
Abstract
Cryo-electron microscopy (cryo-EM) is one of the most powerful experimental methods for macromolecular structure determination. However, accurate DNA/RNA structure modeling from cryo-EM maps is still challenging especially for protein-DNA/RNA or multi-chain DNA/RNA complexes. Here we propose a deep learning-based method for accurate de novo structure determination of DNA/RNA from cryo-EM maps at <5 Å resolutions, which is referred to as EM2NA. EM2NA is extensively evaluated on a diverse test set of 50 experimental maps at 2.0–5.0 Å resolutions, and compared with state-of-the-art methods including CryoREAD, ModelAngelo, and phenix.map_to_model. On average, EM2NA achieves a residue coverage of 83.15%, C4’ RMSD of 1.06 Å, and sequence recall of 46.86%, which outperforms the existing methods. Moreover, EM2NA is applied to build the DNA/RNA structures with 10 to 5347 nt from an EMDB-wide data set of 263 unmodeled raw maps, demonstrating its ability in the blind model building of DNA/RNA from cryo-EM maps. EM2NA is fast and can normally build a DNA/RNA structure of <500 nt within 10 minutes.
Similar content being viewed by others

Introduction
Nucleic acids (NA) including DNA and RNA play important roles in many biological processes by themselves or interacting with proteins. Therefore, determining the 3D structures of nucleic acids is valuable to understand their functions1,2,3,4. Cryogenic electron microscopy (cryo-EM) has become the main-stream experimental approach for macromolecular structure determination5,6,7. Various methods have been developed for data processing and structure determination with cryo-EM technology in the past decade8,9,10,11,12. Many atomic structures of macromolecular complexes have been determined from cryo-EM maps and deposited in the Protein Data Bank (PDB)13. However, structure determination from cryo-EM maps remains a challenging task. Due to the higher intrinsic heterogeneity and dynamics of DNA/RNA than proteins1, such a challenge is especially critical for modeling nucleic acids from the maps of protein-DNA/RNA complexes14. Compared with the progress in protein modeling from cryo-EM maps15,16,17,18,19,20,21,22,23,24,25,26,27,28, the advancement in DNA/RNA modeling from cryo-EM maps has fallen far behind. As such, the structures for DNA/RNA are often omitted during model building of protein-DNA/RNA complexes from cryo-EM maps29,30,31,32.
Several methods have been developed to model DNA/RNA structures from cryo-EM maps, though they face different limitations either in automation or accuracy. RCrane33 is developed as an interactive tool to help researchers build nucleic acids from scratch. Phenix.map_to_model10 is developed to build initial structure models for both nucleic acids and proteins, though it can only build fragments for high-resolution regions. ModelAngelo28 is powerful in solving protein structures and the latest version also supports modeling DNA/RNA. However, it struggles to build a complete structure and assign the correct nucleotide types especially for the regions with poor resolutions. Recently, DeepTracer34 is extended to include model building of DNA/RNA in addition to proteins. However, it only detects P atoms, which is insufficient to determine other atom positions. CryoREAD35 is another tool developed to model DNA/RNA using deep learning. However, its detection for the positions of functional groups instead of atoms limits the accurate placement of individual atoms.
For RNA structure modeling, DRRAFTER14 is developed for addressing the maps of large protein-RNA complexes based on a manually-determined initial helix position. The extended version, auto-DRRAFTER36, is proposed to build the full-length RNA-only structures guided by accurate secondary structures determined from experiments. However, auto-DRRAFTER relies on the secondary structure of RNAs, and its performance is limited by the input of predicted RNA secondary structures. Moreover, auto-DRRAFTER is time- and resource-consuming, and may not necessarily assemble the correct full-length RNA structure due to its Monte-Carlo search nature. Very recently, we have proposed EMRNA37, a deep learning-based framework for the accurate determination of full-length all-atom RNA structures from cryo-EM maps. However, EMRNA is designed for modeling single-chain RNA-only maps, and cannot handle the maps of multi-chain DNA/RNA targets or protein-NA complexes37. Therefore, structure modeling for both DNA and RNA from cryo-EM maps of protein-DNA/RNA or multi-chain DNA/RNA complexes is needed.
In this work, we propose a deep learning-based method to automatically build all-atom nucleic acid structures from cryo-EM maps of protein-DNA/RNA or multi-chain DNA/RNA complexes at <5 Å resolutions, named EM2NA. EM2NA first detects the nucleic acid densities in a raw cryo-EM map through a deep learning network. The identified nucleic acid map region is fed to another network for predicting the nucleotide information, including the positions of P, C4’, and N1 or N9 atoms as well as their nucleotide types. Then, the DNA/RNA backbones are automatically built from the predicted atom positions by integrating a backbone tracing algorithm and considering the nucleotide geometry. The nucleotide types are assigned by sequence alignments and refined by the detection of possible base-pairs in the built backbone. The full-atom structure is finally constructed by aligning template nucleotide conformations onto the backbone. EM2NA is extensively evaluated on a diverse test set of 50 experimental maps at 2–5 Å resolutions, and compared with state-of-the-art approaches. EM2NA is also applied to blindly build the DNA/RNA models from an EMDB-wide data set of 263 unmodeled experimental maps.
Results
The workflow of EM2NA
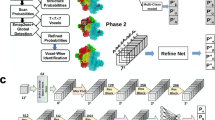
Figure 1 shows an overview of the EM2NA framework. EM2NA starts with the detection of DNA/RNA regions in a raw cryo-EM map through a Swin-Conv-UNet (SCUNet) architecture that has simultaneous local and nonlocal learning capabilities. The stage-1 network is trained to segment a cryo-EM map into NA, protein, and background. After the segmentation, the DNA/RNA density region is used as input of the stage-2 network to predict the backbone atom probabilities and nucleotide types. The two networks adopt the same SCUNet architecture and only differ in the output channels. The neural networks are trained with 322 experimental density maps. The backbone atom probabilities output from the stage-2 network are then converted to three-dimensional (3D) points by detecting the local maxima using a mean-shift algorithm. Next, the combined P and C4’ points are traced to multiple backbone paths by solving a Vehicle Routing Problem (VRP). For each path, the C4’ points are first extracted and the P points are placed for every two C4’ atoms to form a “ ⋯ (C4’-P-C4’) ⋯ ” order. The N1 or N9 points are then assigned for each C4’ point by querying the first-nearest neighbor. In addition, two more P points are added at both terminals. The direction of each path can be easily determined by considering the nucleotide geometries. After the path direction determination, we utilize the Smith-Waterman algorithm to assign the sequence for each of the built backbones. The sequence assignment is further improved by considering the base pairing between nucleotides in double helices. With the backbone path and assigned nucleotide types, the full-atom structure is built by aligning template nucleotide conformations onto the P-C4’-N1/N9 backbone. A post-refinement is recommended, using a third-party program like phenix.real_space_refine, to relax the structure and improve side-chain conformations and map-model correlations.

a The workflow of map segmentation for protein and nucleic acid (NA). The input for EM2NA is a cryo-EM density map and optional DNA/RNA sequences. The input map is first passed to stage-1 network to segment the nucleic acid region. b The workflow for DNA/RNA model building. The segmented nucleic acid map region from the stage-1 network is passed to the stage-2 network for predicting the backbone atom probabilities and nucleotide types at each voxel. The output atom probability map is converted to the backbone points by shifting integer grids to local maximum coordinates. The points are then traced to multiple paths by solving a Vehicle Routing Problem. With input sequences, a path-sequence alignment is utilized to assign nucleotide types to the backbone traces, where the base pairing feature is also considered for double helical parts. Finally, the full-atom DNA/RNA structure is constructed from the backbone traces. A post-refinement is recommended to refine the model, using e.g. phenix.real_space_refine. c The Swin-Conv (SC) UNet architecture for the stage-1 network in a and the stage-2 network in b. The input map is cut into overlapped boxes of 48 × 48 × 48 Å 3, which are fed to the network. The predicted boxes are assembled to the final map as output.
