
Abstract
G protein-coupled receptor 4 (GPR4) belongs to the subfamily of proton-sensing GPCRs (psGPCRs), which detect pH changes in extracellular environment and regulate diverse physiological responses. GPR4 was found to be overactivated in acidic tumor microenvironment as well as inflammation sites, with a triad of acidic residues within the transmembrane domain identified as crucial for proton sensing. However, the 3D structure remains unknown, and the roles of other conserved residues within psGPCRs are not well understood. Here we report cryo-electron microscopy (cryo-EM) structures of active zebrafish GPR4 at both pH 6.5 and 8.5, each highlighting a distribution of histidine and acidic residues at the extracellular region. Cell-based assays show that these ionizable residues moderately influence the proton-sensing capacity of zebrafish GPR4, compared to the more significant effects of the triad residues. Furthermore, we reveal a cluster of aromatic residues within the orthosteric pocket that may propagate the signaling to the intercellular region via repacking the aromatic patch at the central region. This study provides a framework for future signaling and functional investigation of psGPCRs.
Similar content being viewed by others


Introduction
G protein-coupled receptors (GPCRs) represent a diverse group of seven transmembrane proteins constituting the largest superfamily within the human genome and many other genomes. GPCRs play a pivotal role in sensing a wide array of extracellular stimuli, including light, neurotransmitters, hormones, and proteins. Upon stimulation, GPCRs undergo conformational changes, initiating downstream signaling pathways that culminate in specific cellular response1,2,3. Remarkably, ~35% of approved drugs in the pharmaceutical market target GPCRs4. Of the 826 GPCRs identified in humans, only three receptors, namely GPR4, GPR65, and GPR68, are characterized as proton-sensing GPCRs (psGPCRs) since they are activated in response to the extracellular acidic environment. Beyond their responsiveness to protons, psGPCRs have previously been implicated in sensing certain lysolipids5 and membrane stretching force6, albeit with mechanisms that remain to be debated and explored.
psGPCRs have been implicated in several well-documented pH-dependent physiological activities7. Local acidity is a typical feature of many pathological processes, including inflammatory, and neurological diseases, and solid tumor8,9. Elevated fermentative metabolism and poor perfusion are major causes of local acidity, which has been hypothesized to promote disease progression10. Notably, studies have suggested that psGPCRs are highly expressed in tumor cells and the tumor microenvironment (TME)11,12,13,14. This observation is particularly significant given that the pH in the TME typically registers 0.5–1 unit lower than in normal tissues, primarily attributed to the Warburg effect. This metabolic phenomenon, characterized by increased glucose uptake and lactic acid production in rapidly proliferating tumor cells, contributes to the acidification of the extracellular milieu. In addition, it is noteworthy that GPR4 is widely expressed in distinct tissues, including immune, brain, respiratory, and digestive systems, whereas expression of GPR65 and GPR68 seems to be tissue-restricted.
There are several studies that explored the mechanism of psGPCR activation. Early studies implicated a cluster of extracellular histidine residues as primary pH sensors15,16,17,18, whereas more recent data found that proton-sensing is primarily accomplished by a triad of acidic residues within the transmembrane domain19. Interestingly, the phylogenetic analysis suggested that these psGPCRs evolved from GPR132, which lacks one buried acidic residue and is not considered as proton-sensing receptor; on the contrary, no phylogenetic feature can be found for the distributed histdines19. Nevertheless, the entire pathway still shows limitations due to the lack of experimental structures. In this study, we report the cryo-electron microscopy (cryo-EM) structures of active GPR4 at both pH 6.5 and 8.5, each coupled to an engineered Gs protein. These structures, combined with cell-based assays, offer valuable insights into the proton-sensing mechanisms and signal transduction pathways within the psGPCR family.
Results
Structure determination
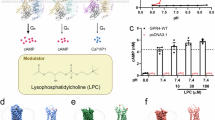
For cryo-EM studies we employed the NanoBiT tethering strategy to facilitate the formation of stable GPR4–Gs complexes (Supplementary Fig. 1)20. To facilitate the binding of a single chain variable fragment (scfv16) (see “Methods”), we modified Gαs based on mini-Gαs, replacing its αN with the equivalent region from Gαi21. We screened GPR4 species from human, rabbit, and zebrafish using these strategies and found that zebrafish GPR4 (named zGPR4 hereafter) yielded a homogeneous complex with the engineered Gs protein. zGPR4 share >60% sequence identity with human GPR4 (hGPR4), with even higher in the Gs protein-interacting region. Specifically, there is only one residue difference in the intercellular half of the TM5-TM6 region between hGPR4 (S211ICL3) and zGPR4 (N219ICL3). Hence, the zGPR4–Gs complex offers insights into how hGPR4 may recognize its downstream G protein and initiate signaling pathways. We prepared the zGPR4–Gs complex samples under two conditions, one at pH 6.5 and the other at pH 8.5. Although the basic condition is expected to generate an inactive receptor, we successfully purified a stable zGPR4–Gs complex potentially due to the NanoBiT method designed to enhance GPCR–G protein stability.
Density maps were reconstructed to resolutions of 3.3 Å and 3.1 Å for pH 6.5 and pH 8.5, respectively (Fig. 1a, b and Supplementary Table 1). Although overall map quality for the pH 8.5 structure is slightly better than the map of the pH 6.5 structure in the transmembrane region, it is relatively poorer in the extracellular loops (ECLs), likely due to a relatively higher entropic nature of these loops at neutral pH (Fig. 1c, d and Supplementary Fig. 2). The two structures are almost identical in the receptor’s transmembrane region and the G protein (Fig. 1e, f). There is slight orientational difference between G protein subunits Gα and Gβγ potentially attributed to the two buffers used perturbing the interactions between the G protein subunits (Fig. 1e), this should not be physiologically relevant as the G protein subunits may not experience similar pH change intracellularly. Also, the receptor–Gαs interface is quite identical under pHs of 6.5 and 8.5, suggesting the intracellular G protein-coupling is anyhow insensitive to pH change. As most of the ECL residues could be modeled in the pH 6.5 structure (named zGPR46.5 hereafter), we will mainly discuss this model unless otherwise specified.

