
Abstract
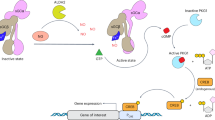
During neuronal pruning, phagocytes engulf shed cellular debris to avoid inflammation and maintain tissue homeostasis. How phagocytic receptors recognize degenerating neurites had been unclear. Here, we identify two glucosyltransferases Alg8 and Alg10 of the N-glycosylation pathway required for dendrite fragmentation and clearance through genetic screen. The scavenger receptor Draper (Drpr) is N-glycosylated with complex- or hybrid-type N-glycans that interact specifically with galectins. We also identify the galectins Crouching tiger (Ctg) and Hidden dragon (Hdg) that interact with N-glycosylated Drpr and function in dendrite pruning via the Drpr pathway. Ctg and Hdg are required in hemocytes for expression and function, and are induced during dendrite injury to localize to injured dendrites through specific interaction with exposed phosphatidylserine (PS) on the surface membrane of injured dendrites. Thus, the galectins Ctg and Hdg bridge the interaction between PS and N-glycosylated Drpr, leading to the activation of phagocytosis.
Similar content being viewed by others


Introduction
The mature nervous system is constructed of relatively precise neuronal connections, requiring progressive and regressive events. During development, neurons extend neurites to form connections, but excess ones are eliminated without apoptosis in a process known as developmental pruning1. Neuronal pruning occurs in many types of neurons, both in vertebrates and invertebrates2. Defective pruning may contribute to neurodegenerative and autoimmune diseases, such as Alzheimer’s, Parkinson’s, amyotrophic lateral sclerosis, and multiple sclerosis1,3,4.
Similar to apoptosis, the conserved phagocytic receptors CED-1 in Caenorhabditis elegans, Draper (Drpr) in Drosophila, and MEGF10 in mammals are also employed in neuronal pruning to regulate cytoskeletal rearrangement and membrane extension of the phagocytes5,6,7,8. These receptors contain an EMI domain and multiple atypical EGF repeats in the extracellular domain. During apoptosis and neuronal pruning, the lipid phosphatidylserine (PS) that is normally found in the cytoplasmic leaflet of the plasma membrane is externalized to the outer leaflet where it functions as the “eat me” signal for phagocytosis9,10. Phagocytic receptors directly or indirectly recognize PS via a process that usually requires context-dependent tethering or auxiliary molecules to induce phagocytosis. In mammals, the bridging molecules such as MFG-E8, Protein S, and Growth arrest-specific gene 6 link PS and these phagocytic receptors to activate phagocytosis11,12,13. In C. elegans, TTR-52, a transthyretin-like protein, is secreted from non-apoptotic cells and interacts with both PS and CED-1 to act as a bridging molecule between apoptotic cells and phagocytes14. The Drosophila Drpr receptor interacts indiscriminately with PS, phosphatidylethanolamine (PE) and phosphatidylinositol (PI) in vitro15, implying a requirement for specific tethering molecules during PS recognition. Pretaporter and DmCaBP1 that interact with the extracellular domain of Drpr are located in the endoplasmic reticulum (ER) and they relocate to the cell surface during apoptosis16,17. However, Drosophila lacking both DmCaBP1 and Pretaporter were found to develop normally and showed no obvious phagocytic defects16,17. Drpr is required for phagocytosis of mushroom body γ neuron axons during pruning, as well as for injured axons and dendrites7,8,18. Indeed, the Drosophila CXC3L1 homolog Orion serves as a bridging molecule between Drpr and PS19,20. Orion localizes to dendritic processes post-injury, and functions to promote phagocytosis of fragmented dendrites19. In sum, different tethering molecules or their combinations bridge the interaction between PS and phagocytic receptors in neuronal pruning.
Glycosylation is the most common posttranslational modification of proteins in eukaryotic cells. N-linked glycosylation that involves conjugating polysaccharides onto the arginine residue regulates a broad range of protein structures and functions, including protein folding, trafficking, and degradation, as well as signal-receptor interactions and cell-cell adhesion21. Mutations in genes involved in the N-glycosylation pathway are often associated with neurological defects such as mental retardation, seizures and hypotonia, although most of the pathological mechanisms remain unclear22. During N-glycosylation, the glycans attached at the NxS/T motifs of proteins in the ER are further diversely modified in the Golgi into high-mannose, hybrid or complex types according to carbohydrate moieties and branching patterns23. Lectins are carbohydrate-binding proteins that interact with glycoproteins through their carbohydrate-recognition domains (CRDs) to regulate glycoprotein activity24. Among the lectin superfamily, the CRDs of galectins specifically bind β-galactosides in the hybrid and complex N-glycan types25. The galectins thus far identified in mammals have been classified into a prototype with one CRD, the tandem-repeat type with two CRDs, and the chimera type with one CRD and an N-terminal peptide25. All three types of galectins form multimers, so they can aggregate glycoproteins such as cell surface receptors for activation26. Accordingly, galectins function in many different cellular activities through their interaction with N-glycosylated proteins25. Galectins also bind proteins through non-glycan binding activities27. In the nervous system, galectin-3 (Gal-3) is induced and secreted from microglia in response to inflammation and degeneration, and through binding to non-glycosylated proteins to promote neurodegeneration28,29.
Here, we used Drosophila class IV dendritic arborization (C4da) neurons as a model to study dendrite degeneration including fragmentation and engulfment in two stages, developmental pruning in the pupal stage and injury-induced degeneration in the larval stage. During metamorphosis, major trunks of dendrites are detached from the soma at 4-5 hours APF, dendritic segments are fragmentated at 10-12 hours APF, and finally the fragmentated debris is cleared at 16-18 hours APF30. Similar processes were observed in the dendrite injury model. By laser ablation at proximal sites of dendrites, the detached segments show signs of degeneration, such as blebbing and fragmentation, at 3 hours after injury (AI) and broken down by 6 hours AI. At 12 hours AI, dendrite debris were not observed, indicating the completion of clearance at the time point30. In these systems, innervated epidermal cells serve as non-professional phagocytes, engulfing degenerating dendrites through the Drpr signaling pathway30. Moreover, the macrophage-like hemocytes facilitate fragmentation of degenerating dendrites in a dendrite injury model30. In a genetic screen for defective dendrite pruning, we identified mutants in the N-glycosylation pathway. We found that N-glycosylation is required in epidermal cells and that epidermally expressed Drpr is N-glycosylated. We observed that four N-glycosylation sites in the extracellular domain of Drpr are required for Drpr localization and function. These four sites are modified with hybrid- and complex-type N-glycans that are recognized by galectins31, leading to our further identification and analyzes of two galectins of the tandem-repeat CRD type in this phagocytic process. The expression of these two galectins was undetectable with dendrites being intact. Upon dendrite ablation, both galectins were mainly induced in hemocytes and localization to the injured dendrites were also observed. Thus, they are named Crouching tiger (Ctg, encoded by CG5335) and Hidden dragon (Hdg, encoded by CG11372 and previously known as Dmgal32) in the study. We further characterized the interaction of Ctg and Hdg with the N-glycosylated Drpr and PS, revealing that PS specifically interacts with both these galectins. Thus, Ctg and Hdg regulate dendrite degeneration during dendrite injury and developmental pruning through the Drpr signaling pathway.
Results
N-glycosylation in epidermal cells is required for dendrite pruning
We have assembled a suite of mutants in which various steps of glycosylation are disrupted and performed a screen for mutants displaying defective dendrite pruning. We identified 70 homozygous mutants that could survive to the pupal stage, and their CD4-tdGFP-labeled dendrites of C4da neurons were examined at 16–18 hours after pupa formation (APF). Among these mutants, the Alg10wgd and xitA mutants displayed a severe pruning defect (Fig. 1a–c). Most of the primary and secondary dendrites in these two mutants were detached from the soma (Fig. 1a–c, red arrows indicate severed sites), with severed dendrites remaining largely unfragmented and not being cleared. These phenotypes are distinct from the type of mutants defective in initial dendrite severing, as shown by expressing the dominant-negative ecdysone receptor EcR-DN (Fig. 1d)33. Moreover, breakup of dendritic microtubules, a critical step prior to dendrite severing, was prominent in the Alg10wgd mutant (Fig. S1a), indicating that the dendrite-severing step is not affected. Thus, we postulated that dendrite fragmentation and clearance are affected in the Alg10wgd and xitA mutants. The xitA allele carries a point mutation in the Alg8 gene34. Alg8 and Alg10 encode glucosyltransferases in the N-glycosylation pathway, catalyzing the transfer of terminal glucoses onto the lipid-linked oligosaccharides that are further transferred to protein substrates35, implying that N-glycosylation may play a role in regulating dendrite pruning.

a–l Confocal images showing dendrite pruning phenotypes of C4da neurons at 16-18 hours APF. All image sizes are 300 µm x 300 µm and the scale bar in (a) is 50 µm. White arrows indicate cell bodies. Red arrows indicate severing sites. a–c C4da neurons labeled by GFP expressed from ppk-CD4-tdGFP in control (a), Alg10wgd (b), and xitA (c) lines. d–f, k ppk-GAL4 control (e) or driving expression of EcR-DN (ppk > EcR-DN) (d), Alg10-RNAi#1 (f), or Alg1-RNAi#1 (k). Dendrites are labeled by GFP expressed from UAS-mCD8GFP. g–i, l A58-GAL4 control (g) or driving expression of Alg10-RNAi#1 (h), Alg10 in Alg10wgd (i), or Alg1-RNAi#1 (l). Dendrites are labeled by GFP expressed from ppk-CD4-tdGFP. j Alg10wgd MARCM neuron generated in the line 5-40-GAL4, UAS-mCD8GFP, sop-FLP; Tub-GAL80, FRT2A/Alg10wgd FRT2A. m Three composite bar graphs (separated by dashed lines) showing quantification for average unpruned dendrite length (mean ± SEM) from confocal images (n≥ 5 larvae for each genotype). The number in the end of each label indicates the number of samples qualified. Statistical significance was determined relative to the control (first genotype in each graph) by Student’s t test, two tailed with * representing p < 0.05, ** for p < 0.01, and *** for p < 0.001.
To test if N-glycosylation is required by C4da neurons during dendrite pruning, we performed tissue-specific RNAi knockdown experiments. We detected normal pruning in ppk-GAL4-driven Alg10-RNAi knockdown C4da neurons, similar to the ppk-GAL4 control (Fig. 1e, f). Consistently, Alg10wgd MARCM clones generated for C4da neurons presented no pruning defects (Fig. 1j), suggesting that Alg10 is not required by neurons for dendrite pruning. Next, we tested the requirement for Alg10 in epidermal cells that are the major phagocytes involved in dendrite fragmentation and clearance30. Whereas the epidermal A58-GAL4 control presented normal pruning (Fig. 1g), A58-GAL4-driven Alg10-RNAi knockdown recapitulated the Alg10wgd mutant defects, with this outcome observed for two independent Alg10 RNAi lines (Fig. 1h, m) in which Alg10 mRNA expression had been knocked down (Fig. S1b). Consistent with the requirement for Alg10 in epidermal cells, we observed that A58-GAL4-driven Alg10 expression rescued Alg10wgd lethality and pruning defects (Fig. 1i). To further confirm the involvement of N-glycosylation in dendrite pruning, we examined the phenotype elicited by Alg1-RNAi knockdown. Alg1 encodes a mannosyltransferase that catalyzes the addition of the first mannose to lipid-linked oligosaccharides35. Epidermal rather than C4da neuronal knockdown of two different Alg1-RNAi lines (Fig. S1b for the knockdown effect) elicited dendrite pruning defects, which were indistinguishable to those arising from Alg10 knockdown (Fig. 1k, l). Total length of remaining dendrites in all these lines was quantified for statistical comparison (Fig. 1m). Thus, these analyzes support that N-glycosylation in epidermal cells is required for fragmentation and clearance of C4da dendrites.