Performance in recovering DNA/RNA structures
We first evaluated the ability of EM2NA in recovering the DNA/RNA structures from cryo-EM maps and compared it with three state-of-the-art methods, including CryoREAD, ModelAngelo, and Phenix (phenix.map_to_model) on the test set of 50 NA-included maps (Supplementary Data 1). Here, the performance is measured by two metrics, residue coverage and C4’ RMSD of the built model, which are calculated using phenix.chain_comparison. The calculation is based on finding the nearest C4’ atom pairs between the built and PDB models in ignorance of nucleotide identity within a certain distance cutoff (e.g. 3 Å by default). The residue coverage measures the percentage of matched nucleotides among all PDB nucleotides, and the C4’ RMSD represents the root mean square deviation between the matched C4’ atoms. It is noted that phenix.map_to_model only built the DNA/RNA models for 44 of 50 maps. Therefore, the evaluation for Phenix is based on the 44 cases. Figure 2a–d shows the box plots and head-to-head comparison of the corresponding results by EM2NA and the other three methods.

a Box plots of the residue coverages for the built models. b Head-to-head comparison of the residue coverages between EM2NA and the other methods. c Box plots of the C4' RMSDs for the built models. d Head-to-head comparison of the C4' RMSDs between EM2NA and the other methods. The box plots indicate the means (cross), medians (median-line), first and third quartiles (bounds of boxes), and ± 1.0 standard deviations (whiskers). Mean values are marked near the bar. e An example of the built model for the Adenosylcobalamin-bound riboswitch dimer in form-1 (EMD-40262, PDB-8SA2, reported resolution 3.10 Å). The panel shows the density map (gray), PDB structure (green) and the models built by four methods (blue). The residue coverage/C4' RMSD are listed below each model. Source data are provided as a Source Data file.
It can be seen from the figure that EM2NA outperforms the other three methods in both residue coverage and C4’ RMSD (Fig. 2a, c). For the residue coverage of built models, EM2NA achieves a high average value of 83.15%, compared with 47.04% for CryoREAD, 74.23% for ModelAngelo, and 40.87 % for Phenix.map_to_model (Fig. 2a). Among the test set of 50 maps, EM2NA obtains a higher coverage than CryoREAD for 48 cases, ModelAngelo for 41 cases, and Phenix for 49 cases, respectively (Fig. 2b). For the atomic accuracy of built models, EM2NA gives a low average C4’ RMSD of 1.06 Å, compared with 2.28 Å for CryoREAD, 1.17 Å for ModelAngelo, and 1.10 Å for phenix.map_to_model, respectively (Fig. 2c). The low C4’ RMSD of Phenix here (Fig. 2c) is because that phenix.map_to_model tends to build the models for only those high-quality density regions in a map9, which thus leads to a lower residue coverage in the built model (Fig. 2a). Specifically, EM2NA yields a better C4’ RMSD than CryoREAD for all 50 cases, ModelAngelo for 38 of 50 cases, and Phenix for 23 of 44 cases, respectively (Fig. 2d).
Besides the C4’ RMSD, we also calculated the base atom recall for each method. The base atom recall measures the percentage of the base atoms that are built near the reference PDB base atoms within a certain distance. On average, EM2NA achieves the base atom recalls of 49.99%, 76.03%, and 87.25% for the cutoffs of 1, 2 and 4 Å, respectively, compared with 16.96%, 49.08%, and 81.07% for CryoREAD, 35.87%, 65.43%, and 83.30% for ModelAngelo, and 32.14%, 47.03%, and 63.87% for Phenix (Supplementary Data 2).
Figure 2e shows an example of Adenosylcobalamin-bound riboswitch dimer in form-1. The bounded dimer forms a symmetric conformation. It can be seen from the figure that EM2NA has the best coverage and C4’ RMSD among the four methods. Specifically, EM2NA achieves a residue coverage/backbone of 87.40%/1.23 Å, compared with 50.20%/2.32 Å for CryoREAD, 68.50%/1.37 Å for ModelAngelo, and Phenix for 26.20%/1.27 Å, respectively. Although CryoREAD recovers many nucleotides from the map, it builds the backbone structure with a very large deviation to the PDB structure, causing a very low coverage. ModelAngelo achieves the second-best coverage but also misses some nucleotides. Phenix is able to place the nucleotide positions well but fails to recover most of the nucleotides. Especially, almost all the nucleotides are missing in two regions highlighted by the dashed rectangles. In addition, EM2NA, ModelAngelo, and Phenix can build good side-chain formations, but CryoREAD gives poor side-chain conformations (Fig. 2e).
Performance in assigning nucleotide types
In addition to recovering a high percentage of the DNA/RNA structure, assigning correct nucleotide types to the recovered residues, is of same importance. Therefore, we also evaluated the performance of assigning nucleotide types to the built models by EM2NA, CryoREAD, ModelAngelo and Phenix (phenix.map_to_model). Two metrics (sequence match/recall) are used to measure the accuracy. After matching the C4’ atom pairs between the built model and reference PDB structure using a distance cutoff, a built nucleotide is considered as ‘correct’ if it has the same nucleotide type as the matched reference nucleotide. Then, the sequence match measures the portion of the ‘correct’ nucleotides relative to the built nucleotides, while the sequence recall measures the percentage of the ‘correct’ nucleotide relative to all the nucleotides in the reference PDB structure.
Figure 3a–d shows the sequence matches and recalls of the built models by EM2NA and the other three methods. It can be seen from the figure that EM2NA performs the best among the four methods in terms of both sequence match and recall. When only considering the residues of the built model, EM2NA achieves an average sequence match of 55.65%, compared with 50.77% for CryoREAD, 27.62% for ModelAngelo, 38.83% for Phenix, respectively (Fig. 3a). Interestingly, ModelAngelo performs the worst here among the four methods (Fig. 3a), although it yields a high residue coverage of 73.65% (Fig. 2a). Among the 50 tested maps, EM2NA achieves a higher sequence match than CryoREAD for 30 cases, ModelAngelo for 44 cases, and Phenix for 45 cases, respectively (Fig. 3b). When considering all the residues in the PDB structure. EM2NA achieves an average sequence recall of 45.95%, which is much higher than 24.20% for CryoREAD, 20.42% for ModelAngelo, and 16.75% for Phenix, respectively (Fig. 3c). Among the 50 maps, EM2NA yields a higher sequence recall than CryoREAD for 48 cases, ModelAngelo for 47 cases, and Phenix for 50 cases, respectively (Fig. 3d).

a Box plots of sequence matches for the built models. b Box plots of sequence recalls for the built models. The box plots indicate the means (cross), medians (median-line), first and third quartiles (bounds of boxes), and ± 1.0 standard deviations (whiskers). Mean values are marked near the box. c Head-to-head comparison of sequence matches. d Head-to-head comparison of sequence recalls. e, f An example of the built model for the SARS-CoV-2 replication-transcription complex bound to GTP, in a pre-catalytic state. The density map (gray) and PDB structure (blue for protein, green for RNA) are shown in e, f shows the comparison of EM2NA, CryoREAD, ModelAngelo, and Phenix, with nucleotide types (A/G/C/U) labeled in different colors (red/green/blue/yellow) and the protein chains from the PDB model colored in transparent gray. Source data are provided as a Source Data file.
