
Abstract
Conventional immunotherapy exhibits low response rates due to the immunosuppressive tumor microenvironment (TME). To overcome this limitation, this study introduces ZFPG nanoparticles (ZFPG NPs) with ZnFe2O4@Pt cores and glucose oxidase (GOx) shells. The ZFPG NPs possess five-enzyme activities, good sonosensitivity, and remarkable magnetic targeting properties, which facilitate sono-metallo-immunotherapy for prostate cancer treatment in male mice. Specifically, the magnetic targeting ability effectively improves their accumulation in tumors while still showing enrichment in the liver and kidneys. The multienzyme cascade catalysis and sonosensitivity of these NPs effectively deplete glutathione and glucose, and enhance the generation and utilization of H2O2, thereby inducing multiple ROS bursts. Furthermore, these comprehensive effects up-regulate the HMOX1 to promote the Fe2+ and lipid peroxides accumulation, thereby inducing immunogenic ferroptosis. This strategy facilitates anti-tumor immunity by ameliorating the immunosuppressive TME and inhibiting lung metastatic progression. This joint warfare strategy offers a powerful solution to address conventional immunotherapy limitations.
Similar content being viewed by others

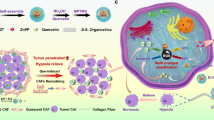
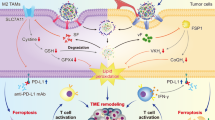
Introduction
Immunotherapy, which aims to augment the host immune system’s ability to combat specific diseases, is ushering in a transformative era in the landscape of cancer treatment1. Nonetheless, the clinical application of immunotherapy faces several formidable challenges, including a low immune response rate, an immunosuppressive tumor microenvironment (TME), and significant immune-related adverse events2. The deliberate induction of apoptosis through anticancer drugs stands as a prominent strategy for eliciting anti-tumor immunotherapeutic responses. Unfortunately, it is commonly regarded as an immunotolerant form of cell demise because the phagocytic clearance of apoptotic tumor cells by macrophages can enhance immunosuppression within the TME and prevent stimulation of the host immune system3. Therefore, activating non-apoptotic cell death pathways has emerged as a robust paradigm for eliminating tumor cells, boosting tumor immunogenicity, and triggering the anti-tumor immune response4.
Ferroptosis, characterized by iron accumulation and lipid peroxidation (LPO), represents a newly discovered programmed cell death mechanism that is distinct from apoptosis, necrosis, and autophagy5. Investigative studies have revealed that an excess of iron ions plays a crucial role in inducing the conversion of M2 macrophages into M1 macrophages and suppressing myeloid suppressor cells (MDSCs)6,7, thus effectively alleviating the immunosuppressive TME and realizing the metal ions-mediated metallo-immunotherapy. Fe-based nanozymes with peroxidase (POD)-like properties have exhibited considerable potential for effective immunogenic ferroptosis8. However, the efficacy of ferroptosis-mediated metallo-immunotherapy is severely hindered by the overexpression of glutathione (GSH) and insufficient levels of H2O29. Therefore, it becomes imperative to enhance the therapeutic effect of Fe-based nanozymes by depleting the GSH and augmenting H2O2 concentration in the TME. Promisingly, glucose oxidase (GOx) efficiently catalyzes the conversion of glucose into H2O2 and glucuronic acid, which is conducive to enhancing the Fe-based nanozyme-mediated Fenton reaction rate. Moreover, the construction of bimetallic nanozyme can control the reaction energy barrier of chemical intermediates through the electronic interaction between two adjacent heterometallic atoms, which can further enhance the POD-like activity10. Therefore, the combination of GOx and bimetallic Fe-based nanozymes can facilitate the POD-like activity-mediated hydroxyl radical (•OH) generation, thereby achieving the synergistic integration of starvation therapy (ST)/chemodynamic therapy (CDT). Additionally, nanozymes with single POD-like activity have limited affinity and insufficient utilization of H2O2. Nanoplatforms with catalase-like (CAT-like) and oxidase-like (OXD-like) activities achieve the conversion of H2O2 to •O2−, which enhances the utilization rate of H2O2 to amplify reactive oxygen species (ROS) generation, thereby accelerating the occurrence of immunogenic ferroptosis.
In addition to inducing immunogenic ferroptosis to achieve effective metallo-immunotherapy, immunogenic cell death (ICD) has also demonstrated efficacy in cancer immunotherapy by exposing a plethora of tumor antigens11. However, most small-molecule ICD inducers, such as doxorubicin12 and oxaliplatin13, are hindered by issues related to nonspecific enrichment and rapid drug metabolism. Fortunately, the utilization of exogenous controlled ICD immunotherapies mediated by functional nanomaterials, such as Fe-based nanoparticles inducing magnetothermal therapy (MTT) and photothermal therapies (PTT), has partially mitigated the drawbacks associated with small molecule ICD inducers14,15. Moreover, Fe-based nanomaterials with superparamagnetic properties are expected to achieve specific accumulation at tumor sites and even provide magnetic resonance imaging (MRI) of the tumor tissue upon magnetic field (MF) irradiation. For all that, concerns about hyperthermia safety still limit their clinical applicability. In contrast, mild ultrasound (US) can penetrate deep into dermal tissues to stimulate ROS generation, enabling the treatment of deep-seated tumors without inducing hyperthermia-related complications16,17. Given the aforementioned factors, the development of a bimetallic Fe-based sonosensitive nano platform with magnetic targeting capability and multienzyme activities for both metallo-immunotherapy and sono-immunotherapy would represent a strategic approach that achieves comprehensive benefits.
In this work, we rationally design biodegradable ZnFe2O4@Pt@PEG-GOx nanoparticles (ZFPG NPs) featuring five enzyme activities similar to POD, CAT, OXD, GOx, and glutathione oxidase (GSHOx) for magnetic targeting-enhanced and MRI-guided sono-metallo-immunotherapy, as illustrated in Fig. 1. The nano platform exhibits exceptional superparamagnetism for magnetic targeting to tumors under MF irradiation. Subsequently, ZFPG NPs deplete the energy substrate of tumors via GOx property, resulting in a “starvation effect” and an increase in H2O2 concentration to augment the generation of toxic •OH. Furthermore, the ZFPG NPs efficiently convert H2O2 to •O2− via the CAT-like and OXD-like activities. This multienzyme cascade catalysis effectively increases the production and utilization of H2O2, thus inducing the outbreak of ROS. Moreover, the ZnFe2O4@Pt heterojunction facilitates rapid electron transfer from ZnFe2O4 (ZFO) to Pt under US irradiation, which enhances 1O2 generation. The multi-mode synergistic effect of ZFPG NPs, especially along with their GSH depletion ability, can induce GPx4 inactivation-mediated excessive accumulation of LPO, ultimately leading to efficient ferroptosis. Notably, this multimodal therapy effectively reverses the immunosuppressive TME to enhance the infiltration of CTLs, which generates anti-tumor immunity to kill the residual tumor cells and inhibits their metastasis. These findings highlight the valuable potential of ZFPG NPs in advancing sono-metallo-immunotherapy for cancer treatment.

a Schematic diagram of the synthesis mechanism of ZFPG nanoplatform; b the anti-tumor mechanism diagram of ZFPG-mediated cascade enzymatic reaction synergistically enhanced sono-metallo-immunotherapy. ZFPG ZnFe2O4@Pt@PEG-GOx, ICD immunogenic cell death, DAMPs damage-associated molecular patterns, DCs dendritic cells, TNF-α tumor necrosis factor-α, IFN-γ interferon-γ, CDT chemodynamic therapy, SDT sonodynamic therapy, ST starvation therapy).
Results and discussion
The synthesis and characterization of the ZFPG NPs
The preparation of the ZFPG NPs involved several steps. Firstly, the ZFO NPs were synthesized using a modified solvothermal method. Subsequently, the in situ growth of Pt NPs was performed on the spherical surface of the ZFO NPs. To further enhance the biocompatibility and aqueous stability, PEGylation was concurrently realized upon the addition of Silane-PEG2k-COOH as a surfactant additive. The silane group could hydrolyze and condensate with the -OH group on the surface of ZnFe2O4@Pt in the aqueous phase environment, thus anchoring Silane-PEG2K-COOH on the surface of the nanomaterials. Finally, GOx was grafted onto the surface of the material through amidation reactions between the –COOH and –NH2 groups under the activation effect of EDC/NHS. The process and mechanism diagram in the formation of the ZFPG NPs are depicted in Fig. 2a and S1.

a Schematic diagram of the preparation of the ZFPG NPs; low-magnification TEM photographs of b ZFO NPs, c ZnFe2O4@Pt NPs, and d ZFPG NPs; e high-magnification TEM photograph of ZFPG NPs (1. Locally enlarged high-resolution TEM images of Pt component, 2. Locally enlarged high-resolution TEM images of ZnFe2O4 component); f HAADF-STEM and element mapping images of Zn, Fe, O, and Pt in the ZFPG NPs; g hydrodynamic size distribution of ZFO, ZFP, and ZFPG measured by DLS (PDI polydiseperse index); h zeta potential of G1-ZFO, G2-ZnFe2O4@Pt, G3-ZFP, and G4-ZFPG (n = 3 independent samples, mean ± SD); i XRD pattern of ZnFe2O4@Pt NPs; XPS spectrum of j Fe 2p and k Pt 4f in ZFPG NPs; and l magnetic hysteresis loop ZFPG NPs at the temperature of 300 K. Source data are provided as a Source Data file.
The as-prepared ZFO NPs exhibited spherical morphology with uniform size and high dispersity, as characterized by transmission electron microscopy (TEM) (Fig. 2b). The average diameter of the ZFO NPs was determined to be 67.11 nm, and dynamic light scattering (DLS) revealed a hydrated particle size of 133.22 nm with a polydispersity (PDI) of 0.153 (Figs. 2g and S2). Subsequently, ZnFe2O4@Pt NPs were prepared by in situ growth of Pt on the surface of ZFO NPs by a one-step reduction method. It was evident that a small amount of Pt NPs was tightly anchored on the ZFO matrix (Fig. 2c). Moreover, the ZnFe2O4@Pt0.2 exhibited the best carrier mobility and sono-catalytic performance when the feed ratio of H2PtCl6·6H2O to ZFO was 0.2 (Fig. S3). Furthermore, the high-resolution TEM (HR-TEM) image of ZFPG NPs showed that the crystal plane spacing of ZnFe2O4 was 0.24 nm, which corresponded to the (222) crystal face of ZnFe2O4 (PDF# 22-1012) with spinel structure. The lattice spacing of Pt NPs was 0.22 nm, corresponding to the (111) crystal face of Pt (PDF#04-0802) with the face-centered cubic structure (Fig. 2e). In addition, the powder X-ray diffraction (XRD) pattern of ZnFe2O4@Pt NPs exhibited peaks in good agreement with the pristine ZnFe2O4 (JCPDS No. 22-1012), further confirming the spinel structure of ZnFe2O4. The absence of crystalline Pt doping could be attributed to the low Pt content in the NPs (Fig. 2i). After PEGylation and GOx-grafting, there were no apparent morphological changes, but the hydrated particle size of ZnFe2O4@Pt-PEG (ZFP) NPs and ZFPG NPs gradually increased (152.93 nm and 185.24 nm), indicating successful surface functionalization (Fig. 2d, g). The variation of zeta potential after each step confirmed the successful in situ growth of Pt NPs, PEGylation, and GOx-grafting (Fig. 2h). Moreover, the gradually increasing weight loss rates of ZFP NPs and ZFPG NPs compared to ZnFe2O4@Pt NPs confirmed the successful PEGylation and GOx-grafting, validating the successful formation of ZFPG NPs (Fig. S4). The UV-vis spectra of ZFPG NPs exhibited the characteristic absorption peak of GOx, which further proved the successful GOx-grafting (Fig. S5). According to the standard curve of GOx, by measuring the ungrafted GOx in the supernatant after the reaction, when the feed ratio of GOx to ZFP NPs was 0.2, the loading content of GOx in ZFPG was 5.56% (Figs. S6 and 7). Furthermore, the elemental mapping of ZFPG NPs clearly showed the coexistence of Zn, Fe, O, and Pt elements within the composite, suggesting the successful formation of the ZFPG NPs (Fig. 2f). X-ray photoelectron spectroscopy (XPS) was conducted to analyze the surface chemical composition of ZFPG NPs. As shown in Fig. 2j, the binding energies of Fe (II) in the 2p1/2 and 2p3/2 orbitals were 709.27 eV and 722.07 eV, respectively, while those of Fe (III) in the 2p1/2 and 2p3/2 orbitals were 710.76 eV and 724.26 eV, respectively. By peak-fitting of Fe 2p XPS, the area under the Fe (III) peak was about 2.66-fold larger than the area under the Fe (II) peak. The presence of a small amount of Fe (II) may be due to the reduction of part of Fe (III) by NaBH4 during the in situ growth of Pt. The binding energies of Pt (0) in the 4f7/2 and 4f5/2 orbitals were 71.25 eV and 74.49 eV, respectively, while those of Pt (II) in the 4f7/2 and 4f5/2 orbitals were 72.36 eV and 75.58 eV, respectively, indicating the successful in situ growth of Pt NPs on the surface of ZFO NPs (Fig. 2k). The XPS analysis of the ZFPG NPs indicated that the atomic ratio of Fe and Zn elements was close to 2, which aligned with the anticipated distribution of Fe and Zn in ZFO. Additionally, the Pt content in the ZFPG NPs was determined to be 3.45% (Fig. S8). Finally, the magnetic properties of the ZFPG NPs were investigated using a vibrating sample magnetometer (VSM) at 300 K in an applied magnetic field ranging from −20 kOe to 20 kOe (Fig. 2l). The hysteresis loop demonstrated the superparamagnetic behavior of the ZFPG NPs, with a recorded saturation magnetization (Ms) of 71.49 emu g−1. The exceptional superparamagnetic property established the capability of the ZFPG NPs to undergo MF-induced enrichment, thereby providing a foundation for in vivo magnetic targeting delivery (Fig. S9).
