
Abstract
Induced pluripotent stem cells (iPSCs) have emerged as a powerful tool in biomedical research, enabling the study of cellular function and early disease mechanisms within patient-specific genetic contexts. Traditionally, iPSCs have been used to model monogenic diseases, where highly penetrant variants produce robust cellular phenotypes detectable in few cell lines. Recent advances in scalability and standardisation now enable systematic comparisons across many donors. This development is particularly relevant for complex diseases, which are driven by numerous genetic variants with small individual effects and therefore require population-scale designs to resolve genotype–phenotype relationships. However, several limitations of iPSC technology continue to challenge the reliability and reproducibility of such studies, constraining their translational relevance. Here, we review the challenges and opportunities of using iPSCs to model complex diseases, structured around three key themes: detecting subtle effects, modelling environmental context, and expanding genetic diversity.
Similar content being viewed by others

Introduction
Complex diseases arise from interactions between genetic variation and environmental factors and include a range of common diseases, such as cardiovascular, neurological, and musculoskeletal diseases, that together constitute a major global health burden. Many of these conditions are age-related, with cellular pathology emerging long before clinical symptoms manifest. Capturing such early disease mechanisms requires human model systems that can integrate genetic variation, cellular complexity, and environmental factors.
Genome-wide association studies (GWAS) have identified numerous genetic variants associated with complex diseases [1, 2], highlighting the significant contribution of genetic factors to complex disease phenotypes. However, translating these genetic associations into mechanistic insights remains a central challenge, since GWAS signals alone rarely resolve how genetic risk manifests in specific cellular contexts, necessitating complementary functional model systems to bridge association and mechanism [3]. Traditionally, modelling systems have provided valuable snapshots of advanced disease stages, but fail to fully capture the tissue-specific, developmental, and environmental contexts in which pathogenic mechanisms occur.
Induced pluripotent stem cells (iPSCs), somatic cells reprogrammed into a pluripotent state [4], offer a unique opportunity to overcome several of these limitations. iPSCs can be differentiated into various cell types while retaining the genetic background of the donor [5], thereby enabling systematic studies of the contribution of genetic variation to disease-relevant cellular phenotypes. Moreover, the process of iPSC differentiation enables researchers to follow cellular trajectories from pluripotency towards mature phenotypes, providing a window into pathogenic mechanisms that precede clinical symptoms.
Despite these advantages, several technical and biological factors, such as induced variability and limited genetic diversity, may constrain the fidelity of iPSC-based complex disease models. Continued methodological refinement will therefore be essential to improve their robustness and translational potential.
This review focuses on three foundational aspects that are critical for advancing iPSC-based modelling of complex diseases: large-scale and standardised iPSC derivation, incorporation of environmental factors, and representation of genetic diversity. We discuss how progress across these domains can transform the iPSC technology into a versatile platform to bridge the gap between genetic risk and cellular mechanisms of disease.
Challenges and opportunities in iPSC-based disease modelling
Genome-wide association studies (GWAS) have identified hundreds of thousands of variants associated with complex traits and diseases [6], yet translating these associations into causal cellular mechanisms remains a major challenge. Resources such as the Genotype-Tissue Expression (GTEx) consortium [7] have provided comprehensive maps of genetic regulation across post-mortem tissues, but such static tissue snapshots cannot capture dynamic gene regulation, cellular heterogeneity, or developmental transitions. A cellular system that recapitulates human physiology is therefore required to interpret complex disease risk at a functional level.
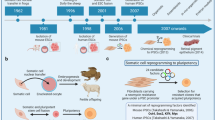
iPSC models can address some of these limitations by enabling experimental access to genetically defined human cell types. iPSC lines can be generated from large cohorts of genetically diverse individuals, enabling association analyses between genetic variation and cellular phenotypes in defined human cell types. These cellular traits can be chosen to reflect molecular or functional processes relevant to the clinical phenotypes identified by GWAS, thereby providing an experimentally accessible intermediate between genotype and disease. Moreover, iPSCs can be differentiated into otherwise inaccessible lineages, such as neurons, cardiomyocytes, and hepatocytes, thereby providing a system to connect genetic variation to molecular and cellular phenotypes (Fig. 1). iPSCs can be used to generate monocultures for high-throughput perturbations, co-cultures that capture intercellular communication, or three-dimensional organoids that can recapitulate aspects of tissue architecture and physiology. For instance, iPSC-derived neuronal cells can self-organise into cerebral organoids expressing brain region and layer-specific markers [8]. Organoids derived from Alzheimer’s disease patients recapitulate hallmark features of the disease, including neuronal hyperexcitability, Aβ aggregation, and tau hyperphosphorylation [9, 10], illustrating the capacity of iPSC-derived systems to capture disease-relevant mechanisms. However, studies comparing primary and iPSC-derived organoids have shown that brain organoids predominantly reflect mid-gestation foetal stages rather than adult tissue, with limited synaptic and metabolic maturity [11, 12], constraining modelling late-onset diseases. Advances such as vascularization [13, 14], immune cell co-cultures [13], and microfluidic perfusion [15] have increased the physiological fidelity of iPSC-derived brain organoids, revealing more complex disease phenotypes. Yet even these models remain developmentally young, underscoring the need for approaches that better represent the biological context of ageing-related disease mechanisms.