a–d Cryo-EM maps showing the disk-shaped micelle (left) and cartoon representation (right) of the zGPR4–Gs complexes under pH 6.5 (a, c) and pH 8.5 (b, d). e, f Superposition of the two structures at pH 6.5 and 8.5 on side view (e) or extracellular view (f). The disulfide bond is shown in stick.
Receptor–G protein interface
At the receptor–Gαs interface, there are several polar residues on the receptor side interaction with the α5 helix of Gαs (Fig. 2a, b). For example, N219ICL3 and E2266.29 (Ballesteros–Weinstein numbering in superscript22) of zGPR4 form a hydrogen bond or salt bridge with D364H5.13 and R368H5.17 (superscript refers to common G protein numbering23) of α5, respectively. Notably, the highly conserved R1233.50 of zGPR4 engages in both a salt bridge with E375H5.24 and a π-cation interaction with the landmark residue of α5, Y374H5.23. The side chain of Y374H5.23 shifts toward ICL1, establishing hydrogen bond connections with Q531.60, N582.37, and E592.38. Additionally, there is a conserved hydrophobic interaction network between L371H5.20 and L376H5.25 of α5 helix and zGPR4 residues V1273.54, I2125.61, V2165.65, I2306.33, and L2336.36. Mutagenesis data confirmed the importance of these residues. The previously characterized essential residue for G protein-coupling, R1233.50, if mutated, nearly abolished the capability of zGPR4 to couple to downstream Gs protein (Fig. 2c). Other tested mutants (Q531.60A, N582.37A, E592.38A, R137ICL2A, N219ICL3A, E2266.29A) can shift the pH50 downward to different extents (0.2–0.5 log units) (Fig. 2c), suggesting these mutations compromise the receptor’s sensitivity to proton concentration by 1.6–3.2 folds. hGPR4 mutations yielded consistent results with those observed for zGPR4 (Fig. 2d), indicating that the chimera complex of zGPR4–Gs indeed mirrors how hGPR4 interacts with its downstream G protein.

a, b The receptor–α5 interface in zGPR46.5. Interacting residues are shown as sticks and are labeled. c, d Mutagenesis analysis of residues in zGPR4 (c) and hGPR4 (d) on the potency of G protein-coupling by the GloSensor cAMP assay. The maximum and minimum activation levels of WT GPR4 were set to 100 and 0%, respectively. The ΔpH50 was calculated by dividing the potency (pH50) of the mutant by the pH50 of WT. Data are shown as means ± SEM from at least three independent experiments performed in triplicate. Source data are provided as a Source Data file.
Our zGPR4–Gs structures reveal a non-canonical Gs-coupling model for zGPR4, with the intercellular tip of TM5-TM6 gently moving outward to accommodate the Gαs protein ( ~ 4 Å compared to an inactive zGPR4 model from AlphaFold) (Fig. 3a, b). This differs from canonical GPCR–Gs complexes, where TM6 undergoes more pronounced movements (12–16 Å) (Fig. 3c). However, the C-terminal tip of the α5 helix in the zGPR4–Gs complex assumes a similar hook structure to that observed in canonical Gs-coupled receptors, albeit moving 5–6 Å toward TMs 1 and 2 due to the smaller intracellular pocket in zGPR4 (Fig. 3c, embedded). In this regard, the zGPR4–Gs structure bears closer resemblance to previously characterized GPCR–Gi complexes (Supplementary Fig. 3a, b), including the recently determined GPR132-Gi complex structure24 (Supplementary Fig. 3c). Our structure, in conjunction with recently solved complex structures with non-canonical Gs-coupling model (e.g., GPR174, EP2, CCKAR), suggests that a larger outward movement of the intercellular TM5-TM6 is not a prerequisite for Gs protein recruitment (Fig. 3d)25.

a, b Superposition of the active zGPR4–Gs structure with its inactive model (AlphaFold). c The two zGPR4 models are further compared with the β2AR (PDB ID: 7BZ2) and A2BR (PDB ID: 7XY7) structures. d Alignment of zGPR4–Gs (current study) with other non-canonical Gs-coupled structures, including GPR174 (PDB ID: 7XV3), EP2 (PDB ID: 7CX2) and CCKAR (PDB ID: 7EZK). Color codes are indicated at the top-left corner. For clarity, only α5 helix is shown in each Gs-coupled structure.
Structural features
In the zGPR46.5 structure, we were able to model the conserved disulfide bond between TM3 and ECL2 (C983.25–C17645.50) (Fig. 1f). Although the last modeled residue at the N-terminus (C17) is proximal to C266 of ECL3, weak density precluded confirmation of a disulfide bond between them. Nevertheless, sequence alignment and structural prediction indicated a conserved disulfide linkage between the two positions for psGPCRs (Supplementary Fig. 4). Through cAMP assays, we found either C17S or C266S on zGPR4 had no significant impact on proton potency or efficacy (Supplementary Figs. 5 and 6), in contrast to the reduced activities for the corresponding mutations on GPR6816. zGPR4 features two additional cysteines (C11, C264) neighboring the aforementioned cysteines compared to hGPR4 (Supplementary Fig. 4), potentially adding complexity to the aspect of disulfide linkage in zGPR4. Alternatively, with two potential disulfide linkages between ECL3 and the N-terminus in zGPR4, breakage of a single disulfide bond may not be essential to destroying the structural rearrangement for receptor activation.
