
Abstract
Tumor necrosis factor-alpha (TNF-α) plays paradoxical roles in cancer, promoting tumor survival via NF-κB activation while also inducing necroptotic cell death. Here, we identify the intracellular pH (pHᵢ) as a decisive switch that reprograms TNF-α signaling from a prosurvival inflammatory response toward tumoricidal necroptosis. In both hepatocellular and cervical carcinoma models, alkalizing the pHᵢ with bicarbonate inhibits TNF-α-mediated IκB phosphorylation and NF-κB nuclear translocation while simultaneously activating the TNF-α-driven necroptotic cascade, which is characterized by the sequential phosphorylation of RIPK1, RIPK3, and MLKL. Mechanistically, bicarbonate induces the depolarization of the mitochondrial membrane potential, reactive oxygen species (ROS) generation, and a cyclophilin D-dependent permeability transition—effects that are synergistically amplified by TNF-α. Genetic knockdown of RIPK1 or RIPK3 abrogates necroptosis, whereas the pharmacological inhibition of ROS suppresses necroptosis and concurrently restores NF-κB nuclear translocation. ROS are identified as the key mediators determining the switch between these opposing TNF-α signaling pathways. In vivo, bicarbonate and TNF-α treatment suppresses tumor growth, enhances necroptotic signaling, and inhibits NF-κB activation. Thus, our work indicates that intracellular alkalization acts as a modifiable regulator to redirect TNF-α from a toxic, proinflammatory cytokine into a potent and selective tumoricidal agent.

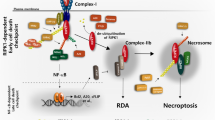
Graphical Abstract: Alkalization of the pHi by bicarbonate reprograms TNF-α signaling from a prosurvival NF-κB axis to a cytotoxic necroptotic pathway. a TNF-α alone activates NF-κB signaling, promoting inflammation and tumor growth. b Bicarbonate alkalizes the pHi and induces mitochondrial dysfunction and reactive oxygen species (ROS) production, which is further amplified by TNF-α. Elevated ROS levels inhibit NF-κB signaling and activate RIPK1-dependent necroptosis. This ROS-mediated switch redirects TNF-α signaling from a proinflammatory, tumor-promoting response to a cytotoxic, necroptotic pathway, resulting in the suppression of both tumor growth and inflammation. This diagram highlights the translational potential of combining bicarbonate with TNF-α for cancer therapy.
Similar content being viewed by others


Introduction
Tumor necrosis factor-alpha (TNF-α) is a pleiotropic cytokine that orchestrates inflammation, immunity, and cell fate decisions [1,2,3]. Its signaling bifurcates into two opposing arms: the activation of the nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) pathway, which promotes survival and inflammation, and the induction of programmed cell death pathways such as apoptosis and necroptosis [4, 5]. Although TNF-α has potent antitumor potential, its clinical utility is limited by two major obstacles. First, TNF-α preferentially activates NF-κB-driven prosurvival programs in cancer cells, and strategies capable of redirecting TNF-α toward necroptosis remain poorly defined[6,7,8]. Second, systemic inflammatory toxicity restricts its therapeutic application [7,8,9]. An effective TNF-α-based therapy therefore requires a means to suppress NF-κB activation while promoting necroptotic signaling specifically within tumor cells.
Intracellular pH (pHᵢ) is a fundamental regulator of cellular metabolism, signaling, and survival [10, 11]. Even modest perturbations in pHᵢ influence enzyme activities, mitochondrial function, ion transport, and signaling pathways. We previously demonstrated that experimentally increasing the pHᵢ disrupts mitochondrial proton gradients, decreases the mitochondrial membrane potential (ΔΨm) and pH gradient (ΔpH), suppresses oxidative phosphorylation (OXPHOS), and triggers a sustained mitochondrial permeability transition (MPT) [12]. These changes initiate a cascade of energetic stress, including AMP-activated protein kinase activation, impaired autophagic flux, and lysosomal dysfunction, which we hypothesize creates a cellular context permissive for the redirection of TNF-α signaling from NF-κB toward necroptosis.
We employ bicarbonate, a physiologically abundant and clinically safe anion, to experimentally manipulate the pHᵢ. Bicarbonate is the second most common anion in human plasma and is a central component of acid–base homeostasis. Clinically, sodium bicarbonate is widely used to correct metabolic acidosis [13,14,15]. Importantly, in our previous clinical Targeting Intratumoral Lactic Acidosis–TACE (TILA–TACE) trials involving 453 hepatocellular carcinoma patients [16, 17], 600 mM bicarbonate was locally delivered into the tumor-feeding artery. This high local concentration was well tolerated, produced substantial improvements in objective responses, and did not cause systemic alkalosis. These human data indicate that tumor-targeted bicarbonate concentrations far exceeding physiological plasma levels are clinically feasible and therapeutically effective.
Here, we show that bicarbonate markedly redirects TNF-α signaling in cancer cells. Bicarbonate dramatically suppresses TNF-α-induced NF-κB activation while promoting mitochondrial reactive oxygen species (ROS) accumulation, persistent MPT, and activation of the RIPK-dependent necroptotic pathway. This dual modulation increases the antitumor efficacy and provides a conceptual framework for leveraging pHᵢ alkalization as a clinically actionable strategy to unlock the therapeutic potential of TNF-α. The central concept is illustrated in the Graphical Abstract, which depicts how bicarbonate-mediated alkalization shifts TNF-α signaling from a prosurvival NF-κB pathway toward mitochondrial ROS-driven necroptosis and tumor suppression.
Materials and methods
Regents and antibodies
Necrostatin-1 (Nec-1) (HY15760, MCE, USA), manganese (salen) chloride (EUK8) (HY-W001583, MCE, USA), N-acetylcysteine (NAC) (HY-B0215, MCE, USA), recombinant human TNF alpha (#HZ1014, Proteintech, USA), calcein-AM (#C3099, Thermo Fisher Scientific, USA), CoCl2 (409332, Sigma‒Aldrich, USA), TMRE (#T699, Thermo Fisher Scientific, USA), Hoechst 33342 (#C1022, Beyotime, China), MitoSoxTM Red (#M36008, Thermo Fisher Scientific, USA), DCFH-DA (#D6883, Sigma, USA), an Annexin V-FITC Apoptosis Detection Kit (#1062L, Beyotime, China), NIM811 (HY-P0025, MCE, USA), and Lipofectamine® RNAi MAX Reagent (13778075, Thermo Fisher Scientific, USA) were used.
GAPDH (#60004-1-lg, Proteintech, USA), NF-κB p65 (#8242, CST, USA), phospho-RIPK1 (Ser166) (#44590, CST, USA), RIPK1 (#3493, CST, USA), RIPK1 (#610458, BD, USA), phospho-RIPK3 (Ser227) (#93654, CST, USA), RIPK3 (#10188, CST, USA), RIPK3 (#17563-1-AP, Proteintech, USA), phospho-MLKL (Ser358) (#91689, CST, USA), MLKL (#14993, CST, USA), phospho-IκB (Ser32) (#2859, CST, USA), IκB (#4814, CST, USA), ubiquitin (P4D1) (#3936, CST, USA), FADD (#2782, CST, USA), PARP (46D11) (#9532, CST, USA), Caspase 8 (1C12) (#9746, CST, USA), γ-H2AX (#83307-2-RR, Proteintech, USA), and H2AX (#68888-1-Ig, Proteintech, USA) antibodies, as well as Corallite 488-conjugate secondary antibodies (#SA00013-2, Proteintech, USA) were used.
Animals
Six-week-old female NOD/SCID mice were purchased from SLAC Laboratory Animal Co., Ltd. (Shanghai, China) and housed at Zhejiang Chinese Medical University Laboratory Animal Research Center. All animal experiments were approved and performed in complete compliance with the guidelines approved by the Animal Ethical and Welfare Committee of Zhejiang Chinese Medical University (Approval number: IACUC-20230918-11). All the mice were fed standard laboratory chow and provided water ad libitum.
Cell lines
The human cancer cell lines SK-HEP-1, HeLa, A549, and RKO were obtained from the Cell Bank of Type Culture Collection of the Chinese Academy of Science (Shanghai, China) and cultured in RPMI-1640 medium supplemented with 10% FBS. The cells were maintained in a humidified incubator at 37 °C with 5% CO2. SK-HEP-1-NC and SK-HEP-1-shCypD cells were previously established and preserved in our laboratory [12].
All the cell lines used in this study were authenticated by short tandem repeat (STR) profiling and were confirmed to match their respective reference profiles. All the cultures were free of Mycoplasma contamination.
siRNA transfection
Receptor-interacting serine/threonine kinase 1 (RIPK1) or receptor-interacting serine/threonine kinase 3 (RIPK3) expression was knocked down in SK-HEP-1 cells by transfecting siRNAs targeting RIPK1 or RIPK3 (Sangon Biotech, Shanghai, China) according to the manufacturer’s instructions. Briefly, the cells were transiently transfected with the appropriate siRNA using Lipofectamine® RNAi MAX Reagent in serum-free Opti-MEM. Cells were collected and assayed for RIPK1 or RIPK3 expression 72 h later. The siRNA oligo sequences used are listed in Supplementary Table S2.
