
Abstract
The hypothalamus plays an important role in aging, but it remains unclear regarding the underlying epigenetics and whether this hypothalamic basis can help address aging-related diseases. Here, by comparing mouse hypothalamus with two other limbic system components, we show that the hypothalamus is characterized by distinctively high-level DNA methylation during young age and by the distinct dynamics of DNA methylation and demethylation when approaching middle age. On the other hand, age-related DNA methylation in these limbic system components commonly and sensitively applies to genes in hypothalamic regulatory pathways, notably oxytocin (OXT) and gonadotropin-releasing hormone (GnRH) pathways. Middle age is associated with transcriptional declines of genes which encode OXT, GnRH and signaling components, which similarly occur in an Alzheimer’s disease (AD)-like model. Therapeutically, OXT-GnRH combination is substantially more effective than individual peptides in treating AD-like disorders in male 5×FAD model. In conclusion, the hypothalamus is important for modeling age-related DNA methylation and developing hypothalamic strategies to combat AD.
Similar content being viewed by others

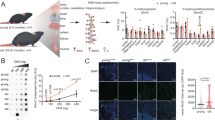

Introduction
Aging-related neurodegenerative diseases such as AD have become major health problems among the elderly, but clinical solutions are still missing partly due to the incomplete knowledge of this disease. Interestingly, recent research has suggested that the functioning of neurogenic brain regions in early life could have long-term effects on aging-related neuronal degeneration and loss1,2,3,4. As is known, there are three major types of neurogenic brain regions, including the hippocampal dentate gyrus5,6, the subventricular zone7,8,9,10, and the mediobasal hypothalamus11,12,13, which are responsible for adult neurogenesis related to the limbic system functions of the hippocampus, olfactory bulb (OB), and hypothalamus, respectively. Recently, our research has led to an understanding that the hypothalamus plays a regulatory role in systemic aging and systemic physiology11,12,13,14,15, while the underlying basis involves the actions from hypothalamic neuropeptides such as GnRH and OXT15,16,17,18. However, to date there is still a lack of research comparing the hypothalamus and other limbic system components in the context of addressing aging or aging-related diseases. To fill this gap, we designed this research project to comparatively study the hypothalamus versus the hippocampus and OB in the context of aging. For our approach, we resorted to DNA methylation epigenetic analysis, given the close relationships between aging and DNA methylation established in research19,20,21,22,23,24,25,26,27,28,29,30,31,32,33. We were inspired to employ this approach, also because we aimed to explore if age-related DNA methylation in the hypothalamus could be informative for developing hypothalamic peptide and/or endocrine approaches to address aging-related diseases such as AD. Hence, we carried out this project and obtained multifaceted data supporting the hypothesis that the hypothalamus is important for modeling age-related DNA methylation epigenetics and developing a related hypothalamic strategy based on the OXT-GnRH combinational therapy to combat AD.
Results
Age-related DNA methylation in the hypothalamus vs. hippocampus and OB
To profile age-related hypothalamic DNA methylation in standard C57BL/6 mice, we studied the hypothalamus which was compared to two other limbic system components: the hippocampus and OB. We employed bisulfite oligonucleotide-captured sequencing (BOCS), a technique established for profiling CG-rich genomic regions. This approach covers all annotated promoters, CG islands, CG shore regions (±2 kb from each CG island), and CG shelf regions (±2 kb from each shore and ±4 kb from each CG island)34,35. The hypothalamus, hippocampus, and OB were isolated from standard male C57BL/6 mice at the age of 2 months vs. 12 months, representing young and middle age, respectively. Studying the transition from young age to middle age can help identify early changes that could be more causally significant for subsequent aging-related diseases. All data from this BOCS method were confirmed to meet the standard through quality check (Suppl. Figure 1 ̶ 3). Then, we computed the average methylation levels of CG and CH sites (H stands for A, T, or C) for each tissue. As shown in Fig. 1A and B, it immediately gained our attention that compared to the hippocampus and OB, DNA methylation levels were distinctly highest in the hypothalamus of young mice, but the difference tended to be less distinct in middle-aged mice. These overall relationships were also seen in many individual chromosomes, as demonstrated in chromosome-wise analysis of these brain regions of young mice (Fig. 1C, D) and middle-aged mice (Fig. 1E, F). Taken together, apparently due to different dynamics of CG methylation and CH demethylation, the pattern of DNA methylation in the hypothalamus distinctly differs from the patterns in the other two brain regions at young age, but this difference declines when approaching middle age.

DNA samples were generated from the hypothalamus (HT), hippocampus (HC), and OB of 2-month-old versus 12-month-old male C57BL/6 and were profiled for cytosine methylation of CG-enriched genomic regions via BOCS and analyzed for average levels of CG and CH sites methylation. A, B Average percentage levels of CG site methylation (A) and CH site methylation (B). C–F Average percentage levels of CG site methylation (C, E) and CH site methylation (D, F) in individual chromosomes. The Y chromosome was not included due to its short size. Data are presented as mean values ± SEM. Statistics: Exact p-values are labeled as text for comparisons between two groups (A, B) and across chromosomes (C–F). Significance is indicated by *p < 0.05 and **p < 0.01, with comparisons conducted at the individual chromosome level among three groups (C–F). n = 3 mice per group (A–F), two-way ANOVA with tissue and age as factors, and post-hoc Tukey test (A, B), and Kruskal-Wallis and post-hoc Dunn test (C–F). Source data are provided as a Source Data file.
Region-related DNA differential methylation at young age and the changes by middle age
We then computed age-related differentially methylated cytosines (DMCs) in each brain region between young and middle age but found only a small number of sites with statistical significance. We also examined how differential methylation between brain regions could change during the transition to middle age. We found that DMCs between the hypothalamus and the other two brain regions were more heavily attributed to hypermethylation than to hypomethylation; in contrast, DMCs between the hippocampus and OB were similarly attributed to hypermethylation and hypomethylation. These findings are summarized in the Volcano plot (Fig. 2) and detailed with chromosome-wide analysis in the Manhattan plot (Suppl. Figure 4, Suppl. Dataset 1). By comparing the two age groups, we found that DMCs between the hypothalamus and either of the other two brain regions were more than 60% lost by middle age; in contrast, DMCs between the hippocampus and OB in young mice were only about 40% lost by middle age. Certainly, cellular heterogeneity among different brain regions can importantly account for DMCs between regions, but we expect that composition of major cell types in a given tissue remains stable by middle age. Thus, we adopted a deconvolution method to test this point, as this approach has been established in the literature to computationally analyze cell compositions in tissue samples36,37,38,39,40. Our deconvolution analysis led to the projection of 7 major cell types in each brain region (Suppl. Fig. 5); the potential identities of these major cell types are only suggested, based on previous single-cell analysis of these brain regions in the literature41,42,43. Our analysis supports the point that middle age does not significantly change the composition of these major cell types. Hence, while we do not exclude a contribution from smaller populations or subtypes, major cell types do not seem to be a driving factor for changes in region-related differential methylation among these brain tissues as animals approach middle age.

