
Abstract
Blockade of the glucagon receptor (GCGR) has been shown to improve glycemic control. However, this therapeutic approach also brings side effects, such as α-cell hyperplasia and hyperglucagonemia, and the mechanisms underlying these side effects remain elusive. Here, we conduct single-cell transcriptomic sequencing of islets from male GCGR knockout (GCGR-KO) mice. Our analysis confirms the elevated expression of Gcg in GCGR-KO mice, along with enhanced glucagon secretion at single-cell level. Notably, Vgf (nerve growth factor inducible) is specifically upregulated in α cells of GCGR-KO mice. Inhibition of VGF impairs the formation of glucagon immature secretory granules and compromises glucagon maturation, lead to reduced α-cell hypersecretion of glucagon. We further demonstrate that activation of both mTOR-STAT3 and ERK-CREB pathways, induced by elevated circulation amino acids, is responsible for upregulation of Vgf and Gcg expression following glucagon receptor blockade. Thus, our findings elucidate parts of the molecular mechanism underlying hyperglucagonemia in GCGR blockade.
Similar content being viewed by others

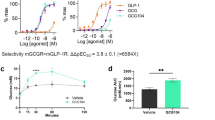
Introduction
Glucagon, a 29 amino acid peptide, was identified a century ago as a hyperglycemic factor derived from the pancreas1. Over the past century, extensive studies have elucidated glucagon’s critical roles in regulating glucose homeostasis, amino acid metabolism, and lipid metabolism2,3. Nevertheless, the full spectrum of glucagon’s functions remains to be fully understood.
Glucagon is secreted by pancreatic α cells and acts as a counter-regulatory hormone to insulin. It binds to the glucagon receptor (GCGR) in hepatocytes, thereby enhancing glycogenolysis and gluconeogenesis, which promotes hepatic glucose production4. Given the central role of the glucagon-GCGR pathway in glucose homeostasis, modulating this pathway has been proposed as a potential therapeutic strategy for diabetes treatment. Indeed, studies have shown that glucagon receptor-knockout (GCGR-KO) mice exhibit resistance to streptozotocin-induced type 1 diabetes and high-fat diet (HFD)-induced type 2 diabetes5,6,7. In recent years, small molecule antagonists, antisense oligonucleotides, and monoclonal antibodies targeting GCGR have been developed and demonstrated efficacy in improving glycemic control in both rodent models of diabetes and human patients with diabetes8,9,10,11,12,13. These findings provide compelling evidence that disrupting GCGR signaling could serve as a viable therapeutic approach for diabetes treatment.
Despite the promising results of GCGR-inhibition in animal models and clinical trials, no such therapies have been approved for clinical use due to adverse effects. The primary side effects associated with GCGR blockade include elevated serum aminotransferase levels, hyperaminoacidemia, α-cell hyperplasia, and hyperglucagonemia11,12,14,15. Hyperglucagonemia may result from α-cell hyperplasia or increased glucagon secretion. To minimize these undesired effects, it is imperative to elucidate the mechanisms underlying hyperglucagonemia before translating GCGR antagonism into clinical practice. Our previous work using a zebrafish model system revealed that GCGR deficiency not only induces α-cell hyperplasia but also upregulates glucagon gene (Gcg) expression in α cells16. However, due to technical limitations in measuring glucagon secretion in zebrafish16, we were unable to determine whether GCGR deficiency increases glucagon secretion at the single-cell level.
In this study, we conducted single-cell transcriptomic analysis of α cells isolated from GCGR-KO mice. Our results unequivocally demonstrate that Gcg gene expression is upregulated and glucagon secretion is also significantly increased in α cells of GCGR-KO mice. Notably, we identified VGF (nerve growth factor inducible) as a key regulator of α cell hypersecretion, whose expression is controlled by both mTOR-STAT3 and ERK-CREB signaling pathways. Collectively, our findings provide insights into the side effects observed following GCGR inhibition and may facilitate the optimization of GCGR-targeting strategies for diabetes therapy.
Results
Disruption of the glucagon receptor significantly alters both gene expression and the interactive network of α cells
To comprehensively examine the functional and physiological changes in α cells following GCGR blockade, we conducted single-cell RNA sequencing (scRNA-seq) on islets isolated from 12–14-week-old male wild-type (WT) and GCGR-knockout (KO) mice (Fig. 1a). After quality filtering (Supplementary Fig. 1), we obtained transcriptomic profiles of 12,588 single cells, including 5667 cells from WT mice and 6921 cells from GCGR-KO mice. Major cell populations within the pancreatic islet were identified based on their characteristic gene expression profiles, including endocrine cells (α, β, δ, and Pp), exocrine cells (acinar and ductal), and other known cell types (mesenchymal, endothelial, immune, and stellate) (Supplementary Fig. 2).

a Schematic representation of the experimental workflow for single-cell RNA-sequencing (scRNA-seq) of WT and GCGR-KO mouse islet. b t-SNE visualization of scRNA-seq data from a total of 4399 α cells. 8 clusters are labeled with different colors as indicated on the right. c Analysis of cell number and distribution across different clusters. d Heatmap illustrating the number of interactions between various cell types. The colored scale bar indicates an increase (red) or decrease (blue) of interaction number in GCGR-KO islets compared to WT islets. e Bubble plot depicting the significantly altered ligand-receptor pairs contributing to signaling pathways from α, β, δ, pp cells to α cells. Red or blue in the color scale bar indicates an increase or decrease signal intensity in GCGR-KO islets relative to WT islets. f, g Effects of somatostatin-14 (SST-14) and insulin on glucose-stimulated glucagon secretion from WT (circles) and GCGR-KO (squares) islets. Islets were incubated in the presence of 1 mM glucose and varying concentrations of SST-14 or insulin for 1 h. Data are collected from 3 independent experiments and expressed as a percentage of maximal secretion, with secretion in the presence of 1 mM glucose alone defined as 100%. Data presented in (f, g) are mean ± SEM and analyzed by two-way ANOVA, followed by Bonferroni post-hoc test. p-values < 0.05 are displayed. Source data are provided as a Source Data file.
To elucidate the functional characteristics of α cells, we performed unsupervised clustering of all α cells identified in our initial analyses, resulting in the identification of a total of eight distinct clusters. (Fig. 1b, c). Among these, one subcluster exhibited high expression of somatostatin (Sst), while another showed increased proliferative capacity in GCGR-KO α cells (Supplementary Fig. 3a–c). Additionally, GCGR-KO α cells expressed elevated levels of Slc38a5, consistent with previous reports17,18 (Supplementary Fig. 3d). Analysis of intercellular interactions revealed an increased number of interactions between α cells and other endocrine cell types in GCGR-KO islets compared to WT islets (Fig. 1d). Further investigation on signal transduction pathways among the three major pancreatic endocrine cell types demonstrated that the probability of interaction between key regulators of glucagon secretion, such as ligand-receptor pairs Sst-Sstr2, Sst-Sstr3, Ins1-Insr, and Ins2-Insr, were significantly reduced in GCGR-KO α cells (Fig. 1e and Supplementary Fig. 3e, f). Experimentally, we confirmed that the inhibitory effects of somatostatin bioactive forms (SST-14) and insulin on glucagon secretion was markedly diminished in GCGR-KO islets (0.1 μM SST-14: 48.2% inhibition; 0.1 μM insulin: 25.5% inhibition) compared to WT islets (0.1 μM SST-14: 75.7% inhibition; 0.1 μM insulin: 50.5% inhibition) (Fig. 1f, g).
Collectively, these findings indicate that disruption of the glucagon receptor not only modifies the gene expression profile of α cells but also alters the pancreatic islet microenvironment.
Disruption of the glucagon receptor increases glucagon expression and granule populations in α cells
Our previous studies have shown that GCGR deficiency increases glucagon expression in zebrafish α cells16. To determine whether this phenomenon extends to mice, we analyzed Gcg expression levels in our mouse islet scRNA-Seq data. Consistent with prior observations, Gcg expression was significantly upregulated in α cells from GCGR-KO mice (Fig. 2a, b).

a Violin and t-SNE plots illustrating the expression levels and distribution of Gcg mRNA in α cells. b Violin plot depicting Gcg expression across different α-cell subclusters. In a and b, the whiskers indicated the maximum and minimum values, the center indicated the median and the bound of the box indicated upper and lower quartiles. c Representative confocal microscopy images of pancreatic sections stained with anti-glucagon antibody. Scale bar, 50 μm. d Quantification of the average single α-cell area (WT, n = 4 mice; GCGR-KO, n = 4 mice). e Quantification of glucagon fluorescence intensity per α cell (WT, n = 4 mice; GCGR-KO, n = 4 mice). f, g Representative TEM images of α-cells from WT and GCGR-KO islets. Yellow dotted lines indicate cell boundaries, and gray or black dots indicate the glucagon granules. Scale bar, 5 μm. h Quantification of the number of glucagon granules per α cell, normalized to cell area. (WT, n = 3 mice; GCGR-KO, n = 3 mice). i Quantification of individual glucagon granule area (WT, n = 3 mice; GCGR-KO, n = 3 mice). j Density curve of individual glucagon granule areas (WT, n = 2313 granules; GCGR-KO, n = 3289 granules). Data presented in (d, e, h, i) are mean ± SEM, analyzed by an unpaired two-tailed t-test. p-values < 0.05 are displayed. Source data are provided as a Source Data file.