Drpr is modified with hybrid- or complex-type N-glycans
Given the requirement for N-glycosylation in epidermal cells for dendrite pruning, we explored if Drpr is a substrate for N-glycosylation since it is also required in epidermal cells for dendrite pruning30. Drpr hosts 16 predicted NxS/T motifs for N-glycosylation36. To determine if Drpr is N-glycosylated, first we performed a Western blot analysis using Drpr antibodies to probe pupal lysates. The Drpr signal appeared as a broad band in the control lysates (Fig. 2b, lane 1). In contrast, when the lysates were treated with PNGase F to remove all types of protein-attached N-glycans, the Drpr signal appeared at lower molecular-weight positions (lane 2). Similar downshifts of the Drpr signal were also detected in lysates prepared from Alg10wgd and xitA pupae (lanes 3 and 4). To further confirm that Drpr is N-glycosylated, we transfected the drpr-HA transgene into S2 cells. Western blot analysis using HA antibody also revealed a downshift in Drpr-HA signals upon PNGase F treatment compared to the mock control (Fig. 2c). These analyzes suggest that Drpr is N-glycosylated.

a Schematic diagram showing the full-length Drpr protein with its various domains (EMI domain, green; EGF repeats, blue; and transmembrane domain, yellow) and N-glycosylation sites, as determined by MS. Blue stars denote paucimannose or high-mannose modifications and red stars denote hybrid and/or complex N-glycan modifications. b Western blot showing N-glycosylated Drpr at higher molecular-weight positions in w1118 control pupal lysates, and at lower molecular-weight positions in PNGase F-treated lysates, or in lysates prepared from Alg10wgd and xitA homozygous pupae. c Western blot showing Drpr-HA signals in transfected S2 cells, which shift to lower molecular-weight positions upon PNGase F treatment. DrprNQ4-HA signal migrated to the position between those of Drpr-HA and PNGase F-treated Drpr-HA. b, c the experiments were repeated at least 3 times independently with similar results. d Schematic diagrams showing three major types of N-glycans: high mannose, hybrid and complex. Blue squares: N-Acetylglucosamine (GlcNAc), green circles: mannose, and yellow circles: galactose. e Overlaid extracted ion chromatograms of the N-glycopeptides identified by LC-MS/MS for each of the four sites (N183, N358, N504, N630) of Drpr and DrprNQ4. The 5 most abundant (or 3 for N630) glycoforms along with those identified as carrying HexNAc≥3 were plotted and listed in the order of signal intensities.
Next, we performed mass spectrometry (MS) to establish the N-glycan profile for immunoprecipitated Drpr-HA prepared from transfected S2 cells. Among the 16 potential N-glycosylation sites, we detected 11 sites (marked with asterisks in Fig. 2a) as having N-glycans containing the high mannose type [HexNAc(n2)Hex(n)] attached to them (Fig. 2d, e and see Supplementary Data 1 for details). Interestingly, some sites—for example, N183, N358, N504 and N630 (marked with red asterisks in Fig. 2a)—were N-glycosylated with a glycosyl composition containing an additional HexNAc [HexNAc(n > 2)Hex(n)] in the LC-MS/MS analysis, which could represent hybrid, complex, or both types of N-glycans (Fig. 2d, e, and S2). Thus, Drpr is a substrate for N-glycosylation.
Whereas the hybrid and complex (H/C) N-glycan types are abundant in mammals, they account for only 1% of total N-glycans in Drosophila37, implying that protein modification with H/C-type N-glycans might be tightly regulated. To investigate the significance of N-glycan modification of Drpr, we generated a drprNQ4 mutant in which the four Asn sites were replaced with Gln (i.e., N183Q, N358Q, N504Q and N630Q), rendering these sites inaccessible for N-glycosylation. Western blot analysis of lysates prepared from transfected cells revealed that DrprNQ4-HA signal migrated slightly faster than the Drpr-HA signal (Fig. 2c). Moreover, MS indicated a complete absence of N-glycosylation at these four sites of DrprNQ4-HA (Fig. 2e and Supplementary Data 1), confirming that these four sites are N-glycosylated.
N-glycosylation of Drpr is required for cell surface localization and dendrite pruning
Next, we examined the effect of mutating the four Asn sites (NQ4) on Drpr distribution and function. Co-localization of A58-GAL4-driven Drpr-GFP and the cell adhesion protein DE-cadherin revealed that Drpr is enriched at the epidermal cell surface (Fig. 3a). However, DrprNQ4-GFP mostly remained inside cells, failing to localize to the cell surface (Fig. 3b). We also found mislocalization of Drpr in the Alg10wgd mutant with accumulation inside cells, although cell surface localization of Drpr was still present (Figs. 3c, d, and S3a). As Alg10 has a strong maternal contribution34,38, we examined Drpr localization in the pupal stage, and found no Drpr localization at cell surface in Alg10wgd (Fig. S3c). To assess if DrprNQ4 could still function in dendrite pruning, we performed rescue experiments on a drpr mutant. In the drprΔ5 mutant, the dendrite pruning defect was prominent at 14 hours APF, i.e., when control dendrites were completely pruned (Fig. 3e, f, i). A similar and stronger pruning defect was observed in the drprindel null mutant at the same stage (Fig. 4e, h). However, the defects in the drpr mutants were not as severe as in the Alg10wgd mutant (dendrite length = 1,377 ± 182.2 μm, compared to 859.4 ± 67.1 μm in drprindel, p = 0.0019 by Student’s t test, two tailed). Taken together, this suggests that substrates other than Drpr could also be modified by N-glycans to contribute to dendrite pruning. The pruning defect in drprΔ5 could be rescued by expressing the drpr-GFP transgene in epidermal cells (Fig. 3g, i). However, the pruning defect was not rescued upon expressing drprNQ4-GFP in epidermal cells (Fig. 3h, i). These results indicate that N-glycan modifications at these four sites of Drpr are essential for the membrane localization of this protein and its function in dendrite pruning.

a, b Confocal images showing localizations of Drpr-GFP (a) or DrprNQ4-GFP (b), with their expression driven by A58-GAL4 in epidermal cells at larval stages. GFP signals in green and DE-cadherin in red in merged images. Scale bar in (a), 50 µm. c, d Confocal images showing endogenous Drpr signals in w1118 (c) or Alg10wgd (d) larval epidermal cells. Scale bar in (c), 50 µm. a–d repeated at least 3 times independently with similar results. e–h Confocal images showing ppk-CD4-tdGFP dendrites at 14 hours APF in control (e), drprΔ5 (f), or the UAS-drpr-GFP (g) or UAS-drprNQ4-GFP (h) transgene driven by A58-GAL4 in the drprΔ5 mutant. White arrows indicate cell bodies. Scale bar in (e), 50 µm. i Bar graph showing quantification for average dendrite length (mean ± SEM) at 14 hours APF (32≥n ≥ 9 for each genotype). One-way ANOVA followed by Tukey’s post-hoc test was used to determine statistical significance, with ** for p < 0.01, *** for p < 0.001, and ns for no significant difference relative to control or to drprΔ5. p < 0.0001 in (i).

a Detection of hdg/CG11372, ctg/CG5335, CG11374, CG13950, RpL19, and Atg7 mRNA expressions by RT-PCR in larval lysates prepared from the w1118, hdgwgv, ctgwgd and hdgwgv ctgwgd lines. The experiment was repeated twice independently with similar results. b Schematics showing the designs to generate the hdgwgv and ctgwgd alleles by CRISPR-Cas9. Yellow and gray boxes represent coding regions and the untranslated regions of CG11372 and CG5335, respectively. In hdgwgv, part of intron 1 and exons 2 and 3 were replaced by GAL4-VP16 and 3xP3-RFP sequences. In ctgwgd, sequences from exon 1 to exon 3 were replaced by 3xP3-RFP and stop codons. The 3xP3-RFP cassette (red/blue boxes) in both alleles were removed by recombination of flanking LoxP sequences (gray circles). The attp and attpX sequences (orange triangles) for chromosomal landing and stop codons are also shown. c–f Confocal images showing dendrites of C4da neurons at 14 hours APF for the ppk-CD4-tdGFP control (c), hdgwgv ctgwgd (d), drprindel (e), and hdgwgv ctgwgd drprindel (f) lines. White arrows indicate cell bodies. Scale bar in (c), 50 μm. g, h Quantification of unpruned dendritic lengths (mean ± SEM) at 14 hours APF for C4da neurons labeled by GFP from co-expressed UAS-mCD8-GFP, 32≥n≥ 25 in each genotype (g) or ppk-CD4-tdGFP, 88≥n≥ 36 in each genotype(h). Statistical significance was determined by one-way ANOVA with Tukey’s post-hoc test, with * representing p < 0.05, ** for p < 0.01, *** for p < 0.001, and ns for no significant difference. p = 0.0001 in (g), and p < 0.0001 in (h).
Ctg and Hdg galectins are involved in dendrite pruning
The H/C-types of N-glycans include the disaccharide N-acetyllactosamine (LacNAc, Galβ1,4GlcNAc) that provides the recognition site for the evolutionary-conserved sugar-bonding proteins galectins25. With the H/C-type N-glycans identified in Drpr, we investigated if galectins are also required for dendrite pruning. We identified six genes—CG11372/Dmgal, CG5335, CG11374, CG13950, CG14879, and Pex2339—in the Drosophila genome as containing the carbohydrate recognition domain (CRD) of galectins. Among them, the first four galectins contain two tandem-repeat CRDs and the latter two are prototype galectins with only a single CRD (Fig. S4a). Whereas CG11372/Dmgal and CG5335 mRNAs were detected in both larvae and pupae, those of CG11374 and CG13950 were not (Figs. 4a and S4b). Therefore, we decided to focus on CG11372/Dmgal and CG5335, which we name hidden dragon (hdg) and crouching tiger (ctg), respectively. Null mutants for hdg and ctg were generated by means of CRISPR-Cas9 technology40 in which the respective protein-coding sequences were replaced by a GAL4-VP16 cassette (to generate hdgwgv) or a stop codon-containing cassette (to generate ctgwgd) (Fig. 4b). We confirmed that expression of hdg and ctg mRNAs were abolished in the respective mutants (Figs. 4a and S4b). As ctg is located within the sixth intron of Atg7, we also examined if mRNA levels of Atg7 were affected in the ctgwgd mutant, but observed that Atg7 expression in this mutant line was equivalent to that of the control line (Figs. 4a and S4b).
Next, we assessed if the galectin mutants exhibited any defects in dendrite pruning. Dendrites of the ctgwgd or hdgwgv single mutants were pruned normally, i.e., equivalent to the wild-type control (Fig. S4c). However, we observed prominent dendrite pruning defects in the hdgwgv ctgwgd double mutant at 14 hours APF (Fig. 4c, d, g). Notably, this pruning defect could be rescued by a genomic transgene of ctg or hdg (ctg-GR or hdg-GR), indicating that these two galectins function redundantly in dendrite pruning (Fig. 4g). The phenotype of the hdgwgv ctgwgd double mutant was similar to that of either drprindel or drprΔ5 mutant, albeit weaker (Figs. 4e, h, and S4f–h. To determine if hdg and ctg function in the drpr pathway, we constructed hdgwgv ctgwgd drpr triple mutants and explored their pruning phenotypes. The pruning phenotypes of the triple mutants were similar to that of the respective drpr mutant (Figs. 4f and S4g), with no statistically significant difference in unpruned dendritic length between the triple mutants and single drpr mutants (Figs. 4h and S4h). Thus, our genetic analysis indicates that ctg and hdg function in the drpr pathway, with both potentially acting as Drpr-binding galectins during the dendrite pruning process.