Multienzyme-like activities of the ZFPG Nanozymes
The ZFPG NPs exhibit intrinsic multienzyme activities, resembling those of POD, CAT, OXD, GOx, and GSHOx. Relevantly, the mutual reinforcement of these enzymatic reactions resulted in the amplification of oxidative stress levels within tumors, as depicted in Fig. 3a.

a Scheme of the multi-catalytic mechanism of ZFPG NPs; b Lineweaver–Burk plotting of ZFO nanozymes (n = 3 independent samples, mean ± SD); c differential and worse charge of Fe3O4 and ZFO nanozymes; d relative free energy of the two catalysts during the catalytic process of H2O2; e dissolved O2 in PBS after co-incubation of H2O2 (10 mM) with ZFP and ZFO for 5 min (Inset: photograph of ZFP solution co-cultured with H2O2); f ESR spectra of •O2– trapped in the ZFO, ZFP, and ZFPG groups; g GOx activity of ZFG NPs measured by TMB probe; h ESR spectra of •OH trapped in the H2O2 alone, ZFO + H2O2, ZFO + H2O2, and ZFPG + Glu groups; i pH value of different concentrations of ZFPG NPs co-incubated with Glu (1 mg mL−1); j concentration-dependent GSH (3 mM) depletion by ZFPG NPs; and k, l time-dependent GSH (1 mM) depletion by ZFPG NPs (Inset: color change image of DTNB probe before and after ZFPG reaction with GSH). Source data are provided as a Source Data file.
The POD-like activity of the ZFO cores was detected through 3,3′,5,5′-tetramethylbenzidine (TMB) probes, which could react with ROS to produce blue products with an absorption peak at 652 nm. The gradually increasing slopes of the reaction curve indicated a clear dependence of the TMB oxidation rate on the substrate concentration of H2O2 (Fig. S10a). To reveal the POD-like enzymatic catalysis mechanism, a steady-state kinetics analysis was performed by changing the concentration of H2O2 at a fixed concentration of ZFO NPs, which was consistent with the classic Michaelis–Menten kinetics (Fig. S10b). According to the corresponding Lineweaver–Burk plots, the enzyme kinetic parameters, including the maximum reaction rate (Vmax) and Michaelis–Menten constant (Km), were calculated (Fig. 3b). The values of Km (0.773 mM) and Vmax (1.72 × 10−7 M s−1) indicated that ZFO NPs exhibited great affinity for H2O2 and could quickly convert it into •OH. The Vmax of ZFO was 2.78-fold higher than that of classical Fe3O4 nanozyme, along with a much lower Km, indicating that ZFO had a higher catalytic reaction rate and enzyme affinity18. To reveal the mechanism underlying the great POD-like property of bimetallic ZFO NPs, the spin-polarized density functional theory (DFT) calculation was conducted on the Vienna ab initio simulation package (VASP) to study the decomposition process of H2O2 on the surface of Fe3O4 and ZFO catalysts19,20. The structural models of Fe3O4 and ZFO, along with the adsorption site of H2O2 and •OH on these two catalysts, were shown in Fig. S11. By comparing the differential and worse charge of these two catalysts, it could be seen that the Zn site lost less charge than the Fe site because the valence state of Zn was relatively lower than Fe (Fig. 3c). The Zn atom with a lower state had more valence electrons, and the electrons were more easily transferred to the empty orbital. In the adsorption process of H2O2 and •OH, H2O2 provided empty orbitals while •OH provided single electrons. Therefore, the introduction of Zn could help ZFO to adsorb more H2O2 and release •OH, enhancing its POD-like activity. The free energy results of the two catalysts during the catalytic process of H2O2 further showed that the introduction of Zn enhanced the adsorption of H2O2, thereby strengthening the activation of H2O2. Moreover, the energy barrier of the decisive step of generating •OH in the decomposition process of H2O2 catalyzed by ZFO was smaller than that of Fe3O4, making it easier to induce the generation of •OH (Fig. 3d). Consequently, bimetallic ZFO NPs exhibited stronger POD-like activity than Fe3O4.
Additionally, the CAT-like property of the Pt component in the ZFPG NPs was validated through the enhanced dissolved O2 concentration after the co-incubation of ZFP NPs with H2O2 solutions (Fig. 3e). Specifically, compared to the H2O2 group (10 mM) and ZFO + H2O2 (10 mM) group, there was a much higher content of dissolved O2 (40 mg mL−1) in the ZFP + H2O2 (10 mM) group after 5 min incubation, which also resulted in a large number of bubbles at the bottom of the reaction vessel. Upon verification of the production of O2, the OXD-mimicking activity of the Pt component was examined using TMB as a substrate similar to the POD-like activity assessment. The Pt component of ZFP NPs could provide electrons to the surrounding O2 molecules to form •O2− radicals with strong oxidation capacity, thus oxidizing TMB to OxTMB in the absence of H2O2 (Fig. S12). Moreover, the OXD-like property of ZFP and ZFPG NPs was confirmed through the ESR assessment (Fig. 3f)21.
GOx can convert the energy substrate glucose (Glu) into H2O2 and gluconic acid, which enhances the H2O2 concentration and accelerates the Fe2+-mediated Fenton reaction rate in the TME, as well as inducing the starvation effect of cancer cells. Therefore, the generation of H2O2 and the decrease of pH mediated by gluconic acid are key indicators of Glu decomposition by GOx. By co-incubating ZnFe2O4@PEG-GOx (ZFG) NPs (100 μg mL−1), Glu (1 mg mL−1), and TMB solution, the ZFG+Glu+TMB group exhibited a strong characteristic absorption peak at 652 nm. This result indicated that GOx could effectively convert Glu into H2O2 and further decompose into ·OH under the action of ZFO (Fig. 3g). To further verify the cascade catalytic effect between POD-like and GOx properties of ZFPG NPs, we used ESR tests to verify •OH generation in the H2O2, ZFO + H2O2, ZFP + H2O2, and ZFPG + Glu groups. The signal of •OH could be detected in the ZFO + H2O2, ZFP + H2O2, and ZFPG + Glu group, which indicated the enzyme cascade catalytic property of ZFPG NPs (Fig. 3h). Moreover, the decreased pH value confirmed the generation of gluconic acid after 3 h of co-incubation between different concentrations of ZFPG and Glu solutions (Figs. 3i and S13). These results suggested that GOx could utilize the energy substrate Glu to boost the cascade catalytic reactions of ZFPG NPs, resulting in enhanced ROS generation.
The abundance of GSH in tumor cells as an endogenous antioxidant has been demonstrated to capture ROS and dilute the anti-tumor effect. Therefore, the depletion of GSH could effectively enhance ROS accumulation. We used 5,5′-dithiobis-(2-nitrobenzoic acid) (DTNB), a reagent that could react to GSH to generate yellow products with an absorption peak at 412 nm, to evaluate the consumption of GSH by the ZFPG NPs. Upon increasing the concentration of ZFPG NPs and extending the incubation time, there was a noticeable enhancement in GSH depletion. Particularly, after the co-incubation of ZFPG NPs (200 μg mL−1) with GSH (0.5 mM) solution for 24 h, complete consumption of GSH was observed (Fig. 3j–l). Moreover, we used DTNB probes to compare the GSH consumption capacity of ZFO, ZFP, and ZFPG NPs. After these materials (200 μg mL−1) were co-cultured with GSH (1 mM) for 6 h, there was no significant difference in UV-vis spectra (Fig. S14). Moreover, the ratio between Fe(III) and Fe(II) decreased from 2.66 to 0.3 after the co-incubation of ZFPG and GSH, indicating that the GSH consumption capacity of the ZFPG NPs was mainly ascribed to the Fe3+ in the ZFO component (Fig. S15). The GSHOx-like property promoted the degradation of ZFPG NPs and the release of metal ions. As shown in Fig. S16, ZFPG NPs suffered significant degradation and 67.87% of iron ions were released after co-incubation of ZFPG in a pH 5.5 solution with 10 mM GSH for 24 h, which could facilitate the maintenance of a relatively high level of ROS within tumor cells during therapeutic interventions.
Enhanced sonosensitivity of the ZFP Schottky heterojunction
Another crucial pathway to generate ROS was attributed to the sonodynamic effect of ZFPG NPs. 1,3-diphenylisobenzofuran (DPBF) was utilized as the singlet oxygen (1O2) probe to detect the US-triggered ROS generation. The sustained generation of 1O2 by ZFO, ZFP, and ZFP + H2O2 under US irradiation was demonstrated in Figs. 4a and S17, which showed a gradual decline in the characteristic absorption peak (425 nm) of the DPBF probe with increasing US stimulation time. As shown in Fig. 4b, after 4 min of US irradiation (1.5 W cm−2, 1.0 MHz, 50% duty cycle), the absorbance of DPBF in the ZFP + H2O2, ZFP, and ZFO groups decreased by 55.36%, 48.89%, and 33.01%, respectively. In addition, we further verified the 1O2 generation ability of different treatments through the ESR spectrometer. As shown in Fig. 4c, the results were consistent with the examination using the DPBF probe, where the ZFP + H2O2 + US demonstrated the strongest 1O2 generation capacity. These results demonstrated that the Schottky heterojunction of ZFP NPs enhanced their sonodynamic efficiency, and the CAT-like activity of ZFP NPs enhanced the interaction between O2 and the ZFP NPs under US treatment, thereby promoting the conversion of O2 to 1O2 in the ZFP + H2O2 group.

a Time-dependent 1O2 generation of ZFP + H2O2 under US irradiation; b DPBF degradation rate of different groups after 4 min US irradiation (G1: PBS, G2: ZFO + US, G3: ZFP + US, G4: ZFP + H2O2 + US, n = 3 independent samples, mean ± SD); c ESR spectra of US triggered 1O2 generation in different groups using TEMP as the trapping agent of 1O2; d Nyquist plots of ZFO and ZFP NPs obtained from electrochemical impedance spectrum (EIS) measurements; e tauc plot of ZFO NPs; f VB-XPS spectra of ZFO NPs; g, h transient absorption spectra; i time profiles of normalized transient absorption for ZFO catalyst after 360 nm laser excitation; j, k transient absorption spectra; and l time profiles of normalized transient absorption for ZFP catalyst after 360 nm laser excitation. Source data are provided as a Source Data file.
The electrochemical impedance spectrum (EIS) was used to describe the interface and conductive properties of nanomaterials. The smaller semicircle in the Nyquist plots of the ZFP NPs indicated the decreased charge-transfer resistance, leading to enhanced catalytic activity of ZFP NPs upon external triggering (Fig. 4d). Moreover, the band gaps of ZFO and ZFP were calculated to be 2.26 eV and 2.13 eV, respectively, using the modified Kubelka-Munk function and plotted in the Tauc plot (Figs. 4e and S18). The narrower band gap of ZFP facilitated the separation of US-triggered electrons and holes, leading to a significant improvement in ROS generation. The valence band (VB) value was measured to be 0.5 eV according to the VB-XPS spectrum (Fig. 4f), and the band gap structure of ZFP was shown in Fig. S19. The ZFP NPs could generate 1O2 under US excitation because their conduction band (CB) edge (−1.76 eV) is more negative than the O2/•O2− redox potential (−0.33 eV).
Finally, we used femtosecond time-resolved transient absorption (fs-TA) spectroscopy to investigate the carrier dynamics in ZFP NPs. We employed a 360 nm laser as the pump light to evaluate the sono-generated carrier dynamics of ZFO and ZFP NPs. Under laser excitation, the ZFO and ZFP NPs exhibited continuous and broadband absorption in the detection light range of 450–700 nm, with the maximum absorption peaks at 630 nm and 600 nm, respectively (Fig. 4g, j). The absorption bands of ZFO and ZFP exhibited a gradual decrease from 1.7 ps to 1000 ps, indicating the reduction in active charge caused by the recombination of shallow electrons and holes22. After 1000 ps, the deceleration in the spectral evolution of charge carriers suggested a relatively sluggish recombination process between holes and deeply trapped electrons23 (Fig. 4h, k). The dynamical decay curves of ZFO NPs were well-fitted by a bi-exponential function, with the two components, τ1 (0.67 ps, 96.78%) and τ2 (250.47 ps, 3.22%). The fastest τ1 lifetime corresponds to hot electrons cooling to the bottom of the conduction band24 and τ2 is ascribed to the band-to-band recombination25. Notably, ZFP NPs exhibited a similar τ1 value (1.19 ps, 82.88%), indicating that the hot electron cooling process was similar to that observed in ZFO NPs. The difference was that the τ2 value (151.36 ps, 17.12%) of ZFP NPs was significantly lower than that of ZFO NPs and that the weight of the process significantly increased26. This obvious change in the process could be attributed to the fact that the ZFP heterojunction promoted the rapid injection of electrons from the trap state into the metal center, thus improving the efficiency of electron separation and transfer27 (Fig. 4i, l). These findings confirmed that the formation of a heterojunction between Pt and ZFO significantly enhanced the separation of electron-hole pairs, thereby augmenting the sonosensitive performance of ZFO NPs.