We next investigated whether the glucagon protein level and glucagon granule number were altered in GCGR-KO mice. Immunostaining of pancreatic sections using a glucagon-specific antibody revealed that both the size of α cells and the total glucagon fluorescence intensity of each single α cells were significantly increased in GCGR-KO mice (Fig. 2c–e). Ultrastructural analysis via transmission electron microscopy (TEM) further demonstrated an increase in the number and size of glucagon granules in α cells from GCGR-KO mice (Fig. 2f–j). Collectively, these results confirm that disruption of the glucagon receptor in mice significantly enhances glucagon expression and glucagon granule populations in α cells.
Disruption of glucagon receptor increases α-cell glucagon secretion
Given the observed increases in both mRNA and protein levels of glucagon in α cells from GCGR-KO mice (Fig. 2), we then examined whether glucagon secretion was similarly elevated. We isolated pancreatic islets from WT and GCGR-KO mice and assessed the glucagon secretion under varying glucose concentrations. As shown in Fig. 3a, GCGR-KO islets exhibited higher basal glucagon secretion at 7 mM glucose. Upon stimulation with low glucose (1 mM), glucagon secretion increased significantly in both WT and GCGR-KO groups. Notably, when normalized 1 mM glucose stimulated glucagon secretion to the baseline (1 mM to 7 mM glucose) of each group, the fold-change in glucagon secretion was significantly higher in the GCGR-KO group compared to the WT group (2.4 vs 1.8, p < 0.001) (Fig. 3b). These results suggest that GCGR-KO islets are more sensitive to low glucose-induced glucagon secretion.

a Measurement of glucagon secretion from isolated WT and CGGR-KO mouse islets. Data are derived from 3 biological replicates. b Fold change in glucagon secretion for each group. Data from (a) were normalized to the baseline (7 mM glucose) for each respective group. c Representative confocal microscopy images of WT and GCGR-KO α-cells before and after stimulation with 1 mM glucose + 10 μM adrenaline (adr). Glucagon granules are visualized by NPY-mCherry expression. Scale bar, 5 μm. d Average exocytosis as a function of time for WT and GCGR-KO α-cells as in (c). For WT, n = 8 cells/4 mice; for GCGR-KO, n = 9 cells/3 mice. Cells were stimulated with 1 mM glucose + 10 μM adrenaline from 1 to 5 min. e Variance in Fluo4 intensity when WT or GCGR-KO islets were perfused with different stimulation (as indicated). The arrow indicates α cells that started spiking after application of 1 mM glucose. Scale bar, 50 μm. f Pancreatic α-cell Ca2+ oscillation in response to a series of stimulations. In the heatmap, each column represents a time point, and each row represents one cell. The color scale ranges from red (high intensity) to blue (low intensity) (upper panel). The average Ca2+ oscillation of α-cells in response to a series of stimulations is shown in the time course (lower panel). Calcium influx responses for each α cell were normalized to the initial fluorescence intensity (20 mM glucose). For WT, n = 53 cells/3 mice; for GCGR-KO, n = 71 cells/3 mice. g Comparison of Ca2+ oscillation intensity in WT and GCGR-KO α-cell in response to indicated stimulations. The whiskers indicated the maximum and minimum values, the center indicated the median and the bound of the box indicated the upper and lower quartiles. Data presented in panels (a, b, d, g) are mean ± SEM. Data in (a, d, g) were analyzed using two-tailed unpaired t-tests. Data in (b) were analyzed by two-way ANOVA with Bonferroni’s post hoc test. p-values < 0.05 are displayed. Source data are provided as a Source Data file.
Secondly, we monitored glucagon granule exocytosis in α cells by tracking the trafficking of NPY-mCherry, which is driven by the pre-proglucagon promoter and serves as a well-established secretory granule marker19 (Supplementary Fig. 4). Following transduction with an adenovirus carrying the NPY-mCherry construct for 72 h, we induced glucagon granule exocytosis using a combination of 1 mM glucose and 10 μM adrenaline. Confocal microscopy was employed to record the exocytotic events of individual α cells. As illustrated in Fig. 3c, d, GCGR-KO α cells exhibited significantly higher levels of glucagon granule exocytosis under stimulating conditions, supporting the hypothesis that glucagon exocytosis is markedly enhanced in α cells from GCGR-KO mice.
Thirdly, given that glucagon secretion is a Ca2+-dependent process triggered by elevated cytoplasmic Ca2+ concentrations, we evaluated changes in cytoplasmic Ca2+ levels in α cells. Islets isolated from WT and GCGR-KO mice were loaded with the cytoplasmic Ca2+ indicator Fluo-4 AM, and cytoplasmic Ca2+ concentration responses to stimulation were recorded by confocal time-lapse imaging (Fig. 3e). Based on previous studies, cells responding to adrenaline stimulation were identified as α cells20,21. As shown in Fig. 3f–g, both WT and GCGR-KO α cells exhibited rapid calcium influx responses, but the frequency and magnitude of cytoplasmic Ca2+ concentration oscillations were notably more pronounced in GCGR-KO α cells. Collectively, these findings suggest that glucagon secretion is significantly increased in the α cells from GCGR-KO mice.
Disruption of the glucagon receptor specifically leads to increased VGF levels in α cells
To further investigate the mechanism underlying α cell glucagon hypersecretion following glucagon receptor knockout, we analyzed differentially expressed genes (DEGs) in α-cells between the WT and GCGR-KO groups. Gene Ontology (GO) analyses revealed that upregulated genes were enriched in pathways associated with endoplasmic reticulum stress response, protein exit from endoplasmic reticulum, hormone transport, and hormone secretion (Supplementary Fig. 5a and Supplementary Data 1). Among the 12 genes related to hormone secretion, Vgf (nerve growth factor inducible) showed a highly significant increase in expression in GCGR-KO α-cells (Log2FC = 1.38; p = 2.21E-203) (Fig. 4a). Moreover, when comparing the expression of these 12 genes across all pancreatic cell types between WT and GCGR-KO mice, Vgf was the only gene that specifically increased in α-cells of GCGR-KO mice (Fig. 4b and Supplementary Fig. 5b–i).

a Violin plot showing the expression levels of Vgf in WT and GCGR-KO α cells. The whiskers indicated the maximum and minimum values, the center indicated the median and the bound of the box indicated the upper and lower quartiles. b Violin plot illustrating Vgf expression across all cell types. c Representative confocal microscopy images of VGF expression in pancreas from WT and GCGR-KO mice. Scale, 50 μm. d Quantification of VGF fluorescence intensity in α-cells from WT (n = 4 mice) and GCGR-KO (n = 4 mice) mouse pancreas sections. e Immunofluorescence staining of VGF and glucagon in αTC1-6 cells. Scale bar, 10 μm. f, g Representative confocal microscopy images of αTC1-6 cells transfected with sh-Control or sh-VGF adenovirus. In (f), cells were stained with anti-glucagon (green) and anti-VGF (red) antibodies. In (g), cells were stained with anti-glucagon (green) and anti-GM130 (red) antibodies. Scale bar, 10 μm. h Western blot analysis of the glucagon protein levels in αTC1-6 cells after VGF knockdown. Cells were stimulated with 1 mM glucose +10 μM adrenaline for 2 h. i Measurement of glucagon secretion from αTC1-6 cells at 1 mM glucose after VGF knockdown. Data are derived from 3 independent experiments. j Immunofluorescence staining of SCG2, CHGA and SCG3 in αTC1-6 cells. Scale bar, 10 μm. k Western blot analysis the SCG2, CHGA and SCG3 protein levels in αTC1-6 cells after VGF knockdown. l Quantification of SCG2, CHGA and SCG3 protein levels in (k). Values were normalized to GAPDH, and data are derived from 3 independent experiments. Data presented in (d, i, l) are mean ± SEM and were analyzed using two-tailed unpaired t-tests. p-values < 0.05 are displayed. Source data are provided as a Source Data file.
To validate the scRNA-seq results, we performed VGF immunostaining on pancreatic sections from WT and GCGR-KO mice. We demonstrated a considerable increase in VGF signals specifically in the α-cells of GCGR-KO mice (Fig. 4c, d). To investigate the subcellular localization of VGF, we also co-stained αTC1-6 cells with anti-glucagon and anti-VGF antibodies. As shown in Fig. 4e, most glucagon granules co-localized with VGF. To explore whether VGF expression could increase α-cell glucagon secretion, we constructed an adenovirus vector expressing VGF under the control of the glucagon promoter (Supplementary Fig. 6a, b) and treated islets isolated from WT mice with control or VGF-expression adenovirus for 72 h. Interestingly, we found that increased VGF expression was accompanied by increased glucagon secretion (Supplementary Fig. 6c–e).