LacNAc-dependent interaction of Drpr with Ctg or Hdg
Ctg and Hdg both harbor two LacNAc-binding CRDs (Fig. 5a, b). Thus, we assessed whether Ctg and Hdg bind N-glycan-modified Drpr and whether the interactions depend on N-glycan modification on Drpr. We first tested the interaction between Ctg and Drpr by performing co-immunoprecipitation (Co-IP) on S2 cells transfected with drpr-HA and GFP-ctg. Western blot using anti-GFP antibodies revealed co-transfected GFP-Ctg in the HA immunoprecipitates from Drpr-HA-expressing S2 cell lysates (lane 4 in Fig. 5c). We used an unrelated carbohydrate-binding protein GFP-Lectin24Db as a negative control, which was not detected in the same assay (lane 6 in Fig. 5c). Next, we tested for interaction between Hdg and Drpr by means of the same assay. In the HA immunoprecipitates from Drpr-HA-expressing S2 cell lysates, we detected a much stronger co-precipitated FLAG-Hdg signal than the control FLAG-GFP on the Western blot using anti-FLAG antibodies (Fig. 5d). Together, these results support that Ctg and Hdg interact with Drpr.

a, b Schematic diagrams showing full-length Ctg (a), and full-length Hdg and N-terminus-truncated HdgΔN (b). c, d S2 cells were transfected with drpr-HA and GFP-ctg or GFP-Lec24Db (c), or with drpr-HA and FLAG-hdg or FLAG-GFP (d). Cell lysates immunoprecipitated (IP) with HA antibodies were subjected to Western blot (IB) analyzes by GFP, HA or FLAG antibodies. e, f S2 cells were transfected with GFP-ctg and drpr-HA or drprNQ4-HA (e) or with FLAG-hdgΔN and drpr-HA or drprNQ4-HA (f). Cell lysates were subjected to immunoprecipitation by HA antibodies and to Western blot analyzes using HA, GFP and FLAG antibodies. Bar graphs showing quantification of GFP-Ctg, e or FLAG-HdgΔN, f level normalized to respective Drpr-HA or DrprNQ4-HA level, with averages (mean ± SEM) shown from Western blots. Statistical significance was determined by Student’s t test, two tailed with ** representing p < 0.01. p = 0.0076 in (e), and p = 0.00227 in (f). g, h Western blots showing transfected S2 cell lysates immunoprecipitated by HA antibodies for Drpr-HA in the presence of 0, 10, 100 and 1000 μM of 3’-O-sialic acid-LacNAc. The HA immunoprecipitates were immunoblotted with anti-GFP for GFP-Ctg (g) or anti-FLAG for FLAG-HdgΔN (h). c–h the experiments were repeated at least 3 times independently with similar results.
To determine if the interactions between Drpr and Ctg or Hdg are N-glycan-dependent, first we assessed if DrprNQ4 interacted with Ctg and Hdg by means of Co-IP/Western blot experiments. As expected, we detected reduced levels of Ctg co-precipitating with DrprNQ4 relative to with wild-type Drpr (Fig. 5e). Levels of Hdg associating with DrprNQ4 remained similar to those determined for wild-type Drpr (Fig. S5a). Hdg contains an N-terminal extension of 132 amino acids to the first CRD that is not present in Ctg or other galectins (Fig. 5b). Accordingly, we tested N-terminal-truncated HdgΔN and N-terminal-only HdgN (amino acids 1-132) fragments for their interactions with Drpr. Both fragments still interacted with Drpr in Co-IP experiments (lanes 7 and 8 in Fig. S5b). We then assessed HdgΔN for N-glycan-dependent interaction with Drpr. Similar to Ctg, HdgΔN exhibited reduced interaction with DrprNQ4 compared to wild-type Drpr in our Co-IP experiments (Fig. 5f).
In a second set of experiments, we challenged the interaction between galectins and Drpr in the presence of the LacNAc derivative 3’-O-sialyl-LacNAc. With increasing concentrations of 3’-O-sialyl-LacNAc, amounts of Ctg or HdgΔN co-immunoprecipitating with Drpr-HA gradually declined, supporting that Ctg and HdgΔN interact with Drpr through the LacNAc moiety (Fig. 5g, h). Thus, Ctg and HdgΔN interact with Drpr via the H/C-type N-glycan modification. Taken together, these two sets of experiments strongly support that Ctg and Hdg bind to the LacNAc-modified H/C-type glycans on Drpr.
Ctg and Hdg are involved in injury-induced dendrite degeneration
In the third-instar larval stage, laser-mediated ablation at proximal sites of C4da dendrites induces fragmentation and clearance of detached dendrites in a process that requires epidermal expression of Drpr30,41. In similar experiments, we observed that ablated dendritic branches were degenerating at 5.5-6.5 hours post-laser injury, including clearance of higher-order branches (Fig. 6b), or clearance of higher-order branches plus fragmentation of lower-order segments (Fig. 6c). Approximately 23.5% of ablated dendrites remained intact at this time-point (Fig. 6a, d). Thus, the dendrotomy assay could be temporally controlled and the phenotypes could be examined in a sensitive time window. We examined if the galectins Ctg and Hdg are involved in the degenerating process. At 5.5-6.5 hours post-injury, the proportion of ablated branches in the hdgwgv ctgwgd double mutant that remained intact increased to 40.5% while the fragmented ones were greatly reduced to 21.6% (Fig. 6d). The impaired dendrite degeneration in the hdgwgv ctgwgd mutant was partially restored by the genomic ctg-GR or hdg-GR transgene, and dendrite fragmentation was almost restored to the level of the control upon introducing both genomic transgenes (Fig. 6d). This outcome indicates that hdg and ctg are involved in the clearance of higher-order branches and fragmentation of lower-order segments in the dendrotomy assay.

a–c Confocal images of C4da neurons labeled by ppk-CD4-tdTom expression, showing an example of intact dendrites without degeneration (a), an example of higher-order branch clearance (b), or an example of higher-order dendrite clearance plus lower-order segment fragmentation (c) at 5.5-6.5 hours post dendrotomy. Red arrows indicate the injury sites. The areas encompassed by yellow dashed lines are shown as zoomed-in images at right. Scale bar in (c), 50 µm. d Quantification of dendrites that remained intact (white bars), higher-order branch clearance (gray bars) or lower-order segment fragmentation (black bars) at 5.5–6.5 hours post dendrotomy. The number in each bar graph indicates the number of samples qualified. Chi-square analyzes were used to determine statistical significance, with *** representing p < 0.001, ** representing p < 0.01, and ns for no significant difference.
To investigate how glycosylation and galectins may impact dendrite degeneration and phagocytosis in the late stages of developmental pruning and the injury model, we examined dendrite phenotypes in hdgwgv ctgwgd double mutant and Alg10wgd mutant, and compared them to the previously reported drprindel mutant phenotypes42. For late-stage developmental pruning phenotypes, we examined mutants at 22 hours APF. We found that substantial amounts of dendrites remained in the Alg10wgd mutant and unengulfed dendrite debris left in the drprindel mutant, while dendrites in the hdgwgv ctgwgd double mutants were almost completely eliminated as in the control (Fig. S6a–e). The result of this comparison suggests that these two galectins have earlier roles in dendrite degeneration, and likely more N-glycosylation substrates in addition to Drpr contribute to dendrite degeneration and phagocytosis during developmental pruning. We then analyzed phenotypes for ablated dendrites at 18-21 hours post-injury, and found that they were almost completely cleared in the double mutant, similar to the ablated dendrites in the control larvae (Fig. S6f,g,j,k). In the Alg10wgd mutants, most of the ablated dendrites were either intact without fragmentation (8/21) or fragmented with some clearance (9/21) (Fig. S6h, k). In the drprindel mutant, most of the ablated dendrites (8/10) show the fragmentation phenotype (Fig. S6i, k), consistent with previous reports30,42. Quantification of the lengths of ablated dendrites relative to their innervating fields suggests that that both Alg10 and Drpr have roles in phagocytosing fragmented dendrites while Alg10 alone is required for fragmentation at this late stage of the injury model (Fig. S6j).
Ctg and Hdg localize at injured dendrites
To examine expression and localization of Ctg and Hdg during the pruning process, we generated GFP-ctg and GFP-hdg knock-in flies in which GFP was tagged at the N-terminus of Ctg and Hdg, respectively (Fig. 7a). We detected expression of GFP-ctg and GFP-hdg mRNAs at both larval and pupal stages (Fig. S7), similar to their endogenous gene expression profiles (Figs. 4a and S4b). Due to strong auto-fluorescence, expression of GFP-Ctg and GFP-Hdg was indiscernible at pupal stages. Therefore, we examined their expression patterns in our dendrotomy model at the larval stage. In dendrites labeled by CD4-tdTomato and not subjected to dendrotomy, basal GFP-Hdg levels were detected in epidermal cells of the GFP-hdg line (Fig. 7b). GFP-Hdg localized to laser-ablated dendrites, with levels being higher than background signal (Fig. 7c, e, white arrowheads in zoomed-in image). GFP-Hdg localization was detected initially at 1-1.5 hours after injury and the signal lasted for 2 hours. Similarly, GFP-Ctg was expressed at basal levels when dendrites were intact (Fig. 7f), but localized to injured dendrites upon dendrotomy (Fig. 7g, white arrowheads in zoomed-in image). However, GFP-Ctg localization at injured dendrites was detected only at 3-3.5 hours after injury, and it also lasted for 2 hours. In the same dendritic arbors, these galectin signals were also detected at unablated branches, cell bodies, and axons after injury, with levels lower than those observed for the injured branches (Fig. 7c, g, dashed circles showing cell bodies). None of these signals were detected in control dendritic arbors in neighboring segments not subjected to dendrotomy (Fig. 7b, f). Thus, dendritic injury induces GFP-Hdg and GFP-Ctg to localize to the ablated dendrite with enriched levels at the severed branches.

a Schematics showing the designs of the GFP-hdg (top) and GFP-ctg (bottom) knock-in alleles. In both alleles, the GFP sequence was inserted after the Met start codon with two linkers SR and RSITSYNVCYTKLSAS. b–d, f–h Confocal images showing signals of GFP-Hdg (b–d) at 1.5 hours post-dendrotomy, and GFP-Ctg (f–h) at 3.5 hours post-dendrotomy in body segments without dendrotomy (b, f), with dendrotomy (c, g), or with dendrotomy in drprΔ5 (d, h). Dendrites were marked by ppk-CD4-tdTOM expression and boxed areas have been enlarged at right. Red arrows indicate ablation sites, white arrowheads indicate GFP signals at ablated branches, and white dashed circles indicate cell bodies. Scale bars in (b) and (f) are 50 µm. e, i Quantification of GFP intensities at intact and injured dendrites. Cut and uncut branches from the injured dendrites were separately scored. The epidermal GFP intensity was subtracted from the dendritic GFP signal intensities, which then was normalized to the epidermal GFP intensity to be shown as ratios (mean ± SEM). 34≥n ≥18 for each genotype in (e), and 46≥n ≥20 for each genotype in (i). One-way ANOVA with Tukey’s post-hoc test was performed to determine statistical significance by comparing to the intact dendrite or by comparing between cut and uncut branches of the same dendrites, and shown as * representing p < 0.05, ** for p < 0.01, *** for p < 0.001, and ns for no significant difference. p = 0.0041 in (e), and p < 0.0001 in (i).
Given the interactions of these two galectins with glycosylated Drpr (Fig. 5), we tested if the dendritic localizations of GFP-Ctg and GFP-Hdg are dependent on Drpr. We found that GFP-Ctg and GFP-Hdg no longer localized at dendrites of both cut and uncut branches, and cell bodies upon dendrotomy in the drprΔ5 mutant (Fig. 7d, e, h, i). Thus, Drpr is involved in recruiting or maintaining GFP-Ctg and GFP-Hdg localizations to injured dendrites.
