
Abstract
beta1-adrenergic receptor (β1-AR) belongs to G protein-coupled receptors, regulating cardiac physiological and pathological process through complex signaling pathways. Physiologically, the activation of β1-AR produces positive chronotropic, positive inotropic and positive dromotropic effects in the heart. However, excessive or sustained activation of β1-AR can cause myocardial injury, arrhythmias, and heart failure. The β1-AR in the heart exhibits tissue-specific distribution patterns and subcellular localization features adapted to its function within cardiomyocytes. Upon ligand binding, the β1-AR undergoes conformational changes and transmits signaling through G protein-dependent pathways (β1-AR/Gs and β1-AR/Gi) as well as a G protein-independent pathway (β1-AR/β-arrestin) to regulate cardiac activity. Subsequently, the β1-AR can either dissociate from G protein to undergo desensitization and terminate signal transduction, or it can be endocytosed into the cell, transported to the lysosome to be degraded, or returned to the plasma membrane to continue its function. Additionally, it has been found that β1-AR can cause or exacerbate heart disease when abnormal changes occur in its distribution density, localization, and mediated downstream signaling pathways. Therefore, β1-AR represents an important pharmacotherapeutic target for the treatment of cardiac diseases. Among the relevant therapeutic agents, β1-AR blockers designed specifically against β1-AR have evolved to the third generation. This review comprehensively analyzes β1-AR from perspectives including its research history, expression, and distribution in the heart, protein structure, signaling pathways, and associations with cardiac diseases.
Similar content being viewed by others


Facts
-
Under physiological conditions, β1-AR is a core protein that regulates cardiac function. In disease states, the expression and function of β1-AR undergo dramatic changes. In chronic heart failure, the expression level of β1-AR decreases from the normal proportion of 80% to approximately 60%. One of the reasons is the enhanced degradation pathway of the receptor.
-
The signaling pathways of β1-AR include G protein-dependent and G protein-independent signaling pathways. The G protein-dependent pathways consist of the β1-AR/Gs pathway and the β1-AR/Gi pathway. The G protein-independent signaling pathway is predominantly the β1-AR/β-arrestin signaling pathway.
-
β1-AR blockers can improve cardiac function by acting on the sympathetic nervous system to inhibit β1-AR-mediated signaling pathways.
Open Questions
-
In cardiac diseases, what are the impacts of alterations in the expression level and distribution of β1-AR on the heart?
-
What are the molecular mechanisms underlying the therapeutic targeting of β1-AR in cardiac diseases?
-
What are the improved strategies for next-generation β1-AR blockers in the treatment of heart diseases?
Introduction
As a vital organ of the human body, the heart continuously pumps blood, ensuring normal physiological activities. In the intricate regulation of cardiac activities, the function of the sympathetic nerve is of great significance. Excitation of the sympathetic nervous system can release norepinephrine (NE). Subsequently, NE activates the beta-adrenergic receptors (β-ARs) in cardiomyocytes, inducing excitation-contraction coupling in the heart [1]. The heart expresses three types of β-ARs, specifically the beta1-adrenergic receptor (β1-AR), the beta2-adrenergic receptor (β2-AR), and the beta3-adrenergic receptor (β3-AR) [2]. Among these, up to 80% are β1-ARs [3]. As the most abundant subtype in the heart, β1-ARs are essential for maintaining normal cardiac function.
The signaling pathways of β1-AR are complex and diverse. It regulates cardiac function through G protein-dependent pathways (β1-AR/Gs and β1-AR/Gi) and the non-G-protein pathway (β1-AR/β-arrestin). Given the pivotal role of β1-AR in cardiac function regulation, it has been used as a well-established therapeutic target for cardiovascular diseases. β1-AR blockers competitively bind to the ligand-binding sites of β1-AR, antagonizing NE. Due to the lack of intrinsic activity, β1-AR blockers prevent the signal transduction of β1-AR when bound to the ligand, decreasing the response of the heart to sympathetic stimulation, which in turn leads to the therapeutic effects in cardiovascular diseases.
In this review, we summarize the research history of β1-AR, with a particular focus on elucidating its molecular structure and signaling pathways. We delve into the roles played by β1-AR in the pathogenesis of various cardiac diseases. Moreover, we discuss the current status of the application and research of β1-AR blockers. This review aims to offer a theoretical foundation for the development of therapeutic strategies for cardiac diseases.
The discovery process of β1-AR
The concept of “receptor” first appeared in the early 20th century. In 1905, John Newport Langley analyzed the effects of nicotine and curare on the contraction of striated muscle, discovering that these two drugs acted on the “accessory substance” of the cell membrane to transmit stimuli that determined the biological effects of the cells [4]. In 1908, Paul Ehrlich referred to the special chemical groups in the protoplasm that interacted with drugs as “receptors” [5]. In 1913, Henry Hallett Dale discovered the interaction of epinephrine and ergotoxin in cats with destroyed spinal cords. Epinephrine alone elevates coronary blood pressure, whereas its coadministration with ergotoxin reduces coronary blood pressure. At the time, however, Henry Hallett Dale did not analyze why this phenomenon occurred from a receptor perspective [6]. It was not until 1948 that Raymond Parker Ahlquist first confirmed evidence for the existence of adrenergic receptors and categorized them into alpha and beta types [7]. Subsequently, James Whyte Black developed the first clinically applicable β-AR blockers based on this theory in 1962: propranolol [8]. In 1967, Anthony M. Lands and his colleagues subdivided β-AR into two subtypes, β1-AR and β2-AR. They also elucidated the differences in the actions of the two subtypes. β1-AR is mainly related to cardiac function and manifests itself in enhanced myocardial contractility, while β2-AR is primarily associated with bronchodilatation and vasodilatation [9]. In 1974, Bo Ablad proposed the coexistence of β1-AR and β2-AR subtypes in the human heart when analyzing the effects of selective and nonselective β1-AR antagonists on different catecholamines [10]. In 1987, researchers clarified that the human β1-AR cDNA is composed of 2400 base pairs through cDNA libraries and named the cDNA of β1-AR as ADRB1 [11]. In 1990, it was further determined that the human β1-AR gene was localized on 10q24-q26 [12]. Although research on β1-AR started early, it was not until 2008 that researchers first analyzed the protein structure of β1-AR in turkeys due to the challenges posed by the high activity and low stability of the receptor itself. This is the first protein structure of β1-AR [13]. In 2021, researchers resolved the protein structure of the human β1-AR [14]. The above research efforts have continued to refine the understanding of β1-AR and laid the foundation for β1-AR research.
Expression and distribution of β1-AR
The expression and distribution patterns of β1-AR in the heart
In the physiological state, β1-AR accounts for approximately 80% of the total cardiac β-AR [3]. Radioligand binding assays [15, 16] and in vivo positron emission tomography experiments [17] have confirmed that the β1-AR in the human heart is relatively uniformly distributed in the myocardial tissue. Despite the relatively homogeneous distribution of β-AR, the distribution of the subtypes varies. The distribution density of β1-AR in various regions of the heart, from highest to lowest, is as follows: sinoatrial node, ventricles, and atria [3, 18]. Studies have found that the density of β1-AR in the sinoatrial node is 4.2 times higher than that in the atrial myocardium [19]. The differential expression of β1-AR between the sinoatrial node and the atrium demonstrates its special role in regulating heart rate and rhythm. Approximately 60% of the β-AR in both the right atrial appendage and left ventricle is β1-AR [20]. Enrichment of β1-AR in specific regions allows the heart to respond more rapidly to the sympathetic nervous system, resulting in positive chronotropic (accelerated heart rate), positive inotropic (enhanced myocardial contraction), and positive dromotropic (accelerated conduction velocity) effects.
The expression level of β1-AR is not constant, but is adjusted according to the state of the heart. Analysis of heart tissue from young and aged rats revealed that the mRNA levels of β1-AR decreased with age [21] (Table 1). A study on cardiac β1-AR changes revealed that β1-AR decreased more significantly with age in women than in men. A curvilinear relationship between age and receptor density stabilized around age 40 in women, whereas there was no statistically significant difference in the β1-AR density in the hearts of men among different age groups. This suggests that age and estrogen may be related to the amount of β1-AR expression in the heart [22] (Table 1). Additionally, a significant reduction in the number of β1-AR has been observed in various cardiac diseases, including dilated cardiomyopathy, aortic valve disease, ischemic cardiomyopathy, mitral valve disease, and tetralogy of Fallot [23]. Radiographic autoradiography showed that the density ratio of β1-AR between endocardium and epicardium was 1.0 ± 0.2 in the normal heart but decreased to 0.46 ± 0.09 and 0.52 ± 0.11 in patients with ischemic and idiopathic dilated cardiomyopathies, respectively. The change in the above ratio resulted from the fact that β1-AR in the epicardium remained relatively stable while β1-AR in the endocardium was significantly reduced. This suggests that β1-AR exhibits significant heterogeneity in transmural distribution in cardiac disease [24].
Heart failure is the end stage of many cardiovascular diseases. The percentage of β1-AR in cardiac tissues of heart failure patients can be reduced from 80% to 60% of normal [25, 26]. To elucidate the reasons for the reduced β1-AR expression in heart failure, existing studies have provided explanations in terms of both gene transcription and receptor degradation (Fig. 1).

Reduced gene transcription capacity and enhanced protein degradation both lead to the downregulation of β1-AR expression. (Created with BioRender.com).
In myocardial tissues of heart failure patients, both messenger ribonucleic acid (mRNA) and protein contents of β1-AR were reduced. Among them, the mRNA content of β1-AR was reduced by about 50% [27] (Table 1). Protein expression of β1-AR decreased from 73 ± 6.1 fmol/mg in normal heart samples to 35 ± 6.7 fmol/mg in heart failure samples [28] (Table 1). This result suggests that the gene transcription capacity of the β1-AR is reduced in heart failure, leading to decreased receptor synthesis [28]. However, it has also been pointed out that the decrease in β1-AR expression in heart failure is not due to a decrease in transcriptional capacity, leading to reduced receptor expression. It is caused by decreased stability of β1-AR mRNA, making it more susceptible to degradation, ultimately resulting in a reduction in β1-AR expression [3].
Degradation of the receptor is also an important factor contributing to the reduction of β1-AR expression. It was found that β1-AR exposed to the membrane surface of cardiomyocytes can be cleaved by elastase. Elastase is an inflammatory protease released by activated neutrophils that accumulates at sites of cardiac injury or inflammatio,n thereby hydrolyzing β1-AR [29, 30]. In addition to elastase, a disintegrin and metalloproteinase 17 (ADAM17) reduces the amount of β1-AR by cleaving the N-terminal specific position of β1-AR [31, 32]. ADAM17, a redox-sensitive enzyme, shows increased expression in the heart tissue of myocarditis and dilated cardiomyopathy [33]. Trypsin can also cleave β1-AR in cells. In Chinese hamster ovary (CHO)-Pro5 cells, the full-length β1-AR NH2-terminus is cleaved specifically at R31 ↓ L32. In cardiomyocytes, trypsin cleaves β1-AR, resulting in the formation of ~40-kDa NH2-terminal and ~30-kDa COOH-terminal fragments [34]. Moreover, G protein-coupled receptor kinases (GRKs) in the heart activate the endocytosis mechanism by phosphorylating β1-AR. With the assistance of β-arrestin, the receptor is endocytosed into the cytosol and enters the lysosome for degradation. The endocytosis of β1-AR is a complex process, which we will describe in the context of the signaling pathway of β1-AR in this paper. Analysis of left ventricular myocardial samples from patients with heart failure revealed significantly increased expression of GRK2 and GRK5. This suggests increased endocytosis of β1-AR in heart failure [35]. Recent studies have shown that lysosomal leucine aminopeptidase (LyLAP) is highly expressed in pancreatic ductal adenocarcinoma (PDA) cells with active endocytosis. Through hydrolysis of hydrophobic peptides, LyLAP preserves lysosomal membrane integrity essential for membrane protein catabolism. When LyLAP is deficient, the hydrophobic peptide segments of the undegraded membrane proteins disrupt the lysosomal membrane, leading to lysosomal dysfunction and subsequently triggering cell death [36]. During chronic heart failure, does increased LyLAP activity in cardiomyocyte lysosomes promote enhanced β1-AR degradation? This question remains to be further investigated. Furthermore, although microtubule depolymerization also reduces cell-surface β1-AR levels, its effect is comparatively modest [37].
It can be seen that the downregulation of β1-AR in heart failure results from the convergence of multiple pathological factors. This β1-AR suppression represents a compensatory protective mechanism to mitigate receptor-mediated cardiotoxicity. While this adaptive response may confer transient cardioprotection, it ultimately heralds a more severe disease trajectory.
The differential distribution of β1-AR in cardiomyocytes
β1-AR, as a transmembrane receptor, exhibits not only region-specific distribution patterns across cardiac compartments but also distinct spatial organization within cardiomyocyte membrane microdomains (Fig. 2).

The distribution of β1-AR in the heart is mostly concentrated around the sinus node. In cardiomyocytes, β1-ARs localize to structures with double-membrane architectures, including T-tubules, SR, Golgi apparatus, and the nuclear envelope. (Created with BioRender.com).
β1-AR is widely found in the cytoplasmic membrane and “T-tubular network” [38]. In the cytoplasmic membrane, β1-AR is concentrated in “non-caveolar cell surface membranes and internal membranes”, while only a very small amount of β1-AR is found in “caveolae and lipid rafts” [39, 40]. The “T-tubular network” forms specialized membrane contacts between the sarcolemma and sarcoplasmic reticulum (SR), creating microdomains where β1-AR orchestrates rapid transduction of sympathetic nervous signals to the cardiomyocyte.
Beyond its plasmalemmal localization, β1-AR exhibits distinct membrane compartmentalization across multiple intracellular organelles. Research identifies that there is functional β1-AR within the SR. Activation of these SR-localized β1-AR promotes cardiac relaxation via protein kinase A (PKA) dependent phosphorylation of phospholamban (PLN/PLB) [41]. The membrane-impermeant β1-AR antagonist sotalol selectively blocks plasmalemmal β1-AR signaling, attenuating myocardial contractility without affecting PLN phosphorylation or cardiac relaxation. The membrane-permeable β1-AR antagonist propranolol concomitantly inhibits both plasmalemmal and SR-localized β1-AR populations, thereby suppressing the phosphorylation of PLN and impairing both cardiac contractility and relaxation [42]. In a mouse model of isoproterenol (ISO) induced heart failure, the association of β1-AR with calcium pumps on the SR was enhanced when β1-AR was decreased on the cytoplasmic membrane. This phenomenon may result from either: β1-AR endocytosis followed by relocation to the SR or aggregation of newly synthesized β1-AR in the SR [43]. In addition to the SR, β1-AR is also present in the Golgi apparatus, where it can likewise be activated. Lipophilic ligands, such as dobutamine, passively diffuse across the plasma membrane to activate Golgi-localized β1-AR. Hydrophilic ligands, such as norepinephrine, rely on the organic cation transporter 3 (OCT3) located on the cell membrane to activate β1-AR in the Golgi apparatus [44]. Furthermore, Golgi-localized β1-AR activates the process of phosphatidylinositol hydrolysis via phospholipase C (PLC) and promote cardiac hypertrophy. Consequently, blocking OCT3 to restrict norepinephrine entry into cells or blocking β1-AR in the Golgi apparatus may serve as a prophylactic measure for endogenous ligand-dependent cardiomyocyte hypertrophy [45].
In addition to organelles, studies have also analyzed enriched cell nuclei. The results demonstrate the presence of β1-AR on the nuclear envelope of cardiomyocytes [46], where it exhibits biological functionality. ISO treatment activates adenylate cyclase (AC) in enriched nuclear preparations from rat hearts, with dose-dependent augmentation of cyclic adenosine monophosphate (cAMP) production [47]. In another study, it was found that treatment of isolated nuclei from rat ventricular myocytes with ISO increased the synthesis of 18S ribosomal RNA (18S rRNA). 18S rRNA is associated with protein synthesis. Meanwhile, the mRNA expression of nuclear factor κB (NFκB) was reduced. NFκB is implicated in cell proliferation and other processes [48]. This suggests that the β1-AR of the cell nuclear membrane may be directly involved in gene regulatory processes.
In summary, the subcellular localization of β1-AR in cardiomyocytes is not merely a static spatial distribution but rather dynamically engages in complex cellular signaling networks. Selective inhibition of β1-AR signaling in specific compartments can enable precise modulation of cardiac function.