Down-regulation of VGF impairs biogenesis of glucagon granules and suppresses glucagon hypersecretion
To further elucidate the role of VGF in glucagon hypersecretion, we employed adenovirus-mediated shRNA targeting Vgf (Supplementary Fig. 7a). The shRNA effectively reduced Vgf expression in both the αTC1-6 cell line and cultured primary islets (Supplementary Fig. 7b, c). When the αTC1-6 cell line was transduced with adenovirus expressing sh-VGF, we observed that the glucagon granules formed larger and concentrated foci near the nuclear periphery, which were distinct from their distribution in the sh-Control group (Fig. 4f). This congregation pattern of glucagon granules in sh-VGF-treated cells resembled the Golgi network. To test this possibility, we co-immunostained glucagon and the Golgi marker GM130 in αTC1-6 cells. Indeed, we observed a significant accumulation of glucagon in a subcellular compartment co-stained with the Golgi marker GM130 after VGF inhibition (Fig. 4g). Given the altered subcellular localization of glucagon granules in VGF-deficient α cells, we investigated whether VGF loss also impacted glucagon maturation. As shown in Fig. 4h, mature glucagon levels were significantly decreased in VGF-deficient α cells, while proglucagon levels remained unchanged between the two groups. Moreover, glucagon secretion was reduced in VGF-knockdown αTC1-6 cells (Fig. 4i). These observations indicate that VGF is required for granule biogenesis and glucagon maturation in α cells.
VGF is a member of the granin family, and the coordination among its family members plays a crucial role in the biogenesis of secretory granules22. When analyzing our scRNA-seq data, we observed that three additional granin family members: Scg2 (Secretogranin II), Chga (Chromogranin A) and Scg3 (Secretogranin III), were upregulated in α cells following glucagon receptor disruption (Supplementary Fig. 8a). During secretory granule biogenesis, SCG2 and CHGA condense to form large aggregates, while SCG3 functions as a trans-membrane sorting adaptor22. Immunofluorescence assays in αTC1-6 cells revealed colocalization of SCG2, CHGA and SCG3 with glucagon granules (Fig. 4j). Notably, VGF suppression led to significant reductions in SCG2 and CHGA levels but did not affect SCG3 expression (Fig. 4k, l and Supplementary Fig. 8b–d). These data suggest that VGF suppression impairs the formation of large aggregates of proglucagon and granin proteins, thereby reducing glucagon granule biogenesis.
To investigate whether VGF suppression affects glucagon levels in GCGR-KO islets, we isolated islets from GCGR-KO mice and transfected them with adenovirus expressing sh-Control or sh-VGF. Subsequent immunostaining for VGF and glucagon revealed a significant decrease in glucagon fluorescence intensity upon VGF depletion (Fig. 5a–c). Additionally, we also investigated the glucagon secretion using perfused GCGR-KO pancreatic islets. In the sh-Control group, there was an increase in glucagon secretion when the glucose concentration switched from 7 to 1 mM. However, in the sh-VGF group, glucagon secretion was blunted in response to lower glucose (Fig. 5d, e).

a Representative confocal microscopy images of GCGR-KO islets transfected with sh-Control or sh-VGF adenovirus. Scale bar, 50 μm. b, c Quantification of α-cell VGF and glucagon fluorescence intensity in (a). For sh-Control, n = 9 islets; for sh-VGF, n = 8 islets. d Dynamic glucagon secretion profiles from perfused GCGR-KO mouse islets transfected with sh-Control or sh-VGF adenovirus. Islets were perfused with KRBH solution containing either 7 mM glucose or 1 mM glucose. Data are presented as mean values ± SEM. e Corresponding area under the curve (AUC) of glucagon secretion from ex vivo perfused islets of GCGR-KO mice. f Schematic diagram of VGF knockdown in vivo. GCGR-KO mice were intraperitoneally injected with AAV-sh-Control or AAV-sh-VGF. Samples were harvested and assessed 14 days post-injection. g Representative confocal microscopy images of VGF expression in pancreas from mice injected with AAV-sh-Control or AAV-sh-VGF. Scale bar, 50 μm. h Quantification of VGF fluorescent intensity in α-cells from (g). For sh-Control, n = 3 mice; for sh-VGF, n = 3 mice. i Average serum glucagon levels in WT (n = 3), GCGR-KO (n = 3), GCGR-KO injected with AAV-sh-Control (n = 4), and GCGR-KO injected with AAV-sh-Control (n = 4) mice. j Dynamic glucagon secretion profiles from perfused islets isolated from GCGR-KO mice injected with AAV-sh-Control or AAV-sh-VGF. Data are presented as mean values ± SEM. k Corresponding area under the curve (AUC) of glucagon secretion from ex vivo perfused islets in (j). Data presented in (b, c, e, h, i, k) are mean ± SEM. Data in (b, e, h, k) were analyzed using two-tailed unpaired t-tests. Data in (c) were analyzed by Mann Whitney test. Data in (i) were analyzed by one-way ANOVA with Bonferroni’s post hoc test. p-values < 0.05 are displayed. Source data are provided as a Source Data file.
To further explore the in vivo effects of VGF suppression on glucagon secretion, we generated an AAV-pan vector expressing VGF shRNA, which has high transfection efficiency in the pancreas23, with non-targeting shRNA serving as a control (AAV-sh-Control) (Fig. 5f). After two weeks of AAV vector exposure, we confirmed a significant reduction in VGF signal in the α-cells of GCGR-KO mice injected with AAV-sh-VGF (Fig. 5g, h). Importantly, serum glucagon levels were significantly lower in AAV-sh-VGF-injected mice compared to controls (Fig. 5i). Although serum insulin levels were slightly reduced in AAV-sh-VGF-injected mice, serum GLP1 levels remained unaffected (Supplementary Fig. 9). Perfusion experiments using islets isolated from these mice revealed that while the sh-Control group showed increased glucagon secretion in response to decreasing glucose concentration from 7 mM to 1 mM, this response was diminished in the sh-VGF group (Fig. 5j, k). Taken together, these data indicate that VGF is a key mediator of glucagon hypersecretion in α cells of GCGR-KO mice.
Hyperaminoacidemia induces VGF upregulation in pancreatic α cells
We sought to identify the inducer of VGF upregulation. Given that α cell adaptations in GCGR antagonism/knockout are primarily driven by circulating factors, particularly elevated amino acids, such as glutamine and alanine17,18,24, we focused on serum and hyperaminoacidemia. We treated αTC1-6 cells with serum collected from WT or GCGR-KO mice for 72 h and found that VGF and proglucagon protein levels were significantly increased in GCGR-KO serum-treated cells (Fig. 6a, b). Similarly, WT islets treated with GCGR-KO mouse serum exhibited higher VGF expression levels (Fig. 6c, d). Moreover, compared with WT serum-treated group, GCGR-KO serum-treated islets showed increased glucagon secretion under both normal (7 mM glucose) and low glucose (1 mM glucose) conditions (Fig. 6e).

a Western blot analysis of VGF and pro-glucagon protein levels in αTC1-6 cells after incubation with WT or GCGR-KO serum for 72 h. b Quantification of VGF and pro-glucagon protein levels from (a). Values were normalized to GAPDH, and data were generated from 3 independent experiments. c Representative confocal microscopy images of pancreatic islets after 72 h incubation with WT or GCGR-KO serum. Scale bar, 50 μm. d Quantification of VGF fluorescence intensity in α cells of islets following serum treatment (WT serum, n = 11 islets; GCGR-KO serum, n = 13 islets). e Glucagon secretion levels from islets treated with WT or GCGR-KO serum. Serum-incubated islets were stimulated at different glucose concentrations for 1 h. Data were generated from 3 independent experiments. f Schematic diagram of serum amino acid measurement. Serum was collected from 6 WT or GCGR-KO mice. g Quantification of serum amino acid levels in WT and GCGR-KO mice. h Western blot analysis of VGF and pro-glucagon protein levels in αTC1-6 cells after incubation with 4 mM glutamine and alanine for 72 h. i Quantification of VGF and pro-glucagon protein levels in (h). Values were normalized to GAPDH, and data were generated from 4 independent experiments. j Representative confocal microscopy images of pancreatic islets after 72 h incubation with 4 mM glutamine and alanine. Scale bar, 50 μm. k Quantification of VGF fluorescence intensity in α cells of islets following amino acid treatment (Control, n = 8 islets; Gln+Ala, n = 8 islets). l Glucagon secretion levels from islets following 72 h treatments with indicated conditions and subsequent stimulated with different glucose concentrations for 1 h. Data were generated from 3 independent experiments. Data presented in (b, d, e, g, i, k, l) are mean ± SEM. Data in (b, d, e, g, i, k) were analyzed using two-tailed unpaired t-tests. Data in (l) were analyzed by two-way ANOVA with Bonferroni’s post hoc test. p-values < 0.05 are displayed. Source data are provided as a Source Data file.
We then surveyed the serum levels of 20 common amino acids in our GCGR-KO mice (Fig. 6f). Consistent with previous studies17,18,25, glucagon receptor blockade leads to hyperaminoacidemia, and glutamine and alanine were the top two changed amino acids (Fig. 6g and Supplementary Data 2). To test whether hyperaminoacidemia induces VGF upregulation, we treated αTC1-6 cells and WT islets with high levels of glutamine and alanine. This treatment significantly increased VGF and proglucagon levels in both αTC1-6 cells and WT islets (Fig. 6h–k). Furthermore, glucagon secretion from high-glutamine- and alanine-treated islets was increased under both normal (7 mM glucose) and low glucose (1 mM glucose) conditions (Fig. 6l). Importantly, VGF suppression using sh-VGF adenovirus abolished the glutamine- and alanine-induced glucagon hypersecretion (Fig. 6l).