Figure 3e, f shows a comparison of the built models by different methods on an example of SARS-CoV-2 replication-transcription complex bound to GTP, in a pre-catalytic state (EMD-26645 PDB 7UOB). On this target, EM2NA builds a very good model with a sequence recall of 72.49%, compared with 27.54% for CryoREAD, 26.07% for ModelAngelo, and 21.76% for Phenix. Further examination revealed that the low sequence recall of ModelAngelo is due to its low sequence match in the built model, while the low sequence recalls of CyroREAD and Phenix are because of their low residue coverages in the built models even though their built models have moderate residue coverage. Especially, Phenix wrongly recovers some nucleotides in the protein region and only builds a small part of the DNA/RNA structure with a very low coverage of 31.90% on this target, resulting in a low sequence recall (Fig. 3).
Examples of the EM2NA-built models
Figure 4 shows several examples of the EM2NA-built models on the targets with various sequence lengths, global folds, map resolutions, and molecule components including NA-only systems and protein-DNA/RNA complexes. In the sub-figures, the PDB models are superposed with the experimental density map. The EM2NA models are superposed with the density map and the PDB models for comparison.

In each panel, the PDB structures are colored in green, the EM2NA-built models are colored in blue, and the density maps are colored in transparent gray. The PDB structures are superposed with the density maps. The EM2NA-built models are superposed with the density maps and the PDB structures. The values of C4' RMSD, residue coverage, and sequence match are labeled below each model. a PDB 7OZ3, EMD-13119 at reported resolution 4.46 Å, S. agalactiae BusR in complex with its busA-promotor DNA. b PDB 7XD3, EMD-33134 at reported resolution 4.05 Å, the relaxed pre-Tet-S1 state of wild-type Tetrahymena group I intron. c PDB 7U2B, EMD-26311 at reported resolution 4.10 Å, a case for human mt-SerRS in complex with mt-tRNA(GCU-TL). d PDB 8DR0, EMD-27667 at reported resolution 2.42 Å, the closed state of RFC:PCNA bound to a 3' ss/dsDNA junction. e PDB 8T4S, EMD-41039 at reported resolution 2.60 Å, a complex of MERS-CoV Nsp1 protein bound to the human 40S Ribosomal subunit.
Figure 4a shows an example of EMD-13119 (PDB 7OZ3) at a reported resolution 4.46 Å. The complex is S. agalactiae BusR with its busA-promotor DNA. In this target, two DNA chains, forming a DNA duplex, are bound to four protein chains. The reported intermediate resolution of 4.46 Å is enough for EM2NA to model the target with very high coverage of 98.90%. The EM2NA-built model also has a good C4’ RMSD of 1.24 Å and a sequence match of 50.60% on this target.
Figure 4b shows a Tetrahymena ribozyme example of EMD-33134 (PDB 7XD3) at reported resolution 4.05 Å. The density map has a moderate resolution among widely-solved Tetrahymena ribozymes. On this target, the EM2NA-built model yields a C4’ RMSD of 1.26 Å, a coverage of 85.80%, and a sequence match of 60.06%. Further examination shows that there is a severe resolution heterogeneity in the map. As such, EM2NA misses some nucleotides in the loop region as the map resolution around the hairpins is poorer than the other region, making it hard to directly model the atomic structure from the map without prior knowledge.
For another map with similar resolution in Fig. 4c (PDB 7U2B, EMD-26311 at reported resolution 4.10 Å), EM2NA achieves a high coverage of 92.50%, an C4’ RMSD of 1.27 Å, and a sequence match of 61.20%. This target includes a human mt-SerRS in complex with mt-tRNA. Although the t-RNA chain in this map is a small system compared to other DNA/RNA structures, EM2NA still succeeds in detecting the DNA/RNA region and build the model with a high coverage (Fig. 4c).
In addition, EM2NA is also capable of modeling the nucleic acids from protein-abundant complexes, as shown in Fig. 4d. The target is a closed state of RFC:PCNA bound to a 3’ss/ds DNA junction, includes 2480 amino-acids but only 40 nucleotides, indicating most of the density region belongs to the protein. Another challenge of this target is that the DNA chains are bound to six protein chains and buried inside the surface, making it a challenge target in detecting and modeling the nucleotides. Nevertheless, EM2NA still builds the model of the target with a high coverage of 92.50%, an C4’ RMSD of 1.12 Å, and a sequence match of 78.40%.
Moreover, EM2NA is also good at modeling large ribosomal targets, such as target EMD-41039 PDB 8T4S (Fig. 4e). The complex is formed by a MERS-Cov Nsp1 protein and a human 40S Ribosomal unit. There are 5453 amino-acids and 1671 nucleotides in the PDB structure. The C4’ RMSD for the EM2NA built model is as low as 0.62 Å and the coverage is as high as 96.40%, and the sequence match is 53.90%. Since the map has a high resolution of 2.60 Å, EM2NA has recovered nearly all nucleotides.
Accuracy estimation of the built model
We introduced a local confidence score (LC-score) to estimate the modeling accuracy of a nucleotide in the EM2NA-built model. The local confidence score is defined as the interpolated densities of the nearest voxels in the predicted C4’ probability map around the C4’ atom of a nucleotide in the built model, as shown in Fig. 5a. The local confidence score does not take nucleotide identity into account. The scores are further smoothed along the chain with a window of weights 1:4:9:4:1 centered at each nucleotide. In addition, we also calculate the displacement of a built nucleotide, which is defined as the distance between its C4’ atom and the nearest C4’ atom in the reference PDB structure.

a Local confidence score is defined as the predicted atom probability at the C4' position. b The nucleotide displacements versus their local confidence scores for n = 8523 C4' atoms that represent the corresponding nucleotides on the main test set of 50 maps. The thick dots indicate the means and error bars/whiskers indicate ± 1.0 standard deviations. c The distribution (Fill Area, left y-axis) and accumulated percentages (Line+Symbol, right y-axis) of local confidences for the built nucleotides in helical and non-helical regions. d Box plots of local confidence scores for n = 5758 helical and n = 2765 non-helical nucleotides. The box plots indicate the means (cross), medians (median-line), first and third quartiles (bounds of boxes), and ± 1.0 standard deviations (whiskers). Mean values are marked near the box. e Examples of the EM2NA-built models colored by local confidence scores. The superimposed PDB structure is colored in green. From left to right are EMD-23600, PDB 7LYS, CasPhi-2 (Cas12j) bound to crRNA and DNA; EMD-23721, PDB 7M9A, ADP-AlF3 bound TnsC structure from ShCAST system; EMD-34832, PDB 8HJ4, anti-CRISPR protein AcrIIC5 bound to Nme1Cas9-sgRNA; and EMD-26856, PDB 7UXA, human tRNA Splicing Endonuclease Complex bound to pre-tRNA-ARG. As indicated in dashed rectangles, non-helical nucleotides tend to be built with lower confidences. Source data are provided as a Source Data file.
Figure 5b shows the local displacement versus the local confidence score for all the nucleotides of the built models on the test set of 50 maps (8523 nucleotides). The scores are merged into bins of 0.05. It can be seen from the figure that the local confidence of a nucleotide shows a good correlation with its local displacement. Therefore, the local confidence score can be used estimate the modeling accuracy of a nucleotide in the built model. Generally, a nucleotide with a confidence score of >0.4 approximately corresponds to a medium modeling accuracy of < 2.0 Å displacement, and a local confidence of >0.65 has a high modeling accuracy of <1.0 Å displacement. A lower local confidence score means a larger deviation from the true nucleotide position or a weak density around the nucleotide in the map, which may need further examination.
