
Abstract
Macroautophagy/autophagy is a key catabolic-recycling pathway that can selectively target damaged organelles or invading pathogens for degradation. The selective autophagic degradation of the endoplasmic reticulum (hereafter referred to as ER-phagy) is a homeostatic mechanism, controlling ER size, the removal of misfolded protein aggregates, and organelle damage. ER-phagy can also be stimulated by pathogen infection. However, the link between ER-phagy and bacterial infection remains poorly understood, as are the mechanisms evolved by pathogens to escape the effects of ER-phagy. Here, we show that Salmonella enterica serovar Typhimurium inhibits ER-phagy by targeting the ER-phagy receptor FAM134B, leading to a pronounced increase in Salmonella burden after invasion. Salmonella prevents FAM134B oligomerization, which is required for efficient ER-phagy. FAM134B knock-out raises intracellular Salmonella number, while FAM134B activation reduces Salmonella burden. Additionally, we found that Salmonella targets FAM134B through the bacterial effector SopF to enhance intracellular survival through ER-phagy inhibition. Furthermore, FAM134B knock-out mice infected with Salmonella presented severe intestinal damage and increased bacterial burden. These results provide mechanistic insight into the interplay between ER-phagy and bacterial infection, highlighting a key role for FAM134B in innate immunity.
Similar content being viewed by others

Introduction
Macroautophagy (hereafter autophagy) is an intracellular catabolic-recycling pathway that promotes survival in response to a diverse range of stress conditions including nutrient starvation or invasive pathogens1. Autophagic cargo can include proteins, lipids, damaged organelles, or even intracellular pathogens. These cytoplasmic components are sequestered inside double-membrane vesicles called autophagosomes. Fully formed autophagosomes then fuse with lysosomes leading to the degradation of the sequestered cargo by resident hydrolytic enzymes. The basic macromolecules obtained from cargo degradation are subsequently transported back to the cytoplasm for reutilization. Autophagy levels are regulated by several Atg (autophagy-related) proteins, involved in all steps of the autophagic process from initiation to lysosomal fusion2. Autophagosome cargo selection is often tightly regulated, specifically targeting damaged cellular components or invasive intracellular pathogens for degradation3. Selective autophagy is achieved by autophagy receptors that link the cargo targeted for degradation with the growing autophagosomal membrane. Autophagy receptors usually contain LC3-interacting regions (LIR), an evolutionary conserved sequence that binds to members of the Atg8/LC3/GABARAP family4. During autophagy initiation, LC3 is lipidated and covalently bound to the growing autophagosomal membrane. Thus, by interacting with LC3, autophagy receptors selectively tether cargo to the sequestering autophagosome (Fig. 1A)3,4. The selective autophagic degradation of the endoplasmic reticulum (ER), termed ER-phagy, has been shown to be necessary to mitigate ER stress and maintain ER homeostasis. ER-phagy controls ER size and morphology, as well as inducing the degradation of misfolded protein aggregates that can be toxic for the cell3,5. Several ER-phagy cargo receptors have been described, including FAM134B6 and TEX2647,8, among others5. Recent reports have begun to describe the different molecular mechanisms by which ER-phagy receptors are regulated in the promotion of ER-phagy9,10,11. The first ER-phagy receptor identified, FAM134B, is an ER transmembrane protein containing a reticulon-homology domain that allows it to sense and induce ER membrane curvature and budding through protein clustering12,13. FAM134B activity requires its oligomerization, which is highly regulated by post-translational modifications, including phosphorylation and acetylation, and is a requisite step in FAM134B-driven ER-phagy9,10,14. Interestingly, FAM134B has also been implicated in the cellular response against viral infection. FAM134B-dependent ER-phagy has been shown to limit SARS-COV-2, Ebola, Zika, and dengue virus replication15,16,17,18. Moreover, multiple pathogens have developed strategies to inhibit ER-phagy by specifically hijacking or cleaving FAM134B16,17,19. Targeting FAM134B leads to ER remodeling, which is thought to benefit invading viruses, creating a favorable environment for replication17,19. However, the links between ER-phagy and bacterial infection; as well as the mechanisms pathogens have evolved to evade the effects of ER-phagy remain poorly understood. In this study, we characterize an unidentified mechanism of bacterial-mediated inhibition of ER-phagy. Specifically, we found that Salmonella enterica serovar Typhimurium (Salmonella), inhibits ER-phagy by specifically targeting the activity of the ER-phagy receptor FAM134B, leading to a pronounced increase in Salmonella burden after invasion. Salmonella was chosen due to its well-characterized intracellular growth cycle, membrane remodeling ability, capacity to avoid lysosomal/autophagic clearance, as well as continuing to be a major cause of foodborne infections worldwide20,21. We show that Salmonella infection prevents FAM134B oligomerization, which is required for efficient ER-phagy. Conversely, Salmonella-mediated ER-phagy blockage could be bypassed by promoting FAM134B oligomerization, which recovered ER-phagy levels. We provide evidence that Salmonella targets FAM134B through the bacterial effector SopF, preventing oligomerization and ER-phagy activation. Furthermore, in vivo analysis of FAM134B knock-out (KO) mice infected with Salmonella revealed intestinal damage and increased bacterial levels in the spleen, intestine, and feces. Our results provide mechanistic insight into the interplay between ER-phagy and bacterial infection, highlighting a key role for FAM134B in innate immunity.

A Schematic for ER-phagy and the ss-RFP-GFP-KDEL probe. During ER-phagy, ER-bound FAM134B binds to LC3 family proteins through a LIR and is sequestered into nascent autophagosomes. ss-RFP-GFP-KDEL is processed by lysosomal hydrolases to generate a free RFP fragment. Lysosomal acidity quenches the GFP signal. LIR, LC3-interacting region; ss, signal sequence (Created in BioRender. Russell, R. (2025) https://BioRender.com/k22m745). B WT and FIP200 KO HEK293A cells stably expressing the ss-RFP-GFP-KDEL reporter were starved for AA for 6 h; starved for 6 h followed by ST infection for the final 1 or 3 h in AA starvation media; or infected in nutrient-rich media with ST for 1 or 3 h. ER-phagy was measured by ss-RFP-GFP-KDEL processing. Non-selective autophagy activity was measured by LC3B lipidation and p62/SQSTM1 degradation. Phospho- and total S6K levels were determined to ascertain AA starvation. Actin was used as a loading control. AA, amino acids; ST, Salmonella Typhimurium; S.E, Short Exposure; L.E, Long Exposure. C Normalized Free RFP-Actin ratio from WT cells in (A). Data are presented as mean values ± SD of three biological experiments. ANOVA,***P < 0.001. NR, Nutrient Rich. D Cells from (B) were either AA starved for 6 h or starved for 3 h followed by ST infection for 3 h in AA starvation media, fixed and imaged by confocal microscopy. Representative images are shown. ss-RFP-GFP-KDEL, yellow; Free RFP, red. Scale bar 5 μm. E Average number of RFP puncta per cell from (D) was quantified. Data are presented as mean values ± SD of five biological experiments. The average number of RFP puncta was calculated from a minimum of 100 cells. ANOVA, ***P < 0.001. F HEK293A WT cells were AA starved for 2 h or starved for 2 h in the presence of ST. Fam134B, Tex264, p62/SQSTM1, and Actin levels were determined by western blot. G Normalized Fam134B-Actin, Tex264-Actin, and p62/SQSTM1 ratios from (F) were quantified. Data are presented as mean values ± SD of five biological experiments. ANOVA, ***P < 0.001, ns, no significance. Source data are provided as a Source Data file.
Results
Salmonella Typhimurium infection blocks ER-phagy
To investigate the possible impact of Salmonella infection on ER-phagy, we generated a HEK293A cell line stably expressing a previously published doxycycline-inducible ER-phagy reporter containing an ER signal sequence, followed by the fluorescent proteins RFP and GFP, and the ER retention sequence KDEL (ss-RFP-GFP-KDEL)7. Upon doxycycline treatment and ER-phagy induction, the fluorescent ER-associated reporter is sequestered inside autophagosomes and cleaved upon lysosome fusion. Unlike GFP, RFP is relatively resistant to lysosomal pH and hydrolases22. As such, a ~25-kDa fragment corresponding to RFP can be detected by western blot upon activation of ER-phagy. Similarly, the same reporter can be used to measure ER-phagy by fluorescent microscopy due to the quenching of GFP fluorescence at lysosomal acidic pH (Fig. 1A). As a result, ER-phagy also can be monitored as an increase in RFP positive structures (lysosome-associated ER-phagy probe) compared to the dual positive probe signal. We tested the reporter by activating ER-phagy through amino acid starvation7 or Torin-1 (a potent autophagy inducer) treatment. As a negative control, we confirmed that ss-RFP-GFP-KDEL did not get cleaved in autophagy-deficient FIP200 KO cells (Supp Fig. 1A).