Synergistic induction of ferroptosis/SDT of ZFPG NPs
Encouraged by the sonosensitive property and multi-enzyme activities of ZFPG NPs, we evaluated the therapeutic efficacy of ZFPG NPs in vitro. All cells were identified by short tandem repeat (STR) and examined for mycoplasma to ensure the quality of all cells before cell experiments. Confocal laser scanning microscope (CLSM) images showed a time-dependent uptake of the Cy5.5-labeled ZFPG NPs, suggesting the efficient internalization of the ZFPG NPs by cancer cells (Fig. 5a, b). To reveal the biocompatibility of ZFP NPs, the cell viability of RWPE-1 cells was assessed after co-incubation with ZFP NPs through a typical CCK-8 assay. The cell survival rate reached 88.24% after 48 h co-incubation at a high concentration of 200 μg mL−1, indicating an acceptable level of biocompatibility for potential clinical applications (Fig. 5c). Moreover, the enzymatic dynamic properties of ZFP NPs were confirmed in Fig. S20. Similarly, the cytotoxicity of free GOx, ZFP, and ZFPG was measured to evaluate whether ZFPG has a cascade catalytic effect. As shown in Fig. S21, the cytotoxicity of ZFPG was much higher than free GOx and ZFP, indicating the cascade catalytic effect of ZFPG. Subsequently, the US-triggered cytotoxicity of ZFPG NPs was evaluated in vitro using various cell lines, mainly including RWPE-1, PC-3, RM-1, 22RV1, DU145, and LNCaP cell lines (Figs. 5d, e and S22). It was noted that the US alone and ZFP treatment had minimal impact on cell viability. Conversely, the groups treated with ZFPG and ZFP + US groups exhibited a significant decrease in cell viability. Notably, the ZFPG + US group demonstrated the strongest cytotoxicity among all these groups, indicating a synergistic anti-tumor effect resulting from ZFPG-mediated enzyme dynamic therapy (EDT) and sonodynamic therapy (SDT). The average cell viability of all prostate cancer cells after ZFPG + US treatment was about 20% compared to 50% in normal prostate cells (RWPE-1). Moreover, the powerful antitumor effects of the ZFPG nano platform were further visualized through fluorescence microscopy using calcein-AM (green fluorescence, Live cells) and PI (red fluorescence, Dead cells) double-staining, which demonstrated a similar tendency to the CCK-8 assay (Figs. 5f and S23 and S24). Additionally, the Annexin-V FITC apoptosis detection kit also confirmed the strongest antitumor effect of the ZFPG + US treatment (Figs. 5g and S25). Scratch assay was further conducted to evaluate whether the ZFPG-mediated therapy could restrain the migration of cancer cells. Fig. 5h illustrated that the ZFPG + US group exhibited the most potent restraining effect, indicating that synergistic CDT/ST/SDT treatment effectively enhanced the inhibition of tumor cell migration.

Endocytosis effect (a) and semiquantitative analysis of intracellular fluorescence intensity (b) after co-incubation of Cy 5.5-labeled ZFPG NPs with PC-3 cells for different times (Red channel: Cy 5.5-labeled ZFPG NPs, blue channel: DAPI, scale bar: 50 μm, n = 3 biologically independent samples, mean ± SD, statistical significance was calculated by one-way ANOVA test); c cell viability of RWPE-1 cells after 24 h and 48 h co-incubation with ZFP NPs (n = 4 biologically independent samples, mean ± SD); d PC-3 and e RM-1 cell viability after six groups of treatment with various concentrations of nanomaterials (G1: control, G2: US, G3: ZFP, G4: ZFPG, G5: ZFP + US, G6: ZFPG + US, n = 4 biologically independent samples, mean ± SD, statistical significance was calculated by one-way ANOVA test); f Live/death staining of PC-3 cells after different treatments (Green channel: Calcein-AM, Red channel: PI, material concentration: 100 μg mL−1, scale bar: 100 μm, n = 3 biologically independent samples with similar results); g flow cytometry apoptosis assay of PC-3 cells after different treatments (material concentration: 200 μg mL−1, G1: Control, G2: US, G3: ZFP, G4: ZFPG, G5: ZFP + US, G6: ZFPG + US); h scratch assay of PC-3 cells after various treatments at different time points (G1: Control, G2: US, G3: ZFP, G4: ZFPG, G5: ZFP + US, G6: ZFPG + US, material concentration: 100 μg mL−1, scale bar: 50 μm, n = 3 biologically independent samples with similar results); i intracellular ROS levels of PC-3 cells were detected by DCFH-DA probe (geen) after different treatments (material concentration: 100 μg mL−1, scale bar: 50 μm, n = 3 biologically independent samples with similar results); and j flow cytometry of intracellular ROS generation of PC-3 cells after various treatment with a material concentration of 100 μg mL−1 (G1: Control, G2: US, G3: ZFP, G4: ZFPG, G5: ZFP + US, G6: ZFPG + US). Source data are provided as a Source Data file.
To investigate the influence of multienzyme activity and sonodynamics of ZFPG NPs on the level of intracellular ROS. A typical 2’,7’-dichlorodihydrofluorescein diacetate (DCFH-DA) probe was employed to visualize the ROS generation in PC-3 cells (Figs. 5i and S26). There was a slight up-regulated green fluorescence signal in the ZFP groups compared to the Control and US groups, which could be attributed to the chemodynamic property of Fe ions released by ZFP NPs. Moreover, the ZFPG and ZFP + US treated cells demonstrated significantly enhanced green fluorescence. Notably, the highest fluorescence intensity was observed in the ZFPG + US group, the ROS content in the ZFPG + US group was 1.61- and 1.35-fold higher than that in the ZFPG and ZFP + US groups, confirming the synergistic effect of multienzyme properties and sonosensitivity in augmenting intracellular ROS levels. Additionally, the flow cytometry technique was employed to quantify the ROS production in PC-3, 22RV1, DU145, and LNCaP cells with different treatments using DCFH-DA staining. The result was consistent with the visual observation of intracellular ROS generation by fluorescence microscope (Figs. 5j and S27 and S28). The significant increase in ROS generation validated the importance of multienzyme activity and sonosensitivity of ZFPG NPs in intracellular ROS modulation.
Mitochondrial membrane potential (MMP) was subsequently monitored with a JC-1 assay kit. Compared to the slight mitochondrial damage upon US irradiation and ZFP treatment, severe damage was observed in the ZFPG, ZFP + US, and ZFPG + US groups. Notably, the ZFPG + US group showed the worst mitochondrial damage due to the synergistic effects of the ZFPG NPs, evidenced by increased JC-1 monomers and reduced JC-1 polymers (Fig. 6a, b). Moreover, the release of cytochrome c (Cyt-C) from mitochondria further validated the effectiveness of the synergistic effect. Fig. 6c, d depicted that the ZFPG and ZFP + US treatments enhanced intracellular Cyt-C release compared with the Control, US, and ZFP groups, while the fluorescence intensity of the ZFPG + US group exhibited 4.12- and 2.24-fold higher than that in the ZFPG and ZFP + US groups respectively, indicating that mitochondria suffered the most severe oxidative damage after the ZFPG + US intervention. Subsequently, we visualized the intracellular GSH levels after various treatments using the Na-8 probe. As shown in Fig. 6e, f, the intracellular GSH level in the ZFP and ZFPG groups significantly decreased in a concentration-dependent manner. There was a lower green fluorescence intensity in the ZFPG group than the ZFP group with the same treated concentration, demonstrating the cascade catalysis effect of ZFPG NPs could enhance the GSH consumption-ability. The GSH-responsive ability resulted in the biodegradation of nanomaterials, Fig. 6g revealed that the ZFPG NPs underwent significant degradation in PC-3 cells within 24 h. There were no undegraded NPs in cells after 5 days of co-incubation, indicating nearly complete degradation of the ZFPG NPs. The exceptional biodegradability of the ZFPG NPs promoted the release of iron ions, thereby facilitating ferroptosis in cancer cells. We further explored the activity of GSH-related GPx4. The western blot results demonstrated the most significant decrease in the expression of intracellular GPx4 protein after the ZFPG (200 μg mL−1) treatment (Fig. 6h). GPx4 is a negative regulator of ferroptosis, so the down-regulation of GPx4 was conducive to LPO accumulation. LPO staining results exhibited a higher LPO accumulation in the ZFPG group than the ZFP group, indicating that the multienzyme activities of ZFPG NPs amplified the LPO accumulation, thereby inducing robust ferroptosis of cancer cells (Fig. 6i, j). Moreover, ZFPG-mediated ferroptosis could effectively destroy mitochondrial morphology. Abnormal mitochondria lacking crista were observed (red arrow), while normal mitochondrial morphology exhibited distinct crista (blue arrow) (Fig. S29).

a Fluorescence images and b intensity of mitochondrial membrane depolarization of PC-3 cells after various treatments with a material concentration of 100 μg mL−1 (G1: Control, G2: US, G3: ZFP, G4: ZFPG, G5: ZFP + US, G6: ZFPG + US, Red channel: JC−1 polymer, Green channel: JC−1 monomer, n = 3 biologically independent samples, mean ± SD, scale bar: 50 μm); c fluorescence images to display the intracellular Cyt-C release in PC-3 cells after different treatments with a material concentration of 100 μg mL−1 (Green channel: Cyt-C, Blue channel: DAPI, scale bar: 50 μm); d quantitative Cyt-C fluorescence intensity (G1: Control, G2: US, G3: ZFP, G4: ZFPG, G5: ZFP + US, G6: ZFPG + US, n = 3 biologically independent samples, mean ± SD, statistical significance was calculated by one-way ANOVA test); e, f fluorescence images and intensity to display the intracellular GSH level in PC-3 cells co-incubated with ZFP and ZFPG NPs for 24 h (scale bar: 50 μm, n = 3 biologically independent samples, mean ± SD, statistical significance was calculated by one-way ANOVA test); g bio-TEM images of ZFPG after co-incubation with PC-3 cells at different time points (white arrows: degraded ZFPG NPs, scale bar: 200 nm, n = 3 biologically independent samples with similar results); h western blot analysis of the expression of GPx4 based on sample processing in PC-3 cells after ZFP and ZFPG treatments with different concentrations (n = 3 biologically independent samples with similar results); and i, j fluorescence images and intensity to display the intracellular LPO level in PC-3 cells co-incubated with ZFP and ZFPG NPs (scale bar: 50 μm, n = 3 biologically independent samples, mean ± SD, statistical significance was calculated by one-way ANOVA test). Source data are provided as a Source Data file.
Enhanced expression of immune signaling molecules through ZFPG NPs
In vitro investigations assessed if ZFPG-induced ferroptosis/SDT enhanced the expression of immune signaling molecules, which could strengthen the antigen presentation of DCs for the immune response against tumors. Surface-exposed calreticulin (CRT) can trigger macrophages and DCs to present antigens to cytotoxic T lymphocytes (CTLs)28. In this regard, the expression of CRT on PC-3 cells following different treatments was visualized and semi-quantitatively analyzed (Fig. S30). The fluorescence images revealed that there was no significant change in the fluorescence intensity of cells treated with the US and ZFP groups compared with the Control group. However, the fluorescence intensity of cells treated with the ZFPG and ZFP + US groups was dramatically increased. Notably, the ZFPG + US intervention demonstrated the highest expression of CRT in cancer cells, which was 2.09- and 1.55-fold of that observed in the ZFPG and ZFP + US groups, respectively, indicating that ZFPG + US intervention was the most effective in inducing CRT expression. Another late ICD biomarker, high mobility group box 1 (HMGB1) protein, was also examined in this study. The fluorescence images showed that increased HMGB1 release was observed in the ZFP, ZFPG, ZFP + US, and ZFPG + US groups, indicating migration of HMGB1 from the nucleus or cytoplasm to the extracellular environment (Fig. S31). During the occurrence of ICD, heat shock protein 90 (HSP90) is exposed on the cell membrane and interacts with CD91 to facilitate the maturation of DCs29. As shown in Fig. S32, HSP90 was significantly upregulated in the ZFPG, ZFP + US, and ZFPG + US groups (increased by 10.27, 13.37, and 19.12 folds, respectively), ascribed to the ZFPG-mediated enhanced thermal damage and oxidative stress from US cavitation. Furthermore, the released adenosine triphosphate (ATP) can bind ionic and metabolic purine receptors on antigen-presentation cells (APCs) to stimulate the maturation and chemotactic induction of APCs30. The release of ATP from dying cells was evaluated by an ATP assay kit. As shown in Fig. S33, the intracellular ATP levels decreased by 31.63%, 48.98%, and 78.64% upon the treatment of ZFPG, ZFP + US, and ZFPG + US, respectively, and this trend was consistent with the increase in extracellular ATP. Overall, ZFPG NPs mediated-ferroptosis/SDT effectively triggered the release and overexpression of DAMPs, potentially enhancing immune stimulatory or subverting immune suppressive effects through the activation of CTLs for sono-metallo-immunotherapy.
