
Abstract
As an abundant agricultural and forestry biomass resource, hemicelluloses are hard to be effectively degraded and utilized by microorganisms due to the constraints of membrane and metabolic regulations. Herein, we report a synthetic extracellular metabolic pathway with hemicellulose-degrading-enzymes controllably displayed on Escherichia coli surface as engineered bacterial consortia members for efficient utilization of xylan, the most abundant component in hemicellulose. Further, we develop a hemicellulose/O2 microbial fuel cell (MFC) configuring of enzyme-engineered bacterial consortia based bioanode and bacterial-displayed laccase based biocathode. The optimized MFC exhibited an open-circuit voltage of 0.71 V and a maximum power density (Pmax) of 174.33 ± 4.56 µW cm−2. Meanwhile, 46.6% (w/w) α-ketoglutarate was produced in this hemicellulose fed-MFC. Besides, the MFC retained over 95% of the Pmax during 6 days’ operation. Therefore, this work establishes an effective and sustainable one-pot process for catalyzing renewable biomass into high-value products and electricity in an environmentally-friendly way.
Similar content being viewed by others

Introduction
Lignocellulose, which is produced through photosynthesis by plants, is a huge solar energy reservoir. As a green and sustainable alternative to fossil energy, it has attracted broad interest in exploring lignocellulose biomass for high-added-value products and biofuel1,2. The biological hydrolysis and fermentation process of the lignocellulosic biorefinery are regarded as the most promising and eco-friendly approach compared to physical and chemical methods3,4. As the most abundant component in lignocellulose, cellulose can be easily metabolized by most microorganisms owing to only glucose units5. Whereas, hemicellulose, accounting for 20–40% of lignocellulose, is hard to effectively degrade and utilized by many microorganisms due to the heterogeneous structure of hemicellulose and the constraints of effective membrane transporters as well as the metabolic regulations in vivo6,7. Thus, the low utilization efficiency of hemicellulose greatly affects lignocellulosic biorefinery8.
Currently, hemicelluloses have been used to produce valuable chemicals such as ethanol, furfural, and xylitol9. Unfortunately, these processes still experience challenges of low conversion activity and poor stability of cells. For example, the yield of hemicelluloses-bioethanol is lower than that of bioethanol fermented from cellulose owing to the poor utilization efficiency of polysaccharides and pentose by microorganisms during fermentations10. Besides, bioelectric energy has been heralded as one of the most potential renewable energies in the future11,12. To realize the generation of energy from hemicelluloses biomass, great efforts via recruitment of microbial community (microbial fuel cells, MFCs) have been devoted13. However, diverse biological properties in one system result in the instability in bacterial community, which severely limits its large-scale application14. To address these challenges, a strategy is highly desirable to realize the efficient production of biofuels and biochemicals directly from hemicellulose instead of its monosaccharides.
As an efficient and green method, enzymatic catalysis has been employed to hydrolyze polysaccharides and oxidize monosaccharides. However, the high cost and poor stability greatly limit their large-scale applications15. Meanwhile, microbial fermentation is another main biological approach to produce biofuels and biochemicals. The cell membrane barrier and intracellular metabolic regulation always restrict the highly efficient substrate utilization, especially polysaccharides16. Enzymes can be displayed on the microbial surface and the resulted whole-cell biocatalyst was obtained by large-scale fermentation and directly applied in biocatalytic reactions without tedious protein purification process, which enables the implemention of complex biochemical reactions even metabolic pathways without the constraints of cell membrane and metabolic regulation17,18,19. Similar to secreted proteins, displayed enzymes could be expressed and folded in periplasm and exported directly across the cell membrane of E. coli to the culture medium using Sec secretory system20. The displayed enzymes could be assembled and anchored onto the outer membrane by fusing with an anchoring motif, which could facilitate their ability to be recycled and regenerated. In contrast, secreted proteins will be folded in cytoplasm or periplasm, depending on the sec or signal sequence used, and then secreted and dispersed in the culture media without geographical restrictions of the cell surface, failing to be recycled or regenerated21.
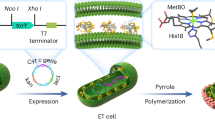
Herein, we report on an artificial hemicellulose-degrading pathway catalyzed by the engineered bacterial consortia whose enzymes from various origins are recruited to achieve the optimal overall reaction rate in a controlled manner, for the high-efficiently converting renewable biomass into electric energy and high-value chemical simultaneously in an environmentally-friendly way. The engineered Escherichia coli (E. coli) (upstream pathway members) degrade xylan, the most abundant biopolymer in hemicellulose22, into monosaccharides, while the following recombinant E. coli (downstream pathway members) oxidize monosaccharides into α-ketoglutarate accompanied by generating the reduced form of nicotinamide adenine dinucleotide (NADH) (Fig. 1). Thus de novo MFC is designed to achieve one-pot efficient production of high-added-value chemical of α-ketoglutarate and powerful electricity from hemicellulose biomass in a “one-stone-two-birds” manner, demonstrating a model to efficiently utilize biomass in a sustainable way.

The final product is α-ketoglutarate. TtGH8 β−1,4 xylanase, SXA β-D-xylosidase, XDH D-xylose dehydrogenase, XylC xylonolactonase, XylD xylonate dehydratase, XylX 2-keto-3-deoxy-D-xylonate dehydratase, KGSADH 2,5-dioxopentanoate dehydrogenase.
Results and discussion
Bacterial surface displaying enzymes (engineered bacterial consortia) involved in the saccharification of xylan (upstream pathway)
Herein, the utilization of xylan experienced two stages, firstly saccharified into monosaccharides (mainly D-xylose), which are then oxidized to produce α-ketoglutarate and release electrons. For the initial phase, hemicellulases hydrolyze β-linkage in the xylan backbone to release monosaccharides. A marine symbiont Teredinibacter turnerae was selected as the bacterial endo-β−1,4 xylanases (TtGH8) source. TtGH8 shows a wide variety of glycoside hydrolases activities, including β-1,4 xylanases, especially the highest activity on mixed-linkage β-1,3 and β-1,4 xylanases23, which could expand the panel of substrate used in our system. β-D-xylosidase (SXA) from Selenominas ruminantium exhibits good thermo-stability as well as superior activity towards 1,4-β-D-xylooligosaccharides24,25. Besides, SXA also has α-arabinofuranosidase activity towards arabino xylanases26. The synergy of these two hemicellulases could break down xylan into monosaccharides. Unfortunately, E. coli BL21 (DE3) can metabolize xylose under the action of xylose isomerase (XylA) and xylulose kinase (XylB)27. Therefore, in order to drive all of the generated xylose by hemicellulose into the designed extracellular metabolic pathway and avoid monosaccharides waste, xylA, and xylB genes in E. coli BL21 (DE3) were knocked out.
During the first stage, when applying two types of hydrolase-displaying on the surface of bacterial cell, polysaccharide generated from lignocellulose pretreatment could be immediately degraded into monosaccharide by these displayed enzymes in a highly efficient way without passing through cell membrane28. TtGH8 and SXA were displayed on cell surface separately or simultaneously as fusion protein using N-terminal region of ice nuclear protein from P. borealis (InaPb) as anchoring motif 29. The connection manner of cell surface-displayed TtGH8-SXA was optimized by introducing linkers Gly-Ser, Gly-Ser-Gly-Gly-Ser-Gly, and (Ala-Pro)7 between TtGH8 and SXA, respectively. Results show that the engineered strains harboring GS and GSGGSG linkers had similar activities. However, (AP)7 linker influenced the functions of cell surface-displayed TtGH8-SXA, and the whole-cell activity reduced 50% compared to that of strain displayed TtGH8-SXA with GS linker. Therefore, Gly-Ser was employed as the linker between TtGH8 and SXA. Surface-displaying enzymes demonstrated obvious expression levels, which were confirmed by Western Blotting (Supplementary Fig. 1) and confocal imaging (Supplementary Fig. 2). Next, biochemical activity assays were conducted to validate biological functions of surface-displayed enzymes by using commercial xylan as substrate using 3,5-dinitrosalicylic acid (DNS) method. The optimized protein expression conditions for displayed enzymes were 0.5 mM isopropyl-β-D-thiogalactopyranoside (IPTG) and 30 °C (Supplementary Fig. 3). However when IPTG concentration was over 1.0 mM, the whole-cell activities of engineered strains were declined due to the imbalance of transcription and secretion30. According to the results of the Quantitative Immunoassay (Supplementary Fig. 4), the number of cell surface-displayed proteins was estimated. Strains displayed different amounts of these enzymes, ~25560, 25300, and 18020 enzyme molecules for TtGH8, SXA and TtGH8-SXA, respectively. The lower number of TtGH8-SXA displayed on the surface per cell suggests the decreased display efficiency for protein with a larger molecule (108 kDa). On the basis of the number of enzymes displayed on cell surface, the effects of single display system and dual-display system on saccharification of hemicellulose were investigated. As can be seen from Supplementary Fig. 5, the strain displaying fusion protein TtGH8-SXA exhibited a 30.37% lower activity towards xylan than the mixture of two strains displaying TtGH8 and SXA individually with molar ratio of 1:1 at the same levels of displayed enzyme molecules, probably due to the lower protein numbers of TtGH8-SXA on cell surface compared to the sum of TtGH8 and SXA single-displayed numbers. Then, the ratio of cell density of two engineered strains was optimized to efficiently hydrolyze xylan into monosaccharides. As indicated in Fig. 2a, different ratios of strain E. coli-TtGH8 to E. coli-SXA resulted in different hydrolysis efficiency with the best ratio of 3:7. TtGH8 and SXA possess different functions during the degradation process of xylan. TtGH8 is responsible for degrading xylan into xylooligosaccharide as well as a small amount of D-xylose and L-arabinose23. The xylooligosaccharide could be further hydrolyzed into D-xylose and L-arabinose under the action of SXA. So, the amounts of required SXA were higher than those of TtGH8 for full hydrolyzation of xylan into monosaccharides. The hydrolysis efficiency of upstream bacterial consortia including E. coli-TtGH8 and E. coli-SXA towards xylan at different concentrations was examined. To quantify the proportion and amounts of pentose in the hydrolysate after strains’ treatment, the resultant D-xylose and L-arabinose were determined by HPLC method, accounting for 97.43% and 2.57%, respectively. Although the efficiency of saccharification was increased with the decreasing xylan concentration from 1 g/L to 0.1 g/L, the yields of pentose monomers also reduced accordingly (Supplementary Fig. 6). When corncob xylan was 1 g/L, the hydrolysis efficiency was 65.43% (w/w) after 6 h reaction (Supplementary Fig. 6), which was about 1.42-fold higher than in vitro process31 and 1.65-fold higher than in vivo process reported previously32. When the concentration of xylan continued to increase, the production of pentose failed to rise proportionally. Anyway, this simple bacterial consortium could generate soluble monosaccharides from xylan ready for the subsequent oxidative degradation.