The zGPR46.5 structure features a partially ordered ECL2 that covers the orthostatic pocket. A cluster of bulky residues within ECL2, including F175, F177, E178, K179 and Y180, obstructs the pathway to the canonical orthostatic pocket (Fig. 4a). The F175 and Y180 form an aromatic triangle with a TM7 residue F2737.32. Remarkably, the F17745.51 that inserted into the pocket is highly conserved among the proton-sensing receptors, indicating an indispensable role for this residue for the structural integrity of these receptors. Both the zGPR46.5 active structure and its inactive model reveal a predominantly covered orthostatic pocket that rich in bulky aromatic residues (Fig. 4a), reminiscent of lipid-binding GPCRs and olfactory receptors. The orthosteric pocket volume of zGPR4 is only 166 Å3, relatively smaller than the size of adenosine A2A receptor (A2AR, 272 Å3)26 and β2 adrenergic receptor (β2AR, 313 Å3)27, and much smaller than these peptidic GPCRs (neurokinin 1 receptor, 651 Å3; angiotensin II receptor type 1, 1346 Å3)28,29 (Supplementary Fig. 7). Since GPR4 senses one of the smallest stimuli (proton) for its activation, such a compact structure within the pocket may facilitate a Van der Waals interaction network between TMs, transmitting the signal of protonation from the extracellular to the intercellular for conformational change. Although in both maps the electron density for the extracellular half is relatively poor compared to the intercellular half, the key motifs within the signaling transduction pathway are relatively clear, including the toggle switch, the PIF motif, and the DPxxY motif. The P5.50I3.40F6.44 motif is unique in GPR4 within proton-sensing receptors (Supplementary Fig. 8). In GPR4, the position of F6.44 is replaced by a short residue, V2416.44; in contrast, the encountering residue of V2416.44 in TM5 is a bulky residue (W2025.51) that is usually occupied by a hydrophobic aliphatic residue (like V/L) in other class A GPCRs. W5.51 and V6.44 are highly conserved across different species of GPR4, confirming the significance of these positions for the signal propagation pathway, and evolving in tandem to adapt to environmental changes.

a Side view of the zGPR4 orthostatic pocket showing a bunch of aromatic residues. b Extracellular view of the zGPR4 showing the titratable H/D/E residues at the ECLs. Some key interacting residues are also shown. a, b The ECL2 is colored gray for clarity.
Extracellular regions were suggested to sense the protons thus trigging the conformational change for activation. Previous work have identified the protonation of H792.66, H165ECL2, and H2697.36 residues of hGPR4 as critical for its activation18. Similarly, the residue corresponding to H79 in zGPR4 (H872.66) has also proved to be essential30. Nevertheless,we found in our active structure of zGPR46.5 the histidine residues are far apart: H173ECL2 of ECL2 is 5–6 Å away from H872.66 or H882.67, and even farther from H93ECL1, H2777.36 and H1634.63 (Fig. 4b). Among them, H93ECL1 and H1634.63 appear to play essential structural roles together with the aromatic residues W9123.50 and W1855.34. Also, there are many acidic residues in the extracellular region, and D241.31, D89ECL1, D168ECL2, and E17845.52 are located around the previously suggested histidine cluster (Fig. 4b).
Mutagenesis studies of featured residues
Our mutagenesis data demonstrate reduced proton potency by 0.2–0.3 log units for zGPR4 mutations H872.66F and H173 ECL2F, and negligible effect for H2777.36F (Fig. 5a), consistent with previous results18. Among these acidic residues at the extracellular region, Mutations at E17845.52 (E17845.52Q, E17845.52A) can each decrease proton potency by 0.2–0.3 log unit while maintaining the maximal efficacy (Emax), suggesting that E17845.52 facilitates the sensing of protons (Fig. 5b). Located centrally in ECL2, E17845.52 is surrounded by several polar residues including K17945.53, Y1063.33, R2556.59 and H2777.36, indicating this acidic residue likely contributes to charge-charge interactions that was not fulfilled by a glutamine or an alanine. Interestingly, we found both D89ECL1N and D89ECL1A mutations significantly increase proton sensitivity (0.4–0.5 log unit), implying that this acidic residue somehow disturbs proton-sensing (Fig. 5b). Similar as D89ECL1, we found a double mutant D164ECL2N&E165ECL2Q can also increase proton sensitivity (0.25 log unit) (Fig. 5c). These four acidic residues at the extracellular region are absolutely conserved among psGPCRs (Supplementary Fig. 4). Mutations of additional three acidic residues, D24NTN, D169ECL2N, E183ECL2Q, do not induce significant change on proton potency, whereas the former two can increase the efficacy compared to WT receptor (Fig. 5c). These results indicate that most extracellular acidic residues do not contribute essentially for proton-sensing.

a–e Mutagenesis analysis of residues in zGPR4 on the proton potency by the GloSensor cAMP assay. Mutagenesis analysis of histidine residues in ECLs (a), highly conserved acidic residues in ECLs (b), partially conserved acidic residues (c), the Na coordinating residues (d), and the aromatic residues within the orthosteric pocket (e). The maximum and minimum activation levels of WT GPR4 were set to 100 and 0%, respectively. The ΔpH50 was calculated by dividing the pH50 of the mutant by the pH50 of WT. Data are shown as means ± SEM from at least three independent experiments performed in triplicate. f Mapping the tested mutations on the zGPR4 structure. Mutations with positive effect (ΔpH50 > 0.2 unit) are colored blue; mutations with negative effect (ΔpH50 < −0.2 unit) are colored red; mutations with negligible effect (0.2 > ΔpH50 > −0.2) are colored gray. Source data are provided as a Source Data file.
An acidic triad (E4.53, D2.50, D7.49) within the core of 7TM is important for the proton-induced activation, and mutations of these residues to glutamine or asparagine (to mimic their neutral form) induce upward shift in EC5019. The latter two are shown to be directly coordinated with a Na+ (together with S3.39) in the inactive state of typical GPCRs, whereas in the active state the sodium was extruded as a result of conformational change31. Interestingly, in GPR4 as well as other proton-sensing GPCRs, the canonical N7.49PxxY7.53 motif is replaced by a D7.49PxxY7.53 motif (Supplementary Fig. 4). To investigate the function of D2907.49 we conducted two mutations, D290N and D290A. The results of cAMP assays showed that while both mutants were expressed at similar levels as the WT receptor (Supplementary Fig. 6), the D2907.49N shifted the pH50 upward from 7.7 to 8.1 (Fig. 5d), consistent with the corresponding mutations reported previously19. Meanwhile, the basal activity of D2907.49N is also significantly elevated compared to WT. In contrast, the D2907.49A mutant compromised both the proton potency and Emax. These results indicate that the D7.49N mutation may loosen the restraint by sodium, making the receptor more easily activated; on the contrary, the D7.49A mutation may disturb the local structure as well as the conformational transition from inactive to active. Furthermore, mutations of the key sodium coordination site by D712.50A or D712.50N can each significantly decrease the proton potency and Emax (Fig. 5d), this can be attributed to the similar reason as the D7.49A mutant. Similarly, a subsidiary sodium coordination residue, N2867.45, can shift the pH50 downward when substituted by alanine (Fig. 5d), indicating it may function by affecting the pKa (where Ka is the acid dissociation constant) of its nearby sodium coordination residue, e.g., the D2907.49.