Amino acid starvation for 6 h is sufficient to induce ER-phagy. To determine if Salmonella infection impacted ER-phagy, we added Salmonella to the amino acid starvation media for the final 1 or 3 h of starvation. Infection with Salmonella for either 1 or 3 h significantly reduced the production of the free RFP fragment when compared to 6 h of amino acid starvation in the absence of Salmonella (Fig. 1 B C), indicating inhibition of ER-phagy upon infection. RFP processing was inhibited by Salmonella when normalized to either actin (Fig. 1C) or total ss-RFP-GFP-KDEL reporter (Supp Fig. 1B). We expanded our analysis of ER-phagy regulation by Salmonella in the HCT116 cell background, which were engineered to stabily express ss-RFP-GFP-KDEL and observed a similar repression of ER-phagy (Supp Fig. 1C and Supp Fig. 1D). Moreover, both HEK293A and HCT116 cells expressing a HaloTag-based pulse-chase reporter for ER-phagy measurement23, showed decreased ER-phagy levels upon Salmonella infection, further corroborating a role for Salmonella in repressing ER-phagy (Supp Fig. 1E and Supp Fig. 1F). Interestingly, Salmonella infection failed to block non-selective autophagy as shown by the degradation of p62/SQSTM1, a cargo receptor regularly used as an index for general autophagic degradation and LC3B-II production, the faster electrophoretic form of LC3B that is produce during autophagy initiation by LC3B lipidation24,25 (Supp Fig. 1G and Supp Fig. 1H). Similarly, the general autophagy reporter mCherry-eGFP-LC3B24, showed no significant differences (RFP puncta formation) between infected and non-infected cells during starvation (Supp Fig. 1I and Supp Fig. 1J). Furthermore, the commercial autophagy detection kit, CYTO-ID, confirmed that non-selective autophagy was not inhibited by Salmonella infection during starvation, suggesting Salmonella specifically targets ER-phagy (Supp Fig. 1K). Importantly, Bafilomycin A1 (BafA1) treatment, a potent inhibitor of autophagosome maturation and cargo degradation, completely blocked free RFP production when cells were starved in the presence of Salmonella, indicating that Salmonella did not increase the turnover rate of autolysosomes (Supp Fig. 1L). The ss-RFP-GFP-KDEL probe can be used to visualize ER-phagy by fluorescence microscopy, where ER-phagy induction is observable through detection of RFP puncta formation. Infection with Salmonella resulted in a significant decrease in ER-phagy induction compared to cells that were not infected (Fig. 1D, E), which was consistent with our western blot analysis. Similar results were observed when we repeated the experiment using the ss-RFP-GFP-KDEL probe in HCT116 cells (Supp Fig. 1M, N), and the HaloTag-based ER-phagy reporter in HEK293A and HCT116 (Supp Fig. 1O, P). We next measured the protein levels of endogenous ER-phagy cargo receptors known to be involved in starvation-induced ER-phagy, namely TEX264 and FAM134B. Because these receptors link the ER to the autophagosome membrane they are ultimately degraded making their protein levels inversely correlated with ER-phagy levels6,7. We measured the endogenous level of each receptor during ER-phagy inducing conditions in the absence and presence of Salmonella and found both TEX264 and FAM134B levels to be significantly higher upon infection, indicating that their degradation is blocked upon Salmonella infection (Fig. 1F, G). Conversely, non-selective p62/SQSTM1 autophagic degradation was similar between infected and non-infected samples. Similar results were observed when these experiments were repeated in HeLa cells (Supp Fig. 1Q). Additionally, Salmonella infection was also able to prevent the degradation of the ER-phagy receptors: FAM134A, FAM134C and RTN3L, further demonstrating the ability of Salmonella to block ER-phagy (Supp Fig. 1R, S). Collectively, these findings indicate that Salmonella infection specifically inhibits ER-phagy, but not non-selective autophagy.
FAM134B is targeted by Salmonella to block ER-phagy
The ER-phagy receptor proteins FAM134B and TEX264 have been previously reported to be targeted by invasive pathogens to disrupt ER morphology and promote infection19. To investigate this possibility, we generated CRISPR KO cell lines of FAM134B and TEX264 (Supp Fig. 2A). As expected, deletion of FAM134B and TEX264 severely decreased ER-phagy (Fig. 2A), which is in line with prior reports7. Consistent with our previous findings, WT cells infected with Salmonella showed a significant decrease in the levels of free RFP compared to uninfected cells during ER-phagy inducing conditions. However, Salmonella infection failed to show a significant difference in ER-phagy levels between infected and uninfected FAM134B KO cells. In contrast, Salmonella was still capable of repressing ER-phagy in TEX264 KO cells (Fig. 2A, B). Immunofluorescence microscopy using the same experimental setup showed a dramatic decrease in RFP puncta formation when ER-phagy was induced in Salmonella-infected WT and TEX264 KO cells. However, RFP puncta formation in FAM134B KO cells infected with Salmonella during ER-phagy induction failed to decrease to the same extent, displaying significant differences (Fig. 2C, D). To further analyze the requirement of FAM134B for Salmonella-mediated ER-phagy repression we created stable cell lines expressing an ER-phagy HALO reporter in WT and FAM134B KO cells. Using this reporter, we confirmed the inability of Salmonella to inhibit ER-phagy in the FAM134B KO background (Supp. Fig. 2B), consistent with the proteolytic cleavage of our ss-RFP-GFP-KDEL reporter (Fig. 2A). These results suggest that Salmonella infection primarily targets FAM134B to inhibit ER-phagy.

A WT, FAM134B KO, TEX264 KO and FIP200 KO HEK293A cells stably expressing the ss-RFP-GFP-KDEL were either kept in nutrient rich media, starved for AA for 6 h or starved for 3 h followed by ST infection for 3 h in AA starvation media. ER-phagy was measured by ss-RFP-GFP-KDEL processing. Actin was used as a loading control. AA, amino acids; ST, Salmonella Typhimurium. B Normalized Free RFP-Actin ratio from cells in (A). Data are presented as mean values ± SD of seven biological experiments. ANOVA, *P < 0.05; ***P < 0.001; ns, no significance. C Cells from (A) were either AA starved for 6 h, or AA starved for 3 h, followed by ST infection for 3 h in AA starvation media, fixed and imaged by confocal microscopy. NR, Nutrient Rich. Representative images are shown. ss-RFP-GFP-KDEL, yellow; Free RFP, red. Scale bar 5 μm. D Average number of RFP puncta per cell from (C) were quantified. Data are presented as mean values ± SD of 5 biological experiments. The averages were calculated from a minimum of 100 cells. ANOVA, ***P < 0.001. E WT, FAM134B KO, TEX264 KO and FAM134B KO cells transfected with WT FAM134B were infected with ST. Bacterial content was determined through a colony-forming unit (CFU). Data are presented as mean values ± SD of three biological experiments. ANOVA, **P < 0.01; ***P < 0.001. F FAM134B KO cells transfected with either FAM134B WT or FAM134B LIR mutant were kept in nutrient rich media, starved for AA for 6 h or starved for 3 h followed by ST infection for 3 h in AA starvation media. ER-phagy was measured by ss-RFP-GFP-KDEL processing. Actin was used as a loading control. G Cells from (F) were fixed and imaged by confocal microscopy. Representative images are shown. ss-RFP-GFP-KDEL, yellow; Free RFP, red; Fam134B, far red. Scale bar 5 μm. H Normalized Free RFP-Actin ratio from cells in (F) and average number of RFP puncta per cell from (G) were quantified. Data are presented as mean values ± SD of five biological experiments. The average number of RFP puncta was calculated from a minimum of 100 cells. ANOVA, ***P < 0.001; ns, no significance. Source data are provided as a Source Data file.
Next, we sought to determine if Salmonella-mediated ER-phagy inhibition impacted intracellular bacterial viability. To this end, we performed a colony-forming unit (CFU) assay in WT, FAM134B KO, TEX264 KO and FAM134B KO cells transfected with FAM134B WT. Analysis of Salmonella viability at 4 h post-infection revealed that FAM134B KO cells had significantly higher number of viable internalized bacteria, suggesting either a defect in the clearance of Salmonella or increased growth when compared to WT and TEX264 KO cells. Furthermore, FAM134B KO cells reconstituted with FAM134B recovered similar levels of Salmonella viability as parental cells (Fig. 2E), indicating FAM134B, but not TEX264, is important for Salmonella growth and survival.
ER-phagy receptors couple the ER to the autophagosomal membrane through LIRs (Fig. 1A). We next sought to determine if the impact of FAM134B on Salmonella growth was due to ER-phagy induction or another uncharacterized autophagy-independent function. To this end, we transfected FAM134B KO cells with either FAM134B WT or FAM134B LIR mutant and performed the ER-phagy reporter RFP processing assay. Intriguingly, we observed no significant difference between infected and uninfected ER-phagy induced cells when the FAM134B LIR mutant was expressed. Conversely, cells expressing FAM134B WT showed a significant decreased in free RFP production when infected with Salmonella (Fig. 2F, H). Consistent with our western blot analysis, we also observed that the decrease in the number of RFP puncta in the FAM134B LIR mutant cells triggered by Salmonella infection was not as pronounced as the one in infected cells expressing FAM134B WT (Fig. 2G, H). When we quantified Salmonella viability by CFU in HCT116 cells, results showed that the expression of the FAM134B LIR mutation resulted in a significant increase in the number of Salmonella compared to cells expressing FAM134B WT (Supp Fig. 2C). Together, these findings suggest that FAM134B plays a crucial role in limiting Salmonella burden and that this role is connected to FAM134B ability to induce ER-phagy.
FAM134B oligomerization is hindered by Salmonella infection
FAM134B promotes ER membrane scission and ER-phagy through its ability to oligomerize9. Therefore, we hypothesized that Salmonella infection might prevent ER-phagy by repressing FAM134B oligomerization. To test this hypothesis, FAM134B-FLAG was immunoprecipitated (IP) after 3 h of ER-phagy induction in the presence and absence of Salmonella infection. Consistent with previous reports, FAM134B oligomers were relatively resistant to denatured conditions and are observed by western blot in a distinct, slow migrating band9,14. Notably, Salmonella infection significantly reduced the formation of FAM134B oligomers (Fig. 3A). Similar results were observed when we repeated the experiment in HCT116 cells (Supp Fig. 3A). Furthermore, amino acid starvation-induced FAM134B puncta formation was dramatically reduced by Salmonella infection, revealing Salmonella blocks ER membrane scission, which is driven by FAM134B oligomerization (Fig. 3B). These results indicate Salmonella may block ER-phagy by preventing the oligomerization of FAM134B. If Salmonella represses FAM134B by preventing oligomerization, then forcing FAM134B oligomerization would be predicted to bypass Salmonella-mediated ER-phagy reduction. To this end, we generated a FAM134B G216R mutant, a naturally occurring mutation that resides in FAM134B reticulon-homology domain and has been described to dramatically enhance FAM134B oligomerization9,26. We transfected either FAM134B WT or FAM134B G216R in FAM134B KO cells and measured ER-phagy through the RFP processing assay. As expected, cells transfected with FAM134B WT exhibited reduced ER-phagy under stimulated conditions when infected with Salmonella (Fig. 3C, D). However, cells transfected with FAM134B G216R showed no significant difference in ER-phagy rates in the presence or absence of Salmonella, indicating that Salmonella inhibits ER-phagy upstream, or at the level, of FAM134B activation. Analysis of ER-phagy by immunofluorescence under the same conditions, showed that FAM134B G216R largely prevented ER-phagy repression by Salmonella, compared to FAM134B WT (Fig. 3E, F). Additionally, Salmonella infection failed to decrease FAM134B G216R oligomerization (Supp Fig. 3B), showing a dramatic increase in self-interaction and oligomerization as previously reported9. Together, these experiments support a model in which Salmonella regulates ER-phagy through repression of FAM134B activity.