Although Drpr clearly localized to epidermal cell boundaries, we also detected Drpr signal beneath dendrites (arrowheads in Fig. S8a). The Drpr signals were absent in the drprΔ5 mutant (Fig. S8b), as well as by drpr-RNAi knockdown driven by A58-GAL4 but not by ppk-GAL4, implying that these Drpr signals are derived from epidermal cells (Fig. S8c, d). We examined if Drpr localization depends on the Ctg and Hdg by performing Drpr immunostaining on the hdgwgv ctgwgd double mutant. Whereas Drpr is required for Ctg and Hdg localization at dendrites post dendrotomy, neither galectin is required for Drpr localization (Fig. S8e). Since Drpr also localizes at epidermal cell boundaries (Fig. S8a), where neither Ctg nor Hdg are enriched, our results indicate that Drpr recruits Ctg and Hdg to dendrites via a specific process that may require other factors apart from their protein-protein interactions.
Hdg and Ctg are required in hemocytes for dendrite degeneration
Next, we investigated the type of cells that express GFP-Hdg and GFP-Ctg during dendrotomy. We utilized tissue-specific GAL4s to drive the UAS-GFP-RNAi transgene to knock down GFP-hdg and GFP-ctg expression. By using ppk-GAL4 for C4da neuronal knockdown or A58-GAL4 for epidermal knockdown, we found that GFP-Hdg and GFP-Ctg were still distributed in epidermal cells and localized to ablated branches upon dendrotomy (Fig. 8a, b, d, e). Clusters of hemocytes reside in the dorsal region of body hemisegments and require neuronal activity for their proliferation and adhesion43. We explored if they could be the cell source of GFP-Hdg and GFP-Ctg expression. By employing hemocyte-specific Hml-GAL4 to drive GFP-RNAi, we found that both GFP-Hdg and GFP-Ctg signals were suppressed, including those epidermally-distributed signals and dendrotomy-induced dendritic signals (Fig. 8c, f). We quantified the signal intensities at epidermal levels and showed that GFP signal intensities were significantly reduced by the Hml-GAL4 driver (Fig. 8g), suggesting that hemocytes are the major cell source for the expressions of GFP-Hdg and GFP-Ctg during dendrotomy.

a–f Confocal images of C4da neurons labeled by ppk-CD4-tdTom in the GFP-hdg knock-in line at 1.5 hours post-dendrotomy (a–c), or in the GFP-ctg knock-in line at 3.5 hours post-dendrotomy (d–f). UAS-GFP-RNAi transgene was driven by ppk-GAL4 (a, d), A58-GAL4 (b, e) or Hml-GAL4 (c, f). Scale bar in (a), 50 µm. g Two composite bar graphs (separated by dashed line) show quantification for relative GFP intensities (mean ± SEM) at the epidermal cell level to a non-GFP control background. 51≥n ≥24 for each genotype. One-way ANOVA followed by Tukey’s post-hoc test was used to determine statistical significance, with ** for p < 0.01, *** for p < 0.001, and ns for no significant difference relative to GFP-hdg or GFP-ctg. p < 0.0001 in GFP-hdg dataset, and p < 0.0001 in GFP-ctg dataset. h–j Confocal images of C4da neurons at 14 hours APF in control hdg+ ctg+, hdgwgvctgwgd double mutant, or double mutant carrying UAS-RFP-hdg or UAS-RFP-ctg transgene driven by Hml-GAL4 (h), ppk-GAL4 (i) or A58-GAL4 (j). Scale bar in (h), 50 µm. White arrows indicate cell bodies and arrowheads indicate unpruned dendrites. k Bar graphs show quantification for dendritic length (mean ± SEM) from confocal images. 36 ≥ n ≥ 17 for each genotype. Statistical significance relative to the control (First bar in each graph denotes dendrite marker) was determined by one-way ANOVA followed by Tukey’s post-hoc test, with * representing p < 0.05, ** representing p < 0.01, *** for p < 0.01, and ns for no significance. p = 0.0008 in Hml-Gal4 rescuing dataset, p = 0.0019 in ppk-GAL4 rescuing dataset, p = 0.0012 in A58-GAL4 rescuing dataset.
We further performed two sets of experiments to show that hemocytes-expressing GFP-hdg and GFP-ctg are involved in promoting dendritic degeneration during dendrotomy. In the first set of experiments, we tested if hemocyte-specific expression of hdg or ctg could rescue the dendrite degenerating phenotype in the hdgwgv ctgwgd double mutant. The percentage of degenerating dendrites (encompassing higher-order branch clearance and lower-order segment fragmentation) was 48% in the hdgwgv ctgwgd; Hml-GAL4 mutant (Fig. S9a). Hemocyte-specific expression of RFP-hdg or RFP-ctg significantly enhanced the percentages of degenerating dendrites to 80% or 79%, respectively (Fig. S9a). In the second set of experiments, we performed hemocyte-specific Hml-GAL4-driven GFP-RNAi knockdown in homozygous GFP-hdg or GFP-ctg knock-in lines. Upon dendrotomy, the percentages of degenerating dendrites in both knockdown lines were reduced, from 85% in the Hml-GAL4 control to 67% for GFP-hdg knockdown and 62% for GFP-ctg knockdown (Fig. S9b). These results support that hemocyte expression of GFP-ctg and GFP-hdg promotes dendritic degeneration upon dendrotomy.
Next, we examined if hemocyte expression of Hdg and Ctg could rescue the pruning defect of the hdgwgv ctgwgd double mutant at the pupal stage. Indeed, Hml-GAL4-driven expression of either RFP-hdg or RFP-ctg in hemocytes of the hdgwgv ctgwgd line restored the normal pruning process (Fig. 8h, k). We also examined if expression of Hdg and Ctg by other cell types elicited the same effect. However, we failed to detect any restoration of dendritic pruning in the hdgwgv ctgwgd mutant by the neuronal ppk-GAL4- or the epidermal A58-GAL4 driver (Fig. 8i–k). In conclusion, hemocytes appear to be the major type of cells that express Hdg and Ctg to promote dendritic degeneration during dendrite injury and developmental pruning.
Differential Hdg and Ctg inductions in hemocytes upon dendrotomy
The GFP signals of the GFP-hdg and GFP-ctg knock-in lines were ubiquitously distributed (Fig. 7b, f), with no specific or enriched signals being observed in hemocytes even upon dendrotomy (Fig. 7c, g). It is possible that galectins expressed in hemocytes are secreted extracellularly, which overwhelms the signals in hemocytes. We then examined the possible transcription regulation of hdg and ctg by dendrotomy. The expressions of GAL4-VP16 in the knock-in lines hdgwgv (Fig. 4b) and ctgwgv (Fig. S10g) presumably reflect the transcriptional expression patterns of hdg and ctg, respectively. The GFP expression levels from UAS-10XGFP driven by GAL4-VP16 in hdgwgv (Fig. S10e, f) and ctgwgv (Fig. S10c, d, h) were not changed by dendrotomy. We then examined whether Hdg and Ctg expressions in hemocytes could be induced by dendrotomy in the hemocyte-overexpression system. We used Hml-GAL4 to drive UAS-RFP-hdg or UAS-RFP-ctg expression in hemocytes, and UAS-NLS-GFP to label the hemocytes. Then, we analyzed expression and localization of RFP-Hdg and RFP-Ctg at C4da dendrites in two adjacent abdominal hemisegments, i.e., one intact and the other subjected to dendrotomy. In the hemisegment hosting intact dendrite, we identified NLS-GFP-positive resident hemocytes (Fig. 9a), but RFP-Hdg signals were undetectable in the hemocytes and C4da neurons (zoomed-in boxes in Fig. 9a). However, in the hemisegment housing ablated dendrites, RFP-Hdg was detected at low levels in both hemocytes and C4da neurons 0.5 hours post-dendrotomy (Fig. 9b). Higher levels of signals were detected in hemocytes and neuronal cell bodies, and localization to ablated branches were prominent at 1 hours post-dendrotomy (zoomed-in boxes and white arrowheads in Fig. 9d). However, RFP-Hdg signals were still almost undetectable in the neighboring hemisegment with intact dendrites even 1 hours post-dendrotomy (Fig. 9c, e). Indeed, throughout the entire larval body, we only found RFP-Hdg signals in the segment housing the injured dendrite, suggesting that the induction and distribution of RFP-Hdg is restricted to the injured dendrite.

a–d Confocal images showing C4da neurons labeled by ppk-CD4-tdGFP expression, and hemocytes with Hml-GAL4-driven RFP-hdg and NLS-GFP expressions. Confocal images were taken at 0-0.5 hours(a, b) or 1 hour (c, d) post-dendrotomy in two adjacent hemisegments of the same larva, with (a, c) for neurons not subjected to dendrotomy, and (b, d) for neurons subjected to dendrotomy. Red arrows indicate the injury sites and white arrowheads indicate RFP signal at dendrites. The areas within white dashed lines are shown in zoomed-in images at bottom, with “1” for cell body, and “2” for representative hemocytes. Scale bar in (a), 50 µm. e Quantitative analysis of RFP signal intensities (mean ± SEM) in hemocytes of Hml > RFP-hdg larvae at 0–0.5 hours and 1 hour with or without dendrotomy. 79≥n≥40 for each genotype. Statistical significance relative to the intact control was determined by one-way ANOVA followed by Tukey’s post-hoc test, with *** for p < 0.001, and ns for no significance. P < 0.0001 in (e).
Next, we tested induction of RFP-Ctg in hemocytes by means of the same experimental approach. However, unlike for RFP-Hdg, we detected strong RFP-Ctg signals in hemocytes and weak RFP-Ctg signals in neuronal bodies in the absence of dendrotomy (Fig. S11a). Upon dendrotomy, we observed dendritic RFP-Ctg localization (arrowheads in Fig. S11b), but the signal intensity in hemocytes was not different to that in hemocytes near intact dendrite (Fig. S11c). Thus, unlike RFP-Hdg, the RFP-Ctg expression level in hemocytes is not regulated by dendrotomy, although its localization to dendrites is regulated.
We then tested if Hdg and Ctg could be induced and localized to dendrites in other cell types upon dendrotomy. Employing A58-GAL4 to drive UAS-RFP-hdg expression in epidermal cells, we detected no RFP-Hdg signal in epidermal cells, hemocytes, neuronal bodies or dendrites with and without dendrotomy (Fig. S12a–c). Also, when UAS-RFP-ctg was driven by A58-GAL4, RFP-Ctg distributed in the epidermal cells (Fig. S12e). However, we were not able to discern dendritic localization of RFP-Ctg from the high levels of epidermal RFP-Ctg signals. Similarly, expression of RFP-Ctg but not RFP-Hdg could be detected in C4da neurons when driven by ppk-GAL4 (Fig. S12f, g). Thus, RFP-Hdg and RFP-Ctg expressions are differentially regulated.
Phosphatidylserine exposure induces the dendritic localization of Ctg and Hdg and subsequent dendritic debris clearance
We investigated how Hdg and Ctg are recruited to dendrites upon dendrotomy. During phagocytosis, the phospholipid phosphatidylserine (PS), also known as the “eat-me” signal, is exposed at cell surfaces to bind and activate phagocytic receptors9. Therefore, we tested if the dendritic localizations of Ctg and Hdg could be induced upon PS exposure. The lipid flippase P4-ATPase internalizes PS from exoplasmic to cytoplasmic membrane leaflets. Ectopic exposure of PS can be induced by inactivating CDC50, the chaperone required for proper folding and localization of P4-ATPases42. In the control without CDC50 inactivation, we did not detect overexpressed RFP-Hdg in hemocytes (Fig. 10a). Upon C4da neuron-specific knockout of CDC50 in ppk-Cas9, CDC50-gRNAs larvae42, we observed upregulated overexpression of Hml-GAL4-driven RFP-Hdg, with enhanced and ubiquitous levels in epidermal cells and hemocytes (asterisks in Fig. 10b). RFP-Hdg localization to dendrites was also discernible (arrowheads in Fig. 10b). We also examined if the expression and localization of RFP-Ctg signals were regulated by CDC50 knockout in neurons. In our control line, the signal for overexpressed RFP-Ctg was mainly confined to hemocytes (asterisk in Fig. 10c). In CDC50 knockout neurons, RFP-Ctg signal was ubiquitously upregulated in epidermal cells and clearly localized to dendritic branches (arrowheads in Fig. 10d). These results support that PS exposure induced by CDC50 inactivation is sufficient to upregulate RFP-Hdg expression in hemocytes, and for localization of both RFP-Hdg and RFP-Ctg to dendrites.

