
Abstract
Migratory functionalization of C–H bonds through metal migration from carbon to carbon under transition metal catalysis is a process of significant academic and industrial interest. Herein, a palladium-catalyzed migratory cyclization of α-bromoalkene derivatives ArXCBr=CH2, in which X denotes a phosphorus (P(O)R), silicon (SiR2), sulfur (SO2), carbon (C(O)), nitrogen (NTs), or oxygen-based moiety, affording various benzoheterocyclic compounds has been developed. Mechanistic investigations have demonstrated that the cyclization reaction proceeds through an unexpected cascade, with trans-1,2-palladium migration between sp2 carbons being a key step of catalytic cycle. To the best of our knowledge, this type of metal migration has not been reported previously.
Similar content being viewed by others
Introduction
Migratory functionalization of C–H bonds through metal migration from carbon to carbon under transition metal catalysis is a process of significant academic and industrial interest1,2,3,4,5,6,7,8,9,10,11,12. It provides a nonclassical means of selectively installing a functional group at a remote C–H position using simple precursors, thus enabling the direct synthesis of challenging structures not accessible through traditional cross-couplings. Most notably, migratory functionalization of alkenes or alkyl halides through a 1,2-metal shift along a sp3 chain and cross-coupling has been well developed for remote C–H bond functionalization (Fig. 1)8,9,10,11,12. Migratory functionalization via cis-1,2-palladium migration between sp2 carbons has been rarely reported13,14,15,16,17,18,19. Another metal migration frequently exploited is a 1,4-metal shift in many transition metal-catalyzed tandem reactions1,4,5.

1,2-Metal migration between sp3 or sp2 carbons in catalytic cycles.
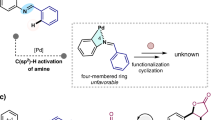
Indenone and five-membered benzoheterocycles are ubiquitous structural motifs found in a wide array of natural products, pharmaceuticals, and agrochemicals (Fig. 2a)20,21. In particular, benzophosphole is widely used in developing functional materials22,23,24,25,26,27, biologically active compounds28,29,30,31,32,33, and chiral ligands34,35,36,37. The transition metal-catalyzed intramolecular coupling of aryl halides or aryl metals with alkenes has become a versatile tool in heterocycle synthesis (Fig. 2b, top)38,39,40,41,42,43. However, this traditional strategy requires the prior introduction of a halo or metal group at the ortho position of the aryl ring. Under this premise, we wondered whether an alternative synthetic route based on a cascade-type process involving 1,n-metal migration from an alkenyl carbon atom to an aryl carbon atom might be designed, thus setting the stage for carbometallation of a C=C double bond (Fig. 2b, bottom). Hayashi reported the Rh-catalyzed cyclization of arylpropargyl alcohols via a 1,4-Rh shift–aryl rhodation sequence, which validates our hypothesis44,45,46. The very wide availability of mono-substituted olefins ArX–CH=CH2 and their facile conversion into ArX–CBr=CH2 by simple bromination and elimination should make this new strategy very synthetically useful. At the outset of our investigations, however, it was unclear whether such a strategy could be implemented as elimination from ArX–CBr=CH2 to give alkynes could be envisaged as being highly feasible.

a Indenone and five-membered benzoheterocycles. b Strategies to synthesize benzocycles. c This work: Pd-catalyzed migratory cyclization via an unexpected reaction cascade.