A FAM134B KO HEK293A cells transfected with FAM134B-FLAG were starved for AA for 3 h in the absence or presence of ST. FLAG was immunoprecipitated. Data are presented as mean values ± SD of five biological experiments. Student’s t test, ***P < 0.001. AA amino acids; ST, Salmonella Typhimurium. B WT cells were incubated with BafA1 and starved for AA for 3 h in the presence or absence of ST. Fam134B puncta formation was observed by confocal microscopy. NR, Nutrient Rich; BafA1, Bafilomycin A1. Data are presented as mean values ± SD of five biological experiments. The average number of Fam134B puncta was calculated from a minimum of 100 cells. Representative images are shown. Fam134B, red; DAPI, blue; ST, green. Scale bar 5 μm. ANOVA, *** P < 0.001. C FAM134B KO cells were transfected with either FAM134B WT or FAM134B G216R and starved for AA for 6 h or starved for 3 h, followed by ST infection for 3 h in AA starvation media. ER-phagy was measured by ss-RFP-GFP-KDEL processing. Actin was used as a loading control. D Normalized Free RFP-Actin ratio from cells in (C). Data are presented as mean values ± SD of five biological experiments. ANOVA, ***P < 0.001; ns, no significance. E FAM134B KO were transfected with either FAM134B WT or G216R and starved for AA for 6 h or starved for 3 h followed by ST infection for 3 h in AA starvation media. Representative images are shown. ss-RFP-GFP-KDEL, yellow; Free RFP, red; Fam134B, far red. Scale bar 5 μm. F Average number of RFP puncta per cell from (E) were quantified. Data are presented as mean values ± SD of five biological experiments. The average number of RFP punctuation was calculated from a minimum of 100 cells. ANOVA, ***P < 0.001. Source data are provided as a Source Data file.
The Salmonella effector SopF blocks ER-phagy
In order to promote invasion and replication, Salmonella expresses two type-III secretion systems, which deliver multiple bacterial effectors into the host cells27. We hypothesized that one of these effectors might be involved in Salmonella-mediated ER-phagy inhibition. To test our hypothesis, we repeated the ss-RFP-GFP-KDEL processing assay infecting them with different Salmonella effector mutants. Among the mutants tested, Salmonella defective for the phosphoinositide-binding effector SopF showed the most complete and consistent loss of ER-phagy regulation (Supp Fig. 4A). Analysis of ER-phagy by western blot and immunofluorescence in our reporter cells showed that SopF-deficient Salmonella was unable to supress ER-phagy compared to WT Salmonella infected controls under stimulated conditions (Fig. 4A–D). Together, these experiments indicate that SopF is necessary for Salmonella-induced ER-phagy repression. Next, we sought to determine if the expression of the SopF effector was sufficient to inhibit ER-phagy. To this end, ER-phagy reporter cells were transfected with FLAG-SopF or control vector. We observed by western blot, that SopF expression was sufficient to inhibit ER-phagy under stimulated conditions (Fig. 4E, F). Consistently, RFP puncta formation was significantly decreased in cells transfected with HA-SopF compared to control cells (Supp Fig. 4B, C). Furthermore, SopF expression dramatically blocked endogenous FAM134B degradation by ER-phagy under stimulated conditions (Supp Fig. 4D). Together, these data indicate the bacterial effector SopF is required for Salmonella to inhibit ER-phagy.

A WT HEK293A cells expressing ss-RFP-GFP-KDEL were AA-starved for 6 h or 3 h, then infected with ST WT or ST ΔsopF for 3 h. ER-phagy was measured via ss-RFP-GFP-KDEL processing. Actin = loading control. NR nutrient rich; AA amino acids; ST = Salmonella Typhimurium. B Normalized Free RFP-Actin ratio from (A). Data are presented as mean values ± SD of five biological experiments. ANOVA, ***P < 0.001; ns no significance. C WT HEK293A cells expressing ss-RFP-GFP-KDEL were AA-starved for 6 h or 3 h + ST WT/ΔsopF infection for 3 h, fixed, and imaged. ss-RFP-GFP-KDEL = yellow; Free RFP = red. Scale bar = 5 μm. D Average RFP puncta/cell from (C). Data are presented as mean values ± SD of five biological experiments. ANOVA, ***P < 0.001. E WT cells from (A) transfected with FLAG-SopF or mock plasmid, AA-starved for 6 h or 3 h + ST WT infection for 3 h. ER-phagy measured. Actin = loading control. F Normalized Free RFP-Actin ratio from (E). Data are presented as mean values ± SD of three biological experiments. ANOVA, ***P < 0.001. G HEK293A FAM134B KO cells with ss-RFP-GFP-KDEL transfected with FAM134B WT, G216R, or FLAG-SopF, then AA-starved for 6 h. Vinculin = loading control. H Normalized Free RFP-Vinculin ratio from (G). Data are presented as mean values ± SD of five biological experiments. ANOVA, **P < 0.01; ***P < 0.001.
Because forcing FAM134B oligomerization with the G216R mutant could bypass Salmonella-mediated ER-phagy blockage, we sought to determine if SopF effects on ER-phagy could also be prevented by FAM134B G216R. Indeed, FAM134B KO cells co-expressing FLAG-SopF and FAM134B G216R were able to significantly induce ER-phagy when stimulated compared to cells transfected with FLAG-SopF and FAM134B WT (Fig. 4G, H). These data suggest Salmonella targets FAM134B oligomerization through SopF.
Recently, the crystal structure of SopF was solved revealing it to be a member of the ADP-ribosyltransferase superfamily28. Indeed, SopF has been shown to catalyze the transfer of ADP-ribose from nicotinamide adenine dinucleotide (NAD+) to the v-ATPase subunit ATP6V0C leading to the inhibition of bacterial autophagy, but not general autophagy or other types of selective autophagy28,29. To test if SopF could directly interact with and target FAM134B, we co-immunoprecipitated FLAG-SopF and endogenous FAM134B, detecting a possibly transient interaction between both proteins (Fig. 5A). To further validate FAM134B and SopF interaction, we performed a TurboID assay, which relies on biotin-based proximity labeling and can detect transient interactions more reliably than co-IP. Briefly, SopF was tagged with a more promiscuous form of BirA, an Escherichia coli-derived biotin ligase. After incubation with biotin, proteins in the near vicinity of the TurboID-tagged SopF become biotinylated, enabling their identification after streptavidin pull-down30. We observed that FAM134B was dramatically enriched after streptavidin pull-down when TurboID-SopF was expressed compared to the TurboID control, indicating SopF comes in close proximity to FAM134B (Fig. 5B).
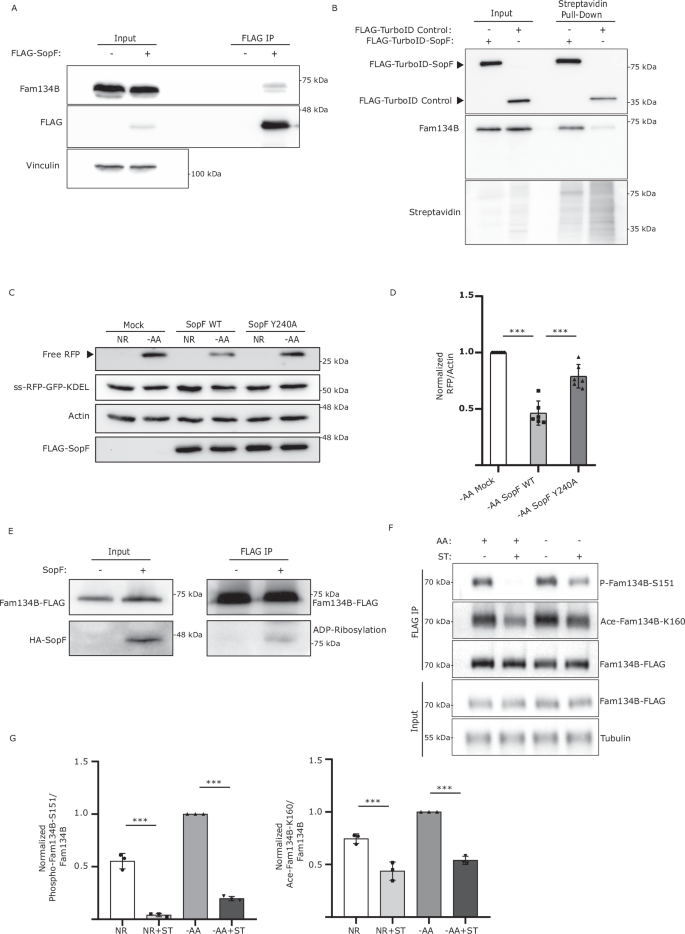
A WT HEK293A cells were transfected with FLAG-SopF or mock plasmid. FLAG-SopF was immunoprecipitated. Representative data from 3 biological experiment. B WT HEK293A cells transfected with FLAG-TurboID-SopF or control plasmid, incubated with Biotin, and subjected to streptavidin pull-down, representative images of 3 biological experiment. C WT HEK293A cells expressing ss-RFP-GFP-KDEL transfected with FLAG-SopF WT, Y240A, or mock plasmid, AA-starved for 6 h. ER-phagy was measured. Actin = loading control. D Normalized Free RFP-Actin ratio from (C). Data are presented as mean values ± SD of six experiments. ANOVA, ***P < 0.001. E FAM134B KO cells transfected with FAM134B-FLAG + HA-SopF or mock plasmid. FLAG was immunoprecipitated. ADP-ribosylation was detected using a pan-ADP ribose binding reagent. Representative image of five biological experiments. F HeLa cells were transfected with FAM134B-FLAG, AA-starved for 3 h and/or ST-infected for 3 h. FLAG was immunoprecipitated. Fam134B Ser151 phosphorylation and Lys160 acetylation were detected using specific antibodies. Tubulin = loading control. G Normalized Phospho-Fam134B-S151/Total Fam134B and Ace-Fam134B-K160/Total Fam134B ratios from (F) Data are presented as mean values ± SD of three biological experiments. ANOVA, ***P < 0.001. Source data are provided as a Source Data file.