To further investigate the impact of DNA/RNA structure geometry on modeling accuracy, we compare the local confidence scores of the built nucleotides between helical and non-helical parts. This is done by first annotating the PDB structure to “helices" and “other" using x3dna-dssr38 and then querying the nearest PDB nucleotide for each built nucleotide. Figure 5c shows the frequency distributions and accumulated percentages at different local confidence for the built nucleotides. It can be seen from the figure that EM2NA tends to perform better on the helical nucleotides than those from non-helical parts. This is understandable because the double-helices in a DNA/RNA structure tend to be solved at a higher resolution. There are the most nucleotides at the local confidence score of 0.88 for helical parts, while the corresponding value is 0.2 for non-helical parts. As such, about 85% and 70% of built helical nucleotides have a medium (LC-score > 0.4) and high (LC-score > 0.65) modeling accuracy, respectively, compared to about 45% and 25% with for built nonhelical nucleotides (Fig. 5c). On average, EM2NA achieves an average local confidence score of 0.69 and 0.42 for helical and non-helical regions, respectively (Fig. 5d). In addition, we also investigated the impact of map local resolutions on local confidences. It can be seen from Supplementary Fig. 1 that as local resolution becomes worse, the average confidence score decreases. This can be understood because lower map resolution increases the uncertainty of atom position determination.
Figure 5e shows four examples of the EM2NA-built models that are colored according to the local confidence score. The corresponding PDB models are superposed to the built model in green. It can be seen from the figure that all the built models have a high accuracy with an C4’ RMSD of ~ 1 Å or better and an average confidence score of > 0.6. Normally, the helical nucleotides are built with a higher confidence than those non-helical ones, and loops or terminals are built less well compared to the double helices. However, it should be emphasized again that a low confidence score does not necessarily mean a wrong structure, but just needs further check.
Application to EMDB-wide unmodeled maps
Besides the main test set, we also applied EM2NA to build the DNA/RNA structures on an EMDB-wide data set of unmodeled experimental maps. The data set is constructed by searching the EMDataResource and filtering with “Single-Particle”, “has DNA" or “has RNA". Those maps with corresponding PDB structures or resolution >5.0 Å are excluded. The filtered data set includes approximately 300 unmodeled maps with resolution ≤5.0 Å. We then applied EM2NA to model the DNA/RNA structures for these maps. It should be mentioned that some maps may not contain DNA/RNA due to a wrong label. Therefore, for a few maps, EM2NA does not detect any DNA/RNA density regions. Finally, EM2NA successfully builds a DNA/RNA model ranging from 10 to 5347 nt for 263 maps (Supplementary Data 3).
Despite lacking of the corresponding PDB structures, we are able to estimate the local quality of the built models in a reference-free manner by local confidence score. Figure 6a shows the distribution and accumulated percentage as a function of the local confidence score for all the modeled nucleotides on both the EMDB and Main test sets. It can be seen from the figure that the frequencies in the local confidence interval of [0.80, 0.90) surpasses those in the other intervals. About 70% of the built nucleotides have a high accuracy with a local confidence score of > 0.6. It is also noted that the local confidence distribution for the EMDB test set is very close to that for the main test set and the average local confidence for the EMDB test set is slightly better (0.63 for EMDB test set versus 0.60 for the main test set). Such a trend suggests that the built models for the EMDB data set of unmodeled maps have a comparable accuracy to those for the main test set with known PDB structures. In addition, our EM2NA method is also robust to the maps with different resolutions and DNA/RNA lengths, and the average confidence score of the built model does not show a good correlation with map resolution (Fig. 6b) and the total length of the DNA/RNA structure (Fig. 6c).

a The distribution (Fill Area, left y-axis) and accumulated percentages (Line+Symbol, right y-axis) of local confidences for n = 64601 individual nucleotides built from n = 263 maps in the EMDB data set. b Average confidence score of each model as a function of map resolution. c Average confidence score of each model as a function of the number of the nucleotides in the built DNA/RNA model. d–f Examples of the EM2NA-built models. The built model (red) is superposed to density map (transparent gray) in each panel. The nucleotides in the built models are also colored from white (low confidence) to blue (high confidence). In d two RNA chains (57 nucleotides) are built from the map (EMD-3377, reported resolution 4.71 Å). The average confidence is 0.75. In e two DNA chains (290 nucleotides) are built from the map forming a DNA-duplex (EMD-8140, reported resolution 3.87 Å). The average confidence is 0.84. In f a large ribosome (5347 nucleotides) is built from the map (EMD-29405, reported resolution 3.10 Å) with an average confidence of 0.63. Dashed circles indicate the less confident nucleotides. Source data are provided as a Source Data file.
Figure 6d–f shows three examples of the built models. For EMD-3377, EM2NA models two RNA chains (a total of 57 nucleotides) in 10 minutes from the map with an intermediate resolution of 4.71 Å. The average confidence score is 0.75 for this target (Fig. 6d). Only one or two nucleotides at the terminal show lower confidences. For EMD-8140, the built model includes two DNA strands forming a DNA-duplex (Fig. 6e). EM2NA builds the model of this target using only 6 minutes. The local confidence estimation shows that nearly all the nucleotides are built with a high confidence, and the average confidence is 0.84 for this target. The side chain conformations are also built very well, in which the base pairs can be observed clearly. As such, it is easy to recognize the model as a typical nucleosome structure (a circular DNA-duplex surrounding the proteins chains), which has been widely studied by researchers. For EMD-29405, this is the largest target containing up to 5347 nucleotides in the EM2NA model (Fig. 6f). It takes approximately 8 hours for EM2NA to build such a large system. Most of the built nucleotides have a high accuracy with a good confidence of > 0.6. Only a few terminals or loops show lower confidence, as they are more flexible than the buried regions (Fig. 6f). On average, the built model gives a good confidence of 0.63 for this target.
Although the EMDB entries do not have a labeled corresponding PDB structure, they may have related/similar structures in the PDB database. Therefore, we further searched the EMDB and identified the studies that the EMDB IDs are involved. After careful inspection, 80 maps match a higher resolution reference structure, on which the C4’ RMSD and residue coverage are calculated accordingly. As shown in Supplementary Data 4, EM2NA can still build good models with a C4’ RMSD of 1.29 Å and a residue coverage of 75.20% on average, although the results are not as good as those for the cases that have a PDB structure. The performance drop for the unmodeled maps can be understood for two reasons. On one hand, researchers often build more than one maps for the same target and would build the PDB structure for the best-quality map in real cryo-EM applications. On the other hand, some reference PDB models used for evaluations may not be exactly the same target as those for the maps.
Computational efficiency
The computational efficiency of EM2NA is also investigated. Figure 7 shows the running times of the deep learning stage, the atomic modeling stage, or the summation of both as a function of the total length of the built model on the total of 313 maps (50 maps in the main test set plus 263 maps in EMDB test set). The ‘deep learning time’ refers to the time consumption for segmenting proteins and nucleic acids, and predicting the atom probability maps and nucleotide types. It is running before the atomic modeling stage. The ‘atomic modeling time’ means the time consumption of the following steps after we generate the predicted atom probability maps and nucleotide maps. The deep learning stages (stage-1,2) are performed on one Nvidia A100 GPU (40 GB) for acceleration, and the atomic modeling stage is run on a single core of Intel(R) Xeon(R) Gold 6240 CPU @ 2.60GHz. As shown in Fig. 7, the running time for deep learning has a linear relationship with the map sizes, and the running time for atomic modeling has a square relationship with the number of built nucleotides, where the latter dominates the total running time for large DNA/RNA structures. Overall, the total running time has a square relationship with the length of the built DNA/RNA structure. Normally, EM2NA can segment a map of up to 600 MB within 25 minutes and build a DNA/RNA structure model of less than 500 nt within 10 minutes. For those systems that have smaller map sizes and few nucleotides, EM2NA can complete the model building within 5 minutes. Running EM2NA with more advanced GPUs will take a shorter time, depending on what GPU the user is actually using. Running on different CPUs will not significantly influence the running time.

