocrine genes in endothelial cells. J. Virol. 94, e01692 (2020).
Yao, S. et al. MiRNA-891a-5p mediates HIV-1 Tat and KSHV Orf-K1 synergistic induction of angiogenesis by activating NF-kappaB signaling. Nucleic Acids Res. 43, 9362–9378 (2015).
Li, T. & Gao, S. J. Metabolic reprogramming and metabolic sensors in KSHV-induced cancers and KSHV infection. Cell Biosci. 11, 176 (2021).
Singh, R. K. et al. Metabolic reprogramming of Kaposi’s sarcoma-associated herpes virus infected B-cells in hypoxia. PLoS Pathog. 14, e1007062 (2018).
Wan, Q. et al. Hijacking of nucleotide biosynthesis and deamidation-mediated glycolysis by an oncogenic herpesvirus. Nat. Commun. 15, 1442 (2024).
Choi, U. Y. et al. Oncogenic human herpesvirus hijacks proline metabolism for tumorigenesis. Proc. Natl. Acad. Sci. USA 117, 8083–8093 (2020).
Privatt, S. R. et al. Comparative polar and lipid plasma metabolomics differentiate KSHV infection and disease states. Cancer Metab. 11, 13 (2023).
Gruffaz, M. et al. Signatures of oral microbiome in HIV-infected individuals with oral Kaposi’s sarcoma and cell-associated KSHV DNA. PLoS Pathog. 16, e1008114 (2020).
Dai, L. et al. Kaposi sarcoma-associated herpesvirus and staphylococcus aureus coinfection in oral cavities of HIV-positive patients: a unique niche for oncogenic virus lytic reactivation. J. Infect. Dis. 221, 1331–1341 (2020).
Little, R. F. & Uldrick, T. S. Are there clues to oral Kaposi sarcoma-associated herpesvirus shedding and Kaposi sarcoma oncogenesis in the oral microbiome? J. Infect. Dis. 221, 1226–1228 (2020).
Zhang, X. et al. A novel vaccine candidate based on chimeric virus-like particle displaying multiple conserved epitope peptides induced neutralizing antibodies against EBV infection. Theranostics 10, 5704–5718 (2020).
Montes-Mojarro, I. A., Fend, F. & Quintanilla-Martinez, L. EBV and the pathogenesis of NK/T cell lymphoma. Cancers 13, 1414 (2021).
Wai, C. M. M. et al. Immune pathway upregulation and lower genomic instability distinguish EBV-positive nodal T/NK-cell lymphoma from ENKTL and PTCL-NOS. Haematologica 107, 1864–1879 (2022).
Ho, J. W. Y. et al. Comprehensive profiling of EBV gene expression and promoter methylation reveals latency II viral infection and sporadic abortive lytic activation in peripheral T-cell lymphomas. Viruses 15, 423 (2023).
Leoncini, L. Epstein-Barr virus positivity as a defining pathogenetic feature of Burkitt lymphoma subtypes. Br. J. Haematol. 196, 468–470 (2022).
Magrath, I. Epidemiology: clues to the pathogenesis of Burkitt lymphoma. Br. J. Haematol. 156, 744–756 (2012).
Lim, S. T. et al. AIDS-related Burkitt’s lymphoma versus diffuse large-cell lymphoma in the pre-highly active antiretroviral therapy (HAART) and HAART eras: significant differences in survival with standard chemotherapy. J. Clin. Oncol. 23, 4430–4438 (2005).
Campo, E. et al. Lymphoma classifications, how to develop a future unified taxonomy. J. Clin. Oncol. 27, 3177–3182 (2024).
Nagasaki, J. et al. The critical role of CD4+ T cells in PD-1 blockade against MHC-II-expressing tumors such as classic Hodgkin lymphoma. Blood Adv. 4, 4069–4082 (2020).
Cader, F. Z. et al. Mass cytometry of Hodgkin lymphoma reveals a CD4(+) regulatory T-cell-rich and exhausted T-effector microenvironment. Blood 132, 825–836 (2018).
Roemer, M. G. M. et al. Major histocompatibility complex class II and programmed death ligand 1 expression predict outcome after programmed death 1 blockade in classic hodgkin lymphoma. J. Clin. Oncol. 36, 942–950 (2018).
Li, C. et al. EBNA2-deleted Epstein-Barr virus (EBV) isolate, P3HR1, causes Hodgkin-like lymphomas and diffuse large B cell lymphomas with type II and Wp-restricted latency types in humanized mice. PLoS Pathog. 16, e1008590 (2020).
Worth, A. J., Houldcroft, C. J. & Booth, C. Severe Epstein-Barr virus infection in primary immunodeficiency and the normal host. Br. J. Haematol. 175, 559–576 (2016).
Carbone, A., Gloghini, A. & Dotti, G. EBV-associated lymphoproliferative disorders: classification and treatment. Oncologist 13, 577–585 (2008).
Healy, J. A. & Dave, S. S. The Role of EBV in the pathogenesis of diffuse large B cell lymphoma. Curr. Top. Microbiol. Immunol. 390, 315–337 (2015).
Malpica, L. et al. EBV-positive diffuse large B-cell lymphoma, not otherwise specified: 2022 update on diagnosis, risk-stratification, and management. Am. J. Hematol. 97, 951–965 (2022).
Munz, C. Redirecting T cells against Epstein-Barr virus infection and associated oncogenesis. Cells 9, 1400 (2020).
Zhang, W. et al. Hepatitis virus-associated non-Hodgkin lymphoma: pathogenesis and treatment strategies. J. Clin. Transl. Hepatol. 11, 1256–1266 (2023).
Wang, C., Xiao, Q., Zhang, X. & Liu, Y. HIV associated lymphoma: latest updates from 2023 ASH annual meeting. Exp. Hematol. Oncol. 13, 65 (2024).
Chen, Y. et al. Estimates of the global burden of non-Hodgkin lymphoma attributable to HIV: a population attributable modeling study. EClinicalMedicine 67, 102370 (2024).
Wang, C. et al. TAF family proteins and MEF2C are essential for Epstein-Barr virus super-enhancer activity. J. Virol. 93, e00513–e00519 (2019).
Molina, E. et al. MYC directly transactivates CR2/CD21, the receptor of the Epstein-Barr virus, enhancing the viral infection of Burkitt lymphoma cells. Oncogene 42, 3358–3370 (2023).
Bristol, J. A. et al. Latent Epstein-Barr virus infection collaborates with Myc over-expression in normal human B cells to induce Burkitt-like Lymphomas in mice. PLoS Pathog. 20, e1012132 (2024).
Ikeda, M., Hayes, C. K., Schaller, S. J. & Longnecker, R. Latent membrane proteins from EBV differentially target cellular pathways to accelerate MYC-induced lymphomagenesis. Blood Adv. 6, 4283–4296 (2022).
Dang, W. et al. IGFBP7-AS1 is a p53-responsive long noncoding RNA downregulated by Epstein-Barr virus that contributes to viral tumorigenesis. Cancer Lett. 523, 135–147 (2021).
Giehler, F. et al. Epstein-Barr virus-driven B cell lymphoma mediated by a direct LMP1-TRAF6 complex. Nat. Commun. 15, 414 (2024).
Shareena, G. & Kumar, D. Epigenetics of Epstein Barr virus - a review. Biochim. Biophys. Acta Mol. Basis Dis. 1869, 166838 (2023).
Kretzmer, H. et al. DNA methylome analysis in Burkitt and follicular lymphomas identifies differentially methylated regions linked to somatic mutation and transcriptional control. Nat. Genet. 47, 1316–1325 (2015).
Oliveira, L. O. D., Costa, I. B. & Quaresma, J. A. S. Association between Epstein-Barr virus LMP-1 and Hodgkin lymphoma LMP-1 mechanisms in Hodgkin lymphoma development. Rev. Med. Virol. 34, e2561 (2024).
Chen, Z. et al. Chronic active Epstein-Barr virus infection of T/NK-cell type mimicking classic Hodgkin lymphoma: clinicopathologic and genetic features of 8 cases supporting a variant with “Hodgkin/Reed-Sternberg-like” cells of NK phenotype. Am. J. Surg. Pathol. 43, 1611–1621 (2019).
Tiacci, E. et al. Pervasive mutations of JAK-STAT pathway genes in classical Hodgkin lymphoma. Blood 131, 2454–2465 (2018).
Etzel, B. M. et al. Mutation analysis of tumor necrosis factor alpha-induced protein 3 gene in Hodgkin lymphoma. Pathol. Res. Pract. 213, 256–260 (2017).
Saleem, A. & Natkunam, Y. Extranodal NK/T-cell lymphomas: the role of natural killer cells and EBV in lymphomagenesis. Int. J. Mol. Sci. 21, 1501 (2020).
Sommermann, T. et al. Functional interplay of Epstein-Barr virus oncoproteins in a mouse model of B cell lymphomagenesis. Proc. Natl. Acad. Sci. USA 117, 14421–14432 (2020).
Zhang, J. et al. LMP1 and EBNA2 constitute a minimal set of EBV genes for transformation of human B cells. Front. Immunol. 14, 1331730 (2023).
Wang, L. et al. The ubiquitin sensor and adaptor protein p62 mediates signal transduction of a viral oncogenic pathway. mBio 12, e0109721 (2021).
Fish, K. et al. Rewiring of B cell receptor signaling by Epstein-Barr virus LMP2A. Proc. Natl. Acad. Sci. USA 117, 26318–26327 (2020).
Messick, T. E. et al. Structure-based design of small-molecule inhibitors of EBNA1 DNA binding blocks Epstein-Barr virus latent infection and tumor growth. Sci. Transl. Med. 11, EAAU5612 (2019).
Zeng, M., Jia, Q. & Chen, J. Enhanced prognostic evaluation of diffuse large B-cell lymphoma: A comprehensive surveillance study incorporating Epstein-Barr virus infection status and immunohistochemical markers. J. Med. Virol. 96, e29834 (2024).
Piris, M. A., Medeiros, L. J. & Chang, K. C. Hodgkin lymphoma: a review of pathological features and recent advances in pathogenesis. Pathology 52, 154–165 (2020).
Navari, M. et al. Pathobiologic roles of Epstein-Barr virus-encoded MicroRNAs in human lymphomas. Int. J. Mol. Sci. 19, 1168 (2018).
Murer, A. et al. MicroRNAs of Epstein-Barr virus attenuate T-cell-mediated immune control in vivo. mBio 10, e01941–01918 (2019).
Wang, Y. et al. A polymorphism in the Epstein-Barr virus EBER2 noncoding RNA drives in vivo expansion of latently infected B cells. mBio 13, e0083622 (2022).
Li, Z. et al. The Epstein-Barr virus noncoding RNA EBER2 transactivates the UCHL1 deubiquitinase to accelerate cell growth. Proc. Natl. Acad. Sci. USA 118, e2115508118 (2021).
Marquitz, A. R., Mathur, A., Edwards, R. H. & Raab-Traub, N. Host gene expression Is regulated by two types of noncoding RNAs transcribed from the Epstein-Barr virus BamHI A rightward transcript region. J. Virol. 89, 11256–11268 (2015).
Harold, C., Cox, D. & Riley, K. J. Epstein-Barr viral microRNAs target caspase 3. Virol. J. 13, 145 (2016).
Vereide, D. T. et al. Epstein-Barr virus maintains lymphomas via its miRNAs. Oncogene 33, 1258–1264 (2014).
