
Abstract
Adenosine receptors play pivotal roles in physiological processes. Adenosine A3 receptor (A3R), the most recently identified adenosine receptor, is expressed in various tissues, exhibiting important roles in neuron, heart, and immune cells, and is often overexpressed in tumors, highlighting the therapeutic potential of A3R-selective agents. Recently, we identified RNA-derived N6-methyladenosine (m6A) as an endogenous agonist for A3R, suggesting the relationship between RNA-derived modified adenosine and A3R. Despite extensive studies on the other adenosine receptors, the selectivity mechanism of A3R, especially for A3R-selective agonists such as m6A and namodenoson, remained elusive. Here, we identify tRNA-derived N6-isopentenyl adenosine (i6A) as an A3R-selective ligand via screening of modified nucleosides against the adenosine receptors. Like m6A, i6A is found in the human body and may be an endogenous A3R ligand. Our cryo-EM analyses elucidate the A3R-Gi complexes bound to adenosine, 5’-N-ethylcarboxamidoadenosine (NECA), m6A, i6A, and namodenoson at overall resolutions of 3.27 Å (adenosine), 2.86 Å (NECA), 3.19 Å (m6A), 3.28 Å (i6A), and 3.20 Å (namodenoson), suggesting the selectivity and activation mechanism of A3R. We further conduct structure-guided engineering of m6A-insensitive A3R, which may aid future research targeting m6A and A3R, providing a molecular basis for future drug discovery.
Similar content being viewed by others


Introduction
Adenosine functions as an extracellular signaling molecule by activating adenosine receptors, besides its fundamental role as a building block of RNA. Adenosine receptors belong to the class A G-protein coupled receptors (GPCRs), and are expressed in various types of tissues1. Adenosine-mediated receptor activation engages in various physiological responses, including immunity, sensory conception, learning, and memory. Furthermore, the involvement of adenosine receptors has been reported in various diseases, including mental disorders and inflammation2. In those diseases, the roles of each receptor subtype are diverse; for example, activation of A1R is fundamental for early ischemic damage, and inhibition of A2AR is effective in treating Parkinson’s disease3. Therefore, the development of selective agents targeting adenosine receptors has garnered keen attention.
Of the four adenosine receptors, A3R was most recently identified4. A3R is widely distributed throughout the body, including brain, heart, testes, lungs and liver in humans5. Furthermore, A3R has a broad distribution in human inflammatory cells, including eosinophils, neutrophils, and mast cells3,6,7,8. A3R activation reportedly plays dual roles, offering both neuroprotection and neurodegeneration, cardioprotection and cardiotoxicity, and anti-inflammatory and proinflammatory effects in humans9. Moreover, it is particularly noteworthy that A3R is often overexpressed in tumors10. The activation of A3R in tumor cells is associated with anticancer effects, and A3R-selective agonists such as namodenoson and piclidenoson are currently in clinical trials and showing promising results11. Thus, the role of A3R in disease has become a major subject of extensive biomedical exploration.
Recently, A3R signaling has been linked to epitranscriptomics, an emerging concept characterized by diverse RNA modifications12. To date, over 170 species of RNA modifications have been identified across all three domains of life13, with approximately 50 species found in mammals14. These RNA modifications are vital for the regulation of post-transcriptional gene expression, influencing RNA stability15, cellular localization16, translation efficiency17, and fidelity18. Among these modifications, N6-methyladenosine (m6A) is one of the most abundant and has been implicated in development, metabolism, and cancer biology by regulating mRNA stability19,20. In addition to its role in intracellular gene expression regulation, we previously discovered that m6A can be released into the extracellular space as a result of RNA catabolism12. Moreover, using a GPCR assay, we demonstrated that m6A is a selective ligand for A3R, with a potency 10 times stronger than that of adenosine. Notably, elevated extracellular m6A concentrations effectively induced inflammation and type I allergy in mice12. However, the detailed relationship between A3R and modified adenosine has yet to be explored.
Although many structures of the other adenosine receptors have been reported, the structure of A3R has remained enigmatic21,22,23,24,25,26,27,28,29,30. Consequently, it is still unclear how nonselective and selective agonists, including drugs and modified adenosine, activate A3R. Here, we perform modified nucleoside screening and discover that beside m6A, tRNA-derived i6A is also an A3R-selective agonist. Subsequent cryo-EM structural analyses elucidate the structures of the A3R-Gi complex bound to two nonselective and three selective agonists, revealing the structural basis of agonist recognition by A3R.
Results
Screening of modified nucleosides against human A3R
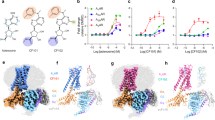
Using a limited set of modified nucleosides, we previously showed that m6A selectively activates A3R12. To fully understand the functional roles of RNA-derived modified nucleosides with A3R, we tested 42 species of modified nucleosides, representing the largest number of mammalian-related modified nucleosides currently available, with A3R as well as the other adenosine receptors by TGFα shedding assays31 (Fig. 1a and Supplementary Table 1). Consistent with the previous study12, m6A showed the greatest potency only for A3R compared with other modified nucleosides and adenosine (calculated intrinsic activity (RAi) values of m6A relative to adenosine = 1300%) (Fig. 1b). In addition to m6A, we found that N6-isopentenyladenosine (i6A), a tRNA-specific RNA modification, selectively activates A3R (RAi = 59%) (Fig. 1c). Moreover, N6, N6-dimethyladenosine (m6,6A), which is the dimethyl variant of m6A exclusively found in 18S rRNA, showed very weak activation of A3R (RAi = 7%) (Fig. 1d). Interestingly, N6-threonylcarbamoyladenosine (t6A), another tRNA-specific RNA modification at the N6 position, did not show any potency on A3R (Fig. 1e). These results suggest that the chemical properties of the modification at the N6 position profoundly impact the receptor activation potency.
It should be noted that we observed that 1-methyladenosine (m1A), which is one of the major and abundant RNA modifications12, can also induce A3R activation. However, since synthetic m1A contains a significant amount of m6A as a by-product of chemical synthesis32, we did not include m1A in the results. Additionally, 2-methyladenosine (m2A) showed weak and non-selective potency towards all four adenosine receptors, although m2A is only found in Escherichia coli and plants, unlike m6A, i6A, m6,6A, and t6A, which are found in humans.

a Heatmaps showing the activation of adenosine receptor subtypes by modified nucleosides, as measured by the TGF-α shedding assay. The color scale represents % receptor activation compared to TPA (12-O-tetradecanoylphorbol 13-acetate)-mediated receptor activation, which induces the maximum alkaline phosphatase(AP)-fused TGFα-shedding response independently of the receptor, and the adenosine receptor-dependence of signals were calculated by subtracting the response in mock-transfected cells from the response in adenosine receptor-expressing cells. The tested compounds’ concentrations were 100 nM for hA1R and hA3R, 1 µM for hA2AR, and 5 µM for hA2BR. Values are shown as an average of three independent experiments. Comparison of AP-TGFα-shedding response curves for hA3R between modified nucleosides (b: m6A, c: i6A, d: m6,6A, and e: t6A) and adenosine. Response curves for mock-transfected cells are shown in the same graph. Ligand-induced AP-TGF-α release ratio into conditioned media is quantified. Symbols and error bars represent mean ± SEM, respectively, of 3–6 independent experiments with each performed in triplicate. Source data are provided as a Source Data file.
