
Abstract
The discovery of CRISPR-Cas systems has paved the way for advanced gene editing tools. However, traditional Cas discovery methods relying on sequence similarity may miss distant homologs and aren’t suitable for functional recognition. With protein large language models (LLMs) evolving, there is potential for Cas system modeling without extensive training data. Here, we introduce CHOOSER (Cas HOmlog Observing and SElf-processing scReening), an AI framework for alignment-free discovery of CRISPR-Cas systems with self-processing pre-crRNA capability using protein foundation models. By using CHOOSER, we identify 11 Casλ homologs, nearly doubling the known catalog. Notably, one homolog, EphcCasλ, is experimentally validated for self-processing pre-crRNA, DNA cleavage, and trans-cleavage, showing promise for CRISPR-based pathogen detection. This study highlights an innovative approach for discovering CRISPR-Cas systems with specific functions, emphasizing their potential in gene editing.
Similar content being viewed by others

Introduction
Clustered regularly interspaced short palindromic repeats (CRISPR)-CRISPR-associated (Cas) systems, which are adaptive immune systems in bacteria and archaea, have been successfully transformed into a variety of powerful genome editing tools over the past decade1,2. Ongoing reports of newly discovered CRISPR-Cas systems and subtypes have shown that not only do bacteria and archaea possess these adaptive immune systems, but viruses (particularly phages) also harbor a vast number of undiscovered CRISPR-Cas systems3,4,5,6,7,8,9,10,11,12,13,14,15.
Among the various DNA-targeting single-effector systems, specific Cas12 effectors, including Cas12a16, Cas12i17, CasΦ7, and Casλ8, are known for not only their ability to cleave double-stranded DNA (dsDNA) targets but also their RNase activity, enabling them to process their own precursor CRISPR RNA (pre-crRNA) specifically (Supplementary Table 1). These Cas12 effectors can directly utilize compact CRISPR RNA (crRNA) arrays, which are significantly easier to construct than Cas9 single-guide RNA arrays; this attribute allows the potential use of these effectors for multiplexed genetic modifications, which are crucial for elucidating and modulating gene interactions and networks that underpin complex cellular function. In a previous study, an LbCas12a effector was developed into an effective platform for multiplexed genome engineering that is capable of targeting endogenous genes, with up to 25 individual crRNAs delivered on a single plasmid when a stabilizer tertiary RNA structure is included18. Additionally, an engineered variant of Cas12i2, which is guided by a short crRNA without the need for a transactivating crRNA (tracrRNA), has been developed into a versatile and high-performance editing tool for in vivo gene therapy19. Given these advancements, functional prioritization of Cas12 candidates with the ability to self-process their pre-crRNA is important because these candidates can enhance the applicability and efficiency of gene editing technologies.
Discovery and functional screening of these CRISPR systems require the accurate identification of nearby Cas proteins, especially system effectors. Specifically, two main strategies are employed to identify Cas proteins: (1) The first strategy is straightforward and involves identifying Cas homologs based on amino acid sequence similarity. Bioinformatics pipelines, such as NCBI’s Prokaryotic Genome Annotation Pipeline (PGAP)20 and CRISPRCasTyper21, use this strategy, relying on BLAST alignments22 and/or HMM profile searches23 against known Cas protein sequences. However, this sequence similarity approach can miss potential distant homologs. A recent study has uncovered 188 rare and previously unrecognized CRISPR-associated gene modules by employing FLSHclust, an algorithm designed for a deep terascale clustering of proteins based on sequence similarity24. This study indicates that while Cas proteins demonstrate ‘LEGO-like’ modularity in their sequence arrangements, they maintain conserved key functional domains that can be used for remote homolog searching; however, these alignment-based methods can not be applied directly for functional recognition. (2) The second strategy involves identifying proteins based on structural similarity besides amino acid sequence similarity4. This strategy is based on the ‘structure-function’ paradigm prevalent in biology and biochemistry, which states that the functions of proteins are determined by their structures. Therefore, evolving protein structure prediction technologies are expected to play crucial roles in CRISPR-Cas discovery and functional screening. The success of AlphaFold2 demonstrated that its sophisticated transformer-based models can effectively learn relevant representations from multiple sequence alignments (MSAs) of protein homologs, identify evolutionarily conserved patterns and coevolving residues, and subsequently reconstruct the three-dimensional (3D) structure of a protein25. In contrast to the two-stage model of AlphaFold2, RoseTTAfold uses rotation- and translation-invariant SE(3) transformer modules in its three-track design, incorporating 1D, 2D, and 3D representations26. However, most of these technologies are dependent on a comprehensive reference dataset that includes a sufficient number of known homologs. In the case of Cas protein identification (especially for effectors of some type V and VI subtypes), the number of known homologs is limited, posing a great obstacle for constructing a reference dataset and for the subsequent discovery and functional investigation of Cas proteins using structure prediction tools. Overall, the systematic discovery and functional screening of CRISPR-Cas systems with limited homologs and a lack of ground-truth structural information are highly important but challenging.
Large-language models (LLMs) have utilized their powerful representation learning ability in diverse fields, including life sciences. The impressive performance of these foundation models, such as the transformer-based LLM ESM-2, in protein modeling has demonstrated their potential ability to extract and parse essential representations from protein sequences alone, which allows for the reconstruction of protein structures without the need for MSAs27. Undeniably, these cutting-edge AI technologies have enhanced our understanding of protein ‘sequence-structure’ connections. Protein foundation models are expected to help model Cas systems with limited Cas homologs for the following advantages: (1) The powerful representation capacity of pretrained protein LLMs can be leveraged for Cas modeling by fine-tuning without extensive task-specific-training data, where the labeled Cas types are limited in this domain; and the Cas discovery and functional recognition problem can be formulated into a learning-based classification problem as a complement to the traditional alignment-based methods, and (2) the structural characteristics of Cas are expected to be captured by protein LLMs from sequences, thus alleviating the issue of a lack of ground-truth structural information in Cas identification, further facilitating the directly functional recognitions of Cas from sequence alone. However, the full potential of the protein foundation models to address these problems is still undetermined.
To this end, we develop an effective and unified AI framework, named CHOOSER (Cas HOmlog Observing and SElf-processing scReening), utilizing protein foundation models for alignment-free discovery of CRISPR-Cas systems with self-processing pre-crRNA capability. CHOOSER is formulated as a learning-based classification system that can be used to detect potential CRISPR-Cas systems and to directly functionally screen type V Cas12 homologs that possess pre-crRNA self-processing abilities, which can serve as a significant complement to the alignment-based methods. In brief, CHOOSER is designed to address the two pivotal questions in CRISPR-Cas system identification and functional screening: (1) An integrative AI strategy is developed to uncover distant Cas homologs by fine-tuning a pretrained large-language model, ESM-2. (2) The specific functions of Cas12 enzymes, namely, their ability to self-process pre-crRNA, can be directly predicted by leveraging the representations produced by the foundation model. Specifically, CHOOSER successfully identifies 3477 potential CRISPR-Cas systems, enhancing the number of known type II, type V, and type VI systems. Among these, CHOOSER detects 39 Cas12 candidates that have previously been overlooked by the existing alignment-based CRISPR-Cas mining tools, such as CRISPRCasTyper. Of these 39 candidates, 11 homologs of Casλ are identified, all of which are predicted to be able to self-process pre-crRNA. Subsequent experiments confirm both the pre-crRNA processing activity and the DNase activity of one of the discovered Casλ homologs, EphcCasλ. Overall, our comprehensive study indicates that a proper representation or embedding derived from a protein LLM can be utilized for CRISPR-Cas system identification and functional screening with limited labeled Cas homologs when ground-truth structural information is unavailable. Computational analysis and experimental validation by CHOOSER provide an unprecedented perspective and methodology for discovering CRISPR-Cas systems with specific functions using foundation models, underscoring the potential for transforming identified Cas homologs into genetic editing tools.
Results
The framework of CHOOSER
We developed an effective framework, CHOOSER (Cas HOmlog Observing and SElf-processing scReening), for the discovery and functional screening of CRISPR-Cas homologs with the ability to self-process pre-crRNA via foundation models (Fig. 1), which consists of 4 steps:

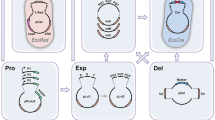
Spring-like symbols indicate CRISPR arrays, while arrowed rectangles indicate ORFs (Cas9 proteins are colored orange, Cas12 proteins are colored blue, Cas13 proteins are colored green, other proteins are colored gray, suspected Cas proteins are colored salmon, and untyped proteins are uncolored.) In the mini-CRISPR array, diamonds indicate directed repeats (DR), and colored oval squares indicate different spacers. In DNA cleavage testing, colored rectangles denote various PAM motifs. Source data are provided as a Source Data file.