Biological mechanism of the ZFPG NPs via RNA sequencing
Androgen-independent PC-3 and DU145 cells and androgen-dependent LNCaP cells after six replicates were treated with PBS and ZFPG + US, and RNA sequencing was employed to investigate the underlying mechanism of ferroptosis/SDT synergistic therapy at the gene level. Compared to the Control group, there were 2207, 6183, and 4705 up-regulated genes and 2931, 6885, and 6091 down-regulated genes in PC-3, DU145, and LNCaP cells treated with ZFPG + US, respectively (Figs. 7a and S34). To further inspect the biological and metabolic pathways in the DEGs of these cells after ZFPG + US treatment, the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway and Gene Ontology (GO) analyses were employed. The KEGG bubble charts in Figs. 7b and S35 revealed that genes related to various pathways, such as MAPK signaling pathway, PI3K-Akt signaling pathway, oxidative phosphorylation, cell homeostasis, ferroptosis, p53 signaling pathway, and glutathione metabolism, significantly changed after ZFPG + US treatment. Additionally, GO analysis confirmed that the DEGs were primarily enriched in biological processes associated with cell cycle, DNA damage, ATP hydrolytic activity, oxidative stress, immune response, and GSH metabolism (Figs. 7c and S36). These results indicated that androgen-dependent and -independent prostate cancer cells showed similar biological and metabolic pathway changes. Gene set enrichment analysis (GSEA) was employed to examine the enrichment of DEGs in gene set units to gain further insight into the biological mechanism of synergistic therapy. The findings revealed that ZFPG + US treatment induced significant gene changes linked to oxidative phosphorylation, antigen processing and presentation, GSH metabolism, and ferroptosis (Fig. 7d). Notably, the protein-protein interaction (PPI) network analysis highlighted the crucial role of the enzyme glyceraldehyde-3-phosphate dehydrogenase (GAPDH) in the interference of the nanosystem with cancer cell glycolysis. It was confirmed that GOx in the nanoparticles effectively consumed energy substrates in the TME (Fig. 7e).

a Volcano plot of DEGs between the Control group and ZFPG + US group (The significance threshold in the volcano plots was set at p < 0.05 and |log2 FC| > 1.2. p-values were adjusted for multiple hypothesis testing and calculated using a negative binomial distribution and the significance test using a two-sided probability); b KEGG term analysis of differential metabolic pathway based on RNAseq after the ZFPG + US treatment (the enrichment analysis of DEGs in KEGG used an overenrichment analysis algorithm, with p-values based on a hypergeometric test); c GO term analysis of differential biological process profiles based on RNAseq after ZFPG + US treatment (The enrichment analysis of DEGs in GO used an overenrichment analysis algorithm, with p-values based on a hypergeometric test); d GSEA enrichment plots of DEGs in the ZFPG + US group; e protein-protein interaction network of the DEGs; f–i heat map of DEGs associated with f oxidative stress, g apoptosis, h ferroptosis, and i immune in PC-3 cells after different treatments; j FASN immunofluorescence staining and fluorescence intensity of PC-3 cells after different treatments (Green channel: FASN, Blue channel: DAPI, scale bar: 50 μm, n = 3 biologically independent samples, mean ± SD, statistical significance was calculated by one-way ANOVA test); k HMOX1 immunofluorescence staining and fluorescence intensity of PC-3 cells after different treatments (Red channel: HMOX1, Blue channel: DAPI, scale bar: 50 μm, n = 3 biologically independent samples, mean ± SD, statistical significance was calculated by one-way ANOVA test). Source data are provided as a Source Data file.
To confirm the specific therapeutic mechanism of ZFPG + US treatment, heat maps of DEGs related to oxidative stress were generated (Figs. 7f and S37). The heat map revealed that the ZFPG + US group exhibited various DEGs, including MAPK, Pi3K-AKT, CCNE1, and other oxidative stress-related genes. It was indicated that ZFPG + US treatment induced severe oxidative stress, which could lead to cell death and changes in related apoptotic pathways. Significant changes in apoptosis-associated genes of these cells were observed after ZFPG + US treatment (Figs. 7g and S38). Up-regulation of BAK1 accelerated mitochondrial voltage-dependent negative ion channel opening, leading to mitochondrial membrane potential loss and Cyt-C release, triggering cancer cell apoptosis31. Additionally, the upregulated CYCS gene could cooperate with BAD to promote apoptosis via the mitochondria-dependent pathway. Moreover, the BAX gene, a proapoptotic member of the Bcl-2 family, enhanced mitochondrial outer membrane permeability, thereby boosting apoptosis32. Conversely, down-regulation of apoptosis suppressor genes CFLAR and BIRC6 activated Capase-9 and SMAC-mediated apoptosis. Therefore, ZFPG + US treatment synergistically triggered various apoptosis-related pathways leading to cancer cell death. Additionally, ferroptosis-related genes were also differentially expressed, among which the up-regulation of GPx4, FTH1, and SLC3A2 might be attributed to the activation of the negative feedback regulation mechanism of cancer cells. Moreover, the up-regulated heme oxygenase-1 (HMOX1) served as a crucial regulatory factor in the ferroptosis process, which could increase transferrin receptors and result in Fe2+ accumulation, thereby facilitating the progression of ferroptosis33. In addition, the ZFPG + US treatment down-regulated the expression of the fatty acid synthase (FASN) gene, which could enhance the occurrence of ferroptosis by altering lipid metabolism and increasing lipid peroxidation (Figs. 7h and S39). The cell staining results further demonstrated that the ZFPG + US treatment could up-regulate the expression of HMOX1 and down-regulate the expression of FASN, respectively (Fig. 7j, k). More importantly, after ZFPG + US intervention, it was also expected to improve the capability of the immune system to recognize and kill cancer cells, as evidenced by immune-related DEG heat maps. The upregulation of HSP90AA1, HSPD1, and HMGB1P6 suggested that the ZFPG + US treatment could induce the ICD of cancer cells. The products encoded by these genes activated the recognition function of immune cells, thereby stimulating both innate immunity and adaptive immunity responses. Besides, the up-regulation of immuno-promoting genes CD81, CD55, and CD24, coupled with the down-regulation of immune escape-promoting genes CD160 further synergistically enhanced the inhibitory effect on cancer cells34 (Figs. 7i and S40). In general, these findings suggested that ZFPG + US treatment effectively disrupted the glycolysis process, reduced intracellular GSH levels, and enhanced intracellular oxidative stress of androgen-dependent and -independent prostate cancer cells, thereby universally inducing immunogenic ferroptosis of prostate cancer cells and stimulating the immune response of the body.
In vivo biodistribution and antitumor efficacy in PC-3 tumor models
The pharmacokinetics of the intravenously injected ZFPG NPs were assessed in PC-3 tumor-bearing male Balb/c nude mice to reveal their in vivo behaviors. We first measured the circulatory half-life (τ1/2) of the ZFPG NPs. The results displayed that the τ1/2 of ZFPG NPs in the body was 3.78 h (Fig. S41). Then the biodistribution of ZFPG NPs in PC-3 tumor-bearing male Balb/c nude mice (n = 3 per group) was investigated using Cy 5.5 fluorescence probes for real-time imaging. Utilizing the superparamagnetism of the ZFPG NPs, the magnetic targeting capability of the NPs was explored by intravenously injecting them (100 µL, 5 mg kg−1) and fixation of NdFeB magnets with a diameter of 1 cm and a thickness of 0.3 cm at the tumor site of each mouse to achieve MF irradiation. The magnetic field strength generated by the NdFeB magnet approximated 2000 gausses. Live imaging revealed a gradual increase in tumor fluorescence over time, with the maximum value reached after 12 h post-injection, indicating that the ZFPG NPs could achieve optimal enrichment at the tumor site after 12 h of MF irradiation. At this time point, the tumor fluorescence intensity in the ZFPG + MF group was significantly greater than that in the ZFPG group, and its fluorescence intensity was 1.74-fold higher than that in the ZFPG group (Fig. 8a, b). Additionally, according to the fluorescence standard curve of Cy-5.5 labeled ZFPG NPs, the maximum enrichment rate of ZFPG at the 12th hour was determined by a fluorescence spectrophotometer as 18.17% ± 2.33% ID/g (Fig. S42). Moreover, ex vivo imaging demonstrated a 2.28-fold increase in tumor fluorescence in the ZFPG + MF group compared to the ZFPG group at 24 h post-injection, confirming the effective magnetic targeting effect of the ZFPG + MF intervention. These results also revealed an off-target effect of ZFPG NPs, with obvious accumulation in the kidney and liver tissues (Fig. S43). The Prussian blue staining results also confirmed this finding (Fig. S44). Furthermore, the in vivo biodistribution of ZFP NPs and ZFPG NPs was assessed by quantifying the iron content (percentage injected dose per gram (% ID/g)) in the tumor and major organs using ICP-MS. As depicted in Fig. 8c, these NPs predominantly accumulated in tumors, liver, and kidney tissues, consistent with the results of fluorescence imaging and Prussian blue staining. Notably, iron content in tumor sites of mice treated with the ZFP + MF and ZFPG + MF groups was significantly higher than that in the ZFP and ZFPG groups, indicating the magnetic targeting ability of ZFP NPs and ZFPG NPs. Exploiting the efficient spin-spin relaxation time (T2) shortening capability of iron oxide analogs, the in vivo T2-weighted MR imaging ability of ZFP, ZFP + MF, ZFPG, and ZFPG + MF after tail vein injection with MF irradiation for 24 h was taken for assessment. Notably, the brightness of tumor sites in mice treated with ZFP and ZFPG was significantly reduced compared with the Control group. More importantly, the brightness of tumor sites in mice treated with ZFP + MF and ZFPG + MF was further reduced (Fig. 8d). According to the gray values determined by the Image J software, the T2 signal intensity of tumor sites in the ZFP + MF and ZFPG + MF groups exhibited a significantly lower level compared to the ZFP and ZFPG groups. This further validated the magnetic targeting ability of NPs and established the potential of these NPs as a promising MRI contrast agent for guiding treatment.

a In vivo imaging of Cy5.5 labeled ZFPG NPs in PC-3-bearing mice with or without MF irradiation (white circle: tumor); b fluorescence intensity of the tumor sites after different treatments at various time points (n = 3 biologically independent samples, mean ± SD, statistical significance was calculated by one-way ANOVA test); c biodistribution of ZFP and ZFPG NPs with or without 24 h of MF irradiation (n = 3 biologically independent samples, mean ± SD, statistical significance was calculated by one-way ANOVA test); d T2-weighted MRI images of mice treated with Control (PBS), ZFP, ZFP + MF, ZFPG, and ZFPG + MF, as well as the gray values of tumor tissues after these treatments (white arrows: tumor, n = 3 biologically independent samples, mean ± SD, statistical significance was calculated by one-way ANOVA test); e the treatment illustration of PC-3 tumor model and experimental design; f body weight curves of mice after different treatments (n = 5 biologically independent samples, mean ± SD); g relative tumor volume curves of mice after different treatments (n = 5 biologically independent samples, mean ± SD); h photograph of tumors after 14 days treatment in different groups (G1: Control, G2: US, G3: ZFP, G4: ZFPG, G5: ZFP + US, G6: ZFPG + US); i representative digital images of H&E, TUNEL, and Ki-67 staining of tumor tissues from different groups (Green channel: TUNEL, Blue channel: DAPI, Brown channel: Ki-67, G1: Control, G2: US, G3: ZFP, G4: ZFPG, G5: ZFP + US, G6: ZFPG + US, scale bars: 50 μm); and j histological changes of major organs after 14 days of treatments were evaluated by typical H&E staining (G1: Control, G2: US, G3: ZFP, G4: ZFPG, G5: ZFP + US, G6: ZFPG + US, scale bar: 100 μm). Source data are provided as a Source Data file.
Inspired by the significant cytotoxicity induced by the ZFPG + US intervention in vitro, we evaluated its anticancer ability in vivo. PC-3 tumor-bearing male Balb/c nude mice were randomly divided into six groups: Control (G1), US (G2), ZFP (G3), ZFPG (G4), ZFP + US (G5), and ZFPG + US (G6) (n = 5 per group), with a tumor volume of 100 mm3. The US irradiation (1.5 W cm−2, 5 min, 1 MHz, 50% duty cycle) was performed on tumors on the first day and the third day after injection of ZFPG NPs (5 mg kg−1) with 24 h MF irradiation, as shown in Fig. 8e. Throughout the treatment process, the body weight and tumor volume of the mice were monitored every two days. Notably, no significant changes in body weight were observed during the 14-day treatment period, confirming the negligible adverse effects of ZFPG NPs (Fig. 8f). The relative tumor volume curve indicated that the ZFPG and ZFP + US groups had a smaller tumor volume compared to the Control, US, and ZFP groups. The ZFPG + US group exhibited the most favorable therapeutic outcome with the smallest tumor volume, confirming the anticipated synergistic effect of the ZFPG + US intervention (Fig. 8g). Further evidence was obtained through digital photographs of solid tumors and tumor weight after the 14-day treatment (Figs. 8h and S45). Moreover, H&E and deoxynucleotidyl transferase-mediated dUTP-biotin nick end labeling (TUNEL) staining of tumor sections showed that the ZFPG + US group exhibited the most significant apoptosis and necrosis. Ki67 antibody staining also confirmed the strongest inhibition of cell proliferation in the ZFPG + US group, further validating the high efficacy of the synergistic treatment induced by ZFPG-mediated ferroptosis/SDT (Fig. 8i). Notably, there were no significant toxicological effects observed in mice after different treatments, as evidenced by blood parameters including blood routine indexes and liver and kidney function (Fig. S46). Further H&E staining assay also displayed no obvious damage in the main organs of mice (Fig. 8j). The comprehensive evaluations demonstrated the good biocompatibility of the ZFPG nanosystems. According to these properties and great therapeutic effects for PC-3 tumor-bearing models, these NPs exhibited great clinical application prospects in treating localized prostate cancer.