Differentially methylated cytosines (DMCs) were analyzed pairwise among the hypothalamus (HT), hippocampus (HC), and olfactory bulb (OB) of 2- and 12-month-old mice, based on q < 0.05 and methylation changes of at least 25%. A–F DMCs between HT and HC (A, B), HT and OB (C, D), and HC and OB (E, F) using Volcano plots (A, C, E) and Venn diagrams (B, D, F). In the Volcano plots, hypermethylation and hypomethylation are shown as green and red dots, respectively. Venn diagrams show the numbers of DMCs, with green arrows pointing upward to indicate hypermethylation and red arrows pointing downward to indicate hypomethylation. The bar graphs display the percentage of tissue-related DMCs that were lost, retained, or gained by middle age, calculated over the total DMCs observed in young mice (B, D, F). All analyses were performed on n = 3 mice per group, statistically via logistic regression using the MethylKit package. Statistical tests were two-sided, with p-values adjusted to q-values using the SLIM method to control for multiple comparisons, with all data (B, D, F) meeting q < 0.05. DMCs are listed as Supplementary Dataset 1. Source data are provided as a Source Data file.
Age-related differential methylated regions in the hypothalamus, hippocampus and OB
Apart from DMC analysis, we also carried out differential methylation regions (DMRs) analysis, which span lengths of DNA sequences and thus can reveal DNA methylation information more globally than locally. We analyzed age-dependent DMRs according to DNA length ranging from 300 to 1000 base pairs (bps). While the analysis yielded variable numbers of DMRs due to changes in DNA lengths, three brain regions consistently exhibited similar patterns, as reflected by an analysis with DNA length of 500 bps (Fig. 3A, Suppl. Dataset 2). Remarkably, age-dependent DMRs in the hypothalamus exhibited a more balanced distribution between hypermethylation and hypomethylation, in sharp contrast to the unbalanced patterns in the other two brain regions, where hypermethylation was stronger than hypomethylation (Fig. 3A). Also, as shown in Fig. 3B, chromosome-wide analysis further confirmed a balanced number of hypothalamic DMRs between hypermethylation and hypomethylation; in contrast, DMRs in the hippocampus and OB were attributed to more hypermethylation than hypomethylation. Subsequently, we explored the genes involved in age-related DMRs in these brain regions. We identified a pool of genes impacted across the hypothalamus, hippocampus, and OB, constituting approximately 12%, 9%, and 7% of the total affected genes in these brain regions, respectively. Overall, we identified 173 genes mapped onto DMRs due to aging across all three brain regions (Fig. 3C). Among these 173 genes, 94, 60, and 34 genes showed more hypomethylation than hypermethylation in the hypothalamus, hippocampus, and OB, respectively, indicating a bias towards hypomethylation in the hypothalamus compared to the other two brain regions. Subsequently, we conducted pathway analysis based on these 173 shared genes. As demonstrated in Fig. 3D, many of these genes were associated with hypothalamic functions, such as circadian rhythm, reproductive function, endocrine gland hormones, feeding, metabolism, cardiovascular function, and immunity, suggesting that DNA methylation changes related to hypothalamic function could be shared with other brain regions.

DMRs in the hypothalamus (HT), hippocampus (HC), and OB between 2 and 12 months of age were calculated per DNA length of 500 bps, with at least 10% of methylation difference with q < 0.05 (false discovery rate, FDR). DMRs were based on CG sites in the sequences. A, B The total number of DMRs in each brain tissue due to hypermethylation and hypomethylation (A) and percentage in individual chromosomes (B). C Venn Diagram of the numbers of the underlying genes per age-related DMRs that are either common or unique among HT, HC, and OB. D A total of 173 common genes per age-related DMRs among HT, HC, and OB were processed with KEGG pathway analysis and grouped according to functions. The left Y axis and bar graphs indicate the numbers of genes, and the right Y axis and red dots represent −log10(p-values). All analyses were based on n = 3 mice per group, statistically via logistic regression using MethylKit package (two-sided test) with p-values adjusted to q-values using the SLIM method to control for multiple comparisons, with all data meeting FDR values of less than 0.05 (A–C) and Fisher’s Exact test (two-sided) (D). DMRs and genes for Fig. 3A, C are listed in Supplementary Dataset 2. Source data are provided as a Source Data file.
Pathway analysis per age-related DMRs in the hypothalamus, OB, and hippocampus
We performed further analysis focusing on individual brain regions to explore the specific molecular pathways affected by age-related changes in DNA methylation. Our investigation involved subsequent mapping of the genes associated with DMRs to a manually curated database of mouse cell signaling and metabolic pathways. Our findings, illustrated in Fig. 4A–C and Suppl. Dataset 3, highlighted the pathways influenced by age-related differential DNA methylation, characterized by the number of genes and their collective significance. Specifically, the pathways identified for the hypothalamus included many components that regulate circadian rhythms and the hypothalamic reproductive peptides OXT and GnRH network (Fig. 4A). Of interest, the pathways identified for the hippocampus and OB were also found to be significantly related to hypothalamic regulatory functions, for example, the circadian entrainment pathway, reproductive OXT pathway, cortisol synthesis and secretion pathway, Cushing syndrome pathway, and aldosterone synthesis and secretion pathway for the hippocampus (Fig. 4B) and the circadian entrainment pathway, OXT signaling pathway, and GnRH secretion pathway for OB (Fig. 4C). Most notably, based on two different computational approaches in Figs. 3 and 4, reproductive neuropeptide OXT and GnRH pathways repeatedly emerged not only in the hypothalamus but also in the hippocampus and OB. In this context, we analyzed how OXT signaling could be related to GnRH signaling using a protein-protein interaction program, showing a good number of co-components in OXT and GnRH signaling pathways (Suppl. Figure 6). Utilizing BOCS metadata, we investigated how aging affects the methylation statuses of genes encoding OXT signaling and GnRH signaling components. As laid out in Fig. 4D, by middle age, many components in OXT signaling and GnRH signaling pathways show age-dependent changes in DNA methylation status in the hypothalamus, hippocampus, and OB. Interestingly, in each brain region, a specific set of OXT and GnRH signaling components was differentially methylated when comparing young and middle-age samples. Hence, aging is associated with diverse DNA methylation changes in the genes encoding molecular components of OXT and GnRH signaling, with these variations being brain region-specific.

A–C Pathway analysis per age-related DMRs (500 bps per length, CG-based DMR analysis) in the hypothalamus (HT) (A), hippocampus (HC) (B), and OB (C), the list was based on p-values in logarithm in the x-axis. The numbers of involved genes in each pathway were indicated with color gradients. The size of each circle indicated the combined scores, computed by taking the log of the p-value from the Fisher’s exact test and multiplying it by the z-score of deviation from the expected rank. Due to space limit, representative pathways are listed along y-axis, while all pathways and genes for Fig. 4A–C are provided as Supplementary Dataset 3. D Components in OXT signaling pathway and GnRH signaling pathway according to age-related DMRs (500 bps per length, CG-based DMR analysis) in the HT, HC, and OB. The size of each circle indicated the levels of hypermethylation or hypomethylation. All analyses were based on n = 3 mice per group, and statistics were based on Fisher’s Exact test (two-sided) (A–C) and logistic regression using MethylKit package (two-sided test) with p-values adjusted to q-values using the SLIM method to control for multiple comparisons (D). Source data are provided as a Source Data file.