Herein, we report the successful realization of this goal through a palladium-catalyzed migratory cyclization of α-bromoalkene derivatives ArXCBr=CH2 1, in which the X moiety is a phosphine oxide, silyl, sulfonyl, carbonyl, amide, or oxygen atom/group, to give benzoheterocycles 2 (Fig. 2c). In this process, two C–H bonds are simultaneously cleaved and coupled to form a new carbon–carbon bond, whereas transition metal-catalyzed C–H/C–H coupling of arenes and alkenes always occurs via an aryl C–H metalation followed by a Heck-type alkenylation process47,48,49,50,51,52,53,54. However, the reaction does not proceed through an oxidative addition–1,4-palladium migration55,56,57,58,59,60,61–arylpalladation sequence as we expected. Detailed mechanistic studies have shown that trans-1,2-metal migration from the α-position to the trans-β-position of the C=C double bond is a key step of the catalytic cycle. This migratory cyclization is synthetically very useful and offers opportunities for the efficient synthesis of indoles and their phosphorus, silicon, sulfur, carbon, and oxygen congeners. Previously, 2,3-unsubstituted benzophosphole was prepared by ring-closing metathesis of phenylstyrylvinylphosphine oxide62,63. As reported herein, the present method enabled the efficient synthesis of benzophosphole oxides, and the asymmetric version of the cascade reaction was also achieved in the presence of a chiral palladium catalyst to give enantio-enriched P-chiral products.
Results
Optimization of the reaction conditions
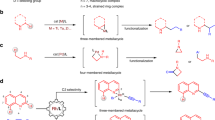
In a first set of experiments, the cyclization of (α-bromoethenyl)diphenylphosphine oxide (1a) was performed in the presence of 5 mol% of palladium catalysts bearing several types of bisphosphine ligands and 2.0 equiv. of CsOPiv (OPiv = pivalate) in 1,2-dichloroethane (DCE) at 80 °C for 12 h (Table 1). The reaction with DPEPhos as a ligand gave a 94% yield of the cyclization product 2a (entry 1). An alternative wide-bite-angle ligand, Xantphos, was not effective for the present reaction (entry 2). Some other bisphosphine ligands, such as dppf, dppe, dppp, dppb, and biphep, gave 2a as the main product in somewhat lower yields (entries 3–7). The reaction performed in toluene afforded 2a in moderate yield, whereas in CH2Cl2 it gave a mixture of 2a and 2a′ in an 85:15 ratio (entries 8 and 9). The use of other bases, such as CsOAc and KOPiv, furnished 2a in moderate yields with medium chemoselectivities (entries 10 and 11). The reaction performed at a lower temperature (60 °C) gave 2a in a slightly lower yield (entry 12).
Pd-catalyzed migratory cyclization: substrate scope
Figure 3 summarizes the results obtained for the migratory cyclization of other ArXCBr=CH2, in which X denotes phosphorus or another element, under the optimized conditions. Cyclizations of (α-bromoethenyl)diarylphosphine oxides 1b–1g, in which the aromatic groups are phenyl moieties substituted with methyl, t-butyl, phenyl, methoxy, fluoro, and trifluoromethyl at the para position, proceeded well, giving the benzophospholes 2b–2g in high yields, irrespective of the electronic properties of the substituents (Fig. 3). (α-Bromoethenyl)diarylphosphine oxides 1h–1k, in which the aromatic groups are phenyl moieties substituted with methyl, methoxy, and trifluoromethyl at the meta position, all proved suitable for this reaction, affording exclusively the benzophospholes 2h–2k in high yields, and the new C–C bonds were formed with high regioselectivity at the less hindered ortho C–H position. Moreover, ortho-substituted phenyl derivative 1l gave 70% yield of the migratory cyclization product 2l, the yield being lower due to some generation of the elimination by-product. The reaction of 1m, bearing a benzo[b]thiophen-5-yl group, gave 67% yield of the corresponding product 2m with high regioselectivity. The migratory cyclizations of alkyl-substituted (α-bromoethenyl)phenylphosphine oxides 1n‒1r also proceeded, furnishing the corresponding products 2n‒2r in moderate yields. The yields of 2n‒2r were moderate due to some generation of the elimination by-product.

Reaction conditions: 1 (0.20 mmol), Pd(OAc)2 (5 mol% Pd), DPEPhos (10 mol%), CsOPiv (0.40 mmol), and DCE (1.0 mL) at 80 °C for 12 h. Isolated yield.