Step 1. Cas homolog discovery using a protein LLM. Prokaryotic-origin Cas single effectors (Cas9, Cas12, and Cas13) were used as training data to fine-tune the protein large-language model ESM-2, and viral-origin homologs were used as testing data to evaluate its performance. The fine-tuned model enables us to identify potential Cas9, Cas12, and Cas13 homologs from other microbial background proteins using protein sequence information alone without extensive task-specific training data.
Step 2. Pre-crRNA self-processing functional screening of Cas12 homologs using the protein LLM. We prioritized Cas12 candidates capable of self-processing their pre-crRNA using embedding derived from ESM-2. Our comprehensive tests indicated that the intermediate encoder outputs of ESM-2, as the representations of the models, are all that is necessary for Cas functional screening after examining a variety of representations from sequences to structures.
Step 3. Phylogenetic analysis and identification of candidate Cas12 enzymes. We first extracted all proteins near the CRISPR arrays that were not annotated as Cas proteins by HMMER. After a preliminary filtering process based on our criteria, these proteins were analyzed using Step 1 of CHOOSER for Cas discovery, with the aim of identifying potential Cas homologs. These suspected Cas proteins were subsequently used to construct a phylogenetic tree in the context of known Cas proteins to determine their subtypes. For type V Cas12 candidates, proteins from clades of interest underwent additional structure alignments and MSAs to identify their RuvC-like domains and potential nuclease active site residues. Finally, these Cas12 candidates were evaluated using Step 2 of CHOOSER to predict whether they were capable of self-processing pre-crRNA.
Step 4. Enzymic activity validation and protospacer adjacent motif (PAM) identification. To rapidly determine whether a Cas12 candidate possessed pre-crRNA processing and DNA cleavage activities, we expressed and purified the candidate protein and then conducted several in vitro experiments. (1) We designed a mini-CRISPR array by concatenating the direct repeat (DR) sequence with a spacer to test for protein pre-crRNA processing activity. (2) We used a PCR product library constructed with 8 randomized nucleotides upstream of the 5′ end of the target spacer to assess DNA cleavage capability and to identify functional PAMs preferentially targeted for depletion by the protein. Once a candidate’s enzymatic activities were confirmed, the candidate was considered suitable for further engineering and development as a gene editing tool.
Cas homolog discovery using a protein LLM
We first constructed the training and testing datasets for the Cas discovery model. To this end, we collected 189,165 prokaryotic metagenome-assembled genomes (MAGs) and 42,535,908 viral metagenome-assembled contigs. These sequences were processed using the CRISPRCasTyper pipeline to compile a dataset of known Cas proteins. As depicted in Fig. 2a, the dataset for prokaryotic-origin proteins consisted of 1299 Cas9 proteins, 722 Cas12 proteins, 136 Cas13 proteins, and 246,282 negative background prokaryotic proteins collected from the CRISPRclass19 dataset28. This dataset was split into training and validation subsets at a ratio of 8:2. Given the rarity of Cas single-effector proteins in the overall pool of proteins of microbial origin, our dataset was highly imbalanced. The ratio of the target Cas9, Cas12, and Cas13 classes to the negative background class was approximately 10:5:1:1,811. For the testing data, we collected a viral-origin protein dataset, as shown in Fig. 2b. Collectively, our datasets contained 188,741 prokaryotic-origin proteins as training data, 59,698 prokaryotic-origin proteins as validation data and 2168 viral-origin proteins as testing data (see “Methods” section). Our reasons for training the model using prokaryotic-origin proteins and testing it on viral-origin proteins were as follows: (1) Based on a prior study revealing that phage-encoded CRISPR‒Cas systems possess all six known Cas types (I~VI)8, we considered it appropriate to assemble a testing dataset of viral-origin proteins. (2) Given the distinctive phage-specific traits of viral-origin Cas homologs, such as CasΦ, which is a phage-specific protein with less than 7% amino acid identity to other type V Cas12 homologs7, our choice of a viral-origin testing dataset is aptly suited for evaluating the model’s performance and generalizability across different species. (3) The discovery of phage-specific type V subtypes suggested that viral metagenomics data may have considerable potential as a valuable reservoir for identifying functional enzymes.

a Prokaryotic-origin Cas single effectors and other background proteins used as training and validation datasets. b Viral-origin Cas single effectors and other viral proteins used as a testing dataset. Source data are provided as a Source Data file. c Schematic of the fine-tuned ESM-2 model for discovering Cas single effectors. d Adjustment of hyperparameters for the focal loss to enhance the performance of the classification model. e, f Data distributions for prokaryotic-origin (e) and viral-origin (f) datasets visualized using the representations extracted by the ESM-2 models before and after fine-tuning. g Suspected Cas homologs identified by our fine-tuned model. Orange symbols with crosses represent Cas9 proteins, blue symbols with X’s represent Cas12 proteins, green symbols with squares represent Cas13 proteins and gray symbols with circles represent other proteins. Pie charts with blue outlines indicate proteins of prokaryotic origin, while those with yellow outlines indicate proteins of viral origin.
We then built an ESM-2-based Cas homolog classification model and compared it with a baseline sequence model, i.e., the long short-term memory (LSTM) network-based model. In this case, Cas homolog discovery was formulated as a supervised multi-class classification task. This task was designed to differentiate homologs of Cas9, Cas12, and Cas13 from a pool of other proteins of microbial origin. For the baseline LSTM model, each protein’s amino acid sequence was padded to a uniform length of 1560 and then tokenized to serve as inputs for the models. We observed that the bidirectional LSTM (BiLSTM) model, despite being configured with an embedding size of 1024, a hidden size of 512, and 36 network layers, was unable to meet the requirements of our multi-class classification task for the highly imbalanced datasets. This inadequacy persisted even after the application of a focal loss function during the training process. These results suggested that conventional language model architectures, such as LSTM, may lack the necessary representation capacity to extract critical features from relatively long protein sequences and severely imbalanced data. On the other hand, in the protein LLM ESM-2, taking advantage of the fine-tuning strategy, we utilized the network’s final layer of encoder output corresponding to the CLS token as a representation for each protein. These representations were then connected to fully connected layers to execute the classification task, as depicted in Fig. 2c (see “Methods” section). Considering the extreme imbalance of our training datasets, we experimented with several loss functions, including balanced cross-entropy (CE) loss and focal loss, and adjusted the hyperparameters in our multi-class classification task (see “Methods” section). As depicted in Fig. 2d, when fine-tuned with protein sequences of prokaryotic origin, the test results demonstrated that the pretrained protein LLM is effective for “few-shot” identification of the distant Cas proteins such as the Cas12i, using the highly imbalanced training data, which only has 3 training samples for this specific Cas12 subtype. Also, it holds the capacity of “zero-shot” identification of viral-specific Cas12 subtypes, i.e., the viral-specific CasΦ, where this subtype does not exist in the training data. Notably, the optimized models successfully detected all viral-origin Cas12 homologs while utilizing a balanced CE loss function. These results clearly demonstrated the effective representation capacity of protein foundation models in Cas homologs discovery. Detailed model performance metrics are provided in Supplementary Data 1.
To verify the effectiveness of the ESM-2 model in capturing essential features for distinguishing Cas single effectors, we visualized the high-dimensional encoder output embeddings from our prokaryotic-origin and viral-origin datasets using Principal Component Analysis (PCA) and t-distributed Stochastic Neighbor Embedding (t-SNE). As depicted in Fig. 2e, f, after fine-tuning the embeddings produced by ESM-2, the model was capable of clearly segregating Cas single effectors from the background proteins of both prokaryotic and viral origins. An expanded visualization of the data distribution of our datasets utilizing representations from the fine-tuned ESM-2 models with varying hyperparameters for the loss function is provided in Supplementary Fig. 1. These results indicated that the fine-tuned pretrained protein language models effectively extracted the essential features necessary to distinguish Cas single effectors.
