
Abstract
The current opioid crisis urgently calls for developing non-addictive pain medications. Progress has been slow, highlighting the need to uncover targets with unique mechanisms of action. Extracellular adenosine alleviates pain by activating the adenosine A1 receptor (A1R). However, efforts to develop A1R agonists have faced obstacles. The equilibrative nucleoside transporter subtype 1 (ENT1) plays a crucial role in regulating adenosine levels across cell membranes. We postulate that ENT1 inhibition may enhance extracellular adenosine levels, potentiating endogenous adenosine action at A1R and leading to analgesic effects. Here, we modify the ENT1 inhibitor dilazep based on its complex X-ray structure and show that this modified inhibitor reduces neuropathic and inflammatory pain in animal models while dilazep does not. Notably, our ENT1 inhibitor surpasses gabapentin in analgesic efficacy in a neuropathic pain model. Additionally, our inhibitor exhibits less cardiac side effect than dilazep via systemic administration and shows no side effects via local/intrathecal administration. ENT1 is colocalized with A1R in mouse and human dorsal root ganglia, and the analgesic effect of our inhibitor is linked to A1R. Our studies reveal ENT1 as a therapeutic target for analgesia, highlighting the promise of rationally designed ENT1 inhibitors for non-opioid pain medications.
Similar content being viewed by others


Introduction
The opioid epidemic has emerged as a pressing global challenge, underscoring the crucial demand for non-addictive and effective pain medications. Despite substantial efforts, advancement in developing non-opioid analgesics with different mechanisms of action have been limited. The involvement of adenosine in antinociception or pain suppression has been well established1,2,3,4,5. The adenosine A1 receptor (A1R), expressed in primary sensory neurons, sensory nerve endings in the spinal cord dorsal horn and supraspinal pain-processing neurons, is mainly responsible for the analgesic effect of adenosine2,4,6. Activation of A1R leads to a reduction in cAMP levels, inhibiting pain transmission at both presynaptic and postsynaptic neurons4. Extensive efforts in developing analgesics targeting A1R activation, as well as other AR subtypes, have been made with limited success. Synthetic A1R agonists, despite their analgesic efficacy2,4, have not advanced to the clinical stages, largely due to their cardiovascular side effects5,7. Alternate approaches, like partial agonists and AR-positive allosteric modulators, show some promises8, but come with their own challenges such as reduced antinociceptive effects9, receptor downregulation, or desensitization-mediated tolerance10. Another strategy involves elevating the extracellular concentration of adenosine instead of a synthetic agonist. The extracellular concentrations of adenosine are maintained at low levels mainly due to its cellular uptake or metabolic degradation11. The main endogenous source of adenosine is through ATP hydrolysis by ectonucleotidases. Increasing extracellular adenosine concentration by means of expressing recombinant ectonucleotidases (PAP or CD73) has demonstrated efficacy against neuropathic pain in preclinical models5. However, further development of this approach has been slow, likely due to the challenges associated with delivering recombinant enzymes. Given the hurdles faced in developing A1R- or enzyme-based therapeutics for pain relief, alternative strategies are needed.
Equilibrative nucleoside transporters (ENTs; Solute Carrier Family 29; SLC29) facilitate bidirectional nucleoside transport across the cell membrane12,13,14,15. Amongst the four subtypes in humans (hENT1-hENT4), hENT1 is the primary adenosine transporter owing to its broad tissue expression profile and high levels of surface expression12,13 while hENT3 expression is limited to organelles such as lysosome and mitochondria and adenosine is not a substrate for hENT4 in physiological pH ranges13,16. The adenosine uptake system has a much greater influence than metabolic transformation in eliminating adenosine from the extracellular space17,18. We hypothesize that by inhibiting hENT1 activity, we could provide a means to increase extracellular adenosine concentrations, resulting in A1R activation in primary sensory neurons, potentially leading to pain relief. This concept of augmenting adenosine action by inhibiting its cellular transport system is analogous to that of inhibiting neurotransmitter transporters, such as the monoamine transporters, to potentiate signaling stimulated by an endogenous neurotransmitter or neuromodulator19,20. This idea of modulating adenosine receptor signaling due to the inhibition of ENT has been explored in several contexts such as hepatic ischemia, lung injury, mucosal inflammation, and cardio-protection21,22,23. Here, we show that a rationally modified hENT1 inhibitor (JH-ENT-01) exhibits analgesic efficacy in animal models of diabetic neuropathy and nerve injury as well as of inflammatory pain, unlike the parent inhibitor dilazep. This suggests that hENT1 is a promising target for treating inflammatory and neuropathic pain, and the rational design of hENT1 inhibitors could set the stage for development of non-opioid analgesics.
Results
Structure-based ENT1 inhibitor design
We previously generated a thermostable variant of hENT1 (hENT1cryst) which contains three mutations and a disordered loop truncation for its enhanced biochemical stability24. These modest engineered components of the transporter are located far away from the inhibitor and substrate binding site and this hENT1cryst construct exhibits binding and transport properties similar to its wild type24 (Supplementary Fig. 1a). We previously crystallized hENT1cryst in complex with the adenosine reuptake inhibitors dilazep or nitrobenzylthioinosine (S-(4-nitrobenzyl)-mercaptopurine riboside; NBMPR), and determined their X-ray structures to 2.3 Å and 2.9 Å resolution, respectively (Supplementary Fig. 1a, b)24. Dilazep has been approved in Europe and Japan as a therapeutic agent for renal disorders, while NBMPR has been used as a research tool to study ENT112,14. hENT1cryst is a monomer, comprised of 11-transmembrane helices (TMs, Supplementary Fig. 1a). The first 6 TMs form one structural unit, which we termed the N-domain, and the final 5 TMs form another bundle which we term the C-domain24 (Supplementary Fig. 1a). These bundles exhibit a pseudo-symmetry, which is similar to that observed in major facilitator superfamily (MFS) transporters25. The central site is accessible to the extracellular side of the membrane and is occupied by inhibitors in our structures, suggesting that both structures represent outward-facing conformations (Supplementary Fig. 1b). The amino acid residues involved in nucleoside recognition reside in the central cavity, suggesting its role as a substrate recognition/binding site, which we term the “orthosteric site”24. The nucleoside moiety of NBMPR nestles snugly within this orthosteric site, leading us to posit that the adenosine recognition by hENT1 will likely be similar to that of nucleoside portion of NBMPR (Supplementary Fig. 1b). Because a substantial portion of NBMPR and dilazep occupy the orthosteric site, they are considered competitive inhibitors26.
We discovered that both inhibitors, rather than solely binding to the orthosteric site, extend and bind to additional sites, which we term “opportunistic sites” (Supplementary Fig. 1b, d). Analysis of the dilazep binding sites suggests that one of the trimethoxyphenyl ring of dilazep binds to the orthosteric site while the remaining portion protrudes toward the extracellular site outside of the central cavity. We refer to this site as “opportunistic site 1” (Supplementary Fig. 1b). In the case of NBMPR, the central cavity of hENT1 is extended to a deep-hydrophobic pocket into which the para-nitrobenzyl group of the adenosine analog snugly fits. Notably, glycine 154 is located in this site of hENT1, while other ENT subtypes contain larger amino acids (e.g. serine in hENT2 and hENT3), rendering NBMPR specific to hENT1 (Supplementary Fig. 1d)24,27. We refer to this site as “opportunistic site 2”.
An overlay of the two inhibitor structures illustrates how they occupy both overlapping and distinct sites within the central cavity of human ENT1 (Fig. 1a). This observation sparked an idea to combine chemical features of dilazep and NBMPR in a manner that could result in simultaneous occupancy of both the “opportunistic sites”, which we anticipated would potentially enhance target potency and specificity. To test this idea, we focused on fusing an additional group onto the dilazep scaffold, as dilazep has proven clinical utility as a human medicine12,14 while NBMPR remains a research tool due to its toxicity deriving from its metabolic breakdown product28,29,30,31. We initially chose the meta-position of the dilazep trimethoxyphenyl ring for installation of a para-nitrobenzyl group, based on the structural superposition, to maximize the chance of the added para-nitrobenzyl group would occupy opportunistic site 1 (Fig. 1a, b). We term this unique hybrid analog of the two distinct adenosine reuptake inhibitors “JH-ENT-01” (Fig. 1b). As illustrated (Supplementary Fig. 2 and Supplementary Methods), a modular synthesis was developed for the preparation of JH-ENT-01.

a Structural overlay of NBMPR and dilazep in the central cavity of hENT1cryst, with shared and distinct inhibitor binding sites labeled. b Chemical structure of dilazep and the rationally designed adenosine reuptake inhibitor, JH-ENT-01. The new inhibitor contains chemical moieties analogous to features of both dilazep and NBMPR. c Inhibitory constants (Ki) from cold-competition displacement of [3H]-NBMPR from hENT1cryst by dilazep of JH-ENT-01 (scintillation proximity assay; n = 6 technical replicates across biological duplicates; mean ± s.e.m. shown). d X-ray crystal structure of JH-ENT-01 complexed to hENT1cryst. e hENT1 and hENT2 dependent [3H]-adenosine (ado) uptake and their inhibition by dilazep and JH-ENT-01 (n = 2–3 biological replicate titrations with technical triplicates, mean ± s.e.m. shown). f Structural overlay of JH-ENT-01 and NBMPR shows a difference of para-nitrobenzyl group binding poses within opportunistic site 2. g Comparison of [3H]-adenosine uptake block in oocytes expressing WT or G154S hENT1 by NBMPR, dilazep, or JH-ENT-01 (n = 3 biological replicates, mean ± s.e.m.; P values indicated from non-paired parametric t-test).