The robustness of the present protocol was further demonstrated by the synthesis of other benzoheterocyclic compounds, in which the X moiety is an atom or group other than phosphine oxide (Fig. 3). It was found that the migratory cyclization of α-bromovinyl ketone 1s, in which the X moiety of the substrate is a carbonyl group, was viable, giving the inden-1-one product 2s in a high yield under the standard conditions. N-Tosylindoles 2t‒2v, in which X is a tosyl-protected amide group, were also efficiently obtained by using the present methodology. When the X moiety of the substrate was an oxygen atom, the migratory cyclization proceeded smoothly to afford benzofuran 2w in 73% yield. Moreover, unsubstituted 1-silaindene 2x was obtained in 63% yield under the present conditions. Benzo[b]thiophene 1-oxide and 1,1-dioxides are an emerging class of heterocyclic compounds with synthetic and medicinal chemistry applications64,65,66,67,68. Fortunately, the reaction of α-bromovinylphenyl sulfoxide 1y gave the benzo[b]thiophene 1-oxide 2y in 83% yield. The cyclizations of α-bromovinyl aryl sulfones 1z–1ab also proceeded smoothly to afford benzo[b]thiophene 1,1-dioxides 2z–3ab in high yields.
Asymmetric synthesis of P-chiral benzophospholes
To explore the potential of this methodology in asymmetric synthesis, a preliminary screening of chiral bisphosphine ligands was carried out. As shown in Fig. 4, reaction of 1a in the presence of a chiral (R)-DM-segphos-palladium catalyst gave 2a in 31% isolated yield with a promising 86% ee, whereby the relatively low yield was due to low conversion of 1a. The asymmetric cyclization of (α-bromoethenyl)diarylphosphine oxide 1b, in which the aromatic group is phenyl moiety substituted with methyl at the para position, gave the benzophosphole 2b in 37% yield with 67% ee. The reaction of 1f, bearing fluoro substitution at the para position of phenyl ring, gave the asymmetric cyclization product 2f in 33% yield with 71% ee. The reaction of 1k, bearing a 2-naphthyl group, gave 44% yield of the asymmetric cyclization product 2k with 57% ee and high regioselectivity. The asymmetric cyclization of 1m, bearing a benzo[b]thiophen-5-yl group, gave 29% yield of the corresponding product 2m with 74% ee and high regioselectivity.

Reaction conditions: 1 (0.20 mmol), Pd(OAc)2 (5 mol% Pd), (R)-DM-segphos (10 mol%), CsOPiv (0.40 mmol), and m-xylene (1.0 mL) at 140 °C for 12 h. Isolated yield. The % ee was determined by HPLC on a chiral stationary phase column.
Mechanistic studies
Subsequently, we conducted preliminary experiments to provide insight into the reaction mechanism. We initially envisaged that the cyclization might proceed via an alkenyl-to-aryl 1,4-Pd migration as a key step. However, when the cyclization reaction of α-bromovinyl di(pentadeuteriophenyl)phosphine oxide (1a-d10) was carried out under the standard conditions, the product was 2a-d9, bearing only nine deuterium atoms at the aryl carbon atoms (Fig. 5a, equation 1). Furthermore, cyclization of α-bromovinyl di(2-deuteriophenyl)phosphine oxide (1a-d2) under our standard conditions proceeded smoothly to give a mixture of 2a-d2 and 2a-d1 in a 2:1 ratio, with one or two deuterium atoms incorporated at the aryl carbon atoms (Fig. 5a, equation 2). No deuterium incorporation at the alkenyl carbon atoms of 2a-d9, 2a-d2, and 2a-d1 clearly demonstrated that no 1,4-palladium migration was involved in the catalytic cycle. The fact that reaction of 1z-d5 afforded benzothiophene sulfone 2z-d4 with no deuterium incorporated at the alkenyl carbon atoms further confirmed this conclusion (Fig. 5a, equation 3). Reaction of ethynyldiphenylphosphine oxide (2a′) in the presence of the DPEPhos-palladium catalyst, NEt3·HBr, and CsOPiv gave the target product 2a in 37% yield (Fig. 5b). As shown in Fig. 5c, cyclization of Z-(β-bromoethenyl)diphenylphosphine oxide (3) gave a 29% yield of 2a. This indicated that the migratory cyclization might involve intramolecular C–H bond activation to form the C–C bond. Under the standard conditions, compound (E)-4 afforded the elimination product 5 in high yield without formation of the cyclization product, whereas no reaction of its (Z)-isomer took place (Fig. 5d).