In summary, in this study, we utilized a fine-tuned ESM-2 model using balanced CE loss to detect suspected Cas single effectors from all suspected proteins near CRISPR arrays. Using our fine-tuned model, we identified putative 3020 type II, 242 type V, and 215 type VI systems that were not reported by alignment-based CRISPRCasTyper. In addition, our model discerned 46 and 47 potential Cas single effectors from prokaryotic and viral data sources, respectively (Fig. 2g). The discovered proteins were considered suspected Cas homologs in subsequent analyses.
Functional screening of Cas12 homologs with self-processing pre-crRNA capability using the protein LLM
Cas12 training data curation and pseudolabeling
In this study, we aimed to establish an in silico strategy to predict whether Cas12 homologs possess the self-processing pre-crRNA functional trait, and this task is formulated as a binary classification problem. The premise to achieve this goal is to functionally label the distinct known Cas12 homologs as the training data for model building. As more subtypes of type V systems have been successively discovered, the nuclease-associated properties of diverse Cas12 homologs have been revealed. As summarized in Supplementary Table 1, certain Cas12 effectors utilize a variety of mechanisms for pre-crRNA processing. To this end, we gathered known Cas12 homologs, with duplicates removed, to construct the training and validation datasets. Cas12a, Cas12c, Cas12i, CasΦ, and Cas12m homologs were labeled pseudopositives for self-processing pre-crRNA, while the remaining Cas12 subtypes were designated as pseudonegatives, as described in the literature and illustrated in Supplementary Table 1. Notably, the newly published Casλ (pseudopositive) and Cas12n (pseudonegative) homologs were excluded during the training process. For the testing dataset, we utilized recently published sequences from the CasPEDIA database29, with duplicates eliminated with sequence similarity less than 70% compared to the training dataset. This included several Casλ and Cas12n homologs. These datasets were curated for subsequent experiments designed to probe the functional traits of Cas12 proteins (see “Methods” section).
Embedding derived from LLMs is likely all you need for self-processing pre-crRNA functional screening of Cas12 homologs
Guided by the ‘sequence-structure-function’ paradigm, we explored a variety of representations, ranging from sequence to structure, as input for the classifiers to distinguish proteins with self-processing pre-crRNA functional properties. Considering the lack of ground-truth structural data for most known Cas proteins and our potential candidates, we first evaluated the reliability of state-of-the-art folding structure prediction tools, such as AlphaFold2, RoseTTAfold2, and ESMfold, in predicting the structures of Cas12 homologs (see “Methods” section and Supplementary Information). The ESMfold-predicted structures were fairly consistent. For the newly discovered Cas12 subtypes such as Casλ and Casπ, which lacked sufficient homologs for MSA, ESMfold outperformed the MSA-based AlphaFold2 and RoseTTAfold2 in both structural alignment and distance map identity (Fig. 3b, c). This could be attributed to the ability of ESMfold to model Cas structures without requiring MSAs from homologs. Therefore, given the aim of our study to identify and screen potential CRISPR-Cas systems, we decided to employ ESMfold-predicted structures to derive our subsequent structure embedding.

a–c Curves of identity between Cα distance maps from cryo-EM structures and their corresponding predicted protein structures, along a range of thresholds \(\theta\): (a) FnCas12a (PDB ID: 6I1K); (b) Casλ (PDB ID: 8DC2); and (c) Casπ (PDB ID: 7YOJ). d Performance of the models in predicting the ability of Cas12 proteins to self-process their pre-crRNA, denoted by F1 scores in the validation (in gray) and testing (in red) datasets. Source data are provided as a Source Data file. e Assorted representations from protein sequences to structures throughout the ESMfold protein structure prediction process. f F1 scores on the testing dataset, illustrating the performance of the models in predicting the ability of Cas12 protein self-processing pre-crRNA, using varied representations. The heatmap, using a color scale from blue to red, displays F1 scores ranging from low to high. Source data are provided as a Source Data file.
We then assessed whether the representation of amino acid sequences and folding structures could discern the trait of self-processing of pre-crRNA in Cas12 homologs (see “Methods” section). For the amino acid sequences, two sequence embeddings were applied: (1) we encoded the whole protein sequences into a fixed-length vector as a representation to feed into the bidirectional LSTM (BiLSTM) networks, configured with 16 layers and a hidden size of 128 (16L), as well as 32 layers and a hidden size of 512 (32L); (2) we used the last hidden layer outputs of protein CLS tokens of the transformer-based pretrained protein LLM ESM-2 models, with parameter sizes of 650 million (650M) and 3 billion (3B), for classification. For the folding structures, we adopted two distinct data processing approaches: (1) inverting the folding structures into embedding using the cutting-edge geometric vector perceptron (GVP)-transformer pretrained model ESM-IF130 and (2) converting the three-dimensional folding structures into two-dimensional distance map matrices. We subsequently deployed a convolutional network, U-Net31, to extract features from the inverse folding embedding and distance maps, further facilitating classification (see “Methods” section).
The F1 scores from classifications using various input embeddings in the testing dataset are displayed in Fig. 3d. Significantly, classifications using ESM-2 embedding significantly outperformed both the BiLSTM models and the structural methods. In detail, the BiLSTM models demonstrated reasonable recall but quite low precision. For instance, the BiLSTM 16L model successfully identified 8 out of 12 Casλ homologs as positives but incorrectly predicted 15 out of 40 pseudonegative Cas12n homologs as positives. In contrast, the structural methods displayed similar recall significantly better precision. For example, ESM-IF1 embedding identified 6 out of 12 Casλ homologs as positive for self-processing pre-crRNA and only misjudged 1 Cas12e homolog as positive. With the ESM-2 embedding methods, we observed both high recall and high precision. Specifically, classification with ESM-2 650M CLS token representations achieved both recall and precision of 1.0, while classification using ESM-2 3B CLS token representations achieved a slightly lower recall. More details can be found in Supplementary Data 3.
Collectively, our comprehensive tests indicated that the embedding derived from LLMs may be all that is necessary for self-processing functional screening of Cas12 homologs when ground-truth structural information is unavailable.
Interpretability analysis of the ESM-2 model for self-processing pre-crRNA functional screening
Considering that previous research has demonstrated that transformer-based language models can extract the structural and even functional properties of proteins using attention mechanisms32, we sought to delve more deeply into the interpretability of the hidden layers of ESM-2 to determine where the identification of Cas12 homologs capable of self-processing pre-crRNA is encapsulated. As depicted in Fig. 3e, ESM-2 adopts a BERT-style encoder-only architecture, and each encoder layer within the ESM-2 model comprises a multihead attention sublayer and a feedforward sublayer. We scrutinized each layer in the ESM-2 models by employing U-Net architectures to extract interpretable features from the encoder output embeddings and the attention weights (see “Methods” section). The CLS token representations of each encoder layer output were also used as input to a classifier tasked with predicting the ability of the Cas12 enzyme to self-process pre-crRNA as previously described. In a manner analogous to the previous experiment, the F1 scores of the probing classifier in our testing dataset served as an indication of the knowledge of this property encoded in the representations. As shown in Fig. 3f, both the 650M and 3B models demonstrated that deeper layers of encoder embeddings or attention weights yielded F1 scores reaching 1.0 in the probing classifier. When used as inputs to the classifier, the CLS token representations, which is the first row of the encoder embedding, also attained F1 scores of 1.0 in the 33rd and 9th layers of the 650M and 3B models, respectively. Taken together, these results indicate that particular properties, in this case, the functional traits of Cas12 homologs, may be present within the intermediate layers of the model.
Phylogenetic analysis and identification of candidate Cas12 enzymes
Using the Cas homolog discovery model of Step 1 of CHOOSER, we identified 39 candidates as potentially Cas12 homologs that had not been detected by existing CRISPR-Cas discovery tools, such as CRISPRCasTyper. To verify these candidates, we conducted several analytical steps as follows:
-
(1)
Phylogenetic analysis. The suspected Cas12 candidates were clustered into five distinct clades within the CasPEDIA’s type V Cas12 phylogenetic tree, as depicted in Fig. 4a (see “Methods” section).