In vivo antitumor efficacy and immune activation in RM-1 tumor models
To further verify the reliability of the ZFPG + US treatment on tumor inhibition, we also assessed their efficacy in RM-1 tumor-bearing C57BL/6J male mice (n = 5 per group) following the treatment protocol in Fig. 9a. The stable body weight in each group of mice during the treatment period indicated that the ZFPG NPs had good biocompatibility (Fig. 9b). The ZFPG + US group exhibited a more pronounced therapeutic efficacy than other groups, evidenced by the relative tumor volume curve and tumor weight (Fig. 9c, d). The results further demonstrated the universal and potent tumor-killing ability of the ZFPG + US treatment. We subsequently evaluated their potential to stimulate an anticancer immune response in a unilateral RM-1 tumor model. The CRT and HSP90 levels were upregulated after the ZFPG, ZFP + US, and ZFPG + US treatments, as indicated by the CRT and HSP90 fluorescence staining images of tumor sections after different treatments. The ZFPG + US group showed the most significant fluorescence staining signal, indicating the highest levels of CRT translocation and HSP90 expression compared to other groups (Fig. 9e, g, and h). The translocation of CRT and the overexpression of HSP90 could be attributed to the fact that ZFPG + US-mediated EDT/SDT enhanced the ROS levels within the tumors, resulting in endoplasmic reticulum stress to promote the translocation of CRT to the cell membrane. Moreover, the damaged DNA and sono-cavitation-induced thermal effects could improve the expression of HSP90. Furthermore, the lowest HMGB1 level was observed in the ZFPG + US group, due to the strongest oxidative stress induced by the ZFPG + US treatment (Fig. 9f, h). The release of DAMPs is conducive to augmenting the immune response activation.

a Schematic illustration of the procedures of ZFPG-mediated SDT; b average body weight curves of mice after varied treatments (n = 5 biologically independent samples, mean ± SD); c relative tumor volume curves of mice after varied treatments (n = 5 biologically independent samples, mean ± SD); d average tumor weight after various treatments (G1: Control, G2: US, G3: ZFP, G4: ZFPG, G5: ZFP + US, G6: ZFPG + US, n = 5 biologically independent samples, mean ± SD, statistical significance was calculated by one-way ANOVA test); e–g the slice fluorescence intensity of e CRT, f HMGB1, and g HSP90 after different treatments (G1: Control, G2: US, G3: ZFP, G4: ZFPG, G5: ZFP + US, G6: ZFPG + US, n = 3 biologically independent samples, mean ± SD, statistical significance was calculated by one-way ANOVA test); and h fluorescence images of tumor slices after CRT, HMGB1, and HSP90 immunofluorescence staining (Red channel: CRT and HSP90, Green channel: HMGB1, Blue channel: DAPI, G1: Control, G2: US, G3: ZFP, G4: ZFPG, G5: ZFP + US, G6: ZFPG + US, scale bar: 100 µm). Source data are provided as a Source Data file.
The influence of ZFPG-mediated ferroptosis/SDT on the immune cell response and related cytokine release was investigated. We first assessed the maturation of DCs in vivo, as it is a crucial indicator of stimulating the body’s immune response, the gating strategies are shown in Fig. S47. In the tumor tissues, the percentage of matured DCs in the ZFPG and ZFP + US groups increased to 30.6% and 35.28%, respectively, representing 1.52- and 1.76-fold higher than the Control group (20.07%). Notably, the ZFPG + US group exhibited the most significant increase in matured DCs at 51.94%, surpassing the ZFPG and ZFP + US groups by 1.7- and 1.47-fold (Fig. 10a). Additionally, to validate the activation of the body’s systemic immune response, we measured the number of matured DCs in single-cell suspensions obtained from lymph nodes and spleen using flow cytometry. Following ZFPG + US treatment, the proportion of matured DCs in the lymph nodes and spleen reached 39.95% and 44.36% respectively, marking a 4.39- and 1.75-fold increase compared to the Control group (Figs. 10d, g and S48 and 49). The strongest enhancement of matured DCs in the ZFPG + US group within the body could be attributed to the synergistic effect of ferroptosis/SDT.

Flow cytometry analysis of matured DCs (a), CD8+ T cells (b), and MDSCs (c) in tumors after various treatments (G1: Control, G2: US, G3: ZFP, G4: ZFPG, G5: ZFP + US, G6: ZFPG + US, n = 3 biologically independent samples, mean ± SD, statistical significance was calculated by one-way ANOVA test); matured DCs (d), CD8+ T cells (e), and MDSCs (f) in lymph nodes after various treatments (G1: Control, G2: US, G3: ZFP, G4: ZFPG, G5: ZFP + US, G6: ZFPG + US, n = 3 biologically independent samples, mean ± SD, statistical significance was calculated by one-way ANOVA test); matured DCs (g), CD8+ T cells (h), and MDSCs (i) in the spleen after various treatments (G1: Control, G2: US, G3: ZFP, G4: ZFPG, G5: ZFP + US, G6: ZFPG + US, n = 3 biologically independent samples, mean ± SD, statistical significance was calculated by one-way ANOVA test); the levels of IFN-γ (j) and TNF-α (k) in the tumor after different treatments (G1: Control, G2: US, G3: ZFP, G4: ZFPG, G5: ZFP + US, G6: ZFPG + US, n = 3 biologically independent samples, mean ± SD, statistical significance was calculated by one-way ANOVA test); l schedule of the prostate cancer lung metastasis experiment; m fluorescence images of metastatic lesions in Luc-RM-1 tumor-bearing black mice after different treatments; n representative images of the Bouin’s Fixative-stained whole lungs (white arrows point to the lesions); and o representative H&E-stained lung tissue sections (G1: Control, G2: US, G3: ZFP, G4: ZFPG, G5: ZFP + US, G6: ZFPG + US). Source data are provided as a Source Data file.
Matured DCs exhibit a robust antigen delivery capability, which contributes to the activation of cytotoxic T cells within the body35. The elevated presence of CD8+ T cells within the tumor represents the most direct evidence of immune response activation, the gating strategies of CD8+ T cells are shown in Fig. S50. As shown in Fig. 10b, the proportion of CD8+ T cells in tumors treated with the ZFPG and ZFP + US groups was increased to 15.61% and 17.58%, respectively, which were 2.01- and 2.26-fold higher than that in the Control group. Remarkably, the highest contents of CD8+ T cells were observed in the ZFPG + US group (25.97%), exhibiting 1.66- and 1.48-fold higher than that in the ZFPG and ZFP + US groups, respectively. It was suggested that ZFPG-mediated synergistic ferroptosis/SDT was more effective than any single therapy in inducing the infiltration of CD8+ T cells within the tumor. The contents of CD8+ T cells in the lymph nodes and spleen were also evaluated. Compared with other groups, the mice treated with the ZFPG + US group exhibited a higher proportion of CD8+ T cells in both the lymph nodes and spleen, which reached 29.04% and 9.0%, respectively (Figs. 10e, h and S48 and 49). These findings demonstrated that ZFPG + US treatment effectively augmented the infiltration of CD8+ T cells within the tumor, thereby achieving potent sono-metallo-immunotherapy.
Moreover, our research demonstrated the significant impact of the treatment method on the number of MDSCs in the tumor (Fig. S51). Compared to the Control group (38.21%), there was no obvious change in the number of MDSCs after the US (36.84%) and ZFP (34.38%) treatment. Conversely, the ZFPG and ZFP + US groups demonstrated a significant reduction in the proportion of MDSCs in tumors, indicating that these two treatments could effectively mitigate the immunosuppressive TME. Moreover, the MDSCs proportion of tumors following the ZFPG + US treatment (11.89%) was significantly lower than that of the ZFPG (27.36%) and ZFP + US (24.56%) treatments, indicating that the ZFPG + US treatment was the most valid in alleviating the immunosuppressive TME (Fig. 10c). Furthermore, the proportion of MDSCs in the lymph nodes (16.17%) and spleen (3.21%) after the ZFPG + US treatment was much lower than those in the Control group (Figs. 10f, i and S48 and 49). These findings indicated that the sono-activated ZFPG NPs could effectively decrease MDSC levels in the tumor and body, thus alleviating the immunosuppressive TME and enhancing the infiltration and activity of CD8+ T cells in tumors36. Furthermore, some immune-related cytokines are crucial in the anti-tumor immune-killing effect. The tumor levels of cytokines were evaluated to further confirm the activation of the immune response37. As shown in Fig. 10j, the ZFPG + US group exhibited the highest level of IFN-γ, which was 1.89- and 1.27-fold higher than that in the ZFPG and ZFP + US groups, respectively. Additionally, the TNF-α in the ZFPG + US group exhibited a peak level, representing a 5.31-fold increase compared to the Control group (Fig. 10k). The obtained results further validated the synergistic effect of ZFPG + US treatment in effectively enhancing cytokine secretion for efficient sono-metallo-immunotherapy. Therefore, the ZFPG immunomodulator could effectively activate the body’s immune response.
To simulate a more realistic biological environment, we utilized Luc-RM-1 cells to construct a C57BL/6J orthotopic prostate cancer model to evaluate these interventions’ therapeutic and immune effects. Figure S52a shows a diagrammatical timeline of the experimental program and implementation. During the treatment process, imaging analysis was performed on each group of mice using the IVIS imaging system on Days 0, 7, and 14 to demonstrate the successful construction of the orthotopic prostate cancer model and to evaluate the tumor growth trend in each group of mice. As shown in Fig. S52b, the fluorescence intensity of the ZFPG and ZFP + US groups was lower than the Control group, indicating that EDT and SDT effectively inhibited the growth of orthotopic prostate tumors. Notably, the ZFPG + US group showed the weakest fluorescence intensity, indicating that the ZFPG + US-mediated combination therapy showed the strongest therapeutic effect. The removed prostate tumor tissue was imaged and weighed at the end of the treatment, and the results were consistent with tumor growth trends (Fig. S52c, d). In addition, we performed H&E, TUNEL, and Ki-67 staining of prostate cancer tissues to further evaluate the therapeutic effects of these interventions (Fig. S52e). Compared with other groups, the ZFPG + US group showed the strongest cancer cell necrosis and apoptosis, as well as the lowest Ki-67 expression, suggesting that ZFPG + US treatment could inhibit cancer cell proliferation and induce cell apoptosis to improve the anti-tumor effect.
To assess the immune effects induced by these interventions, we analyzed mature DCs, CD8+ T cells, and MDSCs in orthotopic prostate tumors after different treatments. As shown in Fig. S53, compared with the Control, US, and ZFP groups, the proportion of mature DCs and CD8+ T cells in the orthotopic prostate tumors after ZFPG, ZFP + US, and ZFPG + US treatments were significantly up-regulated. Among these groups, the ZFPG + US group exhibited the highest proportion of mature DCs (39.78%) and CD8+ T cells (19.05%). Conversely, tumors treated with ZFPG + US exhibited the lowest MDSCs infiltration, which was 59.25% and 66.71% of the ZFPG and ZFP + US groups, respectively. These results again demonstrated that ZFPG + US-mediated combination therapy significantly induced DC maturation and alleviated the immunosuppressive TME, thereby enhancing antigen presentation ability and cytotoxic T lymphocyte infiltration at the tumor site. This ultimately achieves efficient sono-metallo-immunotherapy. Additionally, as the kidney is the main metabolic pathway for ZFPG NPs, we analyzed whether these interventions induced immune responses in the kidneys. As shown in Fig. S54, the ZFP and ZFPG groups did not induce an immune response while ZFP + US and ZFPG + US treatments slightly upregulated the proportion of immune cells in the kidneys, which might be attributed to the activation of the immune system of the body. These results indicated that the short-term enrichment of nanomaterials in the kidney did not elicit a severe renal immune response accompanied by organ damage. Notably, due to the differences in the rate of development, tumor phenotype, and degree of heterogeneity between mouse-derived and human-derived prostate cancer, there may be some differences in the immunotherapy results induced by ZFPG NPs in human-derived prostate cancer. The immune-activating effect of ZFPG NPs in human-derived prostate cancer still needs to be further explored.
The metastasis of malignant tumors is one of the difficulties in oncotherapy. Considering that the two-pronged EDT/SDT mediated by ZFPG + US could effectively activate anti-tumor immunity, we utilized Luc-RM-1 cells to establish lung colonization models to evaluate whether these interventions could inhibit the metastasis of prostate cancer to the lung, the experimental procedure followed the illustration depicted in Fig. 10l. The IVIS system was employed to observe lung metastasis in mice subjected to different treatments on Days 14 and 21, revealing that the ZFPG, ZFP + US, and ZFPG + US treatments could suppress prostate cancer metastasis to the lungs. Notably, the ZFPG + US treatment exhibited the most potent inhibition of metastasis, suggesting that the ZFPG + US mediated sono-metallo-immunotherapy effectively stimulated the body’s immune response, thereby impeding cancer cell metastasis within the body (Fig. 10m). The results of Bouin’s Fixative staining and H&E staining revealed that following intravenous injection of Luc-RM-1 cells, a significant number of lung lesions were observed in the mice treated with Control, US, and ZFP. Conversely, the mice treated with ZFPG, ZFP + US, and ZFPG + US exhibited markedly reduced lung lesions. Notably, no obvious lesions were detected in the lungs of the mice treated with ZFPG + US (Fig. 10n, o). The remarkable findings once again validated the efficacy of the ZFPG + US treatment in activating the immune response of the body, thereby reducing the formation of metastatic lesions and enhancing the killing and inhibitory effects on advanced prostate cancers. Consequently, this treatment holds promise for prolonging advanced patient survival.