a–d Confocal images of C4da neurons labeled by ppk-CD4-tdGFP expression and hemocytes labeled by Hml-GAL4-driven NLS-GFP. Asterisks indicate hemocytes and white arrowheads indicate dendritic RFP signal. Boxed areas have been enlarged at right. a, b Hml-GAL4-driven expression of RFP-hdg or c, d RFP-ctg in hemocytes. a, c Wild-type CDC50 control and b, d CDC50 knockout. a–d the experiments were repeated at least 3 times independently with similar results. e Membrane strips dotted with PE, PC, and PS were immuno-blotted with Flag-tagged Hdg, Ctg or RFP. A schematic showing the position of different lipids on the membrane strips. PE phosphatidylethanolamine, PC phosphatidylcholine, PS phosphatidylserine; and a blank control.
A large amount of shed membrane debris, detected preferentially near terminal branches, is generated acutely upon dendrotomy in CDC50 knockout neurons42. We observed basal levels of dendritic debris in wild-type ablated dendrites (Fig. S13a). We confirmed that dendritic debris was also detected at the branch terminals of CDC50 knockout neurons (arrowheads in Fig. S13b). However, dendritic debris was detected solely at ablated branches (red dashed boxes in Fig. S13a, b) and not at non-ablated branches (yellow dashed boxes) of the same neuron. We used an approach described previously42 to quantify the ratio of debris coverage for ablated branches and found that it was increased in CDC50 knockout neurons relative to wild-type neurons (Fig. S13d). We then tested if the presence of dendritic debris is dependent on Ctg and Hdg. Notably, debris coverage was suppressed by the hdgwgvctgwgd double mutation (Fig. S13c, d). This dendritic debris could be engulfed by epidermal cells in a Drpr-dependent manner42. Thus, we compared the number of ablated versus non-ablated branches (red versus yellow boxes in Fig. S13a–c. Consistently, the ablation/non-ablation branch density ratio was significantly reduced in CDC50 knockout neurons, which was rescued by the hdgwgv ctgwgd double mutation (Fig. S13e). Taken together, enforcing PS exposure by knocking out CDC50 in C4da neurons induces Hdg and Ctg to localize to dendrites. Moreover, Hdg and Ctg are required for terminal branch elimination.
Hdg and Ctg interact with PS
Our finding that PS exposure in CDC50 knockout neurons induced Ctg and Hdg to localize to dendrites prompted us to test if Ctg and Hdg physically interact with PS by means of two in vitro experiments. First, we immunoprecipitated FLAG-tagged Ctg or Hdg from transfected S2 cells (Fig. S14a) for incubation with deuterium-labeled phospholiplids. The precipitated phospholipids were then analyzed by high-performance liquid chromatography-mass spectrometry (HPLC-MS, see Materials and Methods). We detected higher levels of PS bound to Ctg or Hdg than for the controls of FLAG-tagged RFP or beads only (Fig. S14b). However, levels of bound phosphatidylcholine (PC) or phosphatidylethanolamine (PE) to Hdg or Ctg were not significantly different to those of the controls (Fig. S14b). In the second experiment, we tested protein-lipid interactions on a membrane strip dotted with different lipids14. To do so, immunoprecipitated FLAG-Ctg, FLAG-Hdg or FLAG-RFP was incubated with the lipid-dotted membrane. We observed that both Ctg and Hdg were retained at the PS spot, which was not the case for the PC and PE spots (Fig. 10e). No signals were detected for the “beads only” or RFP negative controls. Taken together, these two sets of data strongly support that Ctg and Hdg interact with PS in vitro.
Discussion
N-glycosylation of Drpr is required for dendrite pruning
Although the exact mechanism by which Drpr/CED-1 signaling is activated by the “eat-me” PS signal during phagocytosis is still not fully understood, particularly in various neuronal pruning systems, our findings provide significant insights. Here, we have identified two glucosyltransferases, Alg10 and Alg8, in the N-glycosylation pathway that are required in epidermal cells during pruning. Through point mutations that eliminate N-glycosylation, we found that localization of Drpr to the cell surface and its subsequent role in dendrite phagocytosis are abolished. Therefore, N-glycosylation of Drpr is likely involved in trafficking Drpr to the cell surface to promote dendrite engulfment during pruning. As noted previously34, DE-cadherin also depends on N-glycosylation for its distribution at the plasma membrane of epidermal cells during germband extension in embryonic stages. In addition to functioning as non-professional phagocytes, epidermal cells exert multiple roles in dendrite arborization, such as scaling growth44, space filling45, branch adhesion and stabilization46,47, preventing fasciculation47,48, and trimming terminals49. Thus, epidermal cells play seemingly conflicting roles in the life and death of C4da dendrites.
Hdg and Ctg bridge PS with Drpr during dendrite pruning
Despite the Drpr/CED-1 phagocytic receptor being conserved from C. elegans to human, the molecules required to activate them remain unclear. During phagocytosis, a diverse array of tethering/bridging molecules are required to bridge the phagocytic receptors to the exposed PS signal from apoptotic and damaged cells9. These bridging molecules are context-dependent, according to the apoptotic cell types, phagocytes or phagocytic receptors. The human Drpr/CED-1 homolog MEGF 10 interacts with PS through the C1q linker in synaptic debris clearance6, whereas C. elegans TTR-52 acts as the bridging molecule between PS and CED-1 during apoptosis14. The chimeric-type galectin-3 secreted by mammalian microglia can interact with and activate receptors like Toll-like receptor-4, TREM2 or MerTK in inflammation and phagocytosis50,51,52. In solid phase assays, galectin-3 interacts with anionic PS, potentially through the CRD domain with a sufficient positive charge density53. Thus, secreted galectin-3 may act as a bridging molecule between PS and phagocytic receptors. Herein, we have shown that Hdg and Ctg play a crucial role in developmental dendrite pruning at the pupal stage and in responses to dendrite injury at the larval stage of Drosophila. Upon dendrite severing or injury, Ctg and Hdg are induced and localize to dendrites, which likely promotes clustering of the phagocytic receptor, leading to signaling pathway activation and phagocytosis26,54.
In Drosophila, two ER membrane proteins, Pretaporter and DmCaBP1, have been identified as physically interacting with the Drpr extracellular domain16,17. Upon induction of apoptosis, these proteins relocate to the cell surface to bridge interaction of the apoptotic cells with Drpr and they are also required for apoptosis at embryonic stages. However, Pretaporter and DmCaBP1 play only limited roles in the pruning of mushroom body γ neurons, a model system for studying axonal pruning. During embryonic nervous system development, the tethering receptor Simu acts prior to the Drpr-mediated phagocytosis. Simu recognizes the PS-exposed neurons and form clusters on the surface membrane of the phagocytic glia55. The recently identified CXC3L1 molecule Orion, secreted from nonneural tissues and localized at injured dendrites, serves as a bridging molecule between PS and Drpr19. Major differences could be found between Orion and the two galectins in this study. First, it is suggested that Orion recruits Drpr to injured dendrites, while Hdg and Ctg require Drpr to localize at injured dendrites (Fig. 7d, h), likely through recognition of LacNAc-modified N-glycans at Drpr. Second, Orion is constitutively expressed in peripheral tissues at low levels. Hdg and Ctg, instead, are mainly induced in hemocytes upon dendrotomy (Fig. 8). Third, localization of Orion at dendrites occurs at a later stage (4-6 hours post-dendrotomy) than the localizations of Hdg and Ctg (0.5-3 hours), implying sequential action of these molecules to bridge interaction between PS and Drpr. The immediate response of Hdg and Ctg might enhance the efficiency of the entire phagocytic process.
Two PS-binding proteins Annexin V (AV) and Lactadherin (Lact)56,57 when fused to GFP have been employed to label injured dendrites in Drosophila42. The PS-binding motifs of these proteins include abundant basic amino acids arginine (R) and lysine (K), which together with adjacent peptides are important for PS-binding activity58. Similarly, Hdg and Ctg may interact with negatively-charged PS through the CRD domains that contain several positively-charged amino acids. The CRD domains of Hdg and Ctg can also bind the phagocytic receptor Drpr through their N-glycan modification, thus facilitating the phagocytic process.
Ctg and Hdg are differentially regulated in hemocytes
In mammals, galectin-3 is upregulated in microglia and released into the cerebrospinal fluid during traumatic brain injury or ischemia50,59. Herein, we have also shown some aspect for the regulation of the Drosophila galectins Hdg and Ctg during dendrite injury. Although GFP-hdg and GFP-ctg mRNAs were detected in our knock-in lines (Fig. S7), the fused proteins GFP-Hdg and GFP-Ctg were present at low levels ubiquitously, and except localization to injured dendrites, no obvious difference in the protein levels of GFP-Hdg and GFP-Ctg was detected upon dendrotomy (Fig. 7). Transcription of hdg and ctg are less likely to be induced by dendrotomy, either, as shown by the GFP expression driven by the GAL4-VP16 knock-in (Fig. S10). Interestingly, when driven by the hemocyte-specific Hml-GAL4, the protein level of RFP-Hdg was upregulated upon dendrotomy, whereas the level of RFP-Ctg remained unchanged, with non-induced levels of RFP-Ctg already being high (Figs. 9 and S11). The induction in the RFP-Hdg level was not detected for GFP-Hdg in the knock-in line, which could be explained by the efficient secretion of GFP-Hdg from hemocytes and degradation in epidermal cells (see below). We detected low levels of GFP-Hdg or GFP-Ctg in epidermal cells (Fig. 7b, f), which showed colocalization with the endosomal Rab5 protein, suggesting constant endocytosis and degradation to prevent the accumulation of galectins that might unnecessarily activate phagocytosis (Fig. S15). Furthermore, whereas the galectins mainly localized to the injured dendritic branches, lower-level galectin signals were detected at uninjured branches (Fig. 7c, g), as well as intra-neuronal signals were detected (Figs. 9d and S11, b). We suspect that galectins localized to the surface of uninjured branches and cell bodies might be endocytosed efficiently to prevent unwanted phagocytosis. We also suggested additional layers of regulation for both galectins. For instance, RFP-Hdg signals were not detected from epidermal A58-GAL4 or neuronal ppk-GAL4 driving UAS-RFP-hdg expression (Fig. S12d, f), even with dendrite injury (Fig. S12b). Post-transcriptional regulation of hdg has been suggested, as the hdg mRNA is bound by the hnRNP HRP-48, which could bind protein-coding regions and the 3’UTR of mRNAs and may regulate protein translation, or stability and transport of hdg mRNA60. Also, while RFP-Ctg could be induced for ectopic expressions in epidermis and neurons (Fig. S12e, g), the ectopic expression failed to rescue the pruning defect displayed by the hdg ctg double mutant (Fig. 8h–j). Thus, Ctg protein may present further post-translational regulation such as protein modification and secretion.
