
Abstract
Diffuse-type gastric cancer (DGC) is characterized by strong invasiveness and poor prognosis, frequently associated with peritoneal metastasis. Dysregulation of protein kinase A catalytic subunit beta (PRKACB) has been implicated in various cancers, but its role in DGC remains unclear. This study investigates the expression, function, and molecular mechanisms of PRKACB in DGC metastasis. PRKACB expression was analyzed by immunohistochemistry in tissue samples from DGC and intestinal-type gastric cancer (IGC) patients. Functional assays and a mouse peritoneal metastasis model were employed to evaluate the impact of PRKACB on metastasis. The interaction between PRKACB and RhoA was explored using co-immunoprecipitation, GST pull-down assays, and in situ proximity ligation assays. PRKACB expression was significantly lower in DGC tissues compared to IGC and adjacent non-tumor tissues. Multivariate Cox regression analysis identified low PRKACB expression as an independent prognostic factor for poor overall survival. In DGC cell lines, PRKACB knockdown enhanced cell migration, invasion, pseudopodia formation, and epithelial-mesenchymal transition (EMT), while PRKACB overexpression suppressed these activities. In vivo experiments demonstrated that PRKACB knockdown promoted earlier onset of peritoneal metastasis. Mechanistically, PRKACB interacted with RhoA and promoted its phosphorylation at S188, thereby inhibiting RhoA signaling and its downstream effectors ROCK1 and FAK. Common RhoA mutations in DGC (V38G and N41K) weakened its interaction with PRKACB, leading to reduced phosphorylation and enhanced metastatic potential. Importantly, RhoA inhibitor treatment reversed the pro-metastatic effects induced by low PRKACB expression. In conclusion, low PRKACB expression promotes DGC metastasis through activation of RhoA signaling, and PRKACB downregulation combined with functional RhoA mutations contributes to the aggressive phenotype of DGC. These findings provide new insights into DGC progression and suggest that PRKACB may serve as both a prognostic biomarker and a potential therapeutic target for DGC patients.
Similar content being viewed by others


Introduction
Gastric cancer is one of the most common malignant tumors in the world today, with over one million new cases estimated annually, ranking it fifth in incidence among all malignancies. Each year, approximately 700,000 people die from gastric cancer, making its mortality rate the fourth highest [1]. China is particularly a high-incidence area for gastric cancer worldwide, with nearly 400,000 new cases and almost 300,000 deaths annually, ranking third in both incidence and mortality rates [2]. The high heterogeneity of gastric cancer is a significant factor for its poor prognosis, and the therapeutic outcomes vary among different types of gastric cancer [3]. To better guide clinical diagnosis and treatment, the field has proposed various classification methods for gastric cancer, among which the Lauren classification remains the most commonly used in clinical practice [4]. The principle of the Lauren classification is to divide gastric cancer into intestinal, diffuse, and mixed types based on the morphological structure and immunohistochemical staining of tumor tissues under the microscope [5, 6]. A meta-analysis encompassing 73 studies with a total of 60,000 gastric cancer patients indicated that patients with DGC have a higher risk of death compared to those with IGC (HR: 1.23, p < 0.0001) [7].
The tumor cells of DGC are dispersed under the microscope, do not form glands, and lack the expression of adhesion molecules between tumor cells, especially the expression of the adhesion protein E-cadherin, making DGC prone to spread and metastasis [8, 9]. At the molecular level, DGC exhibits distinct genomic alterations compared to IGC. The Cancer Genome Atlas (TCGA) project proposed a molecular classification dividing gastric cancer into four subtypes, among which the genomically stable (GS) subtype is enriched for the diffuse histological variant and characterized by mutations in RHOA or fusions involving RHO-family GTPase-activating proteins [10]. Notably, RHOA mutations occur in 14-25% of DGC cases but are rarely observed in IGC [11, 12]. RhoA, a member of the Rho GTPase family, plays critical roles in cytoskeletal organization, cell morphology regulation, and motility [13]. RhoA exists primarily in two states: inactive GDP-bound and active GTP-bound forms, cycling between these states through the regulation of guanine nucleotide exchange factors (GEFs), GTPase-activating proteins (GAPs), and guanine nucleotide dissociation inhibitors (GDIs). The phosphorylation of RhoA at serine 188 (S188) negatively regulates its activity by promoting its association with GDIs, thereby inhibiting RhoA signaling [14, 15]. However, the kinase responsible for RhoA-S188 phosphorylation in gastric cancer and its functional significance in DGC metastasis remain poorly understood.
To address this gap, we sought to identify molecules that drive the invasive and metastatic functions of DGC, which may help to better understand the pathogenesis of DGC and provide new avenues for improving the comprehensive treatment outcomes of DGC. In our previous research, proteomic analysis of tissue samples from DGC was conducted, revealing that the expression level of PRKACB is significantly downregulated in metastatic and recurrent DGC [16]. PRKACB, a catalytic subunit of Protein Kinase A (also known as PKA-Cβ), is ubiquitously expressed in tissues and organs throughout the human body. Protein Kinase A (PKA), also known as cyclic-AMP dependent protein kinase A, belongs to the serine/threonine protein kinase family, predominantly located in the cytoplasm. It influences downstream signaling pathway activities and a range of cellular functions by phosphorylating the serine/threonine residues at the C-terminus of protein structures [17,18,19]. Previous studies have reported low expression of PRKACB in several malignancies, including non-small cell lung cancer, malignant gliomas, and leukemia, and its involvement in promoting the malignant phenotype of tumors [20,21,22]. In this study, we investigated the expression, function, and related molecular mechanisms of PRKACB in gastric cancer, with a particular focus on its regulatory role in RhoA signaling and metastasis.
Materials and methods
Clinical samples
A total of 376 gastric cancer tissues and their matched adjacent non-tumor tissue samples were collected for this study from patients who underwent D2 radical gastrectomy at the Department of General Surgery, Zhongshan Hospital, Fudan University. The samples included 154 cases of DGC and 222 cases of IGC. All surgeries achieved R0 resection margins. The postoperative pathology reports identified all cases as gastric adenocarcinoma, and comprehensive clinicopathological and prognostic data were available. Clinicopathological variables collected included TNM stage, Lauren type, age, tumor size, lymphovascular invasion status, and postoperative chemotherapy status.
Public database validation
The prognostic value of PRKACB expression was validated using the Kaplan-Meier Plotter database (http://www.kmplot.com). Overall survival and progression-free survival were analyzed. The hazard ratio with 95% confidence intervals and log-rank P values was calculated automatically by the online tool.
Immunohistochemistry (IHC)
Paraffin-embedded tissue sections were baked at 60 °C for 6–8 h. The sections were dewaxed in xylene and rehydrated through graded alcohols. Endogenous peroxidase activity was blocked using a mixture of 3% hydrogen peroxide and methanol. Heat-induced antigen retrieval was performed using microwave heating in 0.01 M citrate buffer. The sections were incubated with primary antibodies overnight at 4 °C. After rewarming, sections were treated with Primary Antibody Amplifier Quanto and HRP Polymer Quanto. Staining was developed with DAB solution and counterstained with hematoxylin. Staining was scored based on the area and intensity of tumor cell staining. Area scoring was set as follows: 0: <5%; 1: 5–25%; 2: 26–50%; 3: 51–75%; 4: >75%. Intensity scoring was defined as 0: negative; 1: weakly positive; 2: moderately positive; 3: strongly positive. The final score was calculated by multiplying the area score by the intensity score. For Ki67 staining, the percentage of Ki67-positive cells was calculated in five randomly selected high-power fields per section. For quantification of metastatic nodules, Ki67 labeling index was expressed as the percentage of positive nuclei among total tumor cells.