Fig. 4: Analysis of 39 Cas12 candidates. a Phylogenetic tree of all the suspected Cas12 enzymes against the background of the CasPEDIA type V Cas12 dataset. Source data are provided as Source Data files. b Genomic arrangements of representatives of the five clade candidates. Spring-like symbols indicate CRISPR arrays, while arrowed rectangles represent ORFs (suspected Cas12 proteins are colored salmon, and other proteins are colored gray). c Clade 1 candidates shown phylogenetically against the background of Cas12a homologs. Source data are provided as Source Data files. d Clade 2 candidates shown phylogenetically against the background of Cas12m homologs. e MSAs for the direct repeats of Clade 3 suspected CRISPR systems. MSAs are visualized using SnapGene, with a color scale from blue to red representing MSA identity, ranging from low to high. f Structural alignment showing the RuvC-like domain and canonical D-E-D active site residues within the domain of a putative Casλ homolog in Clade 3. A blue rectangle represents a Casλ candidate protein, while the yellow blocks denote the RuvC-like regions of the protein that align with known RuvC proteins.
-
(2)
Genomic loci examination. We examined and illustrated the genomic arrangements of representative loci for each clade in Fig. 4b.
-
(3)
Sequence alignments. We aligned the protein sequences of the candidates against the NCBI ‘nr_clustered’ database (updated 2023-12-28) and against the newly published CRISPR associations identified by the FLSHclust algorithm24 using the ‘BLASTP’ algorithm (details in Supplementary Data 7).
-
(4)
Structural alignments. We compared the predicted folding structures of the candidates with those of RuvC proteins to determine the presence of RuvC-like domains and potential Asp-Glu-Asp (D-E-D) active site residues within those domains (see “Methods” section).
-
(5)
Pre-crRNA self-processing capability prediction. Using the self-processing pre-crRNA screening model of Step 2 of CHOOSER, we estimated the likelihood that each candidate had the ability to self-process pre-crRNA.

In detail, the 7 identified prokaryotic-origin candidates from Clade 1 were categorized into the Cas12a subtype but formed a distinct subcluster that is phylogenetically closer to Cas12a2 (Fig. 4c). These candidates displayed genomic arrangements typical of the type V-A CRISPR-Cas systems, with the Cas1, Cas2, and Cas4 genes located nearby. Sequence alignment indicated that a minority (2 out of 7 candidates) exhibited similarity to ‘Cas12a/Cpf1’ with an e-value of 2e−4, while the remaining candidates showed no significant similarity to any known proteins. Structural alignments had identified a clear RuvC-like domain at the C-terminus of each candidate, and within this domain, the characteristic β-β-α D-E-D motif was also observed (details shown in Supplementary Fig. 3a–d). Six out of these 7 candidates are predicted to have self-processing pre-crRNA capabilities, which is consistent with our understanding of Cas12a enzymes.
The 4 identified viral-origin candidates from Clade 2, together with 17 viral-origin Cas12m homologs identified by our pipeline, were categorized within the Cas12m subtype. However, they formed distinct subclusters that separated from their prokaryotic analogs (Fig. 4d). These 21 viral-origin candidates exhibited a significant increase in protein size, ranging from 623 to 999 amino acids (aa), in contrast to the 519 to 608 aa span of the prokaryotic-origin Cas12m homologs. Notably, while a previous study suggested that only a few Cas12m variants have the canonical RuvC active site residues9, we observed the D-E-D active site residues within the RuvC-like domains of 43% (9 out of 21) viral-origin Cas12m, and of all identified candidates from Clade 2 (Supplementary Fig. 3e, f). Moreover, our analysis indicated that 20 out of the 21 viral-origin Cas12m candidates we identified were predicted to be capable of self-processing pre-crRNA that aligned with the previous studies9,33.
The 11 identified viral-origin candidates categorized as Clade 3, with sequence lengths ranging from 735 to 798 amino acids, were phylogenetically proximate to the Casλ clade (Fig. 4a). Most of these proteins exhibited a clear CRISPR-Cas system arrangement within their genomic loci, as shown in Fig. 4b. Sequence analysis confirmed substantial similarity between all the candidates and the ‘CasLambda’ (PDB ID: 8DC2_A) protein, meeting the significance threshold with e-values of 10−10. The CRISPR repeat sequences of these candidates closely mirrored the direct repeat (DR) sequences documented in prior research8, as presented in Fig. 4e. Structural analysis revealed the RuvC-like domain and the D-E-D catalytic site residues within this domain across the candidates, as shown in Fig. 4f. Considering the known pre-crRNA self-processing ability of Casλ enzymes, the prediction that all candidates possess this trait is consistent with existing knowledge, suggesting that Clade 3 significantly enriches the diversity of the Casλ family, nearly doubling its current catalog.
Candidates from clades 4 and 5 did not correspond to any recognized subtypes but established distinct groups. Specifically, Clade 4 is located phylogenetically between Cas12h and Casλ, while Clade 5 lies between Casf1 and Cas12d. Clade 4 candidates varied widely in size, ranging from 628 to 1052 amino acids, and their genomic loci revealed at least three diverse arrangement types. Clade 5 proteins were notably separated from their associated CRISPR arrays within the genome. Sequence comparisons indicated that most proteins in these clades bore a significant resemblance to ‘transposase’ proteins. This result raises the possibility that clades 4 and 5 may contain transposases. Supplementary Fig. 4 provides an expanded view of these two clades.
Experimental enzymatic activity validation of EphcCasλ
To explore the pre-crRNA processing activities of the identified Casλ proteins, we successfully purified six homologs from Clade 3 and compared their capacity to produce mature crRNAs from cognate pre-crRNAs (Supplementary Fig. 9 and Fig. 5a). Similar to the Casλ1 studied previously8, five additional Casλ enzymes efficient cleavage cognate pre-crRNA at the 3′ end of the spacer region (Fig. 5b). Only one of the six homologs tested to date exhibited no detectable cleavage activity at the same assay condition (IMG/VR-26) (Fig. 5b). Type V CRISPR-Cas systems target DNA sequences preceded by a 2–5 bp Protospacer Adjacent Motif (PAM) for self- versus-non-self recognition. We compared the DNA cleavage activities of Casλ homologs and determined the PAM sequence using an in vitro cleavage assay in which a linear dsDNA engineered with 8 randomized nucleotides positioned upstream of the 5′ end of the target sequence as substrate, incubated with purified Casλ proteins and crRNAs (Fig. 5c). We found that two homologs (IMG/VR-36 and IMG/VR-49) were able to cleave linear target DNA, specifically, IMG/VR-49 exhibits higher DNA cleavage activity compared to IMG/VR-36 (Fig. 5d). Since IMG/VR-49 was discovered in metagenome-assembled contigs from bovine rumen microbial samples and was taxonomically inferred to ‘Enterobacter phage Phc’, we named it EphcCasλ. Deep sequencing un-cleavage dsDNA substrate suggests that EphcCasλ recognizes the TTR PAM (R = A or G) (Fig. 5e), which was consistent with Casλ1. The PAM preference for EphcCasλ nuclease was validated by in vitro cleavage same target with diverse PAM sequences (Fig. 5f). Gel analysis of the cleavage products showed that both EphcCasλ and Casλ1 able to cut dsDNA with TTR PAM sequences. Interestingly, the EphcCasλ nuclease exhibited stronger DNA cleavage activity compared to Casλ1 when targeting the site with the TTA PAM sequence.

a Structure of pre-crRNA substrates consists of a hairpin formed by a direct repeat (DR) sequence followed by a 20 bp spacer. Diamonds symbols indicate directed repeats (DR), and colored oval squares indicate different spacers. b Representative gel of Casλ-mediated pre-crRNA cleavage by six Casλ homologs after 60 min incubation with 5′-FAM labeled pre-crRNA substrates. c Pipeline used to detect dsDNA cleavage and associated PAM recognition by in vitro DNA cleavage assay. Casλ RNP complexes cleave a 5′ PAM library PCR product in vitro, and the uncut part was captured via PCR and subjected to Illumina deep sequencing. Gray rectangles indicate 5′ PAM library PCR products, with blue blocks representing spacers and colored short blocks denoting various PAM motifs. Dark green and dark blue squares represent different barcodes on the sequencing adapters. d Six Casλ homologs cleaved dsDNA in vitro at 37 °C for 1 h. A 500 bp PCR product was cleaved into two 250 bp products. e Analysis of Illumina deep sequencing data showing that the presumed PAM of EphcCasλ was TTR. The weblogo of the presumed PAM that supported target recognition and cleavage was generated using WebLogo (Thymine is colored red, Adenine green, Cytosine blue, and Guanine yellow). Source data are provided as a Source Data file. f EphcCasλ and Casλ1 cleaved TTA/TTG dsDNA in vitro at 37 °C for 3 h. PAM was confirmed to be TTA/TTG. g Trans-cleavage assay conducted with the ssDNA-FAM/BHQ reporter. This reporter is a ssDNA oligonucleotide labeled with a fluorophore (FAM) at one end and a quencher (BHQ) at the other. Initially, fluorescence from FAM is quenched by BHQ due to their proximity (top left). Upon recognizing and binding to a target DNA sequence, the Casλ nuclease becomes activated and can non-specifically cleave nearby ssDNA, including the reporter. As a result, the fluorophore (FAM) is separated from the quencher (BHQ), leading to the emission of fluorescence (bottom right). The activated Casλ is shown in pink, the target DNA sequence in black, the PAM motif in yellow, and the guide RNA in blue. h EphcCasλ exhibited trans-cleavage of DNA at 30 °C, 37 °C, and 44 °C when the PAM was TTA/G. The experiments are shown in (b, d, f) are representative of three independent experiments with similar results.