Baron, M. et al. Epstein-Barr Virus and immune status imprint the immunogenomics of non-Hodgkin lymphomas occurring in immune-suppressed environments. Haematologica 109, 3615–3630 (2024).
Chantziou, A. et al. HIV infection is associated with compromised tumor microenvironment adaptive immune reactivity in Hodgkin lymphoma. Blood Adv. 8, 6215–6231 (2024).
Li, Y. Q. et al. Single-cell analysis reveals malignant cells reshape the cellular landscape and foster an immunosuppressive microenvironment of extranodal NK/T-cell lymphoma. Adv. Sci. 10, e2303913 (2023).
Santisteban-Espejo, A. et al. Prognostic role of the expression of latent-membrane protein 1 of Epstein-Barr virus in classical Hodgkin lymphoma. Viruses 13, 2523 (2021).
Maroui, M. A. et al. Aflatoxin B1 and Epstein-Barr virus-induced CCL22 expression stimulates B cell infection. Proc. Natl. Acad. Sci. USA 121, e2314426121 (2024).
Zhang, J. et al. Long noncoding RNAs involvement in Epstein-Barr virus infection and tumorigenesis. Virol. J. 17, 51 (2020).
Iizasa, H. et al. Role of viral and host microRNAs in immune regulation of Epstein-Barr virus-associated diseases. Front. Immunol. 11, 367 (2020).
De Re, V. et al. Epstein-Barr virus BART microRNAs in EBV- associated Hodgkin lymphoma and gastric cancer. Infect. Agent. Cancer. 15, 42 (2020).
Roemer, M. G. et al. PD-L1 and PD-L2 genetic alterations define classical Hodgkin lymphoma and predict outcome. J. Clin. Oncol. 34, 2690–2697 (2016).
Liu, W. R. & Shipp, M. A. Signaling pathways and immune evasion mechanisms in classical Hodgkin lymphoma. Blood 130, 2265–2270 (2017).
Ma, S. D. et al. PD-1/CTLA-4 blockade inhibits Epstein-Barr virus-induced lymphoma growth in a cord blood humanized-mouse model. PLoS Pathog. 12, e1005642 (2016).
Bi, X. W. et al. PD-L1 is upregulated by EBV-driven LMP1 through NF-kappaB pathway and correlates with poor prognosis in natural killer/T-cell lymphoma. J. Hematol. Oncol. 9, 109 (2016).
Li, Y. et al. Increased coexpression of PD-L1 and IDO1 is associated with poor overall survival in patients with NK/T-cell lymphoma. Leukemia 38, 1553–1563 (2024).
Rana, I., Dahlberg, S., Steinmaus, C. & Zhang, L. Benzene exposure and non-Hodgkin lymphoma: a systematic review and meta-analysis of human studies. Lancet Planet Health 5, e633–e643 (2021).
Muller, A. M., Ihorst, G., Mertelsmann, R. & Engelhardt, M. Epidemiology of non-Hodgkin’s lymphoma (NHL): trends, geographic distribution, and etiology. Ann. Hematol. 84, 1–12 (2005).
Liu, J. et al. Immunosuppressant indulges EBV reactivation and related lymphoproliferative disease by inhibiting Vdelta2(+) T cells activities after hematopoietic transplantation for blood malignancies. J. Immunother Cancer 8, e000208 (2020).
Dolcetti, R., Gloghini, A., Caruso, A. & Carbone, A. A lymphomagenic role for HIV beyond immune suppression? Blood 127, 1403–1409 (2016).
Bende, R. J. et al. Among B cell non-Hodgkin’s lymphomas, MALT lymphomas express a unique antibody repertoire with frequent rheumatoid factor reactivity. J. Exp. Med. 201, 1229–1241 (2005).
Shanahan, F. Nutrient tasting and signaling mechanisms in the gut V. mechanisms of immunologic sensation of intestinal contents. Am. J. Physiol. Gastrointest. Liver Physiol. 278, G191–196 (2000).
Feng, J. et al. Latent membrane protein 1 promotes tumorigenesis through upregulation of PGC1beta signaling pathway. Stem Cell Rev. Rep. 17, 1486–1499 (2021).
Beer, S. et al. EBNA2-EBF1 complexes promote MYC expression and metabolic processes driving S-phase progression of Epstein-Barr virus-infected B cells. Proc. Natl. Acad. Sci. USA 119, e2200512119 (2022).
Chou, Y. C. et al. Requirement for LMP1-induced RON receptor tyrosine kinase in Epstein-Barr virus-mediated B-cell proliferation. Blood 118, 1340–1349 (2011).
Bonglack, E. N. et al. Monocarboxylate transporter antagonism reveals metabolic vulnerabilities of viral-driven lymphomas. Proc. Natl. Acad. Sci USA. 118, e2022495118 (2021).
Dalton, T. et al. Epigenetic reprogramming sensitizes immunologically silent EBV+ lymphomas to virus-directed immunotherapy. Blood 135, 1870–1881 (2020).
Gazon, H. et al. Epigenetic silencing of HTLV-1 expression by the HBZ RNA through interference with the basal transcription machinery. Blood Adv. 4, 5574–5579 (2020).
Atabati, H. et al. Evaluating mRNA expression of tax, B chain of PDGF and PDGF-beta receptors as well as HTLV-I proviral load in ATL patients and healthy carriers. J. Med. Virol. 93, 3865–3870 (2021).
Iwanaga, M. et al. Human T-cell leukemia virus type I (HTLV-1) proviral load and disease progression in asymptomatic HTLV-1 carriers: a nationwide prospective study in Japan. Blood 116, 1211–1219 (2010).
Yamada, K. et al. Human T-cell lymphotropic virus HBZ and tax mRNA expression are associated with specific clinicopathological features in adult T-cell leukemia/lymphoma. Mod. Pathol. 34, 314–326 (2021).
Forlani, G. et al. Dual cytoplasmic and nuclear localization of HTLV-1-encoded HBZ protein is a unique feature of adult T-cell leukemia. Haematologica 106, 2076–2085 (2021).
Zhao, T. et al. HTLV-1 activates YAP via NF-kappaB/p65 to promote oncogenesis. Proc. Natl. Acad. Sci. USA. 119, e2115316119 (2022).
Zuo, X., Zhou, R., Yang, S. & Ma, G. HTLV-1 persistent infection and ATLL oncogenesis. J. Med. Virol. 95, e28424 (2023).
Mahgoub, M. et al. Sporadic on/off switching of HTLV-1 Tax expression is crucial to maintain the whole population of virus-induced leukemic cells. Proc. Natl. Acad. Sci. USA 115, E1269–E1278 (2018).
Rosewick, N. et al. Cis-perturbation of cancer drivers by the HTLV-1/BLV proviruses is an early determinant of leukemogenesis. Nat. Commun. 8, 15264 (2017).
Matsuo, M. et al. Identification and characterization of a novel enhancer in the HTLV-1 proviral genome. Nat. Commun. 13, 2405 (2022).
Rowan, A. G. et al. Evolution of retrovirus-infected premalignant T-cell clones prior to adult T-cell leukemia/lymphoma diagnosis. Blood 135, 2023–2032 (2020).
Yeh, C. H. et al. Loss of FBXW7-mediated degradation of BRAF elicits resistance to BET inhibitors in adult T cell leukemia cells. Mol. Cancer. 19, 139 (2020).
Hleihel, R. et al. Primary cells from patients with adult T cell leukemia/lymphoma depend on HTLV-1 Tax expression for NF-kappaB activation and survival. Blood Cancer J. 13, 67 (2023).
Ahmadi Ghezeldasht, S., Blackbourn, D. J., Mosavat, A. & Rezaee, S. A. Pathogenicity and virulence of human T lymphotropic virus type-1 (HTLV-1) in oncogenesis: adult T-cell leukemia/lymphoma (ATLL). Crit. Rev. Clin. Lab. Sci. 60, 189–211 (2023).
Garcia, J. et al. Naturally occurring T cell mutations enhance engineered T cell therapies. Nature 626, 626–634 (2024).
Kameda, T. et al. CARD11 mutation and HBZ expression induce lymphoproliferative disease and adult T-cell leukemia/lymphoma. Commun. Biol. 5, 1309 (2022).
Watanabe, T. Adult T-cell leukemia: molecular basis for clonal expansion and transformation of HTLV-1-infected T cells. Blood 129, 1071–1081 (2017).
Shah, U. A. et al. North American ATLL has a distinct mutational and transcriptional profile and responds to epigenetic therapies. Blood 132, 1507–1518 (2018).
He, Y. et al. NF-kappaB-induced R-loop accumulation and DNA damage select for nucleotide excision repair deficiencies in adult T cell leukemia. Proc. Natl. Acad. Sci. USA 118, e2005568118 (2021).
Zhi, H. et al. RNF8 dysregulation and down-regulation during HTLV-1 infection promote genomic instability in adult T-cell leukemia. PLoS Pathog. 16, e1008618 (2020).
Marie, P. et al. Gene-to-gene coordinated regulation of transcription and alternative splicing by 3D chromatin remodeling upon NF-kappaB activation. Nucleic Acids Res. 52, 1527–1543 (2024).
Ameur, L. B. et al. Intragenic recruitment of NF-kappaB drives splicing modifications upon activation by the oncogene Tax of HTLV-1. Nat. Commun. 11, 3045 (2020).
Zhong, W. et al. ORP4L is a prerequisite for the induction of T-cell leukemogenesis associated with human T-cell leukemia virus 1. Blood 139, 1052–1065 (2022).
Zhong, W. et al. An acquired phosphatidylinositol 4-phosphate transport initiates T-cell deterioration and leukemogenesis. Nat. Commun. 13, 4390 (2022).
Bangham, C. R. M. Human T cell leukemia virus type 1: persistence and pathogenesis. Annu. Rev. Immunol. 36, 43–71 (2018).
Akkouche, A. et al. In vivo antagonistic role of the human T-cell leukemia virus type 1 regulatory proteins tax and HBZ. PLoS Pathog. 17, e1009219 (2021).
Shichijo, T. et al. Vulnerability to APOBEC3G linked to the pathogenicity of deltaretroviruses. Proc. Natl. Acad. Sci. USA 121, e2309925121 (2024).
Zhang, M. et al. Selective targeting of JAK/STAT signaling is potentiated by Bcl-xL blockade in IL-2-dependent adult T-cell leukemia. Proc. Natl. Acad. Sci. USA 112, 12480–12485 (2015).
Witzens-Harig, M. et al. HTLV-1-associated adult T cell leukemia is highly susceptible to Navitoclax due to enhanced bax expression. Int. J. Cancer. 138, 507–514 (2016).
Tanaka, Y., Mukai, R. & Ohshima, T. HTLV-1 viral oncoprotein HBZ contributes to the enhancement of HAX-1 stability by impairing the ubiquitination pathway. J. Cell. Physiol. 236, 2756–2766 (2021).
Tanaka-Nakanishi, A., Yasunaga, J., Takai, K. & Matsuoka, M. HTLV-1 bZIP factor suppresses apoptosis by attenuating the function of FoxO3a and altering its localization. Cancer Res. 74, 188–200 (2014).
Katsuya, H. et al. The nature of the HTLV-1 provirus in naturally infected individuals analyzed by the viral DNA-capture-seq approach. Cell Rep. 29, 724–735.e724 (2019).
Satou, Y. et al. The retrovirus HTLV-1 inserts an ectopic CTCF-binding site into the human genome. Proc. Natl. Acad. Sci. USA 113, 3054–3059 (2016).