Traditional modelling systems are restricted to peripheral or post-mortem tissues, which have limited translational relevance for complex diseases (upper panel). iPSCs enable access to otherwise inaccessible cell types and multicellular systems derived from easily obtainable tissues (lower panel) and can therefore yield many more specific insights into complex disease mechanisms. iPSC induced pluripotent stem cell. Created with BioRender.
Another important advantage of iPSCs is their capacity to recapitulate key developmental milestones such as primitive streak formation, germ cell specification, and somitogenesis [16,17,18,19,20]. This enables researchers to examine how genetic variation shapes fundamental cellular programmes that influence disease susceptibility, even when clinical pathology arises later in life. For example, iPSC-based models of schizophrenia have revealed early neurodevelopmental abnormalities, including imbalanced excitatory and inhibitory neuronal differentiation, altered progenitor proliferation, and cortical layer disorganization [21], illustrating how disrupted developmental trajectories can contribute to disease risk.
Navigating core challenges in iPSC-based complex disease modelling
Nearly two decades of iPSC-based modelling have yielded important insights into both the strengths and limitations of this system. While iPSCs have shown considerable promise for disease modelling, drug screening, and regenerative medicine, several fundamental challenges remain that require careful experimental consideration (reviewed in refs. [22,23,24,25]).
Some of these limitations stem from the artificial context in which iPSCs are generated. Every technical step that drives somatic cells to pluripotency can introduce variability at genetic [26], epigenetic [27], transcriptomic [28], and proteomic [29] levels, particularly when performed manually. This may cause iPSC-derived cells to deviate from the primary cell types they are intended to model. Across differentiations, iPSC-derived cells generally recapitulate the broad lineage identity of primary cells but often represent immature states and display heterogeneity in subtype composition and functional maturity [30, 31]. Ongoing efforts towards protocol standardisation [32], automated cell culturing [33, 34], and benchmarking against single-cell reference atlases [35], can greatly improve the reproducibility and validity of iPSC-based models, paving the way for a next generation of iPSC research.
Modelling complex diseases introduces additional challenges. First, unlike monogenic disease variants with high penetrance, the genetic variants associated with complex diseases exert subtle to moderate phenotypic effects. Second, complex disease phenotypes are influenced by environmental factors that are difficult to capture in vitro due to their multifactorial nature. Finally, complex diseases are highly polygenic, and to capture the full spectrum of associated genetic variants, it is essential to incorporate global genetic diversity into the model (Fig. 2).

Modelling complex diseases presents several unique challenges, including capturing subtle to moderate cellular effects, modelling environmental influences, and capturing the entire range of associated genetic variants. Various approaches are under development to meet the requirements for addressing these challenges, such as large-scale iPSC derivation, techniques to capture environmental factors, and iPSC biobanks to include more genetic diversity in iPSC models. iPSC induced pluripotent stem cell. Created with BioRender.
Detecting subtle phenotypic effects: balancing scale and resolution
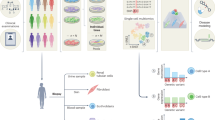
Monogenic disease variants typically confer highly penetrant phenotypic effects that are easily detectable in cellular systems, necessitating only few cell lines. For instance, iPSC-derived neurons carrying SHANK3 haploinsufficiency (a monogenic cause of Phelan-McDermid syndrome) show striking cellular abnormalities, including reduced synapse formation, impaired dendritic arborization, and disrupted network activity [36]. In contrast, complex diseases are influenced by large numbers of genetic variants, each of which typically confers only a small effect on clinical outcomes, limiting their direct biological interpretability. In defined cellular contexts, these genetic variants may exhibit more pronounced molecular effects, although these remain substantially smaller than those related to monogenic mutations. For example, the psychiatric risk variant rs1006737 in the CACNA1C gene has a small clinical effect size (OR ≈ 1.1 [37, 38]) and has subtle effects observable on fMRI [39]. However, on the cellular level, induced neurons derived from homozygous carriers show substantial effects, with a nearly two-fold increase in CACNA1C mRNA expression and 40% higher calcium channel activity [40] (Fig. 3). This illustrates how cellular models can reveal mechanisms that remain difficult to resolve at the clinical level. Extending such studies to population-scale iPSC models would enable the simultaneous investigation of many genetic variants, allowing inter-individual genetic variation, including both GWAS loci and novel variants, to be statistically associated with cellular phenotypes in a manner conceptually analogous to eQTL mapping. Such approaches require sufficiently large sample sizes and rigorous control of technical variability to detect subtle effects.