Furthermore, we also probed the trans-cleavage potential of EphcCasλ on single-stranded DNA (ssDNA). Employing a previously described fluorophore-quencher (FQ) reporter assay34, we examined the nonspecific ssDNA cleavage (trans-cleavage) activity of EphcCasλ (see “Methods” section). We observed that the trans-activation of EphcCasλ was most robust at 37 °C, with diminished signals at 30 °C and 44 °C and no activity at 23 °C or 51 °C (Fig. 5h). These findings indicated that EphcCasλ exhibits temperature dependent ssDNA cleavage activity at an optimal temperature of 37 °C, suggesting its potential utility in CRISPR-Cas-based pathogen detection technologies.
Discussion
The discovery of CRISPR-Cas systems laid the groundwork for the development of CRISPR-Cas-based gene editing technologies. Our research introduces the CHOOSER framework, an effective AI methodology that utilizes protein LLMs, such as ESM-2, to identify and functionally annotate CRISPR-Cas systems. To our knowledge, this study represents a typical application of protein foundation models in the discovery and functional screening of CRISPR-Cas systems. We demonstrated the utility of CHOOSER by applying it to Cas12 enzymes and discovered dozens of potential candidates that previous HMMER-based bioinformatics tools, such as CRISPRCasTyper, had missed. These results demonstrated that protein LLM-based approaches can significantly complement traditional sequence alignment methods for CRISPR-Cas system identification, and they can be utilized directly for Cas functional annotation leveraging the powerful representation capacity of protein foundation models.
Phylogenetic analysis indicated that the identified proteins had marginally longer branch lengths than the known Casλ proteins, and structural prediction alignments revealed relatively low TM-scores of approximately 0.5 (Supplementary Fig. 5). These findings suggested rapid evolutionary changes in the source phages, supporting the notion that prokaryotic viral metagenomic data could be a substantial resource for the future discovery of functional enzymes.
Among the Cas12 candidates identified by CHOOSER, we experimentally confirmed that 5 out of 6 Casλ homologs have significant pre-crRNA processing ability, while two of them exhibit dsDNA cleavage activity. Specifically, a Casλ homolog, EphcCasλ, we discovered that its trans-cleavage activity was temperature dependent, with an optimal temperature of 37 °C. These findings suggested that EphcCasλ is a promising candidate for further development in CRISPR-Cas-based pathogen detection systems.
All these results demonstrate that our models have effectively learned patterns from known protein sequences and can discern remote homologs. However, the current limitation of the CHOOSER framework is its specificity for identifying Cas single effectors, such as Cas9, Cas12, and Cas13. Given that a wide range of microorganisms can evolve and yield multi-domain proteins with reorganized functions, there are likely more Cas remote homologs and diverse Cas single effectors yet to be discovered beyond the known Cas9, Cas12, and Cas13. Additionally, in this study, we have only experimentally validated a single clade of the discovered Cas12 candidates. Despite these limitations, we are confident that the CHOOSER framework can be adapted to discover other types of proteins, provided that appropriate training data for the target protein family is available. This adaptability highlights the potential for expanding the scope of CHOOSER to encompass a broader range of protein families.
Subsequent potential studies based on our results include the following: (1) Although this study initially explored the interpretability of the self-processing pre-crRNA trait of Cas12 enzymes using protein LLMs, the underlying biological mechanisms involved have not been fully elucidated. For instance, the specifics of how the Cas12m homolog processes its own pre-crRNA are unclear33. Moving forward, we aim to delve into the causal links between domain-level and residue-specific functionalities, which could provide a theoretical basis for the functional modification of Cas12 enzymes. (2) We preliminarily validated the self-processing pre-crRNA and DNA cleavage capabilities of EphcCasλ, indicating its promise as a candidate for multiplexed genome editing applications. A thorough investigation of the full characteristics of the EphcCasλ enzyme, such as its cleavage activity in mammalian cells, will be performed in the future. Further engineering modifications to develop this enzyme into an effective CRISPR-Cas-based gene editing tool are expected. (3) This study, through the discovery of 11 variants of the Casλ family, revealed that viral-origin Cas proteins may be undergoing rapid evolution; viral genomes mutate swiftly and are subject to natural selection. This finding provides insight into how enzymes from viral sources could serve as excellent gene editing tools to explore in the future. (4) Our findings aligned with recent research showing that protein LLMs can discern functions of prokaryotic viral proteins35. Moving forward, we would investigate the potential of CHOOSER for other functional discrimination.
Methods
Cas homolog discovery using a protein LLM
Known CRISPR-Cas system identification
All the prokaryotic metagenome-assembled genomes and contigs used for CRISPR-Cas system mining were obtained from a multitude of public databases. These included MGnify36, GMBC37, GEM38, 4D-SZ39, Glacier Microbiomes40, IMG/VR (v3)41, MGV42, Human Virome Database (HuVirDB)43, Gut Phage Database44, INPHARED PHAge Reference Database45, Virus-Host DB46, PLSDB47, and Bacteriophage (ftp://ftp.sanger.ac.uk/pub/pathogens/Phage/)48.
The CRISPR-Cas mining pipeline CRISPRCasTyper (https://github.com/Russel88/CRISPRCasTyper/releases/tag/v1.8.0)21 was utilized to detect known CRISPR-Cas systems since this pipeline annotates Cas proteins using the most recently updated Cas HMM profile database, and it has established a reasonable criterion for identifying known CRISPR-Cas systems with high confidence.
Training and testing dataset curation for building the Cas discovery model
In total, we identified 6639 Cas9 homologs, 2, 342 Cas12 homologs, and 485 Cas13 homologs of prokaryotic origin. After removing redundant proteins with a sequence similarity of no more than 70%, we obtained 1, 299 Cas9, 722 Cas12, and 136 Cas13 nonredundant proteins to serve as the training and validation datasets. The proteins other than the Class 2 Cas single effectors in the prokaryotic published genome-assembled data of Kira S. Makarova 2020 (ftp://ftp.ncbi.nih.gov/pub/wolf/_suppl/CRISPRclass19/)28, a total of 246, 282 proteins after removal of duplicates, served as the negative background proteins during our Cas discovery model training process. To evaluate our models, we combined nonredundant viral-origin proteins, including 164 Cas9 homologs, 290 Cas12 homologs, 47 Cas13 homologs, and 1, 667 negative background proteins, to construct the testing dataset. The published CasΦ (Cas12j) proteins that were not used for training our model are included in the test dataset to test whether the models can discern uncommon subtypes of Cas single effectors.
Obtaining suspected proteins near CRISPR arrays
To discover potential unrevealed CRISPR-Cas systems, we gathered all uncharacterized proteins located in proximity to CRISPR arrays from metagenome-assembled datasets, and we excluded those proteins that were already classified as part of known CRISPR-Cas systems by CRISPRCasTyper. CRISPR arrays were identified using MinCED v0.4.2 (https://github.com/ctSkennerton/minced49). Our dataset included proteins from the putative CRISPR-Cas loci reported by CRISPRCasTyper, which included single cas genes adjacent to a CRISPR array that were identified by HMMER but failed to meet the filtering criteria21. Additionally, we encountered certain uncharacterized large proteins (greater than 600 amino acids) adjacent to CRISPR arrays that lacked recognizable Cas homologs both upstream and downstream, suggesting that they might be components of undiscovered CRISPR-Cas systems missed by current detection methods. We combined all these suspected proteins to discover potential Cas systems.