Polakowski, N. et al. HBZ upregulates myoferlin expression to facilitate HTLV-1 infection. PLoS Pathog. 19, e1011202 (2023).
Sugata, K. et al. HTLV-1 bZIP factor impairs cell-mediated immunity by suppressing production of Th1 cytokines. Blood 119, 434–444 (2012).
Tan, B. J. Y., Sugata, K., Ono, M. & Satou, Y. HTLV-1 persistence and leukemogenesis: a game of hide-and-seek with the host immune system. Front. Immunol. 13, 991928 (2022).
Toulza, F. et al. Human T-lymphotropic virus type 1-induced CC chemokine ligand 22 maintains a high frequency of functional FoxP3+ regulatory T cells. J. Immunol. 185, 183–189 (2010).
Yasuma, K. et al. HTLV-1 bZIP factor impairs anti-viral immunity by inducing co-inhibitory molecule, T cell immunoglobulin and ITIM domain (TIGIT). PLoS Pathog. 12, e1005372 (2016).
Teijaro, J. R. et al. Persistent LCMV infection is controlled by blockade of type I interferon signaling. Science 340, 207–211 (2013).
Wilson, E. B. et al. Blockade of chronic type I interferon signaling to control persistent LCMV infection. Science 340, 202–207 (2013).
Sawada, L. et al. IL-10-mediated signals act as a switch for lymphoproliferation in human t-cell leukemia virus type-1 infection by activating the STAT3 and IRF4 pathways. PLoS Pathog. 13, e1006597 (2017).
Yoshida-Sakai, N. et al. Adult T-cell leukemia-lymphoma acquires resistance to DNA demethylating agents through dysregulation of enzymes involved in pyrimidine metabolism. Int. J. Cancer. 150, 1184–1197 (2022).
Watanabe, T. et al. Reprogramming of pyrimidine nucleotide metabolism supports vigorous cell proliferation of normal and malignant T cells. Blood Adv. 8, 1345–1358 (2024).
Singal, A. G., Kanwal, F. & Llovet, J. M. Global trends in hepatocellular carcinoma epidemiology: implications for screening, prevention and therapy. Nat. Rev. Clin. Oncol. 20, 864–884 (2023).
Rahangdale, L. et al. Human papillomavirus vaccination and cervical cancer risk. BMJ 379, e070115 (2022).
Huang, D. Q. et al. Antiviral therapy utilization and 10-year outcomes in resected hepatitis B virus- and hepatitis C virus-related hepatocellular carcinoma. J. Clin. Oncol. 42, 790–799 (2024).
Shi, Y. & Zheng, M. Hepatitis B virus persistence and reactivation. BMJ 370, m2200 (2020).
Bertoletti, A. & Tan, A. T. Engineering HBV-specific T cells for treatment of HBV-related HCC and HBV infection: past, present and future. Clin. Mol. Hepatol. 30, 728–734 (2024).
Zhang, Y. et al. Therapeutic vaccination with lentiviral vector in HBV-persistent mice and two inactive HBsAg carriers. J. Hepatol. 80, 31–40 (2024).
Gao, P. et al. Immune checkpoint inhibitors in the treatment of virus-associated cancers. J Hematol. Oncol. 12, 58 (2019).
McGlynn, K. A., Petrick, J. L. & El-Serag, H. B. Epidemiology of hepatocellular carcinoma. Hepatology 73, 4–13 (2021).
Gao, X. et al. Hyperbranched poly(beta-amino ester) based polyplex nanopaticles for delivery of CRISPR/Cas9 system and treatment of HPV infection associated cervical cancer. J. Control. Rel. 321, 654–668 (2020).
Cao, B. et al. Development of mesothelin-specific CAR NK-92 cells for the treatment of gastric cancer. Int. J. Biol. Sci. 17, 3850–3861 (2021).
Hillemanns, P. et al. A therapeutic antigen-presenting cell-targeting DNA vaccine VB10.16 in HPV16-positive high-grade cervical intraepithelial neoplasia: results from a phase I/IIa Trial. Clin. Cancer Res. 28, 4885–4892 (2022).
Argenziano, M. E. et al. Epidemiology, pathophysiology and clinical aspects of hepatocellular carcinoma in MAFLD patients. Hepatol. Int. 18, 922–940 (2024).
Brown, Z. J. et al. Management of hepatocellular carcinoma: a review. JAMA Surg. 158, 410–420 (2023).
Cappuyns, S. et al. Critical appraisal of guideline recommendations on systemic therapies for advanced hepatocellular carcinoma: a review. JAMA Oncol. 10, 395–404 (2024).
Polaris Observatory, C. Global prevalence, cascade of care, and prophylaxis coverage of hepatitis B in 2022: a modelling study. Lancet Gastroenterol. Hepatol. 8, 879–907 (2023).
Serti, E. et al. Successful interferon-free therapy of chronic hepatitis C virus infection normalizes natural killer cell function. Gastroenterology 149, 190–200.e192 (2015).
Singal, A. K., Salameh, H., Kuo, Y. F. & Fontana, R. J. Meta-analysis: the impact of oral anti-viral agents on the incidence of hepatocellular carcinoma in chronic hepatitis B. Aliment Pharmacol. Ther. 38, 98–106 (2013).
Global Burden of Disease Liver Cancer, C. et al. The burden of primary liver cancer and underlying etiologies from 1990 to 2015 at the global, regional, and national level: results from the global burden of disease study 2015. JAMA Oncol. 3, 1683–1691 (2017).
Fung, S., Choi, H. S. J., Gehring, A. & Janssen, H. L. A. Getting to HBV cure: the promising paths forward. Hepatology 76, 233–250 (2022).
Boni, C. et al. Combined GS-4774 and tenofovir therapy can improve HBV-specific T-cell responses in patients with chronic hepatitis. Gastroenterology 157, 227–241 (2019).
Wei, L. et al. Efficacy and safety of a nanoparticle therapeutic vaccine in patients with chronic hepatitis B: a randomized clinical trial. Hepatology 75, 182–195 (2022).
Yoshida, O. et al. Intranasal therapeutic vaccine containing HBsAg and HBcAg for patients with chronic hepatitis B; 18 months follow-up results of phase IIa clinical study. Hepatol. Res. 53, 196–207 (2023).
Horng, J. H. et al. HBV X protein-based therapeutic vaccine accelerates viral antigen clearance by mobilizing monocyte infiltration into the liver in HBV carrier mice. J. Biomed. Sci. 27, 70 (2020).
Wang, W. et al. Dual-targeting nanoparticle vaccine elicits a therapeutic antibody response against chronic hepatitis B. Nat. Nanotechnol. 15, 406–416 (2020).
Zhao, H. et al. A therapeutic hepatitis B mRNA vaccine with strong immunogenicity and persistent virological suppression. NPJ Vaccines 9, 22 (2024).
Jiang, Y., Han, Q., Zhao, H. & Zhang, J. The mechanisms of HBV-induced hepatocellular carcinoma. J. Hepatocell Carcinoma 8, 435–450 (2021).
Wong, J. S. et al. Meta-analysis: the efficacy of anti-viral therapy in prevention of recurrence after curative treatment of chronic hepatitis B-related hepatocellular carcinoma. Aliment Pharmacol. Ther. 33, 1104–1112 (2011).
Luo, J. X., Zhang, Y., Hu, X. Y. & Xiang, N. Interferon therapy improves survival in patients with hepatitis B virus-related hepatocellular carcinoma after curative surgery: a meta-analysis. Hepatol. Int. 18, 63–72 (2024).
Terrault, N. A. et al. Update on prevention, diagnosis, and treatment of chronic hepatitis B: AASLD 2018 hepatitis B guidance. Hepatology 67, 1560–1599 (2018).
Lee, K. S. et al. Efficacy and safety of entecavir versus lamivudine over 5 years of treatment: a randomized controlled trial in Korean patients with hepatitis B e antigen-negative chronic hepatitis B. Clin. Mol. Hepatol. 23, 331–339 (2017).
Woo, H. Y. et al. Entecavir+tenofovir vs. lamivudine/telbivudine+adefovir in chronic hepatitis B patients with prior suboptimal response. Clin. Mol. Hepatol. 26, 352–363 (2020).
Choi, J., Jo, C. & Lim, Y. S. Tenofovir versus entecavir on recurrence of hepatitis b virus-related hepatocellular carcinoma after surgical resection. Hepatology 73, 661–673 (2021).
Chung, S. W. et al. Tenofovir is associated with a better prognosis than entecavir for hepatitis B virus-related hepatocellular carcinoma. Clin. Gastroenterol. Hepatol., 23, 300–309 (2024).
Tout, I. et al. Hepatitis B surface antigen seroclearance: Immune mechanisms, clinical impact, importance for drug development. J. Hepatol. 73, 409–422 (2020).
Mon, H. C. et al. Functional cure of hepatitis B in patients with cancer undergoing immune checkpoint inhibitor therapy. J. Hepatol. 82, 51–61 (2024).
Yuen, M. F. et al. Efficacy and safety of bepirovirsen in chronic hepatitis B infection. N. Engl. J. Med. 387, 1957–1968 (2022).
Qi, W. et al. Peg-interferon and nucleos(t)ide analogue combination at inception of antiviral therapy improves both anti-HBV efficacy and long-term survival among HBV DNA-positive hepatocellular carcinoma patients after hepatectomy/ablation. J. Viral Hepat. 27, 387–396 (2020).
Jiang, S. et al. Interim analysis of the PARADISE study: Benefits of add-on peginterferon-alpha in NA-treated patients with CHB. Antiviral Res. 226, 105892 (2024).
Li, C. et al. An E. coli-produced single-chain variable fragment (scFv) targeting hepatitis B virus surface protein potently inhibited virion secretion. Antiviral Res. 162, 118–129 (2019).
Lanzoni-Mangutchi, P. et al. Structure and assembly of the S-layer in C. difficile. Nat. Commun. 13, 970 (2022).
Wang, Y. et al. Chimeric antigen receptors of HBV envelope proteins inhibit hepatitis B surface antigen secretion. Gut 73, 668–681 (2024).
Llovet, J. M. et al. Sorafenib in advanced hepatocellular carcinoma. N. Engl. J. Med. 359, 378–390 (2008).
Cheng, A. L. et al. Efficacy and safety of sorafenib in patients in the Asia-Pacific region with advanced hepatocellular carcinoma: a phase III randomised, double-blind, placebo-controlled trial. Lancet Oncol. 10, 25–34 (2009).
De Matteis, S. et al. Sorafenib in the treatment of virus-related HCC: differences between HCV and HBV. Onco. Targets Ther. 14, 4305–4308 (2021).
Casadei Gardini, A. et al. Sorafenib and regorafenib in HBV- or HCV-positive hepatocellular carcinoma patients: analysis of RESORCE and SHARP trials. Dig. Liver Dis. 49, 943–944 (2017).
Harding, J. J. et al. Phase 1b study of galunisertib and ramucirumab in patients with advanced hepatocellular carcinoma. Cancer Med. 10, 3059–3067 (2021).
Kelley, R. K. et al. A phase 2 study of galunisertib (TGF-beta1 receptor type I inhibitor) and sorafenib in patients with advanced hepatocellular carcinoma. Clin. Transl. Gastroenterol. 10, e00056 (2019).
Shimizu, Y. et al. Plasma and tumoral glypican-3 levels are correlated in patients with hepatitis C virus-related hepatocellular carcinoma. Cancer Sci. 111, 334–342 (2020).
Sawada, Y. et al. Phase I trial of a glypican-3-derived peptide vaccine for advanced hepatocellular carcinoma: immunologic evidence and potential for improving overall survival. Clin. Cancer Res. 18, 3686–3696 (2012).