Determination of the cytosolic pH and mitochondrial pH
The cytosolic pH and mitochondrial pH were determined using pH-sensitive YFP developed by Poburko [18]. Briefly, the cells were transfected with the SypHer (Addgene, #48250) or SypHer mt (Addgene, #48251) plasmids, and stable clones were selected with G418. Stable SK-HEP-1-SypHer cells (pH-sensitive YFP is located in the cytoplasm) or SK-HEP-1-SypHer-mt cells (pH-sensitive YFP is located in the mitochondria) were treated with a control solution (20 mM bicarbonate), TNF-α (25 ng/mL), bicarbonate (60 mM) or the combination (TNF-α + bicarbonate) for 24 h; then, the cells were washed with Hanks’ balanced salt solution (HBSS) (140 mM NaCl, 5 mM KCl, 1 mM MgCl2, 2 mM CaCl2, 20 mM HEPES, and 10 mM glucose; pH 7.4) and incubated with HBSS supplemented with 20 mM bicarbonate, 25 ng/mL TNF-α, 60 mM bicarbonate or the combination (TNF-α + bicarbonate). Afterward, the cells were excited at 405 and 488 nm, and the fluorescence was measured at 441–510 nm and 507–560 nm, respectively. The fluorescence intensity ratios were analyzed and calibrated. For pH calibration, nigericin (5 μg/ml, HY-127019, MCE, USA) and monensin (5 mM, HY-N4302, MCE, USA) were added to the calibration buffer (125 mM KCl, 20 mM NaCl, 0.5 mM MgCl2, 0.2 mM EGTA, and 20 mM HEPES, pH set from 7.0–8.8), incubated with the cells for 10 min, and imaged using the same method.
Viable cell counts
The cells were cultured overnight in 24-well plates at a density of 2 × 104 cells per well or in 12-well plates at a density of 5 × 104 cells per well. The cells were incubated with the control (20 mM bicarbonate), TNF-α (25 ng/mL), bicarbonate (60 mM) or the combination (TNF-α + bicarbonate) for the indicated times. Then, the cells were washed twice with PBS, trypsinized, and collected for counting using a cell counter. Dead cells were excluded by counting cells stained with trypan blue in a hemocytometer under a microscope.
Assessment of the mitochondrial permeability transition and ΔΨm by fluorescence confocal imaging
We used calcein-AM multiplexed with TMRE to measure the MPT and mitochondrial membrane potential (MMP) according to the manufacturer’s protocol. SK-HEP-1 or HeLa cells were seeded on coverslips and incubated overnight. After being treated with the control, TNF-α (25 ng/mL), bicarbonate (60 mM) or the combination (TNF-α + bicarbonate) for 24 h, the cells were incubated with 0.4 μM calcein-AM and 0.1 μM TMRE in FBS-free MEM at 37 °C for 30 min. Afterward, the culture medium was replaced with fresh medium supplemented with 1 mM CoCl2 to quench the cytoplasmic calcein-AM staining. The remaining calcein-AM staining was specific for the mitochondria, and a decrease in calcein-AM fluorescence indicated the MPT. TMRE was used to measure the MMP of the cells. After a 15 min incubation with TMRE, the cells were washed with PBS twice, resuspended in HBSS, and then observed under a Zeiss LSM710 laser confocal microscope (Carl Zeiss, Germany); Zen software was used to process the images.
Western blot and immunoprecipitation
Cells were collected and washed twice with cold PBS and then lysed with M-PERTM Mammalian Protein Extraction Reagent (78501, Thermo Fisher Scientific, USA) supplemented with a protease inhibitor cocktail (HY-K0010, MCE, USA).
Fifty milligrams of tumor tissue was minced, mixed with 1.5 mL of ice-cold M PERTM containing protease inhibitors and then homogenized. The homogenized tissue was subsequently centrifuged (10,000 r/min, 4 °C, 10 min).
The supernatant was collected, and the sample was mixed with loading buffer for Western blotting, as previously described[19]. The protein was heat denatured, and 30 μg was separated on a 10% SDS‒PAGE gel, transferred to a PVDF membrane, incubated with primary and secondary antibodies, and detected using Western Lightning Plus ECL (NEL105001EA, Perkin Elmer, USA).
For immunoprecipitation, the cells were lysed with Cell Complete Lysis Buffer (P0037-100 mL; Beyotime, China) containing protease inhibitors. After centrifugation, the supernatants were incubated with an antibody overnight and then with Protein A/G PLUS-Agarose (sc-2003, Santa Cruz, USA) for 2 h at 4 °C. Immunoprecipitated components were removed and analyzed by Western blotting [19].
Measurement of mitochondrial oxygen consumption
We used an O2K system (OROBOROS) to measure oxidative phosphorylation. Cells were treated with the control, TNF-α (25 ng/mL), bicarbonate (60 mM) or the combination (TNF-α + bicarbonate) and collected by trypsin digestion, after which approximately 3 × 106 cells suspended in 2 ml of culture medium with or without bicarbonate were added to each O2K chamber. Oligomycin (2 µg/mL, HY-N6782, MCE, USA) was used to inhibit ATP synthase activity. Carbonyl cyanide p-(trifluoromethoxy)phenylhydrazone (FCCP, 0.3 μM, HY-100410, MCE, USA) was used to uncouple electron transport and oxidative phosphorylation, while rotenone (an inhibitor of respiratory complex Ⅰ, 0.5 μM, HY-B1756R, MCE, USA) and antimycin A (an inhibitor of respiratory complex Ⅲ, 2.5 μM, sc-202467, Santa Cruz, USA) were used to block electron transfer along the respiratory chain. After each injection of a chemical, oxygen consumption was monitored for approximately 5 min until a relative balance was reached. The data were calculated using the following equations [12]:
Mitochondrial basal respiration = (cellular basal respiration) − (nonmitochondrial respiration rate); cellular basal respiration is the oxygen consumption rate without the abovementioned inhibitors and uncouplers;
Oligomycin-sensitive OCR = (last rate measured before the oligomycin injection) − (minimum rate measured after the oligomycin injection);
Proton leak = (minimum rate measured after the oligomycin injection) − (nonmitochondrial respiration);
Maximal respiration = (maximum rate measured after the FCCP injection) − (nonmitochondrial respiration);
Nonmitochondrial respiration = minimum rate measured after the rotenone/antimycin A injection.
Flow cytometry analysis of cell viability
The cells were harvested by digestion with accutase and centrifuged at 200 × g for 5 min. The cells were subsequently washed with PBS and stained with an Annexin V-FITC and propidium iodide solution for 10 min at room temperature in the dark. The percentage of nonviable cells in each sample was immediately measured using a Beckman Coulter DxFLEX flow cytometer. The detailed gating strategies are provided in Supplementary Fig. S1.
Microscopy imaging of intracellular ROS levels
ROS levels were measured using dichlorodihydrofluorescein diacetate (DCFH-DA) and MitoSOXTM Red mitochondrial superoxide indicator according to the manufacturer’s instructions. Briefly, after various treatments, the cells were loaded with DCFH-DA (at a final concentration of 10 μM for 30 min) or MitoSOXTM Red (at a final concentration of 2.5 μM for 10 min), washed with ice-cold HBSS (Hank’s balanced salt solution, pH 7.2), and then observed under a Zeiss LSM710 laser confocal microscope (Carl Zeiss, Germany) equipped with Zen software to process the images [20].
Immunofluorescence (IF) staining
After various treatments, the cells or tumor tissues were fixed with 4% paraformaldehyde (HK2003, Haokebio, China), permeabilized with 0.2% Triton X-100 (X100-100ML, Sigma‒Aldrich, USA) for 15 min and blocked with 5% goat serum (E510009-0100, Sangon, China) for 1 h. Then, the cells were incubated with primary antibodies at 4 °C overnight. Afterward, the cells were incubated with a Corallite 488-conjugated secondary antibody at 37 °C for 1 h. The nuclei were stained with Hoechst 33342 (#C1022, Beyotime, China) or DAPI (C1341S, Beyotime, China). Images were acquired using a confocal microscope (Zeiss, Germany).
For the analysis of the fluorescence intensity of the pRIPK1 or pRIPK3, the fluorescence intensity ratios were analyzed and calibrated.
For the NF-κB nuclear translocation assay, cells displaying NF-κB nuclear translocation in each image were counted, and the ratio of cells with NF-κB nuclear translocation to total cells was calculated.
Electron microscopy
Samples were prepared for EM using the methods described in our previous reports [21, 22]. Cells were treated and harvested by trypsin digestion, washed once with PBS, and fixed with 2.5% glutaraldehyde (HK2012, HaoKebio, China) in PBS overnight at 4 °C. Tumor tissues were fixed with 2.5% glutaraldehyde in PBS overnight at 4 °C.