Fine-tuning ESM-2 for Cas discovery
With the aim of discovering potential Cas single effectors, we utilized the pretrained ESM-2 model (esm2_t33_650M_UR50D, https://github.com/facebookresearch/esm27) and fine-tuned it for a multi-class classification task. This task involved classifying Cas9s, Cas12s, Cas13s, and background proteins that are not single effectors. We employed the EsmForSequenceClassification class developed by HuggingFace (https://github.com/huggingface/transformers), which uses the CLS token embedding output from the last encoder layer of the ESM-2 model to perform protein classification. We fine-tuned all parameters of both the ESM-2 model and the fully connected layers within EsmForSequenceClassification.
Balanced cross-entropy loss and focal loss
Considering the extreme imbalance among classes in our training dataset, we evaluated several loss functions in addition to the conventional cross-entropy (CE) loss. By multiplying by a weighting factor α, we achieved a balanced CE loss. The weighting factor α is calculated based on the sample number xj in each of the N classes, as shown in Eq. (1):
Moreover, we experimented with the focal loss function, incorporating the weighting factor α and applying a range of focusing parameters γ (γ = 1, 2, and 5), as described in a previous research50.
Embedding visualization
To visualize and compare the embedding representations of the ESM-2 models before and after fine-tuning, we used Principal Component Analysis (PCA) and t-Distributed Stochastic Neighbor Embedding (t-SNE). Initially, PCA was utilized to reduce the dimensionality of the original attribute space of 1280 dimensions. Then we selected the principal components (PCs) that accounted for 80% of the explained variance for further t-SNE dimensionality reduction and visualization.
Functional screening of Cas12 homologs with self-processing pre-crRNA capability using the protein LLM
Training and testing dataset curation for the Cas12 self-processing pre-crRNA functional screening model
For the models for Cas12 screening, we collected all known nonredundant Cas12 proteins and labeled them self-processing pre-crRNA pseudopositives or pseudonegatives according to their subtypes. In detail, 560 pseudopositives (Cas12a, Cas12c, Cas12h, Cas12i, CasΦ, and Cas12m homologs) and 331 pseudonegatives (Cas12b, Cas12e, Cas12f, Cas12g, Cas12k, Cas12l, and TnpB homologs) were included in the training and validation datasets during the Cas12 screening model training process.
After redundancy removal, the Cas12 homologs published in the CasPEDIA29 database included 29 pseudopositives (Cas12a, Cas12c, Cas12h, Cas12i, CasΦ, Cas12m, and Casλ homologs) and 94 pseudonegatives (Cas12b, Cas12e, Cas12k, and Cas12n homologs), which served as the testing dataset.
Evaluating similarities of 3D protein structures
In this study, three state-of-the-art protein structure prediction technologies were employed: AlphaFold2 v.2.1.2, RoseTTAfold2, and ESMfold v.2.0.0. Protein structure alignments were performed using US-align51 v.20220626, with template modeling scores (TM-scores) and root mean square deviation (RMSD) used as measurements of structural similarities of pairwise protein structures.
We also calculated the identity of the α-carbon (Cα) distance map matrices as another metric of protein structural similarity. For a protein composed of n amino acids, the Cα distance maps of the reference structure and the target structure would be two distinct matrices, denoted as X and Y. We set a threshold θ ∈ [0,1] to determine whether each corresponding pair of items, xij in X and yij in Y, were in consensus, as defined in Eq. (2):
The distance map identity of the protein was subsequently calculated via Eq. (3):
Obtaining protein representations extracted by foundation models
For sequence representation extraction, we utilized the ESM-2 pretrained models, specifically esm2_t33_650M_UR50D and esm2_t36_3B_UR50D, to obtain attention weights and output embeddings from each encoder layer, which were subsequently used as inputs for our classifiers.
To generate structural representations, we used the ESMfold-predicted structures in two distinct ways: (1) We used the state-of-the-art geometric vector perceptron (GVP)-transformer pretrained model ESM-IF130 (esm_if1_gvp4_t16_142M_UR50) to transform protein structures into embeddings. (2) We transformed protein structures into two-dimensional distance map matrices using BioPython 1.81 (https://biopython.org/52). These structural representations were also used as inputs for our classifiers.
Building binary classifiers for functional screening of Cas with self-processing pre-crRNA capability using various representations
For each of the amino acid sequence inputs, we encoded the whole protein sequences into a fixed-length vector as a representation to feed into the bidirectional LSTM (BiLSTM) networks. BiLSTM networks were configured with a hidden size of 128 across 16 hidden layers (denoted as ‘16L’) and a hidden size of 512 across 32 hidden layers (denoted as ‘32L’) to facilitate binary classification tasks. The implementations were executed using PyTorch v1.13.1.
For the amino acid sequence, we also used the ESM-2’s final layer of encoder output corresponding to the CLS token as the LLM-derived representations, followed by the use of fully connected layers for the classification tasks.
Regarding the protein 3D structure inputs, we converted structures into inverse folding embeddings and distance matrices, respectively, as structural representations. Then we applied U-Net networks for feature extraction from these representations and performed binary classifications.
Interpretability analysis of the ESM-2 model for self-processing pre-crRNA functional screening
Within each layer of the ESM-2 encoder, we examined three distinct types of representations: (1) the encoder output for the CLS token, (2) the encoder output embeddings, and (3) the multihead attention weights. For the CLS token representation, we utilized fully connected layers for classification as previously described. Regarding the encoder output embeddings and the multihead attention weight matrices, we employed U-Net architectures to extract features, which were then processed through fully connected layers for classification purposes.
Phylogenetic analysis and identification of candidate Cas12 enzymes
Phylogenetic tree reconstruction
For the phylogenetic analysis of Cas9, Cas12, and Cas13, we used the single effectors identified in this study along with homologs provided in the CasPEDIA database. Multi-sequence alignments were generated using MAFFT53 v.7.508 with 1,000 iterations and filtered to remove columns composed of gaps in 95% of the sequences. The phylogenetic tree was inferred using IQTREE54 v.1.6.12 via automatic model selection and 1,000 bootstraps and visualized using iTOL55 v5 (https://itol.embl.de/itol.cgi), as described in previous research8.
Determination of RuvC-like domains
For the suspected Cas12 candidates, we identified their RuvC-like domains through structural alignment with known RuvC proteins. Specifically, we retrieved the sequences of 522 RuvC proteins from the UniProt56 database as of December 12, 2022. We then employed US-align to conduct structural alignments between each Cas12 candidate and the RuvC proteins. The noncontinuous segments of the Cas12 candidates that aligned with any of the RuvC proteins were subsequently identified as RuvC-like domains.
Determination of putative active site residues of RuvC-like domains
The suspected Cas12 candidate sequences were aligned with reference Cas12 proteins to identify the conserved active site residues D-E-D within the RuvC-like domains. Multi-sequence alignments using MAFFT were visualized by SnapGene v.6.0.2. Moreover, the spatial conformation of the RuvC-like domains and their conserved active site residues were visualized using PyMOL v.2.5.2.
Experimental enzymatic activity validation of EphcCasλ
Expression and purification of proteins
Casλ homologs with 6xHis tag overexpression plasmids were transformed into chemically competent E.coli BL21 (Vazyme, C504-02) and incubated overnight at 37 °C on LB-Kan agar plate (50 μg/mL Kanamycin, Sangon, A600286-0005). Single colony was picked to inoculate 3 ml (LB, 50 μg /mL Kanamycin) starter cultures which were incubated at 37 °C for 8 h. Then 500 ml LB-Kan medium(50 μg /mL Kanamycin) was inoculated with 1 ml starter culture and grown at 37 °C to an OD600 of 0.8, cooled down on ice, and gene expression was subsequently induced with 1 mM IPTG followed by incubation overnight at 16 °C. Cells were harvested by centrifugation and resuspended in buffer A (20 mM Tris-HCl, pH 8.0, 500 mM NaCl, 10% Glycerol, 20 mM imidazole), subsequently lysed by sonication, followed by lysate clarification by centrifugation. The soluble fraction was loaded on a 1 ml HisPurTM Ni-NTA Resin (Thermo Fisher, 88221). Bound proteins were washed with wash buffer and subsequently eluted by gradient imidazole (50 mM to 1 M). The eluted proteins were concentrated to 1 mL before loading on a Superdex 200 increase 10/300 GL column (Cytiva) Peak fractions were concentrated to 500 μL and concentrations were determined using a NanoDrop 2000 Spectrophotometer (Thermo Fisher).
