
Abstract
Cancer-associated fibroblasts (CAF) play a crucial role in tumor progression and immune regulation. However, the functional heterogeneity of CAFs remains unclear. Here, we identify antigen-presenting CAFs (apCAF), characterized by high MHC II expression, in gastric cancer (GC) tumors and find that apCAFs are preferentially located near tertiary lymphoid structures. Both in vivo and in vitro experiments demonstrate that apCAFs promote T cell activation and enhances its cytotoxic and proliferative capacities, thereby strengthening T cell-mediated anti-tumor immunity. Additionally, apCAFs facilitate the polarization of macrophages toward a pro-inflammatory phenotype. These polarized macrophages, in turn, promote the formation of apCAFs, creating a positive feedback loop that amplifies anti-tumor immune responses. Notably, baseline tumors in immunotherapy responders across various cancer types exhibit higher levels of apCAFs infiltration. This study advances the understanding of CAFs heterogeneity in GC and highlights apCAFs as a potential biomarker for predicting immunotherapy response in pan-cancer.
Similar content being viewed by others
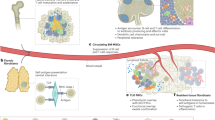


Introduction
Gastric cancer (GC) is a prevalent neoplasm globally, ranking fifth in incidence and fourth in mortality1. Current therapeutic interventions encompass surgical resection, chemotherapy, and radiotherapy, but these treatments fail to demonstrate sufficient effectiveness for patients with advanced GC2. Immune checkpoint blockade (ICB) therapy targeting immune checkpoints, such as programmed death 1 (PD1)/PD1 ligand (PD-L1), can restore the body’s durable anti-tumor immune response, thereby introducing novel prospects for malignant tumor treatment3. The Checkmate-649 study disclosed that ICB therapy combined with chemotherapy can significantly prolong the survival rate of GC patients4. However, subgroup analysis indicated a lack of survival benefit in the addition of ICI to chemotherapy for GC tumors exhibiting low or no PD-L1 expression5, which was closely related to the immunosuppressive tumor microenvironment (TME)6. Therefore, finding approaches to surmount the immunosuppressive microenvironment and enhance the efficacy of ICI therapy is an urgent necessity to ameliorate the prognosis of GC patients.
Cancer-associated fibroblasts (CAF) constitute the most abundant cells in TME and have a substantial impact on tumor growth, invasion, and drug resistance7. The effect of CAFs on tumor immunity has also gained significant attention. CAFs not only directly impede the anti-tumor function of T cells through the PD-L1/PD1 coordinated receptor axis, but also upregulate PD-L1 expression in tumor cells and promote immune evasion8,9. Nonetheless, following the targeted elimination of α-SMA+ CAFs in mouse models of pancreatic cancer, tumor growth was not suppressed, but rather accelerated, and there was also an increase in immunosuppressive cell infiltration within TME, suggesting that CAFs also play a crucial role in maintaining the anti-tumor immune response10.
Here, antigen-presenting CAFs (apCAF), characterized by high MHC II expression, is identified in GC, with its spatial localization and immune modulatory roles extensively characterized, particularly in promoting T cell-mediated anti-tumor immune responses. Furthermore, the potential of apCAFs as a biomarker for predicting immunotherapy response in pan-cancer is unveiled. This study deepens our understanding of the immune regulatory functions of CAFs, offering insights into the development of targeted therapeutic strategies and identifying a promising biomarker for precise application of immunotherapy.
Results
Identification of antigen-presenting CAFs in patients with GC
In order to comprehensively characterize the subpopulation composition of CAFs in GC, we collected scRNA-seq data of 74 samples, obtained from 38 patients in three public scRNA-seq datasets11,12,13, including non-atrophic gastritis, chronic atrophic gastritis, intestinal metaplasia, adjacent tumor tissue, and primary tumor (Fig. 1A). After rigorous quality control and major cell annotation, we obtained a total of 3246 high quality CAFs, which consist of 8 subclusters (Fig. 1B). Next, we explored the frequency of each cluster across various samples and sample types (Supplementary Fig. 1A, B), and evaluated marker genes and pathway activities for each subcluster (Fig. 1C). Based on the characteristic genes of CAFs subclusters reported in prior studies14,15, and the profiles of highly expressed genes and active pathways, we annotated Cluster 0, which is characterized by elevated expression of matrix remodeling genes (COL3A1, VCAN), as mCAFs. Clusters 1, 2, and 3, which exhibit high expression of inflammatory genes (CXCL14, CXCL2, CCL11), were classified as iCAFs. Clusters 4, 6, and 7, distinguished by high expression of epithelial-like genes (KRT19, CLDN18), myogenic genes (MYH11, TAGLN), and nerve-related genes (S100B, PLP1), were designated as eCAFs, myCAFs, and nerve-CAFs, respectively. Cluster 5, which showed high expression of CD74, HLA-DRA, HLA-DRB1 and HLA-DPA1 (genes related to MHC class II molecules) and strong activity in the antigen processing and presentation pathway, was annotated as apCAFs (Fig. 1D). Given the presence of apCAFs in gastric cancer and the uncertainty regarding its role, we selected apCAFs as the subject for further investigation. First, we utilized multiplex immunofluorescence staining to identify apCAFs in FFPE sections of GC (Fig. 1F). Then, through flow cytometry, we identified the presence of apCAFs (CD45-EPCAM-CD3-PDPN+MHC II+ cells) in fresh surgical samples of GC (Fig. 1G). Furthermore, by performing immunofluorescence staining, flow cytometry, and RNA-seq on the same tumor tissue to assess apCAFs, we found that the results from immunofluorescence and flow cytometry were highly consistent with the apCAFs signature derived from single-cell sequencing data (Supplementary Fig. 1C). This consistency further validates the reliability of our observations regarding apCAFs and reflects the robustness of these evaluation methods. Altogether, these results indicated the presence of apCAFs in TME of GC.

A Schematic overview of analytical workflow and experimental design to discover the presence of apCAFs in patients with GC. Created in BioRender. Song, J. (2024) https://BioRender.com/n91k593. B UMAP plot depicting and graph-based clustering of CAFs from 38 patients. C Dotplot displaying the expression of marker genes for CAFs subclusters. D UMAP showing all identified CAFs subtypes based on marker genes. E Heatmap showing pathways enriched in CAFs subclusters. F Representative images of multiplex immunofluorescence characterizing the apCAFs in FFPE sections (green: CD74; red: PDGFRA). Multiplexed immunofluorescence assays are performed twice on tumor samples following assay optimization. G Flow cytometry of apCAFs and their expression profile in fresh gastric tumors.
apCAFs were enriched in tumor tissues and associated with favorable prognosis in GC
Next, we investigated whether the occurrence of apCAFs was driven by the TME. First, tissue microarrays comprising 71 GC tumor tissues and 71 normal tissues were utilized for multiple immunofluorescence staining. The results revealed the significantly higher proportion of apCAFs in tumor tissues compared to normal tissues (Fig. 2A). Subsequently, paired samples of tumor tissues and normal tissues from the same patients with GC were performed flow cytometry analysis, and we discovered the significant enrichment of apCAFs in tumor tissues (Fig. 2B). Analysis of RNA-seq data from the Asian Cancer Research Group (ACRG) cohort also confirmed increased apCAFs signature expression in tumor tissues (Supplementary Fig. 1D). These findings indicated the enrichment of apCAFs in GC tumor tissues, suggesting that the tumor microenvironment drived the formation of apCAFs.

A Representative multiplex immunofluorescence images and the ratio of apCAFs in tumor (n = 71) and normal tissues (n = 71) (green: CD74; red: PDGFRA). Error bars represent the mean ± SEM. Statistical significance was determined using Mann–Whitney two-sided test. Source data are provided as a Source Data file. B The ratio of apCAFs quantified in tumor and normal tissues by flow cytometry (n = 10). Statistical significance was determined using two-sided paired t-test. Source data are provided as a Source Data file. C Clinical characteristics associated with apCAFs immunofluorescence staining in FUSCC cohort (n = 140). Statistical significance was determined using two-sided Chi-square test. Source data are provided as a Source Data file. D Survival analysis of patients with different infiltration levels of apCAFs in FUSCC cohort (n = 140). Statistical significance was determined using log-rank test. Source data are provided as a Source Data file. E Survival analysis of patients with different infiltration levels of apCAFs assessed through apCAFs signature in ACRG cohort (n = 300). Statistical significance was determined using log-rank test. Source data are provided as a Source Data file.