Taken together, these data indicate that increased circulating glutamine and alanine resulting from GCGR blockade drive VGF upregulation and glucagon hypersecretion in α cells.
STAT3 and CREB coordinate the hyperaminoacidemia-induced VGF upregulation
To elucidate the mechanism of glutamine and alanine-induced VGF upregulation in α cells, we used the databases including AnimalTFDB26, GTRD27, and TFBIND28 to predict the transcription factors that could bind to the VGF promoter. By examining the intersecting data from these databases, we identified six candidate transcription factors: STAT3, CREB, ELK1, SRF, PBX1, and SP1 (Fig. 7a). We constructed a luciferase reporter driven by the Vgf promoter and found that glutamine and alanine treatment significantly increased Vgf promoter activity (Fig. 7b). By using siRNA to knockdown each of these six transcription factors, we observed that only STAT3 and CREB suppression significantly reduced the Vgf promoter activity and Vgf expression when αTC1-6 cells were treated with glutamine and alanine (Fig. 7c and Supplementary Fig. 7d–j).

a Venn diagrams showing transcription factors that can bind to the VGF promoter, as identified in the animalTFDB3.0, GTRD and TFBIND databases. b Amino acids increased VGF promoter luciferase activity in αTC1-6 cells incubated with 4 mM glutamine and alanine for 72 h (n = 6). c Screening for transcription factors involved in amino acid-induced VGF promoter activity. αTC1-6 cells were transfected with indicated siRNA and incubated with amino acids for 72 h. Data were generated from 3 independent experiments. d Western blot analysis of p-STAT3-S727, p-STAT3-Y705 and total STAT3 protein levels in αTC1-6 cells incubated amino acids for 72 h. e Quantification of relevant protein levels in (d). Data were generated from 3 independent experiments. f Representative confocal images of p-STAT3-S727 expression in the pancreatic sections from WT or GCGR-KO mice. Scale bar indicates 50 μm. g Western blot analysis of p-STAT3-S727, STAT3, VGF and pro-glucagon protein levels in αTC1-6 cells treated with S3I-201 (STAT3 inhibitor). h Quantification of relevant protein levels in (g). Data were generated from 3 independent experiments. i. Representative confocal microscopy images of VGF and glucagon expression in αTC1-6 cells treated with amino acids alone or plus S3I-201 for 72 h. Scale bar, 10 μm. j VGF promoter activity in αTC1-6 cells transfected with empty vector, wild-type STAT3 or constitutively active STAT3 mutant (STAT3-S727D) for 72 h. Data were generated from 3 independent experiments. k Diagram of STAT3 binding sites at VGF promoter. l Chip-qPCR analysis of STAT3 binding activity at VGF promoter. Data were generated from 3 independent experiments. m Western blot analysis of protein levels after treatment with amino acids or amino acids plus rapamycin in αTC1−6 cells for 72 h. n Quantification of relevant protein levels in (m). Data were generated from 3 independent experiments. Data presented in (b, c, e, h, j, l, n) are mean ± SEM. Data in (b, e, l) were analyzed using two-tailed unpaired t-tests. Data in (c, h, j, n) were analyzed by one-way ANOVA with Bonferroni’s post hoc test. p-values < 0.05 are displayed. Source data are provided as a Source Data file.
CREB drives a master transcriptional network and regulates Vgf and Gcg expression29,30. We confirmed increased p-CREB and p-ERK levels in αTC1-6 cells upon high-level glutamine and alanine treatment (Supplementary Fig. 10a–c). Knockdown of CREB decreased the Vgf and Gcg promoter activity induced by high-level glutamine and alanine (Fig. 7c and Supplementary Fig. 10d). These data suggest that CREB regulates Vgf and Gcg promoter activity during hyperaminoacidemia-induced glucagon hypersecretion in α cells.
We also found that STAT3 controls Vgf promoter activity in α cells (Fig. 7c). Since STAT3 activation involves phosphorylation at either tyrosine 705 (Y705) or serine 727 (S727) residues31, we investigated which active form of STAT3 was involved. As shown in Fig. 7d, e, phosphorylated STAT3-S727 (p-STAT3-S727), but not STAT3-Y705 (p-STAT3-Y705), significantly increased in αTC1-6 cells after high-level glutamine and alanine administration. In mice, p-STAT3-S727, but not p-STAT3-Y705, was dramatically increased in GCGR-KO α cells (Fig. 7f). Conversely, treatment of αTC1-6 cells with the STAT3 inhibitor S3I-201 markedly decreased amino acid-induced STAT3 phosphorylation and VGF levels but not proglucagon levels (Fig. 7g, h). Interestingly, S3I-201 treatment also induced the accumulation of glucagon around the nucleus (Fig. 7i), similar to VGF-knockdown αTC1-6 cells (Fig. 4f). Additionally, S3I-201 treatment suppressed amino acids-induced VGF upregulation and glucagon hypersecretion in cultured islets (Supplementary Fig. 11). Moreover, overexpression of constitutively active STAT3 mutants (STAT3-S727D), but not wild-type STAT3, was sufficient to induce Vgf promoter activity (Fig. 7j). By visual search in the JASPAR database, we noticed two potential STAT3 binding sites in the promoter region of Vgf (Fig. 7k). Our ChIP–qPCR analysis confirmed that STAT3 displayed high binding affinity with these two sites, which was dramatically increased upon amino acid treatment (Fig.7l). All these data support p-STAT3-S727 as a key mediator of hyperaminoacidemia-induced VGF upregulation.
Given that mTOR is the main sensor of amino acids and a key modulator of α-cell adaptations of GCGR antagonism25, we tested whether mTORC1 is necessary for STAT3 activation under hyperaminoacidemic conditions. Indeed, rapamycin, an inhibitor of mTOR, suppressed the amino acid-induced upregulation of p-STAT3-S727 and decreased VGF levels (Fig. 7m, n).
Collectively, these data suggest that mTOR-STAT3 and ERK-CREB pathways coordinate hyperaminoacidemia-induced VGF upregulation in α cells under GCGR blockade.
Discussion
In this study, we utilized scRNA-seq to examine the cellular and molecular consequences of glucagon receptor disruption in murine α cells. Our analyses revealed a significant upregulation in the expression of Gcg and Slc38a5, along with a modest proportion of proliferating α cells (Figs. 1, 2, Supplementary Figs. 2, 3). Additionally, we observed notable changes in the islet microenvironment of GCGR-KO mice, characterized by impaired inhibitory effects of insulin and somatostatin on glucagon secretion (Fig. 1e–g). We also found that the number and size of glucagon granules were markedly increased in α cells isolated from GCGR-KO mice (Fig. 2f–j). Functional assays demonstrated a substantial enhancement of glucagon secretion in islets from GCGR-KO mice compared to wild-type controls (Fig. 3a, b), with increased glucagon granule exocytosis and elevated cytoplasmic Ca2+ concentrations at the single-cell level (Fig. 3c–g). Collectively, these findings provide compelling evidence that GCGR disruption leads to a substantial increase in glucagon secretion from α cells.
Numerous studies have revealed that GCGR blockade leads to α-cell hyperplasia7,17,18,24,32,33. However, none of the studies investigated glucagon secretion at the single-cell level in GCGR-deficient α cells. While some pancreas perfusion studies have shown increased glucagon secretion following GCGR disruption34, it has been challenging to determine whether this increase is due to α-cell hyperplasia or enhanced secretion from individual α cells.
Our current study reveals that GCGR blockade induces α-cell hypersecretion beyond α-cell hyperplasia. Together with our previous findings16, we demonstrate that GCGR blockade not only induces α-cell hyperplasia but also increases glucagon expression and secretion at the single-cell level. Both α-cell hyperplasia and single α-cell hypersecretion contribute to hyperglucagonemia in circulation (Fig. 8a).

a Upon glucagon receptor blockade, the organism increases its demand for glucagon. Several factors (particularly elevated amino acids), arise from the GCGR-deficient liver and act on pancreatic islets, leading to α cell hyperplasia and enhanced α cell secretion. All these changes collectively contribute to hyperglucagonemia. b Increased circulating amino acids (especially glutamine and alanine) activate the mTOR-STAT3 and ERK-CREB signaling pathways. STAT3 enhances VGF transcription, while CREB promotes both VGF and GCG expression. Consequently, glucagon granule biogenesis and glucagon secretion are significantly increased. Conversely, blocking amino acid-induced VGF expression by inhibiting mTOR or STAT3 activation reduces levels of the glucagon granule component VGF, thereby decreasing glucagon granule biogenesis and glucagon secretion.