We then conducted a cold competition scintillation proximity assay by using purified thermostabilized hENT1 construct (hENT1cryst) that was used for structural studies. We found that JH-ENT-01 retains good apparent binding potency to detergent purified hENT1cryst compared to dilazep (Fig. 1c). When compared with apparent Ki from a single-site fit, JH-ENT-01 exhibits a higher displacement ability than that of dilazep (Fig. 1c). We subsequently determined the structure of JH-ENT-01 in complex with hENT1cryst at 2.7 Å using X-ray crystallography (Fig. 1d, Table 1, Supplementary Figs. 1c and 3). Notably, we found that its binding mode is consistent with our anticipated structure-guided design that JH-ENT-01 with the introduced para-nitrobenzyl group does indeed occupy the opportunistic site 2 of hENT1, as evident by strong Fo-Fc signal in both unmodelled and simulated-annealing OMIT maps (Supplementary Fig. 3b). To test whether JH-ENT-01 has increased specificity toward hENT1, we performed a radiotracer uptake assay using Xenopus laevis oocytes expressing wild type hENT1 and hENT2 (Fig. 1e). We monitored [3H]-adenosine uptake by hENT1 and hENT2 and its block by dilazep and JH-ENT-01. hENT3 and hENT4 were not tested because hENT3 is mainly expressed in lysosomes and mitochondria and hENT4 does not transport adenosine at physiological pH13. Due to the limited solubility of JH-ENT-01 we could not reach full block of hENT2 uptake activity, which prevented accurate measurement of its apparently high IC50 (Fig. 1e). However, it is clear that JH-ENT-01 maintains good selectivity for hENT1 over hENT2. We then introduced the G154S mutation into the hENT1 background, owing to the fact it is a key subtype specificity determinant for NBMPR (serine is present at this position in hENT2 and hENT3), and thus an important feature of opportunistic site 224,27 (Supplementary Fig. 1d, f). We found that JH-ENT-01 preferentially inhibited wild type hENT1 over G154S by about two-fold, while dilazep did not show any preference between WT and G154S hENT1, suggesting that JH-ENT-01 enhances the selectivity for hENT1 compared to dilazep, by occupying opportunistic site 2 (Fig. 1g). In the structure of hENT1 in complex with JH-ENT-01, the binding pose of the refined coordinates for the para-nitrobenzyl group is slightly different from that of NBMPR from the previous crystal structure (Fig. 1f). The lack of substantial change in apparent affinity and specificity for JH-ENT-01 may be attributed to the less-than-optimal binding of the para-nitrobenzyl group of JH-ENT-01 to the opportunistic site 2 (Fig. 1f). Additionally, the other trimethoxyphenyl ring of JH-ENT-01, which lacks the para-nitrobenzyl group, may bind to the orthosteric site, given the symmetric nature of dilazep – however we do not see evidence of such an orientation in our crystal structure. Taken together, this initial structure-guided modification of dilazep has resulted in a derivative with distinct binding properties. Most importantly, this hybrid analog retains good apparent affinity and specificity for hENT1 (Fig. 1c, e, g).
ENT1 expression in mouse DRG and spinal cord and human DRG
Pain is originated in primary sensory neurons which project to the spinal cord dorsal horn and make synaptic contacts with postsynaptic neurons32. To explore a possible role of ENT1 in pain regulation, we first examined the expression of ENT1 transcript (Slc29a1) in mouse dorsal root ganglion (DRG) and spinal cord using a highly sensitive and selective RNAscope in situ hybridization (ISH) technique. We found that Slc29a1 mRNA is expressed by the majority of DRG sensory neurons, with a strong labeling in small-sized DRG neurons (Fig. 2a). Furthermore, we found that Slc29a1 mRNA expression was abundant in the spinal cord dorsal horn (Fig. 2b). Double immunostaining revealed co-localization of ENT1 and A1R in the dorsal horn and ventral horn (Fig. 2c), suggesting a potential signaling pathway involving ENT1 and A1R in the spinal cord. Consistently, single-cell analysis revealed broad expression of SLC29A1 transcript in mouse and human sensory neurons including TRPV1+ nociceptors, as well as mouse and human spinal cord neurons (Supplementary Fig. 4). By using RNAscope ISH, we also found that SLC29A1 mRNA is expressed in human DRG neurons, and furthermore, SLC29A1 mRNA is highly co-localized with that of ADOR1 (A1R) mRNA in 76% of human sensory neurons (Fig. 2d). These data suggest that ENT1 is present in the mouse and human nociceptive pathway, including both primary and secondary order neurons, and may play a role in nociception. Taken together with the fact that mouse and human ENT1 share 79% sequence identity, with virtually 100% identity in the orthosteric and opportunistic sites in the central cavity, mouse models for pain are well suited for interrogating potential analgesic activities of hENT1 inhibitors.

a, b RNA scope in situ hybridization showing ENT1 transcript (SLC29A1) expression in the DRG (a) and spinal cord dorsal horn (b) of mice. The dorsal horn top edge is indicated by the dotted line. Scales, 50 μm in a and 100 μm in b. c Double immunostaining showing co-localization of ENT1 and A1R in the dorsal horn and ventral horn (box size, 250 × 250 μm). Right panels, enlarged images from the boxes. Scale, 25 μm. Experiments in a–c were repeated 2–3 times. d Left, RNAscope in situ hybridization images showing co-localization of SLC29A1 and ADOR1 (A1R) in human DRG (hDRG) neurons. The same DRG sections were co-stained with NeuN antibody (neuronal marker) and DAPI (nuclei marker). Arrows indicate neurons that are positive for both ENT1 and A1R. Scale, 50 μm. Right, quantification showing the ratio and percentage of SLC29A1 or/and ADOR1 positive and negative DRG neurons from four human donors.
Effects of ENT1 inhibitors on cardiovascular function
Because the major limitation of full A1R agonists is the cardiac side effects5,7, it is necessary for us to investigate the effects of dilazep and JH-ENT-01 on cardiac function in mice. We performed electrocardiograms (ECG) experiments to check cardiac side effects of JH-ENT-01 and dilazep on mice following either intravenous (i.v., Fig. 3a, b) or intraperitoneal (i.p., Fig. 3c, d) administration of these inhibitors. We found a significant decrease in the heart rates at the first hour following IV administration of dilazep, but not JH-ENT-01 (Fig. 3a, b, 30 mg/kg). In contrast, we found no significant decrease in heart rates following IP administration of either dilazep nor JH-ENT-01 at 10 mg/kg and 30 mg/kg (Fig. 3c, d).

a, b Effects of intravenous (i.v.) injection of dilazep and JH-ENT-01 (30 mg/kg) on heart rates in ECG test, with representative ECG traces before the treatment and at 10 min, 30 min, and 60 min after the vehicle and drug treatment in a, quantification of heart rates as beats per min in b (n = 6 animals from both sexes). Two-way ANOVA followed by Bonferroni posthoc comparison. *P < 0.05, **P < 0.01, ***P < 0.001; ns, not significant. Figure 3b was created in BioRender. He, W. (2024) BioRender.com/f39n75. c, d Effects of intraperitoneal (i.p.) injection of dilazep and JH-ENT-01 (10 and 30 mg/kg) on heart rates in ECG test, with representative ECG traces before the treatment and at 10 min, 30 min, and 60 min after the vehicle and drug treatment in c, and quantification of heart rates as beats per min in d (n = 3 animals from both sexes). All data were expressed as mean ± s.e.m. Figure 3d was created in BioRender. He, W. (2024) BioRender.com/o52y585.
JH-ENT-01 reduces inflammatory pain
We tested whether JH-ENT-01 could modulate pain. Formalin-induced acute inflammatory pain is widely used to assess putative analgesics, and the 2nd phase response is mediated by central sensitization. Intrathecal (i.t.) route may target both DRG and spinal cord mechanisms of pain33. We tested the analgesic effects of JH-ENT-01 using different doses (1, 10, and 100 nmol) and included dilazep (100 nmol) as a comparison. We found that formalin-induced phase 1 pain was not affected by these inhibitors in both sexes at all administered doses (Fig. 4a–c), suggesting that ENT1 inhibitors may not significantly affect nociception and motor function under normal conditions. Notably, the intrathecal (i.t.) injection of JH-ENT-01 led to a dose-dependent reduction in the 2nd phase pain in both males (n = 4, Fig. 4a) and females (n = 4, Fig. 4b), as well as when data from males and females were combined (n = 8, Fig. 4c). This result suggests no obvious sex difference in ENT1-mediated pain suppression. Interestingly, dilazep elicited a significant inhibition of phase 2 pain only when data from both sexes were combined and at a high concentration (100 nmol) (Fig. 4c). To test whether the analgesic effect was associated with motor impairments, we performed a rotarod test before and 60-min after i.t. delivery of 100 nmol dilazep and JH-ENT-01. Animals became less active within 30 min of the administration of ENT1 inhibitors. However, animals showed normal motor function after 60 min after the treatment in the rotarod test, exhibiting no significant changes in the falling latency (Fig. 4d). ENT1 inhibitors, including dilazep, have been shown to produce mild antihyperalgesic effects in models of inflammatory pain in guinea pigs34. It is worthwhile to note that spinal cord synaptic plasticity, also known as central sensitization, plays a critical role in both inflammatory and neuropathic pain32,35. This data is consistent with our hypothesis that JH-ENT-01 may regulate pain via spinal cord mechanisms.