SopF mutations preventing its binding to the N-ribose (E325A) or the nicotinamide (Y224A, Y240A) group of NAD+ largely blocked SopF ADP-ribosylation activity28. Consistently, SopF Y240A failed to prevent ER-phagy activation as observed by a significant increase in RFP production compared to SopF WT. (Fig. 5C, D). Additionally, SopF mutants E325A and Y224A also showed increased ER-phagy compared to SopF WT (Supp Fig. 4E and Supp Fig. 4F). Moreover, expression of SopF WT, but not the ADP-ribosylation mutant SopF Y240A, reduced the formation of FAM134B oligomers (Supp Fig. 4G). To test if SopF could ADP-ribosylate FAM134B we used a pan-ADP-ribose binding reagent capable of detecting both mono and poly-ADP ribosylation. A band was observed when FAM134B was IP in the presence of SopF, which was absent in the control IP, suggesting SopF may directly ADP-ribosylate FAM134B (Fig. 5E).
Recent studies have reported the importance of FAM134B acetylation at Lys160 and phosphorylation at residues Ser149, Ser151 and Ser153 in promoting its oligomerization and ER-phagy activation9,10. We hypothesized that one of the ways Salmonella could inhibit FAM134B oligomerization was by preventing these post-translational modifications. Mass spectrometry analysis of FAM134B in the absence of SopF, showed phosphorylation in Ser151, which was not detected when SopF was co-expressed (Supp Fig. 4H). Consistently, Salmonella infection significantly decreased both FAM134B Ser151 phosphorylation and Lys160 acetylation, in both nutrient rich and starvation conditions (Fig. 5F, G), indicating Salmonella directly targets FAM134B regulation and its ability to oligomerize. Further studies will be required to determine if Salmonella-mediated inhibition of FAM134B oligomerization is driven by SopF directly ADP-ribosylating FAM134B or upstream regulators. Together these results indicate that SopF ADP-ribosylation activity is required for ER-phagy inhibition.
FAM134B restricts Salmonella growth
To better understand the nature of FAM134B-mediated resistance to intracellular Salmonella, we next looked at the factors that could contribute to the FAM134B KO defect, such as bacteria vesicle escape, clearance, and growth. First, we infected WT and FAM134B KO cells with GFP Salmonella and quantified growth post-invasion for 4 hrs. To distinguish between external and internalized Salmonella, cells were stained with a LPS antibody before permeabilization, allowing selective labeling and exclusion of external bacteria in our quantifications. We observed a similar level of bacterial internalization in both WT and FAM134B KO at 1 h (Fig. 6A), indicating similar levels of infection rate. Interestingly, we observed a significant difference in growth rate between WT and FAM134B KO beginning at 2 h post infection that persisted through the remaining time points. Escape from Salmonella-containing vesicles (SCV) is known to increase the growth rate of intracellular Salmonella, which is significantly higher in the cytosol. However, SCV escape in WT cells typically occurs well after 4 h post infection31 and results in the growth of rod-shaped Salmonella that are more spread out than those growing in the SCV. Given the early timepoint of divergence in growth rates and the morphology of internalized Salmonella, it is highly unlikely that FAM134B KO defects are a result of early escape from the SCV. Nevertheless, we quantified the amount of cytosolic Salmonella compared to the total population using a chloroquine resistance CFU assay. Chloroquine accumulates in high concentration within endosomes, preferentially targeting vacuolar, rather than cytosolic Salmonella, rendering SCV-containing Salmonella transcriptionally inactive and non-replicative31. Consistent with other studies, we observed that cytosolic Salmonella accounted for 10% of total bacteria in infected WT cells at similar time points31 (Supp. Fig. 5A). However, there was no significant difference in cytosolic Salmonella between infected WT and FAM134B KO cells, indicating FAM134B is not involved in Salmonella escape from SCVs (Supp. Fig. 5A). We next sought to measure the impact of FAM134B on autophagic bacterial clearance, termed xenophagy. To estimate the relative contribution of FAM134B in Salmonella clearance compared to other forms of autophagy, we performed a CFU assay in WT and FAM134B KO cells treated with the VPS34 inhibitor, VPS34-IN1. The class-III phosphatidylinositol-3-phosphate kinase complex, which is formed by the catalytic subunit PIK3C3/VPS34, among others, plays an essential role in autophagy activation32, thus, VPS34 inhibition blocks general autophagy24. As expected, inhibiting VPS34 dramatically increased Salmonella levels in both WT and FAM134B KO cells. However, VPS34-IN1-treated FAM134B KO cells showed a significant increase in Salmonella burden compared to treated WT cells, suggesting FAM134B may have an additional role in preventing Salmonella growth, not directly related to xenophagy (Supp. Fig. 5B). Autophagosomal degradation of Salmonella is mediated by LC3 recruitment to bacteria and autophagy-deficient cells have been shown to be more permissive for Salmonella growth33. However, the impact of ER-phagy in Salmonella clearance is unclear. Strikingly, FAM134B KO cells contained less LC3B-positive Salmonella compared to WT cells, suggesting a role for FAM134B and ER-phagy in Salmonella clearance (Fig. 6B, C). LC3B targeting to Salmonella is modulated by various xenophagy adapters such as TBK1, which is involved in phosphorylating different xenophagy receptors and recruiting them to the surface of Salmonella for degradation34. Interestingly, we found no significant difference in TBK1 recruitment to Salmonella in WT and FAM134B KO cells (Supp Fig. 5C, D), suggesting FAM134B effects on Salmonella replication might be either downstream of TBK1, or not directly related to xenophagy.

A WT and FAM134B KO HEK293A cells were infected with ST for 1 h followed by Gentamicin wash-off. Cells were fixed at indicated time points. ST, Salmonella Typhimurium. Representative images are shown. ST, green; LPS, far red; DAPI; blue. Scale bar 10 μm. Data are presented as mean values ± SD of five biological experiments. The number of ST were calculated from a minimum of 100 cells. ANOVA, *P < 0.05; **P < 0.01; ***P < 0.001. B HEK293A WT and FAM134B KO cells were infected for 1 h. Autophagic capture of ST was analyzed by immunostaining for LPS and LC3B. Representative images are shown. ST, green; LC3B, red; LPS, far red; DAPI; blue. Scale bar 5 μm. C Percentage of ST colocalizing with LC3B in WT and FAM134B KO cells from (B). Data are presented as mean values ± SD of five biological experiments. The percentage of colocalization was calculated from a minimum of 100 cells. Student’s t test, ***P < 0.001. D HEK293A WT and FAM134B KO cells were infected with either ST WT or ST ΔsopF for 1 h. Autophagic capture of ST was analyzed by immunostaining for LPS and LC3B. Representative images are shown. ST, green; LC3B, red; LPS, far red; DAPI; blue. Scale bar 5 μm. E The ratio between the number of ST colocalizing with LC3B and total internalized ST from (D) is presented. Data are presented as mean values ± SD of five biological experiments. LC3B-ST colocalization was calculated from a minimum of 100 cells. ANOVA, ***P < 0.001; ns, no significance. F HEK293A WT and FAM134B KO were transfected with either FLAG-SopF or a mock plasmid followed by WT ST infection. Bacterial content was determined through colony forming unit (CFU). Data are presented as mean values ± SD of three biological experiments. ANOVA, **P < 0.01; ns, no significance. Source data are provided as a Source Data file.
To determine if SopF is required for the suppression of LC3-positive puncta in FAM134B KO cells, we quantified the localization of LC3B to SopF-deficient Salmonella. We observed the previously reported increase of LC3B colocalization to ΔsopF Salmonella compared to WT Salmonella in our control cells35. However, SopF deletion exerted no significant difference in LC3B colocalization in FAM134B KO cells (Fig. 6D, E). Similarly, SopF overexpression significantly increased WT Salmonella levels in control cells, while overexpression of SopF in FAM134B KO cells showed no significant difference in Salmonella viability compared to mock transfected FAM134B KO cells (Fig. 6F), further indicating that SopF effects on Salmonella growth are linked to FAM134B. Moreover, it reinforces our working model that SopF is inhibiting ER-phagy to influence ER morphology and promote intracellular Salmonella survival. This hypothesis was further corroborated by electron microscopy images showing increased ER area in cells stabily expressing SopF compared to control cells (Supp. Fig. 5E), indicating SopF-mediated inhibition of ER-phagy reconfigures ER morphology. The mechanisms that specifically link FAM134B-dependent ER-phagy to xenophagy are unclear at this time, however, these results suggest an additional, yet undiscovered, pathway linking FAM134B to Salmonella restriction.