DNA methylation and transcription of OXT and GnRH genes in aging or AD-like model
Our earlier work separately related GnRH and OXT to systemic aging or physiology15,16,17,18, altogether suggesting that these peptides might have a close relationship in influencing brain health in conditions such as aging, an idea which is validated through the DMR analysis presented above. Hence, we decided to directly examine the methylation statuses of Oxt and Gnrh1 genes between young and middle-aged mice. Since the hypothalamus exclusively produces both peptides during adulthood, we focused on analyzing DNA methylation data from the hypothalamic samples. We found that a list of cytosines in gene body and promoter region of Oxt became hypomethylated, while a smaller list of cytosines became hypermethylated in the hypothalamus by middle age (Fig. 5A, Suppl. Figure 7A). There was also a list of cytosines in Gnrh1 gene body and promoter, which underwent hypomethylation or hypermethylation by middle age (Fig. 5B, Suppl. Figure 7B). Subsequently, we examined the mRNA levels of Oxt and Gnrh1 in the hypothalamus of mice at young age (2 months) compared to middle age (15 months). Through qRT-PCR method, we obtained data showing that mRNA levels of Oxt and Gnrh1 both dramatically dropped in the hypothalamus during the transition from young to middle age (Fig. 5C). Given our interest in aging-related disease, we employed 5xFAD mice, an AD-like model associated with aging, to assess if similar changes might occur in the hypothalamus prior to the onset of AD-like phenotype. Indeed, the transcription levels of Oxt and Gnrh1 were also evidently downregulated in the hypothalamus of 5xFAD mice compared to wildtype controls (Fig. 5D). These results lend support to our prediction that declines in OXT and GnRH pathways are co-involved in AD-like conditions which are associated with aging.

A, B Differentially methylated cytosines (DMCs) were analyzed in BOCS metadata covering Oxt gene (A) and Gnrh1 gene (B) for the hypothalamic samples of mice between 2 months and 12 months of age. The graphs present the DMCs with at least 1% methylation difference, with genomic positions of cytosines labeled on the Y axis. C, D The mRNA levels of Oxt and Gnrh1 measured in hypothalamic tissues from male C57BL/6 mice under middle age (15 months) vs. young age (2 months) (C) and from male 5xFAD mice vs. male littermate wildtype (WT) controls of 8 months of age (D). Statistics: For panels A and B, analyses were performed using logistic regression with the MethylKit package, with q-values < 0.05 indicating significance (two-sided test), and n = 3 mice per group. Data in panels C and D are presented as mean values ± SEM. au arbitrary unit. Statistical significance was assessed using two-tailed unpaired t-test, with p-values reported, and n = 5 mice per group. Source data are provided as a Source Data file.
DNA methylation and transcription of Adcy family members in aging or AD-like model
In addition to Oxt and Gnrh1, we noted that our age-related DMR analysis across the hypothalamus, hippocampus, and OB shown in Fig. 4 contained several adenylate cyclases (Adcy), a family of proteins that co-mediate OXT and GnRH signaling (Suppl. Fig. 6). By modulating cAMP levels, Adcy members link OXT and GnRH stimulation to downstream protein kinases that regulate various events ranging from gene expression and neurotransmitter release to synaptic plasticity. Abnormal Adcy expression is associated with many neurological disorders, including AD and depressive disorders44,45. For instance, loss of Adcy5 results in Parkinson’s disease-like disorders46, and Adcy7 is linked to familial major depression in both mice and humans47,48. Hence, our methylation analysis further narrowed down to the promoter regions of Adcy members, given the important role of promoters in gene expression, focusing on the hippocampus because of its critical role in cognitive regulation. We found that total methylation levels in the promoter regions of several Adcy genes were significantly lower in middle-aged mice compared to young mice, while Adcy genes often underwent hypomethylation in the proximal promoter regions by middle age (Fig. 6A). In this context, we examined hippocampal mRNA levels and found that middle age was associated with significant downregulation across many Adcy genes (Fig. 6B). Also, we examined the hippocampus of 5xFAD model and observed that hippocampal mRNA levels of several Adcy members were lower in this AD-like model compared to littermate controls (Fig. 6C). Taken together, transcription of Oxt and Gnrh1 in the hypothalamus, as well as transcription of Adcy members in the hippocampus, are commonly defective in both the aging and the 5xFAD model.

A Diagrams depict the promoter regions and subregions of Adcy genes in the hippocampus, showing age-related methylation differences. Comparisons are based on average methylation levels in each promoter region or subregion between middle-aged (12 months) and young (2 months) mice. The promoters are categorized into CG island, distal subregion, and proximal subregion. Red arrows indicate decreased methylation, while green arrows indicate increased methylation in 12-month-old mice compared to 2-month-old mice. B, C The mRNA levels of Adcy genes in hippocampal tissues were measured from male C57BL/6 mice at middle age (15 months) versus young age (2 months) (B), and from male 5xFAD mice versus male littermate wildtype (WT) controls at 8 months of age (C). Data are presented as mean ± SEM. au arbitrary unit. Statistical significance was assessed using a two-tailed unpaired t-test. p-values are reported in the graphs, with n = 3 mice per group for panel A and n = 5 mice per group for panels B, C. Source data are provided as a Source Data file.
Therapeutic significance of OXT-GnRH combinational treatment in AD mouse model
While this work began with DNA methylation analysis based on aging model, our analysis led to identification of OXT and GnRH pathways, which we confirmed at the gene transcriptional level to be significantly altered in an AD-like model which is associated with aging. In the literature, a few studies recently suggested an effect of OXT in attenuating amyloid β (Aβ) deposition or cognitive impairment in ddY or APP/PS1 mice or Sprague Dawley rats49,50,51,52,53,54. Since these studies were based on experimental conditions with relatively mild cognitive disorders, the effects of OXT in treating AD still remain to be defined. Regarding GnRH, researchers have studied its potential in treating diseases such as cancers, and recently, it was shown to have an effect in improving cognition in Down syndrome and dementia55,56,57, but the effect of GnRH on AD is unclear and it remains completely unknown if OXT and GnRH might be used in combination to synergistically and effectively combat against AD.
Hence, we studied a 9-month-old 5xFAD mouse model with strong manifestation of AD-like phenotypes, focusing to compare OXT-GnRH combinational treatment vs. individual peptide treatment. We employed nasal administration, a method which has often been used in research to deliver peptides into the brain for treating neurological diseases58,59,60,61,62,63,64. To help discern the effects of combinational vs. individual treatment, OXT and GnRH were administered nasally at relatively low doses (50 ng and 5 ng, respectively) for a relatively short treatment duration (2 months). After the completion of therapy, animals were assessed with a battery of non-invasive neurobehavioral assays, including an open field, grip strength, y-maze, novel object recognition, social interaction, and Morris Water Maze for various aspects of physical and cognitive functions. As demonstrated in Fig. 7, OXT-GnRH combinational treatment led to very robust and strong effects against various neurological disorders in this AD-like model, while the effects from single peptide treatment were modest or non-significant. At the end, we assessed how OXT-GnRH therapy could affect Aβ levels in this model. Indeed, 5×FAD model had a very large amount of Aβ plaques throughout various brain regions, represented by hippocampal CA1, CA3 and dentate gyrus (DG), entorhinal cortex, and mediobasal hypothalamus. In agreeing with the physiological effects, OXT-GnRH combinational treatment led to a nearly complete reversal of Aβ deposition (Fig. 8). Compared to the combinational treatment, single peptide treatments were much less effective, except that GnRH treatment was also sufficient to reduce Aβ plaques in the mediobasal hypothalamus, while Aβ plaques in fornix bundle in this region were not amended by any treatment. Taken together, compared to single peptide treatment, OXT-GnRH combinational therapy increased the sensitivity and effectiveness in treating neurological disorders and reducing Aβ plaques in male 5×FAD model.