After confirming the enrichment of apCAFs in tumor tissues of GC, we aimed to uncover the clinical implications of apCAFs infiltration. Using multiple immunofluorescence staining, we quantified the degree of apCAFs infiltration within our FUSCC cohort. Notably, patients in the high apCAFs group showed a lower occurrence of intravascular tumor thrombus, with the majority being clinically staged at stage I or II. In contrast, the low apCAFs group displayed a higher rate of intravascular tumor emboli, and the majority of these patients were clinically staged at stage III (Fig. 2C). Survival analysis revealed that patients in the high apCAFs group have a significantly longer overall survival time and recurrence free survival time compared to the low apCAFs group (Fig. 2D, Supplementary Fig. 2A, B). This finding was further corroborated by data from the ACRG cohort (Fig. 2E). Together, these data suggested that apCAFs was a prognostic marker in tumor progression and survival in GC patients.
apCAFs exhibit a predominant localization around TLS
The spatial features of tumor microenvironment is highly associated with tumor progression and anti-tumor immune response16, we proceeded to explore the spatial distribution of apCAFs within the tumor microenvironment. Firstly, spatial transcriptomic sequencing was performed on the primary tumor of GC using FFPE sections (Fig. 3A). The distribution of B cells and T cells was assessed with microenvironment cell populations (MCP)-counter, which estimated cell type abundances from the transcriptome profiles17. Interestingly, apCAFs signature was predominantly expressed in regions enriched with B cells and T cells (Fig. 3B), as identified by pathologists as locations of tertiary lymphoid structures (TLS). We also found the significant positive correlation between the expression of apCAFs signature and TLS signature in the spatial transcriptome (Fig. 3C). We further corroborated the spatial relationship between TLS and apCAFs using multiple immunofluorescence staining. We observed that apCAFs aggregated near TLS, which was consistent with analysis results from the spatial transcriptome (Fig. 3D). We also observed this phenomenon in another spatial transcriptome section from different patient (Fig. 3E). Notably, patients from high apCAFs group possessed more TLS within tumor tissues (Fig. 3F). These results reflected the close association between apCAFs and TLS, and its specific spatial localization suggested that it may play a role in the anti-tumor immune response.

A H&E staining of tissue section for spatial transcriptoms. B Abundance estimation of various cell populations in spatial transcriptomics. C Abundance estimation and relationship of tertiary lymphoid structures (TLS) signature and apCAFs signature in spatial transcriptomics. Correlation was evaluated using the two-sided Spearman rank correlation coefficient. D Representative images of multiplex immunofluorescence characterizing the distribution of TLS and apCAFs (green: CD74; red: PDGFRA; white: CD19; purple: CD3). Multiplexed immunofluorescence assays are performed twice on tumor samples following assay optimization. E H&E staining and the abundance estimation and relationship of TLS signature and apCAFs signature in another spatial transcriptomics section from different patient. Correlation was evaluated using the two-sided Spearman rank correlation coefficient. F Representative H&E staining images of tissue sections (left) and the proportion of TLS-positive tumors (right) in high apCAFs group (n = 70) and low apCAFs group (n = 70). Statistical significance was determined using two-sided Chi-square test. Source data are provided as a Source Data file.
apCAFs modulate immune profile and augment T cell-mediated anti-tumor immune response
Given the close association of apCAFs with favorable prognosis and TLS, we subsequently investigated whether manipulating apCAFs could reprogram the immune landscape in GC. We employed flow cytometry to isolate apCAFs and MHC II- CAFs from murine transplant tumors, and co-implanting them with tumor cells subcutaneously in mice (Fig. 4A). We found that the apCAFs group exhibited slower tumor growth rate and lower tumor weight compared to control group (only tumor cells group) and the MHC II- CAFs group (Fig. 4B, Supplementary Fig. 3A), indicating the tumor-suppressive effect of apCAFs. To analyze the impact of apCAFs on the tumor immune landscape, we conducted scRNA-seq on tumors from control group, apCAFs group and MHC II- CAFs group. After quality control, we employed the UMAP algorithm to determine the distribution of cell clusters and annotated their identities based on the expression of known marker genes (Fig. 4C), we found that tumors in the apCAFs group were enriched with a higher number of T cells (Fig. 4D). Then, we used flow cytometry to examine the differences in T cell subpopulations among the three groups. We observed a significant increase in activated CD4+ T cells (CD69+CD4+ T cells), effector CD4+ T cells (IFN-γ+CD4+ T cells) and cytotoxic CD4+ T cells (GZMB+CD4+ T cells) in the tumors from the apCAFs group, although immunosuppressive regulatory T cells (Treg) also increased (Fig. 4E, Supplementary Fig. 3B). The total amount of CD8+ T cells, as well as the proportion of cytotoxic CD8+ T cells (GZMB+CD8+ T cells) and effector CD8+ T cells (IFN-γ+CD8+ T cells and TNF+CD8+ T cells), were also significantly increased in tumors from the apCAFs group (Fig. 4F). Additionally, we found that T cells in the apCAFs group tumors exhibited the highest cytotoxicity and exhaustion scores (Supplementary Fig. 3C, D), indicating a stronger tumor reactivity. ELISA experiments revealed higher levels of IFN-γ and TNF in the tumors from apCAFs group (Fig. 4G, H). In addition, we further investigated the correlation between apCAFs and antitumor immune response in clinical cohort. Through multiple immunofluorescent staining, we discovered the higher abundance of overall T cells (CD3+ cells), cytotoxic T cells (GZMB+CD3+ cells) and exhausted T cells (PD1+CD3+ cells) within tumors from the high apCAFs group in our FUSCC cohort (Fig. 4I, J, Supplementary Fig. 3E, F), demonstrating the positive association between apCAFs and anti-tumor response. In summary, these results illustrated that apCAFs contributed to the formation of the immune-activated tumor microenvironment.

A Schematic representation of apCAFs adoptive transfer in vivo experiments. Created in BioRender. Song, J. (2024) https://BioRender.com/j06i038. B Tumor growth curves showing tumor volume in mice from control group, apCAFs group and MHC II- CAFs group (n = 5 mice/per group). Error bars represent the mean ± SD. Statistical significance was determined using one-way ANOVA, followed by a two-sided Dunnett’s test to compare the specific differences between the groups. Source data are provided as a Source Data file. C Distribution of immune cell clusters in tumors from control, apCAFs and MHC II- CAFs groups (n = 2 mice/per group). D The proportion immune cell clusters in tumors from control, apCAFs and MHC II- CAFs groups (n = 2 mice/per group). E Relative proportion of total CD4+ T, CD69+CD4+ T cells, IFN-γ+CD4+ T cells and GZMB+CD4+ T cells in tumors from control, apCAFs and MHC II- CAFs groups (n = 5 mice/per group). Error bars represent the mean ± SEM. Statistical significance was determined using one-way ANOVA, followed by a two-sided Dunnett’s test to compare the specific differences between the groups. Source data are provided as a Source Data file. F Relative proportion of total CD8+ T, GZMB+CD8+ T cells, IFN-γ+CD8+ T cells and TNF+CD8+ T cells in tumors from control, apCAFs and MHC II- CAFs groups (n = 5 mice/per group). Error bars represent the mean ± SEM. Statistical significance was determined using one-way ANOVA, followed by a two-sided Dunnett’s test to compare the specific differences between the groups. Source data are provided as a Source Data file. G The concentration of IFN-γ within tumors from control, apCAFs and MHC II- CAFs groups (n = 5 mice/per group). The box is bounded by the first and third quartile with a horizontal line at the median and whiskers extend to the maximum and minimum value. Statistical significance was determined using one-way ANOVA, followed by a two-sided Dunnett’s test to compare the specific differences between the groups. Source data are provided as a Source Data file. H The concentration of TNF within tumors from control, apCAFs and MHC II- CAFs groups (n = 5 mice/per group). The box is bounded by the first and third quartile with a horizontal line at the median and whiskers extend to the maximum and minimum value. Statistical significance was determined using one-way ANOVA, followed by a two-sided Dunnett’s test to compare the specific differences between the groups. Source data are provided as a Source Data file. I Representative images of multiplex immunofluorescence showing T cell marker (green: CD3) and cytotoxic molecule (red: GZMB) expression in the high apCAFs group (n = 80) and low apCAFs group (n = 60) from the FUSCC cohort. J Relative proportion of overall T cells (CD3+ cells) and cytotoxic T cells (GZMB+CD3+ cells) in high apCAFs group (n = 70) and low apCAFs group (n = 70) in FUSCC cohort. Error bars represent the mean ± SEM. Statistical significance was determined using Mann–Whitney two-sided test. Source data are provided as a Source Data file.
Next, we further explored the specific effects of apCAFs on T cells. Given the ability of MHC-II molecules to specifically bind to the TCR of CD4+ T cells18, we first assessed the impact of apCAFs on CD4+ T cell differentiation. Subpopulation analysis of CD4+ T cells in three tumor groups revealed a significantly lower proportion of naive CD4+ T cells and a higher proportion of effector CD4+ T cells in the tumors from apCAFs group (Fig. 5A). Moreover, CD4+ T cells in the apCAFs group expressed more cytotoxic molecules (Nkg7, Prf1, Gzma, Gzmb) and immune-activating cytokines (Cxcl13, Ifng, Ccl3, Ccl4, Ccl5) (Fig. 5B). Pseudotime analysis indicated that most CD4+ T cells in the tumors from apCAFs group were in an activated state (Fig. 5C). To further confirm the activation effect of apCAFs on CD4+ T cells, we co-cultured apCAFs with CD4+ T cells in vitro (Fig. 5D). We found that apCAFs significantly promoted the expression of activation markers (CD69), cytotoxic molecules (GZMB), and cytokines (IFN-γ) in CD4+ T cells, although Treg cells also increased (Fig. 5E, Supplementary Fig. 3G). These results indicated that apCAFs could significantly promote the differentiation of CD4+ T cells into an activated state.