Infected FAM134B KO mice are more susceptible to Salmonella infection
We next sought to determine the pathophysiological effects of Salmonella-mediated inhibition of ER-phagy in vivo. To this end, we performed oral gavage Salmonella infection of WT and FAM134B KO mice36 and analyzed bacterial burden and intestinal damage 5 days post infection. Histochemical analysis of hematoxylin and eosin (H&E) stained intestine samples of infected WT mice presented mucosa and submucosa infiltration, along with occasional damage (Fig. 7A). However, infected FAM134B KO mice showed severe wall, mucosa and submucosa infiltration, as well as marked necrotic damage, fibrin formation and edema. Following H&E, mucosa and submucosa samples were stained to detect Salmonella levels, which showed a significant increase in bacterial burden in FAM134B KO compared to WT cells (Fig. 7B, C). These results are consistent with our in vitro data, highlighting FAM134B role in restricting Salmonella growth. Infections were repeated as described above and WT and FAM134B KO Salmonella load was measured by CFU in feces, spleen, and intestine. Consistent with immunofluorescence analysis, we observed significantly higher levels of Salmonella in the tissue and feces of FAM134B KO mice (Fig. 7D, E, F). We next tried to determine if our previous results extended to primary macrophages, key agents involved in the innate and adaptive immune response against Salmonella infection. Thus, we measured FAM134B-mediated ER membrane scission in starved bone marrow derived macrophages (BMDM) obtained from WT mice that were infected with either Salmonella WT or ΔsopF. As expected, ER-phagy activation induced FAM134B puncta formation, which was significantly blocked by Salmonella WT, but not SopF defective Salmonella (Fig. 7G, H), recapitulating our results in endothelial cells. Finally, BMDM obtained from FAM134B KO mice displayed significantly more Salmonella than WT BMDM after infection (Fig. 7I), further underlining the importance of FAM134B in anti-bacterial response in multiple tissues and cell types. Overall, our results indicate that FAM134B is an important factor in controlling bacterial infection in vivo.

A WT and FAM134B KO C57BL/6J mice were infected with WT GFP ST through oral gavage and after 5 days, their small intestine was fixed and stained with H&E. (a) infiltration, (b) necrosis, (c) fibrosis, and (d) edema. Pathology scores of post infected tissues are presented. Data are presented as mean values ± SD of n = 4 mice. ANOVA, *P < 0.05; **P < 0.01. ST, Salmonella Typhimurium. Scale bar 50 μm. B WT and FAM134B KO mice small intestine samples from (A) were stained with DAPI and GFP to detect ST. ST, Red; DAPI, Blue. Scale bar: 5, 20 and 50 μm. C The number of cells infected with ST from (B) were quantified. Data are presented as mean values ± SD of n = 4 mice, ST infection was calculated from a minimum of 350 cells. Student’s t test, ***P < 0.001. WT and FAM134B KO mice were infected with ST through oral gavage and after 5 days their spleen, whole intestine and feces were collected. Bacterial content was determined through colony- forming unit (CFU). Data are presented as mean values ± SD (D, E) n = 7 mice and (F) n = 7 biological experiment. Student’s t test, *P < 0.05; **P < 0.01. G BMDM, bone marrow derived macrophages from WT mice were kept in NR media, AA starved for 2 hr or starve for 1 h followed by ST WT or ST ΔsopF infection in starvation media for an additional 1 h. Starved samples were treated with BafA1. BMDM were then fixed and imaged by confocal microscopy. Representative images are shown. NR Nutrient Rich, AA amino acids, BafA1 Bafilomycin A1. Scale bar 5 μm. H Average number of Fam134B puncta per cell from (G) were quantified. Data are presented as mean values ± SD of five biological experiments. Average Fam134B puncta formation was calculated from a minimum of 100 cells. ANOVA, ***P < 0.001. I BMDM from WT and FAM134B KO mice were infected with ΔinvA ST. Bacterial content was determined through colony-forming unit (CFU). Data are presented as mean values ± SD of three biological experiments. Student’s t test, *P < 0.05. Source data are provided as a Source Data file.
Discussion
The ER is a dynamic and complex intracellular organelle with several critical functions involved in maintaining cell homeostasis and adapting to stress response37. The selective autophagic degradation of the ER, ER-phagy, aims to restore ER homeostasis through the degradation of portions of the ER and in turn regulate the size and morphology of the organelle. Recently, several studies have begun to uncover the link between ER-phagy and intracellular invasive pathogens. Here, we describe the mechanism by which the intracellular pathogen Salmonella enterica serovar Typhimurium specifically prevents the protein receptor FAM134B oligomerization in order to block ER-phagy and increase bacterial viability after infection (Fig. 8).

Fam134B oligomerization leads to membrane scission, Fam134B LIR binds to LC3-family bound isolation membranes that form autophagosomes and triggers ER remodeling and degradation by ER-phagy. Salmonella infection blocks Fam134B oligomerization via the bacterial effector SopF, hindering ER-phagy induction which in turn promotes Salmonella viability (Created in BioRender. Russell, R. (2025) https://BioRender.com/f09j705).
We determined that Salmonella specifically targets FAM134B, but not TEX264, to block ER-phagy and increase bacterial viability, which was intrinsically linked to FAM134B ability to induce ER-phagy. A previous report highlighted how invasive bacteria capitalize on transforming the ER morphology to improve their viability. Upon Legionella pneumophila infection, multiple ER regulatory proteins, including FAM134A, FAM134B, FAM134C, RTN4 and TEX264, are subject to phosphoribosyl-linked ubiquitination, triggering ER remodeling and membrane recruitment to bacterial containing vacuoles19. Further studies will be required to determine if other members of the FAM134 family or other proteins involved in the regulation of ER morphology and stability are also targeted by Salmonella.
We also identified that Salmonella-mediated ER-phagy blockage is linked to the bacterial effector SopF, specifically its ADP-ribosylation activity. Previously identified as an anti-bacterial autophagy inhibitor, SopF specifically disrupts the interaction between v-ATPase and ATG16L1 by ADP-ribosylating the v-ATPase subunit ATP6V0C28,29. In these experiments SopF was shown to inhibit anti-bacterial autophagy, but not canonical autophagy. However, ER-phagy was not tested29. Thus, it is likely that SopF can ADP-ribosylate multiple targets to both prevent Salmonella clearance and promote Salmonella growth. Interestingly, SopF-mediated ER-phagy blockage could be bypassed by expressing FAM134B G216R mutant, which promotes FAM134B oligomerization. Conversely, Salmonella infection decreased both FAM134B Ser151 phosphorylation and K160 acetylation, which positively regulate oligomerization; and mass spectrometry analysis confirmed SopF expression resulted in undetectable FAM134B Ser151 phosphorylation. However, mass spectrometry failed to measure K160 acetylation and ADP-ribosylation in both the presence and absence of SopF, possibly due to a paucity of protease sites and low peptide coverage for some areas of the protein. Thus, it is possible that SopF prevents FAM134B oligomerization by directly ADP-ribosylating FAM134B or indirectly by targeting upstream regulators of FAM134B involved in the formation of multi-protein clusters required for ER-phagy38. Future studies employing a more targeted proteomics approach will be required to uncover the nature of SopF-driven post-translational changes to FAM134B.
Finally, our in vivo results demonstrated the physiological importance of FAM134B in innate immunity. Infected FAM134B KO mice presented increased necrotic damage compared to infected WT mice. Additionally, Salmonella burden was increased in the spleen, intestine and feces of FAM134B deficient mice, as well as infected BMDMs, suggesting a yet uncharacterized but important role for FAM134B in the innate response against invading pathogens. Altogether, our data uncover a previously undiscovered mechanism by which Salmonella seeks to promote bacterial viability by targeting FAM134B-mediated ER-phagy and transform the host environment to suit their growth needs. We believe this study raises several important questions including the interplay between SopF targets in controlling Salmonella viability and the general ability of FAM134B-dependent restriction for other intracellular bacteria.
Methods
Ethics compliance
All procedures involving mice were conducted at the University of Ottawa animal facility, adhering strictly to the guidelines established by the Canadian Council on Animal Care (CCAC). The University of Ottawa Animal Care Committee approved all experimental protocols
Antibodies and reagents
HA-HRP 1:1000 (Cat#2999), Fam134B 1:1000 (Cat#83414), and phospho‐S6K T389 1:1000 (Cat#9234) antibodies were obtained from Cell Signaling Technology. Anti-LC3B: 1:2000 (Cat#PM036 for immunofluorescence) antibody was purchased from MBL. Pan-ADP-ribose binding reagent 1:1000 (MABE1016), Beta-actin 1:10000 (Cat#A5441 clone AC-15) and Vinculin 1:10000 (Cat#V9131), Fam134C 1:1000 (Cat#HPA016492) antibodies, as well as doxycycline hyclate (Cat#24390-14-5), Chloroquine (Cat#C6628-25G) and VPS34 inhibitor VPS34-IN1 (Cat#1383716-33-3) were obtained from Sigma. DYKDDDDK Epitope Tag 1:1000 (Cat#NBP1-06712 for WB), anti-Tex264 1:1000 (Cat#NBP1-89866) and LC3/MAP1LC3B 1:1000 (Cat#NB100-2220 for western blot) antibodies were purchased from Novus Biologicals. Anti-LPS FITC 1:500 (Cat#sc-52223) and TBK1 1:1000 (Cat#SC-398366) antibodies were purchased from Santa Cruz Biotechnology. LPS: 1:500 (Cat#ab128709), and Anti‐S6K 1:1000(Cat#ab32529) were obtained from Abcam. Anti-RFP (Cat#600-401-379) was obtained from Cedarlane. Anti-p62 1:1000 (Cat#GP62-C) was purchased from Progen. Anti-Tex264 1:1000 (Cat#25858-1-AP), Fam134A 1:1000 (Cat#24650-1-AP) and anti-Tubulin 1:10000 (Cat#66362-1-Ig) were purchased from Proteintech. Bafilomycin A1 was obtained from Tocris (Cat#133410U). Digitonin (Cat#10188-874) was obtained from VWR. Alexa Fluor 647 1:1000 (Mouse Cat#A21235, Rabbit Cat#A21244), Alexa Fluor 568 1:1000 (Cat#A11036), HA tag monoclonal antibody (Cat#26183), Streptavidin-HRP 1:1000 (Cat#21130) and Reticulon 3 poly-clonal antibody 1:1000 (Cat#A302-860A) were purchased from Thermo-Fisher. Anti-phospho-Fam134B-Ser151 and anti-Ace-Fam134B-Lys160 are not commercially available and have been previously described9,10. Anti-HALOTag® Monoclonal Antibody 1:1000 (Cat#G9211) and HALOTag® TMR Ligand (Cat#G8252) were purchased from Promega.