Communication between dendrites and hemocytes
Hemocytes, the macrophage-like cells of Drosophila, have been reported previously to attack and engulf severed dendritic branches61. However, genetic ablation of hemocytes in larvae results in normal dendrite pruning during development, though fragmentation is delayed in the dendrite injury model30, indicating that hemocytes are required for efficient phagocytosis. Our findings support that the role of hemocytes in dendritic phagocytosis mainly operates through the expression and secretion of Hdg and Ctg upon dendrite injury. Nevertheless, the signals released from injured neurons to induce hemocytes to upregulate and secrete galectins remain to be established. During development, the TGFβ signal emitted from da neurons regulates the hemocyte homing that promotes their localization near dendrites43. Apoptotic neurons release Spätzle 5 and Insulin-like signal (ILS) to upregulate Drpr expression in phagocytic glia, thereby removing degenerating axons upon acute axon injury in the adult brain62,63. Further studies are required to investigate the involvement of the aforementioned neuron-emitted and injury-induced signals, or other unidentified signals in upregulating Ctg and Hdg in hemocytes upon dendrite injury64,65,66,67,68,69,70.
Methods
Fly stocks
All flies were raised at 25 °C under a 12:12 hour light:dark cycle. All alleles and their sources are listed in the Supplementary Table 1. Mid-third-instar or wandering larvae, pupae at 14 hours after APF, or pupae at 16-18 hours APF were used for experiments.
Generation of various transgenes
mRNA was extracted from third-instar larvae or pupae and then cDNA was synthesized using SuperScript™ VILO™ Master Mix (Invitrogen). mRNA was extracted according to the protocol of Tsai et al.71. The protein-coding regions of Alg10, drpr-RB, CG11372-RA, and CG5335 were amplified by polymerase chain reaction (PCR) from the larval cDNA and cloned into Topo PCR8 vector (Invitrogen)72, respectively. Based on the PCR8-drpr construct, quick-change site-directed mutagenesis was performed at four sites using
drpr183-CTTTTTCGGGGCCCAGTGCTCGGAGAAGTGCCGAT,
drpr358-GGCTGGACTGTCAGCGCACCTGCGAGTGCG,
drpr504-CTGCAATCCGCAGCAGGGAAGCTGCACCTGTGC,
drpr630-GGAGATCCTCCATGGCCAGAAGAGCTGCGATCACATC, and their complements to generate drpr-N183Q-PCR8, drpr-N358Q-PCR8, drpr-N504Q-PCR8, and drpr-N630Q-PCR8. Next, four different peptide fragments hosting the mutations were amplified by PCR using individual primer sets: 183 f -GGCTCAGGCGGATCTTAAA, 183r- ATGCTTTCCATCCGGACA, 358f-TGTCCGGATGGAAAGCAT, 358r-CATAGGCACGTACCATTGACC, 504f-AATGGTACGTGCCTATGTGC, 504r- ATGCAGTTACCGGAATCG, 630f-GATTCCGGTAACTGCATCTGCT, 630r- GTAAAAGATGCGGCCTCCAC. Then, the PCR fragments were ligated with drpr-PCR8 digested by BglII and SacII using NEBuilder® HiFi DNA Assembly Master Mix. All constructs were verified by sequencing. GATEWAY pTW, pTGW or pTWG, pTRW, or pTWH vectors containing the cDNAs were constructed by LR reaction (Life Tech), and transgenic lines were established. For the genomic rescue transgenic stocks (hdg-GR and ctg-GR), P[acman] BAC clone CH322-40A09 for hdg and P[acman] BAC clone CH322-70N19 for ctg were targeted into the attP2 landing site by means of embryo injection69.
For Alg10wgd, the sequence from Base 1 to Base 22 before the ATG stop codon of Alg10 was deleted using two different gRNAs: CGCCTACCATGAATGGGTCC and ATATGATCCTTTGAGGAGTG. For hdgwgv, the sequence from Base 103 to Base 291 before the ATG stop codon of CG11372-RB was deleted using two different gRNAs: AAAATCCTTTGAGCTCCTGA and GTGGACACTGTCTACATACA. For ctgwgd and ctgwgv, the sequence from Base 60 to Base 85 before the ATG stop codon of CG5335 was deleted using two different gRNAs: GAGAACTCCGGACTCTCCCA and ACGTAGGATCCCCCGTCTGC. GFP-hdg had the GFP peptide inserted after the translation start site (TSS) with an SR linker, as well as the RSITSYNVCYTKLSAS linker between GFP and Hdg, using one gRNA: TATGACGGCAATAATACTCC. GFP-ctg also had the GFP peptide inserted after the TSS with an SR linker, as well as the RSITSYNVCYTKLSAS linker between GFP and Ctg, using one gRNA: TGGTCATGTTCTGGAGGTCG. All six of the above-described transgenic lines were generated by Wellgenetics.
Quantification of dendrites
Live confocal imaging of da neurons with UAS-mCD8-GFP driven by ppk-GAL4, ppk-CD4-tdGFP or ppk-CD4-tdTOM was performed at 14–15 hours or 16–18 hours APF. The dorsal side is top-most in all images. The extent of severing defects is represented by the percentage of C4da neurons having larval dendrites still attached to the soma at 16–18 hours APF. The length of unpruned dendrites was measured using Neuron J in a 300 µm x 300 µm region derived from the dorsal field of C4da neurons within abdominal segments 2–4. Plots of average length and SEM were generated in GraphPad Prism software. Dendrite terminals were counted using the DeTerm program73.
Image analysis
Image processing and analyzes were conducted in ImageJ. For RFP or GFP signal intensity in hemocytes, images were taken with the same settings using a 20X or 40X objective and a Zeiss LSM 880 or 900 system. Five individual hemocytes or epidermal cells were selected to measure the mean intensity of RFP or GFP signal. In order to illustrate more clearly RFP or GFP signal at dendrites or hemocytes in the figures, we have adjusted image contrast and brightness in both the control and experiment sets in the same manner. The protocol for quantifying the debris coverage ratio was adapted from42. The respective images were taken using a 40X objective with 1024×1024 pixels. The images were subjected to processing through Gaussian Blur (Sigma: 1), Auto Local Threshold (Phansalkar method, radius: 50), Particles8 (to remove particles smaller than 200 pixels), and Dilate (iterations=2, count=1) to generate a dendrite mask. The debris signal within the dendrite mask was removed and the debris outside the dendrite mask was converted to a debris mask by thresholding with fixed thresholds (40, 255). Five different 780-pixel regions of interest (ROI) not within the dendrite areas were selected to calculate the dendrite coverage ratio by using the following formula: debris pixel area / ROI area·100%. For the dendrite density ratio, ablated or nonablated dendrite areas were defined, and then the total numbers of dendrite tips in each area were counted. The dendrite density ratio was calculated according to the following formula: [dendrite tip number in ablated dendrite area / ablated dendrite area] / [dendrite tip number in nonablated dendrite area / nonablated dendrite area].
For quantifying GFP signal intensities at dendrites, images were taken with the same settings using a Zeiss LSM 880 or 900 system. Three ROIs of injured or uninjured dendritic segments from a larva were selected for quantification. ROIs on epidermal cells near each selected dendritic ROI were also quantified to serve as backgrounds. Dendritic GFP ratios (Fig. 7e, i) were calculated by subtracting background GFP intensity from dendritic GFP intensities, which were further divided by background GFP intensities. Epidermal GFP ratios (Fig. 8g) were calculated by subtracting non-GFP control intensities from epidermal GFP intensities, which were further divided by non-GFP control intensities.
Immunostaining and fluorescence microscopy
Larvae and pupae were dissected in cold PBS, and fixed for 20 minutes in 4% PFA in PBS. After several washes in PBS with 0.2% Triton (PBST), samples were blocked for one hour in PBST containing 5% NGS (Gibco™ PCN5000). The samples were incubated overnight with primary antibody at 4 °C. After three washes with PBST, the samples were incubated with secondary antibody for 1 hour at room temperature. The samples were washed three times with PBST, and then mounted in mounting medium (80% Glycerol with 0.1% DABCO from Sigma #10981). Primary antibodies used in this study were goat anti-HRP conjugated FITC (1:1000), chicken anti-GFP (1:500), rabbit anti-Rab5 (1:500), rabbit anti-Drpr (1:500), guinea-pig anti-Drpr (1:100), mouse anti-Futsch (1:100) and rat anti-DE-cadherin (1:100). Cy3- and Cy5-conjugated secondary antibodies were used at a dilution of 1:500. Z-stack confocal images were obtained using a Zeiss LSM 710 or Zeiss LSM 880 system and processed in Image J74.
N-Glycan removal by PNGase F
Approximately 30 pupae (0–18 hours) were ground and lysed with 100 µl mRIPA buffer containing 1% Nonidet P-40, 0.5% Triton, 50 mM Tris-HCl (pH 7.5), 150 mM NaCl, 1 mM EDTA, and protease inhibitor cocktail (Complete Tablets; Roche) for 30 minutes on ice. After high-speed centrifugation, 18 µl of the supernatant was removed and mixed with 2 µl of 10X denaturing buffer (NEB) for 10 minutes at 100 °C. After adding 3 µl of 10X reaction buffer (NEB), 2 µl N-glycosidase F (NEB)75 and water were added to generate a mixture of 30 µl, which was incubated for 1 hour at 37 °C. Then, the samples were added to SDS loading buffer and boiled for 10 minutes at 100 °C for Western blot analysis.
Immunoprecipitation
Drosophila S2 cells were maintained at 25 °C in Schneider’s medium (Life Technologies) supplemented with 10% fetal bovine serum (Life Technologies) and 1% penicillin/streptomycin (Life Technologies). S2 cells were seeded into 6-well plates (2×106 cells/well) or 10-cm plates (10 × 106 cells) and transfected with the appropriate plasmids using TransIT®-Insect Transfection Reagent (Mirus): pAc-Gal4, pUAS-DraperHA, pUAS-DraperNQ4HA, pUAS-FLAG-CG11372, pUAS-FLAG-CG11372N, pUAS-FLAG-CG11372ΔN, pUAS-GFP-CG5335 or pUAS-FLAG-CG5335. Seventy-two hours after transfection, cells were spun down at 1500 g for 5 minutes and lysed with mRIPA buffer containing 1% Nonidet P-40, 0.5% Triton, 50 mM Tris-HCl (pH 7 s.5), 150 mM NaCl, 1 mM EDTA, and protease inhibitor cocktail (Complete Tablets; Roche). The lysate was centrifuged for 30 minutes at 4 °C and the supernatant was kept for subsequent experiments. For immunoprecipitation, the supernatant was incubated with antibody-conjugated beads (Anti-HA affinity Matrix, Roche) or mouse anti-GFP antibody (Roche) that had been preincubated overnight with protein G beads or anti-FLAG M2-conjugated beads at 4 °C. The following day, beads were washed five times with mRIPA buffer and then resuspended in 25 µl of mRIPA buffer with 5 µl of 5X SDS loading buffer (250 mM Tris pH 6.8, 25% glycerol, 10% SDS, 1% b-mercaptoethanol, 0.1% bromophenol blue), and then protein complexes were eluted from the beads by boiling for 10 minutes or by acidification in 40 µl 0.2 M glycine pH 2, before being transferred to 10 µl 1 M Tris pH 7.4.
For Western analysis, 10 µl of each sample was loaded onto 8% or 10% SDS-PAGE gels, transferred to nitrocellulose membranes (BioRad), and probed with the appropriate antibodies: 1:5000 rabbit anti-Draper; 1:5000 rat anti-HA; 1:10000 rabbit anti-GFP; 1:5000 mouse anti-FlAG-M2; 1:20000 mouse anti-tubulin. HRP-conjugated secondary antibodies were used at a dilution of 1:5000. Antibodies were diluted in TBS/0.1% Tween-20/5% BSA. We incubated blots overnight with rocking at 4 °C, washed them three times for 15 minutes, probed them with appropriate HRP-conjugated secondary antibody for 2 hours at room temperature, washed them a further three times for 20 minutes, and finally developed them using chemiluminescence (PerkinElmer, Western Lightning Plus, Chemiluminescent Substrate, NEL103E001EA) with a Protec OPTIMAX film processor. Protein blots were stripped by rocking in stripping buffer (SDS 2%, Tris HCl pH 6.8, 0.8 mL 0.0625 M ß-mercaptoethanol) at 50 oC for 30 minutes, washed in 1X TBST, reblocked in 1X TBST with 5% BSA, and then incubated with primary and secondary antibodies.