a Relative production of reducing sugars using corncob xylan (1 g/L) as substrate catalyzed by engineered bacterial consortia involving in the saccharification of xylan (upstream pathway) with different cell density ratios of E. coli-TtGH8 to E. coli-SXA. 1 mL of 50 mM Tris–HCl buffer (pH 7.0) with 10 mM CaCl2 and 1.0 g/L commercial corncob xylan were incubated with artificial bacterial consortia (OD600 = 10) at 37 °C (n = 3 biologically independent experiments). The bacterial consortia with a ratio of 3:7 produced the highest level of reducing sugars compared to those with other ratios. b Relative production of NADH catalyzed by different bacterial consortia using D-xylonic acid as substrate. When different fusion proteins of XylD-KGSADH, XylD-XylX, and XylD-XylX-KGSADH were separately displayed on the cell surface, the resultant strains were named E. coli-XylDK, E. coli-XylDX, and E. coli-XylDXK, respectively. The total OD600 of these four systems were the same, and the cell density ratios of different strains were the same in one system. 1 mL of 50 mM Tris–HCl buffer (pH 7.0) with 10 mM MgCl2, 20 mM D-xylonic acid and 1 mM NAD+ were incubated with artificial bacterial consortia (OD600 = 10) at 37 °C (n = 3 biologically independent experiments). Strain E. coli-XylDXK as the control. The bacterial consortia system composed of E. coli-XylDK and E. coli-XylX produced the highest level of NADH compared to those with another system. c Relative production of α-ketoglutarate catalyzed by different bacterial consortia involving in oxidation of pentose monosaccharides (downstream pathway) with various cell density ratios among E. coli-XDH, E. coli-XylC, E. coli-XylDK and E. coli-XylX using D-xylose as substrate. 1 mL of 50 mM Tris–HCl buffer (pH 7.0) with 10 mM MgCl2, 10 mM D-xylose, and 1 mM NAD+ were incubated with artificial bacterial consortia (OD600 = 10) at 37 °C (n = 3 biologically independent experiments). The bacterial consortia with the ratio of 1:5:20:25 produced the highest level of α-ketoglutarate compared to those with other ratios. d Relative production of α-ketoglutarate catalyzed by different bacterial consortia with various cell density ratios of upstream pathway to downstream using corncob xylan (1 g/L) as substrate at 37 °C for 6 h. The upstream pathway included E. coli-TtGH8 and E. coli-SXA with the optimal ratio of 3:7. The downstream pathway included E. coli-XDH, E. coli-XylC, E. coli-XylDK, and E. coli-XylX with the optimal ratio of 1:5:20:25. The total OD6000 = 10 of the bacterial consortia was applied. n = 3 biologically independent experiments. The bacterial consortia with a ratio of 3:7 produced the highest level of α-ketoglutarate compared to those with other ratios. Data are presented as mean ± SD. The statistical significance is determined by a two-sided t test, and ***, **, * indicate P < 0.001, 0.01, and 0.05, respectively. Source data are provided as a Source Data file.
Engineered bacterial consortia involving in oxidation of pentose monosaccharides (downstream pathway)
To date, D-xylose metabolism pathway has been found in a few microorganisms, which harbors three catabolic routes, including the Weimberg or Dahms pathway, the xylulose-1-phosphate or ribulose-1-phosphate pathway, and the xylose isomerase or xylose reductase-xylitol dehydrogenase pathway33. E. coli can utilize xylose as a carbon source for growth through the native route mediated by XylA and XylB, as well as pentose phosphate pathway and glycolysis. However, the low efficiency limited the application of this pathway in metabolic engineering34. Weimberg route in Caulobacter crescentus involves conversion of D-xylose to α-ketoglutarate by five steps successively catalyzed by XDH, XylC, XylD, XylX, and KGSADH35 (Fig. 1). This pathway has been proved to be an attractive route for biosynthesis of various chemicals from xylose34,36,37. In our current study, the oxidation of pentose and the transferring of electrons could be realized by employing this efficient pathway, during which 1 molecule of α-ketoglutarate and 4 electrons per pentose unit can be generated.
To identify the biological activity of each enzyme involved in Weimberg pathway, the in vitro activities of the purified enzymes were measured. Protein expression conditions were optimized (Supplementary Fig. 7), and enzymes were purified to homogeneity through Ni2+ column affinity chromatography before SDS-PAGE analysis (Supplementary Fig. 8). In this first step of Weimberg pathway, D-xylose is oxidized into D-xylono-lactone and generates NADH catalyzed by XDH using NAD+ as coenzyme. So, the activity of XDH was monitored by measuring the absorbance at 340 nm, typical absorption peak of NADH. As listed in Supplementary Table 3, the enzymatic activity of XDH was 1195.00 ± 8.60 U/mg. To examine the activity of XylC, D-xylono-lactone was used as substrate to produce D-xylonic acid. The enzymatic activity of XylC was 146.93 ± 2.16 U/mg, which was 7-fold lower than that of XDH (Supplementary Table 3). Subsequently, D-xylonic acid would be transformed into 2-keto-3-deoxyxylic acid by XylD with an activity of 77.00 ± 3.00 U/mg. The activity of KGSADH is 54.00 ± 6.00 U/mg, which was also monitored by measuring the generation of NADH using analogous glutaraldehyde as substrate. Finally, the enzymatic activity of XylX was determined only 35.65 ± 0.25 U/mg (Supplementary Table 3) by a coupled assay with XylD and KGSADH. Thus XylX possessed the lowest activity among these five enzymes involved in Weimberg pathway. Therefore, all of the enzymes had biological functions, but the conversion of 2-keto-3-deoxyxylic acid to 2,5-dioxopentanoate catalyzed by XylX may be a limiting step in the overall route, as proposed in other literatures37.
On the basis of the in vitro analysis of these five members in Weimberg pathway, cell surface display systems were constructed. To confirm the localization of enzymes on the surface of E. coli using InaPb anchoring motifs, Western-Blot analysis of outer membrane fraction and immunofluorescent labeling of cells was conducted. All the distinct bands of expected sizes from outer membrane fractions were found (Supplementary Fig. 9), confirming that the introduced genes in different recombinant plasmids were expressed obviously. Compared with the control strain, green fluorescence was visualized for strain samples by confocal microscope (Supplementary Fig. 10). To ascertain the displayed enzymes possessed functions on the surface of bacteria, the whole-cell was regarded as the catalyst in the enzymatic activity assay. After optimizing the protein expression conditions for displayed enzymes (Supplementary Fig. 11), the whole-cell activities were determined. As depicted in Tables S3 and S4, XDH-displaying E. coli exhibits the highest activity of 1.25 ± 0.04 U/OD600 with kcat value of 11.34 ± 1.21 s−1. The whole-cell activity of E. coli-XylC was significantly lower than that of E. coli-XDH. So, the co-display of XDH and XylC would lead to the imbalance of cascade reaction. Therefore, the maximum cascade reaction rate could be achieved by separately displaying XDH and XylC on the cell surface and regulating the ratio of strain E. coli-XDH and E. coli-XylC. On the contrary, XylX-displaying E. coli shows the lowest activity (0.044 ± 0.003 U/OD600) with kcat value of 0.07 ± 0.006 s−1, which corresponds to the in vitro data. These results reveal that XylX was the rate-limiting enzyme in the whole pathway, so the highest overall reaction rate could be realized by adjusting the level of E. coli-XylX in engineered bacterial consortia.