TUNEL assay
Apoptosis in metastatic nodules was detected using the TUNEL Apoptosis Assay Kit (Beyotime, China) according to the manufacturer’s instructions. Briefly, paraffin-embedded tissue sections were deparaffinized and rehydrated. After proteinase K treatment (20 μg/ml, 15 min at room temperature), sections were incubated with TUNEL reaction mixture at 37 °C for 1 h in the dark. Nuclei were counterstained with DAPI. TUNEL-positive cells were visualized using a fluorescence microscope and quantified in five randomly selected fields per section. The apoptotic index was calculated as the percentage of TUNEL-positive cells among total cells.
Cell lines and cell culture
The cell lines used in this study and their sources are as follows: AGS, GES-1, HGC-27, and MKN-45 were obtained from the Gastric Cancer Center, Zhongshan Hospital, Fudan University; HEK293T and NCI-N87 were purchased from the Shanghai Institute of Cell Biology, Chinese Academy of Sciences; KATO III and SNU-668 were acquired from Nanjing Cobioer Biosciences Co., Ltd. The murine gastric cancer cell line YTN16 was kindly provided by Dr. Masami Yamamoto, Dr. Tetsuya Tsukamoto, and Dr. Sachiyo Nomura and maintained in our laboratory. The cell lines AGS, MKN-45, NCI-N87, KATO III, and SNU-668 were cultured in RPMI 1640 medium supplemented with 10% fetal bovine serum (FBS). The cell lines HEK293T and HGC-27 were cultured in DMEM medium, also supplemented with 10% FBS. All cultures were maintained at a constant temperature of 37 °C with 5% CO2 in a humidified incubator. For bioluminescence imaging experiments, YTN16 cells were stably transfected with a firefly luciferase expression vector (pLV-Luc) and selected with puromycin (2 μg/ml) for two weeks. Single-cell clones with high luciferase activity were selected for subsequent experiments.
Reagents and antibodies
RhoA (Cat#ab54835), phospho-RhoA (S188, Cat#ab41435) were purchased from Abcam. PRKACB (Cat#PH8689), Flag-tag (Cat#M20018), GST-tag (Cat#M20025), HA-tag (Cat#M10003) were obtained from Abmart. PRKACB (Cat#55382-1-AP) was purchased from Proteintech. F-actin (Cat#8878) and EMT Antibody Sampler Kit (Cat#9782) were obtained from Cell Signaling Technology. Flag-tag (Cat#F7425), Flag M2 magnetic beads (Cat#M8823) were purchased from Sigma-Aldrich. ROCK Activity Assay Kit (Cat#ab211175) and phospho-FAK (Cat#ab322920) were obtained from Abcam. Rhosin (Cat#HY-12646A, MedChemExpress). Active-RhoA Pull-down Kit was obtained from NewEastbio.
RNA interference, plasmids and transfection
The small interfering RNA (siRNA) (siPRKACB#1: 5′-CCCATTCTTTGCAGACCAATT-3′; siPRKACB#2: 5′-GUUCUAUGCAGCUCAGAUATT-3′; siRhoA#1: 5′-CAGAUACCGAUGUUAUACU-3′; siRhoA#2: 5′-AAGGCAGAGAUAUGGCAAA-3′), negative control siRNA, and the short hairpin RNA (shRNA) specifically targeting PRKACB were constructed by Sangon Biotech (Shanghai, China). The pcDNA3-Flag-PRKACB and pcDNA3-GST-RhoA plasmid was constructed by Youbio Company (Changsha, Hunan, China). Different fragments and mutants of RhoA were generated by subcloning the corresponding cDNAs into the pcDNA3-HA-vector. Cells were transfected with the plasmids, shRNAs, or siRNAs using Lipofectamine 3000 (Invitrogen) following the manufacturer’s instructions.
Wound healing assay
SNU-668 cells were seeded on six-well plates at 1 × 106 cells per well. After incubation until confluence, the cells were scraped using a pipette tip. Cells were washed twice with PBS and photograph under a light microscope (time = 0 h). They were subsequently incubated in serum-free medium at 37 °C and 5% CO2. Photographs of the scratches were taken under the light microscope at different points in time. Analysis was performed using ImageJ software.
Transwell migration/invasion assays
Tumor cell migration (Corning BioCoatTM Control Insert No ECM, 8 μm pore size)/invasion assays were performed according to the manufacturer’s instructions. In brief, 1.5 × 105 tumor cells were inoculated in 100 μL of basic serum-free medium in the upper chamber, and 600 μL of complete medium was added to the lower chamber. For invasion analysis, the bottom of the transwell chamber was precoated with 70 μL of Matrigel (BD Biosciences, San Jose, CA, USA) for 4 h before adding medium. After incubation at 37 °C and 5% CO2 for 24 or 48 h, non-invasive cells on the upper side of the chamber were wiped off. Cells that migrated/invaded to the submembrane surface were fixed using 4% paraformaldehyde and stained with crystal violet. Cells passing through the filter were photographed with a microscope in five randomly selected areas, and the number of transferred cells was calculated.
Immunofluorescence
Cells were seeded onto sterile coverslips placed in 12-well plates. After growing to an appropriate density, the cells were washed twice with pre-chilled PBS. They were then fixed with 4% paraformaldehyde at room temperature for 30 min. Permeabilization was achieved using 0.1% Triton X-100 at 4 °C for 5–10 min. Blocking was performed with 1% bovine serum albumin (BSA) for 1 h. Subsequently, cells were incubated with 594-conjugated E-cadherin Recombinant monoclonal antibody, 488-conjugated phalloidin, and DAPI in the dark at room temperature for 30 min, followed by washing with pre-chilled PBS. The coverslips were mounted onto slides using a mounting medium. Observation and photography were conducted using a laser scanning confocal microscope.
Orthotopic mouse model
Male BALB/c nude mice, 4–5 weeks of age, purchased from SLAC (Shanghai, China), were used for orthotopic mouse model. Briefly, shPRKACB group and control group KATO III stable transfectant cells were digested and resuspended in sterile PBS at a concentration of 1 × 107 cells/ml. Each mouse was subcutaneously injected with 200 µl of the cell suspension into the right flank. Once the tumor volume reached 1500 mm3, the mice were euthanized. The subcutaneous tumor tissues were excised and cut into small pieces approximately 2 × 2 × 2 mm3 in size and rinsed three times with sterile saline. Following anesthesia, the mice were immediately subjected to surgery under aseptic conditions with a midline abdominal incision about 1 cm in length. Using tweezers, the liver was gently retracted to expose the stomach. The deep part of the stomach was gently pulled out, and 50 µl of saline was slowly injected into the subserosal layer of the stomach to form a mound. Using ophthalmic scissors, a small 3 mm incision was made at the edge of the mound, and the tumor tissue mass was gently pushed into the mound through the small opening. The opening was closed with absorbable fine sutures, and the abdomen was closed in layers. The RhoA inhibitor (Rhosin) was administered intraperitoneally at a dose of 40 mg/kg. The control group received an equivalent volume of the drug solvent DMSO. After 8 weeks, the mice were euthanized, and the number and size of metastatic nodules in the peritoneal cavity were counted.
In vivo bioluminescence imaging
Male C57BL/6 mice (5–6 weeks old) were purchased from SLAC (Shanghai, China) and used for the peritoneal dissemination assay. Briefly, the murine gastric cancer cell line YTN16 stably expressing luciferase reporter (YTN16-Luc) was used to generate stable cell lines expressing either control shRNA or shRNA against PRKACB. For peritoneal dissemination, mice were inoculated intraperitoneally with 1 × 107 cells per mouse (n = 6/group). Tumor burden was monitored weekly by in vivo bioluminescence imaging using the IVIS Spectrum imaging system (PerkinElmer). Bioluminescence signals were analyzed with Living Image software (PerkinElmer). D-luciferin potassium salt (Gold Biotechnology), the substrate for firefly luciferase, was administered intraperitoneally at a dose of 150 mg/kg.