To analyze glycosylation of Drpr by MS, Drpr protein from S2 cell lysate was separated by SDS-PAGE and detected by Coomassie blue staining.
The protein de-glycosylation assay was conducted using PNGaseF (NEB) according to the manufacturer’s guidelines. Each Western blot experiment was conducted at least in triplicate.
N-glycopeptide analysis by LC-MS/MS
The ~120-kDa band of Drpr from S2 cell lysate was excised from the gel and cut into small species, followed by distaining by 50% acetonitrile (ACN) in 25 mM ammonium bicarbonate buffer (ABC buffer) at 37 °C for 15 min for two times, and dehydration by 100% ACN. Proteins were reduced with 10 mM dithiothreitol at 37 °C for 1 h and alkylated with 50 mM iodoacetamide in 25 mM ABC buffer for 1 h in the dark at room temperature. Proteins were treated by sequencing grade trypsin (Promega) at an enzyme-to-substrate ratio of 1:50 at 37 °C overnight. The in-gel digested (glyco)peptides were then extracted from gel pieces by sonicating in 1% formic acid (FA), 50% ACN/1% FA, and 50% ACN for 10 min each time.
The digested peptides were cleaned up using ZipTip C18 (Merck Millipore), dried down and redissolved in 0.1% formic acid (Solvent A). Data were acquired on Orbitrap Fusion Lumos Tribrid mass spectrometer (Thermo Fisher Scientific, San Jose, CA) fitted with an Easy-nLC 1200 system (Thermo Fisher Scientific). For each LC–MS/MS analysis, an equivalent of 1 μg glycoprotein digest was loaded onto an Acclaim PepMap RSLC C18 column (Thermo Fisher Scientific, Lithuania) and separated at a flow rate of 300 nL/min using a gradient of 5% to 40% solvent B (80% acetonitrile with 0.1% formic acid) in 90 min. The mass spectrometer was operated in the Top speed (3 s) data-dependent acquisition mode. Briefly, survey full scan MS spectra were acquired in the Orbitrap from 400 to 1800 m/z at a mass resolution of 120,000. The highest charge state ions within charge state 2–10 were sequentially isolated for MS2 analysis using the following settings: HCD MS2 with AGC target at 5 × 104, isolation window 2 Th, orbitrap resolution 30,000, collision energy 28%. We did two different analysis for Draper and one analysis for DraprNQ4. The raw data was uploaded to MASSIVE (accession no. MSV000094180, password: draperms).
Glycopeptide identification and quantification
Raw files were searched by the Byonic (v1.4.0, Protein Metrics Inc., Cupertino, CA) using the following parameters: search against the Drpr protein sequence with specific cleavages at R and K residues, allowing up to two missed cleavages, with the precursor ion mass tolerance set at 10 ppm and the fragment ion mass tolerance at 20 ppm. Cysteine carbamidomethylation (+57.0215 Da, at C) was set for fixed modification, whereas oxidation (+15.9949 Da, at M) was set for variable common modification. The built-in N-glycan libraries of “182 human no multiple fucose” was used to identify N-glycopeptides. The unfiltered Byonic search results were then fed into the Byologic module of the Byos suite (v5.2.31, Protein Metrics Inc., Cupertino, CA) for quantification based on integration of the peak areas of extracted ion chromatograms (XIC AUC) and apex intensity. Both the Byonic identification results and the Byologic quantification results were exported as.csv files and manually combined by aligning the scan numbers. The positive spectrum matches (PSM) were then filtered by score>200 and PEP2D < 0.001, grouped by the N-glycosylation sites, and sorted by XIC AUC values in the excel sheets. The extracted ion chromatograms of the most abundant glycopeptides and those carrying HexNAc≥3 from the same peptide backbone of the same charge state identified for the four selected sites were then manually plotted at 10 ppm mass tolerance for visual representation.
PS binding assay
S2 cells (10 × 106) were seeded onto a 10-cm plate and transfected with the appropriate plasmids. After 72 hours incubation, the cells were lysed and subjected to immunoprecipitation using anti-FLAG M2 magnetic beads. All subsequent steps were done at 4 oC. For HPLC-ESI-Orbitrap-MS with Deuterium-labeled lipid analysis, the anti-FLAG M2 magnetic beads were washed three times with mRIPA buffer and five times with PBS. After washing, the anti-FLAG M2 magnetic beads were incubated for 15 minutes with 10 µM Deuterium-labeled lipid, 1 mM CaCl2 and PBS, before washing with cold 5X PBS. Then, the supernatant was added to the internal clarithromycin standard and extracted using 300 µL MeOH/CHCl3 (1/2) and sonication for 15 minutes. The supernatant was collected, added to 120 µl 0.9% KCl, dried by speedvac, and then subjected to HPLC-ESI-OrBitrap MS.
For the in vitro protein-lipid binding assay using membrane strips14, 10 × 106 S2 cells were seeded onto a 10-cm plate and transfected with the appropriate plasmids. After 72 hours incubation, the cells were lysed and subjected to immunoprecipitation with anti-FLAG M2 magnetic beads. FLAG-tagged proteins were eluted in glycine pH 2.4. After elution, 5–20 µg of the proteins eluted from the beads were incubated with membrane strips,which have been incubated with 1% non-fat milk for 1 hour at room temperature in dark, 3 hours at 4 degree according to the protocol described in Wang et al.14. After incubation, the membrane strips were washed 3 times 10 minutes each with TBST. Membrane strips were incubated with Anti-FLAG M2 antibody (1:1000) for 3 hours at RT. Followed 3 times 10 minutes wash, the membrane strips were incubated with secondary antibody Goat Anti-mouse HRP 1:10000 for 1 hour at RT. After washing 3 times, the membrane striped were detected by using chemiluminescence (PerkinElmer, Western Lightning Plus, Chemiluminescent Substrate, NEL103E001EA).
LacNAc treatment
S2 cells were seeded into 6-well plates (2 × 106 cells/well) and then transfected with the plasmids. Cells were lysed first, centrifuged for 30 minutes, before keeping the supernatant. Different concentrations of 3’-O’-sialic acid-LacNAc were added into the supernatant with antibody-conjugated beads (Anti-HA affinity Matrix, Roche) and incubated overnight at 4 oC. Beads were washed five times and then proteins were eluted by boiling for 10 minutes, with the resulting samples being used for Western blotting.
Dendrotomy assay
Our protocol for dendrotomy was as described by Li et al.76. For dendrite injury, larvae at 72 hours after egg-laying (AEL) were lightly anesthetized with isoflurane and stuck onto coverslips using double-sided tape. Laser ablation was performed on primary dendrites of C4da neurons in the A5 – A6 segments (and the A3-A4 segments as a control) under a Zeiss LSM980 confocal microscope using a 790-nm two-photon laser. The larvae were maintained on normal food for recovery and then imaged using a Zeiss LSM880 or 900 or 980 confocal microscopes at 1-5 hours post injury to determine galectin signal at dendrites or at 5.5 to 6.5 hours post injury to determine dendritic degeneration. For the CDC50 KO mutant experiments, the larvae were imaged at 3.5 hours post injury.
Statistics and reproducibility
Graphpad Prism was used to perform all statistical analyzes. All data are expressed as mean ± SEM. Statistical significance was set at p < 0.05. One-way ANOVA followed by Tukey’s post-hoc test was used to analyze data comprising three or more genotypes. A Chi-square test was used to analyze data for variance within a group. A Student’s t test, two tailed was used when appropriate. *p < 0.05, **p < 0.01, ***p < 0.001. Each Western blot experiment was conducted at least in triplicate reproducible. The immunostaining or live sample was performed at least in 3 different larvae. No statistical method was used to predetermine sample size. No data were excluded from the analyzes. The experiments were randomized for picking larvae. We were not blinded to allocation during experiments and outcome assessment.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The experimental data that support the findings of this study are included within this paper and its Source Data files. The N-glycopeptide analysis raw data of Drpr was uploaded to MASSIVE under accession no. MSV000094180. Source data are provided with this paper.
References
Luo, L. & O’Leary, D. D. Axon retraction and degeneration in development and disease. Annu Rev. Neurosci. 28, 127–156 (2005).
Schuldiner, O. & Yaron, A. Mechanisms of developmental neurite pruning. Cell Mol. Life Sci. 72, 101–119 (2015).
Soyon Hong, V. F. B.-G. et al. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 352, 712–716 (2016).
Wishart, T. M. Synaptic Vulnerability in Neurodegenerative Disease. J. Neuropathol. Exp. Neurol. 65, 733–739 (2006).
Zhou, Z., Hartwieg, E. & Horvitz, H. R. CED-1 is a transmembrane receptor that mediates cell corpse engulfment in C-elegans. Cell 104, 43–56 (2001).
Chung, W. S. et al. Astrocytes mediate synapse elimination through MEGF10 and MERTK pathways. Nature 504, 394–400 (2013).
Awasaki, T. et al. Essential role of the apoptotic cell engulfment genes draper and ced-6 in programmed axon pruning during Drosophila metamorphosis. Neuron 50, 855–867 (2006).
MacDonald, J. M. et al. The Drosophila cell corpse engulfment receptor Draper mediates glial clearance of severed axons. Neuron 50, 869–881 (2006).
Segawa, K. & Nagata, S. An apoptotic ‘eat me’ signal: phosphatidylserine exposure. Trends Cell Biol. 25, 639–650 (2015).
Arandjelovic, S. & Ravichandran, K. S. Phagocytosis of apoptotic cells in homeostasis. Nat. Immunol. 16, 907–917 (2015).
Hanayama, R., Tanaka, M., Miwa, K., Shinohara, A., Iwamatsu, A. & Nagata, S. Identification of a factor that links apoptotic cells to phagocytes. NATURE 417, 6 (2002).
Uehara, H. & Shacter, E. Auto-oxidation and oligomerization of protein S on the apoptotic cell surface is required for Mer tyrosine kinase-mediated phagocytosis of apoptotic cells. J. Immunol. 180, 2522–2530 (2008).
Lew, E. D. et al. Differential TAM receptor-ligand-phospholipid interactions delimit differential TAM bioactivities. Elife 3, e03385 (2014).
Wang, X. et al. Caenorhabditis elegans transthyretin-like protein TTR-52 mediates recognition of apoptotic cells by the CED-1 phagocyte receptor. Nat. Cell Biol. 12, 655–664 (2010).
Tung, T. T. et al. Phosphatidylserine recognition and induction of apoptotic cell clearance by Drosophila engulfment receptor Draper. J. Biochem. 153, 483–491 (2013).
Okada, R. et al. Apoptosis-dependent externalization and involvement in apoptotic cell clearance of DmCaBP1, an endoplasmic reticulum protein of Drosophila. J. Biol. Chem. 287, 3138–3146 (2012).
Kuraishi, T. et al. Pretaporter, a Drosophila protein serving as a ligand for Draper in the phagocytosis of apoptotic cells. EMBO J. 28, 3868–3878 (2009).
Williams, D. W. et al. Local caspase activity directs engulfment of dendrites during pruning. Nat. Neurosci. 9, 1234–1236 (2006).
Ji, H. et al. The Drosophila chemokine-like Orion bridges phosphatidylserine and Draper in phagocytosis of neurons. Proc. Natl Acad. Sci. USA 120, e2303392120 (2023).
Boulanger, A. et al. Axonal chemokine-like Orion induces astrocyte infiltration and engulfment during mushroom body neuronal remodeling. Nat. Commun. 12, 1849 (2021).