The effect size of complex disease genetic variants depends on the biological scale at which they are measured. On the molecular level, a SNP in the CACNA1C gene confers double the mRNA expression and a 40% higher Ca2+ influx, while the effect on the organismal level is substantially smaller, as measured by fMRI of the prefrontal cortex. SNP single nucleotide polymorphism, CACNA1C calcium voltage-gated channel subunit alpha 1, fMRI functional magnetic resonance imaging. Created with BioRender.
Automated and semi-automated iPSC culturing systems have recently emerged to increase throughput and standardisation by performing tasks traditionally performed manually, such as reprogramming, clone selection, passaging and differentiation [33, 34, 41]. Automation is particularly advantageous for cell types requiring long differentiation times to achieve functional maturity, such as neurons. For instance, long-term co-cultures of neurons, astrocytes, and microglia have recapitulated Alzheimer’s disease phenotypes (amyloid-β plaque formation, neuritic dystrophy, synaptic loss) that are difficult to capture in manually maintained cultures, which are often short-term [41]. Although still developing, automated systems have the potential to combine standardisation with increased throughput, facilitating larger-scale, genetically informative iPSC studies that capture a broader spectrum of genetic effects on cellular phenotypes.
Another strategy to increase throughput involves pooled experimental paradigms (i.e. iPSC villages), in which lines from many donors are co-cultured in a single dish under uniform culture conditions [42]. The primary aim of such designs is to enable efficient discovery of genotype–phenotype relationships by reducing technical variability and increasing statistical power across many donors. In current paradigms, donor-specific molecular phenotypes are computationally resolved rather than analysed as bulk averages. Single-cell genetic demultiplexing assigns transcriptomic or proteomic profiles to individual donors within pooled cultures, enabling genotype–phenotype associations under shared conditions [42,43,44,45]. For instance, Census-seq has been used to quantify donor-specific protein expression in iPSC-derived neuronal villages [42], illustrating how genetic influences on cellular phenotypes can be resolved within pooled cultures. Nevertheless, the composition of iPSC villages should be tightly controlled, since lines can differ substantially in proliferation rates [43], resulting in competitive overgrowth. Moreover, inter-individual variation in differentiation efficiency has been observed, although inter-line signalling within the dish may improve this in some cell types [43], but not others [45]. While iPSC villages offer clear advantages for population-scale studies, the extent to which donor-specific molecular variation, particularly beyond transcriptional profiles, can be disentangled from inter-line interactions within the village remains to be determined.
Beyond the genome: modelling environmental influences on complex diseases
Complex diseases arise from the interplay between genetic predispositions and environmental exposures [46]. Modelling environmental influences in cellular systems has proven challenging due to their dynamic and multifactorial nature. Cellular characteristics can be shaped by both the immediate microenvironment (e.g. extracellular matrix), and by broader macroenvironmental factors such as inflammation, toxin exposure, medication, or diet. Macroenvironmental factors vary widely between individuals in intensity, duration, and individual susceptibility, making these effects difficult to reproduce and standardise in experimental models.
The cellular microenvironment, comprising neighbouring cells, extracellular matrix (ECM), interstitial fluid, and physical forces (e.g. shear stress, stiffness gradients, perfusion) defines the local context in which complex disease mechanisms occur [47]. In the ideal experimental setting, ECM composition would be recapitulated, as well as dynamic processes such as fluid flow, nutrient and oxygen delivery, heterotypic cell-cell interactions (e.g. endothelial, immune, stromal) and mechanical competition for space. Current approaches partially address these requirements, where micropatterning of extracellular matrix substrates or cell-adhesion islands enables control over the cellular layouts [48]. Additionally, microfluidic organ-on-a-chip systems allow controlled perfusion, shear stress, and nutrition gradients in iPSC-derived cells, thereby more closely mirroring in vivo physiology [49, 50]. For example, a recent human heart-on-a-chip study incorporating iPSC-derived cardiomyocytes, endothelial cells and fibroblasts under flow showed alignment of endothelial cells and enhanced maturation of cardiomyocytes relative to static culture [51]. Nonetheless, most current iPSC systems typically model single environmental exposures in isolation rather than the complex, dynamic cellular context that maintains physiological tissue homeostasis [52]. While still evolving, these approaches can substantially improve the detection of gene–environment interactions in complex diseases.