CCK8 assay and colony formation assays
For CCK-8 experiments, cell viability was assayed using Cell Counting Kit-8 (Dojindo, Japan) according to the manufacturer’s instructions. Transfected cells were inoculated into 96-well plates at 3000 cells per well. After incubation for the indicated time points, CCK-8 solution was added to each well and incubated for 60 min. The absorbance was measured at OD 450 nm using a BioTek Gen5 system. For colony formation assays, 12-well plates were inoculated with 2000 cells per well. The medium was changed every 3 days. After 6–10 days of incubation, the medium was removed and colonies were fixed in 4% paraformaldehyde for 30 min at room temperature and stained with 0.1% (w/v) crystal violet for 1 h. Photographs were taken and colonies larger than 50 cells were counted using a microscope.
Real time PCR analysis
Cells were collected, and total RNA was extracted using TRIzol reagent (Invitrogen, Thermo Fisher Scientific) according to the manufacturer’s instructions. Total RNA was reverse transcribed into complementary DNA (cDNA) using the PrimeScript RT kit (Takara). The expression levels of RNA of interest were determined at ABI StepOne Plus (Applied Biosystems, Thermo Fisher Scientific) using SYBR Premix Ex Taq (Takara, Japan). It was normalized to GAPDH. Relative expression of mRNAs was analyzed according to the ΔΔCT method. All experiments were performed in triplicate.
Western blotting
Western blot was performed as described previously. Briefly, whole cell extracts were prepared in SDS buffer containing protease and phosphatase inhibitors (Roche Lifesciences). Equal amounts of proteins were separated by SDS polyacrylamide gel electrophoresis. The proteins were then transferred onto PVDF membranes. After blocking, the membranes were incubated with primary antibodies. The membranes were then washed with TBST and incubated with horseradish peroxidase-conjugated secondary antibodies. Visualization was performed using an enhanced chemiluminescence analysis kit (TIANGEN Biotech, China) and an image analyzer ImageQuant LAS 4000 (GE Healthcare, Chicago, IL, USA).
GST-pulldown
The recombinant plasmids of RhoA and GST were transferred to BL21(DE3) strain to obtain GST-RhoA fusion protein. Appropriate amount of fusion protein GST-RhoA was thawed on ice and added to glutathione-Sepharose beads, and mixed with it. Cells containing Flag-PRKACB were simultaneously lysed, the supernatant was centrifuged, BCA protein quantification was performed, and the sample was added to the purified protein EP tube, incubated overnight at 4 °C. Western Blot was performed to detect the relevant protein tag and verify whether PRKACB could bind to RhoA. For RhoA mutant effector binding assays, GST-RhoA wild-type, GST-RhoA-V38G, and GST-RhoA-N41K fusion proteins were purified as described above. Recombinant His-tagged mDia1 and His-tagged ROCK1 proteins were expressed in E. coli and purified using agarose beads according to the manufacturer’s instructions. For pull-down assays, equal amounts of GST or GST-RhoA fusion proteins (wild-type and mutants) were immobilized on glutathione-Sepharose beads and incubated with purified His-mDia1 or His-ROCK1 proteins in binding buffer at 4 °C for 2 h with gentle rotation. After extensive washing with binding buffer, bound proteins were eluted with SDS loading buffer and analyzed by Western blotting using anti-mDia1 or anti-ROCK1 antibodies. GST and GST-RhoA proteins were detected by anti-GST antibody.
In situ proximity ligation assay
To investigate and visualize the interaction between PRKACB and RhoA, an in situ proximity ligation assay (PLA) was performed. SNU-668 cells were cultured in appropriate conditions until they reached 70-80% confluence. Cells were then seeded onto glass slides and allowed to attach overnight. Subsequent to attachment, cells were fixed with 4% paraformaldehyde for 15 min at room temperature and then permeabilized with 0.1% Triton X-100 for 10 min. Following fixation and permeabilization, the cells were blocked with 3% BSA in PBS for 1 h at room temperature to reduce nonspecific binding. The primary antibodies, anti-PRKACB and anti-RhoA, were applied to the cells and incubated overnight at 4 °C. After primary antibody incubation, cells were washed with PBS and incubated with PLA probes according to the manufacturer’s instructions (DUOLINK® PLA Kit, Sigma-Aldrich, Germany). These probes are conjugated to oligonucleotides that can hybridize and ligate if the proteins of interest are in close proximity. Ligation was followed by amplification using rolling circle amplification (RCA) that produces a fluorescent signal detectable by confocal microscopy. The amplification reaction was carried out at 37 °C for 1.5 h. Post-amplification, cells were washed and mounted with a mounting medium containing DAPI to stain the nuclei. Fluorescent signals indicating PRKACB and RhoA interactions were visualized and captured using a laser scanning confocal microscope (Zeiss, Pleasanton, CA, USA). Images were analyzed to quantify the interactions and assess the colocalization of the detected complexes.
Co-immunoprecipitation and mass spectrometer analysis
SNU-668 cells were transfected using Flag-tagged PRKACB plasmid. Transfected cells were collected and lysed using IP lysis buffer (50 mM Tris HCl, pH 7.5, 0.1% NP-40, 15 mM MgCl2, 150 mM NaCl, and 5 mM EDTA) at 4 °C for 2 h. Equal quantities of proteins were then incubated with normal mouse IgG or anti-flag tag antibody immobilized on Protein G agarose on a rotator at 4 °C incubated for 2 h. Wash the beads using IP lysis buffer, add the appropriate amount of loading buffer, and then boil and centrifuge. Immunoprecipitates were separated in 10% SDS-PAGE gel. After staining the gel by silver staining, the specific bands co-immunoprecipitated with anti-Flag antibody were cut and analyzed by LC-MS/MS on mass spectrometer. RhoA was identified with the peptide sequence QVELALWDTAGQEDYDR, corresponding to amino acids 52–68 of human RhoA protein.
Bioinformatic analysis
The 3D structure of the target protein was constructed using the Modeller 9.23 program. A total of 100 models were built, and the model with the highest DOPE score was selected as the final model structure. Protein-protein docking was performed using the ZDOCK program. All parameters were set to default, and 10 complex conformations were output. The conformation with the highest score was chosen as the docking conformation for subsequent molecular dynamics simulation optimization. Molecular dynamics simulations of the protein-protein complex obtained from the docking were conducted using the Amber18 software package. Finally, analysis was performed using the CPPTRAJ module.
Statistical analysis
Statistical analysis was performed with GraphPad Prism 8.0 software. Comparisons between two groups were analyzed with a two-tailed unpaired Student’s t-test, while multiple group comparisons were conducted using one-way ANOVA followed by Dunnett’s or Tukey’s post hoc tests. The results of statistical analyses are presented as mean ± standard deviation. Correlation analysis of continuous variables between two groups was conducted using Spearman’s correlation analysis. Cox proportional hazards regression analysis was performed to identify prognostic factors for overall survival. Univariate Cox regression was first conducted to screen potential prognostic variables. Variables with P < 0.1 in univariate analysis were subsequently included in the multivariate Cox regression model to identify independent prognostic factors. Hazard ratios (HRs) and 95% confidence intervals (CIs) were calculated. Statistical significance was defined as P < 0.05.