Male 5×FAD mice (9 months old) received 2-month OXT (50 ng) and GnRH (5 ng) treatment individually or in combination vs. vehicle control through daily nasal administration and were subsequently examined for a battery of neurobehavioral assays. Results demonstrated body weight, locomotion in open field test, muscle strength in grip test, cognitive function in Y-maze test, novel object recognition in novel object test, social function in sociality test (A), and spatial learning/memory function in Morris Water Maze (MWM) test (B, C) which consisted of 5-day training session followed by probe test while swimming speed in the probe trial was provided as a technical control. Data are presented as mean ± SEM. The p-values are reported based on one-way ANOVA and Tukey post-hoc test between indicated groups (A, C) and two-way ANOVA and Tukey post-hoc test between vehicle and OXT-GnRH combinational treatment at matched time (B), n = 10 mice per group. Source data are provided as a Source Data file.

Aged male 5xFAD mice received 2-month OXT and GnRH treatment individually or in combination vs. vehicle control, as detailed in Fig. 7. At the end of behavioral tests, subgroups of mice were processed for brain amyloid β (Aβ) immunostaining. A, B Aβ immunostaining in hippocampal subregions CA1, CA3 and dentate gyrus (DG) (A), and Aβ immunostaining in hypothalamic region MBH and entorhinal cortex (B). Images represent 5 mice per group. Red, Aβ; blue, DAPI nuclear staining. Bar, 100 µm. C–G Quantification of Aβ plaques. Data are presented as mean ± SEM. Statistical significance was assessed between the indicated groups, p-values are reported based on one-way ANOVA and Tukey post-hoc test, n = 5 mice per group. Source data are provided as a Source Data file.
Finally, given the observed transcriptional changes of Adcy members in 5xFAD model (Fig. 6), we additionally assessed how OXT-GnRH treatment, compared to individual peptide treatment, might affect promoter methylation of these Adcy genes. Thus, we utilized the Next-Generation Sequencing-based Bisulfite Sequencing PCR (NGS-BSP) method to sequence the promoter subregions of candidate Adcy genes, focusing on subregions between CG island and encoding sequence. We performed target sequencing on hippocampal tissues obtained from the 5xFAD mice following combinational versus individual treatment with OXT and GnRH, as described in Fig. 7. Although either treatment did not cause significant changes in average methylation levels of the promoter subregions which we sequenced, our DMC analysis did lead to identification of several short sequences in which cytosine methylation levels were altered differently by OXT-GnRH treatment compared to single peptide treatment (Suppl. Fig. 8). Admittedly, this research is still limited, but it might point to a future direction for studying loci effects of OXT-GnRH treatment on gene promoter activity and if it could significantly affect the transcription of key genes in contributing to the combinational therapy against AD-like pathologies.
Discussion
In this project, we computationally analyzed age-related DNA methylation in the hypothalamus compared to two other limbic system components in a standard mouse model. This analysis led to the discovery that the hypothalamus is characterized by distinctively high-level DNA methylation at young age and the distinct dynamics of DNA methylation and demethylation when approaching middle age. Thus, compared to the other two limbic system regions, the hypothalamus contains more DNA methylation information during young age, contributing to its unique pattern of DNA methylation prior to middle age. During the course of approaching middle age, this distinction of DNA methylation between the hypothalamus and the other limbic system regions becomes to be declined. Thus, while aligning with the ‘hypothalamic control of aging’ paradigm which we previously proposed12,14,15, we further speculate that hypothalamic regulation of mammalian aging could be causally related to loss of epigenetic information from DNA methylation in the genomes of the hypothalamus. In this context, more brain regions should be compared to the hypothalamus in light of age-related DNA methylation. We also surprisingly found that not only the hypothalamus but also other limbic system regions commonly involve age-related DNA methylation of genes related to various hypothalamic endocrine pathways and notably OXT and GnRH pathways. This finding suggests the importance of the hypothalamus in age-related brain regional DNA methylation, warranting future research. Furthermore, the identification of OXT and GnRH pathways in this computational analysis lends support for investigating whether these neuropeptides could be designed to target aging-related diseases.
Hence, using an AD-like condition as an important example of aging-related diseases, we focused intensively on hypothalamic neuropeptide OXT and GnRH pathways for the potential therapeutic development. Following the observation that multiple components in OXT and GnRH pathways become declined in relevant brain regions at the gene transcriptional level in an AD-like mouse model, we were inspired to evaluate the effects of these two neuropeptides in combination versus individually for treating AD-like pathologies in this mouse model. This effort led to a major finding, that is, OXT-GnRH combinational therapy had a robust effect against the severe symptoms in an aged 5×FAD mouse model, showing significantly greater efficacy than individual peptide treatments. This finding can suggest a clinical strategy of using OXT-GnRH combinational therapy to target AD, which would call for clinical research down the road. In the literature, OXT has recently gained some attention mostly for reducing Aβ deposition49,50,51,52,53,54, while it remains unknown if GnRH could be used to treat AD. It is likely that individual peptides, especially when dose and treatment conditions are stronger, can each have a better effect on AD-like disorders, but strategy of developing combinational treatments should have advantages, for instance, low dose of each peptide helps minimize off-target effects, and some therapeutic effects are expected to require interactions and synergy of the two neuropeptide pathways which might not be achievable by individual peptides.
Our study still has limitations, mostly due to practical constraints during developing this project. For the computational study, we have not compared the hypothalamus with other limbic system components or other brain regions; to do so will be valuable to solidify the model on hypothalamic DNA methylation modeling of aging. Technically, we recognize a limitation that we employed 3 replicates of animals per group in our computational assays although it was often similarly used in the literature65,66,67, hence, to include more samples will likely lead to more information. Moreover, while our computational work was based on an aging model, it would be more informative to include samples from aging-related diseases such as AD. For the therapeutic study, while our work was to initially establish the proof of principle for using OXT-GnRH combination to target AD-like pathologies, future research is still needed to optimize dosage and duration, to assess additional ages and AD conditions, and to profile any off-target effects. In particular, we acknowledge a limitation due to the absence of studies on female model, which needs future research to address. Last but not least, while our therapeutics was based on an AD-like model as an example of aging-related disease, it will be very valuable to assess if this strategy could apply to other aging-related diseases, for example, Parkinson’s disease.
Methods
This study was conducted in compliance with all relevant ethical regulations. The animal research protocol was reviewed and approved by the Institutional Animal Care and Use Committee of the Albert Einstein College of Medicine (Protocols #00001111, #00001385, #00001397, #00001398, #00001399 and their previously approved versions).
Animal models
C57BL/6 mice and 5×FAD mice were obtained from Jackson Laboratory and housed under standard conditions in a 12-h light/12-h dark cycle with free access to food and water. All mice in this study were male. All mice were kept under standard, infection-free housing with 3 to 5 mice per cage. Pathogen-free quality of mouse colonies was ensured through quarterly serology, quarterly histopathologic exams and daily veterinarian monitoring of animal health and care. All mice were fed a standard chow from Lab Diet. Perfusion-induced euthanasia was used for tissue sampling and carbon dioxide inhalation euthanasia was used for all other mice which were generated during the study according to the IACUC approved protocols.