Considering the imbalance of activities between E. coli displaying enzymes, the dual-display and tri-display systems were developed to accelerate the overall reaction rate of the latter half of the pathway. The Western Blotting analysis and confocal imaging micrographs indicate the successful expression and display of fusion enzymes on cell surface (Supplementary Figs. 9 and 10). Unfortunately, the degradation of triple fusion protein XylX-XylD-KGSADH was observed, implying the unstable presentation of much larger protein (157 kDa) on the cell surface. The overall reaction rates from D-xylonic acid to α-ketoglutarate were varied using different display systems as biocatalysts. Among them, the bacterial consortia containing XylD-KGSADH-displaying E. coli (E. coli-XylDK) and XylX-displaying E. coli (E. coli-XylX) exhibited the best catalytic efficiency when using D-xylonic acid as substrate and NADH as indicator (Fig. 2b). Then, the cell density ratios of downstream pathway were optimized to realize the maximum overall reaction rate from D-xylose to α-ketoglutarate. Considering the highest activity of E. coli-XDH in the five whole-cell catalysts, the ratio of E. coli-XDH in the prepared bacterial consortia was controlled at a lower level. The ratios of the other three strains, including E. coli-XylC, E. coli-XylDK, and E. coli-XylX, were varied to realize the highest overall reaction rate from D-xylose to α-ketoglutarate. As shown in Fig. 2c, the ratios of E. coli-XylDK and E. coli-XylX had an obvious synergistic effect on the generation of the final product α-ketoglutarate. Thus, the Weimberg pathway can function in the engineered bacterial consortia.
The integration of the engineered bacterial consortia for producing α-ketoglutarate
To integrate the entire pathway for degrading xylan, according to the above optimized enzyme-displaying strain ratios in the upstream pathway and downstream pathway, these involved enzyme-displaying strains were mixed into bacterial consortia with different ratios of cell densities in the saccharification of xylan into pentose and the oxidation pentose into α-ketoglutarate. The most appropriate ratio was 3:7 for bacterial consortia in upstream route to downstream route with the total OD600 of 10.0 taking the titer of α-ketoglutarate as evaluation criterion (Fig. 2d). These results demonstrate that the imbalanced enzymes activities could be adjusted by optimizing strain ratios.
α-Ketoglutarate is not only an important intermediate metabolite in cells, but also is widely used as antioxidant and nutrient supplements in food and pharmaceutical fields38. Due to the low efficiency and complex process of chemical synthetic route39, α-ketoglutarate is 10 times more expensive than other common organic acids such as citric acid and lactic acid. Moreover, the usage of the toxic chemical reagent cyanide in the chemical reaction process has aroused concerns about environmental pollution. The microbial synthesis method is characterized as cost-effective, highly efficient, and eco-friendly40. Until now, Yarrowia lipolytica and Corynebacterium glutamicum have been engineered as microbial cell factories to produce α-ketoglutarate using various substrates, such as raw glycerol and xylose (Table 1). Compared with these substrates, hemicellulose is considered to be the most promising feedstock due to its global availability and cost-effective benefits. Herein, to the best of our knowledge, the employment of renewable hemicellulose to produce α-ketoglutarate was realized for the first time with the yield of 47% (g/g) within 6 h in vitro one-pot reaction by the engineered bacterial consortia saccharification and oxidation pathway. Our yield reaches the highest level within 6 h, which is significantly shorter than over 90 h for microbial cell factories using cost-ineffective glycerol and xylose as substrates (Table 1). It is worth noting that, as the pivot metabolites among the tricarboxylic acid cycle (TCA cycle), glyoxylate cycle, and amino-acid metabolism, the generation of α-ketoglutarate would be impacted by complex metabolic regulation in vivo. In this study, extracellular pathway circumvented this bottleneck and improved the synthetic efficiency. Thus, the bioproduction of α-ketoglutarate from hemicellulose in this work realized the efficient conversion of agricultural and forestry residues to high-added value compound.
The integration of the engineered bacterial consortia for generating electricity in MFC
Figure 3 describes the mechanism of the two-compartment xylan/O2 MFC separated by a Nafion® membrane. The mediator-less anodic compartment containing multi-walled carbon nanotubes (MWCNTs) covered carbon cloth (CC) electrode, which was modified with engineered bacterial consortia (CC/MWCNTs/bacterial consortia) and fuel of xylan, while the cathodic compartment contained MWCNTs-coated CC electrode, which was modified with E. coli-displayed laccase (Lac) (CC/MWCNTs/E.coli-Lac) and 2,2’-azino-bis (3-ethylbenzothiazoline-6-sulfonic acid) (ABTS) as the redox mediator. Lac was confirmed to display on the cell surface by SDS-PAGE, Western-Blot, and confocal imaging (Supplementary Fig. 12) with good catalytic activity (Supplementary Fig. 13), which catalyzes the O2 reduction at the cathode (Fig. 4a). E.coli-Lac exhibits the maximum activity at pH 5.0 (Supplementary Fig. 13c). The synthetic metabolic pathway on the anode compartment was designed to degrade xylan into α-ketoglutarate, generating four electrons per pentose unit. The reactions can be expressed as:

The system was composed of the enzyme-engineered bacterial consortia-based bioanode and E. coli-Lac-based biocathode.

a CVs of the CC/MWCNTs/E.coli-Lac biocathode in 100 mM Na2HPO4-citric acid buffer (pH 5.0) under N2-saturated atmosphere without ABTS (black line), and in the presence of 0.5 mM ABTS under N2-saturated (red line) and under oxygen-saturated atmosphere (blue line). Scan rate: 10 mV s−1. b CVs of CC in electrolyte solution (black line); CC/bacterial consortia in electrolyte solution containing xylan (1 g/L) and NAD+ (4 mM) (red line); CC/MWCNTs/bacterial consortia in electrolyte solution containing xylan (1 g/L) and NAD+ (4 mM) (blue line); CC/MWCNTs in electrolyte solution containing AQDS (10 mM) (green line). CC/MWCNTs/bacterial consortia in electrolyte solution containing xylan (1 g/L), NAD+ (4 mM) and AQDS (10 mM) (purple line). The electrolyte solution is 100 mM Tris–HCl buffer (pH 7.0) containing 100 mM NaCl, 10 mM MgCl2, and 10 mM CaCl2. Scan rate: 10 mV s−1. c Profiles of potential versus current density (j). MFC consisting of CC/MWCNTs/bacterial consortia bioanode and CC/MWCNTs/E.coli-Lac cathode, which contain 10 mM of AQDS in the anodic chamber (peach pink line). MFC consisting of CC/MWCNTs bioanode and CC/MWCNTs/E.coli-Lac cathode, which contained bacterial consortia and 1 mM of AQDS in the anodic chamber (blue line). MFC consisting of CC/MWCNTs/bacterial consortia and CC/MWCNTs/E.coli-Lac biocathode, which contained 1 mM of AQDS in the anodic chamber (light green line). d Profiles of power density dependent on different bioanodes, which are the same as c. Scan rate: 1 mV s−1. CC carbon cloth. Source data are provided as a Source Data file.
The performance of the biocathode and bioanode was examined by cyclic voltammetry (CV). At the CC/MWCNTs/E.coli-Lac, ABTS exhibited a redox pair in the absence of oxygen (red line), while the cathodic specific current significantly enhanced in the presence of O2 (Fig. 4a, light blue line), indicating that with ABTS as the electron transfer mediator, the bacteria-displayed laccase catalyzes the reduction of oxygen. For bioanode, as shown in Fig. 4b, an onset potential at 0.13 V (vs. Ag/AgCl) for enzyme-displayed E. coli was observed (red line). Interestingly, the onset potential for CC/MWCNTs/bacterial consortia was negatively shifted to 0 V (blue line), suggesting that the generated NADH from NAD+ was catalytically oxidized by MWCNTs41. 9,10-anthraquinone-2,7-disulfonic acid (AQDS) was used as an electron mediator to investigate the impact of electron transfer mechanisms on the performance of bioanode. As shown in Fig. 4b, the two reduction peaks (green curves) at −0.6 V and −0.9 V were the reduction peaks of AQDS with MWCNTs modified CC as electrodes. The oxidation peak of AQDS was around −0.5 V on the purple curve, which was shifted to around −0.4 V due to the influence of CC modified with bacterial consortia. The oxidizing redox peak around −0.1 V on the purple curve was the oxidation peak of NADH produced by the catalysis of bacterial consortia in the presence of AQDS. AQDS can lower the onset potential of NADH which was produced by cell surface-displayed enzymes’ catalysis. The onset potential was negatively shifted to −0.20 V (purple line). Therefore, this mediator was implemented in the electrolyte of the anodic chamber in the following studies.