Overall structure of the A3R-Gi complex
For the structural analysis, we focused not only on the modified adenosine analogues obtained through the screening but also on adenosine, 5′-N-ethylcarboxamidoadenosine (NECA), and namodenoson. NECA is an adenosine analogue that acts as a potent agonist for adenosine receptors, and several structures of adenosine receptors bound to NECA have been reported25,29,33,34. Namodenoson is an A3R-selective agonist with potential efficacy in cancer treatment11. We used these two nonselective (adenosine and NECA) and three (m6A, i6A and namodenoson) selective agonists for our structural analysis, but excluded m6,6A due to its low potency and weak selectivity (Fig. 2a).

a Structures of the agonists used for structural analyses. Atom numbers for the adenosine moiety are colored blue. Modifications at N6, C2, and 5′ positions are highlighted in green, red, and blue, respectively. b Cryo-EM maps of the A3R-Gi complex bound to adenosine, NECA, m6A, i6A, and namodenoson. Densities of each agonist are shown in the top-right corner of each map. c Superimposition of each agonist-bound A3R. d Overall structure of the A3R-Gi complex bound to NECA. e Superimposition of A1R (PDB 6D9H), A2AR (PDB 6GDG), A2BR (PDB 8HDP), and A3R. f Close-up view of ECLs of adenosine receptors. ECL2 and ECL3 show remarkable differences among the adenosine receptors. The disulfide bond between ECL2 and TM3 of A3R is labeled. Receptor-G-protein interactions around α5 (g) and ICL2 (h). The residues involved in the interactions are represented by stick models. Black dashed lines indicate hydrogen bonds.
Initially, we attempted the structural analysis of human A3R, but it showed poor monodispersity in fluorescence size exclusion chromatography (FSEC)35. Thus, we evaluated vertebrate A3R homologues by FSEC. Consequently, we identified sheep A3R as the most suitable candidate for structural analysis (Supplementary Fig. 1a). The transmembrane regions of sheep and human A3R exhibit high sequence homology of 90%36,37,38 (Supplementary Fig. 1b). To examine the ligand binding profile of sheep A3R, we evaluated the agonist efficacies of the human and sheep A3Rs by a TGFα shedding assay31, and found that they are quite similar (Supplementary Fig. 1c and Supplementary Table 2). Moreover, the mRNA distribution of sheep A3R is similar to that in humans, with expression mainly in the lungs and pineal glands5. Considering these factors, we concluded that the functional properties of the human and sheep A3Rs do not differ significantly, and proceeded with the structural analysis of the sheep A3R-Gi complexes.
For sample preparation, we adopted the previously reported Fusion-G system, which combines the NanoBiT tethering system and the fusion of the Gα and Gγ subunits as a single polypeptide39,40,41,42. We co-expressed sheep A3R and the fusion-Gi trimer in HEK293 cells, solubilized the complex with detergent, and then purified it using FLAG affinity chromatography. The complex was stabilized with a single-chain variable fragment (scFv16), isolated via gel filtration chromatography (Supplementary Fig. 1d), and subsequently used for cryo-EM analysis (Supplementary Fig. 2a–o). Finally, we obtained the cryo-EM maps of the A3R complex bound to the two nonselective and three selective agonists at overall resolutions of 3.27 Å (adenosine), 2.86 Å (NECA), 3.19 Å (m6A), 3.28 Å (i6A) and 3.20 Å (namodenoson) (Fig. 2b). Notably, the NECA-bound complex exhibited the highest resolution and all models overlapped quite well, so the NECA-bound model is used for the following discussion of the overall structure (Fig. 2c, d).
Comparing the overall structure of A3R with the cryo-EM structures of other adenosine receptors, A3R overlaps with A1R, A2AR, and A2BR with root-mean-square deviations (R.M.S.Ds) of 0.82, 0.96 and 0.90 Å, respectively, indicating considerable similarity with A1R24,26,33 (Fig. 2e). While the overall structures of the adenosine receptors superimpose well, extracellular loops (ECLs) 2 and 3 exhibit secondary structure-level differences (Fig. 2f). ECL2 of the adenosine receptors has varied sequences across the family, and that of A3R is the shortest among them (Supplementary Fig. 3). From the N- to C-terminal side, ECL2 of the adenosine receptors comprises a short helical region, a disordered region, a sheet-like region forming a disulfide bond with the transmembrane helix (TM) 3, and a comparatively conserved region that contributes to the ligand pocket. ECL2 of A3R forms a relatively shorter helix compared to A1R on the N-terminal side. We were able to model the subsequent disordered region, which is also short and distant from the other regions, implying little relevance to the receptor function. In the following sheet-like region, C833.25 and C16545.50 form a disulfide bond (superscripts indicate Ballesteros–Weinstein numbers43), as in the other adenosine receptors. Furthermore, ECL3 of A3R is the shortest among the adenosine receptors, lacking the N-terminal helical structure observed in the other receptors and instead extending straight towards TM7. Considering their low sequence homology, the loop regions are likely to contribute to the differences in receptor function among adenosine receptors (Supplementary Fig. 3).
Next, we inspected the interaction between A3R and Gi. There are hallmark regions of the interactions between class A Gi-coupling receptors, including A3R, and Gi: ICL2 of a receptor as well as α5 of Gi39,44,45,46,47,48,49,50,51. In detail, around α5 of Gi, several residues form hydrogen bonds, represented by the interaction between R1083.50 and C351H5.23 (superscript indicates the common Gα numbering [CGN] system52) (Fig. 2g). In the ICL2 of A3R, V11634.51 penetrates into the hydrophobic cavity of Gi, which is common in class A GPCRs39,44,45,46,47,48,49,50,51,53,54,55 (Fig. 2h). Furthermore, extensive ionic interactions are formed across ICL2. These extended interactions more closely resemble that between A2BR and Gs, rather than that between A1R and Gi23,24,25,26,33,56 (Supplementary Fig. 4a–d). Moreover, A3R has T11534.50 at the C-terminus of TM3, while most class A GPCRs including the other adenosine receptors have P34.50 at this position. While engaging in the interaction with I344G.H5.16 as in A1R-Gi, T11534.50 also slightly elongates TM3. These features distinguish A3R among the adenosine receptors.
Binding modes of adenosine and NECA
The orthosteric ligand pocket of A3R consists of the extracellular halves of TM3, TM5, TM6, TM7 and ECL2, as in the other adenosine receptors23,24,25,26,33,56 (Fig. 3a, b). The binding modes of adenosine and NECA are remarkably similar in terms of the adenosine moiety (Fig. 3c, d). In detail, the adenine moiety forms hydrogen bonds with N2496.55 and π-stacking interactions with F16745.52, while the ribose moiety forms hydrogen bonds with H2717.43, and there are extensive hydrophobic interactions surrounding the entire ligand. Moreover, the modification at the 5′ position of NECA forms an extra hydrogen bond with T943.36, and the following carbon chain engages in hydrophobic contacts with the W2426.48, which is known as the toggle switch motif essential for class A GPCR activation57 (Fig. 3e). These additional interactions would enable NECA to activate A3R at lower concentrations, which is the same as in the other adenosine receptors25,29,33,34.