Macroenvironmental factors shape cellular biology throughout life, influencing both early development and adult disease risk. In utero exposures, such as maternal stress and nutrition, establish epigenetic and metabolic patterns that can persist for decades [53, 54], often interacting with genetic predispositions to drive complex disease progression. At the population level, environment-wide association studies (EnWAS) have been developed to systematically assess a wide range of environmental exposures in relation to disease outcomes. For example, a seminal EnWAS of type 2 diabetes analysed associations between hundreds of environmental factors (e.g. pollutants and micronutrients) and diabetic status, identifying several exposure-disease links ranging from pesticide derivatives to vitamin intake [55]. Such studies have underscored the breadth of environmental factors that can be associated with diseases, and therefore also the difficulty of capturing such factors in modelling systems.
To date, most iPSC-based studies have focused on modelling single environmental exposures. For instance, cigarette smoke and its components have been studies in a range of iPSC-derived cell types under controlled in vitro conditions. iPSC-derived endothelial cells exposed to tobacco smoke increased oxidative stress, DNA damage/repair response, apoptosis and several other cellular pathways that associate with cellular stress [56]. Similarly, iPSC-derived cardiomyocytes have been exposed to smoke and nicotine-containing aerosol bubbled media and exhibited cardiotoxic metabolic changes [57]. Such work demonstrates the feasibility of using iPSC-based systems to interrogate the cellular consequences of defined macroenvironmental exposures. However, these approaches are inherently limited to environmental factors that can be directly measured and experimentally applied in vitro, and are less suited to capturing broader, cumulative, or indirectly acting exposures such as socioeconomic status, psychosocial stress, or complex lifestyle patterns.
Epigenetic mechanisms form a key interface between environmental exposures and the genome, translating environmental exposures to cellular responses such as DNA methylation and histone modifications [58]. Epigenome-wide association studies (EWAS) have linked a wide range of environmental factors to epigenetic changes across different developmental stages, from prenatal exposures such as folate levels [59], to broader factors including nutrition [60], medication [61], and socioeconomic status [62].
However, translating these associations into mechanistic insights within cellular systems remains challenging, as most EWAS are conducted using easily accessible cell types, such as blood cells, which provide limited insights for disease-relevant tissues. Recent advances in CRISPR Cas9 technology now enable targeted manipulation of DNA methylation at specific genomic loci, opening the door for analysing how environmentally induced epigenetic modifications affect cellular mechanisms [63]. Since some exposure-linked epigenetic markers are conserved across tissues (e.g. smoking [64]) and developmental stages (e.g. air pollution [65]), engineering these epigenetic markers in iPSC-derived cells may help identify how systemic environmental influences contribute to disease-relevant phenotypes.
Enhancing genetic representation in iPSC models
Complex diseases are highly polygenic, involving thousands to potentially millions of genetic variants [66]. Capturing this full spectrum of genetic variation is essential to elucidate the molecular mechanisms underlying disease. However, most genetic studies have focused on individuals of European ancestry, representing only a fraction of global diversity. Expanding research to individuals beyond the European mainland is therefore critical to uncover ancestry-specific mechanisms that are rare or absent in European cohorts. Indeed, recent GWAS in African ancestry populations have identified unique genetic variants associated with various complex traits, such as BMI and Parkinson’s disease [67, 68]. Together, these findings underscore the importance of integrating genetic discovery with functional studies that are explicitly designed to capture genetic diversity, to ensure that inferred disease mechanisms are biologically and ancestrally representative. To this end, initiatives such as the iDA project [69] are developing ancestrally diverse iPSC panels to investigate how disease-associated genetic variants influence cellular mechanisms across populations.
Increasing genetic diversity in iPSC-based studies enhances the biological relevance of the model but also introduces additional sources of variation. Even within ancestrally homogeneous populations, inter-line variability can be substantial, arising from biological differences between donors and technical factors related to reprogramming and differentiation [26, 28]. Incorporating donors from multiple ancestral groups adds a further layer of ancestry-dependent differences in downstream biological mechanisms. These ancestry effects may covary with donor-level variation, complicating the delineation of variant-specific mechanisms from broader ancestry-related regulatory patterns. The resulting increase in heterogeneity can reduce statistical power, particularly in smaller cohorts, highlighting the need for balanced sampling strategies and analytical setups that explicitly model population structure when investigating cross-ancestry cellular phenotypes.
Ancestry-dependent variation has readily been observed during iPSC derivation, indicating that ancestral background has a substantial influence on pluripotency-associated genes [70]. Interestingly, these genes are enriched for pathways related to wound healing and cancer, suggesting that ancestry may influence cytoskeletal remodelling and protein localisation involved in the mesenchymal-to-epithelial transition, a critical step in acquiring pluripotency. Such insights underscore that even fundamental cellular processes may be ancestry-dependent, highlighting the importance of ancestrally diverse iPSC biobanks for both mechanistic discovery and the development of robust, generalisable disease models.