A Distribution (up) and proportion (down) of CD4+ T cells subclusters in tumors from control, apCAFs and MHC II- CAFs groups (n = 2 mice/per group). B Genes expression heatmap of CD4+ T cells in tumors from control, apCAFs and MHC II- CAFs groups (n = 2 mice/per group). C Gene expression dynamics along the CD4+ T cells trajectory. D Schematic representation of apCAFs and CD4+ T cells co-cultured experiments. OVA: ovalbumin. Created in BioRender. Song, J. (2024) https://BioRender.com/l34h504. E The expression of CD69, GZMB and IFN-γ in CD4+ T cells co-cultured with apCAFs or MHC II- CAFs, which was measured by flow cytometry (n = 3 biological replicates for each experiment. Data are representative of 3 independent experiments.). Error bars represent the mean ± SEM. Statistical significance was determined using one-way ANOVA, followed by a two-sided Dunnett’s test to compare the specific differences between the groups. Source data are provided as a Source Data file. F The cytotoxicity scores of CD8+ T cells (n = 889) in tumors from control, apCAFs and MHC II- CAFs groups. Statistical significance was determined using one-way ANOVA, followed by a two-sided Dunnett’s test to compare the specific differences between the groups. G Representative images of CFSE-labeled CD8+ T cells and the proportion of proliferated CD8+ T cells after co-culture with control CD4+ T cells or apCAFs-treated CD4+ T cells (n = 3 biological replicates for each experiment. Data are representative of 3 independent experiments.). Error bars represent the mean ± SEM. Statistical significance was determined using unpaired two-sided t-test. Source data are provided as a Source Data file. H Representative flow cytometry strategy and relative proportion of GZMB+CD8+ T cells, IFN-γ+CD8+ T cells and TNF+CD8+ T cells in total CD8+ T cells after co-culture with control CD4+ T cells or apCAFs-treated CD4+ T cells (n = 3 biological replicates for each experiment. Data are representative of 3 independent experiments.). Error bars represent the mean ± SEM. Statistical significance was determined using Mann–Whitney two-sided test. Source data are provided as a Source Data file. I The proportion of tumor cells killed by CD8+ T cells after co-culture with control CD4+ T cells or apCAFs-treated CD4+ T cells (n = 3 biological replicates for each experiment. Data are representative of 3 independent experiments.). Error bars represent the mean ± SEM. Statistical significance was determined using Mann–Whitney two-sided test. Source data are provided as a Source Data file.
The state of CD4+ T cells can profoundly alter the antitumor immune response by affecting the proliferation and activation of CD8+ T cells18,19, and we found CD8+T cells in tumors from apCAFs group had higher cytotoxicity scores (Fig. 5F), indicating enhanced anti-tumor immune response. To further determine whether apCAFs influence the education of CD8+ T cells by CD4+ T cells, in vitro co-cultures of CD4+ T cells treated with apCAFs and CD8+ T cells were conducted. We found that, compared to untreated CD4+ T cells, CD4+ T cells treated with apCAFs significantly promoted the proliferation of CD8+ T cells (Fig. 5G) and facilitated their differentiation into cytotoxic (GZMB+CD8+ T cells) and effector CD8+ T cells (IFN-γ+CD8+T cells and TNF+CD8+ T cells) (Fig. 5H). Importantly, these CD8+ T cells showed significantly enhanced cytotoxicity against tumor cells (Fig. 5I). These results demonstrated that apCAFs significantly enhance the proliferative and activating effects of CD4+ T cells on CD8+ T cells. Overall, through in vivo experiments, in vitro assays, and clinical cohort analyses, we found that apCAFs significantly promote the activation of both CD4+ T cells and CD8+ T cells, thereby boosting T cell-mediated anti-tumor immune response.
apCAFs and pro-inflammation macrophages exhibit positive feedback regulation
Through cell communication analysis, we discovered that apCAFs exhibited the strongest interaction activity with macrophages (Supplementary Fig. 3H), and recent studies also highlighted the crucial role of the interaction between CAFs and macrophages in the anti-tumor immune response20,21,22. Therefore, we delved deeper into elucidating the impact of apCAFs on macrophages. Tumor-associated macrophages (TAM) can polarize into either pro-inflammatory or anti-inflammatory phenotypes, thereby regulating T-cell response within the anti-tumor immune process23. Using the UMAP algorithm, macrophages derived from the control group, apCAFs group and MHC II- CAFs group were partitioned into three subclusters, we observed a higher proportion of macro_H2-Ab subcluster within the tumors from the apCAFs group compared to the control and MHC II- CAFs groups (Fig. 6A). Macro_H2-Ab subcluster exhibited high expression of antigen presentation molecules related to pro-inflammatory phenotype, while the expression of anti-inflammatory molecules was low (Fig. 6B). This demonstrated its pro-inflammatory function and indicating the correlation between apCAFs and pro-inflammatory macrophages. Interestingly, we found that the distance between apCAFs and pro-inflammatory macrophages was significantly shorter than that between MHC II- CAFs and pro-inflammatory macrophages through multiplex immunofluorescence staining (Fig. 6C). Moreover, spatial transcriptomic analysis also confirmed the close spatial distribution of apCAFs and pro-inflammatory macrophages (Fig. 6D, E, Supplementary 4A, B), and apCAFs signature was positively associated with pro-inflammatory macrophages signature in bulk RNA-seq data (Fig. 6F).

A Distribution (left) and proportion (right) of macrophages subclusters in tumors from control, apCAFs and MHC II- CAFs groups (n = 2 mice/per group). B Expression levels of pro-inflammatory phenotype and pro-inflammatory phenotype related genes among various subclusters of macrophages. C Representative images of multiplex immunofluorescence (green: CD74; red: PDGFRA; white: CD68; purple: CD86) and distance from apCAFs or MHC II- CAFs to pro-inflammatory macrophages. Statistical significance was determined using Mann–Whitney two-sided test. Multiplexed immunofluorescence assays are performed twice on tumor samples following assay optimization. D Spatial feature plot of pro-inflammatory macrophages signature in sample 1. ST: Spatial transcriptomics. E Correlation of signature score of apCAFs and pro-inflammatory macrophages in spatial transcriptomics data of sample 1. Correlation was evaluated using the two-sided Spearman rank correlation coefficient. F Correlation of signature score of apCAFs and pro-inflammatory macrophages in ACRG cohort (n = 300). Correlation was evaluated using the two-sided Spearman rank correlation coefficient. G Heatmap showing ligands activity and regulatory potential of the prioritized ligands in apCAFs to macrophages. H Schematic representation of apCAFs or MHC II- CAFs and macrophages co-cultured experiments. Created in BioRender. Song, J. (2024) https://BioRender.com/r25u487. I The expression of pro-inflammatory genes and anti-inflammatory genes in macrophages after co-culture with apCAFs or MHC II- CAFs (n = 3 in each group). J The proportion of pro-inflammatory macrophages (CD86+ cells and MHC II+ cells) in macrophages after co-culture with apCAFs or MHC II- CAFs from five different patients. Error bars represent the mean ± SEM. Statistical significance was determined using one-way ANOVA, followed by a two-sided Dunnett’s test to compare the specific differences between the groups. Source data are provided as a Source Data file. K The prediction of ligand-receptor interaction activity from macrophage subclusters to apCAFs through “CellChat” algorithm. Pro-inflam Macro: pro-inflammation macrophages; Anti-inflam Macro: Anti-inflammation macrophages. L The prediction of ligand-receptor interactions activity between pro-inflammatory macrophages and apCAFs through “CellCall” algorithm. Pro-inflam Macro: pro-inflammation macrophages; Anti-inflam Macro: Anti-inflammation macrophages. M Schematic representation of control macrophages or pro-inflammatory macrophages and pan-CAFs co-cultured experiments. Pro-inflam Macro: pro-inflammation macrophages. Created in BioRender. Song, J. (2024) https://BioRender.com/h05n316. N The proportion of apCAFs in pan-CAFs from five different patients after co-culture with control macrophages or pro-inflammatory macrophages. Error bars represent the mean ± SEM. Statistical significance was determined using unpaired two-sided t-test. Source data are provided as a Source Data file. Pro-inflam Macro: pro-inflammation macrophages.