Unexpectedly, we found that mutations of the bulky aromatic residues within the orthosteric pocket can consistently increase the proton potency of zGPR4 (Fig. 5e). While the increments by Y1103.37F and Y2767.35F are not significant ( < 0.1 log units), Y1063.33F and Y2486.51F can each increase the pH50 by ~0.3 log unit. Y1063.33 and Y2486.51 face each other in the 7TM central region and Y1103.37 and Y2767.35 are located below and above them (Supplementary Fig. 8). Since Y2486.51 is one helical turn above the toggle switch residue F2456.48, these aromatic residues may contribute to a Van der Waals interaction network within the pocket that propagates the signal of proton-sensing to the intercellular region. These results further indicate that the propagation in the central region may not rely on the hydrophilic properties of these residues; alternatively, these tyrosines may already contribute to a hydrophilic interaction network in the inactive state and their mutations to phenylalanine facilitate the transition to active state (Supplementary Fig. 8).
Implications for activation pathway
There are many histidines and acidic residues at the extracellular region, which possess different charge states at different pH: histidine is neutral at high pH but positively charged when protonated at pH < 7.0, whereas extracellular acidic residues consistently maintain a negative charge within the physiological pH range. Nevertheless, our results indicate that WT GPR4 has a pH50 of ~8, contrasting with lower pH50 values of 7–7.4 for GPR65 and 6.3–6.7 for GPR6819,32. This suggests that GPR4 activation might not rely on protonation of extracellular histidines, given its pKa of around 7. Moreover, studies showed that extracellular histidines in psGPCRs are largely unconserved, and even conserved histidines produce varied pharmacological effects upon mutation19. These findings imply that extracellular histidines are unlikely to be key proton sensors. Notably, we found mutations of extracellular histidines as well as acidic residues moderately influenced zGPR4’s potency or efficacy (Fig. 5f), but we did not identify any single mutation that completely abolish the function of zGPR4–unlike the almost abrogation by R1233.50A in the G protein-coupling region. This suggests that these H/D/E residues may primarily fine-tune the receptor’s transition from inactive to active conformations rather than mediate proton-sensing.
In contrast, mutations at the so-called “DyaD” site (D712.50, D2907.49) influenced receptor function more significantly. Specifically, mutations at D712.50 severely compromised efficacy, while the D2907.49A and D2907.49N mutations produced opposing effects on receptor’s proton-sensing ability. These data are in favor of the conclusion that proton-sensing is primarily mediated by the acidic triad residues within the transmembrane domain19. Notably, we found the Y-to-F mutations withinin the orthosteric pocket enhanced the potencies. This indicated that, instead of a water-mediated hydrophilic network, the central region of psGPCRs mainly used bulky sidechains to propagate the signaling transduction (Supplementary Fig. 8). Remarkably, we found a mimicking mutation (Y2767.35C) of a natural genetic variation of hGPR4 can further increase the pH50 compared to Y2767.35F (Fig. 5e), while the mutation has negligible effect on expression or Emax (Supplementary Fig. 6), suggesting the mutant may function simply by favoring the active state. Other natural genetic variations are mostly located in the central region of the 7TM (Supplementary Fig. 9), and we can expect that some of them may facilitate the transition from inactive to active, while some may hamper the transition.
Discussion
Here we report cryo-EM structure of active zGPR4 in complex with an engineered Gs protein. Compared to the inactive AlphaFold model of zGPR4, the ECLs experience relatively larger conformational changes than the transmembrane region. Generally, GPCR activation involves contraction of the 7TM central region and expansion of the extracellular and intracellular regions, which is required to provide space for accommodating the G protein. Sodium acts as a negative allosteric modulator for many class A GPCRs, coordinating with key positions (including 2.50, 3.39, and 7.49) in the inactive state, and is displaced during the transition to the active state31. Buried within the 7TM central region, the pKa of the acidic triad residues likely shifts to the physiological pH range33, meaning that protonation at lower pH neutralizes their charge, and preventing D2.50 and D7.49 from coordinating with positively charged sodium (or potassium) ions. Consequently, a reduction in pH can disrupt sodium coordination essential for receptor activation. Given the conserved nature of the DyaD site (D2.50 is highly conserved among class A GPCRs, D7.49 is half-conserved in δ-branch receptors), the unique apEx site (E4.53) of the acidic triad within psGPCRs likely plays a significant role in signaling transduction, as only these three receptors are fully activated by protons. The molecular details may still await further investigation.
We observed distinct effects from D-to-N mutations at the DyaD sites: D7.49N increased the potency, contrasting with reduced potency from D2.50N mutation. Previous structural studies have shown that an acidic residue at position 7.49 participates in a direct coordination with the sodium in receptors like cysLT134, PAR135, and PAR236, forming an additional coordination bond compared to receptors with a neutral residue (N7.49)37,38,39. Therefore, D7.49N is suggested to loosen the coordination with the sodium, making the receptor more readily activated. The tight coordination by D7.49 is thought to stabilize the inactive conformation, this could be the reason that many δ-branch receptors exhibit low basal activity40. In contrast, D2.50N as well as D2.50A may have totally disrupted the coordination, as evidenced by diminished sodium regulation in mutagenesis studies of psGPCRs and other GPCRs19,31. The reduction in efficacy for zGPR4 due to D2.50N or D2.50A mutations is consistent with compromised G protein signaling observed in other receptors31. The essential role of D2.50 is also underscored by its high conservation across class A GPCRs, whereas D7.49 is only half-conserved in δ-branch receptors (including psGPCRs), with other subfamilies predominantly featuring N7.49.