In summary, we developed a biodegradable ZFPG nanozyme with magnetic targeting and MR imaging capability for multienzyme-induced sono-metallo-immunotherapy. The ZFPG NPs could be selectively accumulated at the tumor site under MF irradiation and trigger a cascade of catalytic reactions to deplete intracellular substrates and enhance multiple bursts of ROS. Moreover, the ZFP heterojunction promoted the rapid injection of electrons from ZFO to Pt upon US irradiation, which enhanced the electron and hole separation and transfer efficiency and facilitated the generation of 1O2 radicals. The synergistic multienzyme effect and sono-catalytic performance of ZFPG nanomedicine effectively induced the up-regulation of the HMOX1, which could facilitate the Fe2+ accumulation and GPx4 inactivation mediated-excessive accumulation of LPO, ultimately leading to efficient ferroptosis/SDT. More importantly, ferroptosis/SDT effectively enhanced the release and expression of cancer cell immune signaling molecules, thereby inducing the maturation of DCs and enhancing their antigen-presenting ability. Meanwhile, ZFPG NPs combined with US treatment reduced the expression and invasion of MDSCs and enhanced the release of immune-related cytokines to alleviate the immunosuppressive TME. Ultimately, CD8+ T cells effectively infiltrated into the tumor and achieved robust sono-metallo-immunotherapy to inhibit tumor growth and metastasis. This study displayed a paradigm for the rational design of a nanozyme immunomodulator for sono-metallo-immunotherapy.
Methods
Ethics approval and consent to participate
The animal care and treatment protocols used in this paper follow the Guiding Principles for Animal Care and Use (China) and have been approved by the Ethics Committee of Shandong Provincial Hospital (approval no.: 2022-130). The maximal tumor burden permitted by the Ethics Committee of Shandong Provincial Hospital is 2000 mm3. All the tumor burden in our experiment did not exceed this criterion. The patients’ samples were used under the guidelines approved by the Ethics Committee on Biomedical Research involving Human Subjects of Shandong Provincial Hospital (approval no. SWYX2024-183). This research has obtained written informed consent from all participants.
Materials
FeCl3·6H2O (99%), ZnCl2 (99.95% metals basis), C6H5Na3O7·2H2O (99%), NaOAc·2H2O (98%), Ethylene glycol (EG, 99%), H2PtCl6·6H2O (Pt ≥ 37.5%), Hydrogen peroxide (H2O2, 30%), d-(+)-Glucose (99.99%), Reduced Glutathione (GSH, 99%), Cyanine5.5 amine (95%), 5,5’-Dithio bis-(2-nitrobenzoic acid) (DTNB, 98%) were obtained from Shanghai Macklin Biochemical CO., LTD. NaBH4 (≥98%) was obtained from Sinopharm Chemical Reagent Co., Ltd. 1,3-diphenylisobenzofuran (DPBF, 97%), glucose oxidase (GOx, G109029, >180 U/mg), N-(3-Dimethylaminopropyl)-N’-ethylcarbodiimide (EDC, 95%), N-Hydroxysuccinimide (NHS, 99%), 3,3’,5,5’-Tetramethylbenzidine dihydrochloride (TMB, >99.0%) were obtained from Shanghai Aladdin Bio-Chem Technology Co., LTD. Silane-PEG2000-COOH (99%) was obtained from Xi’an Ruixi Biological Technology Co., Ltd. Trypsin-EDTA (0.05%, containing phenol red) was purchased from Thermo Scientific (USA). F12K medium was purchased from Beijing Jinming Biotechnology Co., Ltd. JC-1 probe, Calcein-AM/PI Cell Viability/Cytotoxicity Assay Kit, ROS Assay Kit, and Cell lysate was purchased from Beyotime (China). DAPI, Triton X-100, and Seal with goat serum were purchased from Solarbio (China). Na-8 fluorescent probes were purchased from J&K Scientific (China). Annexin V-FITC Apoptosis Detection Kit was purchased from Boster (China). Fluorescein (FITC) Tunel Cell Apoptosis Detection Kit and Hematoxylin & Eosin dye were purchased from Wuhan Sevicebio Technology Co., Ltd. (China). Collagenase Type 2 and DNase I were purchased from Sigma (USA). ATP content determination kits were purchased from APPLYGEN (Beijing, China). Mouse IFN Gamma and TNF-alpha ELISA Kits were purchased from BOSTER (China) and ABclonal (China), respectively. The antibodies used for western blotting, immunohistochemistry, immunofluorescence staining, and flow cytometry were provided in Supplementary Table 1.
Characterization
Transmission electron microscopy (HT-7700, FEI Titan3 Cubed 60–300, and HITACHI) and scanning electron microscopy (Hitachi, SU-70, Japan) were used to reveal the morphology of the nanoparticles, providing detailed insights into their structural features. To confirm the surface composition of ZFPG NPs, X-ray photoelectron spectroscopy (Thermo Scientific K-Alpha, USA) was employed. Structural analysis was carried out using X-ray diffraction (XRD) patterns, obtained with a Rigaku X-ray diffractometer (MiniFlex600, Japan) utilizing Cu Kα radiation (wavelength: 1.5406 Å). Optical properties were assessed through UV-vis spectroscopy using a SPECORD 200 PLUS instrument (Jena, Germany). The surface charge and stability of the NPs were evaluated via zeta potential analysis on NanoBrook 90PlusPALS (Brookhaven, USA). Additionally, confocal laser scanning fluorescence microscopy (CLSM; LSM710 and LSM880, ZEISS, Germany) was used to capture high-resolution fluorescence images, while fluorescence microscopy images were obtained using an optical microscope (SZ51, BX5, CX21, Olympus, Japan). CytoFLEX S flow cytometer (Brea, CA, USA) was used to collect flow cytometry data. Cell viability data was collected using a lnfinite M200PRO plate reader (Tecan, Swiss). In vivo biodistribution fluorescence images were obtained IVIS Spectrum system (PerkinElmer). The concentration of Fe was determined by the ICP-OES (PerkinElmer 8300).
Synthesis of ZnFe2O4 nanoparticles (ZFO NPs)
First, 2.703 g FeCl3 6H2O, 0.681 g ZnCl2, 0.8 g trisodium citrate, and sodium acetate (7.2 g) were dissolved in 80 mL of glycol. The resulting yellow solution was then transferred to a 100 mL polytetrafluoroethylene high-pressure reactor. It was heated at 200 °C for 12 h. Afterward, the solution was allowed to cool naturally, and then collected these nanomaterials through centrifugation (4000 g, 10 min). Subsequently, the collected nanomaterials were re-dispersed in 50 mL anhydrous ethanol and deionized water, respectively, and centrifugated at 4000 g for 10 min. The process was repeated three times to purify the nanomaterial. Finally, the solution was re-dispersed in ultra-pure water in preparation for further use.
Synthesis of ZnFe2O4@Pt-PEG nanoparticles (ZFP NPs)
Twenty-five milligrams of ZFO NPs were dispersed in 10 mL ultra-pure water, and then 5 mg H2PtCl6·6H2O was dispersed in the aforementioned solution under the assistance of ultrasound. Following this, excess ice NaBH4 solution was gradually added drop by drop (50 μL) into the resulting solution and reacted for 30 min under the action of ultrasound. Subsequently, the material was centrifugally (4000 g, 10 min) collected and washed three times using ultra-pure water. The collected material was then re-dispersed in a mixture of 25 mL ethanol and water (VE:Vw = 1:1), followed by adding 50 mg of Silane-PEG2000-COOH and stirred at room temperature for 8 h. Finally, the material was centrifugally (4000 g, 10 min) washed with ultra-pure water (25 mL) three times to purify the materials for further use.
Synthesis of ZnFe2O4@Pt@PEG-GOx nanoparticles (ZFPG NPs)
Twenty-five milligrams of ZFP NPs were dispersed in a 30 mL PBS buffer solution. Then, 25 mg EDC and NHS were added respectively. The nanomaterials were activated for 10 min in dark light. Next, 5 mg glucose oxidase (GOx) was added, and the reaction was continued for 2 h in dark light. The nanomaterials were centrifugally (4000 g, 10 min) washed with ultra-pure water (25 mL) three times to purify the materials for further use. The final harvested nanomaterials were stored at 4 °C for subsequent experiments.
Thermogravimetric analysis
Take 10 mg ZnFe2O4@Pt NPs, ZFP NPs, and ZFPG NPs in a small crucible, respectively. Then, non-isothermal thermogravimetric analysis (TGA) was performed on the above three materials. The temperature range was set to 40–850 °C, and the heating rate was 10 K/min. TG curves of the three materials were drawn according to the experimental results of TGA, which reflected the change of material mass with temperature, thereby obtaining the modified content of each component on the surface of the nanomaterials.
The POD-like activity of ZFO NPs and Kinetic Assay
The POD-like activity of ZFO NPs (100 μg mL–1) was evaluated using TMB (250 μg mL−1) as substrates in the presence of H2O2 with different concentrations (final concentration 0 mM, 0.25 mM, 0.5 mM, 1 mM, 2 mM, and 4 mM). The NaAc buffer solution (20 mM, pH = 5) was used as a solvent to disperse the above reagents. A UV-vis spectrophotometer was used to assess the activity by recording the variation of absorbance at 652 nm every 30 s for 10 min.
DFT procedure
The calculation of DFT was conducted by employing the VASP 6.1 to investigate the decomposition process of H2O2 on the surface of Fe3O4 and ZnFe2O4. The calculations employed the Projector Augmented Wave (PAW) method with a 400 eV cutoff energy, combined with the Perdew–Burke–Ernzerhof (PBE) function to describe the exchange-correlation effects. To account for van der Waals interactions, the DFT-D3 method was applied as a correction. Surface structures were constructed by cleaving two layers of Fe3O4 (111) and ZnFe2O4 (111) facets, separated by a vacuum layer of 15 Å. The structural models were thoroughly relaxed, adhering to a 10−5 eV energy convergence criterion and a 0.02 eV/Å force convergence criterion. Additionally, the precision of the K-point mesh in the Brillouin zone was set to 0.04 × 2π/Å to ensure accurate calculations. The adsorption energy (Eads) was determined using Formula 1, providing insights into the interaction between H2O2 and the catalyst surface.
The energies Eadsorbate, Esubstrate, and Etotal correspond to the adsorbate, substrate, and adsorption structure, respectively. The thermodynamic data at various temperatures were determined by calculating the free energies using Formula 2,
The G, ZPE, TS, and EDFT denote the free energy, zero point energy, entropic contributions, and calculated DFT energy.
The CAT-like activity assay of ZFP NPs
ZFO and ZFP NPs (200 μg mL−1, final concentration) were added into H2O2 solution with a final concentration of 10 mM, and the dissolved oxygen in the solution was recorded by an oxygen dissolving instrument every 30 s for 5 min.
The OXD-like activity assay of ZFP NPs
ZFO and ZFP NPs solution (100 μg mL−1, final concentration) were co-incubated with TMB solution (250 μg mL−1, final concentration) in PBS buffer solution (pH = 5.5, 0.1 M) for 1 h, respectively, and then UV-vis was used to determine whether TMB was oxidized to oxTMB, thereby determining the OXD-like activity of the materials.
The GOx activity assay of ZFG NPs
(a) The cascaded catalytic effect verifies the generation of H2O2. The ZFO NPs, H2O2 and TMB were dissolved and diluted with PBS buffer solution (pH = 5.5, 0.1 M), respectively. ZFO NPs (final concentration 100 μg mL−1) were co-incubated with H2O2 (final concentration 0.5 mM) and TMB (final concentration 250 μg mL−1) for 30 min, and the absorption curve of ZFO + H2O2 + TMB group was recorded by UV-vis ultraviolet spectrophotometer. Then ZFG NPs (final concentration 100 μg mL−1) were co-incubated with glucose (final concentration 1 mg mL−1) and TMB (final concentration 250 μg mL−1) in a PBS buffer solution (pH = 5.5, 0.1 M) for 30 min. The absorption curves of the ZFG + Glu + TMB group were recorded by UV-vis ultraviolet spectrophotometer. Finally, the absorption peaks of the ZFO + H2O2 + TMB group and the ZFG + Glu + TMB group were similar, indicating that GOx could effectively oxidize glucose to H2O2. (b) Gluconic acid production is assessed by measuring the pH of the solution system. Different concentrations of ZFPG NPs (final concentration 0 μg mL−1, 50 μg mL−1, 100 μg mL−1, 150 μg mL−1, and 200 μg mL−1) were incubated with glucose solution (final concentration 1 mg mL−1) for 3 h, and then the pH value of the solution was detected by pH meter. In addition, by co-incubating ZFPG NPs (final concentration 100 μg mL−1) with different concentrations of glucose solution for 3 h, the pH value of the solution was detected by pH meter.
ESR measurements
(a) •OH detection: 200 μg mL−1 ZFO and ZFP nanozyme was added into NaOAc/HOAc buffer (pH = 4.5) containing 10 mM H2O2 and DMPO upon addition of SOD to remove •O2– from the reaction. The •OH determination of ZFPG in glucose (Glu) solution followed the same procedure, except that H2O2 was replaced by Glu solution (5 mM). (b) •O2– detection: 200 μg mL−1 ZFO, ZFP, and ZFPG nanozyme was added into NaOAc/HOAc buffer (pH = 4.5) containing 1 mM H2O2 and BMPO upon addition of tertiary butanol to remove •OH from the reaction.
The GSHOx-like activity assay of ZFPG NPs
(a) ZFPG NPs concentration-dependent GSH depletion. GSH solution (final concentration 3 mM) was fully mixed with ZFPG NPs solution with different concentrations (0–200 μg mL−1) for 6 h, and 2 mL of the above-mixed solution was centrifuged. The supernatant was collected, then DTNB (10 μL, 10 mg mL−1) was added to the above-collected supernatant. The absorbance curves at different concentrations were recorded by UV-vis spectrophotometer after incubation for 10 min. (b) Time-dependent GSH depletion. GSH solution (final concentration 1 mM) was thoroughly mixed with ZFPG NPs solution (final concentration 200 μg mL−1) at different times. 2 mL of the above-mixed solution was centrifuged at 0 h, 1 h, 3 h, 5 h, 7 h, 10 h, and 24 h, respectively, and the supernatant was collected, and DTNB (10 μL, 10 mg mL−1) was added to it for 10 min. The absorbance curves of different time points were recorded by UV-vis spectrophotometer.