a Superimposition of adenosine-bound A1R (PDB 6D9H), A2AR (PDB 2YDO), A2BR (PDB 8HDP), and A3R. b Superimposition of NECA-bound A2AR (PDB 6GDG), A2BR (PDB 7XY7), A3R. Binding modes of adenosine (c) and NECA (d). Black dashed lines indicate hydrogen bonds. e Comparison of the interactions around the 5′ tail of NECA. f Comparison of the binding modes of adenosine to each adenosine receptor. Representative residues involved in the ligand-receptor interaction are represented by stick models. Residues of A3R are labeled. Key interactions are conserved among adenosine receptors. g Alignment of residues comprising the orthosteric pockets of adenosine receptors.
Most residues in the orthosteric pocket are conserved, and the binding sites and the binding modes of adenosine and NECA are quite similar among the adenosine receptors, including key interactions such as hydrogen bonds with N6.55 and H7.43, π-stacking interactions with F45.52, and extensive hydrophobic contacts23,24,25,26,29,33,34 (Fig. 3f, g). Nevertheless, there are some differences between A3R and the other adenosine receptors. The residue at position 3.32 is leucine in human A3R and methionine in sheep A3R, which are longer than valine in the other adenosine receptors. This may enable A3R to form closer interactions with a ligand. At position 6.52, while the other adenosine receptors feature histidine, A3R possesses serine. The histidine in the other adenosine receptors engages in polar interactions with the 5’ hydroxyl group of the adenosine moiety. However, since serine features a shorter side chain than histidine, S1805.42 does not seem to participate in a direct interaction with adenosine. Especially, at position 45.53 in ECL2, human and sheep A3R respectively have valine and arginine. Swapping these residues slightly affected the agonist potencies of human and sheep A3R (Supplementary Fig. 5a, b). In contrast to A3R, the other adenosine receptors have glutamate, which forms a hydrogen-bonding interaction with ligands. Remarkably, a mutation of this glutamate in A2AR led to a significant decrease in its activity58, whereas mutations to glutamate at this position in A3R greatly reduced the potency of agonists12 (Supplementary Fig. 5c, d and Supplementary Table 3). Despite the absence of hydrogen-bonding interactions with E45.53, adenosine showed higher potency for A3R than A2AR and A2BR. Taken together, A3R may form totally different interactions with agonists around this extracellular portion.
Binding mode of m6A
m6A binds to A3R in the same position as adenosine, exhibiting the common interactions such as hydrogen bonding with N2496.55 and H2717.43, and π-stacking interactions with F16745.52 (Fig. 4a, b). Remarkably, the methyl group at the N6 position forms close hydrophobic interactions with a hydrophobic pocket tightly packed by the aliphatic portions of R16845.53, M1735.35, L2526.58, and L2637.35 (Fig. 4c). In contrast to A3R, the residues at these positions are hydrophilic in the other adenosine receptors (Fig. 3g). Mutations of these A3R residues to the corresponding residues in the other adenosine receptors significantly decreased the potency of m6A12, indicating that these hydrophobic residues are exactly what enable A3R to be selectively activated by m6A. Furthermore, this hydrophobic pocket in A3R exhibits closer contacts with agonists compared to the counterparts in the other adenosine receptors (Fig. 4d and Supplementary Table 4). The distance between a ligand and residues is the shortest in A3R-m6A, with the closest interactions. In addition, the distance in A3R-adenosine is shorter than that in the other adenosine receptors. These close interactions could explain the high potency of agonists for A3R. Taken together, this tightly packed hydrophobic pocket would confer the unique agonist selectivity to A3R.

a Superimposition of adenosine-bound and m6A-bound A3R. b Binding mode of m6A. Black dashed lines indicate hydrogen bonds. c Close-up view of the hydrophobic pocket of the m6A-bound model. d CPK models of the hydrophobic pocket of A3R and those of A1R, A2AR, A2BR, and adenosine-bound A3R (PDB 6D9H, 2YDO, 8HDP, and this study, respectively). The methyl group of m6A is especially tightly packed. e–h Relative RAi values of adenosine, m6A, and m6,6A for human A3R mutants, as determined by the TGFα-shedding assay. RAi values are expressed as fold change of the values for the WT. The LogRAi cutoff value was set to −2. Mutations that reduce (e) and enlarge (f) the size of the hydrophobic pocket. g Mutations that replace the original residue to a hydrophilic residue. h Designed mutants for selectively reducing the potency of m6A. Data are presented as mean values ± SEM from at least three independent experiments performed in triplicate (the n values are represented by circles). Source data are provided as a Source Data file.
To validate the importance of this hydrophobic pocket in the recognition of m6A and its variant m6,6A, we conducted comprehensive mutagenesis studies on the residues of the pocket in human A3R. First, changing the bulkiness of the side chains tended to reduce the potency of all agonists. Small-to-large mutations probably led to the partial occlusion of the pocket (Fig. 4e). Of these mutants, V16945.53F reduced the potency of m6A remarkably more than adenosine. In contrast, large-to-small mutations, especially in M1745.35A, could have led to looser packing in the pocket, resulting in the loss of close interactions with agonists (Fig. 4f). Next, hydrophobic-to-hydrophilic mutations, represented by V16945.53N, I2536.58N, and I2536.58Q, reduced the potency of m6A much more than adenosine (Fig. 4g). Repulsion between hydrophilic side chains and hydrophobic modifications should explain this reduction. The changes of the agonist potencies in the mutants highlight the importance of both the hydrophobicity and size of the residues in the hydrophobic pocket.
Based on the results of these single mutants, we attempted structure-guided engineering of m6A-insensitive A3R. m6A, mainly derived from mRNA and rRNA degradation, works as a signal transducer with A3R, and their relationship has attracted keen attention. m6A-insensitive A3R should greatly aid the future work on the m6A-mediated signaling pathways. Combining the mutants that reduced the potency of m6A to a greater extent than that of adenosine, namely V16945.53F, V16945.53N, I2536.58N, and I2536.58Q, we attempted to create m6A-insensitive A3R (Fig. 4h). Among the four combinations, I2536.58N/V16945.53N and I2536.58N/V16945.53F especially caught our attention. While I2536.58N/V16945.53N significantly reduced the potency of m6A, it retained the potency of adenosine. I2536.58N/V16945.53F reduced the potency of m6A even more than I2536.58N/V16945.53N. Although I2536.58N/V16945.53F also somewhat decreased the potency of adenosine, considering that the concentrations of adenosine and m6A are about 150 nM and 30 nM in human plasma12, this mutant have just the right affinity to almost completely shut off the m6A-mediated pathway under in vivo conditions, minimally affecting the adenosine-mediated pathway. These two mutants would greatly facilitate future discoveries of the physiological functions of m6A.
Binding mode of i6A
Compared to adenosine, the binding position of i6A is clearly different (Fig. 5a). While the hydrogen bonds with H2717.43 and π-stacking with F16745.52 are maintained, the hydrogen bond with N2496.55 is absent (Fig. 5b), probably because of the isopentyl group of i6A. The isopentyl group of i6A extends toward N2496.55 and forms close hydrophobic interactions with M1735.35 and L2526.58 (Fig. 5c). The longer modification of i6A enables especially closer contacts with M1735.35 and L2526.58, suggesting stronger interactions with A3R. Taken together, while losing the conserved hydrogen bond, i6A acquires its relatively weak but selective potency for A3R from the close interactions with the hydrophobic pocket.