Given the close spatial relationship between apCAFs and pro-inflammatory macrophages, we further explored the regulatory interactions between them. We found that ligands produced by apCAFs have the potential to promote the expression of IL1A, IL1B, IL6, and HLA-DQA1 (marker genes of pro-inflammatory phenotype24 in macrophages (Fig. 6G)), suggesting that apCAFs may promote the polarization of macrophages towards the pro-inflammatory phenotype. To clarify the impact of apCAFs on macrophage status, we conducted in vitro co-culture experiments with apCAFs and macrophages, and performed RNA sequencing on the macrophages post-co-culture (Fig. 6H). We found that after apCAFs treatment, the expression of inflammatory cytokines (CD80, CD86, CXCL9, CXCL10, CXCL11, IL1A, IL1B and IL6) and antigen presentation-related molecules (IRF1 and HLA-DRA) in macrophages was upregulated, while the expression of anti-inflammatory molecules (MMP9, CD276, MSR1, SPP1 and TREM2) was downregulated (Fig. 6I), indicating the enhancement of their pro-inflammatory function. Flow cytometry results also showed that after apCAFs treatment, the pro-inflammatory markers (CD86 and MHC-II) were upregulated in macrophages (Fig. 6J), while anti-inflammatory marker (CD163) were downregulated (Supplementary Fig. 4C). These results demonstrate that apCAFs promote the polarization of macrophages towards a pro-inflammatory phenotype.
Interestingly, through the prediction of cell communication, “through the prediction of cell communication, we also found that pro-inflammatory macrophages have the potential to regulate apCAFs through receptor-ligand interactions (Fig. 6K, L), suggesting that they may also influence the phenotype of apCAFs. Through co-culture experiment in vitro, we found that pro-inflammatory macrophages increased the proportion of apCAFs (Fig. 6M, N), indicating that pro-inflammatory macrophages promoted the generation of the apCAFs phenotype. In summary, we have identified a bidirectional regulatory relationship between apCAFs and pro-inflammatory macrophages, and this positive feedback loop may contribute to further inducing their immune-activated state.
Baseline intratumoral apCAFs predicts clinical benefits from immunotherapy
The tumor immune microenvironment is a decisive factor in ICB treatment response. The close association of apCAFs with the immune-activated microenvironment prompted us to delve deeper into the role of apCAFs in ICB treatment response. To further explore the predictive value of apCAFs for immunotherapy, two immunotherapy cohorts of GC with pre-treatment biopsy tissues to perform bulk RNA-seq or multiplex immunofluorescence were included. Through the analysis of the KIM cohort, which included 45 metastatic GC patients treated with anti-PD1 therapy (Fig. 7A), we found that the apCAFs signature was significantly upregulated in patients with high immune signature (Fig. 7B). Patients with high expression of the apCAFs signature showed a significantly increased immunotherapy response rate (Fig. 7C), and the apCAFs signature in responders was significantly higher than in non-responders (Fig. 7D). Next, our cohort consisting of 21 GC patients who received neoadjuvant anti-PD1 plus chemotherapy was included as validation cohort (Fig. 7E, F). We collected pre-treatment biopsy samples for multiplex immunofluorescence staining, and we found that responders have a higher abundance of apCAFs in their pre-treatment tumors (Fig. 7G, H), and the ratio of apCAFs could effectively predict the treatment response of patients (Fig. 7I). The results from these two independent cohorts suggested the potential of apCAFs as a biomarker for immunotherapy response.

A Schematic of GC patients treated with anti-PD1 therapy used as discovery cohort. Created in BioRender. Song, J. (2024) https://BioRender.com/m64n940. B Bar chart representing apCAFs signature in patients with high immune signature (n = 22) or low immune signature (n = 23) in discovery cohort. Error bars represent the mean ± SEM. Statistical significance was determined using unpaied t-test. Source data are provided as a Source Data file. C Bar chart representing apCAFs signature in responders (n = 12) and non-responders (n = 33) in discovery cohort. Error bars represent the mean ± SEM. Statistical significance was determined using unpaired two-sided t-test. Source data are provided as a Source Data file. R: responders; NR: non-responders. D ROC curves showing the value of apCAFs in predicting therapeutic response in discovery cohort. Source data are provided as a Source Data file. E Schematic of GC patients treated with neoadjuvant anti-PD1 plus chemotherapy used as validation cohort. Created in BioRender. Song, J. (2024) https://BioRender.com/a10h486. F Representative CT images of patients with different responses to neoadjuvant anti-PD1 plus chemotherapy in validation cohort. R: responders; NR: non-responders. G Representative images of multiplex immunofluorescence characterizing the apCAFs in patients with different responses in validation cohort (green: CD74; red: PDGFRA). H Bar chart representing the proportionof apCAFs in all cells of tumors with different responses to neoadjuvant anti-PD1 plus chemotherapy in validation cohort (n = 21). Error bars represent the mean ± SEM. Statistical significance was determined using unpaired two-sided t-test. Source data are provided as a Source Data file. R: responders; NR: non-responders. I ROC curves showing the value of apCAFs in predicting therapeutic response in validation cohort. Source data are provided as a Source Data file. J UMAP plots and proportion of CAFs clusters in pre-treatment and post-treatment tissues from patients with different responses in triple negative breast cancer cohort (n = 22). TNBC: triple negative breast cancer; R: responders; NR: non-responders. K Violin plots showing apCAFs signature in CAFs (n = 10825) from pre- and post-treatment tissues of patients with different responses. The box is bounded by the first and third quartile with a horizontal line at the median and whiskers extend to the maximum and minimum value. Statistical significance was determined using Mann–Whitney two-sided test. R: responders; NR: non-responders. L Predictive value of apCAFs to clinical benefits in melanoma patients receiving anti-PD1 therapy (n = 41). Error bars represent the mean ± SEM. Statistical significance was determined using unpaired two-sided t-test. Source data are provided as a Source Data file. R: responders; NR: non-responders. M Predictive value of apCAFs to clinical benefits in melanoma patients receiving anti-PD1 and anti-CTLA4 therapy (n = 32). Error bars represent the mean ± SEM. Statistical significance was determined using unpaired two-sided t-test. Source data are provided as a Source Data file. R: responders; NR: non-responders. N Predictive value of apCAFs to clinical benefits in non-small cell lung cancer patients receiving anti-PD1 therapy (n = 16). Error bars represent the mean ± SEM. Statistical significance was determined using Mann–Whitney two-sided test. Source data are provided as a Source Data file. NSCLC: non-small cell lung cancer; R: responders; NR: non-responders.
Given the promising prospects of immunotherapy in the treatment of malignant tumors, we further investigated whether apCAFs could also predict immunotherapy responses in pan-cancer. First, we obtained scRNA-seq data from pre-treatment and post-treatment biopsy tissues of patients with triple negative breast cancer (TNBC) receiving pembrolizumab (Anti-PD1 antibody) in ClinicalTrials.gov: NCT0319738925. After quality filtering, 10,825 fibroblasts were included in subsequent analysis. Using the UMAP algorithm, we divided fibroblasts into five subgroups, including apCAFs, inflammatory CAFs (iCAFs), matrix-associated CAFs (mCAFs), myofibroblasts (myCAFs), and proliferative fibroblasts (Prolif_CAFs). Interestingly, we found that, after receiving immunotherapy, responders exhibited the significant expansion of apCAFs, while non-responders did not display the noticeable increase in apCAFs (Fig. 7J). We further analyzed changes in the apCAFs signature within fibroblasts and observed that both before and after immunotherapy, responders had higher apCAFs signature expression in fibroblasts compared to non-responders. Furthermore, following immunotherapy, responders showed a further increase in the expression of the apCAFs signature in fibroblasts, which was not observed in non-responders (Fig. 7K). These findings suggested that apCAFs was involved in the tumor’s response to immunotherapy and could serve as potential biomarkers for immunotherapy response in TNBC. Furthermore, we found that responders had a significantly higher apCAFs signature compared to non-responders in melanoma patients receiving anti-PD1 or anti-PD1 combined with anti-CTLA4 treatment, and apCAFs signature could predict immunotherapy response (Fig. 7L, M). Similar results were observed in non-small cell lung cancer (NSCLC) patients receiving anti-PD1 treatment (Fig. 7N). In summary, these findings supported the value of apCAFs as an immunotherapy biomarker in pan-cancer.