Kelley, R. K. et al. Safety, efficacy, and pharmacodynamics of tremelimumab plus durvalumab for patients with unresectable hepatocellular carcinoma: randomized expansion of a phase I/II study. J. Clin. Oncol. 39, 2991–3001 (2021).
Ren, Z. et al. Sintilimab plus a bevacizumab biosimilar (IBI305) versus sorafenib in unresectable hepatocellular carcinoma (ORIENT-32): a randomised, open-label, phase 2-3 study. Lancet Oncol. 22, 977–990 (2021).
Hagiwara, S. et al. Clinical implication of immune checkpoint inhibitor on the chronic hepatitis B virus infection. Hepatol. Res. 52, 754–761 (2022).
Zhang, X. et al. Hepatitis B virus reactivation in cancer patients with positive hepatitis B surface antigen undergoing PD-1 inhibition. J. Immunother Cancer 7, 322 (2019).
Yuan, G. et al. Interaction between hepatitis B virus infection and the efficacy of camrelizumab in combination with apatinib therapy in patients with hepatocellular carcinoma: a multicenter retrospective cohort study. Ann. Transl. Med. 9, 1412 (2021).
Hu, B. et al. IFNalpha potentiates anti-PD-1 efficacy by remodeling glucose metabolism in the hepatocellular carcinoma microenvironment. Cancer Discov. 12, 1718–1741 (2022).
Norberg, S. M. & Hinrichs, C. S. Engineered T cell therapy for viral and non-viral epithelial cancers. Cancer Cell 41, 58–69 (2023).
Zou, F. et al. The CD39(+) HBV surface protein-targeted CAR-T and personalized tumor-reactive CD8(+) T cells exhibit potent anti-HCC activity. Mol. Ther. 29, 1794–1807 (2021).
Meng, F. et al. Immunotherapy of HBV-related advanced hepatocellular carcinoma with short-term HBV-specific TCR expressed T cells: results of dose escalation, phase I trial. Hepatol. Int. 15, 1402–1412 (2021).
Wan, X. et al. Genetically redirected HBV-specific T cells target HBsAg-positive hepatocytes and primary lesions in HBV-associated HCC. Clin. Mol. Hepatol. 30, 735–755 (2024).
Zhu, W. et al. A novel engineered IL-21 receptor arms T-cell receptor-engineered T cells (TCR-T cells) against hepatocellular carcinoma. Signal Transduct. Target Ther. 9, 101 (2024).
Smith, R. P. Objective changes in intrauterine pressure during placebo treatment of dysmenorrhea. Pain 29, 59–66 (1987).
Ioannou, G. N. & Feld, J. J. What are the benefits of a sustained virologic response to direct-acting antiviral therapy for hepatitis C virus infection? Gastroenterology 156, 446–460 e442 (2019).
Offersgaard, A., Bukh, J. & Gottwein, J. M. Toward a vaccine against hepatitis C virus. Science 380, 37–38 (2023).
Bailey, J. R., Barnes, E. & Cox, A. L. Approaches, progress, and challenges to hepatitis C vaccine development. Gastroenterology 156, 418–430 (2019).
Donnison, T. et al. A pan-genotype hepatitis C virus viral vector vaccine generates T cells and neutralizing antibodies in mice. Hepatology 76, 1190–1202 (2022).
Bankwitz, D. et al. Hepatitis C reference viruses highlight potent antibody responses and diverse viral functional interactions with neutralising antibodies. Gut 70, 1734–1745 (2021).
Page, K. et al. Randomized trial of a vaccine regimen to prevent chronic HCV infection. N. Engl. J. Med. 384, 541–549 (2021).
Chhatwal, J., Kanwal, F., Roberts, M. S. & Dunn, M. A. Cost-effectiveness and budget impact of hepatitis C virus treatment with sofosbuvir and ledipasvir in the United States. Ann. Intern. Med. 162, 397–406 (2015).
Ma, L. et al. Direct-acting antivirals and interferon-based therapy on hepatocellular carcinoma risk in chronic hepatitis-C patients. Future Oncol. 16, 675–686 (2020).
Kanda, T. et al. APASL HCV guidelines of virus-eradicated patients by DAA on how to monitor HCC occurrence and HBV reactivation. Hepatol. Int. 13, 649–661 (2019).
Sohn, W. et al. Effect of direct-acting antivirals on disease burden of hepatitis C virus infection in South Korea in 2007-2021: a nationwide, multicentre, retrospective cohort study. EClinicalMedicine 73, 102671 (2024).
Bourliere, M. et al. Sofosbuvir, velpatasvir, and voxilaprevir for previously treated HCV infection. N. Engl. J. Med. 376, 2134–2146 (2017).
Kolamunnage-Dona, R. et al. Sorafenib is associated with a reduced rate of tumour growth and liver function deterioration in HCV-induced hepatocellular carcinoma. J. Hepatol. 75, 879–887 (2021).
Bruix, J. et al. Efficacy and safety of sorafenib in patients with advanced hepatocellular carcinoma: subanalyses of a phase III trial. J. Hepatol. 57, 821–829 (2012).
Bruix, J. et al. Prognostic factors and predictors of sorafenib benefit in patients with hepatocellular carcinoma: analysis of two phase III studies. J. Hepatol. 67, 999–1008 (2017).
Boyault, S. et al. Transcriptome classification of HCC is related to gene alterations and to new therapeutic targets. Hepatology 45, 42–52 (2007).
Himmelsbach, K. et al. New aspects of an anti-tumour drug: sorafenib efficiently inhibits HCV replication. Gut 58, 1644–1653 (2009).
Chen, D. Y. et al. Hepatitis C virus-specific CD4+ T cell phenotype and function in different infection outcomes. J. Clin. Investig. 130, 768–773 (2020).
Sangro, B. et al. A clinical trial of CTLA-4 blockade with tremelimumab in patients with hepatocellular carcinoma and chronic hepatitis C. J. Hepatol. 59, 81–88 (2013).
El-Khoueiry, A. B. et al. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): an open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet 389, 2492–2502 (2017).
Sautto, G. A. et al. Chimeric antigen receptor (CAR)-engineered T cells redirected against hepatitis C virus (HCV) E2 glycoprotein. Gut 65, 512–523 (2016).
Seif, M., Einsele, H., Loffler, J. & CAR, T. Cells beyond cancer: hope for immunomodulatory therapy of infectious diseases. Front. Immunol. 10, 2711 (2019).
Ajani, J. A. et al. Gastric cancer, version 2.2022, NCCN clinical practice guidelines in oncology. J. Natl. Compr. Canc. Netw. 20, 167–192 (2022).
Lordick, F. et al. Gastric cancer: ESMO clinical practice guideline for diagnosis, treatment and follow-up. Ann. Oncol. 33, 1005–1020 (2022).
Al-Haddad, M. A. et al. American Society for Gastrointestinal Endoscopy guideline on endoscopic submucosal dissection for the management of early esophageal and gastric cancers: methodology and review of evidence. Gastrointest. Endosc. 98, 285–305 (2023).
Shechter, O. et al. Epstein-Barr virus (EBV) epithelial associated malignancies: exploring pathologies and current treatments. Int. J. Mol. Sci. 23, 14389 (2022).
Sun, C. et al. A gB nanoparticle vaccine elicits a protective neutralizing antibody response against EBV. Cell Host Microbe 31, 1882–1897 (2023).
Wang, F. H. et al. The chinese society of clinical oncology (CSCO): clinical guidelines for the diagnosis and treatment of gastric cancer, 2021. Cancer Commun. (Lond) 41, 747–795 (2021).
Hirata, Y. et al. Early stage gastric adenocarcinoma: clinical and molecular landscapes. Nat. Rev. Clin. Oncol. 20, 453–469 (2023).
Nie, Y., Zhao, W., Lu, L. & Zhou, F. Predictive biomarkers and new developments of immunotherapy in gastric cancer: a 2023 update. Am. J. Cancer Res. 13, 3169–3184 (2023).
Bossi, P. et al. Nasopharyngeal carcinoma: ESMO-EURACAN clinical practice guidelines for diagnosis, treatment and follow-up(dagger). Ann. Oncol. 32, 452–465 (2021).
Jean-Pierre, V. et al. Main targets of interest for the development of a prophylactic or therapeutic Epstein-Barr virus vaccine. Front. Microbiol. 12, 701611 (2021).
Moutschen, M. et al. Phase I/II studies to evaluate safety and immunogenicity of a recombinant gp350 Epstein-Barr virus vaccine in healthy adults. Vaccine 25, 4697–4705 (2007).
Sokal, E. M. et al. Recombinant gp350 vaccine for infectious mononucleosis: a phase 2, randomized, double-blind, placebo-controlled trial to evaluate the safety, immunogenicity, and efficacy of an Epstein-Barr virus vaccine in healthy young adults. J. Infect. Dis. 196, 1749–1753 (2007).
Plate, A. E., Reimer, J. J., Jardetzky, T. S. & Longnecker, R. Mapping regions of Epstein-Barr virus (EBV) glycoprotein B (gB) important for fusion function with gH/gL. Virology 413, 26–38 (2011).
Cui, X. et al. Rabbits immunized with Epstein-Barr virus gH/gL or gB recombinant proteins elicit higher serum virus neutralizing activity than gp350. Vaccine 34, 4050–4055 (2016).
Cui, X. et al. Immunization with Epstein-Barr virus core fusion machinery envelope proteins elicit high titers of neutralizing activities and protect humanized mice from lethal dose EBV challenge. Vaccines 9, 285 (2021).
Massarelli, E. et al. Combining immune checkpoint blockade and tumor-specific vaccine for patients with incurable human papillomavirus 16-related cancer: a phase 2 clinical trial. JAMA Oncol. 5, 67–73 (2019).
Li, G., Zhou, Z., Wang, Z. & Wang, Z. Assessing Epstein-Barr virus in gastric cancer: clinicopathological features and prognostic implications. Infect. Agent Cancer 18, 11 (2023).
Bang, Y. J. et al. Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2-positive advanced gastric or gastro-oesophageal junction cancer (ToGA): a phase 3, open-label, randomised controlled trial. Lancet 376, 687–697 (2010).
Shitara, K. et al. Efficacy and safety of pembrolizumab or pembrolizumab plus chemotherapy vs chemotherapy alone for patients with first-line, advanced gastric cancer: the KEYNOTE-062 phase 3 randomized clinical trial. JAMA Oncol. 6, 1571–1580 (2020).
Janjigian, Y. Y. et al. First-line nivolumab plus chemotherapy versus chemotherapy alone for advanced gastric, gastro-oesophageal junction, and oesophageal adenocarcinoma (CheckMate 649): a randomised, open-label, phase 3 trial. Lancet 398, 27–40 (2021).
Sanchez-Romero, C. et al. Extranodal NK/T cell lymphoma, nasal type: An updated overview. Crit. Rev. Oncol. Hematol. 159, 103237 (2021).
Li, L. L. et al. Clinicopathological characteristics and prognosis of Epstein-Barr virus-associated gastric cancer. Arch. Virol. 169, 114 (2024).
Pan, Y. et al. Case report: Long response to PD-1 blockade after failure of trastuzumab plus chemotherapy in advanced Epstein-Barr virus-associated gastric cancer. Front. Immunol. 13, 1003859 (2022).
Panda, A. et al. Immune activation and benefit from avelumab in EBV-positive gastric cancer. J. Natl. Cancer Inst. 110, 316–320 (2018).