Cell culture
HEK293A (CRL-1573), HCT116 (CCL-247), and HeLa (CCL-2) were purchased from American Type Culture Collection (ATCC), cell lines were cultured in DMEM (Wisent, Cat#319-015-CL) supplemented with 10% bovine calf serum (VWR Life Science Seradigm, Cat#CA76502-732). For amino acid starvation experiments, the medium was prepared based on the Gibco standard recipe, omitting all amino acids and excluding non-essential amino acids. Dialyzed FBS (Thermo Fisher Scientific, Cat#26400044) was used as a substitute for standard FBS. Cells stably expressing the ER-phagy probe were treated with 0.5 μg/mL doxycycline (Sigma, 324385-1GM) for 24 h to induce expression. After induction, the cells were washed twice with PBS and cultured in either complete DMEM supplemented with 10% bovine calf serum or the amino acid starvation medium7.
Transfection
Transfections were performed using polyethylenimine (PEI, medistore uOttawa) or Polyjet DNA transfection reagent (Cat#SL100688) from FroggaBio. The samples were analyzed 48–72 h post-transfection.
Generation of knock-out cell lines using CRISPR/Cas9
FAM134B and TEX264 KO lines were generated in the HEK293A and HCT116 background utilizing CRISPR/Cas9 using guide RNA sequence GTCTGACACAGACGTCTCAG and GCCACAGTGACGTTGCGGAT, respectively. FAM134B guide RNA sequence was previously used7. FIP200 KO HEK293A cell line was generated using guide RNA sequence AGAGTGTGTACCTACAGTGC.
Generation of stable cell lines
HEK293T cells were transiently transfected using polyethylenimine (PEI, Medistore uOttawa) with a lentiviral vector with the packaging plasmids pCMV-VSV-G (Addgene, Plasmid #8454) and psPAX2 (Addgene, Plasmid #12260). After 2–3 days of culture, the supernatant was harvested and filtered through a 0.45-μm syringe filter (Thermo Fisher Scientific, Cat#13100105). For retrovirus production, HEK293T cells were similarly transfected with the retroviral vector pCMV-GP retro and the envelope plasmid pCMV-VSV-G (kindly provided by Dr. Meng, University of Miami). Viral particles were collected from the supernatant as described above. Target cells were then transduced with either retrovirus encoding HALO-mGFP-KDEL or lentivirus encoding ss-RFP-GFP-KDEL in the presence of 10 μg/mL polybrene (Sigma, H9268-5G). Stable transformants were selected using puromycin (Thermo Fisher Scientific, Cat#A1113803) or blasticidin (Abcam, Cat#ab141452)7.
Plasmids
Plasmids pEGFP-C1-SopF (#137734), pCW57-CMV-ssRFP-GFP-KDEL (#128257), pMRX-IB-HaloTag7-mGFP-KDEL (#184904) and pMRX-INU-FLAG-FAM134B (#128260) were obtained from Addgene. SopF cDNA from pEGFP-C1-SopF was subcloned into FLAG-pcDNA to generate FLAG-SOPF plasmids. FAM134B cDNA from pMRX-INU-FLAG-FAM134B was subcloned into c-FLAG-pcDNA to generate FAM134B-FLAG plasmids. All constructs were generated using fast-cloning as previously described39. The PCR reaction components for template amplification were: 50 μl total volume, 0.5 μl Phusion DNA polymerase (New England Biolabs), 5 μl 10× buffer; 5 μl of 2.5 mM dNTPs; 10 ng of plasmid DNA template; 5 pmol of each primer. The PCR cycling parameters were 98 °C 3 min, (98 °C 10 s, 55 °C 30 s, 72 °C 20 s/kb) × 20 cycles, and 72 °C for 5 min. The PCR products were treated with 1 µl of DpnI at 37 °C for 1 h. SopF cDNA from pEGFP-C1-SopF was cloned using Gibson assembly into pFLAG-TurboID C1 to generate pFLAG-TurboID-SopF. pFLAG-TurboID C1 was a gift from Dr. Laura Trinkle.
Site-directed mutagenesis was performed using an efficient one-step site-directed plasmid mutagenesis protocol as previously described40. The PCR reaction of 50 μl contained 2–10 ng of template, 1 μM primer pair, 200 μM dNTPs and 0.5 unit of Phusion DNA polymerase (New England Biolabs). The PCR cycles were initiated at 95 °C for 5 min to denature the template DNA, followed by 15 amplification cycles. Each amplification cycle consisted of 95 °C for 1 min, non-overlapping Tm −5 °C for 1 min and 72 °C for 10 min or 15 min according to the length of the template constructs. The PCR cycles were finished with an annealing step at overlapping Tm -5 for 1 min and an extension step at 72 °C for 30 min. The PCR products were treated with 1 µl of DpnI at 37 °C for 1 h. The PCR specificity of mutagenesis was analyzed by Sanger sequencing. Primers used for site-directed mutagenesis are described in Table 1.
Bacterial strains
Wild-type and ΔinvA (SL1344) Salmonella enterica serovar Typhimurium strains were a gift from Dr. Subash Sad (University of Ottawa). Salmonella ΔsopF, ΔpipB2 and ΔspiC strain (SL1344) and others were a gift from Dr. John Brumell (University of Toronto). Bacteria were grown in Luria-Bertani (LB) broth (Fisher).
Bacterial infection
Salmonella was grown in 4 ml of LB broth at 37 °C at 250 rpm. Overnight cultures of Salmonella were diluted 30-fold and allowed to grow until reaching an OD600 of 1.5, followed by centrifugation of 10,000 g for 2 min, and the resulting pellet was resuspended in 1 ml of phosphate-buffered saline (PBS). Bacterial stock was then diluted to multiplicity of infection (MOI) of 180 in DMEM supplied with 10% heat-inactivated bovine calf serum for infection or amino acid starvation media. Cells cultured in antibiotic-free medium were infected with Salmonella infection and maintained at 37 °C in a 5% CO2 environment for the specified duration. Prior to analysis, cells were washed once with PBS and lysed directly using 1× denaturing SDS sample buffer.
Western blot and immunoprecipitation
Whole-cell lysates were prepared by direct lysis with 1x SDS sample buffer, followed by boiling for 10 min at 95 °C and resolved by SDS–PAGE. Immunoprecipitation cells were harvested in mild lysis buffer (MLB) containing 10 mM Tris pH 7.5, 10 mM EDTA, 100 mM NaCl, 50 mM NaF, and 1% NP‐40, supplemented with protease and phosphatase inhibitor cocktails (including EDTA from APExBIO). The lysates were then centrifuged at maximum speed 17,000 g for 10 min to remove cell debris. Anti-FLAG affinity gel beads (Sigma) were washed once with MLB and then incubated with cell lysates for 1.5 h, followed by a single wash with MLB containing inhibitors and four quick washes with MLB alone. The beads were subsequently boiled in a 1x denaturing sample buffer for 10 min before being resolved by SDS–PAGE. Imaging was conducted using the ChemiDoc™ Touch imaging system (Bio-Rad).
Immunofluorescence
Cells were seeded onto IBIDI-treated coverslips and allowed to adhere overnight. Following treatments for Salmonella infection MOI of 70 was used for IF experiments, cells were fixed in 4% paraformaldehyde in PBS for 15 min and then permeabilized with 50 μg/ml digitonin in PBS for 10 min at room temperature. Subsequently, cells were blocked using a blocking buffer (1% BSA and 2% serum in PBS) for 45 min, followed by incubation with primary antibodies in the same buffer for 1 h at room temperature. After incubation, samples were washed three times in PBS and once in blocking buffer before being incubated in secondary antibodies for 1 h at room temperature. Slides were washed three times in PBS, stained with DAPI, and mounted. Imaging was conducted using a Zeiss LSM 800 AxioObserver Z1 Confocal Microscope. For staining bacterial localization (inside/outside), cells were first incubated with an anti-LPS antibody for 1 h and followed by secondary antibody incubation for 1 h in a blocking buffer before permeabilization. This was followed by three PBS washes between steps. Confocal microscopy images were analyzed using an automated protocol implemented in ImageJ software to minimize bias. The same protocol was consistently applied to each field of view and across all samples. An average of seven unique fields of view from representative experiments were selected for quantification.
Cyto-ID autophagy detection kit assay
Cells were seeded onto ibidi eight well µ-Slides (Ibidi, cat. 80826) and allowed to adhere overnight. Subsequently, cells were subjected to amino acid starvation with or without Salmonella infection. Following treatment, cells were incubated in DMEM without phenol red containing Cyto-ID autophagy detection stain (Enzo, ENZ-KIT175-0050) for 30 min, then washed with PBS and re-incubated with either complete DMEM without phenol red or amino acid media. Images were acquired and deconvolved using an inverted epifluorescent Zeiss AxioObserver.Z1 microscope.
Biotin-based proximity labeling TurboID assay
Transfected cells with either FLAG-TurboID-SopF or FLAG-TurboID control were incubated with 50 μM biotin (Sigma, B4639) for 1 h. Cells were then rinsed with PBS and lysed with high salt RIPA buffer (50 mM Tris pH 7.5, 500 mM NaCl, 1% NP-40, 0.5% deoxycholate), supplemented with protease and phosphatase inhibitor cocktails (including EDTA from APExBIO). Lysates were centrifugated at maximum speed 17,000 g for 10 min at 4 °C and the supernatant was transferred to a new tube and diluted with an equal volume of no salt RIPA buffer (50 mM Tris pH 7.5, 1% NP-40, 0.5% deoxycholate). Diluted samples were incubated with Streptavidin-agarose beads (Thermo Fisher, 20359) for 4 h at 4 °C. Beads were then pelleted at 1000 RPM for 2 min, washed three times with 250 mM salt RIPA buffer and eluted by adding a bead equivalent volume of 2% SDS with 30 mM biotin, vortexed and incubated at 95 °C for 10 min.