Protein molecular structure of β1-AR
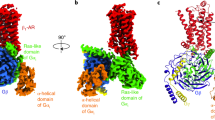
The protein structure of β1-AR includes an extracellular NH2-terminal and intracellular COOH-terminal, seven-transmembrane helices (TM1-TM7), three extracellular loops (ECL1-3), and three intracellular loops (ICL1-3). Among them, the NH2-terminal is associated with ligand recognitio,n and the COOH-terminal is involved in downstream signaling pathways. ECL2 is the longest and most diverse loop [49]. The amino acid residues in ECL2 can affect ligand selectivity and binding affinity by directly interacting with the ligand. ECL2 is not only involved in ligand binding, but also stabilizes the ligand-binding pocket through two disulfide bonds and a sodium ion [13]. ECL3 is involved in the regulation of β1-AR conformational transitions and ligand recognition [50]. Each ICL contains positively charged amino acid residues that bind to negatively charged G proteins. ICL2 and ICL3 are crucial sites for binding G proteins. Specifically, ICL2 mainly enhances the binding between the receptor and G proteins [51]. ICL2 plays a pivotal “switch” role in the activation of β1-AR through the formation of a short α-helix parallel to the membrane surface that interacts with the highly conserved Asp1383.49Arg1393.50Tyr1403.51 (DRY) motif in the third transmembrane helix [13, 52]. Moreover, ICL3 is involved in the selectivity of G protein coupling [53].
The ligand-binding pocket is composed of four transmembrane helices (TM3, TM5, TM6, and TM7) and ECL2 [54]. Ligands interact with β1-AR through hydrogen bonds and hydrophobic interactions [55]. As an endogenous ligand, the binding of NE to β1-AR occurs in three steps. In the first step, NE interacts with two negatively charged amino acid residues: D21745.51 and D3567.32. In the second step, it passes through the “gate” composed of aromatic amino acid residues F21845.52 and F3597.35. In the third step, NE actually enters the orthosteric binding pocket [14]. When the ligand binds to the orthosteric site, transmembrane helices TM5 and TM6 move outward, while TM3 and TM7 shift inward, and β1-AR changes from a non-activated state to an activated state. Upon binding to β1-AR, the G protein (Gs or Gi) dissociates into α and βγ subunits, which then respectively act on downstream effectors.
When β1-AR binds to the Gs protein, the cytoplasmic side of TM6 is rotated outward by approximately 14 Å, TM5 is helically extended, and TM7 is moved inward by about 5 Å. In the β1-AR/Gs complex, the extension of the helix formed by the last four amino acids (Leu374, Tyr377, Leu379, Glu376) of the α5 helix at the C-terminus of the α subunit in Gs can increase the length of the α5 helix by 6 Å, enabling extensive contact with β1-AR. Concurrently, the structural domain of the α5 helix of Gs is rotated by about 96° relative to its own Ras-like GTPase domain. After rotation, the α5 helix is restricted and rotated upon contact with the Gβγ subunit. This rotation opens a tightly sealed deep groove structure within Gs, providing an exit for the release of GDP [55].
When β1-AR binds to the Gi protein, the α5 helix at the C-terminal of the α subunit of Gi rotates approximately 3° relative to its position in the inactive state and moves about 7 Å in the vertical direction. This results in an increased distance in the Ras-like GTPase domain of the Gα subunit, especially the α1, αG, and β6-α5 loops surrounding the GDP-binding pocket region, relative to the center of mass of the Gβγ subunit. Consequently, this leads to partial dissociation between the Ras-like GTPase domain of the Gα subunit and the Gβγ subunit, accompanied by a reduced interaction interface area and a loosened structural integrity [56].
Furthermore, upon activation, the receptor can transmit signals or mediate its own endocytosis through β-arrestin. The binding of β-arrestin to the receptor does not occur through “ligand-binding pocket”, but rather in two distinct binding modes: the “tail conformation” and the “core conformation”.
In the conformation mediated by the ICL3, β-arrestin is not close to the core of the receptor, but is attached to the C-terminal end of the receptor by a long connecting segment to form a “hanging” appearance, hence the term “tail conformation”. This conformation allows for a relatively large spatial distance between β-arrestin and the receptor, providing greater flexibility and facilitating biased agonistic signaling [57]. Deletion of the “finger loop” region in β-arrestin to construct a β-arrestin mutant reveals that the “tail conformation” can mediate receptor endocytosis and β-arrestin signaling, but is not associated with G-protein signaling desensitization [57].
The “core conformation” refers to the tight binding of β-arrestin to regions near the transmembrane helices of the receptor. In this conformation, the finger loop of β-arrestin inserts into the seven-transmembrane core domain of the GPCR. This insertion causes the outward displacement of TM3, TM5 and TM6, preventing the binding of the GPCR to the G protein and thus desensitizing the receptor [58]. When β-arrestin binds to the receptor in this manner, it can interact with the phosphorylated tail of the receptor through the NH2-terminal domain or directly with the core region of the receptor through the finger loop, forming an extensive action interface. The conformational changes in the internal domain of β-arrestin triggered by the “core conformation” are not only a key step in terminating the coupling of G proteins and participating in receptor endocytosis, but also contribute to stabilizing the activated state of β-arrestin, ensuring the efficiency of signaling transduction [59].
Signaling and regulation of β1-AR
The signaling pathways of β1-AR include G protein-dependent and G protein-independent signaling pathways. The G protein-dependent pathways consist of the β1-AR/Gs pathway and the β1-AR/Gi pathway. Gs protein and Gi protein are components of G proteins, respectively mediating the activation and inhibition of AC. The G protein-independent signaling pathway is predominantly the β1-AR/β-arrestin signaling pathway.
β1-AR and the Gs pathway
Upon activation of β1-AR, the activity of downstream proteases, ion channels, and transcription factors can be regulated through the β1-AR/Gs/AC/cAMP/PKA pathway, thereby regulating cardiomyocyte function [60, 61]. The G protein directly coupled to β1-AR can be dissociated into Gα and Gβγ subunits. Gα subunits can be further divided into subtypes such as Gαs, Gαi, Gαq/11, and Gα12/13. Among these, Gαs activates AC, which catalyzes the hydrolysis of ATP to cAMP.
cAMP serves as an intracellular second messenger. It binds to the regulatory subunit of PKA. PKA is an enzyme consisting of a catalytic subunit and a regulatory subunit, which acts as an effector of cAMP [62]. When the intracellular level of cAMP rises, cAMP binds to the regulatory subunit of PKA, leading to the separation of the regulatory subunit from the catalytic subunit and activation of the catalytic subunit. The catalytic subunit regulates cardiac excitation-contraction coupling by phosphorylating specific proteins [63]. This phosphorylation event potentiates the amplitude of calcium transients, enhances contractility, accelerates calcium cycling rates, and elevates pacing rates within cardiomyocytes [64] (Fig. 3).

Upon activation of β1-AR by NE, the receptor couples with Gs protein, leading to dissociation into Gαs and Gβγ subunits. Subsequently, the Gαs subunit activates AC, which catalyzes the hydrolysis of ATP to cAMP. The downstream effectors of cAMP include Epac and PKA. Epac can affect the release of calcium ions from the Golgi apparatus and mitochondria through the PLC/CaMKII pathway. MAO-A on mitochondria inhibits β1-AR signaling and PKA activity on the SR. PKA modulates calcium ion release and reuptake at the SR by phosphorylating RyR, PLN, and SERCA2a, and enhances myocardial contractility by phosphorylating cMyBP-C. In heart failure, β1-AR is over-activated. PKA activity is reduced. ROS production is increased. an increased production of ROS. These molecular alterations exacerbate inflammatory responses and myocardial fibrosis. Enhanced activity of MAO-A further inhibits PKA, thus forming a vicious cycle. As a result, the activity of cMyBP-C decreases, myocardial contractility declines, and myocardial damage continues to worsen. (Created with BioRender.com).
Cardiac excitation-contraction coupling refers to the process in which an electrical signal in the heart triggers the mechanical contraction of cardiomyocytes. It serves as the foundation for the realization of the pumping function of the heart. The action potential activates the L-type calcium channel (LTCC) located on the cardiomyocyte membrane, allowing small amounts of calcium ions to enter the cell from the extracellular space [65]. The inward flow of calcium ions not only directly participates in the contractile mechanism but also triggers the ryanodine receptor (RyR) located on the SR, causing a massive release of calcium ions stored in the SR into the cytoplasm of cardiomyocytes [66]. Increased calcium ions in the cytoplasm bind to troponin C, causing myocardial contraction. The sarcoplasmic/endoplasmic reticulum calcium ATPase isoform 2a (SERCA2a) located on the SR, opens, pumping calcium ions from the cytoplasm back to the SR for storage. As the concentration of calcium ions in the cytoplasm decreases, the cardiac muscle relaxes.
PKA increases the opening of calcium release channels by phosphorylating LTCC [67] and RyR [68], and enhances myocardial contraction by phosphorylating troponin I [69, 70]. PLN is a small-molecule protein located on the SR membrane of cardiomyocytes that modulates the activity of the SR calcium pump [71]. Phosphorylation of PLN by PKA relieves the inhibitory effect of PLN on SERCA2a, thereby facilitating the pumping of calcium ions from the cytoplasm to the SR and rapidly restoring intracellular calcium ion concentration, which shortens myocardial diastole [72]. Consequently, activation of downstream signaling molecules by the β1-AR/Gs/AC/cAMP/PKA pathway could enhance the excitation-contraction coupling process of cardiomyocytes, thereby increasing myocardial contractility [73].
Studies employing techniques such as Förster resonance energy transfer (FRET) have revealed that in the hearts of ISO-induced heart failure mouse models, the interactions between β1-AR and LTCC, as well as between β1-AR and RyR, are significantly diminished, leading to attenuated local PKA signaling and reduced myocardial contractility. Although the interaction between β1-AR and SERCA2a is enhanced, the PKA signaling at this site remains weakened [43]. This suggests that β1-AR, within distinct subcellular localizations and protein complexes, may be subject to specific and independent regulatory mechanisms to accommodate diverse physiological demands or pathological states.
Phosphorylation of proteins associated with the SR by PKA is regulated by monoamine oxidase A (MAO-A) [74] and organic cation transporter 3 (OCT3) [75]. MAO-A is a mitochondrial enzyme that can catabolize catecholamine substances [76]. It has been found that in heart failure, the expression of MAO-A in cardiomyocytes is upregulated, thereby inhibiting β1-AR/PKA signaling at the SR. Inhibition of MAO-A restored local β1-AR signaling at the SR and excitation-contraction coupling function of cardiomyocytes [77]. The uptake of catecholamines by cardiomyocytes requires the involvement of OCT3 [78]. In cardiomyocytes with OCT3 knockout, the phosphorylation of PLN induced by non-membrane-permeable norepinephrine was reduced, whereas the phosphorylation of PLN induced by the membrane-permeable agonist ISO and the activity of PKA were unaffected. This indicates that OCT3 is an essential component for β1-AR/PKA signaling in the SR [41]. Other studies similarly demonstrated that OCT3 synergizes with MAO-A to regulate β1-AR/PKA signaling at the cellular SR. Upon specific knockout of the MAO-A gene in the mouse heart, an increase in the phosphorylation level of proteins associated with the contractile function of cardiomyocytes on the SR was observed. Specifically, the phosphorylation of PLN was enhanced, accompanied by the activation of the β1-AR/PKA signaling pathway at the SR. However, following treatment with cortisol, an inhibitor of OCT3, the enhanced phosphorylation of PLN and the potentiated β1-AR/PKA signaling at the SR resulting from the deficiency of MAO-A can be suppressed [75].
Additionally, PKA can also phosphorylate cardiac myosin binding protein-C (cMyBP-C) in cardiomyocytes. cMyBP-C modulates cardiac muscle contraction through interactions with actin, myosin, and other contractile proteins [79]. When PKA phosphorylates cMyBP-C, the inhibitory effect of cMyBP-C on the myosin head is weakened. This leads to an accelerated cross-bridge cycle, thereby increasing the contractile force of cardiomyocytes. Moreover, the phosphorylated cMyBP-C promotes the dissociation of myosin from actin, which contributes to the acceleration of the transition of cardiomyocytes from the systolic state to the diastolic state. This is essential to maintain the effective pumping function of the heart at higher heart rates [80, 81].
In addition to PKA, the effectors of cAMP also include the Exchange protein directly activated by cAMP (Epac) [73]. Epac phosphorylates downstream proteins [82, 83], including PLN and RyR, via PLC and Ca2+/calmodulin-dependent protein kinase II (CaMKII), thereby affecting calcium release and calcium homeostasis of the SR [84, 85].
It was found that activation of CaMKII upregulated the expression of mitochondrial calcium uniporter (MCU). This upregulation enhanced mitochondrial calcium uptake, thereby maintaining intracellular calcium homeostasis and energy metabolism balance. As a result, it alleviated the cardiac dysfunction and remodeling caused by pressure overload [86]. The Golgi apparatus also possesses the ability to store and regulate calcium ions [87]. It has been verified that, upon stimulation of β1-AR, the Golgi apparatus can release calcium ions through the activation of Epac and CaMKII [88]. Thus, rectifying intracellular calcium ion cycling can improve myocardial contractility, which represents a crucial strategy for the prevention and treatment of heart failure [89].
Myeloid differentiation factor 2 (MD2) is a key protein in endotoxin-induced inflammation that mediates cardiac inflammatory damage, resulting in inflammatory heart failure [90]. ISO was found to activate MD2 in cardiomyocytes through the β1-AR/Gs/AC/PKA/reactive oxygen species (ROS) signaling axis. Blocking the β1-AR/Gs/AC/PKA/ROS signaling axis mitigates ISO-induced inflammatory response, cardiomyocyte hypertrophy, and myocardial fibrosis [91]. Yes-associated protein (YAP) is an important effector molecule in the Hippo signaling pathway. It promotes cell growth and proliferation by activating target genes through translocation into the nucleus [92]. Blockade of the β1-AR/Gs pathway by the β1-AR blocker metoprolol enhances cardiomyocyte proliferation, promotes cardiac regeneration, and improves cardiac function by activating YAP [93]. Recent studies have found that β1-AR reduces the expression of N6-methyladenosine (m6A). This leads to a decrease in the m6A modification level of Yap mRNA, resulting in reduced stability of Yap mRNA and decreased expression of the YAP protein. Ultimately, it inhibits cardiomyocyte proliferation and impedes cardiac regeneration [94].
β1-AR and the Gi pathway
Early findings on the signaling pathway of β1-AR suggested that β1-AR couples exclusively to Gs proteins, but not to Gi proteins [95, 96]. However, as research has progressed, an increasing number of findings have fully convincing demonstrated that β1-AR can also bind to the Gi protein. This was because the C-terminus end of β1-AR contains a PDZ-binding motif. This motif specifically binds to the PDZ domain of postsynaptic density protein 95 (PSD-95), which in turn anchors β1-AR to the cell membrane surface and prevents its coupling with Gi protein [97]. When researchers altered the binding motif of the PDZ domain of β1-AR in mouse cardiomyocytes by swapping the C-termini of β2-AR and β1-AR, they found that the β1-AR carrying the C-terminus of β2-AR could both increase the myocardial beating frequency in response to ISO stimulation at the initial stage of the experiment and decrease the heart rate at the end of the experiment. Meanwhile, this phenomenon was blocked by the Gi protein inhibitor pertussis toxin (PTX). This suggests that altering the PDZ binding motif of β1-AR can enable it to couple with the Gi protein, leading to the occurrence of a biphasic effect. Additional studies have demonstrated that PTX inhibits the activation of extracellular signal-regulated kinases (ERK) that is mediated by β1-AR in Chinese hamster ovary cells (CHO cells). This indicates the involvement of Gi protein [98].
Through co-immunoprecipitation and proximity ligation experiments, researchers discovered that carvedilol, as a blocker of β1-AR, led to the formation of interactions between β1-AR and Gi, whereas NE and ISO did not produce similar effects. This is direct evidence that the β1-AR binds Gi, making it clear that β1-AR has the capability to bind to Gi. This study also found that the use of PTX inhibited carvedilol-induced ERK phosphorylation [99]. Further analysis of the β1-AR/Gi signaling pathway revealed that carvedilol activated the signaling pathway consisting of β1-AR/β-arrestin/epidermal growth factor receptor (EGFR)/ERK [100]. Activation of EGFR provides cardioprotection, thereby counteracting the cardiotoxicity of catecholamines [101]. EGFR internalization and ERK phosphorylation stimulated by carvedilol were completely blocked when β-arrestin or Gi was knocked down [99]. This suggests that carvedilol specifically activates the β1-AR/Gi/β-arrestin/EGFR/ERK signaling pathway to exert cardioprotective effects.