Kim, S. T. et al. Comprehensive molecular characterization of clinical responses to PD-1 inhibition in metastatic gastric cancer. Nat. Med. 24, 1449–1458 (2018).
Wang, F. et al. Safety, efficacy and tumor mutational burden as a biomarker of overall survival benefit in chemo-refractory gastric cancer treated with toripalimab, a PD-1 antibody in phase Ib/II clinical trial NCT02915432. Ann. Oncol. 30, 1479–1486 (2019).
Mishima, S. et al. Clinicopathological and molecular features of responders to nivolumab for patients with advanced gastric cancer. J. Immunother Cancer 7, 24 (2019).
Sun, Y. T. et al. PD-1 antibody camrelizumab for Epstein-Barr virus-positive metastatic gastric cancer: a single-arm, open-label, phase 2 trial. Am. J. Cancer Res. 11, 5006–5015 (2021).
Rha, S. Y. et al. Pembrolizumab plus chemotherapy versus placebo plus chemotherapy for HER2-negative advanced gastric cancer (KEYNOTE-859): a multicentre, randomised, double-blind, phase 3 trial. Lancet Oncol. 24, 1181–1195 (2023).
Janjigian, Y. Y. et al. First-line nivolumab plus chemotherapy for advanced gastric, gastroesophageal junction, and esophageal adenocarcinoma: 3-year follow-up of the phase III checkmate 649 trial. J. Clin. Oncol. 42, 2012–2020 (2024).
Janjigian, Y. Y. et al. The KEYNOTE-811 trial of dual PD-1 and HER2 blockade in HER2-positive gastric cancer. Nature 600, 727–730 (2021).
Shitara, K. et al. Pembrolizumab versus paclitaxel for previously treated, advanced gastric or gastro-oesophageal junction cancer (KEYNOTE-061): a randomised, open-label, controlled, phase 3 trial. Lancet 392, 123–133 (2018).
Bellmunt, J. et al. Pembrolizumab as second-line therapy for advanced urothelial carcinoma. N. Engl. J. Med. 376, 1015–1026 (2017).
Fuchs, C. S. et al. Safety and efficacy of pembrolizumab monotherapy in patients with previously treated advanced gastric and gastroesophageal junction cancer: phase 2 clinical KEYNOTE-059 trial. JAMA Oncol. 4, e180013 (2018).
Liu, X. et al. High PD-L1 expression in gastric cancer (GC) patients and correlation with molecular features. Pathol Res. Pract. 216, 152881 (2020).
Chen, L. T. et al. A phase 3 study of nivolumab in previously treated advanced gastric or gastroesophageal junction cancer (ATTRACTION-2): 2-year update data. Gastric Cancer 23, 510–519 (2020).
Kang, Y. K. et al. Nivolumab in patients with advanced gastric or gastro-oesophageal junction cancer refractory to, or intolerant of, at least two previous chemotherapy regimens (ONO-4538-12, ATTRACTION-2): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 390, 2461–2471 (2017).
Boku, N. et al. Nivolumab in previously treated advanced gastric cancer (ATTRACTION-2): 3-year update and outcome of treatment beyond progression with nivolumab. Gastric Cancer 24, 946–958 (2021).
Caruso, H. G., Heimberger, A. B. & Cooper, L. J. N. Steering CAR T cells to distinguish friend from foe. Oncoimmunology 8, e1271857 (2019).
Baumgardner, J. R. et al. CAR-T therapy and historical trends in effectiveness and cost-effectiveness of oncology treatments. J. Comp. Eff. Res. 9, 327–340 (2020).
Sahin, U. et al. FAST: a randomised phase II study of zolbetuximab (IMAB362) plus EOX versus EOX alone for first-line treatment of advanced CLDN18.2-positive gastric and gastro-oesophageal adenocarcinoma. Ann. Oncol. 32, 609–619 (2021).
Qi, C. et al. Claudin18.2-specific CAR T cells in gastrointestinal cancers: phase 1 trial final results. Nat. Med. 28, 1189–1198 (2024).
Sakamoto, N. et al. Phase I clinical trial of autologous NK cell therapy using novel expansion method in patients with advanced digestive cancer. J. Transl. Med. 13, 277 (2015).
Ishikawa, T. et al. Phase I clinical trial of adoptive transfer of expanded natural killer cells in combination with IgG1 antibody in patients with gastric or colorectal cancer. Int. J. Cancer 142, 2599–2609 (2018).
Yu, M. et al. NSUN6-mediated 5-methylcytosine modification of NDRG1 mRNA promotes radioresistance in cervical cancer. Mol. Cancer 23, 139 (2024).
Wolford, J. E. & Tewari, K. S. Rational design for cervical cancer therapeutics: cellular and non-cellular based strategies on the horizon for recurrent, metastatic or refractory cervical cancer. Exp.Opin. Drug Discov. 13, 445–457 (2018).
Vergote, I. et al. Tisotumab vedotin as second- or third-line therapy for recurrent cervical cancer. N. Engl. J. Med. 391, 44–55 (2024).
Murthy, S. S. et al. Premature mortality trends in 183 countries by cancer type, sex, WHO region, and World Bank income level in 2000-19: a retrospective, cross-sectional, population-based study. Lancet Oncol. 25, 969–978 (2024).
Del Moral-Hernandez, O. et al. TOP2A/MCM2, p16(INK4a), and cyclin E1 expression in liquid-based cytology: a biomarkers panel for progression risk of cervical premalignant lesions. BMC Cancer 21, 39 (2021).
Vallely, A. J. B. et al. Point-of-care HPV DNA testing of self-collected specimens and same-day thermal ablation for the early detection and treatment of cervical pre-cancer in women in Papua New Guinea: a prospective, single-arm intervention trial (HPV-STAT). Lancet Glob. Health 10, e1336–e1346 (2022).
Sivars, L. et al. Cell-free human papillomavirus DNA is a sensitive biomarker for prognosis and for early detection of relapse in locally advanced cervical cancer. Clin. Cancer Res. 30, 2764–2771 (2024).
Najafi, S. Circular RNAs as emerging players in cervical cancer tumorigenesis; a review to roles and biomarker potentials. Int. J. Biol. Macromol. 206, 939–953 (2022).
Bruno, M. T. et al. Possible role of negative human papillomavirus E6/E7 mRNA as a predictor of regression of cervical intraepithelial neoplasia 2 lesions in hr-HPV positive women. Virol. J. 19, 95 (2022).
Almeida, A. M., Queiroz, J. A., Sousa, F. & Sousa, A. Cervical cancer and HPV infection: ongoing therapeutic research to counteract the action of E6 and E7 oncoproteins. Drug Discov. Today 24, 2044–2057 (2019).
Ghanaat, M. et al. Virus against virus: strategies for using adenovirus vectors in the treatment of HPV-induced cervical cancer. Acta Pharmacol. Sin. 42, 1981–1990 (2021).
Clark, E. et al. Veteran women living with human immunodeficiency virus have increased risk of human papillomavirus (HPV)-associated genital tract cancers. Clin. Infect. Dis. 72, e359–e366 (2021).
Joshi, S. et al. Incidence of cervical intraepithelial neoplasia in women infected with human immunodeficiency virus (HIV) with no evidence of disease at baseline: Results of a prospective cohort study with up to 6.4 years of follow-up from India. Int. J. Cancer 144, 1082–1091 (2019).
Mbuya, W. et al. Depletion of human papilloma virus E6- and E7-oncoprotein-specific T-cell responses in women living with HIV. Front. Immunol. 12, 742861 (2021).
Cohen, J. Cancer therapy returns to original target: HIV. Science 365, 530 (2019).
Zhao, X. et al. A safe and potentiated multi-type HPV L2-E7 nanoparticle vaccine with combined prophylactic and therapeutic activity. NPJ Vaccines 9, 119 (2024).
Hasan, Y. et al. A phase 1 trial assessing the safety and tolerability of a therapeutic DNA vaccination against HPV16 and HPV18 E6/E7 oncogenes after chemoradiation for cervical cancer. Int J. Radiat. Oncol. Biol. Phys. 107, 487–498 (2020).
Komdeur, F. L. et al. First-in-human phase I clinical trial of an SFV-based RNA replicon cancer vaccine against HPV-induced cancers. Mol. Ther. 29, 611–625 (2021).
Choi, Y. J. et al. A phase II, prospective, randomized, multicenter, open-label study of GX-188E, an HPV DNA vaccine, in patients with cervical intraepithelial neoplasia 3. Clin. Cancer Res. 26, 1616–1623 (2020).
Barnabas, R. V. et al. Durability of single-dose HPV vaccination in young Kenyan women: randomized controlled trial 3-year results. Nat. Med. 29, 3224–3232 (2023).
Youn, J. W. et al. Pembrolizumab plus GX-188E therapeutic DNA vaccine in patients with HPV-16-positive or HPV-18-positive advanced cervical cancer: interim results of a single-arm, phase 2 trial. Lancet Oncol. 21, 1653–1660 (2020).
Sheth, S. S. et al. Randomized phase II trial of imiquimod with or without 9-valent HPV vaccine versus observation in patients with high-grade pre-neoplastic cervical lesions (NCT02864147). Clin. Cancer Res. 30, 1768–1777 (2024).
Li, X. et al. CRISPR/Cas9 nanoeditor of double knockout large fragments of E6 and E7 oncogenes for reversing drugs resistance in cervical cancer. J. Nanobiotechnol. 19, 231 (2021).
Ehrke-Schulz, E. et al. Claudin18.2-specific CAR T cells in gastrointestinal cancers: phase 1 trial final results. Cancers 12, (2020).
Paolini, F. et al. Intrabodies targeting human papillomavirus 16 E6 and E7 oncoproteins for therapy of established HPV-associated tumors. J. Exp. Clin. Cancer Res. 40, 37 (2021).
Putri, J. F. et al. Mortaparib, a novel dual inhibitor of mortalin and PARP1, is a potential drug candidate for ovarian and cervical cancers. J. Exp. Clin. Cancer Res. 38, 499 (2019).
Sachdev, E. et al. PARP Inhibition In Cancer: An Update On Clinical Development. Target Oncol. 14, 657–679 (2019).
Mann, M., Singh, V. P. & Kumar, L. Cervical cancer: a tale from HPV infection to PARP inhibitors. Genes. Dis. 10, 1445–1456 (2023).
Thaker, P. H. et al. A phase I trial of paclitaxel, cisplatin, and veliparib in the treatment of persistent or recurrent carcinoma of the cervix: an NRG Oncology Study (NCT#01281852). Ann. Oncol. 28, 505–511 (2017).
Qiu, Y. et al. HIV Protease Inhibitors Block HPV16-Induced Murine Cervical Carcinoma And Promote Vessel Normalization In Association With MMP-9 Inhibition and TIMP-3 induction. Mol. Cancer Ther. 19, 2476–2489 (2020).
Park, S. et al. HIV-1 Protease inhibitors slow HPV16-driven cell proliferation through targeted depletion of viral E6 and E7 oncoproteins. Cancers 13, 949 (2021).
Wang, P. et al. HIV gp120/Tat protein-induced epithelial-mesenchymal transition promotes the progression of cervical lesions. AIDS Res. Ther. 20, 82 (2023).
Xu, Q. et al. HIV Tat-conjugated histone H3 peptides induce tumor cell death via cellular stress responses. Hum. Gene. Ther. 34, 42–55 (2023).