Bisulfite oligonucleotide-captured sequencing (BOCS)
The hypothalamus, hippocampus, and OB were obtained from standard, chow-fed male C57BL/6 mice at 2 vs. 12 months of age (n = 3 mice per group). DNA was extracted from these tissues using a Qiagen kit (Cat# 69506). The extracted DNA samples were then sheared into fragments of 150–200 base pairs using sonication, and the fragment sizes were verified using the Agilent TapeStation with D1000 ScreenTape. The fragmented DNA underwent library preparation using the SureSelectXT Methyl-Seq Library Prep Kit, following the manufacturer’s instructions. The generated libraries were subsequently hybridized with the SureSelectXT Mouse Methyl-Seq capture set for 16 h at 65 °C, specifically targeting all CG regions including all annotated promoters, CG islands and shore regions (±2 kb from islands) as well as shelf regions (±2 kb from shores and ±4 kb from islands). The hybridized libraries were captured using DynaBeads MyOne Streptavidin T1 magnetic beads. The captured libraries were eluted from magnetic beads using 0.1 M NaOH. Unmethylated cytosine residues in the captured DNA libraries were then converted to uracil through bisulfite conversion using the EZ DNA Methylation-Gold Kit from Zymo Research. Following bisulfite conversion, DNA was desulfonated and amplified via PCR. The amplified libraries were purified using AMPure XP beads. The purified libraries were indexed, allowing for the multiplexing of samples in a single sequencing run. Quantification of DNA libraries was performed using the TapeStation with D1000 ScreenTape. The libraries were normalized to a concentration of 4 nM, pooled together, and further diluted to a final concentration of 12 pM. The DNA libraries were sequenced on an Illumina MiSeq PE75 for quality control and sequenced on NextSeq500 High PE75 to obtain DNA methylation profiles.
DNA methylation analysis
FASTQ files were accessed from bisulfite sequencing for quality control with FastQC and paired-end reads were trimmed for quality with Illumina’s BaseSpace sequence hub having MethylSeq v2.0.0. Trimming involved removing 3 nucleotides at the 5′ end and 4 nucleotides at the 3′ end, as well as adapter removal using FASTQ Toolkit v2.2. Only reads with a Q-score >30 were used for mapping, reads which did not meet criteria after trimming were discarded. Alignment of trimmed bisulfite converted sequences was carried out using Bismark Bisulfite Mapper v0.14.4/Bowtie 2 v2.2.2 against the mouse reference genome (GRCm38/mm10)68,69. Each sample yielded over 25 million aligned reads, resulting in an average target sequence coverage ranging from 18 to 20×. The aligned BAM files were processed using SAMtools v1.270. After read mapping, methylation calling was carried out using the Bismark methylation extractor. The percentage of methylation was determined by calculating the ratio of methylated cytosines to the total number of cytosines covered in the genome for each specific cytosine position. Differential methylation analysis was primarily conducted in R v4.1.2, using MethylKit v1.20.071. Differentially methylated cytosines were determined from sites passing coverage criteria of minimum 10. Logistic regression was employed to determine differentially methylated sites between tissues, and p-values were adjusted using a false-discovery rate (q-value). Sites with q-value < 0.05 and methylation difference of ≥25% were considered statistically significant. For computing DMRs, criteria were set at a length of 500 bp showing >10% average methylation difference. Gene annotation files for the mouse (GRCm38) were downloaded from Gencode and mapped with DMRs using BEDTools v2.30.071,72. Deconvolution analysis, a method for analyzing cellular heterogeneity based on methylation sequencing data36,73,74,75, was used to computationally assess tissue heterogeneity of cell populations. The computation was based on using DXM (Deconvolution of Subpopulations Existing in Methylation Data), a reference-free approach offering versatility across various Bisulfite Sequencing (BS) methodologies39 which was established in the literature to accurately infer cell-type proportions40. DXM’s initial module “dxm_estimateFracs” was used within the Python environment. Enrichr was used to perform KEGG pathway analysis, allowing the identification and enrichment analysis of genes within specific biological pathways76. Cytoscape, in conjunction with the String module, with confidence >0.4 as the cut-off criteria, was employed to construct protein-protein interaction network, visualization and analysis of interconnections between pathway components77,78. Data were processed and visualized in Python, and computational analyses were conducted using custom scripts or published tools in UNIX, R and Python environments.
Animal treatment and behavioral assays
Male 5xFAD mice under standard housing and feeding were randomly assigned into various treatment groups to receive 50 ng OXT (Cat # 04375-1000IU, Sigma Aldrich) and 5 ng GnRH (Cat # PEP-168, Invitrogen) treatment through daily nasal administration. Following 2-month treatment, animals were sequentially examined with a panel of non-invasive behavioral assays with adequate recovery time between different assays. All behavioral tests were performed in an isolated and designated mouse behavioral room. An anymaze video-tracking system (version 4.99 m, Stoelting) connected with a digital camera and computer was used to record the activities of mice during behavioral procedures. Grip test: Each mouse was positioned on a square grid with a 1-cm mesh, which was then inverted 30 cm above a protective pad. The mouse was then allowed to hang by its paws for a specified period, and the duration for which the mouse remained suspended was recorded. Three repeats were performed for each animal with 30 min rest time between trials. Open field test: A mouse was placed in the lower right corner of a spacious, empty white plastic chamber (40 cm length × 40 width and × 40 cm height) and was allowed to freely move in the chamber while movement and behavior were recorded by the camera for 10 min. Y-maze test: A mouse was placed in the center of Y-maze apparatus and allowed to freely explore for 10 min. The time spent in each arm and the number of arm entries were recorded and analyzed. An entry was considered a success when all four limbs entered an arm. Novel object recognition: A mouse was allowed to freely explore an open-field box (40 cm in length, 40 cm in width, and 40 cm in height) for 10 min prior to experimental sessions. During the familiarization session, the mouse was then allowed to freely explore two similar objects for 10 min. During the test session, one of the two objects was replaced by a novel object for 10 min. The amount of time that the mouse spent exploring each object was recorded. A preference index was calculated using the ratio of the amount of time exploring each object over the total time exploring both objects. Morris water maze (MWM): The test was performed using a round water tank (90 cm in diameter) containing water at temperature of 22 to 23 °C and a non-toxic paint which was added to make opaque and white background. A circular, background color-matched platform with a diameter of 10 cm was placed 25 cm from the wall of the tank and about 1 cm below the water surface which was thus invisible. Animals were trained for 5 days, 4 training sessions each day, and the beginning positions for each training day were randomly chosen. At each training session, mice were placed in a beginning position in the maze and were trained to find the platform within 60 s. Latency to reach the platform of each trial was recorded. On the next day following 5-day training sessions, probe trials were performed by removing the platform and the animal was allowed to swim for 60 s and measured for swimming speed and the latency that the mouse swam to cross the location where the platform was previously located. Sociality: Social interaction was tested in a gray 3-chamber neutral box cage (60 cm in length, 40 cm in width, and 22 cm in height). For adaptation phase, animals were allowed to explore freely for 10 min in the neutral cage for habituating the testing conditions. For social affiliation phase, a new mouse (stranger) in a wire containment cup was placed in a side chamber. The subject mouse was allowed free access to explore each of three chambers for 10 min. For preference testing phase, a second new mouse (new stranger) in a wire containment cup was placed in the opposite side chamber. The subject mouse was allowed to freely explore each of three chambers for 10 min. The time spent in social interaction by sniffing was recorded.