Additionally, carvedilol activates another β1-AR signaling pathway: the β1-AR/Gi/phosphoinositide 3-kinase (PI3K)/protein kinase B (Akt/PKB)/nitric oxide synthase 3 (NOS3)/cyclic guanosine monophosphate (cGMP)/protein kinase G (PKG) pathway. Activation of this pathway enhances myocardial contractility without significantly increasing intracellular calcium ions, avoids myocardial injury caused by calcium overload, reduces cardiomyocyte hypertrophy and apoptosis, and improves both cardiac systolic and diastolic function [102] (Fig. 4).

There are two signaling pathways after binding of β1-AR to Gi protein: 1) β-arrestin/EGFR/ERK pathway; 2) β1-AR/Gi/PI3K/Akt/NOS3/cGMP/PKG pathway. Both pathways exert cardioprotective effects. (Created with BioRender.com).
β1-AR and the β-arrestin pathway
β-arrestin is an adaptor protein that regulates the signaling and trafficking of GPCRs [103, 104]. Binding of β1-AR to β-arrestin produces two effects: desensitization of β1-AR and endocytosis of β1-AR [105]. Desensitization refers to the process in which β1-AR binds to β-arrestin and uncouples from G proteins to terminate β1-AR signaling [106]. Endocytosis refers to the process by which receptors internalize into clathrin-coated vesicles and enter the cell. Subsequently, these receptors are either transported to lysosomes for degradation or recycled to the plasma membrane to resume their functions [107]. β1-AR phosphorylated by GRKs recruits β-arrestin, facilitating the decoupling of the receptor from G proteins and the desensitization to prevent overactivation of the receptor. β-arrestin, in turn, can determine whether β1-AR is internalized [108] by recruiting adaptor proteins such as clathrin [109], adaptor protein 2 (AP2) [110], and guanine nucleotide exchange factor (GEFs) [111]. The phosphorylation pattern of the receptor and the binding of β-arrestin play a pivotal role in intracellular transport, thus determining the ultimate fate of β1-AR [107, 112] (Fig. 5).

Phosphorylated β1-AR dissociates G proteins by recruiting β-arrestin. Subsequently, β1-AR no longer transduces signals through the AC/Gs/cAMP signaling pathway, which is termed “desensitization”. The desensitized β1-AR can still recruit Raf, Src, and EGFR under the action of β-arrestin, thereby activating downstream signaling pathways and exerting protective effects on the heart. Additionally, β-arrestin also mediates the endocytosis of β1-AR. Facilitated by the TGN, β1-AR can either be routed to the lysosomes for degradation or be recycled back to the cell membrane to resume its function. The binding of β-arrestin to β1-AR prevents overactivation of β1-AR. Through endocytosis, this interaction can also maintain a relatively constant number of β1-AR receptors on the cell membrane. (Created with BioRender.com).
Although the desensitized β1-AR no longer transmits information through G protein, it continues to mediate signaling through molecules such as rapidly accelerated fibrosarcoma (Raf) [113], proto-oncogene kinase Src [114] and EGFR [115] after binding to β-arrestin. Raf is a class of mitogen-activated protein kinases that function in intracellular signaling and are involved in the regulation of processes such as cell proliferation, differentiation and apoptosis. Src is involved in the regulation of the cell cycle, adhesion, proliferation and migration [116], and is activated upon receptor desensitization [114]. Activation of Src promotes the activation of Akt [114], a key molecule necessary to mediate cardiac hypertrophy, and activates matrix metalloproteinases (MMPs). MMPs cleave and release the membrane-bound precursor forms of epidermal growth factor (EGF)-like ligands. The released EGF-like ligand then activates EGFR [115]. Activated EGFR catalyzes ERK1/2 phosphorylation through the rat sarcoma viral oncogene homolog (Ras)/Raf/mitogen-activated protein kinase (MEK) cascade reaction [117, 118], thereby antagonizing the stimulation of catecholamine and protecting the heart [101].
Following endocytosis, β1-AR can be directly transported to lysosomes for degradation, thereby completely terminating signaling [119]. Alternatively, a portion of β1-AR can be recycled back to the cell membrane, where it can re-engage in signal transduction and continue to exert its function [101, 115]. The key determinant of whether β1-AR is degraded or continues to function is the “Trans-Golgi Network (TGN)” [120]. TGN also determines the fate of various receptor proteins, including the G-protein-coupled estrogen receptor (GPER) [121], the growth inhibitor type 2 A receptor [122, 123], and the chemokine receptor 5 (CCR5) [124]. However, the mechanisms by which TGN regulates the fate of these proteins remain unclear.
In addition, it was found that even when clathrin-mediated endocytosis is inhibited, β-arrestin can still accumulate in clathrin-coated pits on the cell membrane and activate ERK1/2 through a “long-range activation” mechanism [125]. This indicates that β-arrestin can transmit signals through other mechanisms, although the specific details remain to be elucidated.
As a β1-AR blocker, carvedilol not only biases the activation of β1-AR towards Gi proteins, but also promotes the binding between β1-AR and β-arrestin. Moreover, there exists a synergistic interaction between Gi and β-arrestin, which is relevant to the signaling regulation when carvedilol acts on β1-AR [126]. Studies have revealed that carvedilol, following the activation of β1-AR, promotes signaling through the β-arrestin pathway during Dicer ribonuclease-mediated microRNA maturation, enhancing the ability of the heart to resist injury [127].
The association between β1-AR and cardiovascular diseases
Pathological changes in the heart are the basis of cardiovascular diseases. The β1-AR is precisely closely involved in it, such as increased cardiomyocyte apoptosis [128, 129], activation of endoplasmic reticulum stress response [130], abnormal cardiomyocyte autophagy [131, 132], mitochondrial dysfunction [133], cardiomyocyte hypertrophy [134], as well as myocardial fibrosis and myocardial tissue remodeling [135, 136]. Ultimately, these pathological changes result in cardiac systolic and diastolic dysfunction, and further progress to severe cardiovascular diseases, including myocardial infarction and heart failure. Therefore, maintaining the steady-state of normal β1-AR function is of great significance for preserving the physiological functions of the heart.
β1-AR and dilated cardiomyopathy
Dilated cardiomyopathy is defined as a condition characterized by the degeneration, hypertrophy, and necrosis of cardiomyocytes, which lead to the dilation of the left ventricle or both ventricles and systolic dysfunction. It is also accompanied by myocardial hypertrophy and reduced ventricular systolic function [137]. In dilated cardiomyopathy, a large number of cardiomyocytes undergo hypertrophy, and myocardial tissue exhibits hypertrophic remodeling to adapt to the increased preload and afterload [138, 139].
β1-AR induces cardiac hypertrophy predominantly through the endocytic mechanism [140]. Inhibiting various steps of the β1-AR endocytosis process by using the β1-AR blocker betaxolol, the GRK inhibitor βARK-CK, a dominant negative mutant to suppress β-arrestin, and the Src inhibitor PPI can reverse β1-AR-induced cardiomyocyte hypertrophy [141]. This suggests that the endocytosis of β1-AR is one of the causes leading to cardiac hypertrophy. Additionally, endocytosis of β1-AR activates Akt, which can further promote β1-AR-induced cardiac hypertrophy [140]. Moreover, another study has demonstrated that β1-AR on the Golgi apparatus can regulate cardiac hypertrophy by triggering the hydrolysis of phosphatidylinositol-4-phosphate (PI4P) through phospholipase C epsilon (PLCε). Blocking β1-AR on the Golgi apparatus can inhibit NE-induced cardiac hypertrophy [45] (Fig. 6).

From cardiomyocytes apoptosis to heart failure, β1-AR affects cardiac diseases at different stages through distinct mechanisms. (Created with BioRender.com).
The signaling of β1-AR is related to the stages (early compensation or late decompensation) and types (pressure or volume overload) of cardiac hypertrophy [142]. In two rat models of cardiac hypertrophy induced by pressure overload and volume overload, the density of β1-AR (Table 1), intracellular calcium ion concentration and AC activity were measured during the early compensatory stage (4 weeks) and late decompensatory stage (24 weeks) of cardiac hypertrophy. The results showed that all of the above indices increased at the early stage of volume overload, whereas no significant changes were observed in these indices during the early stage of pressure overload. Cardiac function remained unchanged during the early phase of volume overload, while it significantly decreased during the early stage of pressure overload. In both volumes overload-induced and late-stage pressure overload-induced cardiac hypertrophy, the above indices decreased significantly. This suggests that β1-AR can exert its effects through distinct signaling mechanisms in different types of cardiac hypertrophy.
Furthermore, in cardiac hypertrophy, the reduced responsiveness of β1-AR is not a universal phenomenon among cardiomyocytes, but rather occurs in cardiomyocytes expressing beta-myosin heavy chain (β-MHC) [143]. Cardiomyocytes were isolated after inducing cardiac hypertrophy in mice using transverse aortic constriction (TAC). Compared with β-MHC-negative cardiomyocytes, β-MHC-positive cardiomyocytes exhibited a significant reduction in percentage of cell shortening, relaxation rate, and shortening rate upon stimulation with ISO.
The genetic polymorphisms of β1-AR are involved in the pathogenesis of dilated cardiomyopathy [144] (Fig. 6). In dilated cardiomyopathy, the mRNA abundance of β1-AR in myocardial tissue is significantly reduced [27]. In a study of the β1-adrenergic receptor gene signaling network (β1-GSN), an extensive downstream gene network of β1-AR has been identified. This network is associated with pathological remodeling of the left ventricle in dilated cardiomyopathy, in which metabolic genes (19.3% of the genes, of which 81% were upregulated) are dominant [145]. When analyzing the polymorphic variants Ser49Gly (rs1801252) and Gly389Arg (rs1801253) of β1-AR in patients with dilated cardiomyopathy, it was found that the frequency of the Gly389 allele was significantly higher in patients with dilated cardiomyopathy without ventricular tachycardia than in those with ventricular tachycardia[146]. Moreover, the Gly49 allele of the β1-AR was associated with a lower risk of worsening heart failure than was homozygosity for the Ser49 allele [147]. Yet another study demonstrated that patients with idiopathic dilated cardiomyopathy who carried the Gly49 allele exhibited more severe clinical symptoms and poorer prognoses (impaired cardiac function and higher rates of heart failure-related hospitalization) during follow-up [148]. Polymorphisms are present at codons 49 and 389 of β1-AR, resulting in a Ser49Gly (rs1801252) polymorphism and an Gly389Arg (rs1801253) polymorphism, respectively [149]. A study investigated the effects of the Ser49Gly (rs1801252) polymorphism at codon 49 as well as the Gly389Arg (rs1801253) polymorphism at codon 389 in patients with dilated cardiomyopathy [150]. The results showed that the codon 49 Ser49Gly (rs1801252) polymorphism is the dominant genetic factor influencing β-blocker response and survival in dilated cardiomyopathy. Gly49 carriers respond well to low-dose therapy while Ser49 patients require higher doses. Meanwhile, codon 389 Gly389Arg (rs1801253) has minor, context-dependent effects. This provides a basis for personalized treatment of patients with dilated cardiomyopathy.
β1-AR and cardiac arrhythmias
The incidence of arrhythmia is about 0.5% [151], which triggers a variety of cardiac diseases [152]. Malignant arrhythmias such as ventricular tachycardia (VT) and ventricular fibrillation (VF) are strongly associated with sudden cardiac death (SCD) [153]. Atrial fibrillation is the most common cardiac arrhythmia and can lead to heart failure, stroke [154]. The mechanisms of arrhythmogenesis are complex, involving anatomical structures, the autonomic nervous system, ion channels, and regulatory proteins [155,156,157].
Although atrial fibrillation has notable genetic features, its individualized treatment has not been translated into clinical practice yet [158]. A study that analyzed the impact of the polymorphism variants Gly389Arg (rs1801253) of β1-AR on atrial fibrillation, revealed that the risk of postoperative arrhythmias was significantly increased among patients after cardiac surgery who were carriers of the Gly389 allele. Although atrial fibrillation has notable genetic features, its individualized treatment has not been translated into clinical practice yet [158]. The use of the third-generation β1-AR blocker, bucindolol, can prevent the development of atrial fibrillation and has a more pronounced effect on controlling heart rate in patients who have the Arg389Arg genotype. Patients carrying the Gly389 allele respond better to conventional β-AR blockers (e.g., metoprolol) for heart rate control [159]. In patients with supraventricular tachyarrhythmias, Arg389-homozygotes had the best results with the antiarrhythmic drug Flecainide, with good control of their heart rate. However, the efficacy is poorer in patients carrying the Gly389 allele. Patients who are Gly389-homozygous have worse heart rate control compared to those who are Gly389-heterozygous [160].
The onset of arrhythmias is not merely associated with the genetic polymorphisms of β1-AR, but the functional dysregulation of β1-AR also serves as one of the pathogenic mechanisms contributing to the development of arrhythmias.
In the hearts of mice with knockout of β-AR (including β1-AR, β2-AR, and β3-AR), a significant prolongation of excitatory conduction time and the refractory period was observed. Moreover, a significant reduction in atrial and ventricular arrhythmias was also noted [161].
A study investigating the role of the anti-apoptotic protein Bcl-2-assocaited athanogene 3 (BAG3) in the heart revealed that in adult left ventricular cardiomyocytes, BAG3 modulates cardiomyocyte contraction and action potential duration by interacting with β1-AR and LTCC, thereby reducing the risk of arrhythmias [162]. The administration of metoprolol significantly shortened the duration of norepinephrine-induced atrial fibrillation in mice. This suggests that the prolongation of atrial fibrillation duration by norepinephrine is achieved by activation of β1-AR. Norepinephrine not only increases calcium leakage from the SR but also promotes spontaneous calcium release, whereas metoprolol can inhibit this effect [163]. In a porcine model of ventricular fibrillation, the impact of administration of the β1-AR blocker esmolol during cardiopulmonary resuscitation on the recovery of calcium-circulating proteins and electrical signals was investigated. The results showed that blockade of β1-AR reduces ventricular fibrillation recurrence, attenuates CaMKII overactivation, and phosphorylation of RyR [164].
Desmosomal adhesion refers to intercellular adhesion mediated by desmosomes. β1-AR enhances cardiomyocyte adhesion through the cAMP signaling pathway. When adhesion is weakened due to desmosome disruption (e.g., tryptophan treatment), the velocity of electrical conduction slows down, and the risk of arrhythmia increases. Disruption of desmosomal proteins inhibits the cAMP signaling pathway downstream of β1-AR. Increasing the levels of cAMP restores the electrical conduction function in cardiomyocytes. This observation implies that the activation of the β1-AR/cAMP pathway can mitigate arrhythmias induced by the disruption of desmosomal structures through desmosomal adhesion [165].
Epac, an effector of cAMP, also plays a crucial role in the occurrence and development of arrhythmias. In a mouse model, β1-AR induces calcium leak from the SR through activation of Epac, a process dependent on phosphorylation of the RyR2-S2814 site by CaMKII [130]. It was confirmed by using the highly selective β1-blocker CGP-20712A, the PKA inhibitor PKI, and the highly selective Epac activator 8-pCPT, that continuous stimulation of the β1-AR can be mediated through the Epac-mediated Ca2+/calcineurin (CaN)/nuclear factor of activated T-cells (NFAT) signaling pathway, which downregulates slow current rectifier potassium current density, delays myocardial repolarization, and increases the risk of arrhythmia [166] (Fig. 6).
β1-AR and myocardial infarction
Myocardial infarction is a life-threatening disease characterized by myocardial necrosis resulting from the acute occlusion of coronary arteries. Myocardial ischemia triggers a cascade of reactions, including activation of the sympathetic nervous system and release of adrenaline [167]. Following myocardial infarction, extensive cardiomyocyte apoptosis occurs, accompanied by abnormal proliferation of cardiac fibroblasts and excessive accumulation of collagen fibers. These pathological events collectively lead to myocardial fibrosis and may even result in irreversible structural damage to the heart.
β1-AR is involved in the pathophysiological processes of ventricular remodeling following myocardial infarction.