Then, we studied the performance of MFCs composed of the above-prepared bioanode and biocathode by varying the way of loading engineered strains. The current density ( jmax) of the generated MFC was 371.67 ± 10.36 μA cm−2 when bacterial consortia were immobilized on electrode (Fig. 4c, light green line). However, the jmax of MFC dropped to 297.54 ± 10.24 μA cm−2 when bacterial consortia were dispersed in bioanode chamber (Fig. 4c, blue line). Then the effects of different concentrations of AQDS on the performance of xylan/O2 MFC were investigated. The results show that the highest value of power was achieved when 10 mM of AQDS was used (Fig. 4c, peach pink line). The similar trends were observed in Fig. 4d, and the constructed MFC reached the maximum power output (Pmax) of 68.25 ± 2.38 μW cm−2 when bacterial consortia immobilized on bioanode in the presence of 10 mM of mediator AQDS.
Shewanella oneidensis is one of the most well-known electricigen in nature42. The attempt to engineer S. oneidensis MR-1 using D-xylose as fuel to generate bioelectricity in MFC by introducing D-xylose metabolic route shows restricted power owing to the poor efficiency of this exogenous pathway43. Herein, we reconstituted a D-xylose oxidative pathway and electron transfer chain on the cell surface by displaying Weinberg pathway members. There are two steps that can produce NADH for generating power, which were catalyzed by XDH and KGSADH. Finally, the oxidation of one D-xylose can yield two NADH and four electrons during the whole process, and the final metabolite α-ketoglutarate can be used as the key precursor of several medicine and nutrient substances in food as well as feed44.
The optimization of MFC performance
To boost the power output of MFC, effects of several parameters on electricity generation were investigated. First, the loading amounts of bacterial consortia on the bioanode were optimized. The jmax of MFCs was 671.33 ± 26.21 μA cm−2, corresponding Pmax of 112.87 ± 7.83 μW cm−2 when loading 10 OD600 bacterial consortia (Fig. 5a, peach pink line). The results indicate that the bacterial cell loading exerted effects on power output since the insulated nature of concentrated cells would cause ohmic losses45. Then, MFCs performance fueled by commercial xylan from corncob was tested under the optimal conditions established according to the above experiments. As shown in Fig. 5b, when the concentrations of corncob xylan increased from 0.5 g/L to 1 g/L, the Pmax boosted from 111.82 ± 5.35 μW cm−2 (green line) to 174.33 ± 4.56 μW cm−2 (peach pink line). However, a further increase in concentrations of xylan resulted in 12.22% decrease in power density (blue line), probably due to the poor solubility and high viscosities of commercial xylan prepared in buffer solution. The Pmax of our MFC fueled by 1 g/L xylan is 174.33 ± 4.56 μW cm−2, which is significantly higher than those for xylan-fueled MFCs inoculated with activated sludge (0.609 μW cm−2) or rumen microorganisms (40.5 μW cm−2)13,46.

a Effect of loading amounts of bacterial consortia onto the CC/MWCNTs/bacterial consortia bioanode on the power output. The bioanodes were prepared by dropping 100 μL of the prepared cells with different OD600 values onto the CC/MWCNTs and dried at room temperature. The xylan concentration is 1.0 g/L. The amounts of bacterial consortia were 5 OD (blue line), 10 OD (pink line) and 20 OD (green line), respectively. b Effect of concentrations of commercial corncob xylan on the power output. The bioanodes were prepared by dropping 100 μL of the prepared cells with 10.0 OD600. The concentrations of commercial corncob xylan were 0.5 g/L (blue line), 1.0 g/L (pink line) and 2.0 g/L (green line), respectively. CC carbon cloth. Source data are provided as a Source Data file.
In order to investigate the release and transfer of electrons in bioelectrochemical system, Faraday efficiency (ηF) was determined (Supplementary Fig. 14). The current peaked at 2.07 mA at the onset time and then declined over time. In a batch reaction of 35 h, the cumulative electric charges generated from 50 mL of reaction solution were 58.34 C. The concentration of product α-ketoglutarate determined by HPLC was 3.19 mM, convertible to the yield of 46.6%, suggesting that the same amount of D-xylose was consumed and corresponding to 61.58 C of electricity. In consideration of a conversion efficiency of 97% from NADH to electrons determined in a previous study47, the ηF value was calculated to be 97.7% (58.34 ÷ 61.58 ÷ 0.97 × 100%).
Long-term electricity generation of MFCs fueled by hemicellulose fractions of corncob
As xylan is a predominant component of hemicellulose, we next examined the performance of MFCs powered by real hemicellulose. Biomass corncob pretreated by heating in alkaline solution after neutralizing was used as fuel for power generation48. 1.06 g/L xylan from the pretreated hydrolysates of corncob was fueled, in which D-xylose was not detectable by HPLC. To supply constant fuels to MFC, the above pretreated hemicellulose was supplemented once a day to roughly maintain the same level. Then we examined the performance of the MFC using enzyme-engineered-bacterial consortia-modified-bioanode. As shown in Fig. 6, the Pmax of enzyme-bacterial consortia-bioanode-based MFC was 162.73 ± 3.69 μW cm−2 (Fig. 6, black line). The remarkable power output of the enzyme-bacterial consortia-bioanode-based MFC is much excelled over microbial community based MFC13, possibly because diverse biological properties of various microorganisms in one system may result in the instability in bacterial community. In addition, the long-term generation of electricity was also evaluated. The bacterial consortia-bioanode-based MFC maintained more than 95% of the Pmax after 6 days of operation. On the contrary, poor stability was observed in other biomass-based sugar-fed enzymatic fuel cells49. Besides, the conversion of pretreated hemicellulose from corncob to α-ketoglutarate was also monitored for 6 days. The yield of α-ketoglutarate was 44.3% during the 6 days’ long-term electricity generation of MFC fueled by the pretreated hydrolysates of corncob (Fig. 6, red line). Moreover, the morphology of the bacterial consortia-anode was observed by scanning electron microscope (SEM). After generating power for 6 days, the E. coli consortia and MWCNTs were still closely bound to the CC, while the E. coli were intact (Supplementary Fig. 15). These results indicate that the cells survived and stayed stable on CC during 6 days’ operation. These results suggest that the capabilities of engineered bacterial consortia can efficiently convert hemicellulose biomass into high-value products and electric power.

The time-dependent power density curve (black line) and α-ketoglutarate titers (red line) of enzyme-bacterial consortia-modified-bioanode-based MFC. Electrical outputs (power density) from engineered bacterial consortia were shown. The production of α-ketoglutarate in MFC was measured every 12 h during 6 days. LSV was recorded every 12 h during 6 days. The system's absence of pretreated hemicellulose from corncob was used as negative control (the blue line representing the power density curve and a green line representing α-ketoglutarate titers). n = 3 biologically independent experiments. Data are presented as mean ± SD. Source data are provided as a Source Data file.
In summary, a synthetic pathway that can generate high-value-added chemical of α-ketoglutarate and 4 electrons from hemicellulose in a controllable manner was successfully constructed by integrating six enzyme-displayed strains into a highly efficient bacterial consortia by tuning the expressed enzyme molecules and adjusting ratios of consortia members. The production of α-ketoglutarate with an excellent yield of 0.47 g/g was realized within 6 h by the enzyme-engineered bacterial consortia surface-displayed saccharification and oxidation pathway of xylan, the most abundant hemicellulose type. Then a two-compartment xylan/O2 MFC was assembled using enzyme-bacterial consortia-modified bioanodes and a laccase-displayed strain modified biocathode, realizing the direct conversion of biomass into electricity and α-ketoglutarate. The optimized MFC realized a 46.6% (w/w) yield of α-ketoglutarate, the highest OCV, and the largest Pmax. The Faraday efficiency was 97.7%. Furthermore, this MFC exhibited a considerable power output and long-term stability towards real biomass samples. To the best of our knowledge, this is the first example of using engineered bacteria consortia as biocatalysts for efficient utilization of hemicellulose, representing an important step toward further improving the conversion of biomass to high-value-added chemical and powerful electric energy. Based on these results, we envision that controllably enzyme-engineered bacterial consortia would find a wide range of applications in the fields of metabolic engineering, synthetic biology, enzyme engineering, bioenergy, and enzymatic electrosynthesis.