Results
PRKACB is downregulated in DGC and is associated with tumor progression and poor prognosis
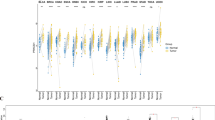
To study the role of PRKACB in gastric cancer, we first need to identify the expression of PRKACB in gastric cancer (including intestinal and diffuse types). We selected 222 samples of IGC and 154 samples of DGC from the gastric cancer sample bank of Zhongshan Hospital affiliated with Fudan University, as well as corresponding adjacent normal gastric mucosa tissue samples, and prepared tissue microarrays. Immunohistochemical staining revealed that PRKACB was mainly expressed in the cytoplasm. Scoring the staining on each point of the tissue microarray, we found no significant difference in PRKACB scores between IGC and its corresponding adjacent tissues (p > 0.05), while the expression of PRKACB in DGC was significantly lower than that in its corresponding adjacent tissues (p < 0.001) (Fig. 1A). Further analysis in DGC showed that in TNM stage III-IV tissue samples, the expression level of PRKACB was significantly lower (p < 0.05) (Fig. 1B). Furthermore, we validated the prognostic value of PRKACB expression using the online Kaplan-Meier Plotter database (http://www.kmplot.com). The results showed that low PRKACB expression was significantly associated with both poor overall survival and progression-free survival in gastric cancer patients (Fig. 1C, D). Univariate and multivariate Cox regression analyses were performed on the clinical cohort of 376 gastric cancer patients. The following variables were included in the analysis: Lauren classification, sex, age, tumor size, T stage, N stage, TNM stage, lymphovascular invasion, PRKACB expression level, and adjuvant chemotherapy status. The results identified two independent prognostic factors for OS. TNM stage III-IV was an independent risk factor for poor survival (HR = 4.37, 95% CI: 2.53–7.55, P < 0.001). Notably, low PRKACB expression was identified as an independent risk factor for poor OS (HR = 3.94, 95% CI: 2.71–5.71, P < 0.001), indicating that patients with low PRKACB expression had ~4-fold increased risk of death compared to those with high expression (Supplementary Table 1 and Supplementary Fig. 1A). In tissue samples with low PRKACB expression, the proportions of T1/2 and T3/4 stages were 30.0% and 70.0%, respectively, and the proportions of N0 and N+ stages were 23.3% and 76.7%, respectively (Fig. 1E). Kaplan-Meier analysis showed that among 154 patients with DGC, overall survival was significantly worse in patients with low PRKACB expression compared to those with high PRKACB expression (p < 0.001) (Fig. 1F). Further, dividing these patients into TNM stages I-II (n = 65) and III-IV (n = 89) and analyzing the two subgroups revealed that in stage I-II patients, PRKACB expression was not significantly correlated with OS (p = 0.070) (Fig. 1G); whereas in stage III-IV patients, OS was significantly worse in those with low PRKACB expression (p = 0.022) (Fig. 1H). These results indicate that PRKACB is lowly expressed in DGC, especially in advanced-stage DGC, and low PRKACB expression suggests poor prognosis.

A Representative immunohistochemical images and statistical graphs of expression levels of PRKACB in adjacent normal tissues, IGC, and diffuse-type gastric cancer. B Representative immunohistochemical images and statistical graphs of expression levels of PRKACB in stage I-II and stage III-IV diffuse-type gastric cancer. C, D Overall survival and progress-free survival of patients with GC based on PRKACB mRNA expression level using probe 202741_at (C) and 235780_at (D) from the kmplot database. E Expression levels of PRKACB in DGC across different T stages and N stages. Kaplan-Meier analysis showing the correlation between PRKACB expression levels and prognosis in overall (F) Stage I-II (G) and Stage III-IV (H) DGC patients. IGC: intestinal-type gastric cancer; DGC: diffuse-type gastric cancer; N: normal; T: tumor; *p < 0.05; ***p < 0.001; ns: not significant.
Low PRKACB expression elevates metastatic ability of DGC cells
We selected cell lines derived from normal gastric mucosal epithelium (GES-1), three cell lines derived from IGC (AGS, HGC-27, and NCI-N87), and three cell lines derived from DGC (KATO III, MKN-45, and SNU-668) for cultivation, and subsequently extracted total protein and RNA. Western blot and qPCR experiments revealed that the protein levels (Supplementary Fig. 2A) and mRNA levels (Supplementary Fig. 2B) of PRKACB in the three DGC cell lines were significantly lower than those in the three IGC cell lines and the normal gastric mucosal epithelial cell line. The two DGC cell lines with the most prominent low expression, SNU-668 and KATO III, were selected for subsequent cell experiments. Small interfering RNAs targeting PRKACB (scramble, siPRKACB#1, and siPRKACB#2) or PRKACB-overexpressing plasmids (Vector and OE) were transfected into SNU-668 and KATO III. RNA and protein were extracted 36 and 48 h later, respectively, and qPCR and Western blot experiments were conducted to detect PRKACB mRNA and protein expression levels to verify transfection efficiency. In the knockdown groups, PRKACB mRNA and protein levels in the siRNA#1 and siRNA#2 groups were significantly lower than those in the scramble group (Supplementary Fig. 2C, D); in the overexpression groups, PRKACB mRNA and protein levels in the OE group were significantly higher than those in the Vector group (Supplementary Fig. 2E, F). After knocking down or overexpressing PRKACB in SNU-668 cells and allowing the cells to cover the culture dish, scratch assays were performed. Photographs were taken at 0, 24, and 48 h at the same location in each culture dish using a light microscope, and Image J software analysis revealed that knocking down PRKACB significantly enhanced the migration ability of SNU-668 cells (Fig. 2A); overexpressing PRKACB significantly reduced the migration ability of SNU-668 cells (Fig. 2B). As KATO III cells are semi-suspended, their migration speed could not be accurately observed under the microscope, so scratch assays were only performed on SNU-668 cells. Transwell chambers were used for cell migration and invasion assays. Results showed that knocking down PRKACB in SNU-668 and KATO III significantly enhanced their migration and invasion abilities (Fig. 2C, D), whereas overexpressing PRKACB in SNU-668 and KATO III significantly reduced their migration and invasion abilities (Fig. 2E, F). To investigate whether PRKACB affects EMT and cell morphology, we performed co-immunofluorescence staining for E-cadherin and F-actin in PRKACB-knockdown SNU-668 cells. The results demonstrated that cells in the PRKACB knockdown group exhibited irregular morphology, became separated from each other, and showed a significant increase in pseudopodia formation. Meanwhile, E-cadherin expression at the cell periphery was markedly reduced and displayed a discontinuous, fragmented pattern, indicating loss of cell-cell adhesion and enhanced invasive capacity (Fig. 2G). Furthermore, we performed PRKACB knockdown or overexpression in SNU-668 and KATO III, respectively, and examined the expression levels of EMT-related proteins by Western blot analysis. The results confirmed that low PRKACB expression promotes EMT in gastric cancer cells, as evidenced by decreased expression of epithelial markers (E-cadherin) and increased expression of mesenchymal markers (N-cadherin, Vimentin) (Fig. 2H, I). These results indicate that knocking down PRKACB significantly enhances the motility of DGC cells and promotes EMT.

A, B After knocking down or overexpressing PRKACB in SNU-668 cells, a scratch assay was performed in a culture dish, and the statistical analysis of cell migration index was conducted using Image J software. Migration index: cell migration area/original scratch area. C–F After knocking down or overexpressing PRKACB in SNU-668 and KATO III, cell migration and invasion assays were performed in transwell chambers, and the number of cells was analyzed statistically using Image J software. G Co-immunofluorescence staining for E-cadherin and F-actin in PRKACB-knockdown SNU-668 cells. Scale bar, 25 µm. H, I After knocking down or overexpressing PRKACB in SNU-668 and KATO III, western blot was conducted to detect EMT markers. scr: scramble; OE: over expression; ns: not significant; *p < 0.05; **p < 0.01; ***p < 0.001.
In vivo experiments demonstrate that low PRKACB expression promotes metastasis in DGC
Stable PRKACB-knockdown KATO III cell lines were established by lentiviral transduction of shPRKACB followed by puromycin selection. To more accurately simulate the real situation of gastric cancer metastasis, we employed a mouse gastric cancer orthotopic implantation model. Stable transfected KATO III cells were first implanted subcutaneously in nude mice to form tumors. Equal-sized small tumor pieces were then implanted into the gastric wall of the nude mice. Eight weeks after confirming successful modeling, the mice were sacrificed, and a thorough exploration of the peritoneal cavity was conducted (Fig. 3A). The number and size of metastatic nodules in each mouse were recorded, revealing that the PRKACB knockdown group had more numerous and relatively larger metastatic nodules (Fig. 3B, C).