These samples were subsequently treated with 1% osmium tetroxide for 1 h, washed with water, and then incubated with 2% uranyl acetate for 30 min, after which they were dehydrated with ethanol and embedded. Thin sections were stained with uranyl acetate and lead citrate and imaged using a TECNAI 10 G2 Spirit Cryo-electron microscope (FEI Company) at 120 kV.
Animal models
All the animal experiments were reviewed and approved by the Animal Ethical and Welfare Committee of Zhejiang Chinese Medical University (Approval number: IACUC-20230918-11). Six-week-old female NOD/SCID mice were purchased from SLAC Laboratory Animal Co., Ltd. (Shanghai, China) and housed in Zhejiang Chinese Medical University Laboratory Animal Research Center.
HCC and CCA tumor xenograft models were established via the subcutaneous injection of SK-HEP-1 cells and HeLa cells (1 × 106 cells per mouse), respectively, into the left flank of mice. When the tumor diameter reached 4 to 6 mm, the mice were randomized into one of four groups: control, TNF-α, bicarbonate and the combination (TNF-α plus bicarbonate).
The mice in each group were intratumorally injected with 100 μL of PBS (control), 100 μL of TNF-α (120 × 104 U/kg) in PBS (TNF-α), 100 μL of 200 mM bicarbonate in PBS (bicarbonate), or 100 μL of TNF-α (120 × 104 U/kg) plus 200 mM bicarbonate in PBS (combination) every 2 days. The volume of the tumors was measured and calculated every 2 days using the formula 0.5 × (length × width2). After treatment, the animals were sacrificed, and tumor tissues were collected for further analysis. For the EM assay, the animals were sacrificed 12 h after treatment, and tumor tissues were collected for further analysis.
Statistical analysis
For each experiment, measurements were performed with n = 3 technical replicates, and each experiment was repeated in three independent experiments (n = 3). The data are presented as the means ± standard deviations (SDs). The data from two groups were compared using two-tailed Student’s t tests. One-way ANOVA was used to compare data among multiple groups. Repeated measures ANOVA (two-way ANOVA) was used to analyze changes in the tumor volume in vivo and the results of the viable cell counts. GraphPad Prism 8 (RRID:SCR_002798) software was used for statistical analyses. The statistical parameters (i.e., exact value of n, what n represents, representation of error bars, and P values) are indicated in each figure legend. In Fig. 6c, we excluded a maximum value of 0.72 g from the control group according to the Grubbs criterion. The details are listed in Table S1.
Results
Bicarbonate activates the tumoricidal activity of TNF-α
We first asked whether a bicarbonate-mediated increase in the intracellular pH (pHᵢ) enhances the tumoricidal activity of TNF-α to test whether intracellular alkalization can potentiate TNF-α-induced cell death (apoptosis or necroptosis).
We previously established a quantitative relationship between the extracellular bicarbonate concentration and the intracellular pH and demonstrated that bicarbonate increased the cytosolic pH to a greater extent than the mitochondrial pH. A greater increase in the cytosolic pH than in the mitochondrial pH results in a reduced ΔpH, a proton gradient across the inner mitochondrial membrane, and the magnitude of intracellular pH changes correlates with bicarbonate concentration. This relationship was recapitulated in the current study (Fig. 1a–c): treatment with bicarbonate (20–160 mM) increased the cytosolic pH from 7.35 to 8.74 and the mitochondrial pH from 7.87 to 8.96, thereby reducing the ΔpH from 0.52 to 0.24.

SK-HEP-1 cells were divided into four groups: control (20 mM bicarbonate), TNF-α, bicarbonate, and combination (TNF-α + bicarbonate). a–c SK-HEP-1-SypHer and SK-HEP-1-SypHer-mt cells treated with bicarbonate for 1 h. The data are presented as the means ± SDs (n = 3 biological replicates). a Medium pH. b Cytosolic and mitochondrial pH. c ΔpH (mitochondrial pH–cytosolic pH). d TNF-α plus bicarbonate increase the cytosolic and mitochondrial pH while reducing the ΔpH after 24 h. The data are presented as the means ± SDs (n = 3 biological replicates). e Bicarbonate dose‒response curves. Cells were treated with 40–120 mM bicarbonate in the presence or absence of 25 ng/mL TNF-α for the indicated intervals. The dashed line represents the curve for the expected additive effect. The data are presented as the means ± SDs (n = 3 biological replicates). f TNF-α dose‒response curves (1–50 ng/mL) for cells cultured in the presence or absence of 60 mM bicarbonate for the indicated intervals. The dashed line represents the curve for the expected additive effect. The data are presented as the means ± SDs (n = 3 biological replicates). g Analysis of cell viability after 24 h. The data are presented as the means ± SDs (n = 3 biological replicates). Unpaired t test: ns, P > 0.05; *P < 0.05; **P < 0.01; and ***P < 0.001.
We measured the cytosolic pH, mitochondrial pH, and ΔpH in cells treated with increasing concentrations of bicarbonate (20, 40, 60, and 80 mM) either alone or in combination with 25 ng/ml TNF-α to determine whether TNF-α itself influences these pH gradients (Fig. 1d). The bicarbonate-induced changes in the cytosolic pH, mitochondrial pH, and ΔpH were highly comparable between the bicarbonate alone and the bicarbonate + TNF-α groups at each concentration tested, indicating that TNF-α does not significantly modify the pH-alkalizing effect of bicarbonate.
Based on previous reports that 25 ng/mL TNF-α induces NF-κB nuclear translocation, we selected this concentration for the bicarbonate dose–response assay. As shown in Fig. 1e, 40 and 60 mM bicarbonate moderately inhibited SK-HEP-1 cell growth, whereas higher concentrations (80–120 mM) induced strong cytotoxicity. TNF-α alone had no discernible effect. The dashed line in Fig. 1e indicates the expected additive effect of TNF-α and bicarbonate, but the observed cell death exceeded this prediction, indicating a synergistic interaction. The strongest synergy occurred with 60 mM bicarbonate and 25 ng/mL TNF-α. At higher bicarbonate concentrations (80–120 mM), cell death was rapid and profound because of severe mitochondrial permeability transition and autophagy blockade, as previously reported [12], leaving little room for further enhancement by TNF-α.
Next, we fixed the bicarbonate concentration at 60 mM and varied the TNF-α concentration. Low-dose TNF-α (1 ng/mL) slightly promoted cell growth, while 5–50 ng/mL had no effect on proliferation. Nonetheless, all tested concentrations synergized with bicarbonate to induce cell death, with 25 ng/mL achieving a saturating response (Fig. 1f).
The flow cytometry analysis further confirmed these findings. TNF-α alone did not induce cell death, and 60 mM bicarbonate induced modest death (8.98% ± 1.11%). However, their combination significantly increased cell death to 37.13% ± 1.69% (Fig. 1g), confirming a synergistic cytotoxic effect.
Together, these results demonstrate that TNF-α alone is noncytotoxic under these conditions, but when combined with the bicarbonate-induced alkalization of pHᵢ, it exerts potent tumoricidal activity. This finding suggests that bicarbonate activates the cell-killing function of TNF-α. The next question is what is the mechanism underlying this activation.
Bicarbonate inhibits TNF-α-induced NF-κB signaling and redirects TNF-α toward RIPK-dependent necroptosis
TNF-α exerts dual and opposing effects on tumor cells: it can activate the NF-κB pathway to promote survival, or it can trigger RIPK-dependent necroptosis to induce cell death [7, 9, 23, 24]. The balance between these two outcomes determines the antitumor potential of TNF-α.
In SK-HEP-1 cells, TNF-α alone induced the phosphorylation of inhibitor of κB (IκB) and robust nuclear translocation of p65 (Fig. 2a, b), confirming the activation of the canonical NF-κB pathway. Bicarbonate alone did not increase IκB phosphorylation or promote p65 nuclear translocation. Compared with TNF-α alone, bicarbonate combined with TNF-α reduced both total IκB and pIκB levels; however, the pIκB/IκB ratio remained higher than that in the control or bicarbonate groups. Correspondingly, the combination treatment produced an intermediate degree of p65 nuclear translocation that was greater than that observed after treatment with control or bicarbonate alone but markedly lower than that observed after treatment with TNF-α alone (Fig. 2b). These findings indicate that bicarbonate significantly attenuates, although it does not fully block, TNF-α-driven NF-κB activation, creating a permissive context for the engagement of alternative signaling pathways.