Methods
Strains and chemicals
All strains and plasmids used in this study are listed in Supplementary Table 1. The primers used are listed in Supplementary Table 2. E. coli DH5α was used for gene recombinant manipulation, and E. coli BL21 (DE3) ΔxylAB was used as a host to express recombinant proteins. Strains of E. coli carrying recombinant plasmids were routinely grown in Luria-Bertani (LB) medium (10 g/L tryptone, 10 g/L NaCl, 5 g/L yeast extract) at 37 °C and 200 rpm. Whenever necessary, an antibiotic (kanamycin, 50 μg/mL) was added. Taq DNA Polymerase and all restriction endonucleases were purchased from Fermentas (St. Leon Rot, Germany). Kits used for molecular cloning were obtained from Sparkjade Biotech Co., Ltd (Qingdao, China). Corncob xylan, to represent hemicellulose, was purchased from Meryer Co. Ltd (Shanghai, China). 4-nitrophenyl-β-D-xylopyranoside (PNPX), ABTS, AQDS, D-xylose, L-arabinose, xylono−1,4-lactone, D-xylonic acid lithium salt and α-ketoglutarate were purchased from Sigma-Aldrich (St. Louis, MO, USA). Platinum (Pt) wire, Ag/AgCl electrode, and Nafion 117 membrane (N117) were purchased from Incole Union Technology Co. Ltd (Tianjin, China). MWCNTs were purchased from Macklin Biochem Technology Co. Ltd (Shanghai, China). Corncob was obtained from the Corn Research Center of Qingdao Agriculture University. Other chemicals were purchased from Solarbio Science & Technology Co., Ltd (Beijing, China).
Plasmids construction
Construction of the knockout plasmids
For genetic operations into the genome of E. coli BL21 (DE3), the pRE112 suicide vector was utilized. To construct plasmids pRE112- ΔxylAB, approximately 862 bp upstream and 857 bp downstream fragments of the xylA and xylB genes were amplified using the upstream primers xylAB-Up-F/R and downstream primers xylAB-Down-F/R, respectively, and inserted into the Kpn I site of pRE112. The plasmid pRE112-ΔxylAB was transferred from E. coli χ7213 to E. coli BL21(DE3) by conjugation. Suitable recombinants were identified by antibiotic resistance screening and sucrose reverse screening. The resulting strain E. coli-ΔxylAB was identified by PCR via the primers xylAB-1-F/R.
Construction of anchoring motif expressing plasmid
The gene encoding N-terminal ice nucleation protein originating from Pseudomonas borealis was amplified from vector pTInaPbN-Xdh by PCR using primers INP-F/INP-R, and inserted into the Nco I/Nde I site of pET-28a (+) to generate plasmid pYJ-00.
Constructions of hemicellulose hydrolytic enzymes expressing plasmids
The gene encoding TtGH8 from Teredinibacter turnerae (GenBank No. CP001614.2), SXA from Selenominas ruminantium (GenBank No. WP_026766185) were codon-optimized for E. coli and synthesized by BGI Co., Ltd (Shenzhen, China). To construct intracellular protein expression vectors pYJ-01 and pYJ-02, TtGH8 and SXA encoding gene and corresponding cloning vector gene were amplified from above cloning plasmids and pET-28a by PCR method using primers pET-28a-F(TtGH8)/pET-28a-R(TtGH8) & TtGH8-1-F/TtGH8-1-R, pET-28a-F (SXA)/pET-28a-R (SXA) & SXA-1-F/SXA-1-R, respectively, and using the ClonExpress Ultra One Step Cloning Kit (Vazyme Biotechnology, Nanjing, China). To construct intracellular protein expression vectors pYJ-03, pYJ-01, and pYJ-02 vectors were used as PCR templates, and pET-28a-TtGH8-F/pET-28a-TtGH8-R & SXA-2-F/SXA-2-R as primers, and two amplified gene fragments were ligated by in-fusion method using the ClonExpress Ultra One Step Cloning Kit. For the construction of plasmids overexpressing enzymes displayed on cell surface, TtGH8 and SXA encoding gene and corresponding cloning vector gene were amplified from above cloning plasmids and pYJ-00 by PCR using primers pET-28a-INP-F (TtGH8)/pET-28a-INP-R (TtGH8) & TtGH8-2-F/TtGH8-2-R, pET-28a-INP-F(SXA)/pET-28a-INP-R(SXA) & SXA-3-F/SXA-3-R, respectively, and using the ClonExpress Ultra One Step Cloning Kit to generate pYJ-04 and pYJ-05. Plasmid pYJ-06 was constructed using pYJ-04 and pYJ-05 as PCR templates, and pET-28a-INP-TtGH8-F/pET-28a-INP-TtGH8-R & SXA-4-F/SXA-4-R as primers by fusion PCR strategy.
Construction of pentose oxidative enzymes expressing plasmids
To construct intracellular protein expression vectors pYJ-07, pYJ-08, pYJ-09, pYJ-10, and pYJ-11, the genes encoding XDH, XylC, XylD, XylX, KGSADH were amplified from the genomic DNA of Caulobacter crescentus NA100035 and corresponding cloning vector gene was amplified from pET-28a by PCR method using primers pET-28a-F (XDH)/pET-28a-R(XDH) & XDH-1-F/XDH-1-R, pET-28a-F (XylC)/PET-28a-R (XylC) & XylC-1-F/XylC-1-R, pET-28a-F(XylD)/pET-28a-R(XylD) & XylD-1-F/XylD-1-R, pET-28a-F(XylX)/pET-28a-R(XylX) & XylX-1-F/XylX-1-R, pET-28a-F(KGSADH)/pET-28a-R(KGSADH) & KGSADH-1-F/KGSADH-1-R, respectively, and using the ClonExpress Ultra One Step Cloning Kit. To construct intracellular protein expression vectors pYJ-12, pYJ-09, and pYJ-11 vectors were used as templates, and pET-28a-XylD-F/pET-28a-XylD-R & KGSADH-2-F/KGSADH-2-R as primers, and two amplified gene fragments were ligated by in-fusion method (the ClonExpress Ultra One Step Cloning Kit). For the construction of plasmids overexpressing enzymes displayed on the cell surface, XDH, XylC, XylD, XylX, KGSADH encoding genes were amplified from above cloning plasmids, and corresponding cloning vector gene was amplified from pYJ-00 by PCR method using primers pET-28a-INP-F(XDH)/pET-28a-INP-R(XDH) &XDH-2-F/XDH-2-R, pET-28a-INP-F(XylC)/pET-28a-INP-R(XylC) & XylC-2-F/XylC-2-R, pET-28a-INP-F(XylD)/pET-28a-INP-R(XylD) & XylD-2-F/XylD-2-R, pET-28a-INP-F(XylX)/pET-28a-INP-R(XylX) & XylX-2-F/XylX-2-R, pET-28a-INP-F(KGSADH)/pET-28a-INP-R(KGSADH) & KGSADH-2-F/KGSADH-2-R, respectively, and using the ClonExpress Ultra One Step Cloning Kit to generate plasmids pYJ-13, pYJ-14, pYJ-15, pYJ-16 and pYJ-17. Plasmid pYJ-18 was constructed using pYJ-15 and pYJ-16 as PCR templates and pET-28a-INP-XylD-F(XylX)/pET-28a-INP-XylD-R(XylX) & XylX-3-F/XylX-3-R primers by fusion PCR strategy (ClonExpress Ultra One Step Cloning Kit). Plasmid pYJ-19 was constructed using pYJ-15 and pYJ-17 as PCR templates, and pET-28a-INP-XylD-F(KGSADH)/pET-28a-INP-XylD-R(KGSADH) & KGSADH-3-F/KGSADH-3-R primers by fusion PCR strategy (ClonExpress Ultra One Step Cloning Kit). Plasmid pYJ-20 was constructed using pYJ-19 and pYJ-16 as PCR templates, and pET-28a-INP-XylD-KGSADH-F/pET-28a-INP-XylD-KGSADH-R & XylX-4-F/XylX-4-R primers by fusion PCR strategy (ClonExpress Ultra One Step Cloning Kit).
Construction of lac-expressing plasmid
For the construction of plasmids overexpressing lac- displayed on the cell surface, the gene encoding laccase from Bacillus subtilis 50 was subcloned into pYJ-00 to generate pYJ-21 by BGI Co., Ltd (Shenzhen, China).
Purification of cytoplasmic enzymes
Cells expressing seven intracellular enzymes, including TtGH8, SXA, TtGH8-SXA, XDH, XylC, XylD, XylX, KGSADH and XylDK (strain TtGH8, SXA, TtGH8-SXA, XDH, XylC, XylD, XylX, KGSADH and XylDK), were separately grown overnight in LB medium with kanamycin, and induced with 0.5 mM IPTG when cells had reached an OD600 of 0.6. After overnight growth at 16 °C, cells were harvested by centrifugation, resuspended in 40 mL 50 mM Tris–HCl (pH 7.0) buffer, broken by ultrasonication, and centrifuged at 12,000 × g for 40 min at 4 °C to remove cell debris, and unbroken cells. The soluble extract was applied to a 5 mL Ni-NTA column that had been equilibrated with 50 mM Tris–HCl (pH 7.0). Then the column was washed consecutively with resuspended buffer and several washing buffer (20 mM Tris–HCl pH 7.0, 300 mM NaCl, 1 mM β-mercaptoethanol) containing increasing amounts of imidazole (10, 20, 30, and 50 mM). The bound protein was eluted with elution buffer (20 mM Tris–HCl pH 7.0, 300 mM NaCl, 200 mM imidazole, 1 mM β-mercaptoethanol). Protein dialysis was conducted in a buffer (10 mM Tris–HCl pH 7.0 containing 100 mM NaCl, 1 mM β-mercaptoethanol) at 4 °C. Protein purity was analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), and protein concentration was determined by Bradford assay.