a The scatter plot of the deep learning time versus the map sizes. b The scatter plot of the atomic modeling time versus the number of the nucleotides in the built model. c The scatter plot of the total running time versus the number of the nucleotides in the built model. Red lines/curves indicate the best fits. Source data are provided as a Source Data file.
Discussion
We have proposed a deep learning-based approach to automatically build DNA/RNA structures from cyro-EM maps, named EM2NA, based on an efficient Swin-Conv-UNet architecture trained with a combination of multiple learning objectives. EM2NA builds the full-atom DNA/RNA structure from the map by integrating several modeling steps: segmenting the DNA/RNA density regions from raw cryo-EM maps, predicting the backbone atom positions and nucleotide types from the segmented DNA/RNA map, iteratively threading possible atom positions into backbone traces, aligning the backbone traces with sequences and detecting base-pairs to assign nucleotide types, and constructing the full-atom structure with template nucleotide conformations. The whole modeling process is fully automated and does not require any human interventions.
EM2NA was extensively evaluated on a benchmark test set of 50 experimental cryo-EM maps. It is shown that EM2NA outperforms three state-of-the-art methods including CryoREAD, ModelAngelo, and Phenix (phenix.map_to_model). For recovering DNA/RNA structures, EM2NA achieves a high residue coverage of 83.15% and low C4’ RMSD of 1.06 Å, which are substantially better than 47.04% and 2.28 Å for CryoREAD, 74.23% and 1.17 Å for ModelAngelo, and 40.87% and 1.10 Å for Phenix. For assigning nucleotide types, EM2NA yields a good sequence recall of 46.86%, compared with 24.20% for CryoREAD, 20.42% for ModelAngelo, and 16.75% for Phenix. In addition, we also applied EM2NA for blind model building on an EMDB-wide data set of 263 unmodeled maps, demonstrating that EM2NA can also build the DNA/RNA structures with good confidences. These results demonstrate the accuracy, robustness, and general applicability of EM2NA. Moreover, EM2NA is fast and can normally build a DNA/RNA model containing <500 nt within 10 minutes, making it a valuable tool in accelerating DNA/RNA structure determination from cryo-EM maps.
Although EM2NA surpasses the existing methods in both modeling automation and performance, it cannot replace humans. For example, assigning the correct nucleotide type for the built model is still a major challenge for EM2NA and other methods. The use of prior knowledge, through human intervention, is still necessary to guide modeling. Besides, it’s also challenging to handle NA’s flexibility given that NA polymer geometry in protein-NA complexes can sometimes differ from standard helix conformation. Furthermore, bulges or flipped-out nucleotides can exist within a natural A-form RNA helical structure. These conformations may not recognized by EM2NA. Nevertheless, EM2NA gives a fast, reliable and automatic interpretation of the map, and removes many barriers in the way of building a model from scratch.
Methods
The network architecture
A Swin-Conv-UNet (SCUNet)39,40 architecture is used to extract the DNA/RNA information from the map. The network consists of three encoder, one transition, and three decoder Swin-Conv (SC) blocks with skip connections between encoders and decoders. Here, the “Swin” represents the Shifted window transformer, which exhibits a good nonlocal modeling capability by computing self-attention of non-overlapping windows while also allowing for cross-window connection41; The “Conv” stands for Convolutional network, which is known for its efficient local modeling capability. Therefore, compared with traditional networks like CNN, the Swin-Conv block has the advantage of both local and nonlocal learning capabilities39. Each Swin-Conv block includes a Swin transformer (SwinT) block that is paralleled with a residual convolutional (RConv) block sandwiched by two 1 × 1 convolutions. The window size of the Swin transformer is set to 3. The 3D convolution layer with kernel size and stride of 2 is used as down-sampling, and the 3D transposed convolution layer with kernel size and stride of 2 is used as up-sampling. The inputs of our network are the density chunks of size 48 × 48 × 48 with a grid interval of 1.0 Å. The outputs are of the same size. Our deep learning detection uses a two-stage pipeline to predict DNA/RNA backbone information. Both stage uses the same network but differs only in the final output channels.
Data sets
The experimental cryo-EM density maps and corresponding PDB structures used in the study were collected from the Electron Microscopy Data Bank (EMDB)42 and Protein Data Bank13. All the EM density maps with resolutions from 2.0 Å to 5.0 Å that have corresponding PDB structures were downloaded from the EMDB. Any EM map (and the associated PDB structure) that met the following criteria were removed: (i) The map has non-orthogonal axis; (ii) The resolution is not given by the FSC-0.143 cut-off; (iii) The reconstruction method is not single-particle analysis. Any PDB structure (and the associated EM map) that met the following criteria were also removed: (i) containing protein only; (ii) having un-assigned nucleotide types; iii) containing backbone atoms only. iv) having severe misfits between map and associated atomic structure. To quantify the criterion (iv), the atomic structures were first simulated to density maps by UCSF Chimera43 and a threshold of 0.60 was selected to filter the maps. For computational efficiency, large ribosomes ( > 3000 nucleotides) are removed because they often contribute too much training data.
All the filtered maps are subject to manual examinations, yielding a total of 424 maps. Afterward, the PDB structures of 424 maps are clustered using MMseqs244. If the longest NA chain from one PDB structure has a sequence identity of > 0.8 to the longest NA chain from another PDB structure, the two PDB structures will belong to one cluster, yielding a final set of 196 clusters. To ensure the diversity of structures and maps, we split the cluster representatives to three categories: ‘NA_only’, ‘large’ and ‘other’. The ‘NA_only’ means the PDB structure contains only nucleic acids and no proteins, the ‘large’ means the structure containing >1000 nucleotides, and the ‘other’ means the others. For each category, we kept 25% of the items as the test set, forming a total of 50 maps. The maps from the other clusters were used to train the networks. Among the training maps, 60% of them is randomly selected to train the stage-1 network and 40% is selected to train the stage-2 networks. For each stage, 80% of the maps are used as the training set and 20% of the maps are used as the validation set (Supplementary Data 5).
It should be noted that here the sequence identity cutoff of 0.8 is used for clustering because it is revealed that the RMSDs between two homologous RNA structures exhibit a relatively large gap before and after the sequence identity of 0.845. Given the limited number of RNA structures in the PDB, such a cutoff selection can achieve a good balance between the number and structural diversity of the RNA structures in the training set. Although the sequence identity of 0.8 is too high to remove the redundancy for the proteins, setting a lower protein sequence identity cutoff (e.g. 0.2) will not significantly change the performance of EM2NA because EM2NA is mainly trained on the NA structures, as shown in Supplementary Fig. 2.