a Superimposition of adenosine- and i6A-bound A3R. b Binding mode of i6A. Black dashed lines indicate hydrogen bonds. c Close-up view of the hydrophobic pocket of the i6A-bound structure. d–f Relative RAi values of adenosine and i6A for human A3R mutants, as determined by the TGFα-shedding assay. The LogRAi cutoff value was set to −2. Mutations that reduce (d) and enlarge (e) the size of the hydrophobic pocket. f Mutations that replace the original residue to a hydrophilic residue. Data are presented as mean values ± SEM from at least three independent experiments performed in triplicate (the n values are represented by circles). The values for adenosine used references in (d–f) are the same as those for adenosine in Fig. 4e–g. Source data are provided as a Source Data file.
We conducted further mutagenesis studies on the hydrophobic pocket to validate its importance in i6A recognition. As in the cases of the other agonists, small-to-large mutations decreased the potency of i6A, which seem to be resulted from the steric occlusion of the endogenous pocket (Fig. 5d). However, I2536.58F greatly enhanced the potency of i6A. This mutation possibly enables the π-stacking with the isopentyl group. In contrast to the small-to-large mutations, the large-to-small mutations enhanced the potency of i6A, suggesting that the additional space in the hydrophobic pocket allows i6A to adopt a more secure orientation (Fig. 5e). Hydrophobic-to-hydrophilic mutations showed various responses, with I2536.58N and I2536.58Q surprisingly enhancing the potency of i6A (Fig. 5f). Taken together, these results suggest that i6A forms interactions with the hydrophobic pocket, but the size of the isopentyl group limits the potency of i6A.
Binding mode of namodenoson
Namodenoson has an adenosine backbone with a 3-iodobenzyl group at the N6 position, a chloro group at the C2 position, and a N-methylcarboxamide group at the 5′ position (Fig. 2a). The adenosine moiety of namodenoson is accommodated in the same position as the other agonists, by hydrogen bonding with N2496.55 and H2717.43, π-stacking interactions with F16745.52, and extensive hydrophobic interactions (Fig. 6a, b). As in the cases with m6A and i6A, the 3-iodobenzyl group at the N6 position of namodenoson wedges into the hydrophobic pocket and exhibits extensive hydrophobic interactions (Fig. 6c). In the past structure-activity relationship (SAR) studies, the 3-iodobenzyl group at the N6 position strongly enhanced the A3R selectivity59. As with m6A, the interactions between the N6 modification and the hydrophobic pocket endow namodenoson with its strong and selective potency towards A3R.

a Superimposition of adenosine- and namodenoson-bound A3R. b Binding mode of namodenoson. Black dashed lines indicate hydrogen bonds. c Close-up view of the hydrophobic pocket of the namodenoson-bound structure. The 3-iodobenzyl group of namodenoson wedges into the hydrophobic pocket. d Close-up view of the interactions around the chloro group of namodenoson. e Close-up view of the interactions around the N-methylcarboxamide group of namodenoson. f Cross section of the orthosteric pocket of A3R. Namodenoson fills the orthosteric pocket comprehensively.
In addition to the modification at the N6 position, a C2 modification position reportedly enhances the potency for A3R60. The chloro group at the C2 position enters a cavity formed by TM1, TM2, and TM7, forming hydrophobic interactions with I722.64 and I2677.39 (Fig. 6d). These additional hydrophobic interactions should further stabilize the ligand-receptor interaction. Like the 5′ tail of NECA, the methylcarboxamide group at the 5′ position of namodenoson forms additional interactions, represented by the hydrogen bond with T943.36 (Fig. 6e). However, compared to NECA, the 5′ tail of namodenoson is shorter, resulting in a weaker hydrophobic interaction with W2426.48, which is closely related to the activation of a class A GPCR. This length might be optimal to efficiently activate A3R, while preventing the nonselective activation of the other adenosine receptors. This inference is consistent with the report that compounds with a N-methylcarboxamide group at the 5′ position exhibited higher selectivity for A3R than those with an N-ethylcarboxamide group59.
Namodenoson exhibited unparalleled potency for A3R among the agonists (Supplementary Fig. 1c and Supplementary Table 2). Compared to the other agonists, namodenoson fills the orthosteric pocket of A3R much more comprehensively (Fig. 6f). Each of the modifications at the N6, 2 and 5′ positions contributes to further the extensive and strong interactions, endowing namodenoson with its exceptional potency (EC50 = 2.41 nM for human A3R) and selectivity for A3R.
Structural insight into the selective recognition of adenosine receptors
To comprehensively understand the selectivity among the adenosine receptors, we compared the structures of the adenosine receptors bound to their selective drugs22,25,27,28 (Fig. 7a–f). While occupying the orthosteric pocket, each drug exhibits a unique extension on the extracellular side. We found that m6A closely interacts with the hydrophobic pocket of A3R, composed of the extracellular portions of TM5, TM6, TM7 and ECL2 (Fig. 7a). In addition to the hydrophobic pocket, the modification at the C2 position of namodenoson interacts with the cavity between TM2 and TM7 of A3R (Fig. 7b). dU172, an A1R-selective agonist, shows a similar binding mode to namodenoson, where it not only interacts with the extracellular sides of TM5, TM6 and TM7, but also penetrates the cavity formed by TM1, TM2 and TM7 of A1R (Fig. 7c). This insertion into the cavity formed by TM1, TM2 and TM7 is also observed in the cases of ZM241385-A2AR and BAY60-6583-A2BR (Fig. 7d, e), indicating that this cavity is one of the major regions contributing to the selectivity among adenosine receptors. Furthermore, an A2AR-selective agonist UK-432097 exhibits a substantial extension of its modification site on the extracellular side, engaging in extensive interactions with TM2, ECL2, TM6, TM7 and ECL3 (Fig. 7f). As observed in the sequence alignment of the adenosine receptors, while the ‘insides’ of the endogenous pocket are remarkably similar, the ‘outsides’ of the endogenous pocket have diverse sequences (Supplementary Fig. 3). As seen in the binding modes of these drugs, the diverse extracellular portions endow drugs with the selectivity for each subtype. Thus, selective drugs of the adenosine receptors may be endowed with two main factors: nonselective binding in the endogenous pocket and selective binding on the extracellular side.

a–f Binding modes of the adenosine receptors and their selective drugs. a m6A-bound A3R. b Namodenoson-bound A3R. c DU172-bound A1R (PDB 5UEN). d ZM241358-bound A2AR (PDB 3PWH). e BAY60-6583-bound A2BR (PDB 7XY6). f UK-432097-bound A2AR (PDB 3QAK). While occupying the orthosteric pocket, each drug exhibits interactions with the extracellular portion of the receptor. These extracellular interactions would be essential for the selectivity among the adenosine receptors.
Discussion
In this study, we identified tRNA-derived N6-isopentenyl adenosine (i6A) as an A3R ligand. We further elucidated the structures of A3R complexes bound to two nonselective and three selective agonists including namodenoson, an A3R-selective agonist in clinical trials. While the binding modes of the agonists are quite similar in terms of the adenosine moiety, the selective agonists form distinct interactions with the hydrophobic pocket of A3R. We further validated our structural insights with functional mutagenesis studies. Based on the results, we created A3R mutants that selectively inhibit the m6A pathway. Our discoveries will greatly aid drug discovery and molecular biological research targeting A3R and modified adenosines.
It should be noted that while unmodified adenosine can be generated by multiple pathways, including anabolic purine synthesis or the catabolic breakdown of ATP or RNA, m6A is exclusively generated through breakdown of RNA. Our previous study has shown that extracellular m6A is mostly derived from lysosomal degradation of ribosome RNA, and extracellular release of m6A is augmented by cytotoxic stimulation, leading to A3R-dependent inflammation and allergy in vivo12. Our cryo-EM analysis thus provides the molecular basis underlying m6A-mediated immune response. In addition, the m6A-insensitive A3R mutants generated in this study paves a way to functionally discriminate the impact of adenosine-dependent and m6A-dependent A3R signaling in cells and in vivo.