SK-HEP-1 cells were treated with the control, TNF-α (25 ng/mL), bicarbonate (60 mM), or combination (TNF-α + bicarbonate) for 24 h or as indicated. a Western blot analysis of pIκBα and IκBα levels and quantification of the pIκBα/IκBα ratio. The data are presented as the means ± SDs (n = 3 biological replicates). b–d, f–h After being cultured for 24 h, the cells in the four groups were collected for IF, WB, IP and EM analyses. b Representative images of immunofluorescence staining showing NF-κB (p65) nuclear translocation. Red arrows indicate p65-positive nuclei. Quantification of nucleus-localized NF-κB. Scale bar: 20 µm. The data are presented as the means ± SDs (n = 3 biological replicates). c Western blot analysis of pRIPK1, RIPK1, pRIPK3, RIPK3, pMLKL, and MLKL levels and the ratios of phosphorylated to total proteins. The data are presented as the means ± SDs (n = 3 biological replicates). d Immunofluorescence staining for pRIPK1 and pRIPK3. Scale bar: 20 µm. The data are presented as the means ± SDs (n = 3 biological replicates). e RIPK1 immunoprecipitation (8 h). Immunoblot showing ubiquitin levels in RIPK1-associated components. Input: total lysate. Co-IP analysis of RIPK1 (f) or RIPK3 (g) and associated proteins (FADD, RIPK1, RIPK3, and MLKL). Input: total lysate. h Representative electron micrographs showing the necroptotic ultrastructure. Unpaired t test: ns, P > 0.05; *P < 0.05; **P < 0.01; and ***P < 0.001.
Because the suppression of NF-κB activity can unmask the cytotoxic activity of TNF-α, we next examined whether bicarbonate shifts TNF-α signaling toward RIPK-dependent necroptosis. Neither TNF-α nor bicarbonate alone significantly increased the phosphorylation of RIPK1, RIPK3, or MLKL. In contrast, the combination treatment prominently induced a coordinated increase in pRIPK1, pRIPK3, and pMLKL levels (Fig. 2c, d), indicating the activation of the necroptotic cascade.
We performed additional dose–response experiments using bicarbonate concentrations ≤30 mM (20–30 mM), which approximate plasma levels, to evaluate whether the effects of bicarbonate are observed at concentrations near the physiological range. At these lower concentrations, bicarbonate caused no significant changes in cell death, NF-κB signaling, or the levels of RIPK-dependent necroptotic markers (Supplementary Fig. S2). These findings indicate that physiological bicarbonate levels are insufficient to overcome cellular buffering systems and do not reproduce the signaling reprogramming observed at higher concentrations.
We elucidated how bicarbonate redirects TNF-α signaling at the receptor-proximal level by analyzing the composition of TNFR1-associated complexes using coimmunoprecipitation. TNF-α increased the levels of ubiquitinated species in the RIPK1 immunoprecipitates, consistent with the assembly/stabilization of the ubiquitin-rich TNFR1 signaling complex. Bicarbonate + TNF-α markedly reduced the ubiquitin signal (Fig. 2e), suggesting the destabilization or deubiquitination of complex I components. RIPK1 pull-down further revealed a modest decrease in the RIPK1–Fas-associated via death domain (FADD) association but a substantial increase in RIPK1–RIPK3 and RIPK1–MLKL interactions in the combination group (Fig. 2f). Direct immunoprecipitation of RIPK3 confirmed the increased interaction between RIPK1 and MLKL (Fig. 2g), providing evidence of necrosome assembly. Transmission electron microscopy corroborated these biochemical findings, showing plasma membrane rupture in cells treated with bicarbonate + TNF-α (Fig. 2h).
We first used Nec-1, a RIP1 kinase inhibitor, to determine whether bicarbonate-induced increase in cytotoxicity depends on RIP kinase activity. Nec-1 reduced pRIPK1, pRIPK3, and pMLKL levels (Fig. 3a) but did not alter TNF-α-induced NF-κB nuclear translocation (Fig. 3b, c), confirming its specificity for the necroptotic pathway. Nec-1 did not affect cell viability in the control, TNF-α-alone, or bicarbonate-alone groups. In contrast, Nec-1 significantly rescued cell viability in the bicarbonate + TNF-α group (39.84% → 68.36%; Fig. 3d), and flow cytometry revealed corresponding reductions in the number of Annexin V/PI–positive cells (Fig. 3e).

SK-HEP-1 cells were treated with the control (20 mM bicarbonate), TNF-α (25 ng/mL), bicarbonate (60 mM), or the combination (TNF-α + bicarbonate) with or without Nec-1 (1 μM) for 24 h. a Western blot analysis of pRIPK1, RIPK1, pRIPK3, RIPK3, pMLKL, and MLKL levels and quantification of the ratios of the phosphorylated proteins to the total proteins. The data are presented as the means ± SDs (n = 3 biological replicates). Unpaired t test. b, c NF-κB (p65) nuclear translocation. Quantification of nucleus-localized NF-κB. Scale bar: 20 µm. The data are presented as the means ± SDs (n=3 biological replicates). Unpaired t test. d Cell–growth curves. The data are presented as the means ± SDs (n = 3 biological replicates). Two-way ANOVA. e Analysis of cell viability. The data are presented as the means ± SDs (n = 3 biological replicates). Unpaired t test. Effects of RIPK1 knockdown (f) or RIPK3 knockdown (g) on pRIPK1, pRIPK3, pMLKL, and total protein levels. h Analysis of cell viability after RIPK1 or RIPK3 depletion. The data are presented as the means ± SDs (n = 3 biological replicates). Unpaired t test. ns, P > 0.05; *P < 0.05; **P < 0.01; and ***P < 0.001.
We performed siRNA knockdown studies to complement the results obtained using pharmacological inhibitors. RIPK1 knockdown markedly reduced pRIPK3 and pMLKL levels and strongly rescued cell viability (Fig. 3f, h), indicating that RIPK1 activity is required for downstream necroptotic signaling. Similarly, RIPK3 knockdown decreased pMLKL levels and suppressed cell death (Fig. 3g, h). RIPK3 depletion also reduced pRIPK1 levels, a reciprocal effect that has previously been reported when RIPK1 autophosphorylation depends on its interaction with RIPK3 [25, 26]. Although mechanistically intriguing, this finding does not alter the conclusion that RIPK3 is essential for necroptosis execution.
We excluded apoptosis and DNA damage-driven cell death as the dominant mechanisms by examining caspase activation, PARP cleavage, and γH2AX expression at the same time points used for the Annexin V/PI analysis (Fig. 1g) and RIPK1/RIPK3/MLKL phosphorylation (Fig. 2c). As shown in Supplementary Fig. S3, compared with the control treatment, bicarbonate + TNF-α treatment induced the robust phosphorylation of RIPK1, RIPK3 and MLKL, whereas only limited cleavage of caspase-8 was detected, and PARP cleavage did not increase. γH2AX is a marker of DNA damage that can be produced in response to multiple forms of cellular stress and cell death. In this system, γH2AX induction in the absence of PARP cleavage and strong caspase activation, together with robust RIPK3/MLKL phosphorylation and pharmacological and genetic rescue of cell death, is consistent with necroptosis-associated DNA damage rather than classical caspase-dependent apoptosis.
Taken together, biochemical, structural, pharmacological, and genetic evidence has shown that bicarbonate suppresses TNF-α-induced NF-κB survival signaling and redirects TNF-α toward RIPK-dependent necroptosis, thereby substantially increasing its tumoricidal activity.
TNF-α increases bicarbonate-induced mitochondrial damage
Our previous study showed that bicarbonate increases both the cytosolic and mitochondrial matrix pH, although the increase in the matrix pH is less pronounced than that in the cytosol[12]. This upward shift in the pH gradient reduces the proton motive force across the mitochondrial inner membrane, leading to a decrease in the ΔΨm and suppression of oxidative phosphorylation. The simultaneous alkalinization of the cytosol and matrix also promotes the opening of the mitochondrial permeability transition pore (mPTP), resulting in a sustained mitochondrial permeability transition and ultimately tumor cell death.
We examined ΔΨm, mitochondrial membrane permeability, and oxidative phosphorylation activity in SK-HEP-1 cells treated with bicarbonate, TNF-α, or their combination for 24 h to determine whether TNF-α modulates these mitochondrial responses. Treatment with bicarbonate alone reduced the ΔΨm, induced the MPT, and suppressed oxidative phosphorylation. Treatment with TNF-α alone increased the ΔΨm but did not alter oxidative phosphorylation or the MPT. In contrast, compared with either treatment alone, the combination of bicarbonate and TNF-α markedly decreased the ΔΨm and oxidative phosphorylation and induced a more extensive MPT (Fig. 4a, c). Transmission electron microscopy showed mitochondrial swelling in the bicarbonate- and bicarbonate + TNF-α-treated cells, whereas the mitochondria in the control and TNF-α-alone groups maintained a normal morphology (Fig. 4b). These findings indicate that TNF-α amplifies bicarbonate-induced mitochondrial dysfunction.