Cell surface display of enzymes and cell surface location analysis
For displaying enzymes on the cell surface of E. coli, recombinant strains were cultured in LB medium to an OD600 of 0.6, 0.5 mM of IPTG was used to induce protein expression. For laccase, cells were further cultured overnight at 16 °C, collected by centrifugation, and incubated in a buffer of Tris–HCl (50 mM, pH 7.0). The other cells were further cultured at 30 °C overnight, which were collected by centrifugation and incubated in Tris–HCl buffer (50 mM, pH 7.0). To determine the successful display of enzymes on the cell surface, outer membrane proteins of recombinant strains and negative control strains were isolated51. The outer membrane protein fractions of related strains were analyzed by 10% (wt/vol) SDS-PAGE.
For the western blot assay, samples were transferred to PVDF membranes at 100 mA for 10 min. Anti-6×HisTag mouse monoclonal antibody (catalog number D191001, Sangon Biotechnology, Shanghai, China) at 1:1000 was added and incubated for 3 h at room temperature, then rinsed three times with phosphate-buffered saline (PBS) with 0.05%(v/v) Tween-20 (PBST). It was then incubated with goat anti-mouse immunoglobulin G (IgG)-horseradish peroxidase (HRP) conjugate (catalog number D110068, Sangon Biotechnology, Shanghai, China) for 1 h at room temperature, washed three times with PBST and then once with PBS. The conjugation of antigen and antibody was detected with W-TMB chromogenic kit (Sangon Biotechnology, Shanghai, China).
For Quantitative Immunoassay, outer membrane proteins of recombinant strains were transferred to low fluorescent background PVDF membranes. Anti-6×HisTag mouse monoclonal antibody at 1:1000 was added and incubated for 3 h at room temperature, then rinsed three times with PBST. It was then incubated with goat anti-mouse IRDye 800CW fluorescent secondary antibodies (catalog number 926-32210, LI-COR, Inc., Lincoln, NE) for 1 h at room temperature, and then washed with PBST, air dried, wrapped in aluminum foil, and stored at 4 °C. LI-COR Odyssey CLx imaging system was used to detect bands on the membrane. The standard curve from known concentrations of purified recombinant XDH run on the same blot was obtained. Specific protein bands are quantified by fluorescent signals (excitation wavelength, 785 nm; emission wavelength, 820 nm) and a linear regression equation generated from recombinant XDH to estimate concentrations of proteins.
For fluorescence imaging, the induced cells were washed with PBS and blocked in PBS buffer containing 1% bovine serum protein for 30 min at room temperature. Next anti-6xHis tag mouse monoclonal antibody was added (1:100) after blocking, incubated overnight at 4 °C, and incubated for 1 h the next day at room temperature. After three washes with PBS, the cells were incubated with FITC-conjugated Donkey anti-Mouse IgG (1:100) for 2 h at room temperature. The cells, after PBS washing, were observed with a Laser Scanning Confocal Microscope (TCSsp5II03040101, Agilent, USA).
Enzymatic activity assay
Enzymatic activity was monitored by measuring the optical density at 600 nm (OD600) for cells using a Cary-60 UV-VIS spectrophotometer (Agilent Technologies, Inc., Santa Clara, CA). Soluble commercial corncob xylan was prepared by water dissolution. TtGH8 activity was assessed on 1 g/L soluble commercial corncob xylan in 50 mM Tris–HCl buffer pH 7.0 with 10 mM CaCl2 at 37 °C. The reducing sugars released by TtGH8 hydrolysis reaction were measured by DNS method with xylose as standard52. SXA enzymatic activity was detected using pNPX as the substrate at 37 °C. The product p-nitrophenol shows a typical peak at 405 nm, which can be monitored by a spectrophotometer26. Enzymatic activity of XDH was assayed by detecting the generation of NADH at 340 nm and 37 °C using 10 mM D-xylose as substrate as well as 1 mM NAD+ as coenzyme. For XylC, it can accelerate the hydrolysis of xylono-1,4-lactone into D-xylonic acid to generate D-xylonic acid, which can reduce the absorbance at 405 nm of p-nitrophenol. Enzyme activity was quantified by measuring the decrease in the absorbance at 405 nm53. Spontaneous hydrolysis of xylono−1,4-lactone was analyzed without catalysts. The enzymatic activity of XylD was assayed according to a modified procedure. In a typical assay, the reaction cocktail contained a certain amount of purified cells and D-xylonic acid. After incubation at 37 °C for a certain period, the samples were mixed with a solution containing 1% semi-carbazide hydrochloride and 1.5% sodium acetate. Finally after incubation at 30 °C for 10 min, the 2-keto-3-deoxyxylic acid produced was quantified by detection of the absorbance at 250 nm typical of semicarbazone37. The enzymatic activity of XylX was assayed spectrophotometrically in a coupled assay with the corresponding previous dehydratase XylD and KGSADH.37,54 The assay was performed in 50 mM Tris–HCl buffer (pH 7.0) with 10 mM MgCl2 containing 20 mM D-xylonic acid and 1 mM NAD+. After the addition of purified enzymes or enzyme-surface-displayed strains, the mixture was incubated at 37 °C. The increasing absorbance at 340 nm caused by NADH produced in the reaction was monitored. Glutaraldehyde was used as the substitute substrate to measure the activity of KGSADH by following the rate of NAD+ reduction by measuring the optical density at 340 nm55. To determine the kinetic constants of cell surface-displayed enzymes, different amounts of substrate were used to initiate a series of enzymatic assays. The data were applied to the Michaelis-Menten kinetic model using GraphPad Prism 5 software (www.graphpad.com). To determine the optimum pH for the E. coli-Lac, all enzymatic activities of the whole-cell catalyst were measured by adding ABTS to the final concentration of 5 mM at 37 °C by varying the pH values of the buffer solution (50 mM), including HAc–NaAc buffer (pH 3–5.5), Na2HPO4–NaH2PO4 buffer (pH 6) and Tris–HCl buffer (pH 7–9). The oxidation activity of E. coli-Lac towards ABTS was calibrated by measuring the absorbance of the supernatant at 420 nm (ε = 36 mM−1 cm−1, referred to ABTS concentration).
Optimization of protein-induced expression conditions
Recombinant strains were cultured in LB liquid medium at 37 °C and 200 rpm. To optimize inducer concentration for overexpressing INP-fused proteins, IPTG at different concentrations of 0.1 mM, 0.5 mM, 1 mM, and 1.5 mM were used when cells grew to an OD600 value of 0.6. Protein expression was induced overnight, and the activities of whole-cell catalyst were determined as above described. In order to investigate the effects of induction temperatures on intracellular and displayed proteins, 0.5 mM of IPTG was added into cultures when cells grew to an OD600 value of 0.6. Protein expression was induced overnight at different temperatures of 16 °C, 25 °C, 30 °C, and 37 °C, and the activities of crude extract or whole-cell catalyst was determined as above described.
Bioproduction of α-ketoglutarate
Recombinant strains were cultured in LB medium to an OD600 of 0.6, 0.5 mM of IPTG was used to induce protein expression. Cells were further cultured at 30 °C overnight, which were collected by centrifugation and incubated in Tris-HCl buffer (50 mM, pH 7.0). To produce α-ketoglutarate, 100 mL of 50 mM Tris–HCl buffer (pH 7.0) with 10 mM MgCl2, 10 mM CaCl2, 1.0 g/L commercial corncob xylan, and 4 mM NAD+ were incubated with artificial bacterial consortia (OD600 = 10) in a flask, which was shaken in an incubator at 150 rpm and 37 °C for 6 h. The produced α-ketoglutarate was determined by HPLC.
Preparation of bioanode and biocathode
The thickness of CC (CeTech, Taiwan, China) is 0.32 ± 0.02 mm, which was cut into pieces of 1.5 × 1.5 cm2 and washed by sonication in water and anhydrous ethanol, respectively. A 100 µL of poly (acrylic acid)-MWCNTs dispersion (0.089 mg/cm2) was cast and dried in air to acquire modified CC/MWCNTs. For the preparation of bioanode, 100 μL of the prepared cells (OD600 = 5.0) were dropped onto the CC/MWCNTs and dried at room temperature to obtain CC/MWCNTs/cells modified electrodes. Then 50 µL of Nafion solution (0.1 wt%) was syringed to the electrode surface to cover the electrode. For the modification of biocathode, 100 µL E. coli-Lac aqueous dispersion (OD600 = 10.0) was coated on the CC/MWCNTs, and subsequently, 50 µL of Nafion solution (0.1 wt%) was dropped onto the surface of the resulting electrode, then dried at room temperature. The thus-prepared electrode was denoted as CC/MWCNTs/E.coli-Lac. The onset potential is defined as the potential at which the current or current density goes above 1 μA cm−2.