In addition to m6A, we discovered that i6A is an A3R ligand. i6A is exclusively found in tRNA including cytosolic tRNASec and tRNASer, mitochondrial tRNASer(UCN), tRNATrp, tRNATyr, tRNAPhe. It is most likely that the extracellular i6A is also derived from the breakdown of these tRNAs, but the catabolism pathway needs to be elucidated in the future study. In addition, the pathophysiological role of i6A-mediated A3R activation and the functional difference with m6A also need to be clarified. Curiously, in the field of plants, i6A and its derivatives can serve as growth hormones through interaction with three cytokinin receptors, which are known as Arabidopsis Histidine Kinase (AHK) 2/3/461. AHK2/3/4 are highly conserved in plants but not in animals. Furthermore, there is no structural similarity between A3R and AHKs, suggesting that i6A might acquire a distinct function during the evolution. Based on our structural analysis and mutagenesis study, we discovered some mutations in the hydrophobic pockets that can greatly suppress adenosine and m6A activity but in turn enhanced i6A-mediated A3R activation. These mutant A3Rs will facilitate the elucidation of the pathological function of the i6A-mediated A3R pathway in future studies.
Previous SAR studies showed that the additional hydrophobic modifications at the N6 position significantly enhanced the A3R selectivity62,63,64. Along with namodenoson, agents such as piclidenoson, MRS5980, MRS5698, and FM101 selectively activate A3R11,65,66 (Supplementary Fig. 6a). All of these agonists have a hydrophobic modification at the N6 position of the adenosine moiety, suggesting that they achieve their A3R selectivity through interactions similar to those observed between namodenoson and A3R. However, some A3R agonists have a larger hydrophobic modification at the C2 position. Compared to namodenoson, these ligands may form more extensive interactions with the cavity formed between TM2 and TM7.
While our manuscript was being reviewed, Cai et al. reported the structures of human A3R bound to piclidenoson and namodenoson67. Comparing these structures with our structure, the orientations of the 3-iodobenzyl group differed in all structures (Supplementary Fig. 6b–d). While their piclidenoson-bound model exhibits the interactions between the 3-iodobenzyl group and the hydrophobic pocket, their namodenoson-bound structure shows a totally different orientation of the 3-iodobenzyl group, which only interacts with V16945.53 and L2647.35. Cai et al. suggested that ECL3 plays a role in the selectivity of A3R. The high selectivity and affinity exhibited by namodenoson and piclidenoson may be due to their ability to interact with multiple regions contributing to the selectivity, including the hydrophobic pocket and ECL3.
In this study, we identified the unique hydrophobic pocket of A3R, which is not involved in ligand-receptor interactions in the other adenosine receptors. This structural insight will greatly facilitate the rational design of selective drugs targeting A3R as well as the other adenosine receptors.
Methods
Screening of A3R-selective nucleosides by TGFα-shedding assay
To measure the activation of the adenosine receptor subtypes (A1R, A2AR, A2BR, A3R), a transforming growth factor-α (TGFα) shedding assay was performed31. Briefly, HEK293A cells were seeded in 6-cm culture dishes. Cells were transfected using a polyethylenimine (PEI) agent (10 μl of 1 mg/ml per dish hereafter; Polysciences) with pCAG plasmids encoding human A1R, A2AR, A2BR, A3R, or mock (400 ng per dish hereafter), together with the plasmids encoding alkaline phosphatase (AP)-tagged TGFα (AP-TGFα; 1 µg) and chimeric Gα subunit proteins (Gαq/o for A1R, Gαq/s for A2AR and A2BR and Gαq/i3 for A3R; 200 ng). These plasmids were gifts from A. Inoue, Tohoku University. After 24 h culture, the transfected cells were harvested using trypsin-EDTA, neutralized with FBS-containing DMEM and collected by centrifugation. Cells were resuspended in Hank’s Balanced Salt Solution (HBSS, GIBCO) containing 5 mM HEPES (pH 7.4) and seeded in a 96-well plate. After 30 min culture, each nucleoside was adjusted with 0.01% bovine serum albumin (BSA)-containing HEPES-HBSS to a concentration comparable to the EC50 of unmodified adenosine for each receptor12 and then added to the cells (final concentrations of 100 nM for A1R, 1 µM for A2AR, 5 µM for A2BR and 100 nM for A3R). After a 1 h incubation, the conditioned media were transferred to an empty 96-well plate. The reaction solution (a mixture of 10 mM p-nitrophenylphosphate (p-NPP), 120 mM Tris-HCl (pH 9.5), 40 mM NaCl, and 10 mM MgCl2) was added to plates containing cells and conditioned media. The absorbance at a wavelength of 405 nm was measured using a microplate reader (Molecular Devices) before and after a 1 h incubation of the plates at room temperature. AP-TGFα release was calculated as follows: (i) relative percentage AP activity in conditioned medium = ΔOD405 CM / (ΔOD405 CM + ΔOD405 Cell), where ΔOD405 CM and ΔOD405 Cell denote changes in OD405 in the conditioned medium and on the cell surface, respectively, before and after a 1 h incubation in the presence of para-nitrophenylphosphate (p-NPP), a substrate for AP, and (ii) percentage AP-TGFα release = (relative percentage AP activity in the conditioned medium in the ligand-stimulated condition) – (relative percentage AP activity in the conditioned medium in the unstimulated condition with vehicle treatment). Finally, we fit the data to the four-parameter sigmoid model (dose-response curve), from which we obtained values of maximal effect (Emax) and half-maximal effective concentration (EC50). Details of each tested nucleoside are provided in Supplementary Table 5.
Expression and purification of A3R-Gi complex
The gene encoding full-length sheep A3R (Uniprot ID: W5QED6) was constructed with the native signal peptide replaced with the HA-signal peptide and the DYKDDDDK Flag epitope tag. LgBiT was fused to the C-terminus of A3R, followed by a 3C protease site and an eGFP-His8 tag. GGSGGGGSGGSSSGG linkers were inserted on both the N-terminal and C-terminal sides of LgBiT. Human Gi1 was fused to the C-terminus of bovine Gγ2, following the GSAGSAGSA linker. Rat Gβ1 was cloned with a C-terminal HiBiT connected with the 15 amino sequence GGSGGGGSGGSSSGG. The resulting constructs were subcloned into the pEG BacMam vector.
The viruses expressing A3R, Gγ2-Gi1, and Gβ1-HiBiT were prepared using the BacMam system. A liter of HEK293S GnTI (N-acetylglucosaminyl-transferase I–negative) cells at a concentration of 3 × 106 cells/mL was co-infected with the prepared viruses expressed at the ratio of 1:1:1.