a–c Effects of intrathecal (i.t.) injection of the ENT1 inhibitors JH-ENT-01 (1, 10, and 100 nmols) and dilazep (100 nmol) on formalin-induced inflammatory pain in phase 1 (0–10 min) and phase 2 (10–45 min) in male mice (a), female mice (b), and males and females combined (c). Top panels show the time course of formalin-induced pain. n = 4 per sex; n = 8 for both sexes. The inhibitors were given 30 min prior to the intraplantar injection of formalin (5%, 20 μl). d Rotarod tests before and 60 min after i.t. delivery of 100 nmol of JH-ENT-01 and dilazep. n = 6 animals per group with mixed sexes. Two-way ANOVA followed by Bonferroni posthoc comparison. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001; ns, not significant. All data were expressed as mean ± s.e.m.
Diverse routes of JH-ENT-01 administration are effective in suppressing inflammatory pain
Although JH-ENT-01 does not have significant cardiovascular side effects in mice when injected intravenously unlike dilazep (Fig. 3b), it is still a concern that JH-ENT-01 can affect cardiovascular function via systemic i.v. injection at high doses. Therefore, we decided to test different routes of ENT1 inhibitors on a persistent inflammatory pain model, induced by Complete Freund’s Adjuvant (CFA).
As we previously reported36, intraplantar CFA injection induced robust mechanical allodynia, as indicated by a reduction in paw withdrawal threshold (PWT) and an increase in paw withdrawal frequency (PWF) to a sub-threshold filament (0.4 g) in von Frey test (Fig. 5a–c). To determine if ENT1 inhibitors would produce accumulating analgesic effects or antinociceptive tolerance, ENT1 inhibitors were injected 1, 3, and 5 days after CFA administration and mechanical pain behaviors were recorded 1, 3, 5, and 24 hours after each injection of ENT inhibitors (Fig. 5a). We found that systemic intravenous injection of JH-ENT-01 (10 mg/ml) elicited a significant increase in PWT (P < 0.001 at 3 h) and decreased PWF significantly (P < 0.001 at 3 h), while dilazep and vehicle had no effects on PWT and PWF (Fig. 5b, c). Notably, we observed accumulating effects over repeated injections of JH-ENT-01. We next administered JH-ENT-01 via subcutaneous (s.c.) injections at 1-, 3-, and 5-days post-CFA (Fig. 5d) and found that JH-ENT-01 (10 mg/ml) elicited a significant increase in PWT (P < 0.01 at 3 h) and a significant decrease in PWF (P < 0.001 at 3 h) after the second and third injections of JH-ENT-01, while dilazep and vehicle did not (Fig. 5d–f).

a–c Effects of intravenous injections of JH-ENT-01 (10 mg/kg, i.v.). a Schematic of drug injections and behavioral testing with PWT shown in b and PWF in c. n = 6 mice/group, equal number male and female). Figure 5a was created in BioRender. He, W. (2024) BioRender.com/f39n675. d–f Effects of subcutaneous injections of JH-ENT-01 (10 mg/kg, s.c.). d Schematic of drug injections and behavioral testing. Figure 5d was created in BioRender. He, W. (2024) BioRender.com/f05p763. e, f PWT shown in e and PWF in f. n = 6 mice/group equal number male and female mice. g–i Effects of intraplantar injections of JH-ENT-01 (10 μg). g Schematic of drug injections and behavioral testing in the CFA model. Created in BioRender. He, W. (2024) BioRender.com/j51a415. h, i PWT (h) and PWF (i) in the CFA model. n = 6 mice/group with equal number male and female mice. Two-way ANOVA followed by Bonferroni posthoc comparison (b, c, e, f, h, i). *P < 0.05, **P < 0.01, ***P < 0.001. All data were expressed as Mean ± SEM. BL baseline; CFA complete Freund’s adjuvant; PWF paw withdrawal frequency; PWT paw withdrawal threshold.
Last, we administered JH-ENT-01 via local intraplanar injection (10 μg) at day 3 post CFA (Fig. 5g) and found that local injection of JH-ENT-01 elicited a rapid increase in PWT (P < 0.001) and decrease in PWF (P < 0.001) at 1 h after drug administration, which lasted longer than 5 hours (Fig. 5h, i). Taken together our data show that JH-ENT-01 exhibits significant inhibition of inflammatory pain via systematic (i.v. and s.c.) and local routes.
JH-ENT-01 alleviates neuropathic pain in different mouse models
Diabetes-induced peripheral neuropathy (DPN) is a major neuropathic pain condition37,38. We tested two widely used mouse models of DPN: the streptozotocin (STZ) model (Fig. 6a) and the db/db genetic model (Fig. 6b). Both models result in a loss of epidermal nerve fibers and generation of neuropathic pain symptoms, including mechanical and cold hypersensitivity39,40. First, we tested potential antinociceptive effects of ENT1 inhibitors in the STZ model, following both local i.t. administration that can target the spinal cord and DRG mechanisms33 and systemic administration via i.v. route. The overall conditions of animals after the STZ injection were reasonably good; all the animals exhibited glucose levels of 300 mg/dl (Supplementary Fig. 5a) or greater and animals may have increased urination and mild weight loss after 4 weeks (Supplementary Fig. 5b). One week after a single STZ injection (150 mg/kg), mice developed robust mechanical allodynia, a cardinal feature of neuropathic pain. We found that JH-ENT-01 (10 nmol, i.t.) elicited a significant increase in PWT (P < 0.001, Fig. 6a). In contrast, i.t. dilazep failed to elicit an increase in PWT in mice with STZ-induced DPN (Fig. 6a). Next, we proceeded with the intravenous administration of JH-ENT-01 and dilazep. We found that i.v. injections of JH-ENT-01 (30 mg/kg) on days 8, 10, and 12 after STZ injection markedly elevated the PWT, with accumulating effects evident after the second and third injections (Supplementary Fig. 5c). Notably, after the third injection, owing to the cumulative effect of the JH-ENT-01, the PWT increase lasted for 24 h (Supplementary Fig. 5c). We observed that 7 out of 16 mice treated with dilazep (30 mg/kg) died (Supplementary Fig. 5c), potentially due to its effects on cardiovascular function (Fig. 3a, b) or an unidentified source of toxicity. In contrast, all mice treated with JH-ENT-01 (30 mg/kg) survived after 3 injections (Fig. 3a and Supplementary Fig. 5c). Moreover, we did not observe any analgesic effect in the remaining mice treated with dilazep (Supplementary Fig. 5c). This systemic treatment of JH-ENT-01 also alleviated STZ-induced cold pain (Supplementary Fig. 5d). The rotarod test showed that repeated i.v. injections of JH-ENT-01 (30 mg/kg) improved the motor function of STZ-induced diabetic mice (Supplementary Fig. 5e). Therefore, JH-ENT-01 is superior to dilazep in terms of safety and analgesic potency. In the genetic model of DPN, db/db mice developed robust cold hypersensitivity, which was also inhibited by intrathecal JH-ENT-01 (30 nmol, Fig. 6b). This treatment of JH-ENT-01 further improved the motor function in db/db mice (Fig. 6c).

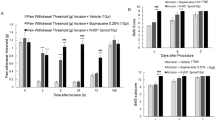
a Mechanical pain (PWT) after intrathecal (i.t., 10 nmol) injections of dilazep (n = 6), JH-ENT-01 (n = 6), and vehicle control (n = 6) in mice with STZ-induced diabetic neuropathy. b Cold pain in acetone test after intrathecal (i.t., 10 nmol) injections of JH-ENT-01 (n = 5) and vehicle control (n = 5) in db/db mice. c Rotarod test showing improved motor function by JH-ENT-01 at 3 hours after the same treatment as shown in b. d, schematic of drug injection and behavioral testing for CCI-induced neuropathic pain and comparison of JH-ENT-01 with gabapentin (30 nmol, i.t.). e, g PWT (e) and cold pain (g) after i.t. injection of JH-ENT-01 and gabapentin. f, h AUC analysis of e, g. (n = 6 mice with equal number male and female; e, g **P < 0.01, ***P < 0.001, **** P < 0.0001, gabapentin vs. JH-ENT-01; f, h *P < 0.05, **P < 0.01, ****P < 0.001, as indicated. i Effects of intrathecal injections (i.t., 30 nmol) of JH-ENT-01 (n = 4) and vehicle (n = 4) in the SNI model. j–l Effects of peri-sciatic injection (j) of JH-ENT-01 (10 nmol) on SNI-induced mechanical pain (k) and cold pain (l). Figure 6j was created in BioRender. He, W. (2024) BioRender.com/a70b516. **P < 0.01, **** P < 0.0001. Two-way ANOVA followed by post-hoc comparison (a, e, g, i, k, l). One-way ANOVA followed by post-hoc comparison (b, f, h); Student’s t test (c, two-sided). Both males and females are included in each group. BL baseline; CCI chronic constriction injury; SNI spared nerve injury; STZ streptozotocin; PWF paw withdrawal frequency; PWT paw withdrawal threshold. All data were expressed as mean ± s.e.m.
To test if our inhibitor has efficacy against additional animal models of neuropathy, we employed two nerve injury models of neuropathic pain, induced by chronic nerve constriction (CCI) and spared nerve injury (SNI). The CCI and SNI models have been widely used for studying neuropathic pain mechanisms and testing pain therapeutics41,42. We found that intrathecal administration of JH-ENT-01 (30 nmol, i.t.) alleviated neuropathic pain in the CCI model by increasing PWT (Fig. 6e, f) and decreasing PWF (Fig. 6g, h). We also found that systemic administration of JH-ENT-01 (30 mg/kg, i.v.) resulted in increases in PWT in the CCI model with similar accumulating analgesic impact as seen in the STZ model (Supplementary Fig. 5f). Intrathecal administration of JH-ENT-01 also significantly reduced the duration of spontaneous pain-like (guarding and licking) behavior in CCI mice (P < 0.001, Supplementary Fig. 5g). We also tested SNI-induced mechanical allodynia by examining PWF. We observed an accumulating analgesic effect following daily i.t. injections of JH-ENT-01. After the third injection, JH-ENT-01 markedly reduced PWF at both 1 h and 3 h post-injection (Fig. 6i).