Beside protons, lysolipids were also suggested as “ligands” for psGPCRs. Our structures of zGPR4 reveal a tiny orthosteric pocket composed of highly conserved aromatic residues within the psGPCR family (Supplementary Fig. 7), which provided a hydrophobic platform to accommodate their potential lipidic ligands. Also, several other receptors (e.g., GPR31 and GPR151) were found to be activated under acidic conditions in vitro, although the GPR31 has been reported as a receptor for endogenous lipid, 12-(S)-hydroxyeicosatetraenoic acid41,42. These findings indicate that proton-sensing may be one of many properties of GPCRs and that other proton-sensing GPCRs may exist that allow detection of a broader pH range.
GPR4 has been linked to several pH-dependent processes under pathological conditions, including modulation of inflammation, angiogenesis and tumor metastasis. In addition, GPR4 has also been identified as a biomarker for neurodegenerative diseases like Parkinson’s and Alzheimer43, and inhibition of GPR4 was shown to reduce neuronal apoptosis and improve memory functions44. These studies underscored the potential therapeutic relevance of antagonists or negative allosteric modulators aimed at inhibiting GPR4 activity or expression across various tissues. Nevertheless, our findings reveal that both hGPR4 and zGPR4 have a pH50 of ~8.0, meaning they are constitutively active at a physiological pH of 7.4. This implies that proton-sensing may not be directly related to their biological function, unlike other psGPCRs such as GPR65 and GPR68, which are activated by changes in physiological pH. That is to say, these psGPCRs may be activated to different levels and thus function differently at various pH conditions. In addition, GPR4 may play some unique roles within the physiological environment in response to other ligands like lysolipids.
In summary, our data support the conclusion that these psGPCRs share a common triad acidic residue essential for proton-sensing19, while the varying amount of histidines and nearby acidic residues likely act as modulators of receptor function. A cluster of aromatic residues within the orthosteric pocket also contributes to the propagation of signaling across the transmembrane domain. The GPR4 structures together with assay results provided a framework for further understanding of the proton-sensing mechanism as well as development of small molecular modulators for these psGPCRs.
Limitations of current study
Our study has some limitations. Firstly, the resolution is insufficient to determine the protonation status of protonatable residues, preventing us from confidently identifying which residues are key for proton-sensing. Second, to obtain the structure, we had to expose both sides of the GPCR to the same pH change, which does not occur under physiological conditions, this limitation constrains our structural interpretations. Lastly, our structure only captures the active state of the receptor. Since proton-sensing relies on (de)protonatable residues with pKa values that differ significantly between inactive and active states, this further complicates our ability to identify specific residues responsible for proton-sensing from our structural data. Nevertheless, our structures may still serve as a valuable resource for comparative analyses of other proton-sensing GPCRs from different species.
Methods
Construct cloning, expression, and purification of zGPR4–Gs complexes
For the receptor, zebrafish GPR4 (residues 2–338), containing an N-terminal thermostabilized apocytochrome b562RIL fusion protein and a C-terminal LgBiT protein, was cloned into pFastBac1 vector, with an N-terminal hemagglutinin signal peptide tag followed by FLAG tag and C-terminal 10 × His tag. For the G protein components, mini-GαsIN, equal to mini-Gs but with its αN replaced by the counterpart of Gi (to facilitate binding of scFv16, which binds to the interface between Gβγ subunits and Gαi subunit)45, was fused to bovine Gγ2 at its N-terminus (Gγ–mini-GsIN). Human Gβ1, followed by SmBiT at its C terminus, was cloned into pFastBac-Dual together with Gγ–mini-GαsIN. Sf9 insect cells were infected with viruses of the receptor (zGPR4-LgBiT) and Gγ–mini-GαsIN–Gβ–SmBiT in the ratio of 1:1 for 72 h at 27 °C.
For the zGPR46.5 complex, the cell pellets were lysed by Dounce homogenization in 20 m MES (pH 6.5), 100 mM NaCl, 5 mM MgCl2, 5 mM CaCl2, 10% glycerol, EDTA-free protease inhibitor cocktail (Bimake), apyrase (25 mU/ml; New England Biolabs), 200 μM Tris(2-carboxyethyl)phosphine (TCEP) (Thermo Fisher Scientific), and scFv16 (15 μg/ml), incubated at 4 °C for 3 h, and then centrifuged at 30,000 rpm for 45 min to collect the membranes. The washed membranes were solubilized in 20 mM MES (pH 6.5), 100 mM NaCl, 5 mM MgCl2, 5 mM CaCl2, EDTA-free protease inhibitor cocktail, 1% (w/v) lauryl maltose neopentyl glycol (LMNG; Anatrace), 0.2% (w/v) cholesteryl hemisuccinate (CHS; Sigma-Aldrich), apyrase (25 mU/mL), and 200 μM TCEP and incubated at 4 °C for 3 h. The supernatant was collected by centrifugation at 30,000 rpm for 45 min and then incubated with TALON immobilized metal affinity chromatography resin (Clontech) with an addition of 20 mM imidazole at 4 °C overnight. The resin was collected by centrifugation at 1000 × g for 5 min, packed into a gravity flow column, and washed with 20 column volumes of buffer containing 20 mM MES (pH 6.5), 100 mM NaCl, 2 mM MgCl2, 2 mM CaCl2, 0.01% (w/v) LMNG, 0.002% (w/v) CHS, 5% glycerol, and 30 mM imidazole and eluted using buffer containing 20 mM MES (pH 6.5), 100 mM NaCl, 2 mM MgCl2, 2 mM CaCl2, 0.01% (w/v) LMNG, 0.002% (w/v) CHS, 5% glycerol, and 300 mM imidazole. The purified zGPR4–Gs complex and scFv16 proteins were mixed in a 1:1 molar ratio at 4 °C overnight and then concentrated with an Amicon Ultra Centrifugal Filter [molecular weight cutoff (MWCO), 100 kDa] and subjected to size-exclusion chromatography(SEC) using a Superdex 6 10/300 GL column (GE Healthcare) with running buffer, containing 20 mM MES (pH 6.5), 100 mM NaCl, 2 mM MgCl2, 0.0025% (w/v) LMNG, and 0.0005% (w/v) CHS. The fractions of monomeric protein complex were pooled and concentrated with an Amicon Ultra Centrifugal Filter (MWCO, 100 kDa) to 12 to 16 mg/ml for cryo-EM studies.