Hajizadeh, F. et al. Silencing of HIF-1alpha/CD73 axis by siRNA-loaded TAT-chitosan-spion nanoparticles robustly blocks cancer cell progression. Eur. J. Pharmacol. 882, 173235 (2020).
Glinsky, G. V. et al. Effects of anticancer agent P-bi-TAT on gene expression link the integrin thyroid hormone receptor to expression of stemness and energy metabolism genes in cancer cells. Metabolites. 12, 325 (2022).
Ferrall, L. et al. Cervical cancer immunotherapy: facts and hopes. Clin. Cancer Res. 27, 4953–4973 (2021).
Loharamtaweethong, K. et al. PD-L1 protein expression and copy number gains in HIV-positive locally advanced cervical cancer. Ther. Adv. Med. Oncol. 12, 1758835920963001 (2020).
Fujinaga, K. P-TEFb as a promising therapeutic target. Molecules. 25, 838 (2020).
Uldrick, T. S. et al. Pembrolizumab induces HIV latency reversal in people living with HIV and cancer on antiretroviral therapy. Sci. Transl. Med. 14, eabl3836 (2022).
Fromentin, R. et al. PD-1 blockade potentiates HIV latency reversal ex vivo in CD4(+) T cells from ART-suppressed individuals. Nat. Commun. 10, 814 (2019).
Rasmussen, T. A. et al. Impact of anti-PD-1 and anti-CTLA-4 on the human immunodeficiency virus (HIV) reservoir in people living with HIV with cancer on antiretroviral therapy: the AIDS malignancy consortium 095 study. Clin. Infect. Dis. 73, e1973–e1981 (2021).
Da Silva, D. M. et al. Immune activation in patients with locally advanced cervical cancer treated with ipilimumab following definitive chemoradiation (GOG-9929). Clin. Cancer Res. 26, 5621–5630 (2020).
Lou, H. et al. Cadonilimab combined with chemotherapy with or without bevacizumab as first-line treatment in recurrent or metastatic cervical cancer (COMPASSION-13): a phase 2 study. Clin. Cancer Res. 30, 1501–1508 (2024).
Voss, F. O., van Beurden, M. & Jordanova, E. S. Topical imiquimod as first-line treatment for vulvar intraepithelial neoplasia. Lancet 399, 1755–1757 (2022).
Knisely, A. et al. Phase 1/2 trial of avelumab combined with utomilumab (4-1BB agonist), PF-04518600 (OX40 agonist), or radiotherapy in patients with advanced gynecologic malignancies. Cancer 130, 400–409 (2024).
Porciello, N. et al. T-cell repertoire diversity: friend or foe for protective antitumor response? J. Exp. Clin. Cancer Res. 41, 356 (2022).
Zhao, Q. et al. Engineered TCR-T cell immunotherapy in anticancer precision medicine: pros and cons. Front. Immunol. 12, 658753 (2021).
Shevyrev, D. V., Tereshchenko, V. P. & Sennikov, S. V. The enigmatic nature of the TCR-pMHC interaction: implications for CAR-T and TCR-T engineering. Int. J. Mol. Sci. 23, 14728 (2022).
Nagarsheth, N. B. et al. TCR-engineered T cells targeting E7 for patients with metastatic HPV-associated epithelial cancers. Nat. Med. 27, 419–425 (2021).
Poorebrahim, M. et al. A costimulatory chimeric antigen receptor targeting TROP2 enhances the cytotoxicity of NK cells expressing a T cell receptor reactive to human papillomavirus type 16 E7. Cancer Lett. 566, 216242 (2023).
Quiros-Fernandez, I. et al. Dual T cell receptor/chimeric antigen receptor engineered NK-92 cells targeting the HPV16 E6 oncoprotein and the tumor-associated antigen L1CAM exhibit enhanced cytotoxicity and specificity against tumor cells. J. Med. Virol. 96, e29630 (2024).
Xu, M. et al. Genome sequencing analysis identifies Epstein-Barr virus subtypes associated with high risk of nasopharyngeal carcinoma. Nat. Genet. 51, 1131–1136 (2019).
Chan, A. T. Nasopharyngeal carcinoma. Ann. Oncol. 21, vii308–312 (2010).
Chan, K. C. A. et al. Analysis of plasma Epstein-Barr virus DNA to screen for nasopharyngeal cancer. N. Engl. J. Med. 377, 513–522 (2017).
Bruce, J. P. et al. Nasopharyngeal cancer: molecular landscape. J. Clin. Oncol. 33, 3346–3355 (2015).
Chen, Y. P. et al. Nasopharyngeal carcinoma. Lancet 394, 64–80 (2019).
Chua, M. L. K., Wee, J. T. S., Hui, E. P. & Chan, A. T. C. Nasopharyngeal carcinoma. Lancet 387, 1012–1024 (2016).
Yilmaz, E. et al. Immunotherapy and biomarker testing in recurrent and metastatic head and neck cancers: ASCO guideline. J. Clin. Oncol. 41, 1132–1146 (2023).
Chen, Y. P. et al. Chemotherapy in combination with radiotherapy for definitive-intent treatment of stage II-IVA nasopharyngeal carcinoma: CSCO and ASCO guideline. J. Clin. Oncol. 39, 840–859 (2021).
Chan, D. C. T. et al. Improved risk stratification of nasopharyngeal cancer by targeted sequencing of Epstein-Barr virus DNA in post-treatment plasma. Ann. Oncol. 33, 794–803 (2022).
Lou, P. J. et al. Performance and operational feasibility of Epstein-Barr virus-based screening for detection of nasopharyngeal carcinoma: direct comparison of two alternative approaches. J. Clin. Oncol. 41, 4257–4266 (2023).
Ma, B. B. Y., Hui, E. P. & Chan, A. T. C. The expanding universe of checkpoint inhibitors for nasopharyngeal cancer. Nat. Med. 27, 1512–1513 (2021).
Okano, M. & Gross, T. G. Advanced therapeutic and prophylactic strategies for Epstein-Barr virus infection in immunocompromised patients. Expert. Rev. Anti. Infect. Ther. 5, 403–413 (2007).
Li, W. et al. Immunotherapeutic approaches in EBV-associated nasopharyngeal carcinoma. Front. Immunol. 13, 1079515 (2022).
Wong, K. C. W. et al. Nasopharyngeal carcinoma: an evolving paradigm. Nat. Rev. Clin. Oncol. 18, 679–695 (2021).
Petrosky, E. et al. Use of 9-valent human papillomavirus (HPV) vaccine: updated HPV vaccination recommendations of the advisory committee on immunization practices. Morb Mortal Wkly. Rep. 64, 300–304 (2015).
Roden, R. B. S. & Stern, P. L. Opportunities and challenges for human papillomavirus vaccination in cancer. Nat. Rev. Cancer 18, 240–254 (2018).
Mohsen, M. O., Zha, L., Cabral-Miranda, G. & Bachmann, M. F. Major findings and recent advances in virus-like particle (VLP)-based vaccines. Semin. Immunol. 34, 123–132 (2017).
Lin, C. L. et al. Immunization with Epstein-Barr Virus (EBV) peptide-pulsed dendritic cells induces functional CD8+ T-cell immunity and may lead to tumor regression in patients with EBV-positive nasopharyngeal carcinoma. Cancer Res. 62, 6952–6958 (2002).
Chia, W. K. et al. A phase II study evaluating the safety and efficacy of an adenovirus-DeltaLMP1-LMP2 transduced dendritic cell vaccine in patients with advanced metastatic nasopharyngeal carcinoma. Ann. Oncol. 23, 997–1005 (2012).
Prasad, M. et al. Epstein-Barr virus-induced ectopic CD137 expression helps nasopharyngeal carcinoma to escape immune surveillance and enables targeting by chimeric antigen receptors. Cancer Immunol. Immunother 71, 2583–2596 (2022).
Zeng, Q., Zhou, Y. & Schwarz, H. CD137L-DCs, potent immune-stimulators-history, characteristics, and perspectives. Front. Immunol. 10, 2216 (2019).
Ruhl, J. et al. Heterologous prime-boost vaccination protects against EBV antigen-expressing lymphomas. J. Clin. Invest. 129, 2071–2087 (2019).
Sharma, S. & Rouce, R. H. Are we there yet? The never-ending quest for an Epstein-Barr virus vaccine. J. Clin. Invest. 129, 1836–1838 (2019).
Taylor, G. S. et al. Dual stimulation of Epstein-Barr Virus (EBV)-specific CD4+− and CD8+−T-cell responses by a chimeric antigen construct: potential therapeutic vaccine for EBV-positive nasopharyngeal carcinoma. J. Virol. 78, 768–778 (2004).
Taylor, G. S. et al. A recombinant modified vaccinia Ankara vaccine encoding Epstein-Barr Virus (EBV) target antigens: a phase I trial in UK patients with EBV-positive cancer. Clin. Cancer Res. 20, 5009–5022 (2014).
Kang, Y. et al. Advances in targeted therapy mainly based on signal pathways for nasopharyngeal carcinoma. Signal Transduct. Target Ther. 5, 245 (2020).
Lin, D. C. et al. The genomic landscape of nasopharyngeal carcinoma. Nat. Genet. 46, 866–871 (2014).
Wang, M. et al. Acquired semi-squamatization during chemotherapy suggests differentiation as a therapeutic strategy for bladder cancer. Cancer Cell 40, 1044–1059 (2022).
Gandhi, M. K. et al. Expression of LAG-3 by tumor-infiltrating lymphocytes is coincident with the suppression of latent membrane antigen-specific CD8+ T-cell function in Hodgkin lymphoma patients. Blood 108, 2280–2289 (2006).
Chua, D. et al. Adoptive transfer of autologous Epstein-Barr virus-specific cytotoxic T cells for nasopharyngeal carcinoma. Int. J. Cancer 94, 73–80 (2001).
Comoli, P. et al. Adoptive transfer of allogeneic Epstein-Barr virus (EBV)-specific cytotoxic T cells with in vitro antitumor activity boosts LMP2-specific immune response in a patient with EBV-related nasopharyngeal carcinoma. Ann. Oncol. 15, 113–117 (2004).
Straathof, K. C. et al. Treatment of nasopharyngeal carcinoma with Epstein-Barr virus-specific T lymphocytes. Blood 105, 1898–1904 (2005).
Chia, W. K. et al. Adoptive T-cell transfer and chemotherapy in the first-line treatment of metastatic and/or locally recurrent nasopharyngeal carcinoma. Mol. Ther. 22, 132–139 (2014).
Zhang, J. et al. Co-expression of PD-1 and PD-L1 predicts poor outcome in nasopharyngeal carcinoma. Med. Oncol. 32, 86 (2015).
Anastasiadou, E. et al. Epstein-Barr virus-encoded EBNA2 alters immune checkpoint PD-L1 expression by downregulating miR-34a in B-cell lymphomas. Leukemia 33, 132–147 (2019).
Ma, B. B. Y. et al. Antitumor activity of nivolumab in recurrent and metastatic nasopharyngeal carcinoma: an international, multicenter study of the mayo clinic phase 2 consortium (NCI-9742). J. Clin. Oncol. 36, 1412–1418 (2018).
Doi, T. et al. Safety and antitumor activity of the anti-programmed death-1 antibody pembrolizumab in patients with advanced esophageal carcinoma. J. Clin. Oncol. 36, 61–67 (2018).
Fang, W. et al. Camrelizumab (SHR-1210) alone or in combination with gemcitabine plus cisplatin for nasopharyngeal carcinoma: results from two single-arm, phase 1 trials. Lancet Oncol. 19, 1338–1350 (2018).