By analyzing scRNA-seq data, we identified a specific increase in Vgf expression in GCGR-KO α cells (Fig. 4a, b). Inhibition of Vgf expression in the GCGR-KO islets significantly reduced glucagon secretion (Fig. 5a–e). Importantly, serum glucagon levels were also significantly reduced when VGF expression was inhibited in vivo (Fig. 5f–k). Further experiments indicated that suppressing the expression of VGF in the αTC1-6 cell line led to the accumulation of glucagon near the Golgi apparatus, as well as reduced glucagon secretion and decreased glucagon peptide maturation (Fig. 4f–i). Moreover, inhibition of VGF also down-regulated SCG2 and CHGA, two other components of pre-hormone aggregates, but not the trans-membrane sorting adaptor SCG3 (Fig. 4k–l and Supplementary Fig. 8). These data demonstrate that VGF is crucial for glucagon hypersecretion of α cells from GCGR-KO mice and plays a role in regulating glucagon granule biogenesis. We further identified the glutamine and alanine (the top two amino acids increased in serum from GCGR-KO mice) as inducers of α cell VGF upregulation (Fig. 6). Elevated amino acid levels regulated VGF increase through mTOR-STAT3 signaling activation (Fig. 7) and induced ERK-CREB signaling activation, contributing to both Gcg and Vgf upregulation (Supplementary Fig. 10).
Taken together, these data suggest that glucagon receptor blockade results in an extreme increase in serum glutamine and alanine. These elevated amino acids activate the mTOR-STAT3 and ERK-CREB pathways, increasing both VGF and glucagon transcription in α cells. Consequently, glucagon granule biogenesis and glucagon secretion are significantly increased in GCGR-deficient α cells (Fig. 8b).
VGF is known to work together with other granin proteins to regulate granule biogenesis in the trans‑Golgi network (TGN)22. It regulates β-cell function by modulating the biogenesis of insulin secretory granules35. Although our knowledge of glucagon granule biogenesis and exocytosis is relatively rudimentary, this study, along with others, suggests that the mechanism of glucagon secretion shares considerable similarities with insulin secretion19,36,37,38. In this study, we reveal that VGF is required for hyperglucagonemia.
Hyperglucagonemia is associated with type 1 and type 2 diabetes, leading to increased hepatic glucose production and hyperglycemia39,40,41. To investigate whether GCG, VGF, SCG2, SCG3, and CHGA contribute to hyperglucagonemia in individuals with diabetes and diabetic mice, we examined gene expression data from α cells in patients with type 2 diabetes (GSE84133)42 and high-fat diet (HFD)-induced obese mice (GSE203151)43. In individuals with type 2 diabetes, we observed elevated expression levels of GCG, VGF, SCG2, SCG3, and CHGA in α cells (Supplementary Fig. 12). Similarly, in HFD-induced obese mice, we found an increase in the expression of Gcg, Scg2, Scg3, and Chga, accompanied by a rise in VGF protein levels (Supplementary Fig. 12). These data suggest that VGF and other granin proteins may be associated with hyperglucagonemia in diabetes.
CREB is well known to regulate Gcg and Vgf transcription29,30,44. In this study, we confirmed that CREB increased both Gcg and Vgf expression in α cells under hyperaminoacidemia conditions (Fig. 7c and Supplementary Fig. 10). Importantly, we found that phosphorylation of serine 727 residue of STAT3, p-STAT3-S727, increased in hyperaminoacidemia-treated α cells, further elevating Vgf transcription in an mTOR-dependent manner. The mTOR complex is a sensor of cellular nutrients, and activation of mTOR signaling in pancreatic α cells leads to cell mass expansion, increased glucagon content, and enhanced glucagon secretion, while suppression of mTOR signaling results in the opposite effect45,46. Blockade of the glucagon receptor results in hyperaminoacidemia in circulation and persistent activation of mTOR signaling in α cells, which works through the liver–α-cell axis17,18,25,47. These studies reveal that mTOR signaling activation is required for α-cell proliferation. Furthermore, we have found that mTOR-STAT3 signal activation plays a role in α cell proliferation24. In this study, we further demonstrated that mTOR-STAT3 is essential for regulating VGF, a glucagon granule component, which is upregulated in GCGR-KO α cells. Despite only a small proportion of proliferating α cells in GCGR-KO islets (Supplementary Fig. 3b and Fig. 2c), most α cells in GCGR-deficient mice were mTOR and p-STAT3-S727 positive and accompanied by elevated VGF levels and hypersecretion of glucagon (Fig. 2a, Fig. 4a, Fig. 7f and Kang et al.24, Solloway et al. 25), suggesting an important role for the mTOR-STAT3-VGF axis in regulating glucagon secretion beyond proliferation. Recent studies have also revealed that chronic hyperglycemia results in sustained activation of mTORC1 in α cells, leading to increased glucagon secretion48. Our data, together with those of Riahi et al.48, reveal a potentially novel function of mTOR in the nutrient-dependent regulation of glucagon secretion.
Indeed, this study has some limitations. Firstly, although we observed that the frequency and magnitude of Ca2+ oscillation were more pronounced in α cells from GCGR-KO mice, the detailed mechanism still needs to be elucidated. Based on our observations (Fig. 3e–g), low glucose, known to open calcium channel and lead to extracellular calcium influx, significantly enhanced the calcium response in GCGR-KO α cells, suggesting increased extracellular calcium influx. However, whether this increase correlates with hyperaminoacidemia requires further investigation. Additionally, adrenaline, which induces cAMP elevation and triggers calcium release from the endoplasmic reticulum, stimulates higher magnitude of calcium oscillation in GCGR-KO α cells, indicating increased calcium release from the endoplasmic reticulum. How hyperaminoacidemia increases ER calcium released in α cells from GCGR-KO mice needs further exploration. Secondly, although we revealed the endogenous function of VGF in hyperglucagonemia of GCGR-KO α cells, VGF polypeptides can be proteolytically cleaved into multiple bioactive peptides by prohormone convertases PC1/3 and PC249, and some of these bioactive peptides, including TLQP-21, TLQP62 and NERP-4, have been confirmed to enhance the β-cell function and attenuate the development of type 2 diabetes50,51,52. Nevertheless, the roles of these bioactive peptides derived from VGF in regulating GCGR-deficient islet microenvironment remain to be explored. Finally, the amino acid transporter SLC38A5 was also dramatically increased in α cells of GCGR-KO mice. While increased SLC38A5 is associated with α cell hyperplasia17,18, its role in the regulating hyperglucagonemia needs further elucidation.
In summary, our studies indicate that glucagon receptor blockade increases organism’s demand for glucagon, resulting in not only α-cell hyperplasia but also enhanced single α-cell glucagon secretion. The elevated circulating amino acids, which result from glucagon receptor blockade, induced mTOR-STAT3 and ERK-CREB pathway activation, and then lead to upregulation of Vgf and Gcg expression. This subsequently results in increased glucagon granule assembly and secretion. Overall, these findings provide a deeper understanding of the physiological changes in α cells during glucagon receptor disruption, revealing potential new targets within this pathway. Such insights may be beneficial for optimizing the clinical application of glucagon receptor antagonism in diabetes treatment. Moreover, elucidating the molecular mechanisms underlying hyperglucagonemia may offer valuable guidance for developing novel diabetes therapeutics.
Methods
Animals
Wild-type (WT) and glucagon receptor knockout (GCGR-KO) mice were bred and maintained in individually-ventilated cages (IVC) with less than five animals per cage under specific pathogen-free (SPF) conditions at the Xiamen University Experimental Animal Center. Mice had free access to a standard diet (65% carbohydrate, 11% fat, 24% protein) and water, the light was on from 8:00 to 20:00, with the temperature kept at 21–24 °C and humidity at 55–70%. The GCGR-KO mice were generated by our laboratory using CRISPR-Cas9 technology, where two sgRNAs were designed to completely delete the glucagon receptor coding sequence32. Only male mice were used in this study, and all experimental procedures were approved by the Animal Care and Utilization Committee of Xiamen University (Protocol No. XMULAC20230225).
Pancreatic islet isolation and preparation of single-cell suspension
Pancreatic islets were isolated from 12-14-week-old male C57BL/6 J mice. After euthanasia, the pancreas was perfused via the common bile duct with 2–4 mL of digestion buffer (Hanks’ Balanced Salt Solution containing type IV collagenase, 1 mg/mL, Gibco, 17104019). The pancreas was then dissected and incubated at 37 °C for 10 minutes, followed by gentle shaking for 3-6 minutes. After digestion, the tissue was suspended in 20 mL RPMI 1640 (Gibco, C11875500) supplemented with 10% FBS and 1% Penicillin/Streptomycin. Density gradient centrifugation was performed, and individual islets with high purity were hand-picked under a dissecting microscope.
To prepare the single-cell suspension, purified islets were digested with 0.25% trypsin-EDTA and filtered through a 40 μm cell strainer. Cell viability was assessed by trypan blue staining followed by microscopy, ensuring that over 90% of cells were viable and singlets.
Single-cell RNA-seq with 10X genomics
We used 4 WT mice and 6 GCGR-KO mice for single-cell RNA-seq. Following preparation of islet single-cell suspensions, the cells were loaded onto 3’ library chips for the Chromium Single Cell 3’ Library (v3) according to the manufacturer’s recommendations (10X Genomics). The RNA libraries were sequenced on MGI2000 platform at BGI-Wuhan, China. Data processing, including sample demultiplexing, alignment, filtering, and UMI counting, was performed using the Cell Ranger Single-Cell Software Suite (v5.0.1; 10x Genomics), with Refdata-gex-mm10-2020-A used for alignment.