We compared the analgesic efficacy of JH-ENT-01 with gabapentin, a commonly used clinical treatment for neuropathic pain43. Notably, in the CCI model, i.t. injection of 10 nmol JH-ENT-01 produced greater inhibition in mechanical pain (PWT, Fig. 6e) and cold pain (Fig. 6g) than the equal amount of gabapentin. Area-under-curve (AUC) analysis revealed significantly larger effects of JH-ENT-01 vs. gabapentin (P < 0.05, Fig. 6f, h). Together, these data suggest that JH-ENT-01 may be more potent than gabapentin in attenuating neuropathic pain.
To test if a different route of administration other than i.t. and i.v. injections would affect pain, we administered JH-ENT1 via peri-sciatic nerve injection (Fig. 6j) and found that JH-ENT-01 reduced mechanical pain (Fig. 6k) and cold pain (Fig. 6l) in the SNI mice.
Collectively, our data shows that JH-ENT-01 exhibits efficacy against three distinct types of animal models of neuropathy via diverse routes of administration without apparent side effects and antinociceptive tolerance.
Pharmacokinetic properties of ENT1 inhibitors
Our results unequivocally demonstrate analgesic efficacy of JH-ENT-01 against inflammatory and neuropathic pain, whereas its parent compound dilazep does not possess such an effect. To further our understanding of the analgesic mechanism of JH-ENT-01 against inflammatory and neuropathic pain, we decided to evaluate its plasma and tissue pharmacokinetic (PK) properties and compare it with those of dilazep. We did not include NBMPR because JH-ENT-01 is designed based on the dilazep scaffold and NBMPR was not considered a clinical candidate due to its immunosuppressant and mutagenic effects deriving from its metabolic breakdown product 6-mercaptopurine28,29,30,31.
Given the fact that intravenous administration of dilazep caused the death of many mice, we decided to test PK properties of ENT1 inhibitors via subcutaneous route. Our results showed that s.c. injections of JH-ENT-01 reduced both inflammatory pain in the CFA model (Fig. 5d–f) and neuropathic pain in the STZ model (Fig. 7a), while dilazep had no effects. For the PK study, 30 mg/kg of dilazep dihydrochloride and equimolar dose JH-ENT-01 were administered subcutaneously to CD-1 male mice. Blood was collected from the heart, followed by a whole-body perfusion with saline. Subsequently, brain, spinal cord, dorsal root ganglia (DRG), and trigeminal ganglion (TG) were collected at several time-points within 24 h. We conducted a general observation of concentration-time data (Fig. 7b and Supplementary Fig. 7) and calculated PK parameters (Table 2) in both plasma and neural tissues. Compared to dilazep, JH-ENT-01 exhibits more desirable PK properties. The time course of dilazep plasma concentrations over a 24-hour period revealed that dilazep reaches its maximum concentration (Cmax) quickly (~0.5 hr), but it is rapidly eliminated. In contrast, while JH-ENT-01 also reaches its Cmax quickly (~0.5 hr), its Cmax (164 μg/ml) is substantially lower than that of dilazep (409 μg/ml). Notably, terminal elimination half-life (t1/2) and mean resident time (MRTinf) are about three times longer for JH-ENT-01 than for dilazep while the clearance (Cl/F) for JH-ENT-01 is lower than that for dilazep, resulting in greater total drug plasma exposure over time as evident from its area under curve (AUC) (Fig. 7b and Table 2). The advantageous PK properties of JH-ENT-01 were also translated into the PK values of brain, spinal cord, DRG, and TG tissues. Namely, the observed tissue PK profiles and magnitude of the distribution (tissue concentration), very much resemble those of the corresponding plasma, suggesting efficient distribution and fast plasma/tissue equilibrium (Fig. 7b, Supplementary Fig. 7, and Table 2). The extended exposure of JH-ENT-01 is consistent with its in vivo analgesic efficacy in terms of duration of pain relief (Figs. 5e, f and 7a, b).

a PWT after subcutaneous (s.c.) injections of dilazep (n = 9, 5 males and 4 females, 3 × 30 mg/kg) and JH-ENT-01 (n = 7, 5 males and 2 females, 3 × 30 mg/kg) in mice with STZ-induced diabetic neuropathy. Arrows indicate three s.c.. injections on days 8, 10, and 12. Note that all mice survived after the s.c. injections. Two-way ANOVA followed by Bonferroni posthoc comparison (P = 0.027 for overall comparison between JH-ENT-01 and dilazep. * P < 0.05, JH-ENT-01 vs. dilazep at 1 h after 3rd injection). b Pharmacokinetic profile of JH-ENT-01 and dilazep in plasma, following a single s.c. injection of JH-ENT-01 (30 mg/kg) and equimolar amount of dilazep (n = 3 male mice per time point). All data were expressed as mean ± s.e.m.
The analgesic effects of JH-ENT-01 depend on A1R
Spinal cord synaptic plasticity, also known as central sensitization, plays a crucial role in the development of neuropathic pain32,44,45. We recorded spontaneous excitatory postsynaptic currents (sEPSCs) in spinal cord slices from STZ-treated diabetic mice (Fig. 8a). When we perfused these spinal cord slices with JH-ENT-01, we observed a significant reduction in the frequency of sEPSC (Fig. 8b–c, P < 0.05), without any noticeable changes in the amplitude of sEPSC (Fig. 8c). Consistent with our observation, ENT1 has been proposed to modulate excitatory synaptic transmission via pre-synaptic modulation in the spinal cord dorsal horn46.

a–c Patch clamp recordings showing the effects of JH-ENT-01 on synaptic transmission (sEPSCs) in spinal cord slices of STZ-treated in mice at 1 week. a Schematic of patch clamp recordings in spinal cord slice. Created in BioRender. He, W. (2024) BioRender.com/d34r338. b sEPSC traces before, during, and after the inhibitor perfusion (3 μM, 3 min). The lower traces are shorter segments of the upper trace, marked as 1 (before the perfusion) and 2 (after perfusion). c Quantification of sEPSC frequency (top) and amplitude (bottom) before and after JH-ENT-01 treatment (n = 13 neurons from 3 male mice, paired Student’s t test, two-sided). d, e Reversal of JH-ENT-01’s analgesia by A1R antagonist DPCPX in STZ-treated mice (d) and CFA-treated mice (e) via intrathecal route. DPCPX (10 nmol) was given 30 min prior to the injection of ENT1 inhibitor (10 nmol). f The reversal of JH-ENT-01’s analgesia on the CFA-induced inflammatory pain is observed only with the A1R-specific antagonist DPCPX (10 nmol), not the A2AR-specific inhibitor ZM241385 (10 nmol), the A2BR-specific inhibitor PSB603 (10 nmol), and the A3R-specific inhibitor MRS3777 (10 nmol). *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. Two-Way ANOVA followed by Bonferroni posthoc comparison. n = 6 males. g sEPSC traces before, during, and after the A1R antagonist DPCPX perfusion (1 μM, green) and DPCPX together with JH-ENT-01 (3 μM, blue). h Quantification of sEPSC frequency (left) and amplitude (right) of g (n = 16 neurons from 3 mice–2 males and 1 female; paired Student’s t test, two-sided). All data were expressed as mean ± s.e.m.
A1R is expressed by the primary sensory neurons of DRG (Fig. 2c, d) and plays an essential role in adenosine-induced analgesia47,48. DPCPX, a selective A1R antagonist8,49, was utilized to determine whether it could inhibit the analgesic action of JH-ENT-01 in the STZ and CFA models. We found that JH-ENT-01 (i.t., 10 nmol) increased the PWT in vehicle-treated STZ mice, but this analgesic action was prevented by i.t. pre-treatment with DPCPX (Fig. 8d). We also found that JH-ENT-01 (i.t. 10 nmol) reduced PWF in CFA mice (Fig. 8e). In order to test if the analgesic effect by JH-ENT-01 is mediated by specific adenosine receptor subtypes, we tested several adenosine receptor subtype-specific inhibitors via i.t. route (10 nmol): DPCPX (A1R-specific antagonist), ZM241385 (A2AR-specific antagonist50), PSB603 (A2BR-specific antagonist51), and MRS3777 (A3R-specific antagonist52) in the CFA model (Fig. 8f). We found that only DPCPX, but not other subtype-specific antagonists, is effective in blocking the analgesic action of JH-ENT-01 (Fig. 8f).
In our spinal cord patch clamp recordings of naive animals, co-administration of JH-ENT-01 with DPCPX did not alter sEPSC frequency (Fig. 8g, h), in contrast to the effect of JH-ENT-01 in spinal cord neurons of neuropathic animals (Fig. 8a–c). DPCPX alone had no effects on sEPSC frequency and amplitude in naive animals, possibly due to the lower basal activity of A1R (Supplementary Fig. 6a, b). Although the mechanism by which ENT1 inhibition regulates A1R activation remains to be established, these results suggest that the analgesic action of the ENT1 inhibitor JH-ENT-01 is linked to A1R, but not other adenosine receptor subtypes. Consistent with our findings, we observed co-localization of ENT1 and A1R in human DRG and mouse spinal cord (Fig. 2c, d).
Discussion
Our structure-guided modification of the hENT1 inhibitor dilazep has demonstrated analgesic efficacy in mouse models of neuropathic as well as inflammatory pain. In contrast, the parent inhibitor dilazep did not show noticeable effects against neuropathic pain, which is known to be more resistant to treatment than inflammatory pain53. Importantly, the modified hENT1 inhibitor does not have any significant impacts on motor and cardiovascular functions, in sharp contrast to dilazep (Fig. 3). Furthermore, JH-ENT-01 does not exhibit any signs of antinociceptive tolerance and, in fact, demonstrates cumulative analgesic effects following repeated injections (Figs. 5b–f, 6e–g, and Supplementary Fig. 5c, f).