The collected cells were resuspended and Dounce-homogenized in 20 mM Tris-HCl, pH 8.0, 100 mM NaCl, 10% glycerol, and 10 μM ligand. After homogenization, 25 mU/mL of apyrase and 10 μM of agonist were added and the lysate was rotated at room temperature for 1 h. Afterwards, the membrane fraction was isolated through ultracentrifugation at 180,000 g for 1 h, and solubilized in 20 mM Tris-HCl, pH 8.0, 150 mM NaCl, 1% n-Dodecyl-β-D-maltoside (DDM), 0.2% Cholesteryl Hemisuccinate (CHS), 10% glycerol, and 10 μM ligand, at 4 °C for 1 hr. The insoluble fraction was removed by ultracentrifugation at 180,000 g for 30 min and the supernatant was then incubated with Anti-DYKDDDDK M1 resin (Sigma-Aldrich) for 1 h. The resin was washed with 20 column volumes of buffer, containing 20 mM Tris-HCl, pH 8.0, 500 mM NaCl, 10% glycerol, 0.1% Lauryl Maltose Neopentyl Glycol (LMNG; Anatrace), 0.01 % CHS, and 1–10 μM ligand. The complex was eluted with buffer, containing 20 mM Tris-HCl, pH 8.0, 150 mM NaCl, 10% glycerol, 0.1% LMNG, 0.01% CHS, 1–10 μM ligand, and 0.15 mg/ml Flag peptide. The eluate was incubated with 0.5 mg of HRV-3C protease (prepared in-house) and 0.5 mg of scFv16 (prepared as described previously51) at 4 °C overnight. The complex was concentrated and separated by size exclusion chromatography on a Superose 6 Increase 10/300 column, in buffer containing 20 mM Tris-HCl, pH 8.0, 150 mM NaCl, 0.01% LMNG, 0.001% CHS, and 1–10 μM agonist. Peak fractions were concentrated for the cryo-EM grid preparation.
Cryo-EM data acquisition
A 3 μL portion of the purified complex was deposited onto a Quantifoil holey carbon grid (R1.2/1.3, Au, 300 mesh), which had been glow-discharged just before use. This grid was then rapidly frozen in liquid ethane using a Vitrobot Mark IV (FEI). Cryo-EM data acquisition was performed with a Titan Krios G3i microscope (300 kV, Thermo Fisher Scientific), outfitted with a BioQuantum K3 imaging filter (Gatan) and a K3 direct electron detector (Gatan). Movies, each with a calibrated pixel size of 0.83 Å per pixel and a defocus range between −0.8 and −1.6 μm, were acquired utilizing Thermo Fisher Scientific’s EPU software for single-particle data collection. Movies were recorded over 2.3 s and divided into 48 frames. The numbers and total exposures of movies are provided in Table 1.
The movies, captured in super-resolution mode, were processed by binning them 2×, followed by dose fractionation and correction for beam-induced motion using RELION 4.0 or cryoSPARC v4.068,69. The contrast transfer function (CTF) parameters were estimated using patch CTF estimation in cryoSPARC.
Although the workflow for each complex differs in its details, the outlines are similar. Thus, the workflow for the NECA-bound complex is described here. The workflows for all complexes are summarized in Supplementary Fig. 2a–e.
A subset of particles was initially identified using the Blob picker from a selection of micrographs, and then subjected to multiple rounds of 2D classification in cryoSPARC. Selected particles were used for training the Topaz model70. For the full dataset, 5,682,938 particles were picked and extracted. Subsequent rounds of hetero-refinement were performed to discard poor-quality particle classes. The refined particles were further refined through 3D classification without alignment in RELION. This process yielded 210,192 high-quality particles in the optimal class, which were reconstructed using non-uniform refinement in cryoSPARC to achieve an overall resolution of 2.86 Å, based on the gold standard Fourier Shell Correlation (FSC = 0.143) criteria. Afterwards, the 3D model was refined with masks on the receptor and the G-protein trimer. The locally refined maps were integrated using phenix.combine_focused_maps for model construction71.
Model building and refinement
The AlphaFold2-predicted sheep A3R model and the cryo-EM structure of the MT1-Gi1 complex (PDB ID: 7DB6) were used as starting templates for modeling the A3R and Gi1 components of the NECA-bound structure, respectively51,72. Initially, these models were positioned into the density map using jiggle fit in COOT, and subsequently, the atomic models were fine-tuned using COOT and refined with phenix.real_space_refine (v1.19), incorporating secondary structure restraints from phenix.secondary_structure_restraints. Restraints for ligands were generated with Grade2 (Smart, O.S., Sharff A., Holstein, J., Womack, T.O., Flensburg, C., Keller, P., Paciorek, W., Vonrhein, C. and Bricogne G. (2021) Grade2 version 1.5.0. Cambridge, United Kingdom: Global Phasing Ltd.). The NECA-bound model was used as the initial model for the other complexes, and the following procedures for the other models were the same as those for the NECA-bound model.
Mutagenesis study to investigate the specificity of the modified adenosines
A total of 34 A3R mutants for V16945.53, I2535.35, M1746.58, and L2647.35 were generated by introducing single point mutations into pCAG plasmids encoding human A3R, using a KOD-Plus-Mutagenesis kit (TOYOBO), and confirmed by Sanger sequencing (Azenta). These plasmids were transfected into cells using a PEI agent with AP-TGFα and Gαq/i3, in the same volume as mentioned above, and the TGFα-shedding assay was performed as described above. Unmodified adenosine, m6A, m6,6A, and i6A were 10-fold serially diluted with 0.01% BSA-containing HEPES-HBSS to give final concentrations in the range of 100 µM to 1 nM. AP-TGFα release percentages were fitted to a four-parameter sigmoidal concentration–response curve, using the Prism 9 software (GraphPad Prism), and the EC50 and Emax values were obtained. Receptor activation is scored using the relative intrinsic activity (RAi), which is defined as the relative Emax/ EC50 value31,73.
Flow cytometry analysis
The cell surface expression of A3R was measured as described previously with modifications54. HEK293A cells were seeded in a 6-cm culture plate at a concentration of 2 × 105 cells per mL (4 mL per dish) and cultured for 1 day before transfection. The cells were transfected with 40 µg of N-terminally 3xFLAG-tagged A3R construct using the Lipofectamine 3000 transfection reagent (ThermoFisher Scientific) and cultured for 24 h. After the cells were rinsed once with D-PBS, 5 mM HEPES (pH 7.4)-containing HBSS was added, and the cells were collected using a cell scraper (IWAKI). After washing with D-PBS, the cell suspension was transferred to a 1.5 mL tube and blocked with FCM buffer (2% Normal Goat Serum, 2 mM EDTA, and 0.1% NaN3 in D-PBS) for 30 min on ice. Following centrifugation, the cell suspension was fluorescently labeled with an anti-Flag epitope (DYKDDDDK) tag monoclonal antibody (Sigma Aldrich) diluted in FCM buffer, for 15 min at room temperature. After washing with D-PBS, the cell suspension was labeled with a goat anti-mouse IgG (H + L) secondary antibody conjugated with Alexa Fluor 488 (ThermoFisher Scientific) diluted in FCM buffer, for 15 min at room temperature. After washing with D-PBS, the cells were resuspended in 500 μL of 2 mM EDTA-containing D-PBS and filtered through a 40-μm filter. The fluorescent intensity of single cells was quantified using a CytoFLEX flow cytometer (Beckman Coulter) equipped with a 488 nm laser, and the geometric mean fluorescent intensity from 10,000 cells per sample was used for analysis. Gating strategies are provided in Supplementary Fig. 7.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Atomic coordinates for the NECA-A3R-Gi, adenosine-A3R-Gi, m6A-A3R-Gi, i6A-A3R-Gi, and namodenoson-A3R-Gi complexes have been deposited in the Protein Data Bank, under accession code 8YH0, 8YH2, 8YH3, 8YH5 and 8YH6, respectively. The associated electron microscopy data have been deposited in the Electron Microscopy Database under accession codes EMD-39278, EMD-39279, EMD-39280, EMD-39281 and EMD-39282, respectively. Source data are provided as a Source Data file. All other data are available from the corresponding author upon request. Source data are provided with this paper.