Analysis of single-cell RNA-seq data
The Seurat R package (v4.0.5) was used for data integration, analysis and visualization. For cell filtering, genes detected in at least 5 cells and cells with more than 200 detected genes were selected. Cells with fewer than 8000 detected genes, less than 15% of mitochondrial gene content, and fewer than 80,000 detected UMIs were retained. Data were normalized using Normalize Data function in Seurat to identify highly variable genes using the “vst” method. The top 3000 differentially expressed genes were selected for principal component analysis (PCA). PC selection was based on an elbow plot, and the top 20 PCs were used for t-distributed stochastic neighbor embedding (t-SNE) construction. Cell clustering was performed using the shared-nearest neighbor (SNN) method. Batch effect correction was conducted using the Harmony R (v0.1.0) package53. Cell types of each cluster were annotated based on expression of the marker genes. Differentially expressed genes (DEG) between the WT and GCGR-KO α cells were identified using the Wilcoxon rank-sum test.
Gene Ontology (GO) analysis was carried out using the ClusterProfiler (v4.0.5) package54, selecting GO terms with a p value < 0.05. Results were visualized using the ggplot2 (v3.3.5) package.
Cell-cell interaction analysis was conducted using the CellChat (v1.4.0) package55. The CellChatDB.mouse database, which contains secreted signaling, ECM-receptor, and cell-cell contact ligand‒receptor (LR) interactions, was used. Seurat objects were directly imported into CellChat and processed according to guidelines. Heatmaps of the number and intensity of interactions between different cell type signaling pathways were generated using the netVisual_heatmap function, while bubble plots of significantly increased and decreased ligand-receptor pairs in GCGR-KO islets compared with WT islets was created using the netVisual bubble function.
Immunostaining of pancreas sections and whole islets
For pancreas section staining, harvested pancreases were fixed in 4% paraformaldehyde overnight at 4 °C, followed by immersion in 30% sucrose solution for 12 h at 4 °C before embedding in O.C.T compound. Ten-micrometer sections were taken with a Leica cryostat microtome. For whole islet staining, purified islets were fixed in 4% paraformaldehyde for 20 min and permeabilized with 0.3% Triton-X100 for 3 h at room temperature. Samples were blocked with PBS containing 5% FBS and 0.1% Tween 20 for 1 h at room temperature and incubated with primary antibodies overnight at 4 °C, followed by incubation with fluorescence-conjugated secondary antibodies for 2 hours at room temperature. All antibodies used in this study are listed in Supplementary Table 1.
Images were captured using a Leica SP8 confocal microscope (LAS X 0.1.14392). Fluorescence intensity was analyzed using ImageJ (v1.54 f) software (National Institutes of Health), with the regions of interest (ROIs) selected using the threshold function. Mean gray values (the ratio of integrated density to area) were automatically calculated for fluorescence intensity measurements.
Transmission electron microscopy (TEM) analysis of mouse α cells
Islets were mounted in 1% agarose, fixed in 0.1 M PBS containing 2% glutaraldehyde for 12 h at 4 °C, and refixed in 1% osmium tetroxide in 0.1 M PBS for 2 h at room temperature. Specimens were dehydrated in a gradient ethanol series and embedded in epoxy resin. Ultrathin sections were prepared using an ultramicrotome (Leica EM UC7), then mounted on copper grids, and stained with uranyl acetate and lead citrate. Images were captured using an H-7800 transmission electron microscope (HITACHI) and analyzed with ImageJ software. Pancreatic α cells were identified by the presence of round dense-core vesicles without any halo, while β cells were characterized by immature secretory granules with light cores and mature secretory granules with dense cores and prominent halos.
Glucagon secretion measurement
Batches of 40 pancreatic islets from each group were pre-incubated in 0.2 ml KRBH containing 0.1% BSA and 7 mM glucose for 1 h at 37 °C. Subsequently, islets were incubated in 0.2 mL KRBH containing 0.1% BSA and 1 mM glucose for 1 h at 37 °C. After each incubation, supernatants were collected. For somatostatin and insulin tolerance tests, islets were divided into 4 groups and pre-incubated with KRBH containing 0.1% BSA and 7 mM glucose for 1 h at 37 °C. After discarding the supernatant, islets were incubated in KRBH containing 0.1% BSA, 1 mM glucose and indicated concentrations of somatostatin (SST-14) or insulin at 37 °C for 1 h. Supernatants were collected post-incubation.
For αTC1-6 cells, culture medium was removed and cells were pre-incubated in 0.5 mL KRBH containing 0.1% BSA and 7 mM glucose for 1 h at 37 °C. Supernatant was discarded, and cells were subsequently incubated in 0.5 mL KRBH containing 0.1% BSA,1 mM glucose, and 10 μM adrenaline (MCE, HY-B0447B) at 37 °C for 1 h. Supernatants were collected and centrifuged at 1000 g, 4 °C for 10 min to remove dead cell debris. After stimulation, samples were lysed with RIPA and stored at −80 °C.
Glucagon secretion was measured using a mouse glucagon ELISA kit (Abmart, AB-J0390A; Cisbio, 62CGLPEG) according to the manufacturer’s protocol, and protein was quantified using a BCA Protein Quantification Kit (Invitrogen, A55865).
Single α cell glucagon granule exocytosis assessment
To label glucagon granules in living α cells, cultured islets were transduced with adenovirus expressing NPY-mCherry under the control of the pre-proglucagon promoter. Approximately 40% of glucagon-positive cells were labeled with red fluorescence (NPY-mCherry) (Fig. S3).
Adenovirus-transfected islets were imaged using a Leica SP8 confocal microscope. Basal exocytosis was imaged in KRBH containing 0.1% BSA and 6 mM glucose, while stimulated exocytosis was imaged in KRBH containing 0.1% BSA, 1 mM glucose and 10 μM adrenaline. Time-lapse recordings spanning the whole cells (z-step: 1 μm, stack of about 15 confocal images with a size of 1024 × 1024 pixels) were taken at 1 min intervals (XYZT imaging). Glucagon granules in z-stack projections at each time point were analyzed using ImageJ software. Granule exocytosis was identified based on the decreased number of granules compared with previous frames, with the exocytosis granule number normalized to the area of each cell.
Calcium imaging and quantification of cytosolic calcium levels
Islets were incubated with Fluo-4AM (5 μM, Invitrogen. F14201) for 1 h in KRBH supplemented with 6 mM glucose and 0.1% BSA at 37 °C in the dark. Following this incubation, islets were then transferred to an imaging chamber for imaging on a Leica SP8 confocal microscope. Time-series images were acquired every 5 s (XYZT imaging, stack of 5 confocal images with a size of 512 × 512 pixels). α cells were identified by their cytoplasmic Ca2+ in response to stimulation with adrenaline.
To visualize intracellular Ca2+ fluctuations, regions of interest (ROIs) were drawn around the individual α cells, and the mean Fluo4-AM fluorescence intensity of each frame was measured using Leica Application Suite X (V2.0.1). Changes in fluorescence intensity were expressed as the ratio of raw fluorescence intensity (F) to the initial value of fluorescence intensity (F0) (F/F0). The baseline (F0) was defined as the average fluorescence intensity of the first 30 frames of each group.
Adenovirus transfection of pancreatic islets and αTC1-6 cell line
Adenoviruses were produced as previously described56. Briefly, oligonucleotides targeting Vgf were annealed and cloned into the Pshuttle-U6-CMV-GFP vector at the SalI and XbaI sites. An empty shuttle vector was used as the control. To generate recombinant adenovirus packaging plasmids, shuttle vectors were linearized by PmeI and transformed into AdEasier competent cells. Recombinant adenoviruses were generated by linearizing the packaging plasmids with PacI and transfecting them into 293 A cells.
For recombinant adenovirus transfection, both pancreatic islets and αTC1-6 cells were incubated with adenovirus-containing medium for 8 h, followed by being cultured in normal medium for an additional 72 h.
Pancreatic islet perfusion
Islets were isolated from three GCGR-KO mice. Batches of 100 size-matched islets transfected with adenovirus expressing control or Vgf-shRNA were preincubated in KRBH buffer containing 0.5% BSA and 7 mM glucose at 37 °C for 30 min. After pre-incubation, islets were perfused with the same buffer for 5 min, followed by changing the perfusion buffer to KRBH containing 0.5% BSA and 1 mM glucose. Perfusate samples were collected every minute and stored at −80 °C. Glucagon content in the samples was determined using a glucagon assay kit (Cisbio, 62CGLPEG).
Adeno-associated virus (AAV) injection
AAV-U6-sh (NC2)-CMV-EGFP-WPRE (AAV-sh-Control, 2.70 × 1013 v.g./mL) and AAV-U6-shRNA (Vgf)-CMV-EGFP-WPRE (AAV-sh-VGF, 9.52 × 1012 v.g./mL) were obtained from Obio Technology (Shanghai, China). AAV was injected intraperitoneally at a dose of 4.8 × 1012 v.g. per mouse two weeks before sample harvesting. Nine- to ten-week-old GCGR-KO mice were used for this experiment.