For the zGPR48.5 complex, the purification process was similar to that of zGPR46.5, except that MES (pH 6.5) was replaced by Tris (pH 8.5). Finally, the fractions of monomeric protein complex were pooled and concentrated with an Amicon Ultra Centrifugal Filter (MWCO, 100 kDa) to 6 to 9 mg/ml for cryo-EM studies.
Cryo-EM grid preparation and data collection
For sample preparation, 3 μl of purified zGPR46.5 and zGPR48.5 were applied onto the glow-discharged holey grids (Ni-Ti Au 300 mesh, NANODIM TECH), respectively. The grids were blotted for 3 s under 100% humidity at 4 °C and then plunge-frozen into liquid ethane using Vitrobot Mark IV (Thermo Fischer Scientific). The cryo-EM data were collected at the Center of Cryo-Electron Microscopy at Zhejiang University. Micrographs were acquired on the Titan Krios microscope (FEI) operated at 300 kV equipped with Selectris energy filter (Thermo Fisher Scientific) and Gatan Falcon 4 detector. EPU software was used for automated data collection according to standard procedures. A calibrated magnification of ×130,000 was used for imaging, yielding a pixel size of 0.931 Å on images. Each micrograph was dose-fractionated to 40 frames at a dose rate of 7.49 e− per pixel per second, with a total exposure time of 6 s, resulting in a total dose of about 52 e− Å−2. The defocus range was set from −1.0 to −2.0 μm.
Cryo-EM image processing
Image processing was conducted using cryoSPARC v4.4.146. Motion correction and contrast transfer function (CTF) parameters estimation were performed with the MotionCorr247 and patch CTF estimation48, respectively.
For zGPR46.5, a total of 8143 micrographs were collected, and 11,069,919 particles were auto-picked and extracted. After several rounds of two-dimensional (2D) classification, 368,456 particles were further subjected to ab initio reconstruction to generate the initial maps. Subsequently, 183,055 particles were selected for non-uniform refinement under C1 symmetry, resulting in a density map with an overall resolution of 3.3 Å, estimated by the gold-standard Fourier shell correlation coefficient (FSC) of 0.143.
Similar procedures were applied to zGPR48.5, a total of 5188 micrographs were collected and 7,819,042 particles were auto-picked and extracted. Following multiple rounds of 2D classification, 558,130 particles were selected for ab initio reconstruction. One subset, comprising 246,730 particles, was then subjected to non-uniform refinement, generating a map with a resolution of 3.1 Å.
Model building and refinement
Structures predicted by AlphaFold49,50 and A2BR–mini-Gs (PDB code 7XY6)51 were used as the starting models for model building and refinement against the electron density map of zGPR46.5–Gs and zGPR48.5–Gs. Models were docked into the EM density maps using Chimera52. Subsequently, iterative manual adjustments and rebuilding were performed in COOT53 and phenix.real_space_refine in Phenix54. The final refinement statistics are provided in Supplementary Table 1. The structure figures were prepared by Chimera and PyMOL (http://www.pymol.org).
Comparison of orthosteric pockets
Structures of four prototype class A GPCRs were taken to compare with GPR4: two small molecule receptors, β2AR (PDB ID: 4LDO) and A2AR (PDB ID: 5G53); and two peptidic receptors, NK1R (PDB ID: 7P00) and AGTR1 (PDB ID: 7F6G). All selected structures are in a fully active state (have cytoplasmic protein partners) and are bound to endogenous ligands (or close analog of endogenous ligand, as in A2AR). Volumes of pockets were measured using the Schrödinger software (Schrödinger Release 2023-4: Schrödinger, LLC, New York, NY, 2023). Structures were processed using the Protein Preparation Wizard tool55. The volumes were calculated using the SiteMap program56,57.
GloSensor cAMP assay
In brief, HEK293 (obtained from certified cell bank at the Chinese Academy of Science and confirmed as negative for mycoplasma contamination) cells in a 6.0 cm dish were cotransfected with 1 μg of zebrafish/human wild-type or mutated GPR4 plasmid and 1 μg of the pGloSensor-22F cAMP plasmid (Promega, USA) using polyethylene (6 μL, Yeasen, China). After incubating overnight, transfected cells were resuspended in DMEM added with 1% dialyzed FBS and then seeded into poly-l-lysine-coated 384-well plates (2 × 104 cells/well) (Costar, USA) for 9–24 h. After removing the culture medium, the cells were incubated with GloSensorTM cAMP reagent (Promega, USA) made in assay buffer (pH 8.4) for 1.5 h at 37 °C. To ensure that assay buffers (30 μL/well) were administered at the correct pH levels, GloSensorTM cAMP reagent was withdrawn from cell plates before adding assay buffer at predefined pH levels. Assay buffers were made using 1x HBSS mixed with different buffer agents for different pH levels, 10 mM HEPES and 10 mM MES for pHs 6.9–8.1, and 20 mM TAPS for pHs 8.2–9.3. Cytation 5 imaging reader (BioTek, USA) was utilized to continually monitor bioluminescence signals for 20–40 min.
Cell surface expression measurement
Cell surface expression of zebrafish/human wild-type or mutated GPR4 was detected by flow cytometry. Briefly, HEK293 cells in a six-well plate were transfected with zebrafish/human wild-type or mutated GPR4 plasmid and cultured for 24 h. The transfected HEK293 cells were harvested by Cell Scraper (Costar, USA) and then washed twice with PBS buffer containing 1% (w/v) BSA. The cells were resuspended by 100 μL PBS buffer containing 1% (w/v) BSA and then followed by the incubation with diluted (0.5 μg per million cells in 100 μL volume) PerCP/Cyanine5.5 anti-FLAG Tag antibody (BioLegend, USA) at 4 °C for 30 min. After washing as described above, fluorescently tagged cells (2 × 104 cells per sample) were detected using an LSR Fortessa flow cytometer (BD Biosciences), and data were analyzed by FlowJo software (TreeStar Inc., Ashland, OR, USA). Quantification was performed using mean fluorescence intensity (MFI) values.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Atomic coordinates and structure factors for the complexes have been deposited in the Protein Data Bank with identification code 8ZFZ (model of Gs-coupled zebrafish GPR4 at pH 6.5) and 9J31 (model of Gs-coupled zebrafish GPR4 at pH 8.5), and the corresponding electron microscopy maps have been deposited in the Electron Microscopy Data Bank under accession codes EMD-60072 (cryo-EM structure of Gs-coupled zebrafish GPR4 at pH 6.5) and EMD-61104 (cryo-EM structure of Gs-coupled zebrafish GPR4 at pH 8.5). All other data are available in the manuscript or supplementary materials. Source data are provided with this paper.