A Representative image of peritoneal metastasis in control and knockdown groups of mice. B Statistical graph showing the number of metastatic nodules in 10 mice per group for both control and knockdown groups. C Statistical distribution of the size of metastatic nodules in 10 mice per group for both control and knockdown groups. D Bioluminescence imaging (IVIS) was used to monitor intraperitoneal tumor metastasis. E Quantification of bioluminescence intensity in mice from control and knockdown groups. Data are presented as mean ± SD (n = 6). F Representative images of TUNEL assay and Ki-67 staining. G Percentage of apoptotic cells quantified based on TUNEL assay. Data are presented as mean ± SD (n = 6). H Quantification of Ki-67 positive cells. Data are presented as mean ± SD (n = 6). scr: scramble; *p < 0.05.
To provide direct temporal evidence of metastasis kinetics, we conducted dynamic observations using longitudinal bioluminescence imaging. We established a peritoneal metastasis model using the murine gastric cancer cell line YTN16 stably expressing luciferase in C57BL/6 mice. Dynamic bioluminescence imaging revealed that mice in the PRKACB knockdown group developed peritoneal metastases at an earlier time point compared to the control group. However, the rate of change in bioluminescence signal intensity showed no significant difference between the two groups, indicating that low PRKACB expression promotes the formation of metastatic foci but does not affect the proliferation rate of established metastases (Fig. 3D, E). To further investigate whether PRKACB affects proliferation and apoptosis within metastatic nodules, we harvested peritoneal metastatic nodules after sacrifice and performed immunohistochemical staining for Ki67 and TUNEL assay. No significant differences in Ki67 labeling index or TUNEL-positive cell percentage were observed between the PRKACB knockdown and control groups (Fig. 3F–H). We knocked down or overexpressed PRKACB in SNU-668 and KATO III, followed by colony formation and CCK8 assays. The results showed no significant changes in the proliferation abilities of SNU-668 and KATO III in the knockdown and overexpression groups compared to the control group (Supplementary Fig. 3A–F). Collectively, these findings demonstrate that PRKACB does not affect the proliferation or apoptosis of gastric cancer cells, but promotes the initiation of metastatic colonization.
PRKACB interacts with RhoA
Based on the molecular characteristics of PRKACB as a protein kinase catalytic subunit, we aimed to identify potential interacting substrate molecules through co-immunoprecipitation (Co-IP) and mass spectrometry. After Co-IP, the gel was silver-stained, and differential bands were excised for mass spectrometry analysis (Fig. 4A). Mass spectrometry analysis of PRKACB immunoprecipitates identified RhoA as a potential interacting protein (Fig. 4B). Considering the molecular function of RhoA, we hypothesized that RhoA might be an effector molecule promoting DGC metastasis due to low PRKACB expression. We first resolved the protein crystal structures of PRKACB and RhoA and found a good structural basis for their direct interaction (Fig. 4C). Next, endogenous Co-IP experiments in SNU-668 and KATO III confirmed their interaction (Fig. 4D). To further verify whether PRKACB can directly bind to RhoA, we performed a GST-pulldown assay with purified proteins in vitro and found that they can directly bind (Fig. 4E). A physiological interaction was observed between endogenous PRKACB and RhoA in SNU-668 cells by PLA (Fig. 4F). This signal is specific since knockdown of PRKACB dramatically reduced the PLA fluorescence intensity (Fig. 4G).

A SNU-668 cells were transfected with PRKACB overexpression plasmid, and 48 h later, the cells were lysed for immunoprecipitation. The immunoprecipitated proteins were separated by PAGE gel and visualized by silver staining. Differential bands were excised for mass spectrometry analysis. B Mass spectrometry spectrum identifying RhoA peptide. C Prediction of protein-protein interaction between PRKACB and RhoA. D Endogenous Co-IP experiments were performed using SNU-668 and KATO III. E GST-pulldown experiments were conducted using GST-RhoA purified protein and intracellularly transfected Flag-PRKACB. F Representative images of SNU-668 cells which were transfected as indicated and applied to in situ proximity ligation assay (PLA, red signal). Scale bar, 20 µm. G The PLA signal (red) in SNU-668 cells of each group was calculated, respectively. scr: scramble; **p < 0.01.
PRKACB influences the metastasis of DGC through phosphorylating RhoA
To investigate whether PRKACB can function through the phosphorylation of RhoA, we transfected small interfering RNA (scramble, siPRKACB#1, and siPRKACB#2) targeting PRKACB into SNU-668 and KATO III. Western blot analysis was used to detect the protein levels of PRKACB, Total-RhoA, Active-RhoA, p-RhoA, Total-FAK, and p-FAK. The results showed that after knocking down PRKACB, the total protein level of RhoA did not change significantly, while p-RhoA significantly decreased, Active-RhoA significantly increased, and the expression of p-FAK downstream of RhoA was upregulated (Fig. 5A). The ROCK1 activity was measured using the ROCK Activity Assay Kit, and it was found that ROCK1 activity was significantly upregulated in the PRKACB knockdown group (Fig. 5B). These results suggest that PRKACB affects the phosphorylation of RhoA, with low expression of PRKACB reducing the phosphorylation level of RhoA, thereby increasing the level of active RhoA and activating downstream ROCK1 and FAK signaling. To determine whether RhoA can be directly phosphorylated by PRKACB, we performed in vitro kinase assays using HA-tagged RhoA proteins to rule out potential steric interference artifacts from the GST tag. The results confirmed that the S188 site of RhoA can be directly phosphorylated by PRKACB (Fig. 5C). To fully characterize the PRKACB-RhoA phosphorylation kinetics, we performed dose-dependent and time-course experiments. The results demonstrated that PRKACB increased RhoA phosphorylation at S188 in a dose- and time-dependent manner (Supplementary Fig. 4A, B). Immunohistochemical staining of 154 DGC tissue samples showed a positive correlation between the expression of PRKACB and p-RhoA (p < 0.001, Spearman’s rho = 0.592) (Fig. 5D).

A After knocking down PRKACB in SNU-668 and KATO III, western blot was conducted to detect the protein levels of PRKACB, Total-RhoA, Active-RhoA, p-RhoA, Total-FAK, and p-FAK(S732). B After knocking down PRKACB in SNU-668 and KATO III, ROCK1 activity was measured using the ROCK Activity Assay Kit. C Phosphorylation of RhoA by PRKACB was assessed in vitro; the experiment was performed three independent times with similar results. D Representative images of high and low expression of PRKACB and p-RhoA in DGC tissues (left panel). The correlation between PRKACB and p-RhoA expression in DGC tissues was analyzed using Spearman’s method (right panel). E–H On the basis of knocking down endogenous RhoA expression in SNU-668 and KATO III, RhoA-S188D and RhoA-S188A mutant plasmids were transfected into the PRKACB knockdown group and the control group, respectively. Western blot was used to detect the protein levels of Total-RhoA, Total-FAK, and p-FAK(S732) (E). ROCK1 activity was measured using the ROCK Activity Assay Kit (F). Transwell assays were performed to evaluate the migration and invasion abilities of SNU-668 and KATO III (G, H). I, J RhoA inhibitor (Rhosin) was used to block the pro-metastatic function of Active-RhoA in a mouse gastric cancer orthotopic implantation model of PRKACB knockdown and control groups. Representative image of peritoneal metastasis in four groups (I). The number of metastatic nodules in each of the four groups was counted and compared for differences (J). scr: scramble; ns: not significant; *p < 0.05; **p < 0.01; ***p < 0.001.
To further clarify that PRKACB activates its downstream pathway by influencing the phosphorylation of the RhoA-S188 site, we first constructed plasmids of RhoA mutants that maintain phosphorylation (S188D) and dephosphorylation (S188A). Then, using small interfering RNA (siRhoA) to knock down the endogenous expression of RhoA in SNU-668 and KATO III, the two mutant plasmids were transfected into the PRKACB knockdown group and control group, respectively. The expression of p-FAK was detected by western blot, and ROCK1 activity was measured using the ROCK Activity Assay Kit. It was found that the activation of the downstream signaling of RhoA was only related to the phosphorylation state of RhoA and no longer related to the expression of PRKACB (Fig. 5E, F). Migration and invasion assays were performed on SNU-668 and KATO III treated with the four aforementioned methods. The results showed that cells with sustained RhoA phosphorylation exhibited weaker migration and invasion capabilities, whereas cells with sustained RhoA dephosphorylation exhibited significantly enhanced migration and invasion capabilities (Fig. 5G, H). These results indicate that the migration and invasion ability and pseudopodia formation of DGC cells are not related to the expression level of PRKACB but are solely related to the phosphorylation state of RhoA. The influence of PRKACB on the migration, invasion, and pseudopodia formation of DGC cells is achieved by affecting the phosphorylation of RhoA.