Discussion
In addition to myCAFs and iCAFs, apCAFs characterized by high expression of MHC II molecules and the upregulation of antigen presentation pathway have been identified in pancreatic cancer26, lung cancer27, and breast cancer28, but it remains unclear whether apCAFs exist in GC. Here, we have uncovered the existence of apCAFs within GC, which correlates with favorable patient outcomes and contributes to the anti-tumor immune response within the tumor microenvironment. Increasing the ratio of apCAFs in GC can activate the anti-tumor immune response, thereby inhibiting tumor growth. In line with our research results, apCAFs in lung cancer directly engage the T-cell receptor (TCR) of effector CD4+ T cells, simultaneously generating C1q, which functions on T-cell C1qbp to rescue them from apoptosis. The specific absence of MHC II or C1q in fibroblasts compromises CD4+ T cell immunity and expedites tumor growth27. In breast cancer, the infiltration of apCAFs is associated with a better prognosis for patients28. However, apCAFs promote the generation of Treg, which could potentially undermine anti-tumor immunity29,30. We also observed that increasing the presence of apCAFs leads to an increase in the number of Treg in TME. The absence of co-stimulatory molecules may be the reason for the increased Treg population induced by apCAFs31. These studies suggest the intricate regulatory role of apCAFs in the anti-tumor immune response. Importantly, we found that after co-culture with apCAFs, the proliferative and activating effects of CD4+ T cells on CD8+ T cells were significantly enhanced, indicating that the overall effect of apCAFs on T cell-mediated anti-tumor immunity is activation. Future studies should explore how to inhibit the production of Treg to further enhance the immune-activating effects of apCAFs. Additionally, apCAFs have been found to be associated with an increase in exhausted T cells in this study, likely due to prolonged T cell activation32. While T cell exhaustion indicates tumor reactivity, the upregulation of terminal exhaustion molecules impairs their anti-tumor immune function33. Immune checkpoint therapy offers the potential to rescue T cells from this exhausted state34,35. Thus, exploring strategies to increase apCAFs levels to activate T cells, while concurrently using immune checkpoint therapy to prevent exhaustion, presents a promising approach to further enhance anti-tumor immune responses.
Interestingly, we observed that apCAFs often appear near TLS, which primarily consist of CD20+ B cells and CD3+ T cells. The formation of TLS in tumors is often associated with improved prognosis and enhanced immune therapy response36. Despite the incomplete elucidation of TLS formation mechanisms, the crucial roles of lymphoid tissue inducer (LTi) cells and lymphoid tissue organizer (LTo) cells in the early stages of TLS formation have been confirmed36,37. In the context of chronic inflammation, the formation and maintenance of TLS primarily depend on stromal cells, predominantly composed of fibroblasts38. In the B16-OVA melanoma model, the PDPN+FAP- CAFs subset within the tumor can act as stromal LTo cells to induce TLS formation39. Moreover, Ludewig and his colleagues demonstrated that targeted deletion of CCL19 expression in lung fibroblasts disrupted T cell infiltration and lymphoid aggregate formation, promoting the progression of lung cancer, which also underscored the significant role of CAFs in the formation of TLS40. The proximity of apCAFs to TLS spatially suggests their potential involvement in TLS formation and maintenance. It’s worth noting that recent study has suggested that CAFs promoted the expression of CXCL13 by T cells through TGF-β secretion, thereby inducing the formation of TLS41, which indicated that CAFs could also facilitate TLS formation through interactions with immune cells. Our research indicates that apCAFs have a strong correlationwith TLS and can activates T cells, which suggests apCAFs may contribute to the formation of TLS. While further research is required to substantiate this hypothesis, our study has offered a fresh perspective on the correlation between CAFs and TLS.
While apCAFs are prominently marked by the high expression of MHC II molecules, existing research studies have consistently concluded that apCAFs alter tumor immunity that extends beyond their role in antigen presentation42. The interaction between CAFs and macrophages represents a crucial pathway for CAFs to exert their influence in TME43. Our research revealed that apCAFs are spatially closer to pro-inflammatory macrophages and exhibit positive feedback regulation with pro-inflammatory macrophages, deepening the comprehension of the immunomodulatory functions attributed to apCAFs and pro-inflammatory macrophages. In accordance with our research findings, a program involving interferon response and antigen cross-presentation mediated by CD4+ T cells, CD8+ T cells, macrophages, and CAFs was identified at the tumor-malignant boundary44. The interaction between CAFs and TAM is recognized as one of the most crucial interactions in TME45, with various types of CAFs and TAM forming distinct cellular niches that influence anti-tumor immune responses46. ECM-producing myCAFs polarizes TREM2+ TAM to create immunosuppressive EcoCellTypes, while detoxification-associated iCAFs is associated with FOLR2+ macrophages, contributing to an immune-protective EcoCellType46. This illustrates the complexity of CAFs-TAM interactions, and our findings regarding the interaction between apCAFs and pro-inflammatory TAM further enhance the understanding of these dynamics. However, the specific mechanisms still require further investigation and it is worth exploring whether apCAFs-mediated immune activation in GC relies on pro-inflammatory macrophages through macrophage-specific depletion in vivo models.
Our research also emphasized the close association between apCAFs and the response to ICB therapy. In our study, we observed that the infiltration of apCAFs before ICB treatment was higher in responders than in non-responders. Additionally, following ICB therapy, a substantial expansion of apCAFs was evident in responders, while non-responders did not exhibit such an increase. These results highlighted the significant role of apCAFs in the response to ICB therapy. Indeed, both TLS and M1-like macrophages are crucial factors in the response to ICB therapy47,48,49. Therefore, it is understandable that apCAFs closely associated with them plays important role in ICB therapy response. Our research also provided promising evidence that apCAFs had the potential to predict ICB response in GC. Through the retrospective clinical cohorts, we found that tumors with high apCAFs infiltration exhibited a higher response rate to immunotherapy in pan-cancer. While the potential of apCAFs as a predictor of immunotherapy efficacy still requires further elucidation through prospective clinical trials, our study has introduced a fresh perspective on predicting immunotherapy response. Using simple assays such as multiplex immunohistochemistry to assess the baseline levels and treatment-induced changes in apCAFs infiltration offers clinically valuable insights into predicting patient responses to immunotherapy. Moreover, the prospect of combining apCAFs with established immunotherapeutic response biomarkers, including MSI status and PD-L1 expression, heralds an advanced stratification approach for pinpointing patients poised to gain from immunotherapy treatments.
In this study, a syngeneic transplantation tumor model was used to investigate the function of apCAFs in vivo. However, this model lacks gastric cancer-specific signaling pathways and microenvironmental pressures, limiting its ability to fully replicate the complexity of the gastric cancer tumor microenvironment. Future research should explore the use of spontaneous gastric cancer models to provide a more physiologically relevant context. Moreover, there is significant heterogeneity in different microregions within GC50, and our study also found that apCAF exhibited a predominant localization around TLS and pro-inflammatory macrophages. However, the reasons behind apCAF’s distribution heterogeneity and its potential relationship with immune-regulatory function of apCAFs remain unclear. Further understanding of this distribution heterogeneity will help elucidate the regulatory mechanisms of apCAFs. In addition, the dynamic changes of apCAFs during immunotherapy and whether increasing apCAFs infiltration can enhance immunotherapy need to be further elucidated.
In conclusion, we have identified the presence of apCAFs in GC and demonstrated that apCAFs can promote anti-tumor immune response. This advances the understanding of heterogeneity and immunoregulatory roles of CAFs. Furthermore, the uncovered positive correlation between apCAFs and immunotherapy response making apCAFs as a potential predictor for immunotherapy response, which helps guiding the application of immunotherapy.
Methods
Patients and tissue sample
All samples were obtained from the Department of Gastric Surgery, Fudan University Shanghai Cancer Center (FUSCC), Shanghai, China. The tissue microarray utilized in this study was sourced from the FUSCC cohort, consisting of 140 samples from patients diagnosed with GC who underwent gastrectomy without neoadjuvant chemotherapy or radiotherapy between November 2008 and June 2010. Patients were followed up every 6 months until November 2015, with overall survival (OS) defined as the time from surgery to death or the end of follow-up, and recurrence free survival (RFS) defined as the time from surgery to recurrence/metastasis or the end of follow-up. Baseline tumor samples from 21 GC patients who received neoadjuvant anti-PD1 plus chemotherapy treatment were collected during pre-treatment gastric endoscopy procedures. Informed consent was obtained from all patients prior to tissue collection, and the study protocol was approved by the Clinical Research Ethics Committee of FUSCC.
Spatial transcriptomics sequencing
Two paraffin-embedded gastric cancer tissue blocks with DV200 >50 were selected for spatial transcriptomic sequencing. The Visium FFPE Spatial platform (10× Genomics) was employed with spatially barcoded mRNA-binding oligonucleotides in the default protocol to capture comprehensive gene expression information for spatial transcriptomics slides. Each capture area (6.5 × 6.5 mm²) contains 5,000 barcode spots with a diameter of 55 µm. H&E image preparation was performed according to the recommended protocol. Raw sequencing reads of spatial transcriptomics were quality checked and mapped by Space Ranger package. The generated gene-spot matrices were analyzed with the Seurat package. Signature scores were calculated based on the average expression levels of relevant genes while spatial feature expression plots were generated through the “SpatialFeaturePlot” function in Seurat.