References
IJzerman, A. P., Jacobson, K. A., Müller, C. E., Cronstein, B. N. & Cunha, R. A. International union of basic and clinical pharmacology. CXII: adenosine receptors: a further update. Pharmacol. Rev. 74, 340–372 (2022).
Effendi, W. I., Nagano, T., Kobayashi, K. & Nishimura, Y. Focusing on adenosine receptors as a potential targeted therapy in human diseases. Cells 9, 785 (2020).
Borea, P. A., Gessi, S., Merighi, S., Vincenzi, F. & Varani, K. Pharmacology of adenosine receptors: the state of the art. Physiol. Rev. 98, 1591–1625 (2018).
Zhou, Q. Y. et al. Molecular cloning and characterization of an adenosine receptor: the A3 adenosine receptor. Proc. Natl. Acad. Sci. USA 89, 7432–7436 (1992).
Gao, Z.-G., Auchampach, J. A. & Jacobson, K. A. Species dependence of A3 adenosine receptor pharmacology and function. Purinergic Signal. 19, 523–550 (2023).
Walker, B. A. et al. Adenosine A3 receptor expression and function in eosinophils. Am. J. Respir. Cell Mol. Biol. 16, 531–537 (1997).
Leung, C. T. et al. The role of activated adenosine receptors in degranulation of human LAD2 mast cells. Purinergic Signal. 10, 465–475 (2014).
Bouma, M. G. et al. Adenosine inhibits neutrophil degranulation in activated human whole blood: involvement of adenosine A2 and A3 receptors. J. Immunol. 158, 5400–5408 (1997).
Gessi, S. et al. The A3 adenosine receptor: an enigmatic player in cell biology. Pharmacol. Ther. 117, 123–140 (2008).
Madi, L. et al. The A3 adenosine receptor is highly expressed in tumor versus normal cells: potential target for tumor growth inhibition. Clin. Cancer Res. 10, 4472–4479 (2004).
Fishman, P. Drugs targeting the A3 adenosine receptor: human clinical study data. Molecules 27, 3680 (2022).
Ogawa, A. et al. N6-methyladenosine (m6A) is an endogenous A3 adenosine receptor ligand. Mol. Cell 81, 659–674.e7 (2021).
Cappannini, A. et al. MODOMICS: a database of RNA modifications and related information. 2023 update. Nucleic Acids Res. 52, D239–D244 (2024).
Chujo, T. & Tomizawa, K. Human transfer RNA modopathies: diseases caused by aberrations in transfer RNA modifications. FEBS J. 288, 7096–7122 (2021).
Wang, X. et al. N6-methyladenosine-dependent regulation of messenger RNA stability. Nature 505, 117–120 (2014).
Roundtree, I. A. et al. YTHDC1 mediates nuclear export of N6-methyladenosine methylated mRNAs. eLife 6, e31311 (2017).
Wang, X. et al. N(6)-methyladenosine modulates messenger RNA translation efficiency. Cell 161, 1388–1399 (2015).
Murphy, F. V., Ramakrishnan, V., Malkiewicz, A. & Agris, P. F. The role of modifications in codon discrimination by tRNA(Lys)UUU. Nat. Struct. Mol. Biol. 11, 1186–1191 (2004).
Aguilo, F. et al. Coordination of m(6)A mRNA methylation and gene transcription by ZFP217 regulates pluripotency and reprogramming. Cell Stem Cell 17, 689–704 (2015).
Lan, Q. et al. The critical role of RNA m6A methylation in cancer. Cancer Res. 79, 1285–1292 (2019).
Cheng, R. K. Y. et al. Structures of human A1 and A2A adenosine receptors with xanthines reveal determinants of selectivity. Structure 25, 1275–1285.e4 (2017).
Glukhova, A. et al. Structure of the adenosine A1 receptor reveals the basis for subtype selectivity. Cell 168, 867–877.e13 (2017).
Draper-Joyce, C. J. et al. Positive allosteric mechanisms of adenosine A1 receptor-mediated analgesia. Nature 597, 571–576 (2021).
Draper-Joyce, C. J. et al. Structure of the adenosine-bound human adenosine A1 receptor–Gi complex. Nature 558, 559–563 (2018).
Chen, Y. et al. Cryo-EM structure of the human adenosine A2B receptor–Gs signaling complex. Sci. Adv. 8, eadd3709 (2022).
Cai, H. et al. Structures of adenosine receptor A2BR bound to endogenous and synthetic agonists. Cell Discov 8, 1–4 (2022).
Xu, F. et al. Structure of an agonist-bound human A2A adenosine receptor. Science 332, 322–327 (2011).
Doré, A. S. et al. Structure of the adenosine A2A receptor in complex with ZM241385 and the xanthines XAC and caffeine. Structure 19, 1283–1293 (2011).
Carpenter, B., Nehmé, R., Warne, T., Leslie, A. G. W. & Tate, C. G. Structure of the adenosine A2A receptor bound to an engineered G protein. Nature 536, 104–107 (2016).
Jaakola, V.-P. et al. The 2.6 angstrom crystal structure of a human A2A adenosine receptor bound to an antagonist. Science 322, 1211–1217 (2008).
Inoue, A. et al. TGFα shedding assay: an accurate and versatile method for detecting GPCR activation. Nat. Methods 9, 1021–1029 (2012).
Liu, H. et al. Development of mild chemical catalysis conditions for m1A-to-m6A rearrangement on RNA. ACS Chem. Biol. 17, 1334 (2022).
García-Nafría, J., Lee, Y., Bai, X., Carpenter, B. & Tate, C. G. Cryo-EM structure of the adenosine A2A receptor coupled to an engineered heterotrimeric G protein. eLife 7, e35946 (2018).
Lebon, G. et al. Agonist-bound adenosine A2A receptor structures reveal common features of GPCR activation. Nature 474, 521–525 (2011).
Hattori, M., Hibbs, R. E. & Gouaux, E. A fluorescence-detection size-exclusion chromatography-based thermostability assay for membrane protein precrystallization screening. Structure 20, 1293–1299 (2012).
Robert, X. & Gouet, P. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 42, W320–W324 (2014).
The UniProt Consortium. UniProt: the universal protein knowledgebase in 2023. Nucleic Acids Res. 51, D523–D531 (2023).
Pándy-Szekeres, G. et al. GPCRdb in 2018: adding GPCR structure models and ligands. Nucleic Acids Res. 46, D440–D446 (2018).
Sano, F. K., Akasaka, H., Shihoya, W. & Nureki, O. Cryo-EM structure of the endothelin-1-ETB-Gi complex. eLife 12, e85821 (2023).
Duan, J. et al. Cryo-EM structure of an activated VIP1 receptor-G protein complex revealed by a NanoBiT tethering strategy. Nat. Commun. 11, 4121 (2020).
Kim, K. et al. Structure of a hallucinogen-activated Gq-coupled 5-HT2A serotonin receptor. Cell 182, 1574–1588.e19 (2020).
Nureki, I. et al. Cryo-EM structures of the β3 adrenergic receptor bound to solabegron and isoproterenol. Biochem. Biophys. Res. Commun. 611, 158–164 (2022).