Prolonged overactivation of β1-AR leads to programmed cardiomyocyte death [128]. After stimulating β1-AR with ISO in mice, the expression of the anti-apoptotic protein B-cell lymphoma 2 (Bcl-2) was significantly decreased in cardiac tissues. In contrast, the expression of the pro-apoptotic proteins Bcl-2-associated X protein (Bax) and Bcl-2 and adenovirus E1B 19-kDa-interacting protein 1 (BNIP1) was notably increased [168]. Treatment of adult rat ventricular myocytes (ARVMs) with NE or ISO increased the proportion of apoptotic cardiomyocytes by 1.72 ± 0.08-fold and 1.81 ± 0.08-fold, respectively. Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) showed a 1.70 ± 0.14-fold proportion of TUNEL-positive cardiomyocytes. The apoptosis of cardiomyocytes could be reversed with CGP20712 A, a selective antagonist of β1-AR. This suggests that activation of β1-AR is a crucial factor in the induction of cardiomyocyte apoptosis [129]. Previous studies have demonstrated that β1-AR promotes cardiomyocyte apoptosis by inhibiting myocyte enhancer factor 2 (MEF2) activity via the cAMP/PKA pathway. Atenolol, a β1-AR blocker, restores MEF2 activity and thus inhibits apoptosis in cardiomyocytes [129, 169]. In addition to the cAMP/PKA pathway, β1-AR also induces cardiomyocyte apoptosis through CaMKII [1]. Blocking L-type calcium channels or inhibiting the activity of CaMKII prevents β1-AR-induced cardiomyocyte apoptosis [170]. Conversely, overexpression of CaMKII in the heart significantly increases cardiomyocyte apoptosis [170]. Moreover, β1-AR induces cardiomyocyte apoptosis by increasing the phosphorylation of inhibitor of kappa B alpha (IκBα) and the expression of tumor necrosis factor-alpha (TNF-α) [171], activating phosphatase and the tensin homolog (PTEN) gene, and downregulating the focal adhesion (FA) signaling pathway [172]. Mammalian sterile twenty-like kinase 1 (Mst1), a Hippo pathway mitogen-activated protein kinase (MAPK) family member, regulates apoptosis and tissue growth. Studies have shown that Mst1 reduces apoptosis in non-cardiomyocytes (such as fibroblasts, macrophages, neutrophils) and necrosis in cardiomyocytes, thereby ameliorating cardiomyopathy. Although Mst1 exerts a pivotal regulatory function during the progression of β1-AR-induced cardiomyopathy, the mechanism of action between Mst1 and β1-AR-activated signaling pathways remains unclear [173] (Fig. 6).
Some researchers constructed myocardial infarction models using β1-AR knockout (β1-ARKO) mice, β2-AR knockout (β2-ARKO) mice, and β1/β2-AR double-KO (DKO) mice. The results showed that β1-AR could adversely affect the heart by activating CaMKII, which was manifested by increased apoptosis of cardiac border cells, expanded infarct area, and a decline in cardiac function indices [174]. Through experiments conducted using the isolated rabbit heart ischemia-reperfusion injury model, it was demonstrated that β1-AR activation leads to the phosphorylation and inactivation of cytochrome c oxidase via PKA signaling, thereby exacerbating myocardial injury (Fig. 6). Blocking β1-AR could reduce the area of myocardial necrosis, reverse the decrease of cytochrome coxidase activity, and maintain the activity of mitochondrial enzyme [175]. Subcutaneous injection of ISO Isoprenaline in male Wistar rats to activate β1-AR in the heart, the results showed that myocardial lesions initiated in the subendocardial region, accompanied by the infiltration of inflammatory cells, collagen deposition, and other phenomena. The extent of lesions was proportional to the dose and duration of ISO administration. Measurement of transforming growth factor-beta 1 (TGF-β1) and brain natriuretic peptide (BNP) in serum revealed that low doses of ISO were effective in inducing changes in the above indicators [136]. Several studies have indicated that GAIP-interacting protein, C-terminus 1 (GIPC1) stabilizes the structure of the β1-AR by directly binding to the C-terminus of the β1-AR. This interaction prevents the receptor from ubiquitination and degradation, thereby maintaining the number of receptors on the cell membrane. In addition, GIPC1 exerts cardioprotective effects by inhibiting the overactivation of the mitogen-activated protein kinase (MAPK) signaling pathway. Overexpression of GIPC1 attenuates ISO-induced myocardial fibrosis and ventricular remodeling in mice [176].
Furthermore, studies have demonstrated that in the myocardial tissues of patients with myocardial infarction, the expression level of β1-AR mRNA is increased [177] (Table 1). However, in the hearts of rats with myocardial infarction, the levels of both protein and mRNA of β1-AR are significantly downregulated [178] (Table 1). The polymorphic variants Gly389Arg(rs1801253) of β1-AR were associated with myocardial infarction [179]. Among them, individuals carrying the Arg389 allele (heterozygotes and homozygous) have a significantly higher risk of developing myocardial infarction compared to those who are Gly389 homozygous [180].
β1-AR and heart failure
Heart failure is a disease caused by dysfunction of the pumping function of the heart. In patients with heart failure, sympathetic overexcitation results in β1-AR desensitization [181]. In the myocardial tissues of patients, there is a decrease in the quantity of β1-AR (Table 1) and an increase in the amount of Gi protein [182]. The increased Gi protein inhibits cAMP formation [3, 183]. In the ISO-induced mouse model of heart failure, the protein level of β1-AR is decreased [43]. When β1-AR is specifically overexpressed in mice, cardiac contractility can be enhanced in the early stage. However, significant cardiomyocyte hypertrophy is also observed. Subsequently, mice develop heart failure with myocardial contractility dropping to 50% and ejection fraction decreasing to 20% [184]. Overexpression of β1-AR may improve cardiac function in the short term, yet the ultimate effect is deleterious. Similarly, in the ISO-induced mouse model of heart failure, upregulation of MAO-A limits activation of β1-AR/PKA signaling. The use of the MAO-A inhibitor clorgyline improves cardiac contractile function by restoring SR-localized β1-AR/PKA/PLN signaling [77]. Synapse-associated protein 97 (SAP97) is a scaffolding protein. It binds to the C-terminus of β1-AR to form the β1-AR-SAP97 signaling complex and regulates downstream signaling pathways [97, 185, 186]. During heart failure, the β1-AR-SAP97 complex is disrupted, leading to a shift of the β1-AR signaling pathway from the cAMP/PKA pathway to the CaMKII pathway. In cardiac-specific SAP97 knockout mice, the deletion of SAP97 promotes the binding of β1-AR and CaMKII and activates the CaMKII pathway. This leads to increased SR calcium leakage, cardiomyocyte apoptosis, and arrhythmias, ultimately impairing cardiac structure and function. Further studies have revealed that GRK5 promotes the dissociation of SAP97 from β1-AR by phosphorylating the PDZ domain of β1-AR, thereby activating the CaMKII pathway. In GRK5 knockout mice, CaMKII activity is inhibited and cardiac remodeling is attenuated [187]. GRK5-mediated β1-AR-SAP97 dissociation is a central mechanism of CaMKII overactivation in heart failure. Inhibition of GRK5 or enhancement of the binding between β1-AR and SAP97 improves cardiac function in heart failure patients by blocking the CaMKII signaling pathway. During the treatment of heart failure with β-blockers (e.g., metoprolol), the inhibition of the β1-AR signaling pathway exerts a protective effect on the heart, alleviating cardiac hypertrophy and reducing the occurrence of arrhythmias. However, molecular mechanism studies have shown that in the signaling pathway mediated by CaMKII, the expression level of the CaMKII protein is not affected by β-AR blockers. CaMKII autophosphorylation is also not affected. Moreover, the phosphorylation status of downstream target proteins, such as RyR, remains unaffected by β-AR blockers [188]. Therefore, although β-AR blockers are used in the treatment of heart failure, they are unable to suppress the pathological cardiac remodeling driven by CaMKII. Targeted inhibition of the CaMKII pathway may be a complementary strategy for the treatment of heart failure in the future (Fig. 6).
In studies of polymorphism variants Gly389Arg (rs1801253) of β1-AR, patients who are Arg389 homozygous were found to have a better response to treatment with β1-blockers (e.g., carvedilol) and a more pronounced improvement in left ventricular function than patients who are carriers of the Gly389 allele [189]. Another study showed that patients carrying the Gly389 allele had a significantly increased risk of heart failure in East Asian populations, whereas they tended to have a reduced risk in Caucasian populations. Patients who are Arg389 homozygous uniformly exhibit a more favorable therapeutic response to selective β1-blockers (e.g., metoprolol), as evidenced by significant improvements in left ventricular ejection fraction and left ventricular end-systolic diameter/volume [190]. Thus, despite the association of β1-AR gene states with heart failure, there are racial differences in their effects. Currently, there is insufficient evidence to support their use as a basis for personalized disease treatment.
β1-AA in cardiovascular diseases
β1-adrenergic receptor autoantibody (β1-AA) is an autoantibody generated against the second extracellular loop of β1-AR. It possesses the function of sustaining activation of β1-AR [191, 192].
Studies have demonstrated that β1-AA promotes myocardial fibrosis [193], inhibits cellular autophagy and induces cardiomyocyte apoptosis [194, 195].
β1-AA is also closely associated with the development and progression of multiple diseases, including dilated cardiomyopathy [196], atrial fibrillation [194, 197], and heart failure [198, 199]. In patients with dilated cardiomyopathy, the positive rate of β1-AA is as high as 30% [200]. After removing β1-AA from the peripheral blood of patients via immunoadsorption, the clinical manifestations of these patients are significantly improved [201].
The aforementioned diseases have been further validated through animal studies. Passive immunization of mice with β1-AA leads to significant reduction in cardiac function and myocardial remodeling [198]. Additionally, passive immunization of rabbits with β1-AA results in obvious arrhythmias [202]. Based on the results of the aforementioned studies, β1-AA exhibits pathogenicity.
The core consensus regarding its pathogenic mechanism is that β1-AA sustained actives of β1-AR [203]. Specifically, in cardiomyocytes, β1-AA induces a prolonged elevation of the cardiomyocyte beating frequency by sustaining β1-AR activation, whereas norepinephrine only causes a transient increase in cardiomyocyte beating frequency [192]. Concurrently, the intracellular cAMP level also increases persistently. Current studies propose two explanations for the sustained activation of β1-AR by β1-AA: 1) β1-AA stabilizes β1-AR in an active conformation to maintain its continuous activation [204]; 2) β1-AA inhibits the endocytosis of β1-AR, thereby enabling its sustained activation [192].
Currently, the primary therapeutic strategy for patients with β1-AA-positive cardiac diseases is immunoadsorption therapy [201, 203]. Additionally, specific cyclic peptides [205] or aptamers [206] are also used to specifically bind to β1-AA and neutralize its activity. Among these approaches, immunoadsorption has been applied in clinical practice and significantly alleviated the symptoms of patients [199]. Furthermore, biased activation of the β2-AR-Gi signaling pathway also inhibited the sustained activation of β1-AR induced by β1-AA [192, 198]. However, the clinical application prospects of this method still require further research.
β1-AR blockers
In the treatment of cardiac diseases, pharmacological intervention has significantly reduced morbidity and mortality [207]. Among the available therapeutic approaches, β1-AR blockers act on the sympathetic nervous system to inhibit β1-AR-mediated signaling pathways, which in turn reduces heart rate and myocardial oxygen consumption, thereby improving cardiac function. Notably, β1-AR blockers exhibit more pronounced therapeutic effects in the treatment of heart failure and the management of cardiac arrhythmias [207].
β1-AR blockers are organic compounds composed of an aryloxypropanolamine side chain, with an aromatic or heteroaromatic ring attached to this side chain [208]. Although different β1-AR blockers vary in their substituents, they generally share a fundamental aryloxypropanolamine structure, which serves as the basis for their pharmacological activity [209]. β1-AR blockers bind competitively and reversibly to the endogenous ligand-binding site of β1-AR. Upon binding, β-blockers and β1-AR form a stable complex. However, the conformation of β1-AR has no change. By occupying the binding site of β1-AR, they prevent catecholamines from interacting with β1-AR, thereby inhibiting G protein activation, blocking AC stimulation, and reducing intracellular cAMP levels. This suppression of downstream molecule activation ultimately blocks or attenuates the positive cardiac effects mediated by the β1-AR signaling pathway, exerting therapeutic effects such as antiarrhythmic action, antianginal activity, and improving heart function [210].
β1-AR blockers are mainly categorized into three generations. First-generation blockers exhibit no selectivity towards β-AR subtypes, such as propranolol and nadolol. Second-generation selective β1-AR blockers include metoprolol, atenolol, bisoprolol, and landiolol. Third-generation selective β1-AR blockers include carvedilol, nebivolol, and labetalol [211].
The first generation of non-selective β-AR blockers not only block β1-AR but also β2-AR, and they have no specificity for either subtype. The blockade of β2-AR, leads to an increase in peripheral vascular resistance, bronchial contraction, inhibition of glycogenolysis, and altered lipolysis. Therefore, in clinical practice, first-generation non-selective β-AR blockers should be applied with caution or contraindicated in patients with diabetes mellitus or pulmonary diseases [207].
Second-generation selective β1-AR blockers exhibit specific antagonism toward β1-AR with only weak affinity for β2-AR. Therefore, they are particularly suitable for patients with heart diseases who also have pulmonary conditions [208].
Third-generation β1-AR blockers exert antagonistic effects not only on β1-AR but also α1-AR. They have minimal impact on glucose and lipid metabolism and possess vasodilatory properties. Consequently, their main side effect is hypotension. Clinically, they are suitable for elderly patients as well as those with cardiovascular diseases and diabetes mellitus [209, 211, 212].
In summary, first-generation β-AR blockers have basic efficacy, such as reducing myocardial contractility and hypotensive effects. However, they are associated with more side effects. Second-generation drugs specifically block β1-AR, improving cardiac selectivity and drug safety more significantly. In addition to targeting β1-AR, third-generation drugs also act on α1-AR and β3-AR, further optimizing therapeutic effects and reducing side effects through synergistic actions of multiple targets and mechanisms [208] (Fig. 7).

The adverse effects of first-generation β-AR blockers, particularly bronchoconstriction and metabolic dysregulation, motivated the development of second-generation selective β1-AR blockers. Subsequently, third-generation selective β1-AR blockers have been designed to possess vasodilatory properties, optimizing their hemodynamic profile. Furthermore, the structural schematic diagram of the representative drugs of three generations of β1-AR blockers (metoprolol, nebivolol, propranolol) is presented. The common structure marked in pink in the figure is aryloxypropanolamine. (Created with BioRender.com).
Propranolol is a first-generation non-selective β-AR blocker [213]. It reduces heart rate, cardiac output, and myocardial contractility by blocking cardiac β1-AR. It can effectively treat hypertension, angina pectoris, and myocardial infarction. However, it also blocks β2-AR, which inhibits hepatic glycogenolysis, delays the recovery of blood glucose and reduces symptoms of hypoglycemia such as palpitations and hand tremors. Therefore, it should be used with caution in patients with hypoglycemia. Blocking β2-AR may also cause bronchospasm. Hence, it is contraindicated in patients with asthma [214]. Although propranolol is inexpensive, it is no longer used as a preferred drug for the treatment of hypertension and other cardiovascular diseases because of its relatively numerous side effects. Clinical studies are now mostly focused on the treatment of infantile hemangiomas [215] and portal hypertension [216].
Landiolol is a second-generation β1-AR blocker that has been extensively investigated in recent years. It is characterized by its ultra-short-acting property and high selectivity [217]. It exerts its effects by competitively antagonizing β1-ARs, thereby inhibiting the action of catecholamines. This mechanism attenuates the heart rate acceleration induced by sympathetic nerve activation, which in turn helps control cardiac arrhythmias and reduces adverse impacts on cardiac function [217]. It is mostly used for the treatment of tachyarrhythmias, especially atrial fibrillation [218], sepsis-associated tachycardia [219, 220], and for preoperative heart rate control in coronary artery and other vascular interventions [221, 222]. Compared with other blockers, landiolol has a more potent chronotropic effect and a weaker hypotensive effect [223]. In recent studies, patients with supraventricular tachycardia demonstrated excellent tolerability to landiolol and had a favorable safety profile [224]. In a canine model of endotoxic shock, a low dosage of landiolol reduces heart rate without affecting hemodynamic recovery in the early stages of shock [225]. The common adverse effects of landiolol include hypotension and bradycardia. In severe cases, it may lead to complete atrioventricular block and sinus node arrest. Therefore, the blood pressure and cardiac rhythm of patients should be strictly monitored during its administration.