Mouse model
The mouse gastric cancer cell line MFC was obtained from Pricella Life Science & Technology Co., Ltd (cat.no. CL-0156). Female 615 mice51 (6 weeks) were obtained from Hangzhou Ziyuan Laboratory Animal Care Co., Ltd, and OT-II mice52 were obtained from Shanghai Model Organisms Center. They were housed in the pathogen-free environment with a temperature of 24 °C, humidity of 50%, and a light-dark cycle of 12 h–12 h. Female mice were used in this study, because no sex-specific differences were observed in clinical cohorts and female mice were mostly used for tumor experiments due to their reduced fighting behaviors which facilitates group housing. All mice were randomly assigned to the control or experimental groups. For the gastric cancer model, 1 × 106 MFC cells resuspended in 200 μL of PBS were injected into the left flank of 615 mice. For co-transplantation experiments, MFC cells (1 × 106) and apCAFs (1 × 105) or MHC II- CAFs (1 × 105) were jointly injected into the left flank of mice. The tumor volume (V) of the mice was measured using a digital caliper to assess the length and width every 2 days, and the volume was calculated as V = (length × width2) / 2. The maximum tumor size allowed is 1000 mm³, and all experiments were conducted in accordance with this limit, which has been approved by Animal Care and Use Committee of FUSCC. Early termination was performed if mice exhibited signs of severe distress, weight loss exceeding 15%, or tumor ulceration. Euthanasia was carried out using CO2 inhalation. All of the mouse experiments were approved by the Animal Care and Use Committee of FUSCC.
Sample preparation
For both human and mouse tumor tissues, fresh samples were washed with PBS and then minced into small pieces. These tissues were digested with collagenase IV (1 mg/ml; Sigma, cat. no. C1889), Dispase II (1 mg/ml; Roche, cat. no. SCM133), and DNase I (0.1 mg/ml; Sigma, cat. no. DN25) with continuous stirring at 37 °C for 1 h. Single-cell suspensions from human tumors were filtered through a 100 μm strainer, while those from mouse tumors were filtered through a 70 μm strainer. Mouse spleens were mechanically dissociated and the resulting single-cell suspensions were filtered through a 40 μm strainer. All single-cell suspensions were washed twice with PBS prior to further experimentation.
Single-cell RNA sequencing
Fresh mouse tumor tissues were collected 15 days after inoculation, and tissue samples were washed with phosphate buffered saline (PBS) and then minced into small pieces. Then, these tissues were digested with collagenase IV (1 mg/ml; Sigma, cat. no. C1889), Dispase II (1 mg/ml; Roche, cat. no. SCM133), and DNase I (0.1 mg/ml; Sigma, cat. no. DN25) with continuous stirring at 37 °C for 1 h. Single cell suspensions were washed twice with flow cytometry buffer, followed by anti-CD45 (Biolegend, cat. no. 103131) staining for 20 min. Sorted by flow cytometry, all immune cells were collected and libraries were constructed according to 10× Genomics single cell sequencing kit following the recommended protocol, and sequenced on Illumina platform.
scRNA-seq data processing and clustering
Raw gene expression matrices were merged and processed by Seurat. To define the fibroblasts in GC, we conducted quality control and performed two rounds of unsupervised clustering. In brief, the first round analysis pertained to distinguishing fibroblasts from other cell types in each dataset separately, based on highly expressing canonical fibroblasts marker genes (COL1A1, PDGFRA, DCN). Mural cells were excluded due to their prominent expression of ACTA2, TAGLN, MYH11, PDGFRB and noticeable distance between the mural cell clusters and fibroblasts clusters in UMAP plot. In order to exclude doublets, we firstly filtered out cells with low quality based on three metrics: (1) cells with fewer than 200 genes or >6000 genes. (2) cells with fewer than 500 or >30000 unique molecular identifiers (UMIs). (3) cells with mitochondrial gene percentages exceeding 20%. Then, “DoubletFinder” package was utilized to estimate potential doublets in each sample and potential doublets were removed. The integration of different sample batches was performed using the harmony method. Scaled data after normalization were used for principal component analysis (PCA) using 2000 highly variable genes. The first 15 principal components were utilized for UMAP embedding, with the first 2 principal components serving as UMAP initialization. Differentially expressed genes were identified using the “FindAllMarkers” to functionally characterize the clusters.
The “AddModuleScore” function from the “Seurat” package was used to compute the average expression of genes in specific gene sets. For the cytotoxic score, eight marker genes for cytotoxic T cells (Nkg7, Gzma, Gzmb, Ifng, Ccl4, Cst7, Prf1, and Ccl3)53 were used to calculate the cytotoxicity score. For the exhausted score, seven marker genes for exhausted T cells (Pdcd1, Ctla4, Havcr2, Lag3, Tigit, Tox and Layn)53 were used to calculate the exhausted score. The final value as the input for further analysis were calculated by \({{\rm{Z}}}=({{\rm{X}}}-\bar{{{\rm{X}}}})/{{\rm{S}}}\), where X is the value from the above procedures. For trajectory analysis, the “monocle” R package was employed to perform pseudotime ordering and infer the state evolution of CD4+ T cells.
Cell communication analysis
To decipher which cell cluster potentially interacts with the cell clusters of interest, the R package “Cellchat”54 and R package “CellCall”55 were employed to systematically infer and visualize the abundance of ligand-receptor interactions among different cell clusters. This toolkit leveraged a database of ligand-receptor interactions to deduce biologically significant cell-cell communication from scRNA-seq data. The R package “NicheNet”56 could predict active ligand and regulatory target gene between interacting cells according to combination of expression data and gene regulatory networks. We applied NicheNet algorithm to infer which ligand and receptor genes potentially impact the cell clusters of interest.
Calculation of the signature in bulk RNA-seq datasets
To identify the apCAFs signature, the “FindAllMarkers” function was used to calculate apCAFs marker genes in the single-cell sequencing dataset. Genes expressed in >75% of apCAFs cells, with expression in <50% of the remaining CAFs cells, and with expression levels in apCAFs cells >1.5 times higher than in other cells were selected as core marker genes for apCAFs. Additionally, characteristic apCAFs features from pan-cancer studies were incorporated57, resulting in a total of 16 genes being included as the apCAFs signature. The TLS signature58 and pro-inflammatory macrophages signature59 are sourced from the previous studies (Supplementary Data 2). We quantified the TLS, pro-inflammatory macrophages and apCAFs abundance in each sample by calculating the average expression of all genes in the signature.
Spatial transcriptomics sequencing analysis
For the integration analysis of spatial transcriptomic data from two different tumor sections, after quality control, we utilized MCP-counter method (which allows the robust quantification of the absolute abundance of eight immune populations from transcriptomic data17) and signatures derived from the integrated scRNA-seq data to assess the abundance scores of each cell type.
Flow cytometry
Before antibody staining, we incubated the cells with anti-Fc receptor antibody to block non-specific staining. For surface staining, single cell suspensions were washed for twice with FACS buffer, followed by antibody staining for 20 min. For cytokine staining, treated with protein transport inhibitor cocktail (Invitrogen, cat. no. C5138) for 6 h, single cell suspensions were then surface stained as above, fixed and resuspended with 10% permeabilization buffer (Biolegend, cat. no. 421002). After flow antibody staining, 7-AAD dye (Biolegend, cat. no. 420403) was used to exclude dead cells. During the flow sorting and analysis process, CD45-EPCAM-CD31-PDPN+MHC II+ cells were defined as apCAFs, CD45-EPCAM-CD31-PDPN+MHC II- cells were defined as MHC II- CAFs, and CD45-EPCAM-CD31-PDPN+ were defined as pan-CAFs. The antibodies used for flow cytometry were included in Supplementary Data 3. The gating strategies used for cell sorting and flow cytometry have been shown in Supplementary Fig. 5 and Supplementary Fig. 6.
CAFs and T cells co-cultured experiment
First, seed the flow-sorted apCAFs or MHC II- CAFs cells into a 96-well plate. Incubate the cells at 37 °C with complete culture medium containing 25 μg/ml ovalbumin (OVA) peptide (OVA 323-339; GenScript; cat.no. RP10610) for 4 h. After incubation, wash the cells three times with PBS. Next, obtain the single-cell suspension by mechanically grinding the spleen of OT-II mice. Filter and wash the suspension, then use mouse CD4+ T cells isolation kit (Biolegend; cat.no.480006) to sort the CD4+ T cells from the suspension. Wash the sorted CD4+ T cells and seed them into the 96-well plate containing apCAFs or MHC II- CAFs at a T cell to CAFs ratio of 1:1. Co-culture the cells for 24 h and proceed with subsequent flow cytometry analysis.