Network training
Detection of the nucleic acid information for experimental density maps is based on the trained networks. Here, we adopted a two-stage object detection framework according to our early attempts. The stage-1 network is used to segment nucleic acid densities out of a raw map, and the stage-2 network is used to detect the nucleotide location and type from the segmented DNA/RNA map. To train the stage-1 network, we label the voxels according to the nearest heavy atoms within 3 Å of the PDB structure. The voxel is labeled ‘0’ if the atom is from proteins, ‘1’ if the atom is from DNA/RNA, and ‘2’ if no heavy atom is within 3 Å or the atom is from other chemical components like water/ions/small molecules. The loss function for stage-1 is a classification loss, which is a combination of CrossEntropyLoss and DiceLoss,
where ypred and yGT stand for the predicted and ground-truth maps for a cropped box, respectively.
To train the stage-2 network, we first define the backbone of nucleic acids as the P, C4’, and N1(N9) atoms, which represent the phosphate, ribose, and base for each nucleotide. The N1 atom is for pyrimidine and the N9 atom is for purine, which represents the first atom in the base connected to the ribose. We select P, C4’ and N1 or N9 atoms as the coarse-grained backbone representations for the following reasons: i) They are from different functional groups (P for phosphate, C4’ for ribose, and N1/N9 for base); ii) The N1 and N9 atoms are equivalent for different bases, as they are the first atoms connected to the ribose; iii) The pseudo-bond between them is around 3-4 Å, and thus it is possible to distinguish these 3 atoms for a map at near-atomic resolution.
The training label is created according to the PDB structure and map resolution. We first extract all nucleotides in the PDB structure, and zone the raw maps within 3 Å of the heavy atoms in the nucleotides. To define a ground truth P atom probability map, we extract the P atoms from the nucleotides. The density value at grid point x is calculated as follows.
where a is the coordinate of the P atom in chain A and the kernel k is defined according to map resolution R as k = (π/(1.2+0.8R))2. The C4’ and N1(N9) probability maps are prepared following a similar manner. Next, to define a ground-truth nucleotide type map, we label the voxels according to the nearest atoms within 3 Å. If the nearest atom is from an A/G/C/U(T), the voxel is labeled to be 0/1/2/3. For other conditions, the voxel is label to be 4. The loss for atom probability prediction is a combination of smooth L1 loss and structural similarity46 (SSIM) loss as follows25.
The loss of nucleotide type prediction is a 4-class Cross-Entropy loss, where the prediction of label-4 is ignored because only the prediction near the backbone is of interest.
To prepare the training data, the grid size of the map is unified to 1.0 Å by trilinear interpolation. The density values of each experimental map are first clipped to between ≥0.0 and ≤99.999% percentile of the density values and then normalized to [0, 1]. The experimental maps and labeled maps are cut into overlapping chunks of size 60 × 60 × 60 with a stride of 30. The experimental chunks with a maximal density value less than 0.0 are excluded from training.
During training, data augmentation is used to prevent overfitting. The input box is augmented by randomly cropping a 48 × 48 × 48 (Å3) box out of a 60 × 60 × 60 (Å3) chunk and 90∘ random rotation is applied on the box. The network is implemented by PyTorch 1.9.0. Adam optimizer is used to update the network weights. We train the nucleic acid/protein density predictor, atom probability predictor (P, C4’, N1/N9), and nucleotide type predictor separately with the same hyper-parameters. The initial learning rate is set to 5e-4 and the L2 regularization coefficient weight_decay is set to 1e-4. During the training, the learning rate will reduce to its half if the average loss of the training process does not descend for 4 epochs. The minimal learning rate is set to 1e-6. We trained each of the networks with a batch size of 56 on two Nvidia Tesla A100 GPU with 40GB memory. The models with the least validation loss are selected as our final models.
Building DNA/RNA backbones
With the predicted P, C4’, and N1(N9) probability maps, the DNA/RNA backbone is constructed by the following steps. The predicted P and C4’ atom probability maps are converted to the atom positions/points by detecting their local maximums using a mean-shift algorithm. The combined P and C4’ positions are threaded to multiple paths by solving a Vehicle Routing Problem (VRP) algorithm47. The VRP allows multiple paths for traveling, which is suitable for building multi-chain DNA/RNA structures here. We estimate the maximal number of backbone fragments \({n}_{path}^{max}\) as
where \({N}_{C{4}^{{\prime} }}\) is the number of C4’ points, and the minimal \({n}_{path}^{min}\) as
and then do a binary-search of the best npath starting from
Namely, if the distance between consecutive points in a path is > 12.0 Å, the npath should be increased because the path can be split into two sub-paths due to the large break. If the distance is <4.0 Å, the npath should be decreased because the two paths should be connected. The searching process is repeated at most 3 times to obtain an appropriate number of backbone paths.
Afterwards, for each path, we extract the C4’ points, and then insert a P point for each of the two consecutive C4’ points. We add two more P points at the beginning and at the end as auxiliary points to help determine the correct path direction. In total, we have n C4’ points and (n + 1) P points in the path and regardless of the path direction, they are arranged as
Next, determining the correct path direction is straightforward by taking the nucleotide bonds and geometries into account. Specifically, for each C4’ point (\({n}_{C{4}^{{\prime} }}^{i}\)), we query its nearest N1/N9 point (\({n}_{N}^{i}\)) and then divide the path above into ‘forward’ and ‘backward’ directions. For the ‘forward’ direction, the P point (\({n}_{p}^{n+1}\)) at the end is removed from the path. The 3-atom representation of a nucleotide can thus be built according to the previous P point (\({n}_{P}^{i}\)) and the current N1/N9 point (\({n}_{N}^{i}\)) and C4’ point (\({n}_{C{4}^{{\prime} }}^{i}\)). For the ‘backward’ direction, the P point (\({n}_{p}^{1}\)) at the beginning is removed and the path is reversed. Similarly, the next P point (\({n}_{P}^{i+1}\)) and the current N1/N9 atom (\({n}_{N}^{i}\)) and C4’ point (\({n}_{C{4}^{{\prime} }}^{i}\)) together defines the nucleotide.
With the 3-atom nucleotide paths for both directions, we can reconstruct them into pseudo-full-atom structures by aligning all template “U” nucleotide conformations to the path based on the P-C4’-N backbone atoms using Arena48 algorithm. Then, the O3’-P bond length between consecutive nucleotides is calculated. By comparing the sum of the bond lengths, the correct direction can be determined because the correct path should have a minimal bond length. This is a critical step in constructing the backbone. According to our observation, the models generated by some tools sometimes exhibit a reversed path direction and an improper nucleotide conformation, as they determine the path direction based on information from sequence alignment, neglecting the nucleotide geometries.
Assigning nucleotide types
The nucleotide types, i.e. A/G/C/U(T), can be assigned to the built DNA/RNA backbone chains/paths with or without providing DNA/RNA sequences. If the target sequences are not provided, we will assign the nucleotide types solely to the deep learning-predicted ones. Namely, the type (A/G/C/U(T)) for each nucleotide is assigned by voting the nearest voxels within 3 Å of each C4’ atom. If the target sequence is provided, after the above assignment, a Smith-Waterman sequence alignment is used to inference the nucleotide types. The scoring matrix is defined by analyzing the base type predictions, including correct prediction and wrong prediction and their frequencies. The gap penalty is set to be −0.50 in the sequence alignment.