Metoprolol also belongs to the second generation of β1-AR blockers. It is commonly used as a combined medication for hypertension [226] and for adjunctive treatment of hypertrophic cardiomyopathy [227], atrial fibrillation [228], and heart failure [229]. Studies have revealed that intravenous administration of metoprolol before reperfusion in patients with acute myocardial infarction can improve left ventricular function. Moreover, the rate of readmission due to heart failure was significantly lower in patients treated with metoprolol [230]. These findings have underscored the importance of using metoprolol for early intervention in myocardial infarction to improve the prognosis. Studies have shown that whether metoprolol is administered to rats acutely via oral intake or chronically through addition to drinking water or intraperitoneal injection, it can produce cardioprotective effects. Nevertheless, metoprolol also increases the risk of ischemic stroke by inhibiting cerebrovascular β1-AR-mediated vasodilation [231]. Furthermore, metoprolol, although not a conventional antiarrhythmic drug, has demonstrated an effect in neonatal foals with irregular tachyarrhythmias into a normal sinus rhythm in clinical cases of treating neonatal foals [232]. Early studies have also demonstrated that metoprolol can prolong the effective atrial action potential refractory period and reduce the fibrillation frequency in a porcine model of atrial fibrillation [233]. The most common adverse effects of metoprolol are bradycardia and hypotension, which are dose-dependent.
Nebivolol, a third-generation β1-AR blocker, is used for the treatment of conditions such as hypertension and heart failure [234]. Nebivolol not only produces cardioprotective effects by blocking β1-AR, but also exhibits anti-inflammatory, antioxidant, and vascular endothelial protection effects [208]. Nebivolol is frequently used in combination with other antihypertensive drugs to treat hypertension [235, 236]. The most common adverse reactions include fatigue (4%-79%), headache (2%-24%), and bradycardia (6%-11%). Nebivolol is potentially superior for patients who are intolerant to traditional β1-AR blockers, such as those with asthma or chronic obstructive pulmonary disease [237, 238].
ABRQβ-006 is a novel therapeutic vaccine designed to target the β1-AR via an active immunization strategy. Its mechanism of action involves the chemical conjugation of ABR-006, a specific peptide derived from the second extracellular loop (ECL2) of the human β1-AR, to Qβ phage virus-like particles (VLPs). Following vaccination, this vaccine elicits the production of high-titer specific antibodies in the body. These antibodies bind to β1-ARs and exert a functional antagonistic effect, thereby inhibiting the downstream pathological signaling pathways mediated by excessive sympathetic nerve activation. In preclinical studies, ABRQβ-006 has demonstrated therapeutic potential for a variety of cardiovascular diseases. Specifically, it effectively reduces systolic blood pressure and ameliorates vascular and myocardial remodeling in hypertensive models. In pressure overload (transverse aortic constriction, TAC) and myocardial infarction (MI) models, it significantly improves cardiac function, inhibits ventricular remodeling, and attenuates myocardial fibrosis and inflammatory infiltration. Notably, its efficacy in MI models is superior to that of metoprolol. These findings indicate that ABRQβ-006 holds broad application prospects in the treatment of hypertension and heart failure [239].
Perspectives
Over the past century, researchers have made a series of important discoveries in analyzing the molecular structure of β1-AR and elucidating the signaling mechanism of β1-AR.
Despite some achievements made in the research around β1-AR, there remain numerous unresolved issues that merit our attention. For instance, the traditional view holds that β1-AR binds to Gs protein rather than Gi protein. However, it has now been confirmed that carvedilol can induce the binding of β1-AR to Gi protein, enabling it to function through a signaling pathway distinct from that of Gs [99]. This finding reveals the complexity and diversity of β1-AR functions. Does NE in vivo also induce the binding of β1-AR to Gi protein? Current research has not yet provided a definitive answer.
As for Gs protein, the conformation of the β1-AR/Gs complex is distinct when bound to agonists of varying efficacy, including full, partial, and weak agonists [240]. Further studies showed that the full agonist-bound β1-AR/Gs complex was the most stable and induced the fastest activation and termination of the β1-AR/Gs signaling pathway. This suggests that there are differences in the efficiency with which the same receptor activates the downstream signaling pathway with different ligands bound.
In addition to Gs and Gi, there are other types of G proteins, such as Gαq/11 and Gα12/13 [241]. It remains unclear whether these aforementioned G proteins can also couple with β1-AR and mediate distinct signaling pathways. The actual diversity of existing G proteins is far greater than previously recognized. The human genome encodes over 20 different Gs, each exhibiting specific tissue distribution and functional characteristics [242]. Consequently, a full consideration of the structural features, expression patterns of each molecule, and their interactions with other cellular components is required to better understand the cellular signaling network of β1-AR.
β1-AR serves as a crucial therapeutic target for various heart diseases. Drugs developed targeting β1-AR are not only applicable in the cardiovascular system but also involved in the treatment of neurological, urological, anti-tumor, and other multifaceted diseases. Therefore, drug development against β1-AR still has a wide range of prospects and important significance. At present, β1-AR blockers have been developed to the third generation. Given that the first-generation blockers are nonselective and restrict patients with concomitant respiratory disease when used to treat cardiovascular disease, the second-generation blockers were developed. Compared to the first-generation blockers, the second-generation blockers exhibit higher selectivity for β1-AR and improve patient tolerance. However, they still cause more side effects such as fatigue, coldness, and fluctuations in blood glucose levels. The development of third-generation blockers aims to further optimize efficacy and reduce side effects. These drugs provide effects not only of β1-AR blockade, but also of vasodilation, anti-inflammation and anti-oxidation, offering more comprehensive cardiac protection. The advancement of new-generation β1-AR blockers necessitates a multi-faceted approach. Primarily, the mitigation of adverse effects should be prioritized through refined molecular design strategies. Secondarily, comprehensive improvements in cardiac and vascular functionality are imperative via biased activation of specific signaling pathways. Additionally, modification of the pharmacokinetic profile is essential, entailing the optimization of drug half-life, distribution, and metabolism. This modification aims to extend the action duration of drugs, decrease the dosing frequency, and improve patient compliance, thereby maximizing clinical efficacy. These efforts can help achieve a more desirable therapeutic effect.
Although this article exclusively focuses on the role of β1-AR in the heart and related research, the distribution and function of β1-AR are far from being limited to cardiac tissues. β1-AR is also distributed in many other organs and tissues, such as the central nervous system, kidneys, and adipose tissues, where it plays important functions. In the central nervous system, it is involved in the regulation of neurotransmitters [243, 244]. In the kidneys, it regulates renin secretion and affects water and salt metabolism [245, 246]. In adipose tissues, it governs lipolysis and is involved in energy metabolism [247]. Therefore, when using β1-AR blockers in the treatment of cardiac diseases, it is necessary to consider the potential effects of the drugs on β1-AR in non-cardiac tissues.
In recent years, with the deepening of research, the application potential of β1-AR blockers has extended beyond the traditional cardiovascular field, presenting broader therapeutic prospects. Non-selective β1-AR blockers, represented by propranolol, have demonstrated remarkable anti-tumor effects in various cancer models [248]. Therefore, the research and development strategies of β1-AR blockers should focus on multi-target design to achieve precision therapy while reducing the side effects of the drugs.
This review summarizes the process of scientific exploration targeting β1-AR, points out the distribution features of β1-AR in the heart, and summarizes the signaling pathways mediated by β1-AR under different conditions, as well as the research progress related to β1-AR and drug applications in various cardiac diseases. It is of great significance to deepen the biological understanding of β1-AR and promote the advancement of the treatment of cardiovascular diseases.
Data availability
No datasets were generated or analysed during the current study.
References
Wang W, Zhu W, Wang S, Yang D, Crow MT, Xiao RP, et al. Sustained beta1-adrenergic stimulation modulates cardiac contractility by Ca2+/calmodulin kinase signaling pathway. Circ Res. 2004;95:798–806.
Lymperopoulos A, Rengo G, Koch WJ. Adrenergic nervous system in heart failure: pathophysiology and therapy. Circ Res. 2013;113:739–53.
Brodde OE. Beta-adrenoceptors in cardiac disease. Pharm Ther. 1993;60:405–30.
Langley JN. On the reaction of cells and of nerve-endings to certain poisons, chiefly as regards the reaction of striated muscle to nicotine and to curari. J Physiol. 1905;33:374–413.
Drews J. Paul Ehrlich: magister mundi. Nat Rev Drug Discov. 2004;3:797–801.
Dale HH. On the action of ergotoxine; with special reference to the existence of sympathetic vasodilators. J Physiol. 1913;46:291–300.
Ahlquist RP. A study of the adrenotropic receptors. Am J Physiol. 1948;153:586–600.
Black JW, Crowther AF, Shanks RG, Smith LH, Dornhorst AC. A New Adrenergic Betareceptor Antagonist. Lancet. 1964;1:1080–1.
Lands AM, Arnold A, McAuliff JP, Luduena FP, Brown TG Jr. Differentiation of receptor systems activated by sympathomimetic amines. Nature. 1967;214:597–8.
Ablad B, Carlsson B, Carlsson E, Dahlöf C, Ek L, Hultberg E. Cardiac effects of beta-adrenergic receptor antagonists. Adv Cardiol. 1974;12:290–302.
Frielle T, Collins S, Daniel KW, Caron MG, Lefkowitz RJ, Kobilka BK. Cloning of the cDNA for the human beta 1-adrenergic receptor. Proc Natl Acad Sci USA. 1987;84:7920–4.
Yang-Feng TL, Xue FY, Zhong WW, Cotecchia S, Frielle T, Caron MG, et al. Chromosomal organization of adrenergic receptor genes. Proc Natl Acad Sci USA. 1990;87:1516–20.
Warne T, Serrano-Vega MJ, Baker JG, Moukhametzianov R, Edwards PC, Henderson R, et al. Structure of a beta1-adrenergic G-protein-coupled receptor. Nature. 2008;454:486–91.
Xu X, Kaindl J, Clark MJ, Hübner H, Hirata K, Sunahara RK, et al. Binding pathway determines norepinephrine selectivity for the human β(1)AR over β(2)AR. Cell Res. 2021;31:569–79.
Brodde OE, Karad K, Zerkowski HR, Rohm N, Reidemeister JC. Coexistence of beta 1- and beta 2-adrenoceptors in human right atrium. Direct identification by (+/-)-[125I]iodocyanopindolol binding. Circ Res. 1983;53:752–8.
Brodde OE, Schüler S, Kretsch R, Brinkmann M, Borst HG, Hetzer R, et al. Regional distribution of beta-adrenoceptors in the human heart: coexistence of functional beta 1- and beta 2-adrenoceptors in both atria and ventricles in severe congestive cardiomyopathy. J Cardiovasc Pharm. 1986;8:1235–42.
Deighton NM, Motomura S, Bals S, Zerkowski HR, Brodde OE. Characterization of the beta adrenoceptor subtype(s) mediating the positive inotropic effects of epinine, dopamine, dobutamine, denopamine and xamoterol in isolated human right atrium. J Pharm Exp Ther. 1992;262:532–8.
Brodde OE, Khamssi M, Zerkowski HR. Beta-adrenoceptors in the transplanted human heart: unaltered beta-adrenoceptor density, but increased proportion of beta 2-adrenoceptors with increasing posttransplant time. Naunyn-Schmiedeberg’s Arch Pharm. 1991;344:430–6.
Rodefeld MD, Beau SL, Schuessler RB, Boineau JP, Saffitz JE. Beta-adrenergic and muscarinic cholinergic receptor densities in the human sinoatrial node: identification of a high beta 2-adrenergic receptor density. J Cardiovasc Electrophysiol. 1996;7:1039–49.
Buxton BF, Jones CR, Molenaar P, Summers RJ. Characterization and autoradiographic localization of beta-adrenoceptor subtypes in human cardiac tissues. Br J Pharm. 1987;92:299–310.
Dobson JG Jr., Fray J, Leonard JL, Pratt RE. Molecular mechanisms of reduced beta-adrenergic signaling in the aged heart as revealed by genomic profiling. Physiol Genom. 2003;15:142–7.
Lindenfeld J, Cleveland JC Jr., Kao DP, White M, Wichman S, Bristow JC, et al. Sex-related differences in age-associated downregulation of human ventricular myocardial β1-adrenergic receptors. J Heart Lung Transpl: Publ Int Soc Heart Transpl. 2016;35:352–61.
Brodde OE, Hillemann S, Kunde K, Vogelsang M, Zerkowski HR. Receptor systems affecting force of contraction in the human heart and their alterations in chronic heart failure. J Heart Lung Transpl: Publ Int Soc Heart Transpl. 1992;11:S164–74.
Beau SL, Tolley TK, Saffitz JE. Heterogeneous transmural distribution of beta-adrenergic receptor subtypes in failing human hearts. Circulation. 1993;88:2501–9.
Bristow MR, Ginsburg R, Umans V, Fowler M, Minobe W, Rasmussen R, et al. Beta 1- and beta 2-adrenergic-receptor subpopulations in nonfailing and failing human ventricular myocardium: coupling of both receptor subtypes to muscle contraction and selective beta 1-receptor down-regulation in heart failure. Circ Res. 1986;59:297–309.
Borovac JA, D’Amario D, Bozic J, Glavas D. Sympathetic nervous system activation and heart failure: Current state of evidence and the pathophysiology in the light of novel biomarkers. World J Cardiol. 2020;12:373–408.
Bristow MR, Minobe WA, Raynolds MV, Port JD, Rasmussen R, Ray PE, et al. Reduced beta 1 receptor messenger RNA abundance in the failing human heart. J Clin Investig. 1993;92:2737–45.
Ungerer M, Böhm M, Elce JS, Erdmann E, Lohse MJ. Altered expression of beta-adrenergic receptor kinase and beta 1-adrenergic receptors in the failing human heart. Circulation. 1993;87:454–63.
Zhu J, Steinberg SF. Beta(1)-Adrenergic Receptor Cleavage and Regulation by Elastase. JACC Basic Transl Sci. 2023;8:976–88.
Port JD. Dissecting Beta-Adrenergic Receptors: The Sum of Many Parts. JACC Basic Transl Sci. 2023;8:989–91.
Zhu J, Steinberg SF. β(1)-adrenergic receptor N-terminal cleavage by ADAM17; the mechanism for redox-dependent downregulation of cardiomyocyte β(1)-adrenergic receptors. J Mol Cell Cardiol. 2021;154:70–9.
Steinberg SF. Redox and proteolytic regulation of cardiomyocyte β(1)-adrenergic receptors - a novel paradigm for the regulation of catecholamine responsiveness in the heart. Front Immunol. 2023;14:1306467.
Xie L, Xue F, Cheng C, Sui W, Zhang J, Meng L, et al. Cardiomyocyte-specific knockout of ADAM17 alleviates doxorubicin-induced cardiomyopathy via inhibiting TNFα-TRAF3-TAK1-MAPK axis. Signal Transduct Target Ther. 2024;9:273.
Zhu J, Steinberg SF. Trypsin cleavage of the β(1)-adrenergic receptor. Am J Physiol Heart Circ Physiol. 2022;322:H486–h91.
Sato PY, Chuprun JK, Schwartz M, Koch WJ. The evolving impact of G protein-coupled receptor kinases in cardiac health and disease. Physiol Rev. 2015;95:377–404.
Jain A, Heremans I, Rademaker G, Detomasi TC, Rohweder P, Anderson D, et al. Leucine aminopeptidase LyLAP enables lysosomal degradation of membrane proteins. Sci (N Y, NY). 2025;387:eadq8331.
Kwan Z, Paulose Nadappuram B, Leung MM, Mohagaonkar S, Li A, Amaradasa KS, et al. Microtubule-Mediated Regulation of β(2)AR Translation and Function in Failing Hearts. Circ Res. 2023;133:944–58.
Bathe-Peters M, Gmach P, Boltz HH, Einsiedel J, Gotthardt M, Hübner H, et al. Visualization of β-adrenergic receptor dynamics and differential localization in cardiomyocytes. Proc Natl Acad Sci USA. 2021;118.
Steinberg SF. beta(2)-Adrenergic receptor signaling complexes in cardiomyocyte caveolae/lipid rafts. J Mol Cell Cardiol. 2004;37:407–15.
Insel PA, Head BP, Ostrom RS, Patel HH, Swaney JS, Tang CM, et al. Caveolae and lipid rafts: G protein-coupled receptor signaling microdomains in cardiac myocytes. Ann N Y Acad Sci. 2005;1047:166–72.
Wang Y, Shi Q, Li M, Zhao M, Reddy Gopireddy R, Teoh JP, et al. Intracellular β(1)-Adrenergic Receptors and Organic Cation Transporter 3 Mediate Phospholamban Phosphorylation to Enhance Cardiac Contractility. Circ Res. 2021;128:246–61.
Liccardo F, Morstein J, Lin TY, Pampel J, Lang D, Shokat KM, et al. Subcellular activation of β-adrenergic receptors using a spatially restricted antagonist. Proc Natl Acad Sci USA. 2024;121:e2404243121.
Xu B, Bahriz S, Salemme VR, Wang Y, Zhu C, Zhao M, et al. Differential Downregulation of β(1)-Adrenergic Receptor Signaling in the Heart. J Am Heart Assoc. 2024;13:e033733.