Emerging large-scale iPSC biobanks are marking a new era in complex disease modelling by providing access to thousands of genetically diverse cell lines. The California Institute for Regenerative Medicine (CIRM) iPSC repository currently includes more than 2500 iPSC lines derived from both healthy donors and patients of African, Asian, European, and Hispanic/Latino ancestries. These lines represent a wide range of complex diseases, including neurodegenerative, neurodevelopmental, cardiovascular and metabolic disorders, offering the opportunity to study cross-population disease mechanisms. Similarly, the WiCell repository is one of the most diverse iPSC biobanks, including ~40% of iPSC lines derived from African American, Arab, Asian, Latino, or Native American individuals. These and other initiatives set the stage for more comprehensive and translationally informative complex disease models.
Conventional GWAS begin with clinical outcomes and subsequently aim to uncover the biological mechanisms underlying associated loci. Population-scale iPSC studies enable the reverse approach by starting from experimentally tractable cellular phenotypes with measurable biological effects and trace these back to their genetic determinants [42, 71]. In such experiments, iPSCs or iPSC-derived cells are profiled for phenotypes of interest (e.g. cell morphology, proliferation) to identify variants that modulate cellular behaviour. This “functional-first” strategy directly links genetic variation to cellular mechanisms and yields variants with well-defined biological effects. These mechanisms can then be examined in clinical cohorts to assess their contribution to disease risk.
Concluding remarks
Despite major progress in identifying genetic associations with complex diseases, translating these into functional mechanisms remains a key challenge. iPSC models provide a versatile framework to connect genetic variation to cellular phenotypes across multiple tissues and disease stages, but their reproducibility and scalability require further refinement. Advances in automation and pooled culture systems can provide the throughput needed for systematic, population-scale studies on complex disease mechanisms. Improving the physiological relevance of iPSC models will depend on better modelling of environmental influences and expanding genetic diversity within iPSC resources. As these developments converge, iPSC-based systems are poised to become a tool for bridging genetic risk and disease biology, offering functional resolution to human genomics.
References
Uffelmann E, Huang QQ, Munung NS, De Vries J, Okada Y, Martin AR, et al. Genome-wide association studies. Nat Rev Methods Prim. 2021;1:59.
Watanabe K, Stringer S, Frei O, Umićević Mirkov M, De Leeuw C, Polderman TJC, et al. A global overview of pleiotropy and genetic architecture in complex traits. Nat Genet. 2019;51:1339–48.
Bruner WS, Grant SFA. Translation of genome-wide association study: from genomic signals to biological insights. Front Genet. 2024;15:1375481.
Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–72.
Takahashi K, Yamanaka S. A decade of transcription factor-mediated reprogramming to pluripotency. Nat Rev Mol Cell Biol. 2016;17:183–93.
Sollis E, Mosaku A, Abid A, Buniello A, Cerezo M, Gil L, et al. The NHGRI-EBI GWAS Catalog: knowledgebase and deposition resource. Nucleic Acids Res. 2023;51:D977–85.
Lonsdale J, Thomas J, Salvatore M, Phillips R, Lo E, Shad S, et al. The Genotype-Tissue Expression (GTEx) project. Nat Genet. 2013;45:580–5.
Yan Y, Arzua T, Logan S, Bai X. Isolation and culture of human-induced pluripotent stem cell-derived cerebral organoid cells. In: Nagy A, Turksen K, editors. Induced Pluripotent Stem (iPS) Cells. New York, NY: Springer US; 2026. p. 483–94. (Methods in Molecular Biology; vol. 2454). https://link.springer.com/10.1007/7651_2020_328.
Bubnys A, Tsai LH. Harnessing cerebral organoids for Alzheimer’s disease research. Curr Opin Neurobiol. 2022;72:120–30.
Gonzalez C, Armijo E, Bravo-Alegria J, Becerra-Calixto A, Mays CE, Soto C. Modeling amyloid beta and tau pathology in human cerebral organoids. Mol Psychiatry. 2018;23:2363–74.
He Z, Dony L, Fleck JS, Szałata A, Li KX, Slišković I, et al. An integrated transcriptomic cell atlas of human neural organoids. Nature. 2024;635:690–8.
Huang J, Zhu Y, Tang J, Liu Y, Lu M, Zhang R, et al. Navigating brain organoid maturation: from benchmarking frameworks to multimodal bioengineering strategies. Biomolecules. 2025;15:1118.
Ji Y, Chen X, Wang Z, Meek CJ, McLean JL, Yang Y, et al. Alzheimer’s disease patient brain extracts induce multiple pathologies in novel vascularized neuroimmune organoids for disease modeling and drug discovery. Mol Psychiatry. 2025;30:4558–75.