We also utilize the helical geometry in the built backbone to refine the nucleotide type assignment. The correctly built backbone (P-C4’-N) enables us to well determine whether two nucleotides can form a base pair with the help of the coarse-grained SS annotation method CSSR49. Specifically, we first initialize a count matrix P with zeros and shape of N × N. Here, N is the number of all nucleotides. Then, we extract all fragments for each chain using shifted windows with length=20 and stride=5. For fragments i, k, we label i to be all ‘A’ and k to be all ‘U’ to see if any possible pairs exist between them using the CSSR method. Afterward, we further check the possible pairs that meet the following criteria: i) if two nucleotides (m, n) are paired, the nucleotides (m + 1, n − 1) or (m − 1, n + 1) are paired too; ii) abs(n − m) > = 4; iii) 14 Å \( < {\text{inter-C4}}^{{\prime} }{\text{-distance}}\,(m,n) < 18\) Å. For the left pairings, we let the count matrix P(m, n) = P(m, n) + 1, and P(n, m) = P(n, m) + 1 for all paired (m, n) in the two fragments. After obtaining the count matrix that records the possible pairing frequency, we extract (m, n)* = argmax(P > 0), then set m, n as being visited, and set P(m, . . . ), P(. . . , m), P(n, . . . ), and P(. . . , n) to be 0. This operation repeats until all nucleotides are visited or no more (m, n)* is paired. Based on the detected pairs, we can refine the sequence assignment based on the fact that the paired nucleotides should satisfy the base pairing principle of A-U/T and C-G. For the paired nucleotides (m, n), if nucleotide-type-of(m) is predicted with a higher probability than nucleotide-type-of(n), we will set nucleotide-type-of(n) to be the complementary nucleotide type of m, or vice versa. After the nucleotide assignment, the final sequence may not agree with the input sequence. In some cases, the assigned sequence is only a substring of the input sequences. If users input DNA/RNA hybrid sequences, EM2NA will automatically determine whether the chains are built into DNA or RNA, according to the best sequence alignment.
Full-atom construction
Based on the built DNA/RNA backbones and assigned nucleotide types, the full-atom structure is built by aligning template nucleotide conformations onto the backbone with the Arena48 algorithm. If the target sequences are not provided, it is necessary to determine if the structure is an RNA (A-form) or DNA (B-form) according to their helices. Then, we can build the correct base conformation for the backbone. To achieve this, we simply compare the detected pairings with the RNA/DNA helix templates generated by 3DNA38. If more than 50% of the base pairs in two fragments are more similar to DNA(B-form), then the backbone fragments are built into the full-atom structure with DNA nucleotides, and vice versa. Such a treatment may not always capture the correct polymer geometry as RNAs in protein-RNA complexes can differ from standard A-form helix. Furthermore, RNA also allows bulges or flipped-out nucleotides to harmoniously exist within a mainly A-form natural helical structure. After a model is built, a post-refinement is recommended to remove severe atomic clashes by using a third-party program like phenix.real_space_refine50 or MD simulations. Our experiment shows that a quick phenix.real_space_refine can slightly improve the RMSD (Supplementary Data 6), though the models used in the study are not refined by default.
Evaluation of the built models
Four metrics, including backbone C4’ RMSD, residue coverage, sequence match, and sequence recall, are used to measure the accuracy of a built DNA/RNA model. The values of these metrics are calculated using the program phenix.chain_comparison9. Specifically, the C4’ RMSD is calculated by querying the nearest representative atom in the target structure within a distance cutoff. The residue coverage measures how many nucleotides are built from the map regardless of nucleotide types relative to the PDB structure, while the sequence match means how many nucleotides in the built model have the same nucleotide types as those in the PDB structure. Sequence recall is the product of residue coverage and sequence match, which measures how many nucleotides in the built model are correctly built from the map in terms of both backbone position and nucleotide type when compared to the PDB structure. All metrics are calculated with a distance cutoff of 3 Å and the C4’ as the representative atom for each nucleotide.
Comparison with related works
We have compared EM2NA with three state-of-the-art methods including CryoREAD, ModelAngelo, and phenix.map_to_model. These methods are all optimized for near-atomic resolution maps. Specifically, CryoREAD is designed for maps with <5 Å resolution, ModelAngelo is designed for maps with <4 Å resolution, and phenix.map_to_model is designed for maps with <4.5 Å resolution. The input sequences are the same for each method. The running command details are listed below.
CryoREAD
We used CryoREAD version 8.2. CryoREAD is downloaded from its authors’ GitHub repository and run according to the instructions on the website. We first ran the deep learning prediction for all the targets by the following command.
-
python3 main.py –mode=0 -F=MAP.mrc -M=best_model_paper –contour=0.5*AUTHOR_RECOMMENDED_CONTOUR –gpu=4 –batch_size=16 –prediction_only
Then, we then ran the modeling command as follows.
-
python3 main.py –mode=0 -F=MAP.mrc -P=SEQ.fasta -M=best_model_paper –contour=0.5*AUTHOR_RECOMMENDED_CONTOUR –gpu=4 –batch_size=16 –rule_soft=0 –resolution RESOLUTION –thread 2
ModelAngelo
We used the ModelAngelo version 1.0.12. The command for modeling is as follows.
-
model_angelo build -v MAP.mrc -pf PROT.fasta -rf RNA.fasta -df DNA.fasta
phenix.map_to_model
We ran phenix.map_to_model in the Phenix suite of version 1.19.2-4158. The command for modeling is as follows.
-
phenix.map_to_model MAP.mrc SEQ.fasta Resolution=RESOLUTION symmetry=C1
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The raw data of the evaluation results are provided in the Article and Supplementary Data. All published data sets used in this paper were taken from the EMDB and PDB (accession codes specified in the figure captions and in Supplementary Data). The atomic models built by EM2NA (50 items in the main test set) has been deposited in Zenodo51. The atomic models for the EMDB 263 items has also been deposited in Zenodo52. Source data are provided with this paper.
Code availability
The EM2NA package is freely available for academic or non-commercial users at http://huanglab.phys.hust.edu.cn/EM2NA/or https://github.com/emberslee/EM2NA/.
References
Ma, H., Jia, X., Zhang, K. & Su, Z. Cryo-EM advances in RNA structure determination. Signal Transduct Target Ther. 7, 58 (2022).
Su, Z. et al. Cryo-EM structures of full-length Tetrahymena ribozyme at 3.1 Å resolution. Nature 596, 603–607 (2021).
Luo, B. et al. Cryo-EM reveals dynamics of Tetrahymena group I intron self-splicing. Nat. Catal. 6, 298–309 (2023).
Liu, D., Thélot, F. A., Piccirilli, J. A., Liao, M. & Yin, P. Sub-3- Å cryo-EM structure of RNA enabled by engineered homomeric self-assembly. Nat. Methods 19, 576–585 (2022).
Nogales, E. The development of cryo-EM into a mainstream structural biology technique. Nat. Methods 13, 24–27 (2016).
Frank, J. Advances in the field of single-particle cryo-electron microscopy over the last decade. Nat. Protoc. 12, 209–212 (2017).
Cheng, Y. Single-particle cryo-EM-How did it get here and where will it go. Science 61, 876–880 (2018).
Zivanov, J. et al. New tools for automated high-resolution cryo-EM structure determination in RELION-3. Elife 7, e42166 (2018).
Terwilliger, T. C., Adams, P. D., Afonine, P. V. & Sobolev, O. V. A fully automatic method yielding initial models from high-resolution cryo-electron microscopy maps. Nat. Methods 15, 905–908 (2018).
Terwilliger, T. C., Adams, P. D., Afonine, P. V. & Sobolev, O. V. Cryo-EM map interpretation and protein model-building using iterative map segmentation. Protein Sci. 29, 87–99 (2020).
De la Rosa-Trevín, J. M. et al. Scipion: A software framework toward integration, reproducibility and validation in 3D electron microscopy. J. Struct. Biol. 195, 93–99 (2016).