Measurement of 1O2 generation
The 1O2 generation from the US irradiation-activated ZFO and ZFP was detected by 1,3-diphenylisobenzofuran (DPBF). Typically, DPBF (10 μM) was added into the ZFO, ZFP, and ZFP + H2O2 dispersion (final material concentration 200 μg mL−1, final H2O2 concentration 1 mM), followed by US irradiation (1.0 MHz, 1.5 W cm−2) for different durations. The UV-vis absorbance of DPBF at 425 nm was then detected. As controls, the 1O2 generation in the presence of DPBF in PBS with US irradiation was also tested for comparison. The ESR measurements involved exposing a mixture solution of 20 μL TEMP, 1 mL ZFP with a concentration of 200 μg mL−1, and 1 mL H2O2 with a concentration of 10 mM to US irradiation for 2 min. The signal of 1O2 was then detected by an ESR spectrometer. Additionally, ESR spectra of US, ZFO, and ZFP were detected under the same conditions without the addition of H2O2.
Cell culture
PC-3 (Cat: CL-0185), LNCaP (Cat: CL-0143), and 22RV1 (Cat: CL-0004) cell lines were bought from Wuhan Procell Life Technology Co., LTD. DU145 (Cat: CX0116) and RM-1 (Cat: CX0051) cell lines were purchased from Boster Biological Technology Co.,Ltd. Luc-RM-1 (Cat: YC-C049-Luc-P) cell line was purchased from Ubigene Biological Technology Co., Ltd. RWPE-1 cell line (Cat: ZQ0351) was bought from Zhong Qiao Xin Zhou Biotechnology Co., Ltd. PC-3 cells were cultured in an F12K medium. DU145 cells were cultured in a MEM medium. RWPE-1 cells were cultured in a keratinocyte medium. RM-1, LNCaP, and 22RV-1 cells were cultured in 1640 medium. All the mediums contained 1% P/S and 10% FBS.
Animals
The 4-week-old healthy male BALB/c nude mice and C57BL/6J black mice, both weighing around 18 g, were obtained from Charles River Laboratories. and kept in the pathogen-free animal room. The animals were kept in ventilated cages and exposed to light for around 10 h. The ambient temperature was controlled at 22 °C, and the humidity was controlled between 50% and 60%.
In vitro cellular uptake of ZFPG NPs
PC-3 cells were inoculated into a laser confocal plate with 6 × 104 cells per plate overnight. Then the medium was replaced with 1 mL Cy 5.5-labeled ZFPG NPs solutions (100 μg mL−1) dispersed in F12K medium and keratin medium, respectively. After co-incubation for different times (0 h, 3 h, 6 h, 9 h, and 12 h), the cells were washed with PBS solution, and DAPI staining solutions (1000 μL dish−1) were added. 30 min later, the cells were photographed with a CLSM.
Cell viability
First, RWPE-1 cells seeded in a 96-well plate at a density of 1 × 105 cells per well were cultured for 24 h. Subsequently, different concentrations of ZFP NPs in the keratinocyte culture medium were added into each well to replace the existing cell culture media. The cells were then incubated for an additional 24 h and 48 h. Following the incubation period, cell viability was assessed using the CCK-8 assay.
The therapeutic efficacy of different materials in cancer cells
Cancer cells were transfected with ZFP and ZFPG NPs (50–200 μg mL−1) for 12 h, and then subjected to 2 min US irradiation (with a power density of 1.5 W cm−2 and a frequency of 1 MHz) or left without irradiation. As control groups, cancer cells treated with PBS and 2 min US irradiation were employed. Subsequently, the cell survival rate was assessed using the CCK-8 method.
Staining of live/dead cells
The concentration of materials used for treating cancer cells was 100 μg mL−1, following the same steps as described above. Subsequently, the cancer cells were incubated with Calcein-AM and propidium iodide. The fluorescence microscope was then employed to observe the apoptosis of the cells. For Calcein-AM, the excitation wavelength was 488 nm, with an exposure time of 200 ms and a gain of 9.3×. On the other hand, the excitation wavelength for PI was adjusted to 594 nm, with an exposure time of 400 ms and a gain of 9.3×.
Flow cytometry analysis
In simple terms, Cancer cells were treated with the above six groups. After treatment, cells were collected and washed with PBS to remove the residue in the cell medium. After being digested by trypsin without EDTA and centrifuged, the cells were collected and washed three times with PBS. Annexin V-FITC and PI were used to stain the cells for 30 min. The samples were then analyzed on a Beckman Coulter Inc. According to the fluorescence signal, the cells were divided into healthy cells, apoptotic cells, and necrotic cells, and the apoptosis rate was calculated.
Effect of ZFPG on proliferation at the cellular level
The PC-3 cells were inoculated into a six-well plate at the rate of 1 × 105 per well. After the cells were overfilled and treated in the above groups for 9 h, the 200-microliter gun tip was used to mark straight lines in the middle of each hole and fix the recording position, and the PBS was washed until there were no floating cells, and then the serum-free F12K was added for culture. The cell migration was recorded after 0 h, 6 h, 12 h, and 24 h.
Detection of intracellular ROS through DCFH-DA probe
Based on the experimental grouping described above, the production rate of ROS was detected using the DCFH-DA probe. After US treatment for 2 min (1.5 W cm−2, 1 MHz, duty ratio 50%), the original culture medium was replaced with a free-medium-included DCFH-DA fluorescent probe (1:1000) and incubated at 37 °C for 30 min. The excess DCFH-DA fluorescent probe was removed by washing the cells three times with a free F12K medium. Finally, the fluorescence microscope was utilized to measure ROS levels in each group. It was set at an excitation wavelength of 488 nm, an exposure time of 350 ms, and a gain of 1.2×.
MMP measurement
MMP changes in PC-3 cells were detected using JC-1 probes. Typically, log-growing cancer cells were inoculated in a 24-well plate (5 × 104 well−1). Then 1 mL ZFP and ZFPG solutions (100 μg mL−1) dispersed in F12K medium were replaced the medium. After co-culture for 12 h, the cells were washed through PBS buffer solution, and the cells were treated as follows: (1) Control, (2) US, (3) ZFP, (4) ZFPG, (5) ZFP + US, and (6) ZFPG + US. The US intensity was 1.5 W cm−2, the frequency was 1.0 MHz, the duty ratio was 50%, and the US time was 2 min. After various treatments, the medium was then incubated for 3 h and replaced with 1 mL JC-1 staining solution. Finally, the cells were cultured at 37 °C and sheltered from light for 30 min, then washed with PBS. The changes in intracellular fluorescence of the JC-1 probe were observed by fluorescence microscope. The excitation wavelength of the JC-1 monomer was 488 nm, the exposure time was 300 ms, and the gain was 1.2×. The excitation wavelength of the JC-1 polymer was 594 nm, the exposure time was 300 ms, and the gain was 7.4×.
The detection of cytochrome C (Cyt-C) release
Immunofluorescence was used to detect the release of Cyt-C in the study. The cells were first treated with the specified group and treatment, then fixed with paraformaldehyde and permeabilized using 0.03% Triton X-100. Subsequently, the cells were co-incubated overnight with an anti-cytochrome c antibody, followed by a 2-h incubation with an Alexa Fluor 488 conjugated secondary antibody. Excess fluorescent secondary antibodies were removed by washing the cells with PBS before staining them with a DAPI staining solution for 10 min. Then employing fluorescence microscope to observe the cells. The DAPI excitation wavelength was 405 nm, with an exposure time of 30 ms and a gain of 0×. On the other hand, the excitation wavelength for the Alexa Fluor 488 conjugated secondary antibody was 488 nm, with an exposure time of 200 ms and a gain of 9.3×.
The GSH depletion assay of ZFPG NPs in PC-3 cells
ZFP and ZFPG were diluted to 0 μg mL−1, 100 μg mL−1, and 200 μg mL−1 with F12K medium, respectively. The Na-8 probe was applied to detect the intracellular concentration of GSH. After processing, the cells were washed with PBS and stained for 30 min with an F12K contained Na-8 probe (1:1000) sheltered from light at room temperature. The changes were cached by a fluorescence microscope. Excitation wavelength: 488 nm, Exposure time: 400 ms, gain: 0×.
GPx4 inhibition assay through western blot
PC-3 cells at the logarithmic growth phase were seeded in a 6-well plate overnight and allowed to adhere. After reaching confluence, cells were grouped and treated as aforementioned. The cells were washed twice with PBS, then lysed on ice for 30 min with RIPA buffer (1:100) containing PMSF, and then centrifuged at high speed. We collected the supernatant and used the BCA assay to determine the protein concentration. The sample was then heated to denature it. Western blotting was performed using a PVDF membrane to obtain the loaded protein. Co-incubated overnight with an anti-GPx4 antibody. After staining, the expression of proteins in different groups was analyzed.
The ZFPG mediated-LPO accumulation assay in PC-3 cells
The Liperfluo-Lipid peroxide probe was used to detect the accumulation of lipid peroxide. The process is the same as GSH detection. The excitation wavelength of the fluorescence microscope was set to 488 nm, the exposure time was 500 ms, and the gain was 0×.
ZFPG-induced immunogenic cell death (ICD) of PC-3 cells under US irradiations in vitro
ZFPG-induced ICDs were identified by in vitro detection of calreticulin (CRT) transposition, HMGB1 release, ATP release, and HSP90 expression. To detect these ICD markers, PC-3 cells were inoculated into a 24-well plate at a density of 5 × 104 cells per well and incubated 12 h. Then ZFP and ZFPG (100 μg mL−1) solution dispersed in the F12K medium was replaced with the previous medium and incubated for 24 h. Following this, the cells were irradiated with US for 2 min, and further incubated for 3 h. Subsequently, the cells were fixed with 4% paraformaldehyde treated with 0.03% triton X-100 and then blocked with 30% goat serum. The cells were incubated with anti-CRT, HMGB1, and HSP90 antibodies, and then added secondary antibodies for a 2-h further incubation. The nuclei were stained with DAPI for 10 min. Finally, a Fluorescence microscope was utilized to observe the samples. The excitation wavelength for DAPI was 405 nm, with an exposure time of 30 ms and a gain of 0×. For the secondary antibody labeled in green, excitation wavelength: 488 nm, exposure time: 300 ms, gain: 6.4×. Lastly, the excitation wavelength for the secondary antibody (red) was 594 nm, with an exposure time of 400 ms and a gain of 0×.
The ATP release was detected by the ATP kit. In simple terms, PC-3 cells at a density of 105 per well were seeded in a 6-well plate overnight and allowed to grow until confluence. After that, ZFP and ZFPG (100 μg mL−1) diluted with blank F12K were added into each well and co-cultured for 6 h. Subsequently, US irradiation was conducted on the cells. Three hours later, the supernatant was collected directly as the sample for detection. According to the instructions of the ATP detection kit (APPLYGEN, Beijing, China), a standard solution and ATP detection working solution were prepared, and the ATP content in the supernatant of the samples was calculated using the standard curve drawn by the standard solution according to the results obtained from the microplate reader.
Biodistribution
Cy 5.5-labeled ZFPG NPs were injected into the tail vein of PC-3 tumor-bearing male nude mice, and fluorescence imaging was performed on mice in the Control (PBS), ZFPG, and ZFPG + MF group at different time points through the IVIS Spectrum system (PerkinElmer), respectively. The dissected mouse tumor tissues and major organs were also imaged using the IVIS Spectrum system (PerkinElmer) to observe the distribution of nanomaterials. The unit for the IVIS imaging is photons emitted per second per centimeter square per steradian (p/sec/cm2/sr).
The therapeutic effect of ZFPG in PC-3 tumor-bearing mouse models
Four-week-old male BALB/c-nude mice were used to establish a subcutaneous tumor model by injecting 100 μL PC-3 cells with a concentration of 5 × 106 cells/mL into the axilla. Once the tumor volume reached approximately 100 mm3, the mice were randomly assigned to six experimental groups: G1: Control, G2: US, G3: ZFP, G4: ZFPG, G5: ZFP + US, and G6: ZFPG + US. The dose of nanomaterials administered to Groups 3, 4, 5, and 6 was standardized at 5 mg kg−1. For Groups 2, 5, and 6, US irradiation was applied to the tumors at 24 and 72 h post-material injection, using the following parameters: 10 min of irradiation at 1.5 W/cm², 1 MHz frequency, and a 50% duty cycle. Tumor volume and body weight were monitored every two days over the 14-day treatment period. Tumor volume was calculated using the formula: volume (V) = length × width2/2. To assess tumor growth, the relative tumor volume (V/V0) was normalized to the initial tumor volume (V0). After 14-day treatment, the mice were euthanized, and tumors were excised from each group for further analysis. Tumor weights were recorded, and blood samples were collected for biochemical and hematological analyses. Moreover, the main organs were collected and fixed for H&E staining.
In vivo sono-immunotherapy of RM-1 tumor-bearing mouse models
To verify the efficacy of ZFPG-mediated sono-immunotherapy, 2 × 106 RM-1 cells were injected subcutaneously into the right axilla of C57BL/6J mice. When the tumor volume reached 100 mm3, we were grouped and treated according to the in vivo treatment protocol described above. Tumor volume was measured every two days until day 14 of the end of treatment. The mice were euthanized after blood collection under deep anesthesia. One part of the tumor was collected in formalin to make the immunofluorescence tissue sections of HMGB1, CRT, and HSP90. The excitation wavelength was 594 nm when photographing CRT and HSP90 sections. The exposure time was 400 ms with a gain of 9.3×. The excitation wavelength was 488 nm when photographing HMGB1 sections. The exposure time was 300 ms with a gain of 6.4×.