Serum amino acid measurement
Serum was collected from six WT or GCGR-KO male mice and analyzed at BGI-Wuhan, China. Test samples were prepared by transferring appropriate amounts of serum into centrifuge tubes, followed by gradient dilution of amino acids mix standards to establish a standard curve. The solvent was added to both the sample and standard solutions, and the mixtures were centrifuged at 13,680 g at 4 °C for 10 minutes to obtain the supernatant for subsequent LC-MS/MS analysis. Analysis was conducted using an LC-MS QTRAP 6500+ system (SCIEX) equipped with a BEH C18 column (2.1 mm × 10 cm, 1.7 μm, Waters) and operated in ESI+ or ESI− mode. Amino acid concentrations were quantified using Skyline software (MacCoss Lab Software), where MRM transitions (ion pairs) were automatically identified and integrated with manual verification as required. Concentrations (μmol/L) were calculated by fitting the integrated peak areas to the standard curve.
Cell culture and Western blotting
The αTC1-6 cell line was purchased from ATCC. Cells were cultured in DMEM (Gibco, C11885500) supplemented with 10% fetal bovine serum (FBS) and 1% Penicillin/Streptomycin, and maintained in a humidified incubator at 37 °C with 5% CO2.
To assess the effect of amino acids on αTC1-6 cells, cells were cultured in MEM (Gibco, 11090-081) supplemented with 5% FBS, 1% Penicillin/Streptomycin, 4 mM glutamine and 4 mM alanine. In some experiments, 30 nM rapamycin (MCE, HY-10219) or 10 μM S3I-201 (MCE, HY-15146) were added along with amino acids. To test the effect of mouse serum, WT or GCGR-KO serum was added to a final concentration of 10%. Cells were cultured for 72 h with medium replenished every 24 h.
For Western blotting analysis, cell lysates were cleared by centrifugation at 10,000 g at 4 °C. Protein concentrations were quantified using a BCA Protein Quantification Kit. Equal amounts of protein from each sample were separated by 10% SDS-PAGE and transferred to a PVDF membrane. After blocking with 5% milk in TBST buffer, the membrane was incubated with primary antibodies followed by appropriate horseradish peroxidase-conjugated secondary antibodies (listed in Supplementary Table 1). For glucagon detection, Tricine-SDS-PAGE was used.
Pancreatic islet culture and serum/amino acids treatment
Mouse islets were isolated from 10 to 12-week-old male mice and cultured in RPMI 1640 containing 10% FBS and 1% Penicillin/Streptomycin. To test the effect of mouse serum, WT or GCGR-KO serum was added to a final concentration of 10%. To determine the effect of amino acids, glutamine and alanine free RPMI 1640 was used, with glutamine and alanine added to the final concentration of 4 mM.
To assay the effect of STAT3 inhibitors, 10 μM S3I 201 was added to the medium. All islets were cultured for 72 h with medium replenished every 24 h. Glucagon secretion from mouse islets was measured as described above.
Luciferase assay
PGL6-VGF-Luc or PGL6-gcg-Luc plasmids and Renilla luciferase expression plasmids were co-transfected into αTC1-6 cells using Lipofectamine™ 2000 (Thermo Fisher, 11668), according to the manufacturer’s instructions. 24 h after transfection, the medium was replenished, and subsequent experiments were conducted.
For amino acid-induced VGF or glucagon expression, the medium was changed to MEM supplemented with 5% FBS, 1% Penicillin/Streptomycin, 4 mM glutamine and 4 mM alanine, and cells were cultured for 72 h. For transcription factor screening, αTC1-6 cells were transfected with siRNA targeting each gene using Lipofectamine™ 2000. 24 h after transfection, the medium was changed as described above, and cells were cultured for 48 h. For STAT3 activity assays, STAT3 or STAT3-mutation plasmids were used for transfection. 24 h after transfection, the medium was replenished and cells were cultured for 48 h. The siRNA target sequences are listed in Supplementary Table 2.
Firefly and Renilla luciferase activities were measured according to the manufacturer’s guidelines (Beyotime, RG088S). Relative values were obtained by normalizing firefly luciferase activity to Renilla luciferase activity.
qRT-PCR
αTC1-6 cells were used to evaluate the efficiency of each siRNA. 72 h after transfection, cells were harvested, and RNA was extracted using TRIzol reagent (Thermo Fisher Scientific, Waltham, MA, USA) according to the manufacturer’s instructions. cDNA was synthesized using M-MLV Reverse Transcriptase (Promega). Quantitative PCR was performed to quantify the expression levels of the tested genes. Primers used are listed in Supplementary Table 3.
Chromatin immunoprecipitation (ChIP)
The JASPAR database was used to predict the candidate binding sites of STAT3. ChIP analysis was performed using a Sonication ChIP Kit (Abclonal, RK20258) according to the manufacturer’s instructions. Briefly, αTC1-6 cells were crosslinked with 1% formaldehyde for 10 min and lysed in SDS lysis buffer. Crosslinked DNAwas sheared into 200–1000 bp fragments by sonication. Sheared chromatin was diluted, and 5% of the diluted supernatant was saved as “input”. The remaining supernatant was immunoprecipitated with p-STAT3-S727 antibodies (CST, 9198). After de-crosslinking, DNA was purified using spin columns. Quantitative PCR was used to quantify the amount of immunoprecipitated DNA. qPCR primers are listed in Supplementary Table 3.
Statistical analysis
All raw data were analyzed using GraphPad Prism 8 software (GraphPad Software v8.3.0, La Jolla, CA, USA). Results are presented as mean values ± SEM. No outlying values were excluded from the datasets used for statistical analysis. All data were first subjected to the Shapiro–Wilk normality test to determine whether parametric or non-parametric analyses would be used. If the data followed a Gaussian distribution, parametric tests were performed: two-tailed unpaired Student’s t-test for two groups, one-way ANOVA for three or more groups, or two-way ANOVA for two-factor assays. Post hoc analyses following ANOVA were conducted using Bonferroni corrections. If the data did not follow a Gaussian distribution, a nonparametric test (Mann–Whitney test to compare two groups or Kruskal–Wallis test with Dunn’s post hoc test to compare three or more groups) was performed. In all cases, differences were considered significant when p < 0.05.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The authors declare that the data supporting the findings of this study are available within the Article, Source Data, and its Supplementary Information. The original scRNA- seq data generated in this study have been deposited in the gene expression omnibus (GEO) database under accession code GSE253271 The previously published data we used in this study are under accession code: GSE8413342, GSE20315143. Source data are provided with this paper.
Code availability
No codes were developed for the analysis of this study. All the sequencing data was analyzed using Seurat.
References
Kimball, C. P. & Murlin, J. R. Aqueous extracts of pancreas: III. some precipitation reactions of insulin. J. Biol. Chem. 58, 337–346 (1923).
Scheen, A. J. & Lefebvre, P. J. Glucagon, from past to present: a century of intensive research and controversies. Lancet Diab. Endocrinol. 11, 129–138 (2023).
Holst, J. J. Glucagon 100 years. Important, but still enigmatic. Peptides 161, 170942 (2023).
Campbell, J. E. & Drucker, D. J. Islet alpha cells and glucagon–critical regulators of energy homeostasis. Nat. Rev. Endocrinol. 11, 329–338 (2015).
Lee, Y., Wang, M. Y., Du, X. Q., Charron, M. J. & Unger, R. H. Glucagon receptor knockout prevents insulin-deficient type 1 diabetes in mice. Diabetes 60, 391–397 (2011).
Conarello, S. L. et al. Glucagon receptor knockout mice are resistant to diet-induced obesity and streptozotocin-mediated beta cell loss and hyperglycaemia. Diabetologia 50, 142–150 (2007).
Gelling, R. W. et al. Lower blood glucose, hyperglucagonemia, and pancreatic alpha cell hyperplasia in glucagon receptor knockout mice. Proc. Natl Acad. Sci. USA 100, 1438–1443 (2003).
Pettus, J. et al. Glucagon receptor antagonist volagidemab in type 1 diabetes: a 12-week, randomized, double-blind, phase 2 trial. Nat. Med 28, 2092–2099 (2022).
Morgan, E. S. et al. Antisense Inhibition of Glucagon Receptor by IONIS-GCGR(Rx) Improves Type 2 Diabetes Without Increase in Hepatic Glycogen Content in Patients With Type 2 Diabetes on Stable Metformin Therapy. Diab. Care 42, 585–593 (2019).
Pettus, J. et al. Effect of a glucagon receptor antibody (REMD-477) in type 1 diabetes: A randomized controlled trial. Diab. Obes. Metab. 20, 1302–1305 (2018).
Kostic, A. et al. A first-in-human pharmacodynamic and pharmacokinetic study of a fully human anti-glucagon receptor monoclonal antibody in normal healthy volunteers. Diab. Obes. Metab. 20, 283–291 (2018).
Kazda, C. M. et al. Evaluation of Efficacy and Safety of the Glucagon Receptor Antagonist LY2409021 in Patients With Type 2 Diabetes: 12- and 24-Week Phase 2 Studies. Diab. Care 39, 1241–1249 (2016).
Okamoto, H. et al. Glucagon Receptor Blockade With a Human Antibody Normalizes Blood Glucose in Diabetic Mice and Monkeys. Endocrinology 156, 2781–2794 (2015).