a Deterium-labeling experiments. b Pd-catalyzed cyclization reaction with ethynyldiphenylphosphine oxide (2a′). c Pd-catalyzed cyclization reaction with (2-bromoethenyl)diphenylphosphine oxide (3). d Pd-catalyzed elimination of compound 4.
DFT calculations and proposed mechanism
Control experiments, in concert with DFT calculations69,70,71(Fig. 6), were used to inform the development of a mechanistic model. The density functional theory (DFT) calculations at the M06L/6-311 + G(d,p)(LANL2TZ for palladium atoms)/SMD(DCE)//B3LYP-D3/6-31G(d)(LANL2DZ for palladium atoms) level was performed to gain a theoretical understanding of the reaction mechanisms (see Supplementary Page S37). We chose the conversion of 1a to 2a as the model reaction, using Pd(OAc)2 as the catalyst and DPEPhos as the ligand. Taking into account the coordination capability of olefins, the free energy profile is initiated by the Pd(0) species INT-1, wherein the ligand and the C=C bond are coordinated to the metal center. Subsequent C‒Br bond oxidative addition to Pd center can occur via a three-membered ring-type transition state 2-ts to form vinyl Pd(II) intermediate INT-3 with an energy barrier of 22.9 kcal/mol, meanwhile releasing 7.5 kcal/mol of energy. In the presence of base, anion exchange of intermediate INT-3 with OPiv- leads to the formation of the thermodynamically stable Pd(II)-OPiv intermediate INT-4. In the generated Pd(II)-OPiv intermediate INT-4, the Pd-OPiv and Pd-ethylene bond lengths are 2.11 and 2.03 Å, respectively, indicating weak Pd-O bonding. An outer-sphere deprotonation with the assistance of base then takes place to give the alkyne-coordinated Pd(II) intermediate INT-6 via transition state 5-ts, which is endergonic 5.0 kcal/mol. The energy barrier for this step is 23.7 kcal/mol. The Pd-C1 bond length and Pd-C2 bond length of Pd-ethylene in the transition state 5-ts are 2.07 and 2.40 Å, respectively, which indicate that Pd center can activate the vinyl moiety. Sequential protonation occurs to form cis-vinyl Pd(II) intermediate INT-8 via transition state 7-ts with the energy barrier of 19.9 kcal/mol. The formed cis-vinyl Pd(II) intermediate 8 can easily isomerize to a trans-one INT-972,73. The intramolecular phenylic C‒H bond activation74,75,76,77,78,79 of diphenylphosphine oxide happens through a concerted metalation-deprotonation (CMD) process via transition state 10-ts to form a six-membered palladacycle INT-11 with 1.4 kcal/mol exergonic, overcoming an energy barrier of 23.2 kcal/mol. Then a C‒C bond reductive elimination generates the final product and regenerates Pd(0) species INT-1 with an energy barrier of 12.7 kcal/mol. Meanwhile, we also explored the alternative blue and green pathways, which involves direct β-hydride elimination and phenylic C‒H bond concerted metalation-deprotonation of intermediate INT-4. These pathways, however, have energy barrier of 27.5 and 44.1 kcal/mol, respectively, which are 3.8 kcal/mol and 20.4 kcal/mol higher than the pathway involving 5-ts, indicating that these pathways are less favorable (see Supplementary Fig. 1).

The energies are reported in kcal/mol and represent the relative free energies calculated using M06L/6-311 + G(d,p)(with LANL2TZ for palladium)/SMD(DCE)//B3LYP-D3/6-31G(d) (with LANL2DZ for palladium).