Gong L, Zhang Y, Zhu Y, Lee U, Luo AC, Li X, et al. Rapid generation of functional vascular organoids via simultaneous transcription factor activation of endothelial and mural lineages. Cell Stem Cell. 2025;32:1200–1217.e6.
Papamichail L, Koch LS, Veerman D, Broersen K, Van Der Meer AD. Organoids-on-a-chip: microfluidic technology enables culture of organoids with enhanced tissue function and potential for disease modeling. Front Bioeng Biotechnol. 2025;13:1515340.
Chin MH, Pellegrini M, Plath K, Lowry WE. Molecular analyses of human induced pluripotent stem cells and embryonic stem cells. Cell Stem Cell. 2010;7:263–9.
Bock C, Kiskinis E, Verstappen G, Gu H, Boulting G, Smith ZD, et al. Reference maps of human ES and iPS cell variation enable high-throughput characterization of pluripotent cell lines. Cell. 2011;144:439–52.
Zheng Y, Xue X, Shao Y, Wang S, Esfahani SN, Li Z, et al. Controlled modelling of human epiblast and amnion development using stem cells. Nature. 2019;573:421–5.
Esfahani SN, Zheng Y, Arabpour A, Irizarry AMR, Kobayashi N, Xue X, et al. Derivation of human primordial germ cell-like cells in an embryonic-like culture. Nat Commun. 2024;15:167.
Sanaki-Matsumiya M, Matsuda M, Gritti N, Nakaki F, Sharpe J, Trivedi V, et al. Periodic formation of epithelial somites from human pluripotent stem cells. Nat Commun. 2022;13:2325.
Räsänen N, Tiihonen J, Koskuvi M, Lehtonen Š, Koistinaho J. The iPSC perspective on schizophrenia. Trends Neurosci. 2022;45:8–26.
Volpato V, Webber C. Addressing variability in iPSC-derived models of human disease: guidelines to promote reproducibility. Dis Model Mech. 2020;13:dmm042317.
Doss MX, Sachinidis A. Current challenges of iPSC-based disease modeling and therapeutic implications. Cells. 2019;8:403.
Cerneckis J, Cai H, Shi Y. Induced pluripotent stem cells (iPSCs): molecular mechanisms of induction and applications. Signal Transduct Target Ther. 2024;9:112.
Guo X, Wang X, Wang J, Ma M, Ren Q. Current development of iPSC-based modeling in neurodegenerative diseases. Int J Mol Sci. 2025;26:3774.
Kilpinen H, Goncalves A, Leha A, Afzal V, Alasoo K, Ashford S, et al. Common genetic variation drives molecular heterogeneity in human iPSCs. Nature. 2017;546:370–5.
Nishizawa M, Chonabayashi K, Nomura M, Tanaka A, Nakamura M, Inagaki A, et al. Epigenetic variation between human induced pluripotent stem cell lines is an indicator of differentiation capacity. Cell Stem Cell. 2016;19:341–54.
Carcamo-Orive I, Hoffman GE, Cundiff P, Beckmann ND, D’Souza SL, Knowles JW, et al. Analysis of transcriptional variability in a large human iPSC library reveals genetic and non-genetic determinants of heterogeneity. Cell Stem Cell. 2017;20:518–532.e9.
Beekhuis-Hoekstra SD, Watanabe K, Werme J, De Leeuw CA, Paliukhovich I, Li KW, et al. Systematic assessment of variability in the proteome of iPSC derivatives. Stem Cell Res. 2021;56:102512.
Strano A, Tuck E, Stubbs VE, Livesey FJ. Variable outcomes in neural differentiation of human PSCs arise from intrinsic differences in developmental signaling pathways. Cell Rep. 2020;31:107732.
Ewoldt JK, DePalma SJ, Jewett ME, Karakan MÇ, Lin YM, Mir Hashemian P, et al. Induced pluripotent stem cell-derived cardiomyocyte in vitro models: benchmarking progress and ongoing challenges. Nat Methods. 2025;22:24–40.
Dobner J, Diecke S, Krutmann J, Prigione A, Rossi A. Reassessment of marker genes in human induced pluripotent stem cells for enhanced quality control. Nat Commun. 2024;15:8547.
Tristan CA, Ormanoglu P, Slamecka J, Malley C, Chu PH, Jovanovic VM, et al. Robotic high-throughput biomanufacturing and functional differentiation of human pluripotent stem cells. Stem Cell Rep. 2021;16:3076–92.
Paull D, Sevilla A, Zhou H, Hahn AK, Kim H, Napolitano C, et al. Automated, high-throughput derivation, characterization and differentiation of induced pluripotent stem cells. Nat Methods. 2015;12:885–92.