Colony-forming unit assay
Cell lines
Cells were infected with Salmonella at a multiplicity of infection (MOI) of 180 for 1 h. Subsequently, the infected cells were rinsed three times and treated with media containing 100 μg/ml Gentamicin for 0.5 h, followed by a 4-h incubation with media containing 50 μg/ml Gentamicin. After incubation, the samples were washed three times with PBS and then lysed using CFU buffer (0.1% Triton X-100 and 0.01% SDS in PBS). The lysates obtained were subjected to serial dilution (1:50, 1:75, and 1:100) and plated onto LB agar plates containing Streptomycin. The plates were incubated at 37 °C for 16–18 h, and the colonies were counted to determine the number of CFU.
Colony-forming unit assay
BMDM
Salmonella ΔinvA was used to quantify intracellular bacterial burden without the issue of cell death acting as a confounding variable. C57BL/6J WT and FAM134B KO BMDMs were seeded in triplicate at a density of 300,000 cells per well. Salmonella ΔinvA was cultured overnight at 37 °C with shaking at 250 RPM in 5 mL of LB broth containing 100 µg/mL streptomycin. The following day, the optical density at 600 nm reached ~2.3, which represents a concentration of 9.2 × 109 CFU/mL. Salmonella ΔinvA was then centrifuged at 9500 RPM for 5 min. The resulting pellet was resuspended in 1 mL of R8 medium. 100 µL of resuspended Salmonella ΔinvA was added to 650 µL of PBS and 250 µL of normal mouse serum (Jackson ImmunoResearch Laboratories) in a 15 mL Falcon tube to facilitate bacterial opsonization. This was incubated with shaking at 37 °C and 210 RPM for 25 min. Following incubation, the opsonized bacteria was centrifuged, washed twice with PBS, and resuspended in R8 medium.
Salmonella ΔinvA was then added to BMDMs at an MOI of 10. Next, the plate was centrifuged at 800 × g for 5 min and incubated at 37 °C for 25 min. After the incubation, the cells were washed twice with PBS containing 50 µg/mL of gentamicin and incubated with R8 medium containing 50 µg/mL of gentamicin. After 1.5 h, the media was replaced with R8 medium containing a reduced concentration of gentamicin (10 µg/mL). Intracellular bacterial replication was quantified by lysing the BMDMs at desired time points using 300 µL of 1% Triton-X lysis buffer. Serial dilutions of the lysates were plated on LB agar containing 100 µg/mL streptomycin to quantify bacterial burden. Plates were incubated overnight at 37 °C and CFU was counted the following day.
Immunohistochemistry staining
Samples were rinsed three times with PBS, treated with 3% H2O2 (in PBS) for 10 min, and washed three times with PBS. Blocked with protein block serum-free (catalog no. X0909 Dako) for 2 h, stained with primary antibody overnight at 4 °C (GFP 1:150 catalog no. Sigma #G1544), washed three times with PBS, incubated with secondary antibody (Alexa Fluor 555 anti-rabbit, catalog no. A31572, 1:1000) for 1 h, washed once with PBS, stained with DAPI (2 mg ml−1, Roche Diagnostics) for 10 min, washed three times with PBS and cover slip-mounted with Fluoromount-G mounting solution (Invitrogen, 00-4958-02). All treatments were done at room temperature unless otherwise stated. Images were acquired using a Zeiss LSM 800 AxioObserver Z1 Confocal Microscope.
In vivo experiments
WT and FAM134B KO C57BL/6J mice were subjected to a 3-h fast from both food and water prior to the oral administration of 20 mg of streptomycin (Millipore Sigma) dissolved in 100 µL of ddH2O. 2 h following the streptomycin treatment, food and water were reintroduced to the mice. The following day, mice were again fasted from food and water for 3 h prior to receiving an oral dose of WT GFP Salmonella (1 × 108 CFU) in 100 µL of saline per mouse. Food and water were reintroduced 2 h after the infection.
5 days post-infection, small intestines were harvested and fixed in 10% formalin for 2 days, then rinsed three times with 30% sucrose. The samples were then dehydrated and paraffin-embedded, sectioned into 4 µm thick slices and mounted onto glass microscope slides. Prior to staining, samples were rehydrated and deparaffinized. Antigen-retrieval for both groups was performed in pH 9.0 EDTA solution, at 110 °C for 12 min in a microwave processor (Histo5, Milestone).
For CFU assays, five days post mice infection, desired organs were collected and homogenized using frosted glass slides (Fisherbrand) and filtered through a 70 µm cell strainer. The filtered spleen cells were centrifuged at 500 × g for 5 min and resuspended in 10 mL R8 medium (RPMI 1640 media (Gibco, Thermo-Fisher Scientific Inc) supplemented with 8% FBS (Gibco) and 55 µM 2-mercaptoethanol (Gibco). Appropriate serial dilutions were made in PBS and 100 µL aliquots were plated onto LB agar plates containing 100 µg/mL of streptomycin. Plates were incubated overnight at 37 °C and CFU were counted the following day.
Mass-spectrometry
Two 15 cm plates expressing FAM134B-FLAG and either HA-SOPF or mock plasmid cultured to 90–100% confluency were starved for amino acids for 1 h and subjected to immunoprecipitation. Beads were eluted with 100 μL Glycine 0.1 M pH3 for 10 min with constant rocking and immediately neutralized with 10 μL 0.5 M Tris-HCl pH 7.4 1.5 M NaCl. Elutes were subjected to mass spectrometry analysis to identify post‐translational modifications. TCEP [Tris(2-carboxyethyl)phosphine hydrochloride; Thermo Fisher Scientific] was added to the samples to a final concentration of 10 mM. Samples were vortexed for 1 h at 37 °C. Chloroacetamide (Sigma-Aldrich) was added for alkylation to a final concentration of 55 mM. Samples were vortexed for another hour at 37 °C. One microgram of trypsin was added, and digestion was performed for 8 h at 37 °C. Samples were dried down and solubilized in 5% ACN-4% formic acid (FA). The samples were loaded on a 1.5 µl pre-column (Optimize Technologies, Oregon City, OR). Peptides were separated on a home-made reversed-phase column (150-μm i.d. by 200 mm) with a 56-min gradient from 10 to 30% ACN-0.2% FA and a 600-nl/min flow rate on a Easy nLC-1200 connected to a Exploris 480 (Thermo Fisher Scientific, San Jose, CA). Each full MS spectrum acquired at a resolution of 120,000 was followed by tandem-MS (MS-MS) spectra acquisition on the most abundant multiply charged precursor ions for 3 s. Tandem-MS experiments were performed using higher energy collision dissociation at a collision energy of 34%. The data were processed using PEAKS X Pro (Bioinformatics Solutions, Waterloo, ON) and a Uniprot database. Mass tolerances on precursor and fragment ions were 10 ppm and 0.01 Da, respectively. Fixed modification was carbamidomethyl (C). Variable selected posttranslational modifications were acetylation (N-ter), oxidation (M), deamidation (NQ), phosphorylation (STY). The data were visualized with Scaffold 5.0 (protein threshold, 99%, with at least two peptides identified and a false-discovery rate [FDR] of 1% for peptides).
Transmission electron microscopy
HEK293A cells were treated with the indicated treatments and fixed overnight in electron microscopy-grade 4% paraformaldehyde (EMS Cat#15713-s) and 3.5% glutaldehyde. To prepare the samples for electron microscopy, the cells were washed in PBS to eliminate the antifreeze solution. The samples are then treated with a mixture of 1.5% potassium ferrocyanide and 2% osmium tetroxide for 1 h, followed by a ddH2O wash. Next, the cells undergo a 20 min incubation in 10 mg/mL TCH solution, another ddH2O wash, and a 30 min treatment with 2% osmium tetroxide, followed by a final ddH2O rinse. The cells then undergo sequential 2 min dehydration in increasing concentrations of ethanol, followed by 5 min final dehydration in propylene oxide. The samples were then transferred to pans containing Durcupan resin and left overnight until the samples sank. The following day, the samples are sandwiched between Aclar sheets coated with a thin resin layer and cured in a 55 °C oven for 3 days. Finally, the samples are sectioned to 70 nm thickness using an ultramicrotome with a diamond knife, mounted on 300-mesh copper grids (EMS cat #G300-Cu), and imaged using a Jeol JEM1400-Flash electron microscope at 80 kV, with magnifications of 8000× and 15k×.
Generation of murine bone marrow-derived macrophages
WT and FAM134B KO C57BL/6J mice were euthanized following the guidelines set by the Canadian Council on Animal Care (CCAC). Bone marrow was harvested from the femur, tibia, and hip bones. The isolated bone marrow cells were plated onto petri dishes (Fisherbrand) that had been pre-coated with 5 ng/mL of macrophage colony-stimulating factor (BioLegend). The cells were then cultured in RPMI 1640 medium supplemented with 8% fetal bovine serum (Gibco) and 50 µg/mL of gentamicin (Gibco). After a 7-day incubation at 37 °C and 5% CO2, the bone marrow-derived macrophages were collected for subsequent experiments.
Statistical analysis
Error bars for western blot analysis represent the standard deviation between densitometry data collected using ImageJ software from unique biological experiments. Statistical analyses were performed using GraphPad Prism 8. Statistical significance was determined using either Student’s t test or ANOVA. Differences with a P value < 0.05 or lower were considered significant. significant. *p < 0.05, **p < 0.01, ***p < 0.001. The number of independent experiments (n), statistical measurements tests utilized, dispersion of measurements, and significance are described in the figure legends and supplementary Table 1. Sample sizing for cellular imaging was chosen to be the minimum number of independent experiments required for statistically significant results and are described in figure legends.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Source data are provided with this paper. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE [1] partner repository with the dataset identifier PXD061148 and https://doi.org/10.6019/PXD061148. Source data are provided with this paper.
References
Yang, Z. & Klionsky, D. J. Eaten alive: a history of macroautophagy. Nat. Cell Biol. 12, 814–822 (2010).