In vitro pre-crRNA processing assay
6-FAM (Fluorescein)-conjugated pre-crRNA substrates were synthesized by GenScript (substrate sequences are provided in Supplementary Data 13). Processing reactions were initiated in a 1:2 molar ratio by combining Casλ proteins and RNA substrate in NEB r2.1 (NEB, B6002V) and subsequently incubated at 37 °C for 30 min, then cooled down on ice before separation on a 20% TBE-Urea-PAGE (Beyotime, R0248S). Gels were visualized and imaged by the Chemidoc MP imaging system.
In vitro PAM depletion assay
A PCR product containing the PAM library was amplified from RTW554 plasmid (Addgene, 160132). Active Casλ ribonucleoprotein (RNP) complexes were assembled by mixing protein and crRNAs (GenScript, sequences are provided in Supplementary Data 13) in a 1:2 molar ratio in NEB r2.1 buffer and incubation at RT for 30 min. In vitro PAM assays were performed in a 30-μL reaction mixture containing 300 ng substrate and 400 ng RNP complex in a final 1× NEB r2.1 buffer (NEB, B6002V). Assays were allowed to proceed at 37 °C for 2 h. Reactions were then treated with RNase A (NEB, T3018L) and proteinase K (NEB, P8107S). Loading dye was added (NEB, B7024) and samples were separated by electrophoresis on a 2% agarose gel. Un-cleaved products were isolated and purified through gel extraction and subsequently amplified by using Phanta Max Super-Fidelity DNA Polymerae (Vazyme, P505-d1) for 25 cycles at the first round PCR (PCR1). And then the products of PCR1 were purified by gel extraction for the second round of PCR (PCR2). For PCR2, DNA was amplified with VAHTSTM DNA adapters set1 for Illumina (Vazyme, N801-01) using KAPA HiFi HotStart ReadyMix (Roche, KK3604) for 6 cycles (crRNA and primers sequences are given in Supplementary Data 13). After that, the products were purified using DNA Clean Beads (Vazyme, N411-01) and sequenced on the Illumina Novaseq 6000 platform.
PAM depletion sequencing data analysis
Amplicon sequencing of the targeted PCR product was used to identify PAM motifs that were preferentially depleted. Paired-end sequencing reads were merged using FLASH57 v.1.2.11 with default parameters. Subsequently, the merged reads were processed as described in a previous study58, and the frequency of each possible 8-nucleotide sequence was calculated. Enriched PAMs were determined by calculating the log2 ratio of the abundance of PAMs relative to that of the control plasmids. These enriched PAMs were subsequently used to generate sequence logos using WebLogo v. 3.7.12 (https://github.com/gecrooks/weblogo).
In vitro DNA cleavage assay
The dsDNA substrates were produced by PCR amplification of pUC57 plasmids. Target cleavage assays were performed in a 30-μL reaction mixture containing 300 ng substrate and 400 ng RNP complex in a final 1x NEB r2.1 buffer (GenScript, target, and crRNA sequences are given in Supplementary Data 13). Assays were allowed to proceed at 37 °C for 1–3 h and samples were analyzed by electrophoresis on a 2% agarose gel.
ssDNA trans-cleavage and florescence detection
For the trans-cleavage assay, 1:2 molar rations of EphcCasλ and crRNA were premixed with the FQ-Labeled reporter (AZENTA, 5′-6-FAM-TTTATTT-BHQ1-3′) in NEB r2.1 buffer and then distributed in 300-μL PCR tubes for the subsequent experiment. A sample containing the activator dsDNA was added to the reporter system and reactions proceeded at different temperatures for 1 h, the florescent signal was detected at an excitation wavelength of 480 nm.
Statistics & reproducibility
No statistical method was used to predetermine the sample size. No data were excluded from the analyses. The experiments were not randomized. The Investigators were not blinded to allocation during experiments and outcome assessment.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The sequencing data generated in this study have been deposited in the NCBI Sequence Read Archive under accession code PRJNA1074107. The model weights of CHOOSER and a minimum dataset to run CHOOSER are available in Zenodo in https://doi.org/10.5281/zenodo.13906238 [https://zenodo.org/records/13906238]59. Source data are provided with this paper.
Code availability
The custom code of CHOOSE, together with the trained models for mining CRISPR-Cas systems and screening Cas12 candidates capable of self-processing pre-crRNA, are available at GitHub repositories (https://github.com/zjlab-BioGene/CHOOSER)60.
Change history
09 January 2025
A Correction to this paper has been published: https://doi.org/10.1038/s41467-025-55913-y
References
Koonin, E. V. & Makarova, K. S. Origins and evolution of CRISPR-Cas systems. Philos. Trans. R. Soc. B 374, 20180087 (2019).
Wang, J. Y. & Doudna, J. A. CRISPR technology: a decade of genome editing is only the beginning. Science 379, eadd8643 (2023).
Shmakov, S. et al. Diversity and evolution of class 2 CRISPR–Cas systems. Nat. Rev. Microbiol 15, 169–182 (2017).
Burstein, D. et al. New CRISPR–Cas systems from uncultivated microbes. Nature 542, 237–241 (2017).
Harrington, L. B. et al. Programmed DNA destruction by miniature CRISPR-Cas14 enzymes. Science 362, 839–842 (2018).
Yan, W. X. et al. Functionally diverse type V CRISPR-Cas systems. Science 363, 88–91 (2019).
Pausch, P. et al. CRISPR-CasΦ from huge phages is a hypercompact genome editor. Science 369, 333–337 (2020).
Al-Shayeb, B. et al. Diverse virus-encoded CRISPR-Cas systems include streamlined genome editors. Cell 185, 4574–4586.e16 (2022).
Wu, W. Y. et al. The miniature CRISPR-Cas12m effector binds DNA to block transcription. Mol. Cell 82, 4487–4502.e7 (2022).
Sun, A. et al. The compact Casπ (Cas12l) ‘bracelet’ provides a unique structural platform for DNA manipulation. Cell Res. https://doi.org/10.1038/s41422-022-00771-2 (2023).
East-Seletsky, A. et al. Two distinct RNase activities of CRISPR-C2c2 enable guide-RNA processing and RNA detection. Nature 538, 270–273 (2016).
East-Seletsky, A., O’Connell, M. R., Burstein, D., Knott, G. J. & Doudna, J. A. RNA targeting by functionally orthogonal type VI-A CRISPR-Cas enzymes. Mol. Cell 66, 373–383.e3 (2017).
Abudayyeh, O. O. et al. RNA targeting with CRISPR–Cas13. Nature 550, 280–284 (2017).
Yan, W. X. et al. Cas13d is a compact RNA-targeting type VI CRISPR effector positively modulated by a WYL-domain-containing accessory protein. Mol. Cell 70, 327–339.e5 (2018).
Xu, C. et al. Programmable RNA editing with compact CRISPR–Cas13 systems from uncultivated microbes. Nat. Methods 18, 499–506 (2021).
Fonfara, I., Richter, H., Bratovič, M., Le Rhun, A. & Charpentier, E. The CRISPR-associated DNA-cleaving enzyme Cpf1 also processes precursor CRISPR RNA. Nature 532, 517–521 (2016).
Zhang, H., Li, Z., Xiao, R. & Chang, L. Mechanisms for target recognition and cleavage by the Cas12i RNA-guided endonuclease. Nat. Struct. Mol. Biol. 27, 1069–1076 (2020).
Campa, C. C., Weisbach, N. R., Santinha, A. J., Incarnato, D. & Platt, R. J. Multiplexed genome engineering by Cas12a and CRISPR arrays encoded on single transcripts. Nat. Methods 16, 887–893 (2019).
McGaw, C. et al. Engineered Cas12i2 is a versatile high-efficiency platform for therapeutic genome editing. Nat. Commun. 13, 2833 (2022).
Tatusova, T. et al. NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res. 44, 6614–6624 (2016).
Russel, J., Pinilla-Redondo, R., Mayo-Muñoz, D., Shah, S. A. & Sørensen, S. J. CRISPRCasTyper: automated identification, annotation, and classification of CRISPR-Cas Loci. CRISPR J. 3, 462–469 (2020).