Luecken MD, Büttner M, Chaichoompu K, Danese A, Interlandi M, Mueller MF, et al. Benchmarking atlas-level data integration in single-cell genomics. Nat Methods. 2022;19:41–50.
Shcheglovitov A, Shcheglovitova O, Yazawa M, Portmann T, Shu R, Sebastiano V, et al. SHANK3 and IGF1 restore synaptic deficits in neurons from 22q13 deletion syndrome patients. Nature. 2013;503:267–71.
Ruderfer, Fanous DM, Ripke AH, McQuillin S, Amdur A, Schizophrenia Working Group of the Psychiatric Genomics Consortium RL, et al. Polygenic dissection of diagnosis and clinical dimensions of bipolar disorder and schizophrenia. Mol Psychiatry. 2014;19:1017–24.
Liu, Blackwood Y, Caesar DH, De S, Geus EJC, Farmer A, et al. Meta-analysis of genome-wide association data of bipolar disorder and major depressive disorder. Mol Psychiatry. 2011;16:2–4.
Bigos KL, Mattay VS, Callicott JH, Straub RE, Vakkalanka R, Kolachana B, et al. Genetic Variation in CACNA1C Affects Brain Circuitries Related to Mental Illness. Arch Gen Psychiatry. 2010;67:939.
Yoshimizu T, Pan JQ, Mungenast AE, Madison JM, Su S, Ketterman J, et al. Functional implications of a psychiatric risk variant within CACNA1C in induced human neurons. Mol Psychiatry. 2015;20:162–9.
Bassil R, Shields K, Granger K, Zein I, Ng S, Chih B. Improved modeling of human AD with an automated culturing platform for iPSC neurons, astrocytes and microglia. Nat Commun. 2021;12:5220.
Mitchell JM, Nemesh J, Ghosh S, Handsaker RE, Mello CJ, Meyer D, et al. Mapping genetic effects on cellular phenotypes with “cell villages”. Genetics. 2023. http://biorxiv.org/lookup/doi/10.1101/2020.06.29.174383.
Neavin DR, Steinmann AM, Farbehi N, Chiu HS, Daniszewski MS, Arora H, et al. A village in a dish model system for population-scale hiPSC studies. Nat Commun. 2023;14:3240.
Cuomo ASE, Seaton DD, McCarthy DJ, Martinez I, Bonder MJ, Garcia-Bernardo J, et al. Single-cell RNA-sequencing of differentiating iPS cells reveals dynamic genetic effects on gene expression. Nat Commun. 2020;11:810.
Jerber J, Seaton DD, Cuomo ASE, Kumasaka N, Haldane J, Steer J, et al. Population-scale single-cell RNA-seq profiling across dopaminergic neuron differentiation. Nat Genet. 2021;53:304–12.
Laville V, Majarian T, Sung YJ, Schwander K, Feitosa MF, Chasman DI, et al. Gene-lifestyle interactions in the genomics of human complex traits. Eur J Hum Genet. 2022;30:730–9.
Warrick JW, Murphy WL, Beebe DJ. Screening the cellular microenvironment: a role for microfluidics. IEEE Rev Biomed Eng. 2008;1:75–93.
Li Y, Jiang W, Zhou X, Long Y, Sun Y, Zeng Y, et al. Advances in regulating cellular behavior using micropatterns. Yale J Biol Med. 2023;96:527–47.
Ronaldson-Bouchard K, Teles D, Yeager K, Tavakol DN, Zhao Y, Chramiec A, et al. A multi-organ chip with matured tissue niches linked by vascular flow. Nat Biomed Eng. 2022;6:351–71.
Palasantzas VEJM, Tamargo-Rubio I, Le K, Slager J, Wijmenga C, Jonkers IH, et al. iPSC-derived organ-on-a-chip models for personalized human genetics and pharmacogenomics studies. Trends Genet. 2023;39:268–84.
Liu Y, Kamran R, Han X, Wang M, Li Q, Lai D, et al. Human heart-on-a-chip microphysiological system comprising endothelial cells, fibroblasts, and iPSC-derived cardiomyocytes. Sci Rep. 2024;14:18063.
Doherty EL, Aw WY, Hickey AJ, Polacheck WJ. Microfluidic and organ-on-a-chip approaches to investigate cellular and microenvironmental contributions to cardiovascular function and pathology. Front Bioeng Biotechnol. 2021;9:624435.
Reichetzeder C. Overweight and obesity in pregnancy: their impact on epigenetics. Eur J Clin Nutr. 2021;75:1710–22.
Dieckmann L, Czamara D. Epigenetics of prenatal stress in humans: the current research landscape. Clin Epigenet. 2024;16:20.