To assess the impact of CD4+ T cells on CD8+ T cells proliferation and function, CD8+ T cells were first isolated from the spleens of 615 mice using mouse CD8+ T cells isolation kit (Biolegend; cat.no.480008), and labeled with CellTrace CFSE dye (1 μM; Thermo Fisher Scientific; cat.no.C34570) for 20 min. After washing the cells three times with complete DMEM medium, they were resuspended in a medium containing mouse T-Activator CD3/CD28 beads (25 μl/ml; Thermo Fisher Scientific; cat.no.11452D) and mouse recombinant IL-2 (100 IU/ml; STEMCELL; cat.no.78081). CD4+ T cells, which had been co-cultured with apCAFs, were collected and washed. CD4+ T cells and CD8+ T cells were then co-cultured at a 1:1 ratio for 72 h, followed by flow cytometry analysis. For assessing CD8+ T cells function, CD4+ T cells, previously co-cultured with apCAFs, were co-cultured with CD8+ T cells cells at a 1:1 ratio for 48 h. Cell stimulation cocktail plus protein transport inhibitors (2 μl/ml; Thermo Fisher Scientific; cat.no. 00-4975-93) was then added to the co-culture system, and flow cytometry analysis was performed after an additional 6 h. To assess the cytotoxicity of CD8+ T cells, tumor cells were first labeled with CellTrace CFSE dye (1 μM; Thermo Fisher Scientific; cat.no.C34570) and then plated in a 96-well plate. CD8+ T cells, which had been co-cultured with CD4+ T cells, were collected and co-cultured with the tumor cells at a ratio of 5:1 (T cells to tumor cells). After 48 h, the cells were stained with 7-AAD dye and analyzed by flow cytometry.
Isolation and co-culture of CAFs with macrophages
The human monocytic cell line THP-1 was purchased from Pricella Life Science & Technology Co., Ltd (cat.no. CL-0233). The macrophages were obtained by treating THP-1 cells with PMA (150 nM; MedChemExpress; cat.no. HY-18739) for 24 h. Fresh GC tissues from patients who underwent surgery in Fudan university shanghai cancer center were washed with phosphate-buffered saline (PBS) in three times and then digested. The obtained single-cell suspension was subjected to flow cytometry sorting, and the obtained apCAFs and MHC II- CAFs were co-cultured with macrophages in a 1:1 ratio for 48 h before flow analysis.
For the co-culture experiment with pro-inflammatory macrophages and pan-CAFs, macrophages were first induced with LPS (10 ng/ml; MedChemExpress; cat.no. HY-D1056) and IFN-γ (20 ng/ml; MedChemExpress; cat.no. HY-P7025) for 48 h. Fresh gastric cancer surgical samples were then used to isolate pan-CAFs via flow cytometry sorting, and they were seeded at a 1:1 ratio into culture plates with the pro-inflammatory macrophages. After 48 h of co-culture, flow cytometry analysis was performed.
Survival analysis
The median value was employed as the threshold to distinguish between the high apCAFs and low apCAFs groups. Survival curves were generated using the Kaplan-Meier method, and p-values were derived from the log-rank test. A multivariable Cox proportional hazards model was utilized to calculate hazard ratios, with survival analysis performed using the R package “survival”.
Multiplex immunofluorescence staining
FFPE sections were subjected to dewaxing and epitope retrieval. After incubation with primary antibodies, slides were dealt with signal amplification and antibody stripping. Then nuclei were stained with DAPI (5 µg/ml; Thermo Fisher Scientific; cat.no. D1306). The stained slides were imaged and scanned using the 3DHISTECH fluorescence imaging scanner and the quantification of cell proportions was performed using HALO pathology analysis system (Indica Lab). All the antibodies were purchased from Abcam and included in Supplementary Data 3.
Statistical analysis
For comparisons of more than two groups, one-way analysis of variance (ANOVA) was used, assuming the data followed a normal distribution. When comparing two independent groups, the unpaired t-test was applied for normally distributed data, while the Mann–Whitney test was used for non-normally distributed data. For paired samples, the paired t-test was used to compare two groups. Differences in proportions between groups were assessed using the Chi-square test. All statistical tests were two-sided unless otherwise specified, and a P-value of <0.05 was considered statistically significant. Error bars in the figures represent the standard deviation (SD) or standard error of the mean (SEM). GraphPad Prism version 9.0.1 and R software version 4.1.2 were used for statistical analysis.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The raw sequencing data for single-cell RNA sequencing of mouse gastric tumors generated in this study have been deposited at the Genome Sequence Archive (GSA) at the National Genomics Data Center (Beijing, China) under accession code CRA022852. The raw sequencing data for transcriptomic and spatial transcriptomic sequencing of human gastric cancer tissues, as well as transcriptomic sequencing of THP-1 cells co-cultured with human antigen-presenting cancer-associated fibroblasts, have been deposited at GSA-Human database under accession codes HRA010368, HRA010384, and HRA010356, respectively. Due to data privacy laws, access to the raw sequencing data is available for non-commercial use under controlled access, and requests for access can be made to the corresponding authors. The expression matrices from single-cell RNA sequencing of mouse gastric tumors generated in this study are available in the Open Archive for Miscellaneous Data (OMIX) database under accession code OMIX007266. The expression matrices from bulk RNA sequencing and spatial transcriptomics sequencing of human gastric cancer tissues are available in the OMIX database under accession code OMIX007961. The expression matrices of THP-1 cells co-cultured with antigen-presenting cancer-associated fibroblasts are available in the Science Data Bank [https://doi.org/10.57760/sciencedb.18610]. Additionally, three publicly available single-cell RNA-seq datasets of human gastric cancer are from the Gene Expression Omnibus (GEO) under accession codes GSE134520, GSE167297, and GSE150290. The immunotherapy single-cell RNA-seq dataset is available from the public database [https://lambrechtslab.sites.vib.be/en/single-cell]. The Asian Cancer Research Group bulk sequencing dataset and non-small cell lung cancer immunotherapy cohort are available from the GEO database under accession codes GSE62254 and GSE126044, respectively. The immunotherapy cohorts for gastric cancer (KIM cohort) and melanoma (Gide cohort) are available from the Tumor Immune Dysfunction and Exclusion (TIDE) database [http://tide.dfci.harvard.edu/download/]. All data supporting the findings of this study are included in the Supplementary Information or are available from the authors. Unique reagents used in this study are also available upon request. The raw numbers for charts and graphs are provided in the Source Data file, where applicable. Source data are provided with this paper.
Code availability
Codes used in this study are available at GitHub (https://github.com/SongJunquan/GC-apCAF).
References
Sung, H. et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 71, 209–249 (2021).
Smyth, E. C. et al. Gastric cancer. Lancet 396, 635–648 (2020).
Morad, G. et al. Hallmarks of response, resistance, and toxicity to immune checkpoint blockade. Cell 184, 5309–5337 (2021).
Janjigian, Y. Y. et al. First-line nivolumab plus chemotherapy versus chemotherapy alone for advanced gastric, gastro-oesophageal junction, and oesophageal adenocarcinoma (CheckMate 649): a randomised, open-label, phase 3 trial. Lancet 398, 27–40 (2021).
Zhao, J. J. et al. Low programmed death-ligand 1-expressing subgroup outcomes of first-line immune checkpoint inhibitors in gastric or esophageal adenocarcinoma. J. Clin. Oncol. 40, 392–402 (2022).
Li, K. et al. Advances in clinical immunotherapy for gastric cancer. Biochim. Biophys. Acta. Rev. Cancer 1876, 188615 (2021).
Rimal, R. et al. Cancer-associated fibroblasts: origin, function, imaging, and therapeutic targeting. Adv. Drug Deliv. Rev. 189, 114504 (2022).
Pinchuk, I. V. et al. PD-1 ligand expression by human colonic myofibroblasts/fibroblasts regulates CD4+ T-cell activity. Gastroenterology 135, 1228–1237 (2008).
Li, Z. et al. Cancer-associated fibroblasts promote PD-L1 expression in mice cancer cells via secreting CXCL5. Int. J. Cancer 145, 1946–1957 (2019).
Ozdemir, B. C. et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell 25, 719–734 (2014).
Zhang, P. et al. Dissecting the single-cell transcriptome network underlying gastric premalignant lesions and early gastric cancer. Cell Rep. 27, 1934–1947.e5 (2019).
Jeong, H. Y. et al. Spatially distinct reprogramming of the tumor microenvironment based on tumor invasion in diffuse-type gastric cancers. Clin. Cancer Res. 27, 6529–6542 (2021).
Kim, J. et al. Single-cell analysis of gastric pre-cancerous and cancer lesions reveals cell lineage diversity and intratumoral heterogeneity. NPJ Precis. Oncol. 6, 9 (2022).
Luo, H. et al. Pan-cancer single-cell analysis reveals the heterogeneity and plasticity of cancer-associated fibroblasts in the tumor microenvironment. Nat. Commun. 13, 6619 (2022).
Chhabra, Y. & Weeraratna, A. T. Fibroblasts in cancer: unity in heterogeneity. Cell 186, 1580–1609 (2023).
Elhanani, O., Ben-Uri, R. & Keren, L. Spatial profiling technologies illuminate the tumor microenvironment. Cancer Cell 41, 404–420 (2023).
Becht, E. et al. Estimating the population abundance of tissue-infiltrating immune and stromal cell populations using gene expression. Genome Biol. 17, 218 (2016).