SK-HEP-1, SK-HEP-1-NC, and SK-HEP-1-shCypD cells were treated with the control, TNF-α (25 ng/mL), bicarbonate (60 mM), or combination (TNF-α + bicarbonate) for 24 h or as indicated. a Representative images of calcein-AM (green) and TMRE (red) fluorescence. Loss of calcein-AM = MPT; loss of TMRE = Δψm depolarization. Scale bar: 20 µm. The data are presented as the means ± SDs (n = 3 biological replicates). Unpaired t test. b Electron micrographs of mitochondrial morphology. c Seahorse analysis of mitochondrial respiration. The data are presented as the means ± SDs (n = 3 biological replicates). Unpaired t test. d CypD knockdown prevents the bicarbonate + TNF-α-induced MPT and Δψm depolarization. The data are presented as the means ± SDs (n = 3 biological replicates). Unpaired t test. e Growth curves of SK-HEP-1-NC and SK-HEP-1-shCypD cells subjected to the four treatments. The data are presented as the means ± SDs (n = 3 biological replicates). Unpaired t-test. f Analysis of cell viability. The data are presented as the means ± SDs (n = 3 biological replicates). Unpaired t test. g, h The cells were pretreated with ± NIM811 (600 nM, 4 h) and then treated with the indicated reagents for 24 h. g NIM811 attenuated the MPT (calcein-AM) and Δψm depolarization (TMRE). The data are presented as the means ± SDs (n = 3 biological replicates). Unpaired t test. h NIM811 rescued cell viability. The data are presented as the means ± SDs (n = 3 biological replicates). Unpaired t test. ns, P > 0.05; *P < 0.05; **P < 0.01; and ***P < 0.001.
We used SK-HEP-1-shCypD cells, a stable line with knockdown of cyclophilin D (CypD), a key regulator of mPTP opening [27], which was previously established in our laboratory[12], to assess whether this amplified MPT is required for the synergistic effects of bicarbonate and TNF-α. As expected, CypD knockdown attenuated the bicarbonate-induced loss of ΔΨm and suppressed mPTP opening (Fig. 4d).
We then compared the viability of SK-HEP-1-NC and SK-HEP-1-shCypD cells over 72 h. After treatment with TNF-α alone, both lines exhibited similar growth (Fig. 4e, left panel). After treatment with bicarbonate alone, CypD-knockdown cells initially grew faster, although the difference was diminished over time (Fig. 4e, middle panel). Strikingly, the combined bicarbonate + TNF-α treatment resulted in a significant decrease in the death of CypD-knockdown cells compared with control cells (Fig. 4e, right panel), indicating that CypD contributes substantially to the cytotoxic effect. The flow cytometry analysis confirmed this result: the death rate of SK-HEP-1-shCypD cells (24.19% ± 0.77%) was markedly lower than that of SK-HEP-1-NC cells (62.98% ± 1.43%) (Fig. 4f).
We tested NIM811, a CypD-selective inhibitor[28, 29], to complement the genetic evidence. NIM811 suppressed bicarbonate + TNF-α-induced mPTP opening (calcein), prevented the collapse of ΔΨm (Fig. 4g), and significantly rescued cell viability (Fig. 4h), closely phenocopying the protective effect of CypD knockdown.
Together, these findings indicate that the cyclophilin-D-dependent opening of the mPTP is essential for the synergistic mitochondrial damage and tumor cell death induced by bicarbonate and TNF-α. The evidence obtained from both genetic (CypD knockdown) and pharmacologic (NIM811) inhibition converges to show that the CypD-mediated MPT is a critical cell death mechanism downstream of the bicarbonate/TNF-α interaction.
ROS levels determine TNF-α-induced necroptosis and persistent MPT
Previous studies have indicated that cellular ROS levels dictate how TNF-α signals: moderate levels of ROS promote NF-κB activation[30], whereas high levels of ROS trigger necroptosis [31, 32] and a persistent mitochondrial permeability transition [33, 34]. We tested whether we would obtain similar results using our system by measuring cytosolic and mitochondrial ROS levels in SK-HEP-1 cells after 24 h of treatment with TNF-α, 60 mM bicarbonate, or their combination. Both single treatments increased mitochondrial and cytosolic ROS levels, whereas the combination treatment resulted in significantly higher ROS levels than either treatment alone (Fig. 5a).

SK-HEP-1 cells were treated for 24 h with the control, TNF-α (25 ng/mL), bicarbonate (60 mM), or a combination (TNF-α + bicarbonate) in the presence or absence of NAC (1 mM) or EUK8 (20 μM). a Representative images of MitoSOX Red (mitochondrial ROS) and DCFH-DA (cytosolic ROS) fluorescence. Scale bar: 20 µm. The data are presented as the means ± SDs (n = 3 biological replicates). b NAC and EUK8 suppressed mitochondrial ROS production induced by bicarbonate + TNF-α. The data are presented as the means ± SDs (n = 3 biological replicates). c Western blot analysis of the levels of pRIPK1, pRIPK3, pMLKL and total proteins, and the quantification of the ratios of the phosphorylated proteins to the total proteins. The data are presented as the means ± SDs (n = 3 biological replicates). d, e Representative images of immunofluorescence staining showing NF-κB (p65) nuclear translocation. Red arrows indicate p65-positive nuclei. Quantification of nucleus-localized NF-κB. Scale bar: 20 µm. The data are presented as the means ± SDs (n = 3 biological replicates). f ROS inhibitors prevented the MPT (calcein-AM). The data are presented as the means ± SDs (n = 3 biological replicates). g ROS inhibitors attenuated Δψm depolarization (TMRE). The data are presented as the means ± SDs (n = 3 biological replicates). h ROS inhibition rescued cell viability. The data are presented as the means ± SDs (n = 3 biological replicates). Unpaired t test. ns, P > 0.05; *P < 0.05; **P < 0.01; and ***P < 0.001.
We evaluated whether ROS control the shift from NF-κB activation to necroptosis using two antioxidant systems: N-acetylcysteine (NAC), which primarily reduces cytosolic ROS levels, and EUK8, a superoxide dismutase/catalase mimetic with strong mitochondrial ROS-scavenging activity [35,36,37]. After 24 h, EUK8, but not NAC, suppressed TNF-α-induced mitochondrial ROS production, whereas both inhibitors markedly reduced mitochondrial ROS production induced by bicarbonate alone or in combination with TNF-α, with EUK8 having the strongest effect (Fig. 5b).
We determined whether increased ROS levels drive the activation of the necroptotic machinery by assessing pRIPK1, pRIPK3, and pMLKL levels following antioxidant treatment. Both NAC and EUK8 suppressed the phosphorylation of all three proteins induced by the combination treatment, with EUK8 again displaying greater potency (Fig. 5c). Neither inhibitor affected NF-κB nuclear translocation in the control or single-treatment groups, but both increased nuclear p65 in the combination group (Fig. 5d–e), indicating that ROS suppression shifts TNF-α signaling away from necroptosis and restores NF-κB activity.
We next examined mitochondrial function. NAC and EUK8 prevented mPTP opening in the control, TNF-α, and bicarbonate groups. In the combination group, NAC completely blocked mPTP opening and fully restored ΔΨm, whereas EUK8 provided partial protection (Fig. 5f–g).
Finally, cell viability assays demonstrated that both inhibitors significantly rescued cells from bicarbonate + TNF-α-induced death, with EUK8 providing the strongest protection (32.40% → 88.24%; Fig. 5h).
Together, these results show that the marked increase in mitochondrial ROS levels induced by bicarbonate + TNF-α is a key determinant that redirects TNF-α signaling from NF-κB activation toward RIPK-dependent necroptosis and persistent MPT. The finding that NAC, which is primarily a cytosolic ROS scavenger, also blocks mPTP opening and necroptosis indicates that ROS from multiple subcellular compartments cooperate to regulate MPT sensitivity and the commitment to necroptotic death. Thus, while mitochondrial ROS act as critical initiators, the overall cellular redox environment ultimately dictates whether TNF-α exerts prosurvival effects or transitions to inducing a necroptotic program.
Synergistic effects of bicarbonate and TNF-α on killing HeLa, A549, and RKO cells
We determined whether the synergistic effects of bicarbonate and TNF-α synergy are generalizable across tumor types by examining HeLa cells under the same four treatment conditions. TNF-α (25 ng/mL) or 50 mM bicarbonate alone modestly inhibited cell proliferation, whereas the combination markedly reduced the number of viable cells at 24–72 h (Supplementary Fig. S4a). Compared with all the other conditions, flow cytometry confirmed a substantial increase in cell death in the combination group (Supplementary Fig. S4b).
Bicarbonate partially suppressed TNF-α-induced NF-κB (p65) nuclear translocation (Supplementary Fig. S4c). In parallel, the combination treatment substantially increased the phosphorylation of RIPK1, RIPK3, and MLKL (Supplementary Fig. S4d–f), indicating the activation of RIPK-dependent necroptosis. The greatest reduction in the MMP was observed in the bicarbonate + TNF-α group (Supplementary Fig. S4g), consistent with the increase in mitochondrial injury.