Patel CJ, Bhattacharya J, Butte AJ. An Environment-Wide Association Study (EWAS) on Type 2 Diabetes Mellitus. Zhang B, editor. PLoS ONE. 2010;5:e10746.
Chu MC, Morimoto C, Kawai C, Miyao M, Tamaki K. Effects of DNA degradation and genotype imputation on high-density SNP microarray in pairwise kinship analysis. Leg Med Tokyo Jpn. 2023;60:102158.
Simms L, Yu F, Palmer J, Rudd K, Sticken ET, Wieczorek R, et al. Use of human induced pluripotent stem cell-derived cardiomyocytes to predict the cardiotoxicity potential of next generation nicotine products. Front Toxicol. 2022;4:747508.
Bollati V, Baccarelli A. Environmental epigenetics. Heredity. 2010;105:105–12.
Joubert BR, Den Dekker HT, Felix JF, Bohlin J, Ligthart S, Beckett E, et al. Maternal plasma folate impacts differential DNA methylation in an epigenome-wide meta-analysis of newborns. Nat Commun. 2016;7:10577.
Zhang Y, Kutateladze TG. Diet and the epigenome. Nat Commun. 2018;9:3375.
Anderson SJ, Feye KM, Schmidt-McCormack GR, Malovic E, Mlynarczyk GSA, Izbicki P, et al. Off-Target drug effects resulting in altered gene expression events with epigenetic and “Quasi-Epigenetic” origins. Pharm Res. 2016;107:229–33.
Cerutti J, Lussier AA, Zhu Y, Liu J, Dunn EC. Associations between indicators of socioeconomic position and DNA methylation: a scoping review. Clin Epigenet. 2021;13:221.
Kang JG, Park JS, Ko JH, Kim YS. Regulation of gene expression by altered promoter methylation using a CRISPR/Cas9-mediated epigenetic editing system. Sci Rep. 2019;9:11960.
Stueve TR, Li WQ, Shi J, Marconett CN, Zhang T, Yang C, et al. Epigenome-wide analysis of DNA methylation in lung tissue shows concordance with blood studies and identifies tobacco smoke-inducible enhancers. Hum Mol Genet. 2017;26:3014–27.
Saenen ND, Martens DS, Neven KY, Alfano R, Bové H, Janssen BG, et al. Air pollution-induced placental alterations: an interplay of oxidative stress, epigenetics, and the aging phenotype? Clin Epigenet. 2019;11:124.
Boyle EA, Li YI, Pritchard JK. An expanded view of complex traits: from polygenic to omnigenic. Cell. 2017;169:1177–86.
Zhang X, Brody JA, Graff M, Highland HM, Chami N, Xu H, et al. Whole genome sequencing analysis of body mass index identifies novel African ancestry-specific risk allele. Nat Commun. 2025;16:3470.
Rizig M, Bandres-Ciga S, Makarious MB, Ojo OO, Crea PW, Abiodun OV, et al. Identification of genetic risk loci and causal insights associated with Parkinson’s disease in African and African admixed populations: a genome-wide association study. Lancet Neurol. 2023;22:1015–25.
Screven LA, Pantazis CB, Andersh KM, Hong S, Vitale D, Lara E, et al. Harnessing diversity to study Alzheimer’s disease: a new iPSC resource from the NIH CARD and ADNI. Neuron. 2024;112:694–7.
Bisogno LS, Yang J, Bennett BD, Ward JM, Mackey LC, Annab LA, et al. Ancestry-dependent gene expression correlates with reprogramming to pluripotency and multiple dynamic biological processes. Sci Adv. 2020;6:eabc3851.
Wells MF, Nemesh J, Ghosh S, Mitchell JM, Salick MR, Mello CJ, et al. Natural variation in gene expression and viral susceptibility revealed by neural progenitor cell villages. Cell Stem Cell. 2023;30:312–332.e13.
Funding
ESvZ was supported by ZonMw Open grant 09120012010099. EAL was supported by Horizon 2020 Marie Sklodowska-Curie CO-FUND 707404. HHA was supported by the RadboudUMC Hypatia grant R0007330.
Author information
Authors and Affiliations
Contributions
ESvZ selected relevant papers and wrote and revised the manuscript. EAL supervised the writing process in the initial stages and gave feedback. JBJvM, RN, JHG, and RAP provided feedback of the finished manuscript. HHA supervised the writing from first draft to final manuscript and revised manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
van Zanten, E.S., Loehrer, E.A., van Meurs, J.B.J. et al. Bridging population and cell: modelling complex diseases with human induced pluripotent stem cells. Eur J Hum Genet (2026). https://doi.org/10.1038/s41431-026-02071-4
Received:
Revised:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41431-026-02071-4