The observation of the analgesic efficacy of JH-ENT-01 in inflammatory and neuropathic pain models, the co-localization of ENT1 and A1R in DRG sensory neurons and spinal cord neurons, and the specificity of the A1R antagonist in reversing JH-ENT-01’s analgesic action is consistent with our initial hypothesis that ENT1 inhibition activates A1R in the spinal cord pain circuit, resulting in pain relief. It is likely that ENT1 inhibition raises extracellular adenosine levels in the spinal cord, and the subsequent A1R activation reduces pain via Gi/o-mediated inhibition of cAMP signaling2. A1R may also control pain through Gβγ-mediated activation of potassium channels in dorsal horn neurons2,4. Further studies are warranted to test the mechanisms of JH-ENT-01-mediated A1R activation using biochemical approaches, as well as genetic approaches (e.g., ENT1 and A1R knockout mice and conditional knockout mice).
Characterization of JH-ENT-01 reveals that it retains high affinity and specificity for hENT1. Notably, JH-ENT-01 exhibits improved PK properties compared to dilazep, a distinction that likely contributes to its in vivo efficacy. However, the fact that JH-ENT-01 is superior to dilazep in analgesic efficacy, even when delivered locally (intrathecally, Fig. 6a and intraplantarly, Fig. 5g–i), suggests that the compound’s affinity and specificity are also key factors. It is well documented that dilazep binds many non-specific targets54,55,56,57. Therefore, it is possible that the analgesic efficacy of JH-ENT-01 is in part due to its increased specificity for ENT1 against non-specific targets. Given that JH-ENT-01’s interaction with the opportunistic site 2 is suboptimal, there is great potential to refine the affinity and specificity of this compound. Such enhancements may further improve its in vivo efficacy. Consistent with this idea, a paper on dilazep derivatives was published during the revision of this manuscript, which postulated that several dilazep derivatives with high affinity are due to the occupancy of the same opportunistic site58. Additionally, several other clinical ENT1 inhibitors with different chemical scaffolds13,26,59,60, as well as more optimally targeting the opportunistic site 1, offer promising avenues for optimization through rational design. Lastly, our JH-ENT-01 sets an important benchmark for the PK properties of ENT1 inhibitors repurposed as analgesics.
To the best of our knowledge, ENT1 inhibitors have previously been shown to have mild antihyperalgesic effects in animal models of inflammatory pain34, but not those of neuropathic pain. Our studies highlight ENT1 as an important target for both inflammatory and neuropathic pain treatment. Given the unique mode of action for the adenosine reuptake inhibitor in treating neuropathic pain, coupled with the well-established biological basis for pain suppression through elevated adenosine levels, we postulate that a rational approach to designing inhibitors targeting ENTs holds promise for the development of non-opioid pain therapeutics. Such therapeutics could offer reduced addiction risks and greater efficacy against neuropathic pain.
The ideal path of therapeutic development against ENT1 would be orally active drugs. An oral formulation of dilazep is an approved treatment for cardiopathy61, so exploring oral administration of JH-ENT-01 is a worthy endeavor. As discussed, the major hurdle to develop full A1R agonists or adenosine reuptake inhibitors as pain therapeutics is potential cardiovascular side effects that may arise from systemic intravenous administrations. We have shown that local routes, such as intrathecal, subcutaneous or peri-sciatic nerve injection of JH-ENT-01 are highly effective in reducing both inflammatory and neuropathic pain in mice. Therefore, therapeutics targeting ENT1 via these local routes could also be developed–possibly within a shorter time frame.
Methods
Our research complies with all relevant ethical regulations: The animal studies were approved by the Institutional Animal Care and Use Committee of Duke University (protocol number: A240-23-12). Animal experiments were conducted in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and ARRIVE guidelines. Non-diseased human DRGs were obtained from four human donors through National Disease Research Interphase (NDRI) with permission of exemption from the Duke IRB (Protocol ID: Pro00051508).
Radiotracer uptake in transporter expressing Xenopus laevis oocytes
[3H]-adenosine was purchased from Moravek (Brea, CA). Defolliculated Xenopus laevis oocytes were obtained from Xenopus 1 Corp. (Dexter, MI). Uptake assays were performed in a manner similar to a previous report62, with the following modifications. A total of 16–20 ng cRNA were injected per oocyte, with expression carried out at 17 °C for 2–4 days. Uptake was performed on 5 oocytes per replicate incubated for 30 min at room temperature in ND-96 buffer (96 mM NaCl, 2 mM KCl, 1 mM MgCl2, 1.8 mM CaCl2, 5 mM HEPES, pH 7.4 with KOH) supplemented with 5% DMSO, inhibitor and 200 nM [3H]-adenosine. The reactions were terminated by washing the oocytes extensively in ND-96 buffer.
Synthesis of JH-ENT-01
Detailed synthesis and characterization of JH-ENT-01 is described in Supplementary Information. JH-ENT-01 used in this study may not fully satisfy all criteria for a high-quality chemical probe due to potential off-target activity and lack of negative control compounds and in-cell validation63.
Expression, purification, and crystallization of hENT1cryst in complex with JH-ENT-01
Protein expression, purification, and crystallization was performed similarly to what was previously reported24 with the following modifications: JH-ENT-01 was included in all purification buffers at 10 μM concentration. Purified JH-ENT-01 bound hENT1cryst was concentrated to 40–50 mg mL−1 and crystallized with lipidic cubic phase24,64. Crystal hits appeared in 35–50% PEG 400, 0.1 M glycine pH 9.0 and 0.5 M NaCl. The plate-like crystals were harvested and frozen from LCP 3–5 days after setting up trays.
Crystal structure determination
Datasets were collected on the NE-CAT remote access 24ID-C beamlines at the Advanced Photon Source at a wavelength of 0.979 Å. Data was indexed and integrated using XDS65. To obtain a complete dataset, BLEND analysis in CCP4 was performed to combine data from five different crystals66,67,68. Molecular replacement was performed using Phaser-MR in Phenix69, using the high-resolution dilazep-bound hENT1cryst structure as a search model (PDB ID: 6OB7). Owing to the high level of isomorphism with the search model, Free-R flags were transferred from 6OB7 to prevent validation set contamination. Iterative rounds of model building in COOT and refinements of the model against X-ray in phenix.refine were performed, resulting in Rwork/Rfree values of 0.22/0.26, and excellent protein geometries (98.9% Ramachandran favored with no outliers).
Scintillation proximity assays
[3H]-NBMPR was purchased from Moravek (Brea, CA). Binding assays were carried out similarly to what was previously described24. Unlabeled JH-ENT-01 or dilazep was added in rounds to SPA mixture containing 85 nM purified GFP-fusion transporter, 10 nM [3H]-NBMPR and 2.0 mg/mL Cu-PVT beads, all in 20 mM Tris-HCl pH 8.0, 150 mM NaCl, 1.0 mM DDM. Following drug addition, a brief equilibration at room temperature was performed, followed by scintillation counting. Apparent cold-displacement Ki’s were determined from fitting the data in GraphPad Prism to a non-linear regression equation to describe competitive binding (one site). Raw data and regression equations/fits are included in the source data.
Animals
CD1 mice (8–10 weeks of both sexes) were purchased from Charles River Laboratories. Mice with leptin receptor mutation (db/db mice, BKS.Cg-m+/+Leprdb/J mice (stock number: 000642) were purchased from Jackson Laboratories (Bar Harbor, ME) and maintained at Duke animal facility. These mutant mice developed diabetic neuropathy and hyperalgesia at the age of 3 months. Mice were group-housed on a 12-hour light/12-hour dark cycle at 22 ± 1 °C with free access to food and water. All mice were randomly assigned to experimental groups. Sample sizes were estimated based on our previous studies for similar types of behavioral, biochemical, and electrophysiological assays and analyzes. Two to five mice were housed per cage. The animal studies were approved by the Institutional Animal Care and Use Committee of Duke University (protocol number: A240-23-12). Animal experiments were conducted in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and ARRIVE guidelines.
Animal models of pain
The Type 1 Diabetes Mellitus model was induced by intraperitoneal administration with 150 mg/kg streptozotocin (STZ, Sigma-Aldrich). Mice with blood glucose levels of 300 mg/dl or greater were considered diabetic. Acute inflammatory pain was induced by intraplantar injection of diluted formalin (5%, 20 μl). Persistent inflammatory pain was induced by intraplantar injection of 20 μl of CFA (no dilution, Sigma)36. Peripheral nerve injury was performed to generate neuropathic pain in anesthetized mice. Anesthesia was induced with 4% isoflurane in 100% oxygen in an anesthetic chamber, then maintained with 2.5 % for surgery. For the CCI surgery, the sciatic nerve was exposed, and 3 ligatures (1 mm between each ligature) were placed around the nerve proximal to the trifurcation with 7–0 prolene suture70. The ligatures were loosely tied until a brief flick of the affected hind limb was observed. For the SNI surgery, the tibial and common peroneal branches of the sciatic nerve were tightly ligated with a 5.0 silk, followed by transection and removal of a 3–5 mm portion of the nerve. The third peripheral branch of the sciatic nerve, the sural nerve, was left intact, and any contact with or stretching of the sural nerve was carefully avoided41.