To better demonstrate that low PRKACB expression exerts its pro-metastatic function through RhoA, we treated PRKACB-knockdown SNU-668, KATO III, and MKN-45 cells with the specific RhoA inhibitor Rhosin and quantified the rescue effects using migration and invasion assays. The results showed that treatment with the RhoA inhibitor abolished the pro-metastatic effects induced by low PRKACB expression, further confirming the functional role of the PRKACB/RhoA axis in promoting metastasis (Supplementary Fig. 4C). Furthermore, to validate these findings in vivo, we used a RhoA inhibitor (Rhosin) to block the pro-metastatic function of Active-RhoA in a mouse gastric cancer orthotopic implantation model of PRKACB knockdown and control groups (Fig. 5I). The results showed that in the two PRKACB knockdown groups, the number of metastatic nodules in the DMSO group was significantly higher than in the Rhosin group; in the two DMSO groups, the number of metastatic nodules in the shPRKACB group was significantly higher than in the scr group; whereas, in the two Rhosin groups, there was no significant difference between the shPRKACB group and the scr group (Fig. 5J). These experimental results indicate that knocking down PRKACB can promote the metastasis of DGC, but this effect can be blocked by the RhoA inhibitor.
High-frequency mutations of RhoA in DGC weaken its interaction with PRKACB
To identify the specific region where RhoA binds to PRKACB, we constructed three truncated RhoA mutant plasmids and found through Co-IP experiments that, in addition to the full-length RhoA, a truncated mutant of RhoA containing amino acids 34–80 could also bind to PRKACB, indicating that the binding region is located within the peptide segment of amino acids 34–80 of RhoA (Fig. 6A). Using amber18 software, we performed molecular dynamics simulations on the protein-protein complexes obtained from molecular docking to predict potential interactions between amino acids at the RhoA and PRKACB binding interface (Fig. 6B). Based on the molecular dynamics simulation results of PRKACB-RhoA binding, we performed site-directed mutagenesis of key amino acid residues at the interaction interface. Co-IP experiments revealed that mutations at Y236, F240, and R257 in PRKACB resulted in weakened or abolished binding to RhoA, accompanied by reduced RhoA phosphorylation levels. Similarly, mutations at V38, N41, and L69 in RhoA attenuated its interaction with PRKACB and decreased RhoA phosphorylation. These experiments demonstrate that R257 in PRKACB and N41 in RhoA play critical roles in the PRKACB-RhoA interaction, and disruption of this binding leads to diminished RhoA phosphorylation (Fig. 6C, D).

A Three RhoA truncation mutants were constructed to locate the interaction region with PRKACB through Co-IP experiments. B The relative positions of amino acid residues at the key interface of PRKACB and RhoA binding, along with the potential interaction forces (yellow dashed lines represent hydrogen bonds), were simulated. C, D Site-directed mutagenesis of key amino acids of RhoA and PRKACB was performed based on the molecular dynamics simulation. Co-immunoprecipitation (Co-IP) experiments were conducted to investigate the effects of RhoA or PRKACB mutations on binding to each other and phosphorylation of RhoA. E–H Four point mutation plasmids, V38G, N41K, W58S, and L69R, were constructed and co-transfected with Flag-PRKACB into SNU-668 cells. Co-IP and western blot experiments were conducted to investigate the effects of RhoA mutations on binding to and phosphorylation by PRKACB in DGC (E). ROCK1 activity was measured using the ROCK Activity Assay Kit (F). Transwell assays were performed to assess the migration (G) and invasion (H) abilities of SNU-668 cells. EV empty vector, FL full length, WCL whole cell lysate, WT wild type, ns not significant. *p < 0.05; **p < 0.01; ***p < 0.001.
Through TCGA data analysis, we found that a high mutation rate of RhoA is specific to DGC (including signet ring cell carcinoma of the stomach) and is not commonly observed in IGC (Supplementary Fig. 5A). We hypothesized that, in addition to the specific low expression of PRKACB, RhoA mutations might also contribute to the higher metastatic potential of DGC compared to IGC. We first reviewed the literature on gene sequencing results of DGC and summarized the most common RhoA mutation sites and types in this cancer subtype (Supplementary Fig. 5B). Based on this information, we speculated that mutations such as V38G, N41K, W58S, and L69R in the RhoA protein might affect its interaction with PRKACB. We then constructed plasmids carrying these four point mutations and co-transfected them with Flag-PRKACB into SNU-668 cells. Using Co-IP experiments to verify the binding between PRKACB and these RhoA mutants, and labeling proteins with tag antibodies to eliminate the influence of endogenous RhoA, we found that RhoA-WT could bind to PRKACB; RhoA-V38G showed reduced binding to PRKACB; RhoA-W58S and RhoA-L69R had significantly reduced binding to PRKACB; and RhoA-N41K could not bind to PRKACB at all (Fig. 6E). Using the ROCK Activity Assay Kit to detect ROCK1 activity in these cell lines, we found that ROCK1 activity was significantly upregulated in cells transfected with the RhoA mutant plasmids (Fig. 6F). Further functional assays revealed that the migration and invasion abilities of cells transfected with the RhoA mutants were significantly enhanced, with the most pronounced increase observed in SNU-668 cells transfected with the RhoA-N41K mutant (Fig. 6G, H).
To determine whether the V38G and N41K mutations alter RhoA conformational dynamics independent of PRKACB, thereby modulating effector interactions, we constructed recombinant proteins of the two mutants and performed GST pull-down assays to assess their interactions with downstream effectors mDia1 and ROCK1. The results demonstrated that neither the V38G nor the N41K mutation affects the binding of RhoA to mDia1 or ROCK1. These findings indicate that the V38G and N41K mutations do not alter RhoA conformational dynamics in a manner that would modulate its interactions with downstream effectors independent of PRKACB (Supplementary Fig. 5C, D). These results suggest that certain RhoA mutations in DGC can affect its binding to PRKACB, leading to decreased phosphorylation levels of RhoA, which further promotes the metastasis of DGC.
Discussion
In this study, we identified PRKACB as a novel tumor suppressor in DGC that functions through direct phosphorylation of RhoA at S188. Compared with IGC, DGC is characterized not only by the distinctive RHOA gene mutations but also by the characteristic low expression of PRKACB. The poor prognosis of DGC with low PRKACB expression is due to the decreased phosphorylation level of RhoA and the increased level of active RhoA, which subsequently promotes metastasis of DGC. Furthermore, certain RhoA mutations can affect its binding to PRKACB, further reducing RhoA phosphorylation level and making DGC more prone to metastasis (Fig. 7).

Compared with IGC, DGC exhibits significantly downregulated PRKACB expression, leading to reduced RhoA phosphorylation, accumulation of active RhoA, and consequent promotion of tumor metastasis. Furthermore, RhoA mutations frequently observed in DGC impair RhoA -PRKACB binding, further diminishing RhoA phosphorylation and exacerbating the metastatic phenotype.