Ballesteros, J. A. & Weinstein, H. [19] Integrated methods for the construction of three-dimensional models and computational probing of structure-function relations in G protein-coupled receptors. Methods Neurosci. 25, 366–428 (1995).
Yuan, Y. et al. Structures of signaling complexes of lipid receptors S1PR1 and S1PR5 reveal mechanisms of activation and drug recognition. Cell Res. 31, 1263–1274 (2021).
Zhuang, Y. et al. Molecular recognition of morphine and fentanyl by the human μ-opioid receptor. Cell 185, 4361–4375.e19 (2022).
Hua, T. et al. Activation and signaling mechanism revealed by cannabinoid receptor-Gi complex structures. Cell 180, 655–665.e18 (2020).
Kato, H. E. et al. Conformational transitions of a neurotensin receptor 1–Gi1 complex. Nature 572, 80–85 (2019).
Oshima, H. S. et al. Optimizing cryo-EM structural analysis of Gi-coupling receptors via engineered Gt and Nb35 application. Biochem. Biophys. Res. Commun. 693, 149361 (2024).
Akasaka, H. et al. Structure of the active Gi-coupled human lysophosphatidic acid receptor 1 complexed with a potent agonist. Nat. Commun. 13, 5417 (2022).
Izume, T. et al. Structural basis for lysophosphatidylserine recognition by GPR34. Nat. Commun. 15, 902 (2024).
Okamoto, H. H. et al. Cryo-EM structure of the human MT1–Gi signaling complex. Nat. Struct. Mol. Biol. 28, 694–701 (2021).
Flock, T. et al. Universal allosteric mechanism for Gα activation by GPCRs. Nature 524, 173–179 (2015).
Rasmussen, S. G. F. et al. Crystal structure of the β2 adrenergic receptor–Gs protein complex. Nature 477, 549–555 (2011).
Iwama, A. et al. Structure and dynamics of the pyroglutamylated RF-amide peptide QRFP receptor GPR103. Nat. Commun. 15, 4769 (2024).
Nagiri, C. et al. Cryo-EM structure of the β3-adrenergic receptor reveals the molecular basis of subtype selectivity. Mol. Cell 81, 3205–3215.e5 (2021).
Zhang, K., Wu, H., Hoppe, N., Manglik, A. & Cheng, Y. Fusion protein strategies for cryo-EM study of G protein-coupled receptors. Nat. Commun. 13, 4366 (2022).
Zhou, Q. et al. Common activation mechanism of class A GPCRs. eLife 8, e50279 (2019).
Guo, D. et al. Molecular basis of ligand dissociation from the adenosine A2A receptor. Mol. Pharmacol. 89, 485–491 (2016).
Gallo-Rodriguez, C. et al. Structure-activity relationships of N6-benzyladenosine-5′-uronamides as A3-selective adenosine agonists. J. Med. Chem. 37, 636–646 (1994).
Kim, H. O. et al. 2-Substitution of N6-benzyladenosine-5′-uronamides enhances selectivity for A3 adenosine receptors. J. Med. Chem. 37, 3614–3621 (1994).
Sakakibara, H. Cytokinins: activity, biosynthesis, and translocation. Annu. Rev. Plant Biol. 57, 431–449 (2006).
Ohno, M. et al. Modulation of adenosine receptor affinity and intrinsic efficacy in adenine nucleosides substituted at the 2-position. Bioorg. Med. Chem. 12, 2995–3007 (2004).
Tchilibon, S. et al. (N)-methanocarba 2,N6-disubstituted adenine nucleosides as highly potent and selective A3 adenosine receptor agonists. J. Med. Chem. 48, 1745–1758 (2005).
Ciancetta, A. & Jacobson, K. A. Structural probing and molecular modeling of the A3 adenosine receptor: a focus on agonist binding. Molecules 22, 449 (2017).
Jacobson, K. A., Tosh, D. K., Jain, S. & Gao, Z.-G. Historical and current adenosine receptor agonists in preclinical and clinical development. Front. Cell. Neurosci. 13, 124 (2019).
Jeong, L. S. et al. Structure-activity relationships of truncated D- and l-4′-thioadenosine derivatives as species-independent A3 adenosine receptor antagonists. J. Med. Chem. 51, 6609–6613 (2008).
Cai, H. et al. Cryo-EM structures of adenosine receptor A3AR bound to selective agonists. Nat. Commun. 15, 3252 (2024).
Zivanov, J. et al. New tools for automated high-resolution cryo-EM structure determination in RELION-3. eLife 7, e42166 (2018).
Punjani, A., Rubinstein, J. L., Fleet, D. J. & Brubaker, M. A. cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296 (2017).
Bepler, T. et al. Positive-unlabeled convolutional neural networks for particle picking in cryo-electron micrographs. Nat. Methods 16, 1153–1160 (2019).
Liebschner, D. et al. Macromolecular structure determination using X-rays, neutrons and electrons: recent developments in Phenix. Acta Crystallogr. Sect. D Struct. Biol. 75, 861–877 (2019).
Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021).
Ehlert, F. J., Griffin, M. T., Sawyer, G. W. & Bailon, R. A simple method for estimation of agonist activity at receptor subtypes: comparison of native and cloned M3 muscarinic receptors in guinea pig ileum and transfected cells. J. Pharmacol. Exp. Ther. 289, 981–992 (1999).
Acknowledgements
We thank K. Ogomori and C. Harada for technical assistance. This work was funded by the JSPS KAKENHI (Grant Nos. 21H05037 to O.N., 22K19371 and 22H02751 to W.S., 23KJ0491 to F.K.S., 24KJ0906 to H.A., JP21H02659 and JP21H05265 to F.-Y.W.), the Lotte Foundation (W.S.), the Takeda Science Foundation (to W.S. and F.-Y.W.), the FOREST program from JST (Grant No. JPMJFR220K to A.O.), the ERATO program from JST (Grant No. JPMJER2002 to F.-Y.W.), the Japan Agency for Medical Research and Development (AMED) (Grant No. JP233fa627001 to O.N.) and the Platform Project for Supporting Drug Discovery and Life Science Research (Basis for Supporting Innovative Drug Discovery and Life Science Research (BINDS)) from AMED, grant numbers JP23ama121002 (support number 3272, O.N.) and JP23ama121012 (supporting number 4837, F.-Y.W.).
Author information
Authors and Affiliations
Contributions
H.S.O. performed the complex purification, the cryo-EM data collection, the single-particle analysis, model building and refinement, and designed the mutants with assistance from F.K.S., H.A., H.H.O., A.I., C.N., and W.S. A.O. performed the screening and the cell-based assays, with assistance from T.K. and F.-Y.W. H.S.O., A.O., F.-Y.W, W.S., and O.N. wrote the manuscript with inputs from all authors. F.-Y.W., W.S., and O.N. supervised the research.
Corresponding authors
Ethics declarations
Competing interests
O.N. is a co-founder and an external director of Curreio, Inc. The remaining authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Kai Cai and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Oshima, H.S., Ogawa, A., Sano, F.K. et al. Structural insights into the agonist selectivity of the adenosine A3 receptor. Nat Commun 15, 9294 (2024). https://doi.org/10.1038/s41467-024-53473-1
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-024-53473-1
This article is cited by
-
Research progress of CD73-adenosine signaling regulating hepatocellular carcinoma through tumor microenvironment
Journal of Experimental & Clinical Cancer Research (2025)
-
Molecular basis of ligand binding and receptor activation at the human A3 adenosine receptor
Nature Communications (2025)