Ye, J., McGinnis, S. & Madden, T. L. BLAST: improvements for better sequence analysis. Nucleic Acids Res. 34, W6–W9 (2006).
Finn, R. D., Clements, J. & Eddy, S. R. HMMER web server: interactive sequence similarity searching. Nucleic Acids Res. 39, W29–W37 (2011).
Altae-Tran, H. et al. Uncovering the functional diversity of rare CRISPR-Cas systems with deep terascale clustering. Science 382, eadi1910 (2023).
Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021).
Baek, M. et al. Accurate prediction of protein structures and interactions using a three-track neural network. Science 373, 871–876 (2021).
Lin, Z. et al. Evolutionary-scale prediction of atomic-level protein structure with a language model. Science 379, 1123–1130 (2023).
Makarova, K. S. et al. Evolutionary classification of CRISPR–Cas systems: a burst of class 2 and derived variants. Nat. Rev. Microbiol. 18, 67–83 (2020).
Adler, B. A. et al. CasPEDIA Database: a functional classification system for class 2 CRISPR-Cas enzymes. Nucleic Acids Research 52, D590–D596 (2024).
Hsu, C. et al. Learning Inverse Folding from Millions of Predicted Structures. http://biorxiv.org/lookup/doi/10.1101/2022.04.10.487779 (2022).
Ronneberger, O., Fischer, P. & Brox, T. U-Net: convolutional networks for biomedical image segmentation. In Medical Image Computing and Computer-Assisted Intervention – MICCAI 2015 (eds. Navab, N., Hornegger, J., Wells, W. M. & Frangi, A. F.) Vol. 9351 234–241 (Springer International Publishing, Cham, 2015).
Vig, J. et al. BERTology Meets Biology: Interpreting Attention in Protein Language Models. Preprint at http://arxiv.org/abs/2006.15222 (2021).
Omura, S. N. et al. Mechanistic and evolutionary insights into a type V-M CRISPR–Cas effector enzyme. Nat. Struct. Mol. Biol. 30, 1172–1182 (2023).
Chen, J. S. et al. CRISPR-Cas12a target binding unleashes indiscriminate single-stranded DNase activity. Science 360, 436–439 (2018).
Flamholz, Z. N., Biller, S. J. & Kelly, L. Large language models improve annotation of prokaryotic viral proteins. Nat. Microbiol. https://doi.org/10.1038/s41564-023-01584-8 (2024).
Richardson, L. et al. MGnify: the microbiome sequence data analysis resource in 2023. Nucleic Acids Res. 51, D753–D759 (2023).
Coelho, L. P. et al. Towards the biogeography of prokaryotic genes. Nature 601, 252–256 (2022).
Nayfach, S. et al. A genomic catalog of Earth’s microbiomes. Nat. Biotechnol. 39, 499–509 (2021).
Zhu, J. et al. Over 50,000 metagenomically assembled draft genomes for the human oral microbiome reveal new taxa. Genom. Proteom. Bioinform. 20, 246–259 (2022).
Liu, Y. et al. A genome and gene catalog of glacier microbiomes. Nat. Biotechnol. 40, 1341–1348 (2022).
Camargo, A. P. et al. IMG/VR v4: an expanded database of uncultivated virus genomes within a framework of extensive functional, taxonomic, and ecological metadata. Nucleic Acids Res. 51, D733–D743 (2023).
Nayfach, S. et al. Metagenomic compendium of 189,680 DNA viruses from the human gut microbiome. Nat. Microbiol. 6, 960–970 (2021).
Soto-Perez, P. et al. CRISPR-Cas system of a prevalent human gut bacterium reveals hyper-targeting against phages in a human virome catalog. Cell Host Microbe 26, 325–335.e5 (2019).
Camarillo-Guerrero, L. F., Almeida, A., Rangel-Pineros, G., Finn, R. D. & Lawley, T. D. Massive expansion of human gut bacteriophage diversity. Cell 184, 1098–1109.e9 (2021).
Cook, R. et al. INfrastructure for a PHAge REference Database: identification of large-scale biases in the current collection of cultured phage genomes. Phage 2, 214–223 (2021).
Mihara, T. et al. Linking virus genomes with host taxonomy. Viruses 8, 66 (2016).
Schmartz, G. P. et al. PLSDB: advancing a comprehensive database of bacterial plasmids. Nucleic Acids Res. 50, D273–D278 (2022).
Pickard, D. et al. Molecular characterization of the Salmonella enterica serovar typhi Vi-typing bacteriophage E1. J. Bacteriol. 190, 2580–2587 (2008).
Bland, C. et al. CRISPR Recognition Tool (CRT): a tool for automatic detection of clustered regularly interspaced palindromic repeats. BMC Bioinform. 8, 209 (2007).
Lin, T.-Y., Goyal, P., Girshick, R., He, K. & Dollar, P. Focal Loss for Dense Object Detection. IEEE Trans. Pattern Anal. Mach. Intell. 42, 318–327 (2020).
Zhang, C., Shine, M., Pyle, A. M. & Zhang, Y. US-align: universal structure alignments of proteins, nucleic acids, and macromolecular complexes. Nat. Methods 19, 1109–1115 (2022).
Cock, P. J. A. et al. Biopython: freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics 25, 1422–1423 (2009).
Katoh, K. & Standley, D. M. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780 (2013).
Nguyen, L.-T., Schmidt, H. A., Von Haeseler, A. & Minh, B. Q. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274 (2015).
Letunic, I. & Bork, P. Interactive Tree Of Life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 49, W293–W296 (2021).
The UniProt Consortium. et al. UniProt: the universal protein knowledgebase in 2023. Nucleic Acids Res. 51, D523–D531 (2023).
Magoč, T. & Salzberg, S. L. FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics 27, 2957–2963 (2011).
Karvelis, T. et al. PAM recognition by miniature CRISPR–Cas12f nucleases triggers programmable double-stranded DNA target cleavage. Nucleic Acids Res. 48, 5016–5023 (2020).
Wenhui, L. CHOOSER model weights and a minimum dataset. Zenodo https://doi.org/10.5281/ZENODO.13906238 (2024).
ZhejiangLab-BioGene. zjlab-BioGene/CHOOSER: v1.0. Zenodo https://doi.org/10.5281/ZENODO.13906792 (2024).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. T24250193 to Q.L., No. 32341008 to Q.L., No. 62002265 to G.H.C.), the National Key Research and Development Program of China (Grant No. 2021YFF1201200 to Q.L., No. 2021YFF1200900 to Q.L., No. 2022YFC2702705 to J.Z.), Youth Foundation Project of Zhejiang Lab (Grant No. K2023PE0AA02 to W.H.L.), Shanghai Pilot Program for Basic Research, Shanghai Science and Technology Innovation Action Plan-Key Specialization in Computational Biology, Shanghai Shuguang Scholars Project, and Shanghai Excellent Academic Leader Project, National Natural Science Foundation of China (Grant No. 62088101), Shanghai Municipal Science and Technology Major Project (Grant No. 2021SHZDZX0100).
Author information
Authors and Affiliations
Contributions
Q.L., X.X.H. and W.H.L. conceived the project. W.H.L. built the CHOOSER framework. W.H.L., W.K.W., R.Z.C., G.H.C., Q.C.C., P.X.M., J.T. and M.H.G. analyzed the data. W.H.L., L.Y.H., Y.Q.L. and G.H.C. built and trained the AI models. J.Z., Q.L., X.Y.J. and Q.X.G. designed experiments, purified proteins, and performed biochemical experiments. Q.L., W.H.L., X.X.H. and J.Z. wrote this manuscript. This manuscript was reviewed and approved by all co-authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Jin Liu, who co-reviewed with Sita Sirisha Madugula and, Yingsi Zhou and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Li, W., Jiang, X., Wang, W. et al. Discovering CRISPR-Cas system with self-processing pre-crRNA capability by foundation models. Nat Commun 15, 10024 (2024). https://doi.org/10.1038/s41467-024-54365-0
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-024-54365-0
This article is cited by
-
Harnessing artificial intelligence to advance CRISPR-based genome editing technologies
Nature Reviews Genetics (2026)
-
Machine learning and deep learning for genomic data: a data-centric approach to CRISPR/Cas9 gene editing
International Journal of Data Science and Analytics (2025)
-
An updated evolutionary classification of CRISPR–Cas systems including rare variants
Nature Microbiology (2025)