Scott, H. & Panin, V. M. N-glycosylation in regulation of the nervous system. Adv. Neurobiol. 9, 367–394 (2014).
Freeze, H. H. et al. Neurological aspects of human glycosylation disorders. Annu. Rev. Neurosci. 38, 105–125 (2015).
Pamela Stanley, H. S. & Taniguchi. N. N-Glycans. Essentials of Glycobiology. 2009: Cold Spring Harbor (NY): Cold Spring Harbor Laboratory Press.
Sharon, N. & Lis, H. History of lectins: from hemagglutinins to biological recognition molecules. Glycobiology 14, 53R–62R (2004).
Liu, F. T. & R. D. Cummings, Galectins. Essentials of glycobiology. 2009: Cold Spring Harbor (NY): Cold Spring Harbor Laboratory Press.
Nabi, I. R., Shankar, J. & Dennis, J. W. The galectin lattice at a glance. J. Cell Sci. 128, 2213–2219 (2015).
Liu, F. T. & Stowell, S. R. The role of galectins in immunity and infection. Nat. Rev. Immunol. 23, 479–494 (2023).
Xue, S. et al. Elevated galectin-3 is associated with aging, multiple sclerosis, and oxidized phosphatidylcholine-induced neurodegeneration. J. Neurosci. 43, 4725–4737 (2023).
Siew, J. J. et al. Galectin-3 aggravates microglial activation and tau transmission in tauopathy. J. Clin. Invest 134, e165523 (2024).
Han, C. et al. Epidermal cells are the primary phagocytes in the fragmentation and clearance of degenerating dendrites in Drosophila. Neuron 81, 544–560 (2014).
Patnaik, S. K. et al. Complex N-glycans are the major ligands for galectin-1, −3, and −8 on Chinese hamster ovary cells. Glycobiology 16, 305–317 (2006).
Pace, K. E. et al. Characterization of a novel Drosophila melanogaster galectin. Expression in developing immune, neural, and muscle tissues. J. Biol. Chem. 277, 13091–13098 (2002).
Kuo, C. T., Jan, L. Y. & Jan, Y. N. Dendrite-specific remodeling of Drosophila sensory neurons requires matrix metalloproteases, ubiquitin-proteasome, and ecdysone signaling. Proc. Natl Acad. Sci. USA 102, 15230–15235 (2005).
Zhang, Y. et al. The glucosyltransferase Xiantuan of the endoplasmic reticulum specifically affects E-Cadherin expression and is required for gastrulation movements in Drosophila. Dev. Biol. 390, 208–220 (2014).
Aebi, M. N-linked protein glycosylation in the ER. Biochim Biophys. Acta 1833, 2430–2437 (2013).
Brunak, S. & Gupta, R. Prediction of glycosylation across the human proteome and the correlation to protein function. Pac. Symp. Biocomputing 7, 310–322 (2002).
Aoki, K. et al. Dynamic developmental elaboration of N-linked glycan complexity in the Drosophila melanogaster embryo. J. Biol. Chem. 282, 9127–9142 (2007).
Dominica, C., Lebois, E. & Selva, E. The Role of N‐glycosylation in Drosophila Development: Characterization of alg10. The FASEB Journal. 22, https://doi.org/10.1096/fasebj.22.1_supplement.1026.2, (2008).
Cooper, D. N. Galectinomics: finding themes in complexity. Biochim Biophys. Acta 1572, 209–231 (2002).
Zirin, J. et al. State-of-the-art CRISPR for in vivo and cell-based studies in Drosophila. Trends in Genetics, 38, 437–453 (2022).
Tao, J. & Rolls, M. M. Dendrites have a rapid program of injury-induced degeneration that is molecularly distinct from developmental pruning. J. Neurosci. 31, 5398–5405 (2011).
Sapar, M. L. et al. Phosphatidylserine externalization results from and causes neurite degeneration in drosophila. Cell Rep. 24, 2273–2286 (2018).
Makhijani, K. et al. Regulation of Drosophila hematopoietic sites by Activin-beta from active sensory neurons. Nat. Commun. 8, 15990 (2017).
Parrish, J. Z. et al. The microRNA bantam functions in epidermal cells to regulate scaling growth of dendrite arbors in drosophila sensory neurons. Neuron 63, 788–802 (2009).
Poe, A. R. et al. Dendritic space-filling requires a neuronal type-specific extracellular permissive signal in Drosophila. Proc. Natl Acad. Sci. USA 114, E8062–E8071 (2017).
Soba, P. et al. The Ret receptor regulates sensory neuron dendrite growth and integrin mediated adhesion. Elife 4, e05491 (2015).
Yang, W. K. et al. Epidermis-Derived L1CAM Homolog Neuroglian Mediates Dendrite Enclosure and Blocks Heteroneuronal Dendrite Bundling. Curr. Biol. 29, 1445–1459.e3 (2019).
Tenenbaum, C. M. et al. Enclosure of dendrites by epidermal cells restricts branching and permits coordinated development of spatially overlapping sensory neurons. Cell Rep. 20, 3043–3056 (2017).
Li, H. et al. Fringe-positive Golgi outposts unite temporal Furin 2 convertase activity and spatial Delta signal to promote dendritic branch retraction. Cell Rep. 40, 111372 (2022).
Burguillos, M. A. et al. Microglia-secreted galectin-3 acts as a toll-like receptor 4 ligand and contributes to microglial activation. Cell Rep. 10, 1626–1638 (2015).
Boza-Serrano, A. et al. Galectin-3, a novel endogenous TREM2 ligand, detrimentally regulates inflammatory response in Alzheimer’s disease. Acta Neuropathol. 138, 251–273 (2019).
Caberoy, N. B. et al. Galectin-3 is a new MerTK-specific eat-me signal. J. Cell Physiol. 227, 401–407 (2012).
Lukyanov, P., Furtak, V. & Ochieng, J. Galectin-3 interacts with membrane lipids and penetrates the lipid bilayer. Biochem Biophys. Res. Commun. 338, 1031–1036 (2005).
Williamson, A. P. & Vale, R. D. Spatial control of Draper receptor signaling initiates apoptotic cell engulfment. J. Cell Biol. 217, 3977–3992 (2018).
Shklyar, B. et al. Caspase activity is required for engulfment of apoptotic cells. Mol. Cell Biol. 33, 3191–3201 (2013).
Köhler, G., Hering, U., Zschörnig, O. & Arnold, K. Annexin V interaction with phosphatidylserine-containing vesicles at low and neutral pH. Biochemistry 36, 8189–8194 (1997).
Shi, J. et al. Lactadherin detects early phosphatidylserine exposure on immortalized leukemia cells undergoing programmed cell death. Cytom. A 69, 1193–1201 (2006).
Caberoy, N. B. et al. Efficient identification of phosphatidylserine-binding proteins by ORF phage display. Biochem Biophys. Res. Commun. 386, 197–201 (2009).
Yip, P. K. et al. Galectin-3 released in response to traumatic brain injury acts as an alarmin orchestrating brain immune response and promoting neurodegeneration. Sci. Rep. 7, 41689 (2017).
McMahon, A. C. et al. TRIBE: Hijacking an RNA-editing enzyme to identify cell-specific targets of RNA-binding proteins. Cell 165, 742–753 (2016).
Williams, D. W. & Truman, J. W. Cellular mechanisms of dendrite pruning in Drosophila: insights from in vivo time-lapse of remodeling dendritic arborizing sensory neurons. Development 132, 3631–3642 (2005).
McLaughlin, C. N. et al. Dying neurons utilize innate immune signaling to prime glia for phagocytosis during development. Dev. Cell 48, 506–522.e6 (2019).
Musashe, D. T. et al. Insulin-like signaling promotes glial phagocytic clearance of degenerating axons through regulation of draper. Cell Rep. 16, 1838–1850 (2016).
Marc, R., Freeman, J. D., Junhyong, K., Eric, J. & Chris Q. D. Unwrapping Glial Biology: Gcm Target Genes Regulating Glial Development, Diversification, and Function. Neuron 38, 567–580 (2003).
Shklover, J. et al. JNK pathway activation is able to synchronize neuronal death and glial phagocytosis in Drosophila. Cell Death Dis. 6, e1649 (2015).
Wucherpfennig, T., Wilsch-Bräuninger, M. & González-Gaitán, M. Role of Drosophila Rab5 during endosomal trafficking at the synapse and evoked neurotransmitter release. J. Cell Biol., 161, 609–624 (2003).
Galko, M. J. & Krasnow, M. A. Cellular and genetic analysis of wound healing in Drosophila larvae. PLoS Biol. 2, E239 (2004).
Logan, M. A. et al. Negative regulation of glial engulfment activity by Draper terminates glial responses to axon injury. Nat. Neurosci. 15, 722–730 (2012).
Venken, K. J. et al. Versatile P[acman] BAC libraries for transgenesis studies in Drosophila melanogaster. Nat. Methods 6, 431–434 (2009).
Schindelin, J. et al. Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682 (2012).
Tsai, Y. W. et al. Glia-derived exosomal miR-274 targets Sprouty in trachea and synaptic boutons to modulate growth and responses to hypoxia. Proc. Natl Acad. Sci. USA 116, 24651–24661 (2019).
Bouveret, R. et al. NKX2-5 mutations causative for congenital heart disease retain functionality and are directed to hundreds of targets. Elife. 4, e06942 (2015).
Kanaoka, Y. et al. DeTerm: Software for automatic detection of neuronal dendritic branch terminals via an artificial neural network. Genes Cells 24, 464–472 (2019).
Schneider, C. A., Rasband, W. S. & Eliceiri, K. W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675 (2012).
Tarentino, A. L., Gómez, C. M. & Plummer, T. H. Jr. Deglycosylation of asparagine-linked glycans by peptide:N-glycosidase F. Biochemistry 24, 4665–4671 (1985).
Li, D. et al. A drosophila in vivo injury model for studying neuroregeneration in the peripheral and central nervous system. J. Vis. Exp. 5, 57557 (2018).
Acknowledgements
We thank M. Freeman, E. Kurant, F.-W. Yu, M. A. Logan, H. Chun, and K. Emoto for sharing reagents and stocks; TRiP (Harvard Medical School), Wellgenetics, Bloomington Stock Center, VDRC, and Kyoto Stock Center for fly stocks; and the Developmental Studies Hybridoma Bank for antibodies. We gratefully acknowledge the Imaging Core of IMB and Imaging Core of NPAS, all at Academia Sinica, for technical assistance, and the Academia Sinica Common Mass Spectrometry Facilities for Proteomics and Protein Modification Analysis (funded by AS-CFII-108-107) for glycopeptide LC-MS/MS data acquisition. This work was supported by grants from NSCT (MOST 112-2811-B-001-063 and MOST105-2811-B-001-065) and Academia Sinica (AS-KPQ-109-BioMed) to C.T.C.
Author information
Authors and Affiliations
Contributions
H.-H.S., H.Li., and C.Y.-Y. contributed dendrite injury experiments. H.-H.S. and Y.-C.H. contributed for glyco-related mutant screen. H.-H.S., H.L., C.-L.A., and Y.-J.C. contributed data qualification. M.-Y.H., H.-M.J., C.-H.L. contributed deuterium lipid HPLC-MS. Y.-J.P., H.-Y.L., and C.-H.L. contributed synthesis LacNAc derivates. C.-H.Y., S.-Y.L., K.H.K. contributed N-glycan analysis. H.-H.S. and C.-T.C. contributed experimental design and wrote the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Ronghu Wu, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Sung, HH., Li, H., Huang, YC. et al. Galectins induced from hemocytes bridge phosphatidylserine and N-glycosylated Drpr/CED-1 receptor during dendrite pruning. Nat Commun 15, 7402 (2024). https://doi.org/10.1038/s41467-024-51581-6
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-024-51581-6