Drug injection
For intrathecal injections, lumbar puncture was made by a Hamilton micro syringe (Hamilton) fitted with a 30-G needle between the lumbar (L5 and L6) spinal levels to deliver reagents (10 µl) into cerebral spinal fluid. For intravenous administration, a volume of 50 to 100 µl of drug or vehicle was injected into the tail vein with a 30-G needle. Subcutaneous injection was given beneath the back skin at a 45-degree angle to the pinched-up skin. For hindpaw injections, drugs were injected using a Hamilton micro-syringe with a 30-gauge needle under 2% isoflurane anesthesia with continuous monitoring. For peri-sciatic nerve injection, mice were lightly anesthetized by inhalation of 1 to 2% isoflurane. The JH-ENT-01 solution (10 nmol in 10 μl) was injected into the sciatic notch of the hindlimb between the greater trochanter and the ischial tuberosity with 27-guage needle connected to a tuberculin syringe. When the needle encountered the body of the ischium, a withdrawal response was observed and then the drug solution was injected over several seconds.
Behavioral tests for sensory and motor function
We habituated mice in the testing environment for 2 d and performed behavioral testing in a blinded manner. We assessed formalin-evoked spontaneous inflammatory pain by measuring the time (in seconds) the mice spent on licking and flinching the affected paws every 5 min for 45 min after intraplantar injection of 5% formalin. The interventional drug used in this experiment was pre-injected 30 minutes before the formalin injection. For assessing mechanical pain, we used a series of von Frey fibers with logarithmically increasing stiffness (0.02–2.56 g, Stoelting), which were applied to the plantar surface of the hind paw. The paw withdrawal threshold (PWT) was calculated using the up-down method. To further determine mechanical allodynia, paw withdrawal frequency (PWF) was measured by applying a sub-threshold von Frey filament (0.16 g for CCI mice and 0.4 g for STZ mice) to the plantar surface of the hind paw for 10 times. Cold pain was assessed in acetone test: we gently applied 20 μl of acetone to the hindpaw using a pipette and the response duration to acetone was scored. Motor function was evaluated by the rotarod test using a rotarod system (IITC Life Science). Mice underwent a 2-day training period (2 trials per mouse) before the test. During rotarod testing, the speed of rotation was accelerated from 5 to 15 rpm over 60 s with a testing period cutoff of 300 s for each trial and 2 total trials performed. The fall latency of each mouse was recorded and averaged. For these behavioral tests, the experimenter was blinded to the treatments given to mice.
Electrocardiogram recording
Mice were anesthetized with urethane (20%, 1.2 g/kg). Subdermal needle electrodes were inserted under the skin of the upper limbs and the left lower limb. The signal was amplified by AM 1800 2 Channel amplifier (A-M systems, Carlsborg, WA, United States) and filtered at 5 kHz. Data was collected and analyzed offline using pCLAMP10 software (Molecular Devices, San Jose, CA, United States). After the recordings, the mice were immediately euthanized by decapitation.
Patch-clamp recording from spinal dorsal horn neurons
As we previously reported71, STZ-treated or naive mice were anesthetized with intraperitoneal injection of urethane (1.5–2.0 g/kg). After a dorsal laminectomy, the lumbosacral segment of spinal cord was removed. The spinal cord was placed into the preoxygenated, ice-cold cutting solution (in mM: sucrose 240, NaHCO3 25, KCl 2.5, NaH2PO4 1.25, CaCl2 0.5, MgCl2 3.5). The mice were then immediately euthanized by decapitation. A transverse spinal cord slice (300–400 µm thick) was cut on a vibrating microslicer (VT1200s, Leica, Wetzlar, Germany). Spinal cord slices were incubated for 30 min in artificial cerebrospinal fluid (ACSF) which contained (in mM): NaCl 126, KCl 3, CaCl2 2.5, MgCl2 1.3, NaH2PO4 1.25, NaHCO3 26, and D-glucose 11) equilibrated with 95% O2 and 5% CO2 gas mixture. The spinal cord slice was placed in a recording chamber and continuously perfused with ASCF equilibrated with 95% O2 and 5% CO2 gas mixture at a flow rate of 2–4 ml/min at room temperature. Whole-cell patch-clamp recordings were made from outer lamina II (IIo) neurons in voltage clamp mode with patch pipette having a resistance of 5–8 MΩ. Lamina II was identified as a translucent band under a microscope (BX51WIF; Olympus) with light transmitted from below. The holding potential was set to −70 mV when recording excitatory postsynaptic currents (EPSCs). The patch pipette solution for EPSC recording contained (in mM): Potassium gluconate 135, CaCl2 0.5, MgCl2 2, KCl 5, EGTA 5, HEPES 5, ATP-Mg salt 5. Drugs were dissolved in ACSF and applied by superfusion without alteration of the perfusion rate and temperature. Signals were amplified using an Axopatch 700B amplifier (Molecular Devices, San Jose, CA) and were filtered at 2 kHz and digitized at 5 kHz. Data were collected and analyzed using pClamp 10.3 software (Molecular Devices, San Jose, CA). EPSCs were analyzed using Minianalysis ver. 6.0.3 (Synaptosoft, Decatur, GA).
Human DRGs
Non-diseased human DRGs were obtained from four human donors through National Disease Research Interphase (NDRI) with permission of exemption from the Duke IRB (Protocol ID: Pro00051508), and the histochemical preparations were as previously reported72,73. Postmortem lumbar DRGs were delivered in ice-cold culture medium within 48–72 h of death. Four human donors were used, including one female 47-year-old Caucasian (f47c), one male 32-year-old Caucasian (m32c), one female 22-year-old Caucasian (f22c), and one female 64-year-old Caucasian (f64c).
ISH and double staining with IHC
Mice were deeply anaesthetized with isoflurane and transcardially perfused with PBS followed by 4% paraformaldehyde. Lumbar spinal cord segments (L4-L5) were isolated, post-fixed, and incubated in a sucrose gradient (20 30%). Tissues were then embedded in OCT medium (Tissue-Tek) and cryosectioned. DRG tissues were sectioned at 14 μm-thick, spinal cords were sectioned at 30 μm. Human DRG samples were bisected, drop-fixed in 4% paraformaldehyde overnight, then similarly cryopreserved and sectioned at 14 μm. ISH was performed using the RNAscope system (Advanced Cell Diagnostics) following the manufacturer’s instructions73. Probes used were murine Slc29a1 (562321-C2) for mouse DRG and spinal cord, and human SLC29A1 (266381-C3) and human ADORA1 (580821-C3) for human DRG, followed by NeuN immunostaining. The primary antibody used was: (1:500, guinea pig, Sigma-Aldrich, cat. # ABN90). The secondary antibody used wasCF488a donkey-anti-guinea pig (1:500, Biotium cat. # 20169). For each experiment, all images were acquired with the same settings, a minimum of 4 sections from each animal were selected, and four human DRG from separate donors were included for data analysis.
Immunohistochemistry (IHC)
Mice were deeply anaesthetized with isoflurane and transcardially perfused with PBS followed by 4% paraformaldehyde. Lumbar spinal cord segments (L4-L5) were isolated and post-fixed overnight and incubated in a sucrose gradient (20–30%). Tissues were then embedded in OCT medium (Tissue-Tek) and cryosectioned into 30 μm-thick spinal cord free-floating sections. The sections were blocked with 5% normal donkey serum in 0.3% PBS-Triton for 1 h at room temperature and then incubated overnight at 4 °C with anti-ENT1 (mouse, 1:500, Santa Cruz Biotechnologies sc-377283) and anti-A1AR (rabbit, 1:500, alomone labs AAR-006) primary antibodies. The sections were then washed 3× in 0.1% PBS-tween20 and incubated with the following secondary antibodies for 2 h at room temperature: CF555 donkey-anti-mouse (1:500, Biotium cat. # 20037) and CF488a donkey-anti-rabbit (1:500, Biotium cat. # 20015). The sections were then washed with 3× in 0.1% PBS-tween20 and mounted in Fluoromount-G mounting medium with DAPI (Southern Biotech, cat. # 0100-20) and observed under a confocal laser scanning microscope (Zeiss 880 inverted confocal, via the Duke Light Microscopy Core).
Animal PK experiment
The PK studies were performed with 6–7-week-old CD-1 male mice (Charles River). All procedures were performed as painlessly as possible, according to IACUC-approved animal protocol (Duke Univ. # A107-23-04). The 30 mg/kg dose of JH-ENT-01 (free base) and 29 mg/kg dilazep (as dilazep dihydrochloride, equimolar to 30 mg/kg JH-ENT-01) were administered subcutaneously as 8 μL per gram of body weight. The injectable formulation, used to solubilize the investigated compounds, consisted of: 5% DMSO, 20% PEG-400, 20% propylene glycol, and 55% saline. The animals were euthanized at 10 min, 30 min, 1.5, 4.5, 8, 12, and 24 h post injection, plasma and tissues collected and kept at −80 °C until the analysis. The animals were induced with 5% isoflurane and maintained anesthetized with 2% isoflurane in oxygen at the flow of 1 L/min (nose mask). After opening the chest, 0.5–1 mL blood was taken from left heart ventricle by 23 G needle/syringe into a vial containing 10 μL of 50 mg/mL K2EDTA in water. Plasma was separated by centrifugation at 2000 × g for 5 min at room temperature. Through the same heart puncture, a 20 G blunted cannula was led into the ascending aorta and secured by aneurysm clip. Incision of the right atrium was made with micro scissors and ~10 mL saline was infused (or until liver visibly cleared out of blood). Additionally, tissues (brain, spinal cord, TG, DRG) was collected and, along with plasma, stored at −80 °C until the analysis.