Brain sections and immunostaining
The mice from the above therapeutic experiment were used for brain Aβ immunostaining (subgroup of brain samples from these mice used for DNA promoter target sequencing). To obtain brain samples, mice were anesthetized with 3–5% isoflurane inhalation and were transcardially perfused with saline over 5 min before brains were collected. Brain hemispheres were post-fixed with 4% PFA and then infiltrated with 20–30% sucrose for immunostaining. Brain sections were generated with a thickness of 20 μm using a cryostat. To facilitate Aβ immunostaining, sections were treated with 88% formic acid to expose the antigen, followed by blocking with a buffer solution containing 5% BSA, 5% goat serum, and 0.1% Triton X-100 in PBS. Subsequently, brain sections were incubated overnight at 4 °C with mouse anti-Aβ primary antibody (anti-β-Αmyloid, BioLegend, #803003, 1:500) in a blocking buffer. Technical controls included appropriate species-matched naive IgGs. Following three washes, the sections were incubated with secondary antibody (Goat anti-mouse IgG antibody Alexa Fluor 555, Thermo Fisher, #A21422, 1:500), and after three washes, sections were covered with a mounting medium containing DAPI for visualizing the nuclei of cells in the sections.
Image capture and quantification
Brain sections were imaged for a target region using the LAS X software-equipped Leica Stellaris 8 confocal microscope. The same confocal setting and parameters were used for all samples in this study. Confocal scanning was set at 600 Hz scan speed, 4 lines average, and 2 frame average. 20×/0.75 dry objective was used for imaging hippocampal subregions, and 10×/0.40 dry objective used for imaging other brain regions. Aβ plaque signals were imaged with an ALEXA channel at excitation wavelength of 553 nm and emission light collected with a wavelength of 558 to 730 nm. DAPI staining signal was imaged at excitation wavelength of 405 nm and emission light collected with a wavelength of 430 to 558 nm. The pinhole size was set at 56.7 μm for both channels. Images were exported as scaled viewer tiff files with lossless compression. ImageJ 1.54 f was used to quantitate image signal of interest. The same settings of ImageJ were used to process all images in this study. RGB images were converted into 8-bit grayscale format for each channel by ImageJ, and the entire region of interest (ROI) was delineated according to the anatomic structure. Aβ plaques in an ROI were subsequently identified using a threshold signal of 122 to 255 scale, and the plaque density of a target brain region was calculated as Aβ signal per area of ROI.
Quantitative RT-PCR
The hypothalamus and hippocampus samples were obtained from standard male C57BL/6 mice at the age of 2 vs. 15 months and from male 5xFAD mice and littermate male WT controls at the age of 8 months for extraction of total RNA using TRIzol reagent (Invitrogen), followed by reverse transcription using High-Capacity RNA to cDNA kit (Thermo Fisher) according to the manufacturer’s instructions. Quantitative real-time PCR was conducted using Power SYBR Green PCR Master Mix (Thermo Fisher) with specific primer sets (see below). β-actin was used as a reference and changes were calculated using 2-ΔΔCt method.
Oxt: 5ʹ-GCTGCCAGGAGGAGAACTAC-3ʹ; 5ʹ-GGCAGCCATCTGCAAGAGAA-3ʹ
Gnrh1: 5ʹ-GCATTCTACTGCTGACTGTGTGTT-3ʹ; 5ʹ-GTTCTGCCATTTGATCCACCT-3ʹ
Adcy3: 5ʹ-TCTTTGACTGCTACGTGGTAGT-3ʹ; 5ʹ-GGCCCGTGAAAAGTTCAGG-3ʹ
Adcy5: 5ʹ-AAGATCCTCGGGGATTGTTACT-3ʹ; 5ʹ-CTCCCGGACCAACGAGATG-3ʹ
Adcy7: 5ʹ-AAGGGGCGCTACTTCCTAAAT-3ʹ; 5ʹ-GTGTCTGCGGAGATCCTCA-3ʹ
Adcy9: 5ʹ-CAACAGCGTGAGGGTCAAGAT-3ʹ; 5ʹ-CATGGAGTCGAATTTGGGGTC-3ʹ
β-actin: 5ʹ-CCTCTATGCCAACACAGTGC-3ʹ; 5ʹ-GCTAGGAGCCAGAGCAGTAA-3ʹ
Statistics and reproducibility
Sample sizes were designed considering the relevant literature for physiological, biochemical and histological experiments12,14,15,79,80, relevant literature for DNA bisulfite sequencing65,66,67, and relevant literature for target bisulfite sequencing67,81. Neurobehavioral tests were performed making group information blind to experimental performers when collecting the data. All physiological and histological results represented repeated observations independently and through complimentary approaches. All data with biological replicates were presented as mean ± S.E.M. The number of biological replicates and applied statistical methods are detailed in figure legends. Data were analyzed for parametric or non-parametric distributions with statistical tools such as Shapiro-Wilk test, D’Agostino-Pearson test, Anderson-Darling test, and Kolmogorov-Smirnov test. Data that followed parametric distribution were analyzed using ANOVA with Tukey’s multiple comparisons when involving more than two groups and two-tailed unpaired Student’s t-test when involving only two groups. Data that did not follow parametric distribution were analyzed including Kruskal-Wallis test and Dunn’s multiple comparisons when having more than two groups and two-tailed Mann-Whitney test when having only two groups. Additional statistics included logistic regression and Fisher’s Exact test contained in applied computational programs. Statistical significance of all data was determined at p < 0.05. For computing differential methylation sites and regions, logistic regression-based modeling was applied in R-based MethylKit v1.20.0, with a false discovery rate set at q < 0.05.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
BOCS datasets for all biological samples generated in this study are available in the NCBI Gene Expression Omnibus (GEO) repository with accession number GSE276875. Target promoter subregional data and raw immunofluorescence images are deposited in publicly accessible platform Figshare (https://figshare.com/s/c2ed30d10acfa8a43f56). Source data are provided with this paper. There is no restriction on data availability in this manuscript, and research materials developed from this study are freely available to research upon request. Source data are provided with this paper.
Code availability
Code/scripts for computational analysis are deposited in the publicly accessible GitHub repository (https://github.com/DongshengCai/Brain-subregional-DNA-methylation-aging).
References
Shohayeb, B., Diab, M., Ahmed, M. & Ng, D. C. H. Factors that influence adult neurogenesis as potential therapy. Transl. Neurodegener. 7, 4 (2018).
Culig, L., Chu, X. & Bohr, V. A. Neurogenesis in aging and age-related neurodegenerative diseases. Ageing Res. Rev. 78, 101636 (2022).
Dean, D. C. et al. Brain differences in infants at differential genetic risk for late-onset Alzheimer disease: a cross-sectional imaging study. Jama Neurol. 71, 11–22 (2014).
Fu, H., Hardy, J. & Duff, K. E. Selective vulnerability in neurodegenerative diseases. Nat. Neurosci. 21, 1350–1358 (2018).
van Praag, H. et al. Functional neurogenesis in the adult hippocampus. Nature 415, 1030–1034 (2002).
Kuhn, H. G., Dickinson-Anson, H. & Gage, F. H. Neurogenesis in the dentate gyrus of the adult rat: age-related decrease of neuronal progenitor proliferation. J. Neurosci. 16, 2027–2033 (1996).