Punjani, A., Rubinstein, J. L., Fleet, D. J. & Brubaker, M. A. cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296 (2017).
Berman, H. M. et al. The Protein Data Bank. Nucleic Acids Res. 28, 235–242 (2000).
Kappel, K. et al. De novo computational RNA modeling into cryo-EM maps of large ribonucleoprotein complexes. Nat. Methods 15, 947–954 (2018).
Baker, M. L. et al. Modeling protein structure at near atomic resolutions with Gorgon. J. Struct. Biol. 174, 360–373 (2011).
Lindert, S. et al. EM-fold: De novo folding of alpha-helical proteins guided by intermediate-resolution electron microscopy density maps. Structure 17, 990–1003 (2009).
Chen, M. & Baker, M. L. Automation and assessment of de novo modeling with Pathwalking in near atomic resolution cryoEM density maps. J. Struct. Biol. 204, 555–563 (2018).
Wang, R. Y. et al. De novo protein structure determination from near-atomic-resolution cryo-EM maps. Nat. Methods 12, 335–338 (2015).
Frenz, B. et al. RosettaES: a sampling strategy enabling automated interpretation of difficult cryo-EM maps. Nat. Methods 14, 797–800 (2017).
Terashi, G. & Kihara, D. De novo main-chain modeling for EM maps using MAINMAST. Nat. Commun. 9, 1618 (2018).
Si, D. et al. Deep learning to predict protein backbone structure from high-resolution Cryo-EM density maps. Sci. Rep. 10, 4282 (2020).
He, J. & Huang, S.-Y. Full-length de novo protein structure determination from cryo-EM maps using deep learning. Bioinformatics 37, 3480–3490 (2021).
Pfab, J., Phan, N. M. & Si, D. DeepTracer for fast de novo cryo-EM protein structure modeling and special studies on CoV-related complexes. Proc. Natl. Acad. Sci. USA 118, e2017525118 (2021).
He, J. & Huang, S.-Y. EMNUSS: a deep learning framework for secondary structure annotation in cryo-EM maps. Brief. Bioinform. 22, bbab156 (2021).
He, J., Lin, P., Chen, J., Cao, H. & Huang, S.-Y. Model building of protein complexes from intermediate-resolution cryo-EM maps with deep learning-guided automatic assembly. Nat. Commun. 13, 4066 (2022).
Zhang, X., Zhang, B., Freddolino, P. L. & Zhang, Y. CR-I-TASSER: assemble protein structures from cryo-EM density maps using deep convolutional neural networks. Nat. Methods 19, 195–204 (2022).
Zhou, X. et al. Progressive assembly of multi-domain protein structures from cryo-EM density maps. Nat. Comput. Sci. 2, 265–275 (2022).
Jamali, K. et al. Automated model building and protein identification in cryo-EM maps. Nature 628, 450–457 (2024).
Nguyen, T. H. D. 5 tri-snRNP. Nature 523, 47–52 (2015).
Greber, B. J. et al. Architecture of the large subunit of the mammalian mitochondrial ribosome. Nature 505, 515–519 (2014).
Chaker-Margot, M., Barandun, J., Hunziker, M. & Klinge, S. Architecture of the yeast small subunit processome. Science 355, eaal1880 (2017).
Li, X. et al. Structure of ribosomal silencing factor bound to mycobacterium tuberculosis ribosome. Structure 23, 1858–1865 (2015).
Keating, K. S. & Pyle, A. M. RCrane: semi-automated RNA model building. Acta Crystallogr. D Biol. Crystallogr. 68, 985–995 (2012).
Nakamura, A. Fast and automated protein-DNA/RNA macromolecular complex modeling from cryo-EM maps. Brief. Bioinform. 24, bbac632 (2023).
Wang, X., Terashi, G. & Kihara, D. CryoREAD: de novo structure modeling for nucleic acids in cryo-EM maps using deep learning. Nat. Methods 20, 1739–1747 (2023).
Kappel, K. et al. Accelerated cryo-EM-guided determination of three-dimensional RNA-only structures. Nat. Methods 17, 699–707 (2020).
Li, T. et al. All-atom RNA structure determination from cryo-EM maps. Nat. Biotechnol. https://doi.org/10.1038/s41587-024-02149-8 (2024).
Lu, X. J., Bussemaker, H. J. & Olson, W. K. DSSR: an integrated software tool for dissecting the spatial structure of RNA. Nucleic Acids Res. 43, e142 (2015).
Zhang, K. et al. Practical blind image denoising via Swin-Conv-UNet and data synthesis. Mach. Intell. Res. 20, 822–836 (2023).
He, J., Li, T. & Huang, S.-Y. Improvement of cryo-EM maps by simultaneous local and non-local deep learning. Nat. Commun. 14, 3217 (2023).
Liu, Z. et al. Swin transformer: Hierarchical vision transformer using shifted windows. In Proceedings of the IEEE/CVF International Conference on Computer Vision, (2021).
Lawson, C. L. et al. EMDataBank unified data resource for 3DEM. Nucleic Acids Res. 44, D396–D403 (2016).
Pettersen, E. F. et al. UCSF Chimera–a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 (2004).
Steinegger, M. & Söding, J. MMseqs2 enables sensitive protein sequence searching for the analysis of massive data sets. Nat. Biotechnol. 35, 1026–1028 (2017).
Capriotti, E. & Marti-Renom, M. A. Quantifying the relationship between sequence and three-dimensional structure conservation in RNA. BMC Bioinform 11, 1–10 (2010).
Wang, Z., Bovik, A. C., Sheikh, H. R. & Simoncelli, E. P. Image quality assessment: from error visibility to structural similarity. IEEE Trans. Image Process. 13, 600–612 (2004).
Helsgaun, K. An extension of the Lin-Kernighan-Helsgaun TSP solver for constrained traveling salesman and vehicle routing problems. Roskilde: Roskilde Univ. 12, 966–980 (2017).
Perry, Z. R., Pyle, A. M. & Zhang, C. Arena: rapid and accurate reconstruction of full atomic RNA structures from coarse-grained models. J. Mol. Biol. 435, 168210 (2023).
Zhang, C. & Pyle, A. M. CSSR: assignment of secondary structure to coarse-grained RNA tertiary structures. Acta Crystallogr. D Struct. Biol. 78, 466–471 (2022).
Afonine, P. V. et al. Real-space refinement in PHENIX for cryo-EM and crystallography. Acta Crystallogr. D Struct. Biol. 74, 531–544 (2018).
Li, T., Huang, S.-Y. EM2NA program. Zenodo https://doi.org/10.5281/zenodo.13865219 (2024).
Li, T. EM2NA models for the EMDB dataset. Zenodo https://doi.org/10.5281/zenodo.12742831 (2024).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (grant Nos. 32161133002 and 62072199) and the startup grant of Huazhong University of Science and Technology.
Author information
Authors and Affiliations
Contributions
S.H. conceived and supervised the project. T.L., H.C., and J.H. designed and performed the experiments. S.H. and T.L. analyzed the data. T.L. and S.H. wrote the manuscript. All authors read and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Arne Elofsson, Luca Gebert and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Li, T., Cao, H., He, J. et al. Automated detection and de novo structure modeling of nucleic acids from cryo-EM maps. Nat Commun 15, 9367 (2024). https://doi.org/10.1038/s41467-024-53721-4
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-024-53721-4
This article is cited by
-
Integrated experimental and AI innovations for RNA structure determination
Nature Biotechnology (2026)
-
AI meets RNA: revolutionizing structure prediction
Science China Life Sciences (2025)