Fabrication of MFC
The dual-chamber hemicellulose/O2 MFC was assembled with anodic compartment containing the artificial bacterial consortia-modified CC/MWCNTs/cells and cathodic compartment containing CC/MWCNTs/E.coli-Lac, separated by a Nafion 117 proton exchange membrane (DuPont, USA) with a diameter of 1.6 cm. The anodic electrolyte consisted of 100 mM Tris–HCl buffer (pH 7.0) containing 100 mM NaCl, 10 mM MgCl2, 10 mM CaCl2, 4 mM NAD+, and xylan. 10 mM AQDS was used as a mediator when necessary. The cathodic electrolyte consisted of 100 mM Na2HPO4–citric acid buffer (pH 5.0) containing 0.5 mM ABTS. Polarization curves were obtained by performing linear sweep voltammetry (LSV) at the scan rate of 1 mV s−1 and 37 °C. The specific current (I) was recorded in real-time. The voltage (V) between the anode and the cathode was set as the Y-axis of polarization curves. The output power (P) was derived via the relationship: P = V × I. The specific current and power were normalized to the geometric area of the anode (1.5 cm × 1.5 cm = 2.25 cm2) to obtain the current density and power density, respectively. The calculations of current density and power density refer to Eq. (4) and Eq. (5), respectively:
Faradaic efficiency (η F) assay
The ηF was determined through amperometry at 0.45 V. The generation of current was monitored during a reaction time in a volume of 50 mL at 37 °C. The reaction system is composed of 100 mM NaCl, 10 mM MgCl2, 10 mM CaCl2, and 4 mM NAD+, 1 g/L commercial corncob xylan in 100 mM Tris–HCl buffer (pH 7.0). The production of α-ketoglutarate was detected by HPLC. The total charge (C) was calculated according to the generated current during the whole time. The ηF was calculated using the equation as follows56:
where I is the current generated, dt is the time to produce current, Cα-KG is the concentration of produced α-ketoglutarate during the whole time, V is the reaction volume, n is the number of electrons generated per D-xylose consumed, and F is Faraday constant = 96,485 C per mole electron.
Long-term electricity generation of MFCs powered by pretreated hemicellulose
To obtain the hemicellulose from lignocellulose, biomass sample corncob was pretreated48. The milled corncob was pretreated by 2.5 M NaOH with a solid-to-liquid ratio of 1:30 (g/mL) at 115 °C for 1 h. After filtration, the filtrate was neutralized with 1 M acetic acid for use. The amounts of xylan were determined by gradient precipitation with ethanol and then freeze-drying6. This pretreated hemicellulose was used as fuel to power MFC for monitoring electricity and α-ketoglutarate generation. 100 μL of the prepared cells (OD600 = 10.0) was dropped onto the 2 mg/mL of MWCNTs-coated CC surface (2.25 cm2) and dried at room temperature. The pretreated corncob was supplemented into anodic electrolyte daily to maintain the constant concentration according to that of α-ketoglutarate. LSV was performed every 12 h, incubating at 37 °C for 6 days. The produced α-ketoglutarate was determined by HPLC.
Morphology observation of the bio-nanocomposite-modified CC
To determine whether the E. coli consortia and MWCNT were successfully attached to the CC surface, scanning electron microscopic images of the modified CC were recorded using JSM-7500F scanning electron microscopy (JEOL, Tokyo, Japan). The CC was first washed three times with PBS, soaked with 2.5% glutaraldehyde, and fixed at 4 °C for 12 h. Next, the fixed CC was cleaned three times with PBS for 5 min each time. Then, the cleaned CC was dehydrated with 30%, 50%, 70%, 90%, and 100% ethanol for 5 min each time. Then the dehydrated CC was further dehydrated with 30%, 50%, 70%, 90%, and 100% tert-butanol/ethanol for 5 min each time. After dehydration, the CC was frozen at −20 °C. Next, the frozen CC was put into the freeze-drying machine (Songyuan Huaxing Biotechnology Co., Ltd, Beijing, China) for freeze-drying. Then, the CC was pasted to the copper platform with conductive glue, followed by gold spraying.
Analytical methods
Cell growth was monitored by measuring the optical density at 600 nm (OD600) for cells using a Cary-60 UV-VIS spectrophotometer. Concentrations of D-xylose, L-arabinose, α-ketoglutarate were detected via Ultimate 3000 HPLC (ThermoFisher, USA) using an Aminex HPX87H column. The mobile phase was 0.05 M H2SO4, and the flow rate was 0.6 mL min−1 at a refractive-index detector at 50 °C37. CV was performed in a three-electrode configuration with CC/MWCNTs/cells as the working electrode, an Ag/AgCl reference electrode, and Pt wire as an auxiliary electrode connecting to a CHI 1000 C potentiostat (CH Instrument, Shanghai, China). The electrochemical reactions were performed at 37 °C.
Statistics and reproducibility
Statistical analyses were mainly performed using Microsoft Excel software (version 2021). Double-tailed t test or one-way ANOVA and a posteriori test were used for variance analysis. The data were expressed as mean ± standard deviation. Each group included at least three independent biological samples. Compared to a reference sample, significance was established with a P value <0.05. No statistical method was used to predetermine the sample size. No data were excluded from the analyses. The experiments were not randomized. The investigators were not blinded to allocation during experiments and outcome assessment.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Data supporting the findings of this work are available within the paper and Supplementary Information files. Source data are provided with this paper.
References
Schulte, L. A. et al. Meeting global challenges with regenerative agriculture producing food and energy. Nat. Sustain. 5, 384–388 (2022).
Qiao, J. et al. Evolving robust and interpretable enzymes for the bioethanol industry. Angew. Chem. Int. Ed. 62, e202300320 (2023).
Chen, Z. et al. Exploitation of lignocellulosic-based biomass biorefinery: a critical review of renewable bioresource, sustainability and economic views. Biotechnol. Adv. 69, 108265 (2023).
Yuan, J. S., Pavlovich, M. J., Ragauskas, A. J. & Han, B. Biotechnology for a sustainable future: biomass and beyond. Trends Biotechnol. 40, 1395–1398 (2022).
Lynd, L. R. et al. Toward low-cost biological and hybrid biological/catalytic conversion of cellulosic biomass to fuels. Energ. Environ. Sci. 15, 938–990 (2022).
Li, H. et al. Effect of structural characteristics of corncob hemicelluloses fractionated by graded ethanol precipitation on furfural production. Carbohyd. Polym. 136, 203–209 (2016).
Choi, J. W., Jeon, E. J. & Jeong, K. J. Recent advances in engineering Corynebacterium glutamicum for utilization of hemicellulosic biomass. Curr. Opin. Biotechnol. 57, 17–24 (2019).
Xiao, M. et al. Cellulosomal hemicellulases: indispensable players for ensuring effective lignocellulose bioconversion. Green Carbon 2, 57–69 (2024).
Vuong, T. V. & Master, E. R. Enzymatic upgrading of heteroxylans for added-value chemicals and polymers. Curr. Opin. Biotech. 73, 51–60 (2022).
Saha, B. C. Hemicellulose bioconversion. J. Ind. Microbiol. Biotechnol. 30, 279–291 (2003).
Mahapatra, D. M., Mishra, P., Thakur, S. & Singh, L. Leveraging artificial intelligence in bioelectrochemical systems. Trends Biotechnol. 40, 535–538 (2022).
Xiao, X. et al. Tackling the challenges of enzymatic (bio)fuel cells. Chem. Rev. 119, 9509–9558 (2019).
Tao, M. N. et al. Enhanced denitrification and power generation of municipal wastewater treatment plants (WWTPs) effluents with biomass in microbial fuel cell coupled with constructed wetland. Sci. Total Environ. 709, 136159 (2020).
Liu, Z. D. et al. Performance and microbial community of carbon nanotube fixed-bed microbial fuel cell continuously fed with hydrothermal liquefied cornstalk biomass. Nat. Commun. 185, 294–301 (2015).
Wu, S. K., Snajdrova, R., Moore, J. C., Baldenius, K. & Bornscheuer, U. T. Biocatalysis: enzymatic synthesis for industrial applications. Angew. Chem. Int. Ed. 60, 88–119 (2021).
Cai, M. et al. Microbial production of L-methionine and its precursors using systems metabolic engineering. Biotechnol. Adv. 69, 108260 (2023).
Fang, S. Q. et al. Controllable display of sequential enzymes on yeast surface with enhanced biocatalytic activity toward efficient enzymatic biofuel cells. J. Am. Chem. Soc. 142, 3222–3230 (2020).
Hasunuma, T. & Kondo, A. Development of yeast cell factories for consolidated bioprocessing of lignocellulose to bioethanol through cell surface engineering. Biotechnol. Adv. 30, 1207–1218 (2012).
Fishilevich, S., Amir, L., Fridman, Y., Aharoni, A. & Alfonta, L. Surface display of redox enzymes in microbial fuel cells. J. Am. Chem. Soc. 131, 12052–12053 (2009).
Van Bloois, E., Winter, R. T., Kolmar, H. & Fraaije, M. W. Decorating microbes: surface display of proteins on Escherichia coli. Trends Biotechnol. 29, 79–86 (2011).