Metastasis is the main cause of death in patients with malignant tumors [23]. The detachment of tumor cells from the primary site involves EMT, which increases cell motility and invasiveness [24, 25]. Collective cell migration is commonly observed at the invasive frontiers of infiltrative cancers (such as DGC), where these leading invading cells pave the way for many followers [26, 27]. In this process, the actin-dependent protrusions of tumor cell pseudopodia are one of the key factors in tumor cell migration and the eventual formation of metastases [28, 29]. In this study, we found that PRKACB can affect the migration and invasion of DGC cells. Further immunofluorescence staining of the cytoskeleton revealed that DGC cells formed more pseudopodia after PRKACB knockdown, which partially explains why these cells are more prone to migration and invasion. Importantly, we demonstrated that PRKACB knockdown not only promotes pseudopodia formation but also induces EMT, as evidenced by decreased E-cadherin expression and increased mesenchymal markers, consistent with recent findings that RhoA activation promotes EMT through regulation of cell-cell adhesion [30]. Furthermore, our bioluminescence imaging experiments provided direct evidence that low PRKACB expression accelerates the onset of metastatic colonization rather than affecting the proliferation rate of established metastases, highlighting the critical role of the PRKACB-RhoA axis in the early steps of metastasis.
DGC is characterized by a lack of cell cohesion and a highly invasive growth pattern. A key point that has influenced our understanding of DGC is the discovery that hereditary DGC carries mutations in the CDH1 gene, which encodes E-cadherin [31]. In addition to hereditary DGC, the sporadic form is also often associated with the loss of E-cadherin, possibly due to somatic mutations or promoter hypermethylation. The characteristic RHOA gene mutations discovered in DGC further expand our understanding of this cancer subtype.
Rho proteins, members of the small GTPase Ras superfamily, play key roles in many cellular biological processes. When RhoA is phosphorylated, it tightly binds to GDI and cannot convert to the active RhoA-GTP state, thereby inhibiting RhoA signaling activity [32, 33]. Several kinases have been reported to phosphorylate RhoA at S188, including PKA and PKG [34]. However, the specific kinase responsible for RhoA phosphorylation in gastric cancer has remained unclear. Our findings demonstrate that PRKACB, as the catalytic subunit of PKA, directly phosphorylates RhoA at S188 in a dose- and time-dependent manner, establishing a novel regulatory mechanism in DGC. Zhang et al. created the first RHOA-driven DGC organoid and mouse models, demonstrating that RHOAY42C/+ functions as an oncogene by activating downstream ROCK and FAK signaling [35].
ROCK is a serine-threonine kinase activated upon binding to GTP-bound RhoA, and serves as a critical regulator of the actin cytoskeleton [36,37,38,39,40]. FAK is a non-receptor protein tyrosine kinase that drives multiple pro-tumor signaling pathways [41,42,43,44]. Through structural analysis of the PRKACB-RhoA interaction interface, we found that common DGC-associated RhoA mutations, particularly those affecting residues within the 34–80 amino acid region, exhibited varying degrees of impaired binding to PRKACB, with the N41K mutation completely abolishing this interaction. Notably, these mutations did not affect RhoA’s ability to engage downstream effectors such as mDia1 and ROCK1, indicating that they selectively disrupt the negative regulatory input from PRKACB while preserving oncogenic signaling capacity. This selective uncoupling provides a molecular explanation for why RhoA-mutant DGCs exhibit enhanced metastatic potential: the mutant RhoA escapes PRKACB-mediated phosphorylation and inactivation, resulting in constitutively elevated RhoA-ROCK-FAK signaling that drives tumor cell invasion and dissemination.
Our findings have important therapeutic implications. PRKACB expression may serve as a potential biomarker for predicting responsiveness to RhoA inhibitor therapy. Furthermore, therapeutic strategies targeting low PRKACB expression represent an important direction for future research. If low PRKACB expression results from epigenetic silencing, such as promoter hypermethylation, demethylating agents may be considered as a potential therapeutic approach to restore gene expression [45]. Alternatively, exploiting synthetic lethality by identifying genes that form synthetic lethal relationships with PRKACB through genome-wide CRISPR/Cas9 screening could provide novel therapeutic targets for cancer cells with low PRKACB expression [46]. From a nucleic acid-based therapeutic perspective, delivery of a functional PRKACB gene via viral vectors (e.g., adeno-associated virus) or non-viral vectors, as well as direct mRNA delivery for translation into functional protein, represents promising strategies [47].
Several limitations of this study should be acknowledged. First, the heterogeneity of human DGC suggests that additional mechanisms may contribute to disease progression in different patient subpopulations. Second, the mechanisms underlying PRKACB downregulation in DGC, whether through genetic, epigenetic, or post-transcriptional regulation, remain to be fully elucidated. Third, while our RhoA inhibitor rescue experiments provide proof-of-concept evidence, the clinical translation of this strategy requires further optimization. In conclusion, our study establishes the PRKACB-RhoA-ROCK-FAK axis as a key pathway driving DGC metastasis and provides potential therapeutic strategies for this aggressive disease.
Data availability
All data required to assess the study’s conclusions are included in this article and/or the Supplementary Materials. Any additional information relevant to the work is available from the authors upon reasonable request.
References
Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global Cancer Statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71:209–49.
Zheng R, Zhang S, Zeng H, Wang S, Sun K, Chen R, et al. Cancer incidence and mortality in China, 2016. J Natl Cancer Center. 2022.
Joshi SS, Badgwell BD. Current treatment and recent progress in gastric cancer. CA Cancer J Clin. 2021;71:264–79.
Smyth EC, Nilsson M, Grabsch HI, van Grieken NCT, Lordick F. Gastric cancer. Lancet. 2020;396:635–48.
Iyer P, Moslim M, Farma JM, Denlinger CS. Diffuse gastric cancer: histologic, molecular, and genetic basis of disease. Transl Gastroenterol Hepatol. 2020;5:52.
Lauren P. The two histological main types of gastric carcinoma: diffuse and so-called intestinal-type carcinoma. Attempt a Histo-Clin Classification Acta Pathol Microbiol Scand. 1965;64:31–49.
Petrelli F, Berenato R, Turati L, Mennitto A, Steccanella F, Caporale M, et al. Prognostic value of diffuse versus intestinal histotype in patients with gastric cancer: a systematic review and meta-analysis. J Gastrointest Oncol. 2017;8:148–63.
Luo W, Fedda F, Lynch P, Tan D. CDH1 gene and hereditary diffuse gastric cancer syndrome: molecular and histological alterations and implications for diagnosis and treatment. Front Pharmacol. 2018;9:1421.
Graziano F, Humar B, Guilford P. The role of the E-cadherin gene (CDH1) in diffuse gastric cancer susceptibility: from the laboratory to clinical practice. Ann Oncol Off J Eur Soc Med Oncol. 2003;14:1705–13.
Cancer Genome Atlas Research N. Comprehensive molecular characterization of gastric adenocarcinoma. Nature. 2014;513:202–9.
Kakiuchi M, Nishizawa T, Ueda H, Gotoh K, Tanaka A, Hayashi A, et al. Recurrent gain-of-function mutations of RHOA in diffuse-type gastric carcinoma. Nat Genet. 2014;46:583–7.
Wang K, Yuen ST, Xu J, Lee SP, Yan HH, Shi ST, et al. Whole-genome sequencing and comprehensive molecular profiling identify new driver mutations in gastric cancer. Nat Genet. 2014;46:573–82.
Ridley AJ, Hall A. The small GTP-binding protein rho regulates the assembly of focal adhesions and actin stress fibers in response to growth factors. Cell. 1992;70:389–99.
Tkachenko E, Sabouri-Ghomi M, Pertz O, Kim C, Gutierrez E, Machacek M, et al. Protein kinase A governs a RhoA-RhoGDI protrusion-retraction pacemaker in migrating cells. Nat Cell Biol. 2011;13:660–7.
Rolli-Derkinderen M, Sauzeau V, Boyer L, Lemichez E, Baron C, Henrion D, et al. Phosphorylation of serine 188 protects RhoA from ubiquitin/proteasome-mediated degradation in vascular smooth muscle cells. Circ Res. 2005;96:1152–60.
Li H, Fu X, Zhao J, Li C, Li L, Xia P. et al. EXOC4 Promotes Diffuse-Type Gastric Cancer Metastasis via Activating FAK Signal. Mol Cancer Res. 2022;20:1021–34.