Lois, C. & Alvarez-Buylla, A. Proliferating subventricular zone cells in the adult mammalian forebrain can differentiate into neurons and glia. Proc. Natl. Acad. Sci. USA 90, 2074–2077 (1993).
Alvarez-Buylla, A. & Lois, C. Neuronal stem cells in the brain of adult vertebrates. Stem Cells (Dayt. Ohio) 13, 263–272 (1995).
Doetsch, F., Caillé, I., Lim, D. A., García-Verdugo, J. M. & Alvarez-Buylla, A. Subventricular zone astrocytes are neural stem cells in the adult mammalian brain. Cell 97, 703–716 (1999).
Alvarez-Buylla, A. & Garcia-Verdugo, J. M. Neurogenesis in adult subventricular zone. J. Neurosci. 22, 629–634 (2002).
Xiao, Y. Z. et al. Reducing hypothalamic stem cell senescence protects against aging-associated physiological decline. Cell Metab. 31, 534–548.e535 (2020).
Zhang, Y. L. et al. Hypothalamic stem cells control ageing speed partly through exosomal miRNAs. Nature 548, 52–57 (2017).
Li, J. X., Tang, Y. Z. & Cai, D. S. IKK beta/NF-kappa B disrupts adult hypothalamic neural stem cells to mediate a neurodegenerative mechanism of dietary obesity and pre-diabetes. Nat. Cell Biol. 14, 999–1012 (2012).
Wang, Z., Wu, W., Kim, M. S. & Cai, D. GnRH pulse frequency and irregularity play a role in male aging. Nat. Aging 1, 904–918 (2021).
Zhang, G. et al. Hypothalamic programming of systemic ageing involving IKK-beta, NF-kappaB and GnRH. Nature 497, 211–216 (2013).
Tang, Y. & Cai, D. Hypothalamic inflammation and GnRH in aging development. Cell Cycle 12, 2711–2712 (2013).
Zhang, G. & Cai, D. Circadian intervention of obesity development via resting-stage feeding manipulation or oxytocin treatment. Am. J. Physiol. Endocrinol. Metab. 301, E1004–E1012 (2011).
Zhang, G. et al. Neuropeptide exocytosis involving synaptotagmin-4 and oxytocin in hypothalamic programming of body weight and energy balance. Neuron 69, 523–535 (2011).
Horvath, S. Erratum to: DNA methylation age of human tissues and cell types. Genome Biol. 16, 96 (2015).
Hannum, G. et al. Genome-wide methylation profiles reveal quantitative views of human aging rates. Mol. Cell 49, 359–367 (2013).
Jones, M. J., Goodman, S. J. & Kobor, M. S. DNA methylation and healthy human aging. Aging Cell 14, 924–932 (2015).
Pal, S. & Tyler, J. K. Epigenetics and aging. Sci. Adv. 2, e1600584 (2016).
Dor, Y. & Cedar, H. Principles of DNA methylation and their implications for biology and medicine. Lancet (Lond., Engl.) 392, 777–786 (2018).
Hou, Y. et al. Ageing as a risk factor for neurodegenerative disease. Nat. Rev. Neurol. 15, 565–581 (2019).
Unnikrishnan, A. et al. The role of DNA methylation in epigenetics of aging. Pharmacol. Therapeutics 195, 172–185 (2019).
Zhang, W., Qu, J., Liu, G. H. & Belmonte, J. C. I. The ageing epigenome and its rejuvenation. Nat. Rev. Mol. Cell Biol. 21, 137–150 (2020).
Sikora, E. et al. Cellular senescence in brain aging. Front Aging Neurosci. 13, 646924 (2021).
Rutledge, J., Oh, H. & Wyss-Coray, T. Measuring biological age using omics data. Nat. Rev. Genet 23, 715–727 (2022).
Seale, K., Horvath, S., Teschendorff, A., Eynon, N. & Voisin, S. Making sense of the ageing methylome. Nat. Rev. Genet 23, 585–605 (2022).
Wang, K. et al. Epigenetic regulation of aging: implications for interventions of aging and diseases. Signal Transduct. Target Ther. 7, 374 (2022).
He, X., Memczak, S., Qu, J., Belmonte, J. C. I. & Liu, G. H. Single-cell omics in ageing: a young and growing field. Nat. Metab. 2, 293–302 (2020).
Horvath, S. & Raj, K. DNA methylation-based biomarkers and the epigenetic clock theory of ageing. Nat. Rev. Genet. 19, 371–384 (2018).
Yang, J. H. et al. Loss of epigenetic information as a cause of mammalian aging. Cell https://doi.org/10.1016/j.cell.2022.12.027 (2023).
Masser, D. R. et al. Sexually divergent DNA methylation patterns with hippocampal aging. Aging Cell 16, 1342–1352 (2017).
Masser, D. R. et al. Bisulfite oligonucleotide-capture sequencing for targeted base- and strand-specific absolute 5-methylcytosine quantitation. Age (Dordr.) 38, 49 (2016).
Song, J. & Kuan, P. F. A systematic assessment of cell type deconvolution algorithms for DNA methylation data. Brief Bioinform. 23, https://doi.org/10.1093/bib/bbac449 (2022).
Rahmani, E. et al. Correcting for cell-type heterogeneity in DNA methylation: a comprehensive evaluation. Nat. Methods 14, 218–219 (2017).
Rahmani, E. et al. Cell-type-specific resolution epigenetics without the need for cell sorting or single-cell biology. Nat. Commun. 10, 3417 (2019).
Fong, J. et al. Determining subpopulation methylation profiles from bisulfite sequencing data of heterogeneous samples using DXM. Nucleic Acids Res. 49, e93 (2021).
Jeong, Y. et al. Systematic evaluation of cell-type deconvolution pipelines for sequencing-based bulk DNA methylomes. Brief Bioinform. 23, https://doi.org/10.1093/bib/bbac248 (2022).
Hajdarovic, K. H. et al. Single-cell analysis of the aging female mouse hypothalamus. Nat. Aging 2, 662–678 (2022).
Yang, Y. T. et al. STAB2: an updated spatio-temporal cell atlas of the human and mouse brain. Nucleic Acids Res. 52, D1033–D1041 (2024).
Tepe, B. et al. Single-cell RNA-Seq of mouse olfactory bulb reveals cellular heterogeneity and activity-dependent molecular census of adult-born neurons. Cell Rep. 25, 2689–2703.e2683 (2018).
Devasani, K. & Yao, Y. Expression and functions of adenylyl cyclases in the CNS. Fluids Barriers CNS 19, 23 (2022).
Mons, N., Segu, L., Nogues, X. & Buhot, M. C. Effects of age and spatial learning on adenylyl cyclase mRNA expression in the mouse hippocampus. Neurobiol. Aging 25, 1095–1106 (2004).
Iwamoto, T. et al. Motor dysfunction in type 5 adenylyl cyclase-null mice. J. Biol. Chem. 278, 16936–16940 (2003).
Joeyen-Waldorf, J. et al. Adenylate cyclase 7 is implicated in the biology of depression and modulation of affective neural circuitry. Biol. Psychiatry 71, 627–632 (2012).
Hines, L. M. et al. A sex-specific role of type VII adenylyl cyclase in depression. J. Neurosci. 26, 12609–12619 (2006).
Takahashi, J. et al. Oxytocin reverses Aβ-induced impairment of hippocampal synaptic plasticity in mice. Biochem Biophys. Res Commun. 528, 174–178 (2020).