Liquid chromatography—tandem-mass spectrometry (LC/MS/MS) assays for measurement of JH-ENT-01 and dilazep
The JH-ENT-01 and dilazep concentration in plasma and tissue was measured by LC/MS/MS assays adapted to required sensitivity/concentration range and the nature/available quantity of the sample. JH-ENT-03 (a JH-ENT-01 analog with an additional ortho-methyl group on the para-nitrobenzyl moiety of JH-ENT-01, in-house made) and hexobendine dihydrochloride (ChemCruz) were used as internal standards, respectively. Agilent 1100/1200 LC, AB/Sciex API5500 QTrap MS/MS instrument, and Analyst® Version 1.6.2 (AB Sciex, Ontario, Canada) were used for all measurements. A typical plasma assay was performed as follows. A 10 μL plasma aliquot of sample was mixed with 20 μL of working internal standard solution (10 ng/mL in MeOH). The sample was vigorously agitated in Fast Prep 120 (Thermo-Savant) at Speed 4.0 for 20 Sec, 2 cycles. After centrifugation at 13 × g for 5 min, 20 μL of supernatant was diluted with 20 μL of mobile phase A (see below), mixed, and an aliquot was injected onto the LC-MS/MS system. The liquid chromatography (LC) conditions were as follows. Column: Eclipse Plus C18, 4.6 × 50 mm, 1.8 μm, at 1 mL/min. Mobile phase A: formic acid in water (0.1%, v/v, pH adjusted to 7 with ammonium hydroxide). Mobile phase B: 100% acetonitrile. Elution gradient (linear): 0–1 min 10–90% B, 1–1.5 min 90% B, 1.5–1.7 min 90–10% B. The analyte and internal standard were measured in positive ion mode. The following MRM transitions of the respective [M + H]+ ions were used. JH-ENT-01: m/z 726.2/195.1, JH-ENT-03 (int.std.): m/z 740.3/195.1, dilazep: m/z 605.2/195.1, and hexobendine: m/z 593.2/195.1. Calibration standards were prepared in commercially available pooled drug-free plasma. In case of tissue samples, the tissues were weighed, homogenized with 2 parts (v/v) water, further diluted 1/5–1/19 by water, then 100 μL of the homogenate and 300 μL of chloroform vigorously agitated by FastPrep (see above). After centrifugation for 5 min at 13 × g, the organic layer was evaporated to dryness by gentle stream of nitrogen and reconstituted with 50 μL of equal volumes of mobile phases A and B; 10 μL was injected into LC/MS/MS system.
PK modeling and calculations
To obtain relevant PK parameters from concentration vs time data, non-compartmental approach within WinNonlin software package (v. 2.1) was utilized.
Statistics
All data were expressed as mean ± SEM. The sample size for each experiment was indicated in figure legends. GraphPad Prism 8.0 software was used to perform statistical analysis. The data were analyzed by two-way ANOVA, followed by Bonferroni’s post-hoc test for multi-group comparison, or by Student t test for two-group comparison. P < 0.05 was considered statistically significant. Statistical significance was indicated as * P < 0.05, ** P < 0.01, *** P < 0.001, **** P < 0.0001.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Atomic coordinates and structure factors for the X-ray structure of JH-ENT-01 bound hENT1 have been deposited in the Protein Data Bank with the PDB ID 8TZI. The source data underlying Figs. 1c, e, g, 2d, 3b, d, 4a–d, 5b, c, e, f, h, i, 6a–c, e–i, k, l, 7a, b, 8c–f, h, and Supplementary Fig. 5a–g, 6b and 7. are provided as a Source Data file. Source data are provided with this paper.
References
Sawynok, J. Adenosine and ATP receptors. Handb. Exp. Pharmacol. 309–328 (2007).
Sawynok, J. Adenosine receptor targets for pain. Neuroscience 338, 1–18 (2016).
Sawynok, J. & Sweeney, M. I. The role of purines in nociception. Neuroscience 32, 557–569 (1989).
Vincenzi, F., Pasquini, S., Borea, P. A. & Varani, K. Targeting adenosine receptors: a potential pharmacological avenue for acute and chronic pain. Int. J. Mol. Sci. 21, 8710 (2020).
Zylka, M. J. Pain-relieving prospects for adenosine receptors and ectonucleotidases. Trends Mol. Med. 17, 188–196 (2011).
Vincenzi, F. et al. A(1) adenosine receptor partial agonists and allosteric modulators: advancing toward the clinic? Front. Pharm. 11, 625134 (2020).
Kiesman, W. F., Elzein, E. & Zablocki, J. A1 adenosine receptor antagonists, agonists, and allosteric enhancers. Handb. Exp. Pharmacol. 25–58 (2009).
Draper-Joyce, C. J. et al. Positive allosteric mechanisms of adenosine A(1) receptor-mediated analgesia. Nature 597, 571–576 (2021).
Schaddelee, M. P. et al. Pharmacokinetic/pharmacodynamic modelling of the anti-hyperalgesic and anti-nociceptive effect of adenosine A1 receptor partial agonists in neuropathic pain. Eur. J. Pharm. 514, 131–140 (2005).
Li, X., Bantel, C., Conklin, D., Childers, S. R. & Eisenach, J. C. Repeated dosing with oral allosteric modulator of adenosine A1 receptor produces tolerance in rats with neuropathic pain. Anesthesiology 100, 956–961 (2004).
Dunwiddie, T. V. & Masino, S. A. The role and regulation of adenosine in the central nervous system. Annu. Rev. Neurosci. 24, 31–55 (2001).
King, A. E., Ackley, M. A., Cass, C. E., Young, J. D. & Baldwin, S. A. Nucleoside transporters: from scavengers to novel therapeutic targets. Trends Pharm. Sci. 27, 416–425 (2006).
Wright, N. J. & Lee, S. Y. Toward a molecular basis of cellular nucleoside transport in humans. Chem. Rev. 121, 5336–5358 (2021).
Baldwin, S. A. et al. The equilibrative nucleoside transporter family, SLC29. Pflug. Arch. 447, 735–743 (2004).
Young, J. D., Yao, S. Y., Baldwin, J. M., Cass, C. E. & Baldwin, S. A. The human concentrative and equilibrative nucleoside transporter families, SLC28 and SLC29. Mol. Asp. Med. 34, 529–547 (2013).
Young, J. D., Yao, S. Y., Sun, L., Cass, C. E. & Baldwin, S. A. Human equilibrative nucleoside transporter (ENT) family of nucleoside and nucleobase transporter proteins. Xenobiotica 38, 995–1021 (2008).
Dunwiddie, T. V. & Diao, L. Extracellular adenosine concentrations in hippocampal brain slices and the tonic inhibitory modulation of evoked excitatory responses. J. Pharm. Exp. Ther. 268, 537–545 (1994).
Pedata, F., Pazzagli, M., Tilli, S. & Pepeu, G. Regional differences in the electrically stimulated release of endogenous and radioactive adenosine and purine derivatives from rat brain slices. Naunyn Schmiedebergs Arch. Pharm. 342, 447–453 (1990).
Aggarwal, S. & Mortensen, O. V. Discovery and Development of Monoamine Transporter Ligands. Adv. Neurobiol. 30, 101–129 (2023).
Hamon, M. & Blier, P. Monoamine neurocircuitry in depression and strategies for new treatments. Prog. Neuropsychopharmacol. Biol. Psychiatry 45, 54–63 (2013).
Zimmerman, M. A. et al. Equilibrative nucleoside transporter (ENT)-1-dependent elevation of extracellular adenosine protects the liver during ischemia and reperfusion. Hepatology 58, 1766–1778 (2013).
Eckle, T. et al. Crosstalk between the equilibrative nucleoside transporter ENT2 and alveolar Adora2b adenosine receptors dampens acute lung injury. FASEB J. 27, 3078–3089 (2013).
Chambers, E. D. et al. Blockade of equilibrative nucleoside transporter 1/2 protects against Pseudomonas aeruginosa-induced acute lung injury and NLRP3 inflammasome activation. FASEB J. 34, 1516–1531 (2020).
Wright, N. J. & Lee, S. Y. Structures of human ENT1 in complex with adenosine reuptake inhibitors. Nat. Struct. Mol. Biol. 26, 599–606 (2019).
Drew, D., North, R. A., Nagarathinam, K. & Tanabe, M. Structures and general transport mechanisms by the major facilitator superfamily (MFS). Chem. Rev. 121, 5289–5335 (2021).
Vlachodimou, A., Konstantinopoulou, K., AP, I. J. & Heitman, L. H. Affinity, binding kinetics and functional characterization of draflazine analogues for human equilibrative nucleoside transporter 1 (SLC29A1). Biochem. Pharm. 172, 113747 (2020).
SenGupta, D. J. & Unadkat, J. D. Glycine 154 of the equilibrative nucleoside transporter, hENT1, is important for nucleoside transport and for conferring sensitivity to the inhibitors nitrobenzylthioinosine, dipyridamole, and dilazep. Biochem. Pharm. 67, 453–458 (2004).
Elion, G. B. Symposium on immunosuppressive drugs. Biochemistry and pharmacology of purine analogues. Fed. Proc. 26, 898–904 (1967).
Benedict, W. F., Baker, M. S., Haroun, L., Choi, E. & Ames, B. N. Mutagenicity of cancer chemotherapeutic agents in the Salmonella/microsome test. Cancer Res. 37, 2209–2213 (1977).
Cass, C. E., Muzik, H. & Paterson, A. R. Combination therapy of mouse leukemia L1210 by 1-beta-D-arabinofuranosylcytosine and 6-[(4-nitrobenzyl)thio]-9-beta-D-ribofuranosylpurine. Cancer Res. 35, 1187–1193 (1975).
Wang, C. et al. Dipyridamole analogs as pharmacological inhibitors of equilibrative nucleoside transporters. Identification of novel potent and selective inhibitors of the adenosine transporter function of human equilibrative nucleoside transporter 4 (hENT4). Biochem. Pharm. 86, 1531–1540 (2013).
Woolf, C. J. & Salter, M. W. Neuronal plasticity: increasing the gain in pain. Science 288, 1765–1769 (2000).