Feng, Y., He, D., Yao, Z. & Klionsky, D. J. The machinery of macroautophagy. Cell Res. 24, 24–41 (2014).
Gatica, D., Lahiri, V. & Klionsky, D. J. Cargo recognition and degradation by selective autophagy. Nat. Cell Biol. 20, 233–242 (2018).
Klionsky, D. J. & Schulman, B. A. Dynamic regulation of macroautophagy by distinctive ubiquitin-like proteins. Nat. Struct. Mol. Biol. 21, 336–345 (2014).
Chino, H. & Mizushima, N. ER-Phagy: quality and quantity control of the endoplasmic reticulum by autophagy. Cold Spring Harb. Perspect. Biol. 15, a041256 (2023).
Khaminets, A. et al. Regulation of endoplasmic reticulum turnover by selective autophagy. Nature 522, 354–358 (2015).
Chino, H., Hatta, T., Natsume, T. & Mizushima, N. Intrinsically disordered protein TEX264 mediates ER-phagy. Mol. Cell 74, 909–921.e6 (2019).
An, H. et al. TEX264 is an endoplasmic reticulum-resident ATG8-interacting protein critical for ER remodeling during nutrient stress. Mol. Cell 74, 891–908.e10 (2019).
Jiang, X. et al. FAM134B oligomerization drives endoplasmic reticulum membrane scission for ER-phagy. EMBO J. 39, e102608 (2020).
Wang, X. et al. A regulatory circuit comprising the CBP and SIRT7 regulates FAM134B-mediated ER-phagy. J. Cell Biol. 222, e202201068 (2023).
Chino, H. et al. Phosphorylation by casein kinase 2 enhances the interaction between ER-phagy receptor TEX264 and ATG8 proteins. EMBO Rep. 23, e54801 (2022).
Bhaskara, R. M. et al. Curvature induction and membrane remodeling by FAM134B reticulon homology domain assist selective ER-phagy. Nat. Commun. 10, 2370 (2019).
Siggel, M., Bhaskara, R. M., Moesser, M. K., Ikić, I. D. & Hummer, G. FAM134B-RHD protein clustering drives spontaneous budding of asymmetric membranes. J. Phys. Chem. Lett. 12, 1926–1931 (2021).
González, A. et al. Ubiquitination regulates ER-phagy and remodelling of endoplasmic reticulum. Nature 618, 394–401 (2023).
Chiramel, A. I., Dougherty, J. D., Nair, V., Robertson, S. J. & Best, S. M. FAM134B, the selective autophagy receptor for endoplasmic reticulum turnover, inhibits replication of Ebola virus strains Makona and Mayinga. J. Infect. Dis. 214, S319–S325 (2016).
Lennemann, N. J. & Coyne, C. B. Dengue and Zika viruses subvert reticulophagy by NS2B3-mediated cleavage of FAM134B. Autophagy 13, 322–332 (2017).
Tan, X. et al. Coronavirus subverts ER-phagy by hijacking FAM134B and ATL3 into p62 condensates to facilitate viral replication. Cell Rep. 42, 112286 (2023).
Zhang, L. et al. AMFR-mediated Flavivirus NS2A ubiquitination subverts ER-phagy to augment viral pathogenicity. Nat. Commun. 15, 9578 (2024).
Shin, D. et al. Regulation of phosphoribosyl-linked serine ubiquitination by deubiquitinases DupA and DupB. Mol. Cell 77, 164–179.e6 (2020).
Sher, A. A., Mustafa, B. E., Grady, S. C., Gardiner, J. C. & Saeed, A. M. Outbreaks of foodborne Salmonella enteritidis in the United States between 1990 and 2015: An analysis of epidemiological and spatial-temporal trends. Int. J. Infect. Dis. 105, 54–61 (2021).
Bakowski, M. A., Braun, V. & Brumell, J. H. Salmonella-containing vacuoles: directing traffic and nesting to grow. Traffic 9, 2022–2031 (2008).
Katayama, H., Yamamoto, A., Mizushima, N., Yoshimori, T. & Miyawaki, A. GFP-like proteins stably accumulate in lysosomes. Cell Struct. Funct. 33, 1–12 (2008).
Yim, W. W.-Y., Yamamoto, H. & Mizushima, N. A pulse-chasable reporter processing assay for mammalian autophagic flux with HaloTag. eLife 11, e78923 (2022).
Klionsky, D. J. et al. Guidelines for the use and interpretation of assays for monitoring autophagy (4th edition).Autophagy 17, 1–383 (2021).
Ichimura, Y. et al. A ubiquitin-like system mediates protein lipidation. Nature 408, 488–492 (2000).
Davidson, G. L. et al. Frequency of mutations in the genes associated with hereditary sensory and autonomic neuropathy in a UK cohort. J. Neurol. 259, 1673–1685 (2012).
LaRock, D. L., Chaudhary, A. & Miller, S. I. Salmonellae interactions with host processes. Nat. Rev. Microbiol. 13, 191–205 (2015).
Xu, Y. et al. ARF GTPases activate Salmonella effector SopF to ADP-ribosylate host V-ATPase and inhibit endomembrane damage-induced autophagy. Nat. Struct. Mol. Biol. 29, 67–77 (2022).
Xu, Y. et al. A bacterial effector reveals the V-ATPase-ATG16L1 axis that initiates xenophagy. Cell 178, 552–566.e20 (2019).
Trinkle-Mulcahy, L. Recent advances in proximity-based labeling methods for interactome mapping. F1000Research 8, 135 (2019).
Knodler, L. A., Nair, V. & Steele-Mortimer, O. Quantitative assessment of cytosolic Salmonella in epithelial cells. PloS One 9, e84681 (2014).
Gatica, D., Chiong, M., Lavandero, S. & Klionsky, D. J. The role of autophagy in cardiovascular pathology. Cardiovasc. Res. 118, 934–950 (2022).
Birmingham, C. L., Smith, A. C., Bakowski, M. A., Yoshimori, T. & Brumell, J. H. Autophagy controls Salmonella infection in response to damage to the Salmonella-containing vacuole. J. Biol. Chem. 281, 11374–11383 (2006).
Thurston, T. L. M., Ryzhakov, G., Bloor, S., von Muhlinen, N. & Randow, F. The TBK1 adaptor and autophagy receptor NDP52 restricts the proliferation of ubiquitin-coated bacteria. Nat. Immunol. 10, 1215–1221 (2009).
Lau, N. et al. SopF, a phosphoinositide binding effector, promotes the stability of the nascent Salmonella-containing vacuole. PLoS Pathog. 15, e1007959 (2019).
Kohno, S., Shiozaki, Y., Keenan, A. L., Miyazaki-Anzai, S. & Miyazaki, M. An N-terminal-truncated isoform of FAM134B (FAM134B-2) regulates starvation-induced hepatic selective ER-phagy. Life Sci. Alliance 2, e201900340 (2019).
Bravo, R. et al. Endoplasmic reticulum and the unfolded protein response: dynamics and metabolic integration. Int. Rev. Cell Mol. Biol. 301, 215–290 (2013).
Foronda, H. et al. Heteromeric clusters of ubiquitinated ER-shaping proteins drive ER-phagy. Nature 618, 402–410 (2023).
Li, C. et al. FastCloning: a highly simplified, purification-free, sequence- and ligation-independent PCR cloning method. BMC Biotechnol. 11, 92 (2011).
Liu, H. & Naismith, J. H. An efficient one-step site-directed deletion, insertion, single and multiple-site plasmid mutagenesis protocol. BMC Biotechnol. 8, 91 (2008).
Acknowledgements
We would like to thank Dr. John Brumell for sharing Salmonella mutants. We would also like to thank Karyn King for providing FIP200 KO cell lines and Dr. Rudolf Mueller for scoring H&E staining. A big thank you to Zaida Ticas, Marjan Khalili and Mufida Alazzabi from the Louise Pelletier HCF at the University of Ottawa for all their help. We would like to acknowledge the assistance of StemCore Laboratories Genomics Core Facility, OHRI, University of Ottawa (RRID:SCR_012601). The authors must acknowledge the Cell Biology and Image Acquisition Core (RRID: SCR_021845) funded by the University of Ottawa, Ottawa, Natural Sciences and Engineering Research Council of Canada, and the Canada Foundation for Innovation. We gratefully acknowledge the IHC tissue samples processing services provided by the Louise Pelletier HCF (RRID: SCR_021737) at the University of Ottawa. The authors acknowledge the Electron Microscopy Core (RRID: SCR_025398) funded by the University of Ottawa, Brain-Heart Interconnectome (BHI) via Canadian First Research Excellence Fund (CFREF). Finally, proteomics analyses were performed by the Center for Advanced Proteomics Analyses, a Node of the Canadian Genomic Innovation Network that is supported by the Canadian Government through Genome Canada. This work was supported by the Canadian Institutes of Health Research (CIHR), funding reference number 181799 (D.G.) and 376756 (R.C.R.), 153034 (R.C.R.), and Natural Sciences and Engineering Research Council of Canada #2023-05587 (R.C.R.). R.M.A. received support from the Ottawa Institute of System Biology and the Centre for Infection, Immunity and Inflammation.
Author information
Authors and Affiliations
Contributions
D.G. and R.M.A. designed in vitro experiments. D.G. and R.M.A. performed in vitro experiments. D.G., R.M.A., R.EH., S.S., and R.R. designed mice experiments. D.G., R.M.A., and R.E.H. performed mice experiments. R.M. scored H&E samples. D.G., R.M.A., and R.R. wrote the manuscript. M.M. provided FAM134B KO mice. B.L. and Q.S. performed Fam134B phosphorylation and acetylation experiments. All authors discussed the results and commented on the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Viktor Korolchuk and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Gatica, D., Alsaadi, R.M., El Hamra, R. et al. The ER-phagy receptor FAM134B is targeted by Salmonella Typhimurium to promote infection. Nat Commun 16, 2923 (2025). https://doi.org/10.1038/s41467-025-58035-7
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-58035-7