According to the DFT calculations, we propose a plausible mechanism for the palladium-catalyzed migratory cyclization in Fig. 7. The mechanism involves C‒Br oxidative addition of vinyl bromide, base-assisted outer-sphere deprotonation, sequential protonation, phenylic C‒H bond concerted metalation-deprotonation and C‒C bond reductive elimination. For comparative analysis, the direct β-hydride elimination pathway and the concerted metalation-deprotonation mechanism for the phenylic C–H bond are investigated using density functional theory; however, both pathways are determined to be energetically disfavored. The key trans−1,2-Pd migration core is driven by the cooperative C‒H activation of the alkene coordinated to Pd. The electron-deficient Pd(II) center polarizes both the α- and β-carbons of the coordinated ethene, significantly weakening the terminal C‒H bond. This electronic perturbation enables HOPiv to participate in an unconventional outer-sphere proton abstraction, bypassing the classical inner-sphere pathway. The resulting Pd‒H intermediate undergoes stereochemical reversal migration and insertion, establishing a migration pathway distinct from the traditional β-hydride mechanism.

The catalytic cycle is proposed according to the control experiments and the DFT calculations.
Synthesis of P-chiral bisphosphine ligands
Chiral bisphosphine ligands are of key importance in transition metal-catalyzed asymmetric synthesis of optically active products34,37,80,81,82,83. Enantio-enriched 2a obtained above was reduced to 1-phenylphosphindane (RP)-6 in the presence of Pd/C and H2 in 94% yield with >99.5% ee after recrystallization from methanol (Fig. 8a). Moreover, P-chiral bisphosphine ligand L1 was easily prepared by treatment of (RP)-6 with the strong base lithium diisopropylamide and CuCl2 in THF, followed by reduction with HSiCl3/NEt3 to give the product in an overall yield of 46%. Asymmetric addition of 4-MeOC6H4B(OH)2 to 2a in the presence of a chiral diene*-Rh catalyst proceeded smoothly to afford 3-arylated phosphindane (SP,RC)-7 in 42% yield with >99.5% ee after recrystallization from methanol (Fig. 8b). Following the above steps, P-chiral bisphosphine ligand L2 was also synthesized in an overall yield of 32%. L1 and L2 were then successfully directly used as chiral ligands in asymmetric hydrogenation to give compound 8 with 92% ee and 97% ee, respectively (Fig. 8c). The Hayashi–Miyaura reaction84,85,86,87,88,89,90,91 of cyclohexanone and PhB(OH)2 in the presence of the Rh/L1 or Rh/L2 catalysts also proceeded smoothly to give the 1,4-addition product 9 with >99% ee and 98% ee, respectively (Fig. 8d).

a, b Asymmetric synthesis of P-chiral bisphosphine ligands. c, d Applications of these ligands in asymmetric synthesis.
Discussion
In summary, we have reported a Pd-catalyzed migratory cyclization of ArXCBr=CH2 to give In summary, we have reported a Pd-catalyzed migratory cyclization of ArXCBr=CH2 to give benzoheterocycles, specifically indoles and their phosphorus, silicon, sulfur, carbon, and oxygen congeners. Detailed mechanistic studies have shown that the trans-1,2-palladium migration from the α-position to the trans-β-position of the C=C double bond is a key step of the catalytic cycle. The applicability of the present method has been showcased through the synthesis of new P-chiral bisphosphine ligands.
Methods
A typical procedure for palladium-catalyzed migratory cyclization of ArXCBr=CH2 (Table 1, entry 1)
An oven-dried sealed tube equipped with a PTFE-coated stir bar was charged with Pd(OAc)2 (2.24 mg, 10 μmol, 5.0 mol% of Pd), 1a (61.2 mg, 0.20 mmol), DPEPhos (10.8 mg, 20 μmol), CsOPiv (93.6 mg, 0.40 mmol) under argon. DCE (1.0 mL) was added successively, and the mixture was stirred at 80 °C for 12 h. The reaction mixture was passed through a short column of silica gel with dichloromethane as eluent and the water stayed in silica gel. The solvent was removed on a rotary evaporator. After 1H NMR analysis of the residue, the crude product was subjected to silica gel chromatography (eluent:dichloromethane/methanol (30/1)) as the eluent to give 2a (42.5 mg, 94% yield, 0.19 mmol) as a green solid.