Hsu, C. et al. Safety and antitumor activity of pembrolizumab in patients with programmed death-ligand 1-positive nasopharyngeal carcinoma: results of the KEYNOTE-028 study. J. Clin. Oncol. 35, 4050–4056 (2017).
Mansfield, A. S. et al. Outcomes With pembrolizumab monotherapy in patients with programmed death-ligand 1-positive NSCLC with brain metastases: pooled analysis of KEYNOTE-001, 010, 024, and 042. JTO Clin. Res. Rep. 2, 100205 (2021).
Nishio, M. et al. KEYNOTE-025: Phase 1b study of pembrolizumab in Japanese patients with previously treated programmed death ligand 1-positive advanced non-small-cell lung cancer. Cancer Sci. 110, 1012–1020 (2019).
Smith, C. et al. Complete response to PD-1 blockade following EBV-specific T-cell therapy in metastatic nasopharyngeal carcinoma. NPJ Precis. Oncol. 5, 24 (2021).
Yang, Y. et al. Camrelizumab versus placebo in combination with gemcitabine and cisplatin as first-line treatment for recurrent or metastatic nasopharyngeal carcinoma (CAPTAIN-1st): a multicentre, randomised, double-blind, phase 3 trial. Lancet Oncol. 22, 1162–1174 (2021).
Lim, D. W. et al. Clinical efficacy and biomarker analysis of dual PD-1/CTLA-4 blockade in recurrent/metastatic EBV-associated nasopharyngeal carcinoma. Nat. Commun. 14, 2781 (2023).
Lin, S. et al. Early change of plasma Epstein-Barr virus DNA load and the viral lytic genome level could positively predict clinical outcome in recurrent or metastatic nasopharyngeal carcinoma receiving anti-programmed cell death 1 monotherapy. BMC Cancer 24, 797 (2024).
Li, D. et al. EpCAM-targeting CAR-T cell immunotherapy is safe and efficacious for epithelial tumors. Sci. Adv. 9, eadg9721 (2023).
Tang, X. et al. CD137 co-stimulation improves the antitumor effect of LMP1-specific chimeric antigen receptor T cells in vitro and in vivo. Onco. Targets Ther. 12, 9341–9350 (2019).
Cho, H. I. et al. A novel Epstein-Barr virus-latent membrane protein-1-specific T-cell receptor for TCR gene therapy. Br. J. Cancer 118, 534–545 (2018).
Antman, K. & Chang, Y. Kaposi’s sarcoma. N. Engl. J. Med. 342, 1027–1038 (2000).
Wu, X. J. et al. One hundred and five Kaposi sarcoma patients: a clinical study in Xinjiang, Northwest of China. J. Eur. Acad. Dermatol. Venereol. 28, 1545–1552 (2014).
Saller, J. et al. Response to checkpoint inhibitor therapy in advanced classic Kaposi sarcoma: a case report and immunogenomic study. J. Natl. Compr. Canc. Netw. 16, 797–800 (2018).
Tashakori, M. & Beckman, A. K. Circulating plasmablastic cells in a patient with HHV-8-associated multicentric Castleman disease and Kaposi sarcoma. Blood 142, 2124 (2023).
Krown, S. E. & Borok, M. Z. Evaluation of treatments for HIV-associated Kaposi sarcoma in Africa. Infect. Agent Cancer 16, 25 (2021).
Volesky-Avellaneda, K. D. et al. Cancers attributable to infections in the US in 2017: a meta-analysis. JAMA Oncol. 9, 1678–1687 (2023).
Casper, C. et al. KSHV (HHV8) vaccine: promises and potential pitfalls for a new anti-cancer vaccine. NPJ Vaccines 7, 108 (2022).
Ramon-Luing, L. A. et al. Valganciclovir modulates the tumor necrosis factor axis molecules expression and CD4+ T-cell subsets in disseminated Kaposi sarcoma patients. Clin. Expert. Immunol. 215, 190–201 (2024).
Volkow, P. et al. Impact of valganciclovir therapy on severe IRIS-Kaposi sarcoma mortality: an open-label, parallel, randomized controlled trial. PLoS One 18, e0280209 (2023).
Gothland, A. et al. Primaquine as a candidate for HHV-8-associated primary effusion lymphoma and Kaposi’s sarcoma treatment. Cancers 14, 543 (2022).
Vaccher, E. & Carbone, A. Simultaneous occurrence of KSHV-associated malignancies in a patient affected by HIV. Blood 137, 3149 (2021).
Chen, J. et al. Echinomycin as a promising therapeutic agent against KSHV-related malignancies. J. Hematol. Oncol. 16, 48 (2023).
Li, W. et al. A SEER data-based nomogram for the prognostic analysis of survival of patients with Kaposi’s sarcoma. J. Cancer Res. Ther. 19, 917–923 (2023).
Margolis, D. M. Advancing toward a human immunodeficiency virus cure: initial progress on a difficult path. Infect. Dis. Clin. North Am. 38, 487–497 (2024).
Sun, Y. & Wang, L. Development of anti-HIV therapeutics: from conventional drug discovery to cutting-edge technology. Pharmaceuticals 17, 887 (2024).
Krown, S. E. et al. Treatment of advanced AIDS-associated Kaposi sarcoma in resource-limited settings: a three-arm, open-label, randomised, non-inferiority trial. Lancet 395, 1195–1207 (2020).
Nyirenda, M. et al. Early progression and immune reconstitution inflammatory syndrome during treatment of mild-to-moderate Kaposi sarcoma in sub-Saharan Africa and South America: incidence, long-term outcomes, and effects of early chemotherapy. J. Acquir. Immune Defic. Syndr. 84, 422–429 (2020).
Zhang, X., Yang, K., Chen, S. & Ji, Y. Tacrolimus ointment for the treatment of superficial Kaposiform hemangioendothelioma and tufted angioma. J. Dermatol. 46, 898–901 (2019).
Mendez-Solis, O. et al. Kaposi’s sarcoma herpesvirus activates the hypoxia response to usurp HIF2alpha-dependent translation initiation for replication and oncogenesis. Cell Rep. 37, 110144 (2021).
Cornejo-Juarez, P. et al. Bone marrow culture yield for the diagnosis of opportunistic diseases in patients with AIDS and disseminated Kaposi sarcoma. Curr. HIV Res. 18, 277–282 (2020).
Ramaswami, R. et al. A pilot study of liposomal doxorubicin combined with bevacizumab followed by bevacizumab monotherapy in patients with advanced Kaposi sarcoma. Clin. Cancer Res. 25, 4238–4247 (2019).
Reid, E. G. et al. Pilot trial AMC-063: safety and efficacy of bortezomib in AIDS-associated Kaposi sarcoma. Cli. Cancer Res. 26, 558–565 (2020).
Host, K. M. et al. Kaposi’s Sarcoma-associated herpesvirus increases PD-L1 and proinflammatory cytokine expression in human monocytes. mBio. 8, e00917 (2017).
Delyon, J. et al. PD-1 blockade with pembrolizumab in classic or endemic Kaposi’s sarcoma: a multicentre, single-arm, phase 2 study. Lancet Oncol. 23, 491–500 (2022).
Zer, A. et al. Phase II single-arm study of nivolumab and ipilimumab (Nivo/Ipi) in previously treated classical Kaposi sarcoma (cKS). Ann. Oncol. 33, 720–727 (2022).
Gambichler, T. & Susok, L. PD-1 blockade for disseminated Kaposi sarcoma in a patient with atopic dermatitis and chronic CD8 lymphopenia. Immunotherapy 12, 451–457 (2020).
Uldrick, T. S. et al. Assessment of the safety of pembrolizumab in patients with HIV and advanced cancer—a phase 1 study. JAMA Oncol. 5, 1332–1339 (2019).
Skoetz, N. et al. Comparison of first-line chemotherapy including escalated BEACOPP versus chemotherapy including ABVD for people with early unfavourable or advanced stage Hodgkin lymphoma. Cochrane Database Syst. Rev. 5, CD007941 (2017).
Atallah-Yunes, S. A., Murphy, D. J. & Noy, A. HIV-associated Burkitt lymphoma. Lancet Haematol. 7, e594–e600 (2020).
Atallah-Yunes, S. A., Habermann, T. M. & Khurana, A. Targeted therapy in Burkitt lymphoma: Small molecule inhibitors under investigation. Br. J. Haematol. 204, 2165–2172 (2024).
Cao, G. D. et al. The oncolytic virus in cancer diagnosis and treatment. Front. Oncol. 10, 1786 (2020).
McGee-Avila, J. K. et al. Cancer treatment disparities in people with HIV in the United States, 2001-2019. J. Clin. Oncol. 42, 1810–1820 (2024).
Rattotti, S. et al. Lymphomas associated with chronic hepatitis C virus infection: a prospective multicenter cohort study from the rete ematologica lombarda (REL) clinical network. Hematol. Oncol. 37, 160–167 (2019).
Tsutsumi, Y. et al. Efficacy and prognosis of antiviral therapy on hepatitis C following treatment of lymphoma in HCV-positive diffuse large-cell lymphoma. Ann. Hematol. 96, 2057–2061 (2017).
Tan, C. R. C. et al. Combination antiretroviral therapy accelerates immune recovery in patients with HIV-related lymphoma treated with EPOCH: a comparison within one prospective trial AMC034. Leuk. Lymphoma 59, 1851–1860 (2018).
Sabattini, E. et al. CD30 expression in peripheral T-cell lymphomas. Haematologica 98, e81–82 (2013).
Kim, M. et al. A phase II study of brentuximab vedotin in patients with relapsed or refractory Epstein-Barr virus-positive and CD30-positive lymphomas. Haematologica 106, 2277–2280 (2021).
Kim, S. J. et al. Efficacy of brentuximab vedotin in relapsed or refractory high-CD30-expressing non-Hodgkin lymphomas: results of a multicenter, open-labeled phase II trial. Cancer Res. Treat. 52, 374–387 (2020).
Tan, D., Diong, C. P., Loh, Y. & Goh, Y. T. Histone deacetylase (HDAC) inhibitors when combined with a proteasome inhibitor are safe and effective in patients with extranodal natural killer/T-cell lymphoma (ENKTL). Ann. Oncol. 27, 1811–1812 (2016).
Kim, S. J. et al. Epstein-Barr virus reactivation in extranodal natural killer/T-cell lymphoma patients: a previously unrecognized serious adverse event in a pilot study with romidepsin. Ann. Oncol. 27, 508–513 (2016).
Tsai, P. F. et al. Interplay between PKCdelta and Sp1 on histone deacetylase inhibitor-mediated Epstein-Barr virus reactivation. J. Virol. 85, 2373–2385 (2011).
Wang, C. et al. Impact of initial chemotherapy cycles and clinical characteristics on outcomes for HIV-associated diffuse large B cell lymphoma patients: the central and western China AIDS lymphoma league 001 study (CALL-001 study). Front. Immunol. 14, 1153790 (2023).
Schommers, P. et al. Incidence and risk factors for relapses in HIV-associated non-Hodgkin lymphoma as observed in the German HIV-related lymphoma cohort study. Haematologica 103, 857–864 (2018).
Kojo, A. et al. Preferential inhibition of mouse hepatic coumarin 7-hydroxylase by inhibitors of steroid metabolizing monooxygenases. Pharmacol. Toxicol. 65, 104–109 (1989).
Wang, C. et al. Selinexor in combination with R-EPOCH for patients with previously untreated HIV-associated diffuse large B cell lymphoma(DLBCL). Blood 142, 6292–6292 (2023).