Nec-1 inhibited the phosphorylation of RIPK1/3/MLKL induced by the combination treatment and significantly increased cell viability (Supplementary Fig. S5a, b) but had minimal effects on cells in the control or single-treatment groups. Nec-1 did not alter NF-κB nuclear translocation (Supplementary Fig. S5c), confirming that its protective effect reflects the inhibition of RIPK signaling.
In A549 cells (Supplementary Fig. S6a, b), TNF-α or bicarbonate alone induced little to no cell death, whereas the combination treatment markedly increased cell death, which is consistent with our observations in SK-HEP-1 and HeLa cells. Mechanistically, the combination treatment significantly increased the phosphorylation of RIPK1, RIPK3, and MLKL, while neither agent alone had a detectable effect. These data indicate that A549 cells undergo RIPK-dependent necroptosis in response to bicarbonate + TNF-α, further supporting the generality of this signaling reprogramming mechanism.
In RKO cells (Supplementary Fig. S6a, c), TNF-α or bicarbonate alone had minimal effects, while the combination treatment resulted in a pronounced increase in cell death, demonstrating that the synergistic cytotoxic effect was preserved. However, unlike the other cell lines tested, the combination treatment did not increase the phosphorylation of RIPK1 or MLKL, suggesting that cell death in RKO cells does not proceed through the canonical RIPK-dependent necroptosis pathway. This finding indicates that while bicarbonate + TNF-α consistently increases tumor cell death, the downstream mechanism may be cell context dependent.
Taken together, these results indicate that the synergistic cytotoxic effects of bicarbonate and TNF-α are reproducible across multiple cancer cell types, although the precise death pathway involved may vary depending on the cellular context. Importantly, the central conclusion of this study that intracellular alkalization reprograms TNF-α responses toward increased tumor cell killing is robust across all tested models. Elucidating the alternative death pathway engaged in RKO cells is beyond the scope of the current study and will be the focus of future investigations.
Extracellular alkalization via NaOH is insufficient to recapitulate the effects of bicarbonate on TNF-α signaling
We determined whether the effects of bicarbonate reflect simple extracellular alkalization by comparing medium in which the pH was adjusted with NaOH to the equivalent pH levels generated with bicarbonate. Adjusting the medium to a pH of 8.0–8.5 with NaOH produced only minimal increases in the cytosolic pH (7.40 → 7.45–7.47) and mitochondrial pH (7.93 → 8.01–8.08) without significantly altering the ΔpH (Supplementary Fig. S7a–c). In contrast, matching pH values using 40–80 mM bicarbonate resulted in pronounced increases in the cytosolic pH, mitochondrial pH, and ΔpH (Fig. 1d). These data show that an identical extracellular pH, when achieved with NaOH, does not produce the pronounced intracellular alkalinization or the increased ∆pH generated by bicarbonate.
We next assessed mitochondrial function under control, TNF-α, NaOH (pH 8.0), NaOH + TNF-α, and bicarbonate + TNF-α conditions. After 24 h, NaOH—either alone or in combination with TNF-α—did not affect mPTP opening, ΔΨm, or mitochondrial ROS levels compared with TNF-α alone. In contrast, the bicarbonate + TNF-α combination triggered robust mPTP opening, marked ∆Ψm loss, and substantial mitochondrial ROS accumulation, thereby recapitulating our primary findings (Supplementary Fig. S7d, e).
Similarly, NaOH alone or in combination with TNF-α did not increase the phosphorylation of RIPK1, RIPK3, or MLKL. In contrast, bicarbonate + TNF-α substantially increased the levels of all three necroptosis markers, indicating that NaOH does not mimic the signaling effects of bicarbonate (Supplementary Fig. S7f).
Collectively, these control experiments demonstrate that the synergistic effects of bicarbonate are not a consequence of simple extracellular alkalization. Instead, they arise from the unique physicochemical and transport properties of bicarbonate, which establish distinct intracellular pH gradients and engage specific signaling cascades.
Iso-osmotic NaCl does not mimic the effects of bicarbonate on TNF-α signaling
We explicitly included iso-osmotic NaCl controls matched to the ionic strength of the bicarbonate conditions to exclude the possibility that osmotic stress contributes to the observed effects. These experiments showed that NaCl did not significantly affect cell death, NF-κB signaling, or RIPK-dependent necroptotic signaling (Supplementary Fig. S8).
TNF-α combined with bicarbonate suppresses tumor growth
Having established that bicarbonate potently redirects TNF-α signaling toward necroptosis in vitro, we next sought to determine whether this combination elicited antitumor activity in vivo.
We established subcutaneous xenograft models using SK-HEP-1 and HeLa cells in NOD/SCID mice to evaluate the in vivo antitumor activity of bicarbonate combined with TNF-α. When the tumors reached ~80 mm³, the mice were randomized into four intratumoral treatment groups: control, TNF-α, bicarbonate, and combination groups. The agents were administered by multipoint injection every other day. Tumors were measured throughout treatment and collected for analysis upon euthanasia.
In the SK-HEP-1 model, treatment with TNF-α alone paradoxically accelerated tumor growth, resulting in significantly larger tumor volumes and weights compared with all other treatments (Fig. 6b, c). In contrast, the combination of bicarbonate + TNF-α markedly suppressed tumor growth and the tumors were significantly smaller than those in the control group. In the HeLa xenograft model, treatment with TNF-α or bicarbonate alone had little effect on tumor growth, whereas the combination treatment significantly reduced both the tumor volume and tumor weight (Fig. 6e, f). These results indicate that bicarbonate converts TNF-α from a tumor-promoting cytokine to a tumor-suppressive cytokine in vivo.

Tumor-bearing NOD/SCID mice received multipoint intratumoral injections of the control, TNF-α, bicarbonate, or a combination of these agents. a, c SK-HEP-1 xenografts. a Representative photographs of the tumors. b Tumor-growth curves. The dashed line indicates the expected additive effect. The data are presented as the means ± SDs (n = 5 biological replicates). Two-way ANOVA. c Tumor weights. The data are presented as the means ± SDs (n = 5 biological replicates). Unpaired t test. One outlier was removed from the control group (see Supplementary Table 1). d, f HeLa xenografts. d Representative photographs of the tumors. e Tumor-growth curves. The dashed line indicates the expected additive effect. The data are presented as the means ± SDs (n = 6 biological replicates). Two-way ANOVA. f Tumor weights. The data are presented as the means ± SDs (n = 6 biological replicates). Unpaired t test. ns, P > 0.05; *P < 0.05; **P < 0.01; and ***P < 0.001.
We next assessed the activation of necroptotic signaling in tumor tissues. Western blotting revealed that the combination treatment substantially increased pRIPK1, pRIPK3, and pMLKL levels in both liver (Fig. 7a) and cervical (Fig. 7b) cancer xenografts. Immunofluorescence staining confirmed elevated pRIPK1 and pRIPK3 levels in the combination group (Fig. 7c, d), verifying the involvement of the necroptotic execution pathway in vivo. Conversely, the combination treatment suppressed TNF-α-induced NF-κB nuclear translocation in both tumor models (Fig. 7e–f).

Western blots showing the levels of pRIPK1, pRIPK3, pMLKL and total proteins in SK-HEP-1 (a) and HeLa (b) xenografts. Quantification of the ratios of the phosphorylated proteins to the total proteins. The data are presented as the means ± SDs (n ≥ 5 biological replicates). c, d Immunofluorescence staining of tumor tissues for pRIPK1 and pRIPK3. NF-κB (p65) nuclear translocation in SK-HEP-1 (e) and HeLa (f) tumors. Scale bar: 20 µm. The data are presented as the means ± SDs (n ≥ 5 biological replicates). g Representative electron micrographs showing mitochondria in SK-HEP-1 tumors. Unpaired t test. ns, P > 0.05; *P < 0.05; **P < 0.01; and ***P < 0.001.
An ultrastructural analysis of tumor sections revealed swollen mitochondria with a reduced crista density specifically in bicarbonate-treated and combination-treated tumors, providing direct morphological evidence of mitochondrial injury. In contrast, the control and TNF-α-alone groups displayed a normal mitochondrial morphology (Fig. 7g).
Collectively, these in vivo data demonstrate that bicarbonate synergizes with TNF-α to suppress tumor growth in vivo. The underlying mechanism involves bicarbonate-mediated attenuation of TNF-α-driven NF-κB activation coupled with its potentiation of RIPK-dependent necroptosis, thereby decisively redirecting TNF-α signaling toward an antitumor outcome.
Discussion
Our findings reveal that the alkalization of the intracellular pH by bicarbonate reprograms TNF-α signaling from a prosurvival NF-κB axis toward a cytotoxic, RIPK-dependent necroptotic pathway. This mechanism addresses two longstanding challenges in TNF-based cancer therapy: its limited tumoricidal efficacy and its potent systemic inflammatory toxicity. By shifting TNF-α away from NF-κB-driven survival and inflammation and toward regulated necroptosis, bicarbonate may fundamentally expand the therapeutic potential of this cytokine.