Irannejad R, Pessino V, Mika D, Huang B, Wedegaertner PB, Conti M, et al. Functional selectivity of GPCR-directed drug action through location bias. Nat Chem Biol. 2017;13:799–806.
Nash CA, Wei W, Irannejad R, Smrcka AV Golgi localized β1-adrenergic receptors stimulate Golgi PI4P hydrolysis by PLCε to regulate cardiac hypertrophy. eLife. 2019;8.
Vaniotis G, Allen BG, Hébert TE. Nuclear GPCRs in cardiomyocytes: an insider’s view of β-adrenergic receptor signaling. Am J Physiol Heart Circ Physiol. 2011;301:H1754–64.
Boivin B, Lavoie C, Vaniotis G, Baragli A, Villeneuve LR, Ethier N, et al. Functional beta-adrenergic receptor signalling on nuclear membranes in adult rat and mouse ventricular cardiomyocytes. Cardiovasc Res. 2006;71:69–78.
Vaniotis G, Del Duca D, Trieu P, Rohlicek CV, Hébert TE, Allen BG. Nuclear β-adrenergic receptors modulate gene expression in adult rat heart. Cell Signal. 2011;23:89–98.
Nicoli A, Dunkel A, Giorgino T, de Graaf C, Di Pizio A. Classification Model for the Second Extracellular Loop of Class A GPCRs. J Chem Inf Model. 2022;62:511–22.
Lawson Z, Wheatley M. The third extracellular loop of G-protein-coupled receptors: more than just a linker between two important transmembrane helices. Biochem Soc Trans. 2004;32:1048–50.
Scarselli M, Li B, Kim SK, Wess J. Multiple residues in the second extracellular loop are critical for M3 muscarinic acetylcholine receptor activation. J Biol Chem. 2007;282:7385–96.
Burstein ES, Spalding TA, Brann MR. The second intracellular loop of the m5 muscarinic receptor is the switch which enables G-protein coupling. J Biol Chem. 1998;273:24322–7.
Wong SK, Ross EM. Chimeric muscarinic cholinergic:beta-adrenergic receptors that are functionally promiscuous among G proteins. J Biol Chem. 1994;269:18968–76.
Rosenbaum DM, Cherezov V, Hanson MA, Rasmussen SG, Thian FS, Kobilka TS, et al. GPCR engineering yields high-resolution structural insights into beta2-adrenergic receptor function. Science. 2007;318:1266–73.
Rasmussen SG, Choi HJ, Rosenbaum DM, Kobilka TS, Thian FS, Edwards PC, et al. Crystal structure of the human beta2 adrenergic G-protein-coupled receptor. Nature. 2007;450:383–7.
Alegre KO, Paknejad N, Su M, Lou JS, Huang J, Jordan KD, et al. Structural basis and mechanism of activation of two different families of G proteins by the same GPCR. Nat Struct Mol Biol. 2021;28:936–44.
Cahill TJ 3rd, Thomsen AR, Tarrasch JT, Plouffe B, Nguyen AH, et al. Distinct conformations of GPCR-β-arrestin complexes mediate desensitization, signaling, and endocytosis. Proc Natl Acad Sci USA. 2017;114:2562–7.
Shukla AK, Westfield GH, Xiao K, Reis RI, Huang LY, Tripathi-Shukla P, et al. Visualization of arrestin recruitment by a G-protein-coupled receptor. Nature. 2014;512:218–22.
Maharana J, Sano FK, Sarma P, Yadav MK, Duan L, Stepniewski TM, et al. Molecular insights into atypical modes of β-arrestin interaction with seven transmembrane receptors. Science. 2024;383:101–8.
Graham RM. Adrenergic receptors: structure and function. Clevel Clin J Med. 1990;57:481–91.
Leone M, Albanèse J, Martin C. Positive inotropic stimulation. Curr Opin Crit Care. 2002;8:395–403.
Taylor SS, Søberg K, Kobori E, Wu J, Pautz S, Herberg FW, et al. The Tails of Protein Kinase A. Mol Pharm. 2022;101:219–25.
Chen D, Wang J, Cao J, Zhu G. cAMP-PKA signaling pathway and anxiety: Where do we go next?. Cell Signal. 2024;122:111311.
Woo AY, Xiao RP. β-Adrenergic receptor subtype signaling in heart: from bench to bedside. Acta Pharmacol Sin. 2012;33:335–41.
Gurney AM, Charnet P, Pye JM, Nargeot J. Augmentation of cardiac calcium current by flash photolysis of intracellular caged-Ca2+ molecules. Nature. 1989;341:65–8.
Huke S, Bers DM. Ryanodine receptor phosphorylation at Serine 2030, 2808 and 2814 in rat cardiomyocytes. Biochem Biophys Res Commun. 2008;376:80–5.
Bates SE, Gurney AM. Ca(2+)-dependent block and potentiation of L-type calcium current in guinea-pig ventricular myocytes. J Physiol. 1993;466:345–65.
Ullrich ND, Valdivia HH, Niggli E. PKA phosphorylation of cardiac ryanodine receptor modulates SR luminal Ca2+ sensitivity. J Mol Cell Cardiol. 2012;53:33–42.
Sulakhe PV, Vo XT, Phan TD, Morris TE. Phosphorylation of inhibitory subunit of troponin and phospholamban in rat cardiomyocytes: modulation by exposure of cardiomyocytes to hydroxyl radicals and sulfhydryl group reagents. Mol Cell Biochem. 1997;175:98–107.
Sulakhe PV, Vo XT. Regulation of phospholamban and troponin-I phosphorylation in the intact rat cardiomyocytes by adrenergic and cholinergic stimuli: roles of cyclic nucleotides, calcium, protein kinases and phosphatases and depolarization. Mol Cell Biochem. 1995;149-150:103–26.
Vafiadaki E, Kranias EG, Eliopoulos AG, Sanoudou D. The phospholamban R14del generates pathogenic aggregates by impairing autophagosome-lysosome fusion. Cell Mol Life Sci: CMLS. 2024;81:450.
Simmerman HK, Jones LR. Phospholamban: protein structure, mechanism of action, and role in cardiac function. Physioll Rev. 1998;78:921–47.
Grimm M, Brown JH. Beta-adrenergic receptor signaling in the heart: role of CaMKII. J Mol Cell Cardiol. 2010;48:322–30.
Shi Q, Malik H, Crawford RM, Streeter J, Wang J, Huo R, et al. Cardiac monoamine oxidase-A inhibition protects against catecholamine-induced ventricular arrhythmias via enhanced diastolic calcium control. Cardiovasc Res. 2024;120:596–611.
Wang Y, Zhao M, Xu B, Bahriz SMF, Zhu C, Jovanovic A, et al. Monoamine oxidase A and organic cation transporter 3 coordinate intracellular β(1)AR signaling to calibrate cardiac contractile function. Basic Res Cardiol. 2022;117:37.
Kaludercic N, Takimoto E, Nagayama T, Feng N, Lai EW, Bedja D, et al. Monoamine oxidase A-mediated enhanced catabolism of norepinephrine contributes to adverse remodeling and pump failure in hearts with pressure overload. Circul Res. 2010;106:193–202.
Wang Y, Zhao M, Shi Q, Xu B, Zhu C, Li M, et al. Monoamine Oxidases Desensitize Intracellular β(1)AR Signaling in Heart Failure. Circul Res. 2021;129:965–7.
Dahl EF, Wright CD, O’Connell TD. Quantification of catecholamine uptake in adult cardiac myocytes. Methods Mol Biol. 2015;1234:43–52.
Nagayama T, Takimoto E, Sadayappan S, Mudd JO, Seidman JG, Robbins J, et al. Control of In Vivo Contraction/Relaxation kinetics by Myosin Binding Protein C: Protein Kinase A Phosphorylation Dependent and Independent Regulation. Circulation. 2007;116:2399–408.
Sadayappan S, Osinska H, Klevitsky R, Lorenz JN, Sargent M, Molkentin JD, et al. Cardiac myosin binding protein C phosphorylation is cardioprotective. Proc Natl Acad Sci USA. 2006;103:16918–23.
Previs MJ, Beck Previs S, Gulick J, Robbins J, Warshaw DM. Molecular mechanics of cardiac myosin-binding protein C in native thick filaments. Sci (N Y, NY). 2012;337:1215–8.
Ruiz-Hurtado G, Morel E, Domínguez-Rodríguez A, Llach A, Lezoualc’h F, Benitah JP, et al. Epac in cardiac calcium signaling. J Mol Cell Cardiol. 2013;58:162–71.
Ai X, Curran JW, Shannon TR, Bers DM, Pogwizd SM. Ca2+/calmodulin-dependent protein kinase modulates cardiac ryanodine receptor phosphorylation and sarcoplasmic reticulum Ca2+ leak in heart failure. Circ Res. 2005;97:1314–22.
Curran J, Hinton MJ, Ríos E, Bers DM, Shannon TR. Beta-adrenergic enhancement of sarcoplasmic reticulum calcium leak in cardiac myocytes is mediated by calcium/calmodulin-dependent protein kinase. Circ Res. 2007;100:391–8.
Pereira L, Métrich M, Fernández-Velasco M, Lucas A, Leroy J, Perrier R, et al. The cAMP binding protein Epac modulates Ca2+ sparks by a Ca2+/calmodulin kinase signalling pathway in rat cardiac myocytes. J Physiol. 2007;583:685–94.
Wang P, Xu S, Xu J, Xin Y, Lu Y, Zhang H, et al. Elevated MCU Expression by CaMKIIδB Limits Pathological Cardiac Remodeling. Circulation. 2022;145:1067–83.
Lissandron V, Podini P, Pizzo P, Pozzan T. Unique characteristics of Ca2+ homeostasis of the trans-Golgi compartment. Proc Natl Acad Sci USA. 2010;107:9198–203.
Yang Z, Kirton HM, MacDougall DA, Boyle JP, Deuchars J, Frater B, et al. The Golgi apparatus is a functionally distinct Ca2+ store regulated by the PKA and Epac branches of the β1-adrenergic signaling pathway. Sci Signal. 2015;8:ra101.
Kho C, Lee A, Hajjar RJ. Altered sarcoplasmic reticulum calcium cycling–targets for heart failure therapy. Nat Rev Cardiol. 2012;9:717–33.
Park SH, Kim ND, Jung JK, Lee CK, Han SB, Kim Y. Myeloid differentiation 2 as a therapeutic target of inflammatory disorders. Pharm Ther. 2012;133:291–8.
Qian JF, Liang SQ, Wang QY, Xu JC, Luo W, Huang WJ, et al. Isoproterenol induces MD2 activation by β-AR-cAMP-PKA-ROS signalling axis in cardiomyocytes and macrophages drives inflammatory heart failure. Acta Pharm Sin. 2024;45:531–44.
Zhao B, Tumaneng K, Guan KL. The Hippo pathway in organ size control, tissue regeneration and stem cell self-renewal. Nat cell Biol. 2011;13:877–83.
Sakabe M, Thompson M, Chen N, Verba M, Hassan A, Lu R, et al. Inhibition of β1-AR/Gαs signaling promotes cardiomyocyte proliferation in juvenile mice through activation of RhoA-YAP axis. eLife. 2022;11.
Guan K, Li Z. β-adrenergic receptor inhibits heart regeneration by downregulating Yap m6A modification. Cell Death Dis. 2025;16:294.
Devic E, Xiang Y, Gould D, Kobilka B. Beta-adrenergic receptor subtype-specific signaling in cardiac myocytes from beta(1) and beta(2) adrenoceptor knockout mice. Mol Pharm. 2001;60:577–83.
Xiao RP, Zhang SJ, Chakir K, Avdonin P, Zhu W, Bond RA, et al. Enhanced G(i) signaling selectively negates beta2-adrenergic receptor (AR)–but not beta1-AR-mediated positive inotropic effect in myocytes from failing rat hearts. Circulation. 2003;108:1633–9.
Xiang Y, Devic E, Kobilka B. The PDZ binding motif of the beta 1 adrenergic receptor modulates receptor trafficking and signaling in cardiac myocytes. J Biol Chem. 2002;277:33783–90.
Martin NP, Whalen EJ, Zamah MA, Pierce KL, Lefkowitz RJ. PKA-mediated phosphorylation of the beta1-adrenergic receptor promotes Gs/Gi switching. Cell Signal. 2004;16:1397–403.
Wang J, Hanada K, Staus DP, Makara MA, Dahal GR, Chen Q, et al. Gα(i) is required for carvedilol-induced β(1) adrenergic receptor β-arrestin biased signaling. Nat Commun. 2017;8:1706.
Kim IM, Tilley DG, Chen J, Salazar NC, Whalen EJ, Violin JD, et al. Beta-blockers alprenolol and carvedilol stimulate beta-arrestin-mediated EGFR transactivation. Proc Natl Acad Sci USA. 2008;105:14555–60.
Noma T, Lemaire A, Naga Prasad SV, Barki-Harrington L, Tilley DG, Chen J, et al. Beta-arrestin-mediated beta1-adrenergic receptor transactivation of the EGFR confers cardioprotection. J Clin Investig. 2007;117:2445–58.
Wang Q, Wang Y, West TM, Liu Y, Reddy GR, Barbagallo F, et al. Carvedilol induces biased β1 adrenergic receptor-nitric oxide synthase 3-cyclic guanylyl monophosphate signalling to promote cardiac contractility. Cardiovasc Res. 2021;117:2237–51.
Moore CA, Milano SK, Benovic JL. Regulation of receptor trafficking by GRKs and arrestins. Annu Rev Physiol. 2007;69:451–82.
Shenoy SK, Lefkowitz RJ. β-Arrestin-mediated receptor trafficking and signal transduction. Trends Pharm Sci. 2011;32:521–33.
Belmonte SL, Blaxall BC. G protein coupled receptor kinases as therapeutic targets in cardiovascular disease. Circ Res. 2011;109:309–19.
Ferguson SS. Evolving concepts in G protein-coupled receptor endocytosis: the role in receptor desensitization and signaling. Pharm Rev. 2001;53:1–24.
Prossnitz ER. Novel roles for arrestins in the post-endocytic trafficking of G protein-coupled receptors. Life Sci. 2004;75:893–9.
Odnoshivkina JG, Averin AS, Khakimov IR, Trusov NA, Trusova DA, Petrov AM. The mechanism of 25-hydroxycholesterol-mediated suppression of atrial β1-adrenergic responses. Pflug Arch: Eur J Physiol. 2024;476:407–21.
Goodman OB Jr., Krupnick JG, Santini F, Gurevich VV, Penn RB, Gagnon AW, et al. Beta-arrestin acts as a clathrin adaptor in endocytosis of the beta2-adrenergic receptor. Nature. 1996;383:447–50.
Laporte SA, Oakley RH, Zhang J, Holt JA, Ferguson SS, Caron MG, et al. The beta2-adrenergic receptor/betaarrestin complex recruits the clathrin adaptor AP-2 during endocytosis. Proc Natl Acad Sci USA. 1999;96:3712–7.
Premont RT, Claing A, Vitale N, Perry SJ, Lefkowitz RJ. The GIT family of ADP-ribosylation factor GTPase-activating proteins. Functional diversity of GIT2 alternative splicings. J Biol Chem. 2000;275:22373–80.
Chen H, Zhang S, Zhang X, Liu H. QR code model: a new possibility for GPCR phosphorylation recognition. Cell Commun Signal: CCS. 2022;20:23.
Zang Y, Kahsai AW, Pakharukova N, Huang LY, Lefkowitz RJ. The GPCR-β-arrestin complex allosterically activates C-Raf by binding its amino terminus. J Biol Chem. 2021;297:101369.
Pakharukova N, Masoudi A, Pani B, Staus DP, Lefkowitz RJ. Allosteric activation of proto-oncogene kinase Src by GPCR-beta-arrestin complexes. J Biol Chem. 2020;295:16773–84.
Tilley DG, Kim IM, Patel PA, Violin JD, Rockman HA. beta-Arrestin mediates beta1-adrenergic receptor-epidermal growth factor receptor interaction and downstream signaling. J Biol Chem. 2009;284:20375–86.
Roskoski R Jr. Src protein-tyrosine kinase structure, mechanism, and small molecule inhibitors. Pharm Res. 2015;94:9–25.
Lavoie H, Gagnon J, Therrien M. ERK signalling: a master regulator of cell behaviour, life and fate. Nat Rev Mol Cell Biol. 2020;21:607–32.
Kolch W. Coordinating ERK/MAPK signalling through scaffolds and inhibitors. Nat Rev Mol Cell Biol. 2005;6:827–37.
Lymperopoulos A. GRK2 and β-arrestins in cardiovascular disease: Something old, something new. Am J Cardiovasc Dis. 2011;1:126–37.