Speiser, D. E. et al. CD4(+) T cells in cancer. Nat. Cancer 4, 317–329 (2023).
Guo, M., Liu, M. Y. R. & Brooks, D. G. Regulation and impact of tumor-specific CD4(+) T cells in cancer and immunotherapy. Trends Immunol. 45, 303–313 (2024).
Garcia Garcia, C. J. et al. Stromal HIF2 regulates immune suppression in the pancreatic cancer microenvironment. Gastroenterology 162, 2018–2031 (2022).
Liu, Y. et al. Identification of a tumour immune barrier in the HCC microenvironment that determines the efficacy of immunotherapy. J. Hepatol. 78, 770–782 (2023).
Timperi, E. et al. Lipid-associated macrophages are induced by cancer-associated fibroblasts and mediate immune suppression in breast cancer. Cancer Res. 82, 3291–3306 (2022).
Bi, K. et al. Tumor and immune reprogramming during immunotherapy in advanced renal cell carcinoma. Cancer Cell 39, 649–661.e5 (2021).
Chen, S. et al. Macrophages in immunoregulation and therapeutics. Signal Transduct. Target Ther. 8, 207 (2023).
Bassez, A. et al. A single-cell map of intratumoral changes during anti-PD1 treatment of patients with breast cancer. Nat. Med. 27, 820–832 (2021).
Elyada, E. et al. Cross-species single-cell analysis of pancreatic ductal adenocarcinoma reveals antigen-presenting cancer-associated fibroblasts. Cancer Discov. 9, 1102–1123 (2019).
Kerdidani, D. et al. Lung tumor MHCII immunity depends on in situ antigen presentation by fibroblasts. J. Exp. Med. 219, e20210815 (2022).
Friedman, G. et al. Cancer-associated fibroblast compositions change with breast cancer progression linking the ratio of S100A4(+) and PDPN(+) CAFs to clinical outcome. Nat. Cancer 1, 692–708 (2020).
Huang, H. et al. Mesothelial cell-derived antigen-presenting cancer-associated fibroblasts induce expansion of regulatory T cells in pancreatic cancer. Cancer Cell 40, 656–673.e7 (2022).
Costa, A. et al. Fibroblast heterogeneity and immunosuppressive environment in human breast cancer. Cancer Cell 33, 463–479.e10 (2018).
Mikami, N. et al. Epigenetic conversion of conventional T cells into regulatory T cells by CD28 signal deprivation. Proc. Natl Acad. Sci. USA 117, 12258–12268 (2020).
Baessler, A. & Vignali, D. A. A. T cell exhaustion. Annu. Rev. Immunol. 42, 179–206 (2024).
Miller, B. C. et al. Subsets of exhausted CD8(+) T cells differentially mediate tumor control and respond to checkpoint blockade. Nat. Immunol. 20, 326–336 (2019).
Zebley, C. C. et al. T cell dysfunction and therapeutic intervention in cancer. Nat. Immunol. 25, 1344–1354 (2024).
Cillo, A. R. et al. Blockade of LAG-3 and PD-1 leads to co-expression of cytotoxic and exhaustion gene modules in CD8(+) T cells to promote antitumor immunity. Cell 187, 4373–4388.e15 (2024).
Schumacher, T. N. & Thommen, D. S. Tertiary lymphoid structures in cancer. Science 375, eabf9419 (2022).
Sato, Y. et al. The roles of tertiary lymphoid structures in chronic diseases. Nat. Rev. Nephrol. 19, 525–537 (2023).
Buckley, C. D. et al. Stromal cells in chronic inflammation and tertiary lymphoid organ formation. Annu. Rev. Immunol. 33, 715–745 (2015).
Rodriguez, A. B. et al. Immune mechanisms orchestrate tertiary lymphoid structures in tumors via cancer-associated fibroblasts. Cell Rep. 36, 109422 (2021).
Cheng, H. W. et al. CCL19-producing fibroblastic stromal cells restrain lung carcinoma growth by promoting local antitumor T-cell responses. J. Allergy Clin. Immunol. 142, 1257–1271.e4 (2018).
Kinker, G. S. et al. Mature tertiary lymphoid structures are key niches of tumour-specific immune responses in pancreatic ductal adenocarcinomas. Gut 72, 1927–1941 (2023).
Tsoumakidou, M. The advent of immune stimulating CAFs in cancer. Nat. Rev. Cancer 23, 258–269 (2023).
Kennel, K. B. et al. Cancer-associated fibroblasts in inflammation and antitumor immunity. Clin. Cancer Res. 29, 1009–1016 (2023).
Jerby-Arnon, L. & Regev, A. DIALOGUE maps multicellular programs in tissue from single-cell or spatial transcriptomics data. Nat. Biotechnol. 40, 1467–1477 (2022).
Mayer, S. et al. The tumor microenvironment shows a hierarchy of cell-cell interactions dominated by fibroblasts. Nat. Commun. 14, 5810 (2023).
Croizer, H. et al. Deciphering the spatial landscape and plasticity of immunosuppressive fibroblasts in breast cancer. Nat. Commun. 15, 2806 (2024).
Vanhersecke, L. et al. Mature tertiary lymphoid structures predict immune checkpoint inhibitor efficacy in solid tumors independently of PD-L1 expression. Nat. Cancer 2, 794–802 (2021).
Sautes-Fridman, C. et al. Tertiary lymphoid structures in the era of cancer immunotherapy. Nat. Rev. Cancer 19, 307–325 (2019).
Xiang, X. et al. Targeting tumor-associated macrophages to synergize tumor immunotherapy. Signal Transduct. Target Ther. 6, 75 (2021).
Sun, C. et al. Spatially resolved multi-omics highlights cell-specific metabolic remodeling and interactions in gastric cancer. Nat. Commun. 14, 2692 (2023).
Staats, J. Standardized nomenclature for inbred strains of mice: eighth listing. Cancer Res. 45, 945–977 (1985).
Barnden, M. J. et al. Defective TCR expression in transgenic mice constructed using cDNA-based alpha- and beta-chain genes under the control of heterologous regulatory elements. Immunol. Cell Biol. 76, 34–40 (1998).
Pan, Y. et al. EHBP1L1 drives immune evasion in renal cell carcinoma through binding and stabilizing JAK1. Adv. Sci. 10, e2206792 (2023).
Jin, S. et al. Inference and analysis of cell-cell communication using CellChat. Nat. Commun. 12, 1088 (2021).
Zhang, Y. et al. CellCall: integrating paired ligand-receptor and transcription factor activities for cell-cell communication. Nucleic Acids Res. 49, 8520–8534 (2021).
Browaeys, R., Saelens, W. & Saeys, Y. NicheNet: modeling intercellular communication by linking ligands to target genes. Nat. Methods 17, 159–162 (2020).
Gao, Y. et al. Cross-tissue human fibroblast atlas reveals myofibroblast subtypes with distinct roles in immune modulation. Cancer Cell 42, 1764–1783.e10 (2024).
Cabrita, R. et al. Tertiary lymphoid structures improve immunotherapy and survival in melanoma. Nature 577, 561–565 (2020).
Zhang, L. et al. Single-cell analyses inform mechanisms of myeloid-targeted therapies in colon cancer. Cell 181, 442–459.e29 (2020).
Acknowledgements
This study was sponsored by National Natural Science Foundation of China (82372874 (L.X.W.), 82002545 (L.C.C.)), Natural Science Foundation of Shanghai (21ZR1414600 (L.X.W.)), and Shanghai Sailing Program (20YF1409200; L.C.C.). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of manuscript.
Author information
Authors and Affiliations
Contributions
L.X.W., L.F.L. and W.Y.N. conceived and designed this study. S.J.Q., W.R.Y., L.C.C. and Z.Z.X. performed experiments. S.J.Q., W.R.Y. and L.X.J. analyzed the data. S.J.Q., W.R.Y. and L.C.C. drafted the manuscript. All authors contributed to the article and approved the submitted version.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Christopher Hanley and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Song, J., Wei, R., Liu, C. et al. Antigen-presenting cancer associated fibroblasts enhance antitumor immunity and predict immunotherapy response. Nat Commun 16, 2175 (2025). https://doi.org/10.1038/s41467-025-57465-7
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-57465-7
This article is cited by
-
Cancer-associated fibroblasts: enablers of tumor drug resistance
Journal of Translational Medicine (2026)
-
Tumor microenvironment dynamics in gastric cancer pathogenesis and therapeutic resistance
Molecular Cancer (2026)
-
Reprogramming the tumor immune microenvironment in cervical cancer: synergy between radiotherapy and immunotherapy
Molecular Cancer (2026)
-
A live tumor fragment platform to assess immunotherapy response in core needle biopsies while addressing challenges of tumor heterogeneity
Journal of Translational Medicine (2026)
-
IL-1β-mediated suppression of CIITA attenuates IFN-γ-induced MHC-II expression on Fibroblasts
Cell Communication and Signaling (2025)