Mechanistically, bicarbonate attenuated TNF-α-induced IκB phosphorylation and NF-κB nuclear translocation while markedly increasing RIPK1, RIPK3, and MLKL phosphorylation. These effects were highly consistent across hepatocellular and cervical cancer models, despite their divergent baseline responses to TNF-α in vitro and in vivo. Such discrepancies are well recognized and reflect the influences of cytokine gradients, stromal interactions, and NF-κB-dependent survival signaling within the tumor microenvironment—factors that are absent in vitro. Importantly, however, the synergistic antitumor effects of bicarbonate and TNF-α remained robust across all the systems, underscoring that intracellular alkalization reliably redirected TNF-α signaling toward necroptosis regardless of baseline sensitivity.
ROS have emerged as central determinants controlling this signaling reprogramming. Moderate ROS levels favor NF-κB activation, whereas high ROS levels promote necroptosis and a persistent MPT [38,39,40]. Both TNF-α and bicarbonate independently increased the levels of cytosolic and mitochondrial ROS, but their combination resulted in a disproportionately larger increase in the level of ROS. This ROS surge amplified necroptotic kinase activation, promoted ΔΨm collapse, and drove sustained mPTP opening. NAC and EUK8, which target cytosolic and mitochondrial ROS, respectively, attenuated RIPK1/RIPK3/MLKL phosphorylation, prevented mPTP opening, rescued ΔΨm, and restored NF-κB activation. These findings indicate that mitochondrial ROS are necessary but not solely sufficient and that ROS from multiple intracellular compartments cooperate to determine whether TNF-α activates NF-κB signaling or triggers necroptosis. Because mPTP opening itself promotes additional ROS release (“ROS-induced ROS release”), a feed-forward loop likely strengthens the necroptotic commitment, although our study does not directly elucidate this secondary amplification mechanism.
A further question is why bicarbonate but not NaOH produces such profound intracellular reprogramming. Although NaOH and bicarbonate can generate the same extracellular pH, only bicarbonate increases the concentration and buffering capacity of the HCO3⁻/CO2 system, which is the dominant regulator of the intracellular pH. Because OH⁻ does not enter cells and does not participate in buffering, NaOH-adjusted medium does not appreciably change the intracellular HCO3⁻/CO2 equilibrium or pH gradient. In contrast, elevated levels of extracellular bicarbonate, which are transported by solute carrier family 4 (SLC4) [41,42,43] carriers, drive significant cytosolic and mitochondrial alkalization. Consistent with this result, NaOH-adjusted medium failed to alter the pHᵢ, ΔΨm, mPTP status, ROS production, or TNF-α signaling, whereas bicarbonate robustly modulated all these processes. Similarly, concerns regarding an increased Na⁺ load or osmolarity were mitigated by previous findings [12] showing that iso-osmotic NaCl does not alter the cytosolic or mitochondrial pH, ΔΨm, or MPT, indicating that ionic/osmotic effects do not account for the mitochondrial activity of bicarbonate.
The mitochondrial response to TNF-α alone also provided insights into the signaling switch. Although TNF-α has been reported to induce mitochondrial depolarization in apoptotic contexts, mitochondrial responses are well known to be biphasic and context dependent. In several systems, such as T cells, neurons, and cardiovascular models [44,45,46,47], TNF-α induces an early phase of mitochondrial hyperpolarization linked to an NF-κB/HIF-1-mediated metabolic adaptation before any apoptotic depolarization occurs. Consistent with this finding, treatment with TNF-α alone caused a mild increase in Δψm, preserved oxidative phosphorylation, and did not induce cell death in our system, indicating a prosurvival state rather than injury. This adaptive mitochondrial hyperpolarization was overridden by bicarbonate, which decreased the Δψm, induced CypD-dependent MPT, and shifted TNF-α signaling toward necroptotic execution. Thus, the TNF-α-induced increase in Δψm likely reflects an NF-κB-dependent survival checkpoint, whereas bicarbonate negates this adaptation and drives necroptosis.
Our results also have implications for TNF-α-associated toxicity. The clinical limitations of TNF-α stem largely from its strong NF-κB-driven inflammatory activity, which induces systemic cytokine release, vascular permeability, and organ injury. In this study, bicarbonate markedly suppressed TNF-α-induced NF-κB activation while enhancing its necroptotic, tumoricidal activity. This dual modulation suggests that bicarbonate may reduce the inflammatory toxicity associated with TNF-α while simultaneously increasing its antitumor potency. However, because our experiments were performed in immunodeficient mice without systemic cytokine or organ toxicity measurements, definitive conclusions regarding a reduction in toxicity cannot be drawn. Dedicated acute toxicity studies in immunocompetent models will be needed to rigorously test this possibility.
Necrostatin-1 (Nec-1) was the first small-molecule inhibitor identified to block RIPK1 kinase activity and has been widely used to investigate RIPK1-dependent necroptosis. We acknowledge that Nec-1 exhibits reported off-target activity, most notably the inhibition of indoleamine-2,3-dioxygenase (IDO). In the present study, Nec-1 markedly suppressed the phosphorylation of RIPK1, RIPK3, and MLKL, consistent with the effective blockade of the necroptotic signaling cascade. Importantly, Nec-1 did not alter TNF-α-induced NF-κB nuclear translocation, indicating that it does not interfere with upstream TNF-α/NF-κB signaling. Functionally, Nec-1 had no effect on cell viability under control, TNF-α-alone, or bicarbonate-alone conditions but selectively rescued cells from death induced by the bicarbonate + TNF-α combination. This conditional protective effect argues against a dominant contribution of off-target activities such as IDO inhibition in our experimental context. We complemented pharmacological inhibition with genetic loss-of-function approaches to further mitigate concerns regarding inhibitor specificity. siRNA-mediated depletion of RIPK1 phenocopied the effects of Nec-1, suppressed RIPK3 and MLKL phosphorylation and strongly rescued cell viability. The concordance between pharmacological inhibition and genetic depletion provides orthogonal evidence that the observed cell death is driven by bona fide RIPK1-dependent necroptosis rather than the off-target effects of Nec-1.
Collectively, our work identifies intracellular alkalization as a previously unrecognized regulator of TNF-α signaling. By modulating the pHᵢ and redox balance, bicarbonate suppresses NF-κB-mediated survival and inflammatory pathways while promoting RIPK-dependent necroptosis through mitochondrial dysfunction and ROS amplification. This combined effect not only increases tumor cell death but also may broaden the therapeutic window of TNF-α. Our in vivo data confirm substantial tumor suppression with bicarbonate + TNF-α cotreatment, supporting the concept that the targeted manipulation of the intracellular pH and mitochondrial dynamics may unlock the full tumoricidal potential of TNF-α and convert a historically toxic cytokine into a more selective anticancer agent.
Data availability
The data supporting the findings reported in this study are available from the corresponding author upon request.
References
Christofferson DE, Li Y, Yuan J. Control of life-or-death decisions by RIP1 kinase. Annu Rev Physiol. 2014;76:129–50.
Brenner D, Blaser H, Mak TW. Regulation of tumour necrosis factor signalling: live or let die. Nat Rev Immunol. 2015;15:362–74.
Kondylis V, Kumari S, Vlantis K, Pasparakis M. The interplay of IKK, NF-κB and RIPK1 signaling in the regulation of cell death, tissue homeostasis and inflammation. Immunol Rev. 2017;277:113–27.
Silke J, Rickard JA, Gerlic M. The diverse role of RIP kinases in necroptosis and inflammation. Nat Immunol. 2015;16:689–97.
Webster JD, Vucic D. The balance of TNF mediated pathways regulates inflammatory cell death signaling in healthy and diseased tissues. Front Cell Dev Biol. 2020;8:365.
Sica A, Invernizzi P, Mantovani A. Macrophage plasticity and polarization in liver homeostasis and pathology. Hepatology. 2014;59:2034–42.
Balkwill F. TNF-alpha in promotion and progression of cancer. Cancer Metastasis Rev. 2006;25:409–16.
Cruceriu D, Baldasici O, Balacescu O, Berindan-Neagoe I. The dual role of tumor necrosis factor-alpha (TNF-α) in breast cancer: molecular insights and therapeutic approaches. Cell Oncol. 2020;43:1–18.
van Loo G, Bertrand MJM. Death by TNF: a road to inflammation. Nat Rev Immunol. 2023;23:289–303.
Zhou Y, Chang W, Lu X, Wang J, Zhang C, Xu Y. Acid-base homeostasis and implications to the phenotypic behaviors of cancer. Genom Proteom Bioinform. 2023;21:1133–48.
Song X, Zhu S, Xie Y, Liu J, Sun L, Zeng D, et al. JTC801 induces pH-dependent death specifically in cancer cells and slows growth of tumors in mice. Gastroenterology. 2018;154:1480–93.
Ying C, Jin C, Zeng S, Chao M, Hu X. Alkalization of cellular pH leads to cancer cell death by disrupting autophagy and mitochondrial function. Oncogene. 2022;41:3886–97.