Takahashi, J. et al. Intracerebroventricular administration of oxytocin and intranasal administration of the oxytocin derivative improve β-amyloid peptide (25-35)-induced memory impairment in mice. Neuropsychopharmacol. Rep. 42, 492–501 (2022).
El-Ganainy, S. O. et al. Intranasal oxytocin attenuates cognitive impairment, β-amyloid burden and tau deposition in female rats with Alzheimer’s disease: interplay of ERK1/2/GSK3β/caspase-3. Neurochem. Res. 47, 2345–2356 (2022).
Selles, M. C. et al. Oxytocin attenuates microglial activation and restores social and non-social memory in APP/PS1 Alzheimer model mice. iScience 26, 106545 (2023).
Koulousakis, P. et al. Exogenous oxytocin administration restores memory in female APP/PS1 Mice. J. Alzheimers Dis. 96, 1207–1219 (2023).
Ye, C. et al. Oxytocin nanogels inhibit innate inflammatory response for early intervention in Alzheimer’s disease. ACS Appl Mater. Interfaces 14, 21822–21835 (2022).
Garrido, M. P., Hernandez, A., Vega, M., Araya, E. & Romero, C. Conventional and new proposals of GnRH therapy for ovarian, breast, and prostatic cancers. Front Endocrinol. (Lausanne) 14, 1143261 (2023).
Sterling, K., Cao, R. & Song, W. Gonadotropin releasing hormone (GnRH): a hormone therapy boosts cognition in Down syndrome and dementia. Signal Transduct. Target Ther. 8, 49 (2023).
Manfredi-Lozano, M. et al. GnRH replacement rescues cognition in Down syndrome. Science 377, eabq4515 (2022).
Patel, D., Patel, B. & Wairkar, S. Intranasal delivery of biotechnology-based therapeutics. Drug Discov. Today 27, 103371 (2022).
Akita, T. et al. Involvement of trigeminal axons in nose-to-brain delivery of glucagon-like peptide-2 derivative. J. Control Release 351, 573–580 (2022).
Finger, E. et al. Adaptive crossover designs for assessment of symptomatic treatments targeting behaviour in neurodegenerative disease: a phase 2 clinical trial of intranasal oxytocin for frontotemporal dementia (FOXY). Alzheimers Res Ther. 10, 102 (2018).
Lindenmaier, Z. et al. Examining the effect of chronic intranasal oxytocin administration on the neuroanatomy and behavior of three autism-related mouse models. Neuroimage 257, 119243 (2022).
Wang, Y. et al. Repeated oxytocin prevents central sensitization by regulating synaptic plasticity via oxytocin receptor in a chronic migraine mouse model. J. Headache Pain. 22, 84 (2021).
Bales, K. L. et al. Long-term exposure to intranasal oxytocin in a mouse autism model. Transl. Psychiatry 4, e480 (2014).
Huang, H. et al. Chronic and acute intranasal oxytocin produce divergent social effects in mice. Neuropsychopharmacology 39, 1102–1114 (2014).
Zocher, S., Overall, R. W., Lesche, M., Dahl, A. & Kempermann, G. Environmental enrichment preserves a young DNA methylation landscape in the aged mouse hippocampus. Nat. Commun. 12, 3892 (2021).
Alberca, C. D., Papale, L. A., Madrid, A. & Alisch, R. S. Hippocampal and peripheral blood DNA methylation signatures correlate at the gene and pathway level in a mouse model of autism. Hum. Mol. Genet. 32, 3312–3322 (2023).
Hadad, N. et al. Absence of genomic hypomethylation or regulation of cytosine-modifying enzymes with aging in male and female mice. Epigenetics Chromatin 9, 30 (2016).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012).
Krueger, F. & Andrews, S. R. Bismark: a flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics 27, 1571–1572 (2011).
Danecek, P. et al. Twelve years of SAMtools and BCFtools. Gigascience 10, https://doi.org/10.1093/gigascience/giab008 (2021).
Akalin, A. et al. methylKit: a comprehensive R package for the analysis of genome-wide DNA methylation profiles. Genome Biol. 13, R87 (2012).
Quinlan, A. R. & Hall, I. M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842 (2010).
Titus, A. J., Gallimore, R. M., Salas, L. A. & Christensen, B. C. Cell-type deconvolution from DNA methylation: a review of recent applications. Hum. Mol. Genet. 26, R216–R224 (2017).
Lee, D., Lee, S. & Kim, S. PRISM: methylation pattern-based, reference-free inference of subclonal makeup. Bioinformatics 35, i520–i529 (2019).
Barrett, J. E. et al. Quantification of tumour evolution and heterogeneity via Bayesian epiallele detection. BMC Bioinf. 18, 354 (2017).
Kuleshov, M. V. et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 44, W90–97, (2016).
Szklarczyk, D. et al. The STRING database in 2021: customizable protein-protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res 49, D605–D612 (2021).
Shannon, P. et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504 (2003).
Tang, Y., Zuniga-Hertz, J. P., Han, C., Yu, B. & Cai, D. Multifaceted secretion of htNSC-derived hypothalamic islets induces survival and antidiabetic effect via peripheral implantation in mice. Elife 9, https://doi.org/10.7554/eLife.52580 (2020).
Zhang, Y., Reichel, J. M., Han, C., Zuniga-Hertz, J. P. & Cai, D. Astrocytic process plasticity and IKKbeta/NF-kappaB in central control of blood glucose, blood pressure, and body weight. Cell Metab. 25, 1091–1102.e1094 (2017).
Ren, J. et al. Intrauterine hyperglycemia exposure results in intergenerational inheritance via DNA methylation reprogramming on F1 PGCs. Epigenetics Chromatin 11, 20 (2018).
Acknowledgements
We gratefully thank Cai lab members for technical support, thank W. Freeman and Oklahoma Nathan Shock Center on Aging for providing kind assistance and Core support, and thank CD Genomics for target methylation sequencing. This research was supported through Einstein institutional resources and partly through Milky Way Research Foundation award and National Institutes of Health R01AG031774 (all supports to D. Cai).
Author information
Authors and Affiliations
Contributions
S.S.U. performed all computational analysis on BOCS metadata and target promoter sequencing data, did statistics for all sequencing data, contributed to data interpretation and provided writing assistance. H.J. performed animal treatment and behavioral study, performed qRT-PCR assays, and contributed to data interpretation. Q.Z. performed histological and immunostaining experiment and contributed to data interpretation. M.W.K. co-performed animal treatment and behavioral assays. Y.C. contributed to qRT-PCR primers and target promoter bisulfite sequencing. A.B.C. contributed to animal treatment study. D.C. conceived the hypothesis and ideas, conceptualized and constructed the project, designed computational and experimental strategies, advised and supervised all experiments, instructed and concluded data analysis, led and concluded data interpretation, and wrote the paper. All authors approved the paper.
Corresponding author
Ethics declarations
Competing interests
D.C. has an invention disclosure with Albert Einstein College of Medicine (OXT-GnRH approaches for addressing AD or aging). Other authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Isabel Castanho and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Usmani, S.S., Jung, HG., Zhang, Q. et al. Targeting the hypothalamus for modeling age-related DNA methylation and developing OXT-GnRH combinational therapy against Alzheimer’s disease-like pathologies in male mouse model. Nat Commun 15, 9419 (2024). https://doi.org/10.1038/s41467-024-53507-8
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-024-53507-8