Kawasaki, Y. et al. Distinct roles of matrix metalloproteases in the early- and late-phase development of neuropathic pain. Nat. Med. 14, 331–336 (2008).
Maes, S. S., Pype, S., Hoffmann, V. L., Biermans, M. & Meert, T. F. Antihyperalgesic activity of nucleoside transport inhibitors in models of inflammatory pain in guinea pigs. J. Pain. Res. 5, 391–400 (2012).
Costigan, M., Scholz, J. & Woolf, C. J. Neuropathic pain: a maladaptive response of the nervous system to damage. Annu. Rev. Neurosci. 32, 1–32 (2009).
Chen, O. et al. MicroRNA let-7b enhances spinal cord nociceptive synaptic transmission and induces acute and persistent pain through neuronal and microglial signaling. Pain 165, 1824–1839 (2024).
Vincent, A. M., Callaghan, B. C., Smith, A. L. & Feldman, E. L. Diabetic neuropathy: cellular mechanisms as therapeutic targets. Nat. Rev. Neurol. 7, 573–583 (2011).
Boulton, A. J. et al. Diabetic neuropathies: a statement by the American Diabetes Association. Diab. Care 28, 956–962 (2005).
Sullivan, K. A. et al. Mouse models of diabetic neuropathy. Neurobiol. Dis. 28, 276–285 (2007).
O’Brien, P. D., Sakowski, S. A. & Feldman, E. L. Mouse models of diabetic neuropathy. ILAR J. 54, 259–272 (2014).
Decosterd, I. & Woolf, C. J. Spared nerve injury: an animal model of persistent peripheral neuropathic pain. Pain 87, 149–158 (2000).
Bennett, G. J. & Xie, Y. K. A peripheral mononeuropathy in rat that produces disorders of pain sensation like those seen in man. Pain 33, 87–107 (1988).
Finnerup, N. B. et al. Pharmacotherapy for neuropathic pain in adults: a systematic review and meta-analysis. Lancet Neurol. 14, 162–173 (2015).
Salter, M. W. & Kalia, L. V. Src kinases: a hub for NMDA receptor regulation. Nat. Rev. Neurosci. 5, 317–328 (2004).
Xu, Z. Z. et al. Neuroprotectin/protectin D1 protects against neuropathic pain in mice after nerve trauma. Ann. Neurol. 74, 490–495 (2013).
Ackley, M. A. et al. Control of glutamatergic neurotransmission in the rat spinal dorsal horn by the nucleoside transporter ENT1. J. Physiol. 548, 507–517 (2003).
Zylka, M. J. et al. Prostatic acid phosphatase is an ectonucleotidase and suppresses pain by generating adenosine. Neuron 60, 111–122 (2008).
Goldman, N. et al. Adenosine A1 receptors mediate local anti-nociceptive effects of acupuncture. Nat. Neurosci. 13, 883–888 (2010).
El-Naggar, A. E., Helmy, M. M., El-Gowilly, S. M. & El-Mas, M. M. Adenosine A1 receptors of the medullary solitary tract arbitrate the nicotine counteraction of neuroinflammation and cardiovascular dysfunction in septic rats. Sci. Rep. 13, 17818 (2023).
Latini, S., Bordoni, F., Corradetti, R., Pepeu, G. & Pedata, F. Effect of A2A adenosine receptor stimulation and antagonism on synaptic depression induced by in vitro ischaemia in rat hippocampal slices. Br. J. Pharm. 128, 1035–1044 (1999).
Kotańska, M. et al. PSB 603 – a known selective adenosine A2B receptor antagonist – has anti-inflammatory activity in mice. Biomed. Pharmacother. 135, 111164 (2021).
Schneider, G. et al. Extracellular nucleotides as novel, underappreciated pro-metastatic factors that stimulate purinergic signaling in human lung cancer cells. Mol. Cancer 14, 201 (2015).
Baron, R., Binder, A. & Wasner, G. Neuropathic pain: diagnosis, pathophysiological mechanisms, and treatment. Lancet Neurol. 9, 807–819 (2010).
Kaochar, S. et al. Inhibition of GATA2 in prostate cancer by a clinically available small molecule. Endocr. Relat. Cancer 29, 15–31 (2022).
Sakumura, T. et al. Dilazep, a nucleoside transporter inhibitor, modulates cell cycle progression and DNA synthesis in rat mesangial cells in vitro. Cell Prolif. 33, 19–28 (2000).
Deguchi, H. et al. Dilazep, an antiplatelet agent, inhibits tissue factor expression in endothelial cells and monocytes. Blood 90, 2345–2356 (1997).
Takenaga, M., Kitagawa, H., Hirai, A., Tamura, Y. & Yoshida, S. Effect of dilazep on phospholipid metabolism and arachidonic acid cascade in human platelets in vitro. J. Jpn. Atheroscler. Soc. 12, 363–370 (1984).
Dilweg, M. A. et al. Exploring novel dilazep derivatives as hENT1 inhibitors and potentially covalent molecular tools. Purinergic Signal. https://doi.org/10.1007/s11302-024-10026-x (2024).
Armstrong, D. et al. Characterization of the adenosine pharmacology of ticagrelor reveals therapeutically relevant inhibition of equilibrative nucleoside transporter 1. J. Cardiovasc. Pharm. Ther. 19, 209–219 (2014).
Hermann, R. et al. Cladribine as a potential object of nucleoside transporter-based drug interactions. Clin. Pharmacokinet. 61, 167–187 (2022).
Sambhi, M. P., Kannan, R., Thananopavarn, C., Ookhtens, M. & Gudenzi, M. Therapeutic tolerance, hemodynamic effects, and oral dose kinetics of dilazep dihydrochloride in hypertensive patients. J. Pharm. Sci. 78, 281–284 (1989).
Wright, N. J. et al. Methotrexate recognition by the human reduced folate carrier SLC19A1. Nature 609, 1056–1062 (2022).
Arrowsmith, C. H. et al. The promise and peril of chemical probes. Nat. Chem. Biol. 11, 536–541 (2015).
Caffrey, M. A comprehensive review of the lipid cubic phase or in meso method for crystallizing membrane and soluble proteins and complexes. Acta Crystallogr. F. Struct. Biol. Commun. 71, 3–18 (2015).
Kabsch, W. XDS. Acta Crystallogr. D. Biol. Crystallogr. 66, 125–132 (2010).
Foadi, J. et al. Clustering procedures for the optimal selection of data sets from multiple crystals in macromolecular crystallography. Acta Crystallogr. D. Biol. Crystallogr. 69, 1617–1632 (2013).
Evans, P. R. An introduction to data reduction: space-group determination, scaling and intensity statistics. Acta Crystallogr. D. Biol. Crystallogr. 67, 282–292 (2011).
Evans, P. R. & Murshudov, G. N. How good are my data and what is the resolution? Acta Crystallogr. D. Biol. Crystallogr. 69, 1204–1214 (2013).
Liebschner, D. et al. Macromolecular structure determination using X-rays, neutrons and electrons: recent developments in Phenix. Acta Crystallogr. D. Struct. Biol. 75, 861–877 (2019).
Chen, G., Park, C. K., Xie, R. G. & Ji, R. R. Intrathecal bone marrow stromal cells inhibit neuropathic pain via TGF-beta secretion. J. Clin. Invest. 125, 3226–3240 (2015).
Furutani, K. et al. Novel proresolving lipid mediator mimetic 3-oxa-PD1n-3 docosapentaenoic acid reduces acute and chronic itch by modulating excitatory and inhibitory synaptic transmission and astroglial secretion of lipocalin-2 in mice. Pain 164, 1340–1354 (2023).
Xu, Z. Z. et al. Inhibition of mechanical allodynia in neuropathic pain by TLR5-mediated A-fiber blockade. Nat. Med. 21, 1326–1331 (2015).
Wang, Z. et al. Central opioid receptors mediate morphine-induced itch and chronic itch via disinhibition. Brain 144, 665–681 (2020).
Acknowledgements
This work used the Northeastern Collaborative Access Team beamline 24ID-C (P30 GM124165 and S10 RR029205) at the APS (DE-AC02-06CH11357). This work was supported by the NIH grants R61NS138215 (S.-Y.L., J.H., R-R.J.), R01GM137421 (S.-Y. L. and J.H.) and R01NS13182 (R.-R.J.) and the DoD grants W81XWH2110756 and W81XWH2210646 (R.-R.J.). Pharmacokinetic studies are in part supported by NCI Comprehensive Cancer Center Core Grant P30CA014236 (I.S.).
Author information
Authors and Affiliations
Contributions
N.J.W. solved the structure and performed in vitro binding under the guidance of S.-Y.L., J.G.F., and N.J.W. performed in vitro transport experiments under the guidance of S.-Y.L. Y.M., W.H., K.F., A.M., O.C., and S.B. performed animal and histochemical studies and ex vivo patch clamp recordings under the guidance of R.-R.J., H.P., C.G.W., and Y.Z. synthesized the compound under the guidance of J.H. P.F. and I.S. performed PK studies. S.Y.L., J.H., R.-R.J., and N.J.W. conceptualized this study. S.-Y.L. and R.-R.J. wrote the draft with input from all authors.
Corresponding authors
Ethics declarations
Competing interests
N.J.W., J.H., R.-R.J., and S.-Y.L. are inventors of the patent application that was filed (028193-0022-WO01). The rest of the authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Rajgopal Govindarajan, Bert Van den Berg and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wright, N.J., Matsuoka, Y., Park, H. et al. Design of an equilibrative nucleoside transporter subtype 1 inhibitor for pain relief. Nat Commun 15, 10738 (2024). https://doi.org/10.1038/s41467-024-54914-7
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-024-54914-7