Guan, H. P. et al. Glucagon receptor antagonism induces increased cholesterol absorption. J. Lipid Res 56, 2183–2195 (2015).
Guzman, C. B. et al. Treatment with LY2409021, a glucagon receptor antagonist, increases liver fat in patients with type 2 diabetes. Diab., Obes. Metab. 19, 1521–1528 (2017).
Kang, Q. et al. Disruption of the glucagon receptor increases glucagon expression beyond alpha-cell hyperplasia in zebrafish. J. Biol. Chem. 298, 102665 (2022).
Kim, J. et al. Amino Acid Transporter Slc38a5 Controls Glucagon Receptor Inhibition-Induced Pancreatic alpha Cell Hyperplasia in Mice. Cell Metab. 25, 1348–1361.e1348 (2017).
Dean, E. D. et al. Interrupted Glucagon Signaling Reveals Hepatic alpha Cell Axis and Role for L-Glutamine in alpha Cell Proliferation. Cell Metab. 25, 1362–1373.e1365 (2017).
Omar-Hmeadi, M., Lund, P. E., Gandasi, N. R., Tengholm, A. & Barg, S. Paracrine control of alpha-cell glucagon exocytosis is compromised in human type-2 diabetes. Nat. Commun. 11, 1896 (2020).
Hamilton, A. et al. Adrenaline Stimulates Glucagon Secretion by Tpc2-Dependent Ca(2+) Mobilization From Acidic Stores in Pancreatic alpha-Cells. Diabetes 67, 1128–1139 (2018).
Panzer, J. K., Tamayo, A. & Caicedo, A. Restoring glutamate receptor signaling in pancreatic alpha cells rescues glucagon responses in type 1 diabetes. Cell Rep. 41, 111792 (2022).
Bartolomucci, A. et al. The extended granin family: structure, function, and biomedical implications. Endocr. Rev. 32, 755–797 (2011).
Zheng, Y. et al. Activation of Pancreatic Acinar FXR Protects against Pancreatitis via Osgin1-Mediated Restoration of Efficient Autophagy. Res. (Wash. D. C.) 2022, 9784081 (2022).
Kang, Q. et al. ErbB3 is required for hyperaminoacidemia-induced pancreatic alpha cell hyperplasia. J. Biol. Chem. 300, 107499 (2024).
Solloway, M. J. et al. Glucagon Couples Hepatic Amino Acid Catabolism to mTOR-Dependent Regulation of alpha-Cell Mass. Cell Rep. 12, 495–510 (2015).
Hu, H. et al. AnimalTFDB 3.0: a comprehensive resource for annotation and prediction of animal transcription factors. Nucleic Acids Res 47, D33–D38 (2019).
Kolmykov, S. et al. GTRD: an integrated view of transcription regulation. Nucleic Acids Res 49, D104–D111 (2021).
Tsunoda, T. & Takagi, T. Estimating transcription factor bindability on DNA. Bioinformatics 15, 622–630 (1999).
Di Rocco, G. et al. Interplay of the E box, the cyclic AMP response element, and HTF4/HEB in transcriptional regulation of the neurospecific, neurotrophin-inducible vgf gene. Mol. Cell Biol. 17, 1244–1253 (1997).
Muller, T. D., Finan, B., Clemmensen, C., DiMarchi, R. D. & Tschop, M. H. The New Biology and Pharmacology of Glucagon. Physiol. Rev. 97, 721–766 (2017).
Hu, X., Li, J., Fu, M., Zhao, X. & Wang, W. The JAK/STAT signaling pathway: from bench to clinic. Signal Transduct. Target Ther. 6, 402 (2021).
Yuan, H. et al. Crispr-Cas9 mediated complete deletion of glucagon receptor in mice display hyperglucagonemia and alpha-cell hyperplasia. Biochem Biophys. Res Commun. 643, 121–128 (2023).
Li, M. et al. Glucagon receptor inactivation leads to alpha-cell hyperplasia in zebrafish. J. Endocrinol. 227, 93–103 (2015).
Svendsen, B. et al. Insulin Secretion Depends on Intra-islet Glucagon Signaling. Cell Rep. 25, 1127–1134.e1122 (2018).
Stephens, S. B. et al. The Prohormone VGF Regulates beta Cell Function via Insulin Secretory Granule Biogenesis. Cell Rep. 20, 2480–2489 (2017).
Asadi, F. & Dhanvantari, S. Pathways of Glucagon Secretion and Trafficking in the Pancreatic Alpha Cell: Novel Pathways, Proteins, and Targets for Hyperglucagonemia. Front Endocrinol. (Lausanne) 12, 726368 (2021).
MacDonald, P. E. & Rorsman, P. Metabolic Messengers: glucagon. Nat. Metab. 5, 186–192 (2023).
Omar-Hmeadi, M. & Idevall-Hagren, O. Insulin granule biogenesis and exocytosis. Cell Mol. Life Sci. 78, 1957–1970 (2021).
Ahren, B. & Larsson, H. Impaired glucose tolerance (IGT) is associated with reduced insulin-induced suppression of glucagon concentrations. Diabetologia 44, 1998–2003 (2001).
Faerch, K. et al. Insulin Resistance Is Accompanied by Increased Fasting Glucagon and Delayed Glucagon Suppression in Individuals With Normal and Impaired Glucose Regulation. Diabetes 65, 3473–3481 (2016).
Brissova, M. et al. alpha Cell Function and Gene Expression Are Compromised in Type 1 Diabetes. Cell Rep. 22, 2667–2676 (2018).
Baron, M. et al. A Single-Cell Transcriptomic Map of the Human and Mouse Pancreas Reveals Inter- and Intra-cell Population Structure. Cell Syst. 3, 346–360.e344 (2016).
Rubio-Navarro, A. et al. A beta cell subset with enhanced insulin secretion and glucose metabolism is reduced in type 2 diabetes. Nat. Cell Biol. 25, 565–578 (2023).
Jin, T. Mechanisms underlying proglucagon gene expression. J. Endocrinol. 198, 17–28 (2008).
Bozadjieva Kramer, N. et al. Glucagon Resistance and Decreased Susceptibility to Diabetes in a Model of Chronic Hyperglucagonemia. Diabetes 70, 477–491 (2021).
Bozadjieva, N. et al. Loss of mTORC1 signaling alters pancreatic alpha cell mass and impairs glucagon secretion. J. Clin. Invest 127, 4379–4393 (2017).
Wewer Albrechtsen, N. J. et al. The Liver-alpha-Cell Axis and Type 2 Diabetes. Endocr. Rev. 40, 1353–1366 (2019).
Riahi, Y. et al. Hyperglucagonaemia in diabetes: altered amino acid metabolism triggers mTORC1 activation, which drives glucagon production. Diabetologia 66, 1925–1942 (2023).
Trani, E. et al. Isolation and characterization of VGF peptides in rat brain. Role of PC1/3 and PC2 in the maturation of VGF precursor. J. Neurochem 81, 565–574 (2002).
Zhang, W. et al. The NERP-4-SNAT2 axis regulates pancreatic beta-cell maintenance and function. Nat. Commun. 14, 8158 (2023).
Petrocchi-Passeri, P. et al. The VGF-derived peptide TLQP-62 modulates insulin secretion and glucose homeostasis. J. Mol. Endocrinol. 54, 227–239 (2015).
Stephens, S. B. et al. A VGF-derived peptide attenuates development of type 2 diabetes via enhancement of islet beta-cell survival and function. Cell Metab. 16, 33–43 (2012).
Korsunsky, I. et al. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Methods 16, 1289–1296 (2019).
Yu, G., Wang, L. G., Han, Y. & He, Q. Y. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 16, 284–287 (2012).
Jin, S. et al. Inference and analysis of cell-cell communication using CellChat. Nat. Commun. 12, 1088 (2021).
Luo, J. et al. A protocol for rapid generation of recombinant adenoviruses using the AdEasy system. Nat. Protoc. 2, 1236–1247 (2007).
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (82470841 to M.L.), Natural Science Foundation of Xiamen City, China (3502Z20227162 to M.L.) and Natural Science Foundation of Fujian Province, China (2023I0002 to M.L.). This work was also supported by grants from Key Laboratory of Tropical Marine Ecosystem and Bioresource, Ministry of Natural Resources (2021ZD01 to M.L.). We thank Tuanlao Wang, Yuhong Chen, Shan Jiang, Yun Hong, and members of the Li lab for constructive discussions. We thank Luming Yao for transmission electron microscopy section preparation.
Author information
Authors and Affiliations
Contributions
M.L. and Q.J. supervised this work, and, as such, had full access to all of the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis. M.L. and J.J. designed the study. J.J., X.B. and Q.K. performed key experiments. M.L., Q.J., J.J., Q.K., X.B., and F.J. participated in the planning of the work and the interpretation of the results. J.J. drafted the manuscript. M.L., F.S.W., and Q.J. revised the paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Oladapo Olaniru and the other, anonymous, reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Jia, J., Bai, X., Kang, Q. et al. Blockade of glucagon receptor induces α-cell hypersecretion by hyperaminoacidemia in mice. Nat Commun 16, 2473 (2025). https://doi.org/10.1038/s41467-025-57786-7
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-57786-7