References
Katritch, V., Cherezov, V. & Stevens, R.C. Structure-function of the G protein-coupled receptor superfamily. Annu. Rev. Pharm. Toxicol. 53, 531–556 (2013).
Wacker, D., Stevens, R. C. & Roth, B. L. How ligands illuminate GPCR molecular pharmacology. Cell 170, 414–427 (2017).
Venkatakrishnan, A. J. et al. Molecular signatures of G-protein-coupled receptors. Nature 494, 185–194 (2013).
Sriram, K. & Insel, P. A. G protein-coupled receptors as targets for approved drugs: how many targets and how many drugs? Mol. Pharm. 93, 251–258 (2018).
Im, D. S. Two ligands for a GPCR, proton vs lysolipid. Acta Pharm. Sin. 26, 1435–1441 (2005).
Wei, W. C. et al. Coincidence detection of membrane stretch and extracellular pH by the proton-sensing receptor OGR1 (GPR68). Curr. Biol. 28, 3815–3823.e3814 (2018).
Imenez Silva, P. H. & Wagner, C. A. Physiological relevance of proton-activated GPCRs. Pflug. Arch. 474, 487–504 (2022).
Eltzschig, H. K. & Carmeliet, P. Hypoxia and inflammation. New Engl. J. Med. 364, 656–665 (2011).
Boedtkjer, E. & Pedersen, S. F. The acidic tumor microenvironment as a driver of cancer. Annu. Rev. Physiol. 82, 103–126 (2020).
Becker, H. M. & Deitmer, J. W. Proton transport in cancer cells: the role of carbonic anhydrases. Int. J. Mol. Sci. 22, 3171 (2021).
Damaghi, M., Wojtkowiak, J. W. & Gillies, R. J. pH sensing and regulation in cancer. Front. Physiol. 4, 370 (2013).
Justus, C. R. & Yang, L. V. GPR4 decreases B16F10 melanoma cell spreading and regulates focal adhesion dynamics through the G13/Rho signaling pathway. Exp. Cell Res. 334, 100–113 (2015).
Justus, C. R. et al. Contextual tumor suppressor function of T cell death-associated gene 8 (TDAG8) in hematological malignancies. J. Transl. Med. 15, 204 (2017).
Wang, H.-X. et al. Overexpression of G-protein-coupled receptors 65 in glioblastoma predicts poor patient prognosis. Clin. Neurol. Neurosurg. 164, 132–137 (2018).
Ludwig, M. G. et al. Proton-sensing G-protein-coupled receptors. Nature 425, 93–98 (2003).
Huang, X. P., Kenakin, T. P., Gu, S., Shoichet, B. K. & Roth, B. L. Differential roles of extracellular histidine residues of GPR68 for proton-sensing and allosteric modulation by divalent metal ions. Biochemistry 59, 3594–3614 (2020).
Wang, J. Q. et al. TDAG8 is a proton-sensing and psychosine-sensitive G-protein-coupled receptor. J. Biol. Chem. 279, 45626–45633 (2004).
Liu, J. P. et al. Each one of certain histidine residues in G-protein-coupled receptor GPR4 is critical for extracellular proton-induced stimulation of multiple G-protein-signaling pathways. Pharm. Res. 61, 499–505 (2010).
Rowe, J. B., Kapolka, N. J., Taghon, G. J., Morgan, W. M. & Isom, D. G. The evolution and mechanism of GPCR proton sensing. J. Biol. Chem. 296, 100167 (2021).
Duan, J. et al. Cryo-EM structure of an activated VIP1 receptor-G protein complex revealed by a NanoBiT tethering strategy. Nat. Commun. 11, 4121 (2020).
Maeda, S. et al. Development of an antibody fragment that stabilizes GPCR/G-protein complexes. Nat. Commun. 9, 3712 (2018).
Ballesteros, J. A. & Weinstein, H. In Methods in Neurosciences, Vol. 25 (ed. Stuart C. Sealfon) 366–428 (Academic Press, 1995).
Flock, T. et al. Universal allosteric mechanism for Galpha activation by GPCRs. Nature 524, 173–179 (2015).
Wang, J. L. et al. Functional screening and rational design of compounds targeting GPR132 to treat diabetes. Nat. Metab. 5, 1726–1746 (2023).
Nie, Y. et al. Specific binding of GPR174 by endogenous lysophosphatidylserine leads to high constitutive G(s) signaling. Nat. Commun. 14, 5901 (2023).
Carpenter, B., Nehmé, R., Warne, T., Leslie, A. G. & Tate, C. G. Structure of the adenosine A(2A) receptor bound to an engineered G protein. Nature 536, 104–107 (2016).
Ring, A. M. et al. Adrenaline-activated structure of β2-adrenoceptor stabilized by an engineered nanobody. Nature 502, 575–579 (2013).
Thom, C. et al. Structures of neurokinin 1 receptor in complex with G(q) and G(s) proteins reveal substance P binding mode and unique activation features. Sci. Adv. 7, eabk2872 (2021).
Zhang, D. et al. Structural insights into angiotensin receptor signaling modulation by balanced and biased agonists. EMBO J. 42, e112940 (2023).
Mochimaru, Y. et al. Extracellular acidification activates ovarian cancer G-protein-coupled receptor 1 and GPR4 homologs of zebra fish. Biochem. Biophys. Res. Commun. 457, 493–499 (2015).