Martin NK, Gaffney EA, Gatenby RA, Gillies RJ, Robey IF, Maini PK. A mathematical model of tumour and blood pHe regulation: the HCO3-/CO2 buffering system. Math Biosci. 2011;230:1–11.
Ibrahim-Hashim A, Cornnell HH, Abrahams D, Lloyd M, Bui M, Gillies RJ, et al. Systemic buffers inhibit carcinogenesis in TRAMP mice. J Urol. 2012;188:624–31.
Martin NK, Robey IF, Gaffney EA, Gillies RJ, Gatenby RA, Maini PK. Predicting the safety and efficacy of buffer therapy to raise tumour pHe: an integrative modelling study. Br J Cancer. 2012;106:1280.
Chao M, Wu H, Jin K, Li B, Wu J, Zhang G, et al. A nonrandomized cohort and a randomized study of local control of large hepatocarcinoma by targeting intratumoral lactic acidosis. Elife. 2016;5:e15691.
Jin K, Zeng S, Li B, Zhang G, Wu J, Hu X, et al. Bicarbonate-integrated transarterial chemoembolization (TACE) in real-world hepatocellular carcinoma. Signal Transduct Target Ther. 2025;10:281.
Poburko D, Santo-Domingo J, Demaurex N. Dynamic regulation of the mitochondrial proton gradient during cytosolic calcium elevations. J Biol Chem. 2011;286:11672–84.
Liu B, Fang M, Hu Y, Huang B, Li N, Chang C, et al. Hepatitis B virus X protein inhibits autophagic degradation by impairing lysosomal maturation. Autophagy. 2014;10:416–30.
Wu H, Wang Y, Ying M, Jin C, Li J, Hu X. Lactate dehydrogenases amplify reactive oxygen species in cancer cells in response to oxidative stimuli. Signal Transduct Target Ther. 2021;6:242.
Han W, Li L, Qiu S, Lu Q, Pan Q, Gu Y, et al. Shikonin circumvents cancer drug resistance by induction of a necroptotic death. Mol Cancer Ther. 2007;6:1641–9.
Li L, Han W, Gu Y, Qiu S, Lu Q, Jin J, et al. Honokiol induces a necrotic cell death through the mitochondrial permeability transition pore. Cancer Res. 2007;67:4894–903.
Moujalled DM, Cook WD, Murphy JM, Vaux DL. Necroptosis induced by RIPK3 requires MLKL but not Drp1. Cell Death Dis. 2014;5:e1086.
Vanlangenakker N, Bertrand MJ, Bogaert P, Vandenabeele P, Vanden Berghe T. TNF-induced necroptosis in L929 cells is tightly regulated by multiple TNFR1 complex I and II members. Cell Death Dis. 2011;2:e230.
Cho YS, Challa S, Moquin D, Genga R, Ray TD, Guildford M, et al. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell. 2009;137:1112–23.
Moriwaki K, Chan FK. RIP3: a molecular switch for necrosis and inflammation. Genes Dev. 2013;27:1640–9.
Lemasters JJ, Theruvath TP, Zhong Z, Nieminen AL. Mitochondrial calcium and the permeability transition in cell death. Biochim Biophys Acta. 2009;1787:1395–401.
Ma S, Boerner JE, TiongYip C, Weidmann B, Ryder NS, Cooreman MP, et al. NIM811, a cyclophilin inhibitor, exhibits potent in vitro activity against hepatitis C virus alone or in combination with alpha interferon. Antimicrob Agents Chemother. 2006;50:2976–82.
Waldmeier PC, Feldtrauer JJ, Qian T, Lemasters JJ. Inhibition of the mitochondrial permeability transition by the nonimmunosuppressive cyclosporin derivative NIM811. Mol Pharmacol. 2002;62:22–9.
Morgan MJ, Liu ZG. Crosstalk of reactive oxygen species and NF-κB signaling. Cell Res. 2011;21:103–15.
Zhang Y, Su SS, Zhao S, Yang Z, Zhong CQ, Chen X, et al. RIP1 autophosphorylation is promoted by mitochondrial ROS and is essential for RIP3 recruitment into necrosome. Nat Commun. 2017;8:14329.
Lu B, Gong X, Wang ZQ, Ding Y, Wang C, Luo TF, et al. Shikonin induces glioma cell necroptosis in vitro by ROS overproduction and promoting RIP1/RIP3 necrosome formation. Acta Pharmacol Sin. 2017;38:1543–53.
Dzeja PP, Holmuhamedov EL, Ozcan C, Pucar D, Jahangir A, Terzic A. Mitochondria: gateway for cytoprotection. Circ Res. 2001;89:744–6.
Aronis A, Komarnitsky R, Shilo S, Tirosh O. Membrane depolarization of isolated rat liver mitochondria attenuates permeability transition pore opening and oxidant production. Antioxid Redox Signal. 2002;4:647–54.
Halasi M, Wang M, Chavan TS, Gaponenko V, Hay N, Gartel AL. ROS inhibitor N-acetyl-L-cysteine antagonizes the activity of proteasome inhibitors. Biochem J. 2013;454:201–8.
Menezes LB, Segat BB, Tolentino H, Pires DC, Mattos LMM, Hottum HM, et al. ROS scavenging of SOD/CAT mimics probed by EPR and reduction of lipid peroxidation in S. cerevisiae and mouse liver, under severe hydroxyl radical stress condition. J Inorg Biochem. 2023;239:112062.
Zeng X, Zhang YD, Ma RY, Chen YJ, Xiang XM, Hou DY, et al. Activated Drp1 regulates p62-mediated autophagic flux and aggravates inflammation in cerebral ischemia-reperfusion via the ROS-RIP1/RIP3-exosome axis. Mil Med Res. 2022;9:25.
Thoma A, Lightfoot AP. NF-kB and inflammatory cytokine signalling: role in skeletal muscle atrophy. Adv Exp Med Biol. 2018;1088:267–79.
Li S, Liu Y, Li D, Zhang K, Zhang Z, Zhang Z, et al. Microalgal astaxanthin ameliorates cypermethrin-induced necroptosis and inflammation via targeting mitochondrial Ca2+ homeostasis and the ROS-NF-κB-RIPK3/MLKL axis in carp hepatocytes (Cyprinus carpio). Fish Shellfish Immunol. 2024;154:109944.
Chen X, Liu C, Yu R, Gan Z, Zhang Z, Chen Z, et al. Interaction between ferroptosis and TNF-α: Impact in obesity-related osteoporosis. FASEB J. 2023;37:e22947.
Gorbatenko A, Olesen CW, Boedtkjer E, Pedersen SF. Regulation and roles of bicarbonate transporters in cancer. Front Physiol. 2014;5:130.
Hwang JM, Kao SH, Hsieh YH, Li KL, Wang PH, Hsu LS, et al. Reduction of anion exchanger 2 expression induces apoptosis of human hepatocellular carcinoma cells. Mol Cell Biochem. 2009;327:135–44.
Wu TT, Hsieh YH, Wu CC, Tsai JH, Hsieh YS, Huang CY, et al. Overexpression of anion exchanger 2 in human hepatocellular carcinoma. Chin J Physiol. 2006;49:192–8.
Shakibaei M, Sung B, Sethi G, Aggarwal BB. TNF-alpha-induced mitochondrial alterations in human T cells requires FADD and caspase-8 activation but not RIP and caspase-3 activation. Antioxid Redox Signal. 2010;13:821–31.
Perl A, Gergely P Jr, Nagy G, Koncz A, Banki K. Mitochondrial hyperpolarization: a checkpoint of T-cell life, death and autoimmunity. Trends Immunol. 2004;25:360.
Lambert CM, Roy M, Meloche J, Robitaille GA, Agharazii M, Richard DE, et al. Tumor necrosis factor inhibitors as novel therapeutic tools for vascular remodeling diseases. Am J Physiol Heart Circ Physiol. 2010;299:H995–1001.
Albensi BC. What is nuclear factor kappa B (NF-κB) doing in and to the mitochondrion?. Front Cell Dev Biol. 2019;7:154.
Acknowledgements
This work was supported in part by a key project (2018C03009) funded by the Zhejiang Provincial Department of Sciences and Technologies (to XH) and China Natural Sciences Foundation projects (81470126 and 82073038 to XH).
Author information
Authors and Affiliations
Contributions
XH, conception, study design, data analysis, and writing the manuscript; CY, experiments, data analysis, and writing the manuscript; DW, experiments and data analysis; SYZ, experiments; CMJ, experiments; and MFY, experiments.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Ying, C., Wang, D., Zeng, Sy. et al. Alkalization of the intracellular pH reprograms TNF-α signaling from inflammatory NF-κB activation to tumoricidal RIP kinase-dependent necroptosis in cancer cells. Acta Pharmacol Sin (2026). https://doi.org/10.1038/s41401-026-01786-w
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41401-026-01786-w