Rylatt DB, Aitken A, Bilham T, Condon GD, Embi N, Cohen P. Glycogen synthase from rabbit skeletal muscle. Amino acid sequence at the sites phosphorylated by glycogen synthase kinase-3, and extension of the N-terminal sequence containing the site phosphorylated by phosphorylase kinase. Eur J Biochem. 1980;107:529–37.
Embi N, Rylatt DB, Cohen P. Glycogen synthase kinase-3 from rabbit skeletal muscle. Separation from cyclic-AMP-dependent protein kinase and phosphorylase kinase. Eur J Biochem. 1980;107:519–27.
Skalhegg BS, Tasken K. Specificity in the cAMP/PKA signaling pathway. Differential expression, regulation, and subcellular localization of subunits of PKA. Front Biosci. 2000;5:D678–93.
Chen Y, Gao Y, Tian Y, Tian DL. PRKACB is downregulated in non-small cell lung cancer and exogenous PRKACB inhibits proliferation and invasion of LTEP-A2 cells. Oncol Lett. 2013;5:1803–8.
Chen TC, Hinton DR, Zidovetzki R, Hofman FM. Up-regulation of the cAMP/PKA pathway inhibits proliferation, induces differentiation, and leads to apoptosis in malignant gliomas. Lab Invest. 1998;78:165–74.
van Oirschot BA, Stahl M, Lens SM, Medema RH. Protein kinase A regulates expression of p27(kip1) and cyclin D3 to suppress proliferation of leukemic T cell lines. J Biol Chem. 2001;276:33854–60.
Talmadge JE, Fidler IJ. AACR centennial series: the biology of cancer metastasis: historical perspective. Cancer Res. 2010;70:5649–69.
Kang Y, Pantel K. Tumor cell dissemination: emerging biological insights from animal models and cancer patients. Cancer Cell. 2013;23:573–81.
Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Investig. 2009;119:1420–8.
Chung YC, Wei WC, Hung CN, Kuo JF, Hsu CP, Chang KJ, et al. Rab11 collaborates E-cadherin to promote collective cell migration and indicates a poor prognosis in colorectal carcinoma. Eur J Clin Invest. 2016;46:1002–11.
Cheung KJ, Gabrielson E, Werb Z, Ewald AJ. Collective invasion in breast cancer requires a conserved basal epithelial program. Cell. 2013;155:1639–51.
Chen WT. Proteolytic activity of specialized surface protrusions formed at rosette contact sites of transformed cells. J Exp Zool. 1989;251:167–85.
Guirguis R, Margulies I, Taraboletti G, Schiffmann E, Liotta L. Cytokine-induced pseudopodial protrusion is coupled to tumour cell migration. Nature. 1987;329:261–3.
Pastushenko I, Blanpain C. EMT transition states during tumor progression and metastasis. Trends Cell Biol. 2019;29:212–26.
Guilford P, Hopkins J, Harraway J, McLeod M, McLeod N, Harawira P, et al. E-cadherin germline mutations in familial gastric cancer. Nature. 1998;392:402–5.
Nam S, Kim JH, Lee DH. RHOA in gastric cancer: functional roles and therapeutic potential. Front Genet. 2019;10:438.
Chang KK, Cho SJ, Yoon C, Lee JH, Park DJ, Yoon SS. Increased RhoA activity predicts worse overall survival in patients undergoing surgical resection for lauren diffuse-type gastric adenocarcinoma. Ann Surg Oncol. 2016;23:4238–46.
Olson MF. Rho GTPases, their post-translational modifications, disease-associated mutations and pharmacological inhibitors. Small GTPases. 2018;9:203–15.
Zhang H, Schaefer A, Wang Y, Hodge RG, Blake DR, Diehl JN, et al. Gain-of-function RHOA mutations promote focal adhesion kinase activation and dependency in diffuse gastric cancer. Cancer Discov. 2020;10:288–305.
Maekawa M, Ishizaki T, Boku S, Watanabe N, Fujita A, Iwamatsu A, et al. Signaling from Rho to the actin cytoskeleton through protein kinases ROCK and LIM-kinase. Science. 1999;285:895–8.
Ohashi K, Nagata K, Maekawa M, Ishizaki T, Narumiya S, Mizuno K. Rho-associated kinase ROCK activates LIM-kinase 1 by phosphorylation at threonine 508 within the activation loop. J Biol Chem. 2000;275:3577–82.
Sebbagh M, Renvoize C, Hamelin J, Riche N, Bertoglio J, Breard J. Caspase-3-mediated cleavage of ROCK I induces MLC phosphorylation and apoptotic membrane blebbing. Nat Cell Biol. 2001;3:346–52.
Sumi T, Matsumoto K, Nakamura T. Specific activation of LIM kinase 2 via phosphorylation of threonine 505 by ROCK, a Rho-dependent protein kinase. J Biol Chem. 2001;276:670–6.
Van Eyk JE, Arrell DK, Foster DB, Strauss JD, Heinonen TY, Furmaniak-Kazmierczak E, et al. Different molecular mechanisms for Rho family GTPase-dependent, Ca2+-independent contraction of smooth muscle. J Biol Chem. 1998;273:23433–9.
McLean GW, Komiyama NH, Serrels B, Asano H, Reynolds L, Conti F, et al. Specific deletion of focal adhesion kinase suppresses tumor formation and blocks malignant progression. Genes Dev. 2004;18:2998–3003.
Parsons JT, Slack-Davis J, Tilghman R, Roberts WG. Focal adhesion kinase: targeting adhesion signaling pathways for therapeutic intervention. Clin Cancer Res. 2008;14:627–32.
Schultze A, Fiedler W. Therapeutic potential and limitations of new FAK inhibitors in the treatment of cancer. Expert Opin Investig Drugs. 2010;19:777–88.
Zhao J, Guan JL. Signal transduction by focal adhesion kinase in cancer. Cancer Metastasis Rev. 2009;28:35–49.
Christodoulidis G, Koumarelas KE, Kouliou MN, Thodou E, Samara M. Gastric cancer in the era of epigenetics. Int J Mol Sci. 2024;25.
Wu Y, Wang Y, Wang Y, Qiu H, Yuan X, Xiong H, et al. Exploring synthetic lethality in cancer therapy: CRISPR-Cas9 technology offers new hope. Biochim Biophys Acta Rev Cancer. 2025;1880:189370.
Magoola M, Niazi SK. Current progress and future perspectives of RNA-based cancer vaccines: a 2025 update. Cancers. 2025;17.
Acknowledgements
We thank the pathologists in Zhongshan Hospital for their work of tissue microarrays construction, IHC staining, and evaluation of tissue samples in our study. This work was supported by grants from the National Natural Science Foundation of China (Grant Nos. 82002527) and the Scientific Research Foundation of Zhongshan Hospital [Grant Number: ZY2024-003].
Author information
Authors and Affiliations
Contributions
Haojie Li, Yuanyuan Ruan, and Jie Sun designed this study. Jie Sun and Junjie Zhao performed most of the experiments and interpreted the data. Chenyu Tian, Chengbo Ji, and Jingdong Liu collected and analyzed the clinical data. Haojie Li and Jie Sun prepared the manuscript. Xu Yang participated in manuscript revision and additional experiments; Xuefei Wang reviewed and edited the manuscript. Sachiyo Nomura provided the YTN-16 cells. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interest.
Ethics approval
Tissue samples and clinicopathological data were obtained from Zhongshan Hospital, Fudan University. Written informed consent from each patient was achieved. The research was approved by the Research Ethics Committee of Fudan University and was performed in accordance with the ethical standards laid down in the 1964 Declaration of Helsinki and its later amendments.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by Professor Stephen Tait
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sun, J., Zhao, J., Yang, X. et al. Loss of PRKACB facilitates metastasis of diffuse-type gastric cancer through RhoA signaling activation. Cell Death Dis 17, 281 (2026). https://doi.org/10.1038/s41419-026-08553-z
Received:
Revised:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41419-026-08553-z