Liu, Z. et al. Combined cell-surface display- and secretion-based strategies for production of cellulosic ethanol with Saccharomyces cerevisiae. Biotechnol Biofuels. 8, 162 (2015).
Naidu, D. S., Hlangothi, S. P. & John, M. J. Bio-based products from xylan: a review. Carbohyd. Polym. 179, 28–41 (2018).
Fowler, C. A. et al. Structure and function of a glycoside hydrolase family 8 endoxylanase from Teredinibacter turnerae. Acta Crystallogr. D. 74, 946–955 (2018).
Jordan, D. B., Li, X. L., Dunlap, C. A., Whitehead, T. R. & Cotta, M. A. Beta-D-xylosidase from Selenomonas ruminantium of glycoside hydrolase family 43. Appl. Biochem. Biotechnol. 137-140, 93–104 (2007).
Jordan, D. B. & Braker, J. D. Beta-D-xylosidase from Selenomonas ruminantium: thermodynamics of enzyme-catalyzed and noncatalyzed reactions. Appl. Biochem. Biotechnol. 155, 330–346 (2009).
Jordan, D. B. & Braker, J. D. beta-D-Xylosidase from Selenomonas ruminantium: role of glutamate 186 in catalysis revealed by site-directed mutagenesis, alternate substrates, and active-site inhibitor. Appl. Biochem. Biotechnol. 161, 395–410 (2010).
Rodriguez, G. M. et al. Engineering xylose utilization in Yarrowia lipolytica by understanding its cryptic xylose pathway. Biotechnol. Biofuels 9, 149 (2016).
Anandharaj, M. et al. Constructing a yeast to express the largest cellulosome complex on the cell surface. Proc. Natl. Acad. Sci. USA 117, 2385–2394 (2020).
Liang, B., Li, L., Mascin, M. & Liu, A. Construction of xylose dehydrogenase displayed on the surface of bacteria using ice nucleation protein for sensitive D-xylose detection. Anal. Chem. 84, 275–282 (2012).
Li, L., Kang, D. G. & Cha, H. J. Functional display of foreign protein on surface of Escherichia coli using N-terminal domain of ice nucleation protein. Biotechnol. Bioeng. 85, 214–221 (2004).
Li, H. L. et al. The hydrolytic efficiency and synergistic action of recombinant xylan-degrading enzymes on xylan isolated from sugarcane bagasse. Carbohyd. Polym. 175, 199–206 (2017).
Zheng, Z., Chen, T., Zhao, M., Wang, Z. & Zhao, X. Engineering Escherichia coli for succinate production from hemicellulose via consolidated bioprocessing. Microb. Cell Fact. 11, 37 (2012).
Li, X. W., Chen, Y. & Nielsen, J. Harnessing xylose pathways for biofuels production. Curr. Opin. Biotechnol. 57, 56–65 (2019).
Rossoni, L. et al. Engineering Escherichia coli to grow constitutively on D-xylose using the carbon-efficient Weimberg pathway. Microbiology 164, 287–298 (2018).
Stephens, C. et al. Genetic analysis of a novel pathway for D-xylose metabolism in Caulobacter crescentus. J. Bacteriol. 189, 2181–2185 (2007).
Liu, M., Ding, Y., Xian, M. & Zhao, G. Metabolic engineering of a xylose pathway for biotechnological production of glycolate in Escherichia coli. Microb. Cell. Fact. 17, 51 (2018).
Tai, Y. S. et al. Engineering nonphosphorylative metabolism to generate lignocellulose-derived products. Nat. Chem. Biol. 12, 247–253 (2016).
Sun, L. et al. Current advance in biological production of short-chain organic acid. Appl. Microbiol. Biotechnol. 104, 9109–9124 (2020).
Zeng, W., Zhang, H., Xu, S., Fang, F. & Zhou, J. Biosynthesis of keto acids by fed-batch culture of Yarrowia lipolytica WSH-Z06. Bioresour. Technol. 243, 1037–1043 (2017).
Stottmeister, U. et al. White biotechnology for green chemistry: fermentative 2-oxocarboxylic acids as novel building blocks for subsequent chemical syntheses. J. Ind. Microbiol. Biotechnol. 32, 651–664 (2005).
Liu, A., Lang, Q., Liang, B. & Shi, J. Sensitive detection of maltose and glucose based on dual enzyme-displayed bacteria electrochemical biosensor. Biosens. Bioelectron. 87, 25–30 (2017).
Zhu, H. & Li, Y. Turning light into electricity, biologically. Green. Carbon 1, 14–19 (2023).
Li, F. et al. Engineering Shewanella oneidensis enables xylose-fed microbial fuel cell. Biotechnol. Biofuels 10, 196 (2017).
Ali, R., Mittal, G., Sultana, S. & Bhatnagar, A. Ameliorative potential of alpha-ketoglutaric acid (AKG) on acute lung injuries induced by ammonia inhalation in rats. Exp. Lung Res. 38, 435–444 (2012).
Gal, I., Schlesinger, O., Amir, L. & Alfonta, L. Yeast surface display of dehydrogenases in microbial fuel-cells. Bioelectrochemistry 112, 53–60 (2016).
Zang, G. L. et al. Direct electricity recovery from Canna indica by an air-cathode microbial fuel cell inoculated with rumen microorganisms. Environ. Sci. Technol. 44, 2715–2720 (2010).
Zhu, Z., Kin Tam, T., Sun, F., You, C. & Percival Zhang, Y. H. A high-energy-density sugar biobattery based on a synthetic enzymatic pathway. Nat. Commun. 5, 3026 (2014).
Oliveira, E. E. et al. Xylan from corn cobs, a promising polymer for drug delivery: production and characterization. Bioresour. Technol. 101, 5402–5406 (2010).
Shi, P. et al. Biomass sugar-powered enzymatic fuel cells based on a synthetic enzymatic pathway. Bioelectrochemistry 144, 108008 (2022).
Durao, P. et al. Proximal mutations at the type 1 copper site of CotA laccase: spectroscopic, redox, kinetic and structural characterization of I494A and L386A mutants. Biochem. J. 412, 339–346 (2008).
Liang, B. et al. Development of bacterial biosensor for sensitive and selective detection of acetaldehyde. Biosens. Bioelectron. 193, 113566 (2021).
Ontanon, O. M. et al. EcXyl43 beta-xylosidase: molecular modeling, activity on natural and artificial substrates, and synergism with endoxylanases for lignocellulose deconstruction. Appl. Microbiol. Biot. 102, 6959–6971 (2018).
Sutter, J. M., Johnsen, U. & Schonheit, P. Characterization of a pentonolactonase involved in D-xylose and L-arabinose catabolism in the haloarchaeon Haloferax volcanii. FEMS Microbiol. Lett. 364, 1–8 (2017).
Wasserstrom, L. et al. Exploring D-xylose oxidation in Saccharomyces cerevisiae through the Weimberg pathway. AMB Express 8, 33 (2018).
Borgstrom, C. et al. Identification of modifications procuring growth on xylose in recombinant Saccharomyces cerevisiae strains carrying the Weimberg pathway. Metab. Eng. 55, 1–11 (2019).
Zhu, Z. & Zhang, Y. P. In vitro metabolic engineering of bioelectricity generation by the complete oxidation of glucose. Metab. Eng. 39, 110–116 (2017).
Yovkova, V., Otto, C., Aurich, A., Mauersberger, S. & Barth, G. Engineering the α-ketoglutarate overproduction from raw glycerol by overexpression of the genes encoding NADP+-dependent isocitrate dehydrogenase and pyruvate carboxylase in Yarrowia lipolytica. Appl. Microbiol. Biotechnol. 98, 2003–2013 (2013).
Tenhaef, N. et al. Microaerobic growth-decoupled production of alpha-ketoglutarate and succinate from xylose in a one-pot process using Corynebacterium glutamicum. Biotechnol. J. 16, 2100043 (2021).
Acknowledgements
This work is financially supported by the National Key Research and Development Program of China (2021YFA0910400, A.H.L.) and the National Natural Science Foundation of China (22278233, B.L.; 22378222, J.M.Y.).
Author information
Authors and Affiliations
Contributions
B.L., S.C., A.H.L., and J.M.Y. conceived and coordinated the study. B.L., A.H.L., S.C., and J.M.Y. wrote the article with input from all other co-authors. B.L., J.Y., C.F.M., and L.W. constructed the engineered strains and tested enzymatic activity and MFCs. Y.R.Z. and Z.C.L. performed electrochemical testing. J.Y. conducted SEM characterizations. L.Z. and J.L. performed biomass pretreatment. B.L., J.Y., C.F.M., and L.W. analyzed the experimental data. All authors contributed to the writing of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interest.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Liang, B., Yang, J., Meng, CF. et al. Efficient conversion of hemicellulose into high-value product and electric power by enzyme-engineered bacterial consortia. Nat Commun 15, 8764 (2024). https://doi.org/10.1038/s41467-024-53129-0
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-024-53129-0