Cheng SB, Filardo EJ. Trans-Golgi Network (TGN) as a regulatory node for β1-adrenergic receptor (β1AR) down-modulation and recycling. J Biol Chem. 2012;287:14178–91.
Cheng SB, Quinn JA, Graeber CT, Filardo EJ. Down-modulation of the G-protein-coupled estrogen receptor, GPER, from the cell surface occurs via a trans-Golgi-proteasome pathway. J Biol Chem. 2011;286:22441–55.
Lelouvier B, Tamagno G, Kaindl AM, Roland A, Lelievre V, Le Verche V, et al. Dynamics of somatostatin type 2A receptor cargoes in living hippocampal neurons. J Neurosci: J Soc Neurosci. 2008;28:4336–49.
Csaba Z, Lelouvier B, Viollet C, El Ghouzzi V, Toyama K, Videau C, et al. Activated somatostatin type 2 receptors traffic in vivo in central neurons from dendrites to the trans Golgi before recycling. Traffic. 2007;8:820–34.
Escola JM, Kuenzi G, Gaertner H, Foti M, Hartley O. CC chemokine receptor 5 (CCR5) desensitization: cycling receptors accumulate in the trans-Golgi network. J Biol Chem. 2010;285:41772–80.
Eichel K, Jullié D, von Zastrow M. β-Arrestin drives MAP kinase signalling from clathrin-coated structures after GPCR dissociation. Nat Cell Biol. 2016;18:303–10.
Dwivedi H, Baidya M, Shukla AK. GPCR Signaling: The Interplay of Gαi and β-arrestin. Curr Biol. 2018;28:R324–r7.
Kim IM, Wang Y, Park KM, Tang Y, Teoh JP, Vinson J, et al. β-arrestin1-biased β1-adrenergic receptor signaling regulates microRNA processing. Circ Res. 2014;114:833–44.
Jane-wit D, Altuntas CZ, Johnson JM, Yong S, Wickley PJ, Clark P, et al. Beta 1-adrenergic receptor autoantibodies mediate dilated cardiomyopathy by agonistically inducing cardiomyocyte apoptosis. Circulation. 2007;116:399–410.
Communal C, Singh K, Sawyer DB, Colucci WS. Opposing effects of beta(1)- and beta(2)-adrenergic receptors on cardiac myocyte apoptosis: role of a pertussis toxin-sensitive G protein. Circulation. 1999;100:2210–2.
Pereira L, Cheng H, Lao DH, Na L, van Oort RJ, Brown JH, et al. Epac2 mediates cardiac β1-adrenergic-dependent sarcoplasmic reticulum Ca2+ leak and arrhythmia. Circulation. 2013;127:913–22.
Wang L, Hao H, Wang J, Wang X, Zhang S, Du Y, et al. Decreased autophagy: a major factor for cardiomyocyte death induced by β1-adrenoceptor autoantibodies. Cell Death Dis. 2015;6:e1862.
Andersson L, Cinato M, Mardani I, Miljanovic A, Arif M, Koh A, et al. Glucosylceramide synthase deficiency in the heart compromises β1-adrenergic receptor trafficking. Eur heart J. 2021;42:4481–92.
Coronado M, Fajardo G, Nguyen K, Zhao M, Kooiker K, Jung G, et al. Physiological Mitochondrial Fragmentation Is a Normal Cardiac Adaptation to Increased Energy Demand. Circ Res. 2018;122:282–95.
Morisco C, Zebrowski DC, Vatner DE, Vatner SF, Sadoshima J. Beta-adrenergic cardiac hypertrophy is mediated primarily by the beta(1)-subtype in the rat heart. J Mol Cell Cardiol. 2001;33:561–73.
Masson S, Arosio B, Luvarà G, Gagliano N, Fiordaliso F, Santambrogio D, et al. Remodelling of cardiac extracellular matrix during beta-adrenergic stimulation: upregulation of SPARC in the myocardium of adult rats. J Mol Cell Cardiol. 1998;30:1505–14.
Flori L, Lazzarini G, Spezzini J, Pirone A, Calderone V, Testai L, et al. The isoproterenol-induced myocardial fibrosis: A biochemical and histological investigation. Biomed Pharmacother. 2024;174:116534.
Gigli M, Stolfo D, Merlo M, Sinagra G, Taylor MRG, Mestroni L. Pathophysiology of dilated cardiomyopathy: from mechanisms to precision medicine. Nat Rev Cardiol. 2025;22:183–98.
Nakamura M, Sadoshima J. Mechanisms of physiological and pathological cardiac hypertrophy. Nat Rev Cardiol. 2018;15:387–407.
Shimizu I, Minamino T. Physiological and pathological cardiac hypertrophy. J Mol Cell Cardiol. 2016;97:245–62.
Morisco C, Marrone C, Galeotti J, Shao D, Vatner DE, Vatner SF, et al. Endocytosis machinery is required for beta1-adrenergic receptor-induced hypertrophy in neonatal rat cardiac myocytes. Cardiovasc Res. 2008;78:36–44.
Endoh M. Novel signalling cascade for cardiac hypertrophy activation by uncoupling and internalization of beta1-adrenoceptors. Cardiovasc Res. 2008;78:5–7.
Sethi R, Saini HK, Guo X, Wang X, Elimban V, Dhalla NS. Dependence of changes in beta-adrenoceptor signal transduction on type and stage of cardiac hypertrophy. J Appl Physiol. 2007;102:978–84.
Pandya K, Porter K, Rockman HA, Smithies O. Decreased beta-adrenergic responsiveness following hypertrophy occurs only in cardiomyocytes that also re-express beta-myosin heavy chain. Eur J Heart Fail. 2009;11:648–52.
Podlowski S, Wenzel K, Luther HP, Müller J, Bramlage P, Baumann G, et al. Beta1-adrenoceptor gene variations: a role in idiopathic dilated cardiomyopathy?. J Mol Med. 2000;78:87–93.
Tatman PD, Kao DP, Chatfield KC, Carroll IA, Wagner JA, Jonas ER, et al. An extensive β1-adrenergic receptor gene signaling network regulates molecular remodeling in dilated cardiomyopathies. JCI insight. 2023;8.
Iwai C, Akita H, Shiga N, Takai E, Miyamoto Y, Shimizu M, et al. Suppressive effect of the Gly389 allele of the beta1-adrenergic receptor gene on the occurrence of ventricular tachycardia in dilated cardiomyopathy. Circ Soc. 2002;66:723–8.
Forleo C, Resta N, Sorrentino S, Guida P, Manghisi A, De Luca V, et al. Association of beta-adrenergic receptor polymorphisms and progression to heart failure in patients with idiopathic dilated cardiomyopathy. Am J Med. 2004;117:451–8.
Wang L, Lu L, Zhang F, Chen Q, Shen W. Polymorphisms of beta-adrenoceptor and natriuretic peptide receptor genes influence the susceptibility to and the severity of idiopathic dilated cardiomyopathy in a Chinese cohort. J Card Fail. 2010;16:36–44.
Maqbool A, Hall AS, Ball SG, Balmforth AJ. Common polymorphisms of beta1-adrenoceptor: identification and rapid screening assay. Lancet. 1999;353:897.
Magnusson Y, Levin MC, Eggertsen R, Nyström E, Mobini R, Schaufelberger M, et al. Ser49Gly of beta1-adrenergic receptor is associated with effective beta-blocker dose in dilated cardiomyopathy. Clin Pharm Ther. 2005;78:221–31.
Khurshid S, Choi SH, Weng LC, Wang EY, Trinquart L, Benjamin EJ, et al. Frequency of Cardiac Rhythm Abnormalities in a Half Million Adults. Circ Arrhythmia Electrophysiol. 2018;11:e006273.
Sossalla S, Vollmann D. Arrhythmia-Induced Cardiomyopathy. Dtsch Arzteblatt Int. 2018;115:335–41.
Blackwell DJ, Schmeckpeper J, Knollmann BC. Animal Models to Study Cardiac Arrhythmias. Circ Res. 2022;130:1926–64.
Benjamin EJ, Wolf PA, D’Agostino RB, Silbershatz H, Kannel WB, Levy D. Impact of atrial fibrillation on the risk of death: the Framingham Heart Study. Circulation. 1998;98:946–52.
Fei YD, Wang Q, Hou JW, Li W, Cai XX, Yang YL, et al. Macrophages facilitate post myocardial infarction arrhythmias: roles of gap junction and KCa3.1. Theranostics. 2019;9:6396–411.
Gawałko M, Balsam P, Lodziński P, Grabowski M, Krzowski B, Opolski G, et al. Cardiac Arrhythmias in Autoimmune Diseases. Circulation J. 2020;84:685–94.
Yang Y, Fan A, Lin H, Wang X, Yang K, Zhang H, et al. Role of macrophages in cardiac arrhythmias: Pathogenesis and therapeutic perspectives. Int Immunopharmacol. 2025;149:114206.
Owais A, Barney M, Ly OT, Brown G, Chen H, Sridhar A, et al. Genetics and Pharmacogenetics of Atrial Fibrillation: A Mechanistic Perspective. JACC Basic Transl Sci. 2024;9:918–34.
Parikh KS, Piccini JP. Pharmacogenomics of Bucindolol in Atrial Fibrillation and Heart Failure. Curr Heart Fail Rep. 2017;14:529–35.
Doki K, Sekiguchi Y, Kuga K, Aonuma K, Homma M. β1-Adrenergic receptor Arg389Gly polymorphism affects the antiarrhythmic efficacy of flecainide in patients with coadministration of β-blockers. Pharmacogenet Genomics. 2016;26:481–5.
Stöckigt F, Brixius K, Lickfett L, Andrié R, Linhart M, Nickenig G, et al. Total beta-adrenoceptor knockout slows conduction and reduces inducible arrhythmias in the mouse heart. PloS one. 2012;7:e49203.
Feldman AM, Gordon J, Wang J, Song J, Zhang XQ, Myers VD, et al. BAG3 regulates contractility and Ca(2+) homeostasis in adult mouse ventricular myocytes. J Mol Cell Cardiol. 2016;92:10–20.
Suita K, Fujita T, Hasegawa N, Cai W, Jin H, Hidaka Y, et al. Norepinephrine-Induced Adrenergic Activation Strikingly Increased the Atrial Fibrillation Duration through β1- and α1-Adrenergic Receptor-Mediated Signaling in Mice. PloS one. 2015;10:e0133664.
Jingjun L, Yan Z, Weijie, Dongdong Z, Guosheng L, Mingwei B. Effect and mechanism of esmolol given during cardiopulmonary resuscitation in a porcine ventricular fibrillation model. Resuscitation. 2009;80:1052–9.
Schinner C, Erber BM, Yeruva S, Waschke J. Regulation of cardiac myocyte cohesion and gap junctions via desmosomal adhesion. Acta Physiol. 2019;226:e13242.
Aflaki M, Qi XY, Xiao L, Ordog B, Tadevosyan A, Luo X, et al. Exchange protein directly activated by cAMP mediates slow delayed-rectifier current remodeling by sustained β-adrenergic activation in guinea pig hearts. Circ Res. 2014;114:993–1003.
Maslov LN, Naryzhnaya NV, Voronkov NS, Kurbatov BK, Derkachev IA, Ryabov VV, et al. The role of β-adrenergic receptors in the regulation of cardiac tolerance to ischemia/reperfusion. Why do β-adrenergic receptor agonists and antagonists protect the heart?. Fundam Clin Pharm. 2024;38:658–73.
Dostanic S, Servant N, Wang C, Chalifour LE. Chronic beta-adrenoreceptor stimulation in vivo decreased Bcl-2 and increased Bax expression but did not activate apoptotic pathways in mouse heart. Can J Physiol Pharm. 2004;82:167–74.
Hashemi S, Salma J, Wales S, McDermott JC. Pro-survival function of MEF2 in cardiomyocytes is enhanced by β-blockers. Cell Death Discov. 2015;1:15019.
Zhu WZ, Wang SQ, Chakir K, Yang D, Zhang T, Brown JH, et al. Linkage of beta1-adrenergic stimulation to apoptotic heart cell death through protein kinase A-independent activation of Ca2+/calmodulin kinase II. J Clin Investig. 2003;111:617–25.
Wang Y, Wang Y, Yang D, Yu X, Li H, Lv X, et al. β₁-adrenoceptor stimulation promotes LPS-induced cardiomyocyte apoptosis through activating PKA and enhancing CaMKII and IκBα phosphorylation. Crit Care. 2015;19:76.
Seqqat R, Guo X, Rafiq K, Kolpakov MA, Guo J, Koch WJ, et al. Beta1-adrenergic receptors promote focal adhesion signaling downregulation and myocyte apoptosis in acute volume overload. J Mol Cell Cardiol. 2012;53:240–9.
Lee GJ, Yan L, Vatner DE, Vatner SF. Mst1 inhibition rescues β1-adrenergic cardiomyopathy by reducing myocyte necrosis and non-myocyte apoptosis rather than myocyte apoptosis. Basic Res Cardiol. 2015;110:7.
Yoo B, Lemaire A, Mangmool S, Wolf MJ, Curcio A, Mao L, et al. Beta1-adrenergic receptors stimulate cardiac contractility and CaMKII activation in vivo and enhance cardiac dysfunction following myocardial infarction. Am J Physiol Heart Circulatory Physiol. 2009;297:H1377–86.
Spear JF, Prabu SK, Galati D, Raza H, Anandatheerthavarada HK, Avadhani NG. beta1-Adrenoreceptor activation contributes to ischemia-reperfusion damage as well as playing a role in ischemic preconditioning. Am J Physiol Heart Circulatory Physiol. 2007;292:H2459–66.
Sun X, Han Y, Yu Y, Chen Y, Dong C, Lv Y, et al. Overexpressing of the GIPC1 protects against pathological cardiac remodelling. Eur J Pharm. 2024;971:176488.
Muslimova E, Rebrova T, Kondratieva D, Korepanov V, Sonduev E, Kozlov B, et al. Expression of the β1-adrenergic receptor (ADRB1) gene in the myocardium and β-adrenergic reactivity of the body in patients with a history of myocardium infraction. Gene. 2022;844:146820.
Du Y, Zhang M, Zhao W, Shu Y, Gao M, Zhuang Y, et al. Let-7a regulates expression of β1-adrenoceptors and forms a negative feedback circuit with the β1-adrenoceptor signaling pathway in chronic ischemic heart failure. Oncotarget. 2017;8:8752–64.
McLean RC, Hirsch GA, Becker LC, Kasch-Semenza L, Gerstenblith G, Schulman SP. Polymorphisms of the beta adrenergic receptor predict left ventricular remodeling following acute myocardial infarction. Cardiovasc drugs Ther. 2011;25:251–8.
Yilmaz A, Kaya MG, Merdanoglu U, Ergun MA, Cengel A, Menevse S. Association of beta-1 and beta-2 adrenergic receptor gene polymorphisms with myocardial infarction. J Clin Lab Anal. 2009;23:237–43.
Böhm M, Flesch M, Schnabel P. Beta-adrenergic signal transduction in the failing and hypertrophied myocardium. J Mol Med. 1997;75:842–8.
Bristow MR, Ginsburg R, Minobe W, Cubicciotti RS, Sageman WS, Lurie K, et al. Decreased catecholamine sensitivity and beta-adrenergic-receptor density in failing human hearts. N Engl J Med. 1982;307:205–11.
Woo AY, Song Y, Xiao RP, Zhu W. Biased β2-adrenoceptor signalling in heart failure: pathophysiology and drug discovery. Br J Pharm. 2015;172:5444–56.
Engelhardt S, Hein L, Wiesmann F, Lohse MJ. Progressive hypertrophy and heart failure in beta1-adrenergic receptor transgenic mice. Proc Natl Acad Sci USA. 1999;96:7059–64.
Nooh MM, Chumpia MM, Hamilton TB, Bahouth SW. Sorting of β1-adrenergic receptors is mediated by pathways that are either dependent on or independent of type I PDZ, protein kinase A (PKA), and SAP97. J Biol Chem. 2014;289:2277–94.
Fu Q, Kim S, Soto D, De Arcangelis V, DiPilato L, Liu S, et al. A long lasting β1 adrenergic receptor stimulation of cAMP/protein kinase A (PKA) signal in cardiac myocytes. J Biol Chem. 2014;289:14771–81.
Xu B, Li M, Wang Y, Zhao M, Morotti S, Shi Q, et al. GRK5 Controls SAP97-Dependent Cardiotoxic β(1) Adrenergic Receptor-CaMKII Signaling in Heart Failure. Circ Res. 2020;127:796–810.