The redundant tumors, lymph nodes, and spleen were placed in PBS for follow-up experiments. The number of mature DCs, CD8+ T, and MDSC cells in the tumor, lymph nodes, and spleen were analyzed by flow cytometry. The CD8+ T cells were stained with anti-CD45 APC-eFluor 780, anti-CD3-PE/Cyanine 7, and anti-CD8-FITC. The DC cells were stained with anti-CD11c-eFluor™ 450, anti-CD80-PerCP/Cyanine5.5, and anti-CD86-APC. The MDSCs were stained with anti-CD11b-FITC, anti-Ly-6G/Ly-6C (Gr-1)-PE. Finally, these two cells were analyzed on a flow cytometer.
Elisa assessment
Each group absorbed 10 μL of suspension-containing cells and inoculated them into 12-well plates after tumor extraction. The suspension was diluted with RPMI-1640 complete medium containing 10% FBS, and lonomycin was diluted to 1 μg mL−1 and PMA to 50 ng mL−1. The 12-well plates were then incubated at 37 °C overnight. On the second day, the suspension was centrifuged, and the supernatant was collected for Elisa’s analysis.
Construction of the orthotopic prostate tumor model
10% chloral hydrate was used to anesthetize male C57BL/6J mice. Then a 1 cm transverse incision was made along the midline of the lower abdomen to find a pink prostate gland. Luc-RM-1 cells (2 × 105) dispersed in 25 μL of PBS were slowly injected into the dorsal lobe of the prostate using an insulin syringe. Subsequently, the abdominal muscles and cortex were sutured with absorbable surgical sutures (4-0). One week later, potassium D-luciferin was injected into the abdomen of the mice, and the establishment and growth of the orthotopic prostate tumors were monitored via the IVIS imaging system. The successfully modeled mice were randomly divided into six groups (n = 4 per group). During the treatment process, nanomedicine (5 mg/kg) was injected into the tail vein of mice on days 0 and 5, respectively, and US irradiation (10 min, 1.5 W/cm2, 1 MHz, 50% duty cycle) was performed on days 1 and 6. After the treatment, the tumor tissues of mice were collected and weighed, then stained with H&E, TUNEL, and Ki67, and finally, the immune cells in the tumor tissues of each group were analyzed by flow cytometry.
Establishment of a lung colonization model
Two million Luc-RM-1 cells were injected subcutaneously into the groin of mice to construct primary tumors. When the tumor size was 100 mm3, the mice were randomly divided into six groups (n = 3 per group), and the primary tumors were treated with the US on the 1st and 3rd days after tail vein injection (5 mg kg−1). After 7 days of treatment, each mouse was injected with Luc-RM-1 tumor cells (100 μL, 2 × 105) through the tail vein. The tumor metastasis of mice was observed by the IVIS system on the 14th and 21st days, respectively. On the 21st day, the lungs of sacrificed mice were taken to observe the tumor metastasis and H&E staining analysis.
Normalized control of fluorescence intensity
When measuring the fluorescence intensity of various experiments in our manuscript, the normalization factor of each experimental group was calculated by using the blank control group as the internal reference. The fluorescence intensity of the blank control and the experimental group was measured under the appropriate exposure time and gain conditions, and the same detection system and measurement parameters were used in the measurement process to ensure the reliability and accuracy of the fluorescence signal. Then the normalized factors of each experimental group were calculated, and the standard fluorescence intensity of each experimental group was calculated according to the normalized factors.
Statistical analysis
The reported values served as averages of at least three biological replicates and represent independent biological experiments with similar results. All data are presented as mean ± standard deviation (SD) analyzed using GraphPad Prism 9.0 (GraphPad Software). Other data were analyzed using OriginPro 2021. The significance of the data was evaluated according to a one-way analysis of variance (ANOVA) test.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The main data supporting the results of this study are available within the paper and the associated Supplementary Information. There is no data from third-party or publicly available datasets. The accession number for the raw data files for the transcriptome sequencing reported in this paper is NCBI PRJNA1181299. All data supporting the findings of this study are available from the corresponding author upon request. Source data are provided with this paper.
References
Porter, L. H. et al. Low-dose carboplatin modifies the tumor microenvironment to augment CAR T cell efficacy in human prostate cancer models. Nat. Commun. 14, 5346 (2023).
Lu, Y. et al. Immunological conversion of solid tumours using a bispecific nanobioconjugate for cancer immunotherapy. Nat. Nanotechnol. 17, 1332–1341 (2022).
Zhou, Y. et al. Blockade of the phagocytic receptor MerTK on tumor-associated macrophages enhances P2X7R-dependent STING activation by tumor-derived cGAMP. Immunity 52, 357–373.e359 (2020).
Wang, X., Hua, P., He, C. & Chen, M. Non-apoptotic cell death-based cancer therapy: molecular mechanism, pharmacological modulators, and nanomedicine. Acta Pharm. Sin. B 12, 3567–3593 (2022).
Feng, Y. et al. A ferroptosis-targeting ceria anchored halloysite as orally drug delivery system for radiation colitis therapy. Nat. Commun. 14, 5083 (2023).
Zhang, L. et al. Integration of AIEgens into covalent organic frameworks for pyroptosis and ferroptosis primed cancer immunotherapy. Nat. Commun. 14, 5355 (2023).
He, S. et al. A semiconducting iron‐chelating nano‐immunomodulator for specific and sensitized sono‐metallo‐immunotherapy of cancer. Angew. Chem. Int. Ed. 135, e202310178 (2023).
Qin, W. et al. Protease‐activatable nanozyme with photoacoustic and tumor‐enhanced magnetic resonance imaging for photothermal ferroptosis cancer therapy. Adv. Funct. Mater. 33, 2209748 (2022).
Zhou, Y., Fan, S., Feng, L., Huang, X. & Chen, X. Manipulating intratumoral fenton chemistry for enhanced chemodynamic and chemodynamic‐synergized multimodal therapy. Adv. Mater. 33, 2104223 (2021).
Ren, W. et al. Isolated diatomic Ni–Fe metal–nitrogen sites for synergistic electroreduction of CO2. Angew. Chem. Int. Ed. 58, 6972–6976 (2019).
Wang, Z. et al. Immunogenic camptothesome nanovesicles comprising sphingomyelin-derived camptothecin bilayers for safe and synergistic cancer immunochemotherapy. Nat. Nanotechnol. 16, 1130–1140 (2021).
Han, Z. Y. et al. Metabolic regulation of tumor microenvironment with biohybrid bacterial bioreactor for enhanced cancer chemo‐immunotherapy. Adv. Funct. Mater. 33, 2302728 (2023).
Li, J. et al. Tumor‐activated size‐enlargeable bioinspired lipoproteins access cancer cells in tumor to elicit anti‐tumor immune responses. Adv. Mater. 32, 2002380 (2020).
Fu, H., Chen, L., Fang, W., Hu, P. & Shi, J. Mild magnetothermal enhanced nanocatalytic immunotherapy for solid tumors by immune cell activation and intratumoral infiltration. Nano Today 52, 101987 (2023).
Jiang, H., Fu, H., Min, T., Hu, P. & Shi, J. Magnetic-manipulated NK cell proliferation and activation enhance immunotherapy of orthotopic liver cancer. J. Am. Chem. Soc. 145, 13147–13160 (2023).
Li, J. et al. Precision cancer sono-immunotherapy using deep-tissue activatable semiconducting polymer immunomodulatory nanoparticles. Nat. Commun. 13, 4032 (2022).
Duan, Y. et al. Reticular chemistry-enabled sonodynamic activity of covalent organic frameworks for nanodynamic cancer therapy. Angew. Chem. Int. Ed. 62, e202302146 (2023).
Huo, M., Wang, L., Chen, Y. & Shi, J. Tumor-selective catalytic nanomedicine by nanocatalyst delivery. Nat. Commun. 8, 357 (2017).
Kresse, G. et al. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169 (1996).
Kresse, G. et al. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 59, 1758 (1999).
Wang, Z. et al. Biomimetic nanoflowers by self-assembly of nanozymes to induce intracellular oxidative damage against hypoxic tumors. Nat. Commun. 9, 3334 (2018).
Ruan, X. et al. A twin S‐scheme artificial photosynthetic system with self‐assembled heterojunctions yields superior photocatalytic hydrogen evolution rate. Adv. Mater. 35, 2209141 (2022).
Shen, R. et al. Heteroatom‐ and bonded Z‐scheme channels‐modulated ultrafast carrier dynamics and exciton dissociation in covalent triazine frameworks for efficient photocatalytic hydrogen evolution. Adv. Funct. Mater. 33, 2301463 (2023).
Chen, Y. et al. Achieving long-lived shallow trapping states in carbon nitride through the n-π*electronic transition for enhanced photocatalytic hydrogen generation. Appl. Catal. B 342, 123453 (2024).
Xiang, X. et al. Ultrafast electron transfer from CdS quantum dots to atomically-dispersed Pt for enhanced H2 evolution and value-added chemical synthesis. Appl. Catal. B 340, 123196 (2024).
Li, C. et al. Covalent organic frameworks with high quantum efficiency in sacrificial photocatalytic hydrogen evolution. Nat. Commun. 13, 2357 (2022).
Luan, X., Yu, Z., Zi, J., Gao, F. & Lian, Z. Photogenerated defect‐transit dual S‐scheme charge separation for highly efficient hydrogen production. Adv. Funct. Mater. 33, 2304259 (2023).
Liu, X. et al. Breaking spatiotemporal barriers of immunogenic chemotherapy via an endoplasmic reticulum membrane-assisted liposomal drug delivery. ACS Nano 17, 10521–10534 (2023).
Basu, S., Binder, R. J., Ramalingam, T. & Srivastava, P. K. CD91 is a common receptor for heat shock proteins gp96, hsp90, hsp70, and calreticulin. Immunity 14, 303–313 (2001).
Sun, L. et al. ATP‐responsive smart hydrogel releasing immune adjuvant synchronized with repeated chemotherapy or radiotherapy to boost antitumor immunity. Adv. Mater. 33, 2007910 (2021).
Yu, X. et al. The receptor kinases BAK1/SERK4 regulate Ca2+ channel-mediated cellular homeostasis for cell death containment. Curr. Biol. 29, 3778–3790 (2019).
Kale, J., Osterlund, E. J. & Andrews, D. W. BCL-2 family proteins: changing partners in the dance towards death. Cell Death Differ 25, 65–80 (2017).
Fang, X. et al. Ferroptosis as a target for protection against cardiomyopathy. Proc. Natl. Acad. Sci. USA 116, 2672–2680 (2019).
Li, Y. M. et al. Receptor‐interacting protein kinase 3 deficiency recruits myeloid‐derived suppressor cells to hepatocellular carcinoma through the chemokine (C‐X‐C motif) ligand 1-chemokine (C‐X‐C motif) receptor 2 axis. Hepatology 70, 1564–1581 (2019).
Chen, Z. et al. Peptide-appended nanosonosensitizers targeting tumor glycolysis for synergistic sonodynamic–immunometabolic therapy of spinal-metastasized tumors. Adv. Mater. 35, 2304246 (2023).
Zhang, P. et al. Targeting myeloid derived suppressor cells reverts immune suppression and sensitizes BRAF-mutant papillary thyroid cancer to MAPK inhibitors. Nat. Commun. 13, 1588 (2022).
Sun, X. et al. Amplifying STING activation by cyclic dinucleotide–manganese particles for local and systemic cancer metalloimmunotherapy. Nat. Nanotechnol. 16, 1260–1270 (2021).
Acknowledgements
The authors acknowledge financial support from the National Natural Science Foundation of China (grant no. 52472284, Y.J.) and the Taishan Scholars Program of Shandong Province (tstp20230658, Q.F.). We would also like to thank the Analytical Center for Structural Constituent and Physical Property of Core Facilities Sharing Platform, Shandong University for the Femtosecond transient absorption spectroscopy system (TAS, 2104573S).
Author information
Authors and Affiliations
Contributions
Y.W. and H.L. contributed equally to this work. Y.W. was mainly responsible for the design of materials and experimental protocols and wrote the original manuscript of this article, H.L. was responsible for the verification of the biological properties of the materials. J.L. and Y.L. revised the original draft. K.Z. was involved in the guidance of the design of the immune verification. H.L. guided theoretical calculations. Q.F. and Y.J. conceived the program design and financial support of the experiments and revised the manuscript. All authors read and approved the final article.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Huihui Wang, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wang, Y., Li, H., Lin, J. et al. Engineering nanozyme immunomodulator with magnetic targeting effect for cascade-enzyodynamic and ultrasound-reinforced metallo-immunotherapy in prostate carcinoma. Nat Commun 16, 1876 (2025). https://doi.org/10.1038/s41467-025-57190-1
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-57190-1
This article is cited by
-
Carbon dot sensitized Cu3P sonozymes for cuproptosis-enhanced and heterojunction-amplified sono-immunotherapy through activating cGAS-STING pathway
Journal of Nanobiotechnology (2025)
-
Cell death signaling and immune regulation: new perspectives on targeted therapy for sepsis
Cellular & Molecular Biology Letters (2025)
-
Review: carbon-dot-based fluorescent nanomaterials in non-invasive tumor therapy
Journal of Materials Science (2025)
-
Nanozymes as Next-Generation Therapeutics: Synergizing Enzyme-Mimetic Activity with Nanoscale Features
Biomedical Materials & Devices (2025)