Chiang, A. K., Tao, Q., Srivastava, G. & Ho, F. C. Nasal NK- and T-cell lymphomas share the same type of Epstein-Barr virus latency as nasopharyngeal carcinoma and Hodgkin’s disease. Int. J. Cancer 68, 285–290 (1996).
Tse, E. & Kwong, Y. L. Epstein Barr virus-associated lymphoproliferative diseases: the virus as a therapeutic target. Exp. Mol. Med. 47, e136 (2015).
Gottschalk, S. et al. Generating CTLs against the subdominant Epstein-Barr virus LMP1 antigen for the adoptive immunotherapy of EBV-associated malignancies. Blood 101, 1905–1912 (2003).
Cho, S. G. et al. Long-term outcome of extranodal NK/T cell lymphoma patients treated with postremission therapy using EBV LMP1 and LMP2a-specific CTLs. Mol. Ther. 23, 1401–1409 (2015).
Bollard, C. M. et al. Sustained complete responses in patients with lymphoma receiving autologous cytotoxic T lymphocytes targeting Epstein-Barr virus latent membrane proteins. J. Clin. Oncol. 32, 798–808 (2014).
Prockop, S. et al. Off-the-shelf EBV-specific T cell immunotherapy for rituximab-refractory EBV-associated lymphoma following transplantation. J. Clin. Investig. 130, 733–747 (2020).
Marjanska, A. & Styczynski, J. Who is the patient at risk for EBV reactivation and disease: expert opinion focused on post-transplant lymphoproliferative disorders following hematopoietic stem cell transplantation. Expert Opin. Biol. Ther. 23, 539–552 (2023).
Liu, L., Liu, Q. & Feng, S. Management of Epstein-Barr virus-related post-transplant lymphoproliferative disorder after allogeneic hematopoietic stem cell transplantation. Ther. Adv. Hematol. 11, 2040620720910964 (2020).
Toner, K., McCann, C. D. & Bollard, C. M. Applications of cell therapy in the treatment of virus-associated cancers. Nat. Rev. Clin. Oncol. 21, 709–724 (2024).
Mahadeo, K. M. et al. Tabelecleucel for allogeneic haematopoietic stem-cell or solid organ transplant recipients with Epstein-Barr virus-positive post-transplant lymphoproliferative disease after failure of rituximab or rituximab and chemotherapy (ALLELE): a phase 3, multicentre, open-label trial. Lancet Oncol. 25, 376–387 (2024).
Nikiforow, S. et al. Tabelecleucel for EBV+ PTLD after allogeneic HCT or SOT in a multicenter expanded access protocol. Blood Adv. 8, 3001–3012 (2024).
Vickers, M. A. et al. Establishment and operation of a good manufacturing practice-compliant allogeneic Epstein-Barr virus (EBV)-specific cytotoxic cell bank for the treatment of EBV-associated lymphoproliferative disease. Br. J. Haematol. 167, 402–410 (2014).
Feng, Y. et al. The expression and clinical significance of programmed cell death receptor 1 and its ligand in tumor tissues of patients with extranodal nasal NK/T cell lymphoma. Sci. Rep. 12, 36 (2022).
Ng, S. B. et al. Epstein-Barr virus-associated primary nodal T/NK-cell lymphoma shows a distinct molecular signature and copy number changes. Haematologica 103, 278–287 (2018).
Nagato, T. et al. Programmed death-ligand 1 and its soluble form are highly expressed in nasal natural killer/T-cell lymphoma: a potential rationale for immunotherapy. Cancer Immunol. Immunother. 66, 877–890 (2017).
Tian, X. P. et al. Novel target and treatment agents for natural killer/T-cell lymphoma. J Hematol. Oncol. 16, 78 (2023).
Tao, R. et al. Sintilimab for relapsed/refractory extranodal NK/T cell lymphoma: a multicenter, single-arm, phase 2 trial (ORIENT-4). Signal Transduct Target. Ther. 6, 365 (2021).
Huang, H. et al. Sugemalimab monotherapy for patients with relapsed or refractory extranodal natural killer/T-cell lymphoma (GEMSTONE-201): Results from a single-arm, multicenter, phase II study. J. Clin. Oncol. 41, 3032–3041 (2023).
Kwong, Y. L. et al. PD1 blockade with pembrolizumab is highly effective in relapsed or refractory NK/T-cell lymphoma failing l-asparaginase. Blood 129, 2437–2442 (2017).
Li, H. et al. CAR-T cells targeting CD38 and LMP1 exhibit robust antitumour activity against NK/T cell lymphoma. BMC Med. 21, 330 (2023).
Wang, X. et al. Expanding anti-CD38 immunotherapy for lymphoid malignancies. J. Exp. Clin. Cancer Res. 41, 210 (2022).
Wang, Y. et al. Facing challenges with hope: universal immune cells for hematologic malignancies. Cancer Biol. Med. 20, 229–247 (2023).
Krakowski, A. J. Patients who sign against medical advice. Psychiatr. J. Univ. Ott. 10, 254–259 (1985).
Wu, J. et al. Chimeric antigen receptor-modified T cell immunotherapy for relapsed and refractory adult Burkitt lymphoma. Front. Immunol. 13, 879983 (2022).
Liu, Y. et al. Sequential different B-cell antigen-targeted CAR T-cell therapy for pediatric refractory/relapsed Burkitt lymphoma. Blood Adv. 6, 717–730 (2022).
Hattenhauer, S. T. et al. Enabling CAR T-cell therapies for HIV-positive lymphoma patients—a call for action. HIV Med. 24, 957–964 (2023).
Nakano, N. et al. Outcomes in human T-cell leukemia virus type I carriers after hematopoietic stem cell transplantation for diseases other than adult T cell leukemia/lymphoma: a Japanese national survey. Lancet Reg. Health West Pac. 40, 100902 (2023).
Ishitsuka, K. & Tamura, K. Human T-cell leukaemia virus type I and adult T-cell leukaemia-lymphoma. Lancet Oncol. 15, e517–e526 (2014).
Cheminant, M. et al. KIR3DL2 contributes to the typing of acute adult T-cell leukemia and is a potential therapeutic target. Blood 140, 1522–1532 (2022).
Gill, P. S. et al. Treatment of adult T-cell leukemia-lymphoma with a combination of interferon alfa and zidovudine. N. Engl. J. Med. 332, 1744–1748 (1995).
Nasr, R., Marcais, A., Hermine, O. & Bazarbachi, A. Overview of targeted therapies for adult T-cell leukemia/lymphoma. Methods Mol. Biol. 1582, 197–216 (2017).
Bazarbachi, A. et al. Meta-analysis on the use of zidovudine and interferon-alfa in adult T-cell leukemia/lymphoma showing improved survival in the leukemic subtypes. J. Clin. Oncol. 28, 4177–4183 (2010).
Cook, L. B. & Taylor, G. P. Treatment of adult T-cell leukaemia/lymphoma: is the virus a target? Curr. Opin. Infect. Dis. 28, 583–588 (2015).
Izutsu, K. et al. An open-label, single-arm phase 2 trial of valemetostat for relapsed or refractory adult T-cell leukemia/lymphoma. Blood 141, 1159–1168 (2023).
Kabiri, M., Sankian, M., Hosseinpour, M. & Tafaghodi, M. The novel immunogenic chimeric peptide vaccine to elicit potent cellular and mucosal immune responses against HTLV-1. Int. J. Pharm 549, 404–414 (2018).
Suehiro, Y. et al. Clinical outcomes of a novel therapeutic vaccine with Tax peptide-pulsed dendritic cells for adult T cell leukaemia/lymphoma in a pilot study. Br. J. Haematol. 169, 356–367 (2015).
Wang, T. T. et al. Current state of therapeutics for HTLV-1. Viruses 16, 1616 (2024).
Panfil, A. R., Green, P. L. & Yoder, K. E. CRISPR genome editing applied to the pathogenic retrovirus HTLV-1. Front. Cell Infect. Microbiol. 10, 580371 (2020).
Tanaka, A. et al. A novel therapeutic molecule against HTLV-1 infection targeting provirus. Leukemia 27, 1621–1627 (2013).
Nakagawa, M. et al. Targeting the HTLV-I-regulated BATF3/IRF4 transcriptional network in adult T cell leukemia/lymphoma. Cancer Cell 34, 286–297.e210 (2018).
Rojo-Romanos, T. et al. Precise excision of HTLV-1 provirus with a designer-recombinase. Mol. Ther. 31, 2266–2285 (2023).
Hoshida, Y. et al. Pathogenesis and prevention of hepatitis C virus-induced hepatocellular carcinoma. J. Hepatol. 61, S79–S90 (2014).
Lo Cigno, I. et al. High-risk HPV oncoproteins E6 and E7 and their interplay with the innate immune response: Uncovering mechanisms of immune evasion and therapeutic prospects. J. Med. Virol. 96, e29685 (2024).
Ma, T. et al. KSHV induces aerobic glycolysis and angiogenesis through HIF-1-dependent upregulation of pyruvate kinase 2 in Kaposi’s sarcoma. Angiogenesis 18, 477–488 (2015).
Acknowledgements
The authors thank Professor Huilai Zhang from the Department of Lymphoma at Tianjin Medical University Cancer Institute and Hospital, Professor Zhian Hu from the Department of Physiology at the Third Military Medical University, and Professor Yang Liang from the Department of Hematologic Oncology at Sun Yat-sen University Cancer Center for their assistance and suggestions during the writing process. This work was supported by Chongqing Science and Health Joint Medical Research Sprint, Key Projects, and Major Projects (2025DBXM002), Chongqing Innovative Medical Device Application Demonstration Project (CQEIC2024MDAD-050), the Key Project of Chongqing Technology Innovation and Application Development Special Project (CSTB2024TIAD-KPX0031), National Natural Science Foundation of China (82370114), Special Project for Performance Incentive Guidance of Scientific Research Institutions (2024JXJL-YFX0022), Natural Science Foundation of Chongqing, China (CSTB2022NSCQ-MSX1150), the Fundamental Research Funds for the Central Universities (2022CDJYGRH-001) and the Chongqing Professional Talents Plan (cstc2022ycjh-bgzxm0048).
Author information
Authors and Affiliations
Contributions
There are five first authors in this manuscript and they have equally contributed to this project. Qing Xiao, Yi Liu, Tingting Li, Chaoyu Wang, and Sanxiu He performed the selection of literature, drafted the paper and prepared the figures. Liuyue Zhai and Zailin Yang collected the related references and organized the tables. We have three corresponding authors in this manuscript. Xiaomei Zhang, Yongzhong Wu, and Yao Liu conceived, designed, and edited the manuscript. All authors have read and approved the article.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Xiao, Q., Liu, Y., Li, T. et al. Viral oncogenesis in cancer: from mechanisms to therapeutics. Sig Transduct Target Ther 10, 151 (2025). https://doi.org/10.1038/s41392-025-02197-9
Received:
Revised:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41392-025-02197-9
This article is cited by
-
Chimeric antigen receptor T‑cell therapy in chronic viral infections: a review
Journal of Translational Medicine (2026)
-
Viral-driven oncogenesis in T/NK-cell lymphomas: parallels and divergences between HTLV-1 and EBV
International Journal of Hematology (2026)
-
KSHV and cancer: understanding the oncogenic machinery for next-generation diagnostic tools and therapies
Archives of Microbiology (2026)
-
Advancements in understanding tumor-resident bacteria and their application in cancer therapy
Military Medical Research (2025)