Dewenter M, Neef S, Vettel C, Lämmle S, Beushausen C, Zelarayan LC, et al. Calcium/Calmodulin-Dependent Protein Kinase II Activity Persists During Chronic β-Adrenoceptor Blockade in Experimental and Human Heart Failure. Circ Heart Fail. 2017;10:e003840.
Muthumala A, Drenos F, Elliott PM, Humphries SE. Role of beta adrenergic receptor polymorphisms in heart failure: systematic review and meta-analysis. Eur J Heart Fail. 2008;10:3–13.
Liu WN, Fu KL, Gao HY, Shang YY, Wang ZH, Jiang GH, et al. β1 adrenergic receptor polymorphisms and heart failure: a meta-analysis on susceptibility, response to β-blocker therapy and prognosis. PloS one. 2012;7:e37659.
Becker NP, Müller J, Göttel P, Wallukat G, Schimke I. Cardiomyopathy - An approach to the autoimmune background. Autoimmun Rev. 2017;16:269–86.
Chen H, Cao N, Wang L, Wu Y, Wei H, Li Y, et al. Biased activation of β(2)-AR/Gi/GRK2 signal pathway attenuated β(1)-AR sustained activation induced by β(1)-adrenergic receptor autoantibody. Cell Death Discov. 2021;7:340.
Shang L, Zhang L, Shao M, Feng M, Shi J, Dong Z, et al. Elevated β1-Adrenergic Receptor Autoantibody Levels Increase Atrial Fibrillation Susceptibility by Promoting Atrial Fibrosis. Front Physiol. 2020;11:76.
Yang N, Sun H, Xi L, Zhang L, Lu Y, Wang Q, et al. Oroxin B Resembles Bisoprolol in Attenuating Beta1-Adrenergic Receptor Autoantibody-Induced Atrial Remodelling via the PTEN/AKT/mTOR Signalling Pathway. Clin Exp Pharm Physiol. 2025;52:e70011.
Wei XQ, Zhang HS, Wei GH, Zhang JG, Du YY, Tan HY, et al. Honokiol Protects against Anti-β1-Adrenergic Receptor Autoantibody-Induced Myocardial Dysfunction via Activation of Autophagy. Oxid Med Cell Longev. 2018;2018:1640804.
Werner S, Wallukat G, Becker NP, Wenzel K, Müller J, Schimke I, et al. The aptamer BC 007 for treatment of dilated cardiomyopathy: evaluation in Doberman Pinschers of efficacy and outcomes. ESC heart Fail. 2020;7:844–55.
Xi L, Sun H, Yang N, Wang Q, Zhang L, Song J, et al. Pioglitazone Alleviates β1-Adrenergic Receptor Antibody-Induced Atrial Fibrillation Susceptivity via Mitigation of PPAR-γ-Mediated Metabolic Inflexibility. Curr Med Chem. 2024.
Cao N, Chen H, Bai Y, Yang X, Xu W, Hao W, et al. β2-adrenergic receptor autoantibodies alleviated myocardial damage induced by β1-adrenergic receptor autoantibodies in heart failure. Cardiovasc Res. 2018;114:1487–98.
Nagatomo Y, Li D, Kirsop J, Borowski A, Thakur A, Tang WH. Autoantibodies Specifically Against β1 Adrenergic Receptors and Adverse Clinical Outcome in Patients With Chronic Systolic Heart Failure in the β-Blocker Era: The Importance of Immunoglobulin G3 Subclass. J Card Fail. 2016;22:417–22.
Mobini R, Fu M, Wallukat G, Magnusson Y, Hjalmarson A, Hoebeke J. A monoclonal antibody directed against an autoimmune epitope on the human beta1-adrenergic receptor recognized in idiopathic dilated cardiomyopathy. Hybridoma. 2000;19:135–42.
Mobini R, Staudt A, Felix SB, Baumann G, Wallukat G, Deinum J, et al. Hemodynamic improvement and removal of autoantibodies against beta1-adrenergic receptor by immunoadsorption therapy in dilated cardiomyopathy. J Autoimmun. 2003;20:345–50.
Li H, Scherlag BJ, Kem DC, Benbrook A, Shen X, Cunningham MW, et al. Inducible cardiac arrhythmias caused by enhanced β1-adrenergic autoantibody expression in the rabbit. Am J Physiol Heart Circ Physiol. 2014;306:H422–8.
Düngen HD, Dordevic A, Felix SB, Pieske B, Voors AA, McMurray JJV, et al. β(1)-Adrenoreceptor Autoantibodies in Heart Failure: Physiology and Therapeutic Implications. Circ Heart Fail. 2020;13:e006155.
Deubner N, Berliner D, Schlipp A, Gelbrich G, Caforio AL, Felix SB, et al. Cardiac beta1-adrenoceptor autoantibodies in human heart disease: rationale and design of the Etiology, Titre-Course, and Survival (ETiCS) Study. Eur J Heart Fail. 2010;12:753–62.
Boivin V, Beyersdorf N, Palm D, Nikolaev VO, Schlipp A, Müller J, et al. Novel receptor-derived cyclopeptides to treat heart failure caused by anti-β1-adrenoceptor antibodies in a human-analogous rat model. PloS One. 2015;10:e0117589.
Patel PA, Hernandez AF. Targeting anti-beta-1-adrenergic receptor antibodies for dilated cardiomyopathy. Eur J Heart Fail. 2013;15:724–9.
Ma S, Jiang M, Wang X, Li B. Clinically approved representative small-molecule drugs for cardiopathy therapy. Eur J Med Chem. 2025;283:117172.
Drygała S, Radzikowski M, Maciejczyk M. β-blockers and metabolic modulation: unraveling the complex interplay with glucose metabolism, inflammation and oxidative stress. Front Pharm. 2024;15:1489657.
Taddei S, Tsabedze N, Tan RS. β-blockers are not all the same: pharmacologic similarities and differences, potential combinations and clinical implications. Curr Med Res Opin. 2024;40:15–23.
Mericskay M, Zuurbier CJ, Heather LC, Karlstaedt A, Inserte J, Bertrand L, et al. Cardiac intermediary metabolism in heart failure: substrate use, signalling roles and therapeutic targets. Nat Rev Cardiol. 2025;22:704–27.
Oliver E, Mayor F Jr., D’Ocon P. Beta-blockers: Historical Perspective and Mechanisms of Action. Rev Espanola de Cardiol(Engl ed.). 2019;72:853–62.
Bennett M, Chang CL, Tatley M, Savage R, Hancox RJ. The safety of cardioselective β(1)-blockers in asthma: literature review and search of global pharmacovigilance safety reports. ERJ open research. 2021;7.
do Vale GT, Ceron CS, Gonzaga NA, Simplicio JA, Padovan JC. Three Generations of β-blockers: History, Class Differences and Clinical Applicability. Curr Hypertens Rev. 2019;15:22–31.
Tiotiu A, Novakova P, Kowal K, Emelyanov A, Chong-Neto H, Novakova S, et al. Beta-blockers in asthma: myth and reality. Expert Rev Respiratory Med. 2019;13:815–22.
Apra C, Dumot C, Bourdillon P, Pelissou-Guyotat I. Could propranolol be beneficial in adult cerebral cavernous malformations?. NeurosurgRev. 2019;42:403–8.
Albillos A, Krag A. Beta-blockers in the era of precision medicine in patients with cirrhosis. J Hepatol. 2023;78:866–72.
Floria M, Oancea AF, Morariu PC, Burlacu A, Iov DE, Chiriac CP, et al. An Overview of the Pharmacokinetics and Pharmacodynamics of Landiolol (an Ultra-Short Acting β1 Selective Antagonist) in Atrial Fibrillation. Pharmaceutics. 2024;16.
Ayabe K, Komiyama T, Takekawa H, Kang H, Tsumagari Y, Ito M, et al. Differential Effects of Landiolol in Patients with Atrial Fibrillation and Atrial Tachycardia. Medicina. 2024;60.
Rehberg S, Frank S, Černý V, Cihlář R, Borgstedt R, Biancofiore G, et al. Landiolol for heart rate control in patients with septic shock and persistent tachycardia. A multicenter randomized clinical trial (Landi-SEP). Intensive Care Med. 2024;50:1622–34.
Vazgiourakis V, Mantzarlis K, Makris D. Landiolol in patients with septic shock. Intensive Care Med. 2025;51:221.
Barwig M, Janisch M, Gessl J, Kübler W, König C, Schwantzer G, et al. Efficacy of bolus injections of landiolol hydrochloride as premedication in coronary artery CT angiography. Insights Imaging. 2025;16:13.
Kowalik K, Silverman M, Oraii A, Conen D, Belley-Côté EP, Healey JS, et al. Landiolol for perioperative atrial tachyarrhythmias in cardiac and thoracic surgery patients: a systematic review and meta-analysis. Br J Anaesth. 2024;133:222–5.
Rao SJ, Kanwal A, Kanwal A, Danilov A, Frishman WH. Landiolol: An Ultra-Short-Acting β-Blocker. Cardiol Rev. 2024;32:468–72.
Michel-Behnke I, Müller M, Stiller B, Kriebel T, Kanaan M, Környei L, et al. Landiolol is effective and safe in paediatric supraventricular tachycardia: evidence from a European prospective multicentre open-label phase III study (LANDI-PED). Europace: European pacing, arrhythmias, and cardiac electrophysiology: journal of the working groups on cardiac pacing, arrhythmias, and cardiac cellular electrophysiology of the European Society of Cardiology. 2025;27.
Uemura K, Kawada T, Zheng C, Li M, Sugimachi M. Low-Dose Landiolol Reduces Heart Rate and Cardiac Oxygen Consumption Without Compromising Initial Hemodynamic Resuscitation in a Canine Model of Endotoxin Shock. Shock. 2019;52:102–10.
Zhao X, Liu T, Yang Q, Yang G, Li X, Tang X, et al. Initial treatment with a single capsule containing half-dose quadruple therapy vs standard-dose dual therapy in hypertensive patients (QUADUAL): a randomized, blinded, crossover trial. BMC Med. 2025;23:56.
Garcia-Pavia P, Bilen O, Burroughs M, Costabel JP, de Barros Correia E, Dybro AM, et al. Aficamten vs Metoprolol for Obstructive Hypertrophic Cardiomyopathy: MAPLE-HCM Rationale, Study Design, and Baseline Characteristics. JACC Heart Fail. 2025;13:346–57.
Hintze TD, Downing JV, Acquisto NM, Downton Mslis K, Yardi I, Moran M, et al. Metoprolol vs diltiazem for atrial fibrillation with rapid ventricular rate: Systematic review and meta-analysis of adverse events. Am J Emerg Med. 2025;89:230–40.
Huang CW, Yu AS, Zhou H, Pak K, Shaw SF, Shi J, et al. Comparative Effectiveness of Bisoprolol, Carvedilol, and Metoprolol Succinate in Patients with Heart Failure and CKD. Clin J Am Soc Nephrology: CJASN. 2025;20:136–8.
Pizarro G, Fernández-Friera L, Fuster V, Fernández-Jiménez R, García-Ruiz JM, García-Álvarez A, et al. Long-term benefit of early pre-reperfusion metoprolol administration in patients with acute myocardial infarction: results from the METOCARD-CNIC trial (Effect of Metoprolol in Cardioprotection During an Acute Myocardial Infarction). J Am Coll Cardiol. 2014;63:2356–62.
Moore CL, Henry DS, McClenahan SJ, Ball KK, Rusch NJ, Rhee SW. Metoprolol Impairs β1-Adrenergic Receptor-Mediated Vasodilation in Rat Cerebral Arteries: Implications for β-Blocker Therapy. J Pharm Exp Ther. 2021;376:127–35.
Leduc L, Abraham M, Slack J. Intravenous administration of quinidine and metoprolol for treatment of atrial fibrillation in 2 neonatal foals. J Vet Intern Med. 2024;38:2783–9.
van den Berg MP, van de Ven LL, Witting W, Crijns HJ, Haaksma J, Bel KJ, et al. Effects of beta-blockade on atrial and atrioventricular nodal refractoriness, and atrial fibrillatory rate during atrial fibrillation in pigs. Jpn Heart J. 1997;38:841–8.
Hanif N, Zamir A, Imran I, Saeed H, Majeed A, Rehman AU, et al. Clinical pharmacokinetics of nebivolol: a systematic review. Drug Metab Rev. 2023;55:428–40.
Desideri G, Pegoraro V, Cipelli R, Ripellino C, Miroddi M, Meto S, et al. Extemporaneous combination therapy with nebivolol/valsartan for the treatment of hypertension: a study of real-world evidence in Europe. Curr Med Res Opin. 2024;40:1211–9.
Desideri G, Cipelli R, Pegoraro V, Ripellino C, Miroddi M, Meto S, et al. Extemporaneous combination therapy with nebivolol/amlodipine for the treatment of hypertension: a real-world evidence study in Europe. Curr Med Res Opin. 2024;40:733–43.
Cheng JW. Nebivolol: a third-generation beta-blocker for hypertension. Clin Ther. 2009;31:447–62.
Fongemie J, Felix-Getzik E. A Review of Nebivolol Pharmacology and Clinical Evidence. Drugs. 2015;75:1349–71.
Ke F, Kuang W, Hu X, Li C, Ma W, Shi D, et al. A novel vaccine targeting β1-adrenergic receptor. Hypertens Res: J Jpn Soc Hypertens 2023;46:1582–95.
Su M, Paknejad N, Zhu L, Wang J, Do HN, Miao Y, et al. Structures of β(1)-adrenergic receptor in complex with Gs and ligands of different efficacies. Nat Commun. 2022;13:4095.
Dupré DJ, Robitaille M, Rebois RV, Hébert TE. The role of Gbetagamma subunits in the organization, assembly, and function of GPCR signaling complexes. Annu Rev Pharm Toxicol. 2009;49:31–56.
Thompson MD, Chidiac P, Jose PA, Hauser AS, Gorvin CM. Genetic variants of accessory proteins and G proteins in human genetic disease. Crit Rev Clin Lab Sci. 2025;62:113–34.
Luo F, Guo NN, Li SH, Tang H, Liu Y, Zhang Y. Reduction of glutamate release probability and the number of releasable vesicles are required for suppression of glutamatergic transmission by β1-adrenoceptors in the medial prefrontal cortex. Neuropharmacology. 2014;83:89–98.
Sugama S, Takenouchi T, Hashimoto M, Ohata H, Takenaka Y, Kakinuma Y. Stress-induced microglial activation occurs through β-adrenergic receptor: noradrenaline as a key neurotransmitter in microglial activation. J Neuroinflamm. 2019;16:266.
Hespel P, Lijnen P, Vanhees L, Fagard R, Amery A. Beta-adrenoceptors and the regulation of blood pressure and plasma renin during exercise. J Appl Physiol (Bethesda, Md: 1985). 1986;60:108–13.
Sandilands AJ, O’Shaughnessy KM. β1-Adrenoreceptor Polymorphisms and Blood Pressure: 49S Variant Increases Plasma Renin But Not Blood Pressure in Hypertensive Patients. Am J Hypertens. 2019;32:447–51.
Konkar AA, Zhai Y, Granneman JG. beta1-adrenergic receptors mediate beta3-adrenergic-independent effects of CGP 12177 in brown adipose tissue. Mol Pharm. 2000;57:252–8.
Carnet Le Provost K, Kepp O, Kroemer G, Bezu L. Trial watch: beta-blockers in cancer therapy. Oncoimmunology. 2023;12:2284486.
Acknowledgements
We would like to express our gratitude to Yani Wang, Xinlei Wang, and Shenghan Xu for their support in the revision of this manuscript. We also wish to thank Ruizhe Wang for his assistance in creating Fig. 7 of this article. Figures were created with BioRender (https://app.biorender.com/).
Funding
The work was supported by the Natural Science Foundation of Shandong Province (ZR2024QH115), Shandong Second Medical University Research and Innovation Project (2023BSQD003), Graduate Student Fund of Shandong Second Medical University (2024YJSCX017), Innovation and Entrepreneurship Training Program for College Students of Shandong Second Medical University (X2025077).
Author information
Authors and Affiliations
Contributions
WCX and JL searched the information and wrote the drafts. JJ and ML helped with editing, checking, and formatting. WXW and MC checked the English. XYZ, XDC, and HC proposed the concept of design, organized, conceived, led the writing, and supervising. All authors have read and agreed to the publication version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval
Ethical approval is not involved.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Xu, W., Li, J., Ju, J. et al. The beta1-adrenergic receptor in the heart. Cell Death Discov. 12, 46 (2026). https://doi.org/10.1038/s41420-025-02907-w
Received:
Revised:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41420-025-02907-w