Katritch, V. et al. Allosteric sodium in class A GPCR signaling. Trends Biochem. Sci. 39, 233–244 (2014).
Howard, M. K. et al. Molecular basis of proton-sensing by G protein-coupled receptors. Cell https://doi.org/10.1016/j.cell.2024.11.036 (2024).
Isom, D. G., Cannon, B. R., Castaneda, C. A., Robinson, A. & Garcia-Moreno, B. High tolerance for ionizable residues in the hydrophobic interior of proteins. Proc. Natl. Acad. Sci. USA 105, 17784–17788 (2008).
Luginina, A. et al. Structure-based mechanism of cysteinyl leukotriene receptor inhibition by antiasthmatic drugs. Sci. Adv. 5, eaax2518 (2019).
Zhang, C. et al. High-resolution crystal structure of human protease-activated receptor 1. Nature 492, 387–392 (2012).
Cheng, R. K. Y. et al. Structural insight into allosteric modulation of protease-activated receptor 2. Nature 545, 112–115 (2017).
Wang, S. et al. D(4) dopamine receptor high-resolution structures enable the discovery of selective agonists. Science 358, 381–386 (2017).
Rappas, M. et al. Comparison of orexin 1 and orexin 2 ligand binding modes using x-ray crystallography and computational analysis. J. Med. Chem. 63, 1528–1543 (2020).
Fenalti, G. et al. Molecular control of δ-opioid receptor signalling. Nature 506, 191–196 (2014).
Zeghal, M., Laroche, G., Freitas, J. D., Wang, R. & Giguere, P. M. Profiling of basal and ligand-dependent GPCR activities by means of a polyvalent cell-based high-throughput platform. Nat. Commun. 14, 3684 (2023).
Mashiko, M., Kurosawa, A., Tani, Y., Tsuji, T. & Takeda, S. GPR31 and GPR151 are activated under acidic conditions. J. Biochem. 166, 317–322 (2019).
Guo, Y. et al. Identification of the orphan G protein-coupled receptor GPR31 as a receptor for 12-(S)-hydroxyeicosatetraenoic acid. J. Biol. Chem. 286, 33832–33840 (2011).
Zhou, S. et al. Identification of 5 potential predictive biomarkers for Alzheimer’s disease by integrating the unified test for molecular signatures and weighted gene coexpression network analysis. J. Gerontol. A Biol. Sci. Med. Sci. 78, 653–658 (2023).
Haque, M. E. et al. The neuroprotective effects of GPR4 inhibition through the attenuation of caspase mediated apoptotic cell death in an MPTP induced mouse model of Parkinson’s disease. Int. J. Mol. Sci. https://doi.org/10.3390/ijms22094674 (2021).
Nehmé, R. et al. Mini-G proteins: Novel tools for studying GPCRs in their active conformation. PLoS ONE 12, e0175642 (2017).
Punjani, A., Rubinstein, J. L., Fleet, D. J. & Brubaker, M. A. cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296 (2017).
Zheng, S. Q. et al. MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods 14, 331–332 (2017).
Zhang, K. Gctf: Real-time CTF determination and correction. J. Struct. Biol. 193, 1–12 (2016).
Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021).
Varadi, M. et al. AlphaFold Protein Structure Database: massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 50, D439–d444 (2022).
Chen, Y. et al. Cryo-EM structure of the human adenosine A(2B) receptor-G(s) signaling complex. Sci. Adv. 8, eadd3709 (2022).
Pettersen, E. F. et al. UCSF Chimera-a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 (2004).
Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. Features and development of Coot. Acta Crystallogr. D. Biol. Crystallogr. 66, 486–501 (2010).
Adams, P. D. et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D. Biol. Crystallogr. 66, 213–221 (2010).
Sastry, G. M., Adzhigirey, M., Day, T., Annabhimoju, R. & Sherman, W. Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. J. Comput Aided Mol. Des. 27, 221–234 (2013).
Halgren, T. A. Identifying and characterizing binding sites and assessing druggability. J. Chem. Inf. Model 49, 377–389 (2009).
Halgren, T. New method for fast and accurate binding-site identification and analysis. Chem. Biol. Drug Des. 69, 146–148 (2007).
Acknowledgements
This work was supported by the basic research program of the Science and Technology Commission of Shanghai Municipality (21JC1402400), the Fundamental Research Funds for the Central Universities (40500-20101-222383), and the National Natural Science Foundation of China grants 32171215 (G.S.), 82172644 (W.L.), 32371344 (H.R.), and 82230002 (M.L.). The authors thank the Instruments Sharing Platform of the School of Life Sciences, East China Normal University, and ECNU Multifunctional Platform for Innovation (011). The authors thank Profs. Zhi-Jie Liu and Tian Hua for their guidance and assistance on EM sample preparation. The authors are grateful to the staff at the Core Facilities of Life Sciences Institute and the Center of Cryo-Electron Microscopy at Zhejiang University for their technical assistance.
Author information
Authors and Affiliations
Contributions
Yitong Ma optimized constructs, expressed and purified the complex proteins for cryo-EM studies, and edited the initial manuscript; Yijie Wang undertook the cAMP assays. Mengyuan Tang undertook cryo-EM data collection, 3D reconstruction and optimized the initial structure models. Yuan Weng helped cell culture and molecular cloning. Ying Chen and Yueming Xu assisted antibody and grid preparation. Shuxiao An did mutations for the cAMP assay. Yiran Wu and Suwen Zhao were involved in the model analysis and discussion. Huanhuan Xu and Dali Li provided reagents and oversaw the data analysis, Mingyao Liu oversaw the project and edited the manuscript. Weiqiang Lu supervised the cAMP assay and edited the manuscript. Heng Ru supervised the cryo-EM data collection and structural modeling, and edited the manuscript. Gaojie Song supervised the project, analyzed the data, and wrote the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Matthew Belousoff and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Ma, Y., Wang, Y., Tang, M. et al. Cryo-EM structure of an activated GPR4–Gs signaling complex. Nat Commun 16, 605 (2025). https://doi.org/10.1038/s41467-025-55901-2
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-55901-2