A typical procedure for palladium-catalyzed asymmetric migratory cyclization of ArXCBr=CH2 (Fig. 4)
An oven-dried sealed tube equipped with a PTFE-coated stir bar was charged with Pd(OAc)2 (2.24 mg, 10 mmol, 5.0 mol% of Pd), 1a (61.2 mg, 0.20 mmol), (R)-DM-segphos (14.5 mg, 20 mmol), CsOPiv (93.6 mg, 0.40 mmol) under argon. m-Xylene (1.0 mL) was added successively, and the mixture was stirred at 140 °C for 12 h. The reaction mixture was passed through a short column of silica gel with dichloromethane as eluent and the water stayed in silica gel. The solvent was removed on a rotary evaporator. After 1H NMR analysis of the residue, the crude product was subjected to silica gel chromatography (eluent:dichloromethane/methanol (30/1)) as the eluent to give (R)-2a (14.0 mg, 31% yield, 0.062 mmol) as a green solid.
Data availability
Detailed experimental procedures, characterization data, NMR spectra of new compounds, HPLC spectra for chiral compounds, detailed computational results, and calculated structures are available within Supplementary Information. Cartesian coordinates of the calculated structures are available from Source Data, which are provided with this paper. All the data supporting the findings of this work are available within the article and its Supplementary Information files or from the corresponding author upon request. Source data are provided with this paper.
References
Ma, S. & Gu, Z. 1,4‐Migration of rhodium and palladium in catalytic organometallic reactions. Angew. Chem. Int. Ed. 44, 7512–7517 (2005).
Shi, F. & Larock, R. C. Remote C–H activation via through-space palladium and rhodium migrations. Top. Curr. Chem. 292, 123–164 (2010).
Croisant, M. F., Van Hoveln, R. & Schomaker, J. M. Formal dyotropic rearrangements in organometallic transformations. Eur. J. Org. Chem. 2015, 5897‒5907 (2015).
Rahim, A., Feng, J. & Gu, Z. 1,4‐Migration of transition metals in organic synthesis. Chin. J. Chem. 37, 929–945 (2019).
Li, M.-Y., Wei, D., Feng, C.-G. & Lin, G.-Q. Tandem reactions involving 1,4‐palladium migrations. Chem. Asian J. 17, e202200456 (2022).
Ju, C. W. & Zhao, D. Transition metal‐catalyzed C‒H bond functionalization involving metal migration. In Handbook of CH-Functionalization, 1‒23 (Wiley, 2022).
Wang, Y., He, Y. & Zhu, S. Nickel-catalyzed migratory cross-coupling reactions: new opportunities for selective C–H functionalization. Acc. Chem. Res. 56, 3475–3491 (2023).
Biswas, S. Mechanistic understanding of transition-metal-catalyzed olefin isomerization: metal-hydride insertion-elimination vs π-allyl pathways. Comment. Inorg. Chem. 35, 301–331 (2015).
Vasseur, A., Bruffaerts, J. & Marek, I. Remote functionalization through alkene isomerization. Nat. Chem. 8, 209–219 (2016).
Sommer, H., Juliá-Hernández, F., Martin, R. & Marek, I. Walking metals for remote functionalization. ACS Cent. Sci. 4, 153–165 (2018).
Kochi, T., Kanno, S. & Kakiuchi, F. Nondissociative chain walking as a strategy in catalytic organic synthesis. Tetrahedron Lett. 60, 150938 (2019).
Massad, I. & Marek, I. Alkene isomerization through allylmetals as a strategic tool in stereoselective synthesis. ACS Catal. 10, 5793–5804 (2020).
Limmert, M. E., Roy, A. H. & Hartwig, J. F. Kumada coupling of aryl and vinyl tosylates under mild conditions. J. Org. Chem. 70, 9364–9370 (2005).
Hansen, A. L. et al. Heck coupling with nonactivated alkenyl tosylates and phosphates: examples of effective 1,2-migrations of the alkenyl palladium(II) intermediates. Angew. Chem. Int. Ed. 45, 3349–3353 (2006).
Ebran, J.-P., Hansen, A. L., Gøgsig, T. M. & Skrydstrup, T. Studies on the heck reaction with alkenyl phosphates: can the 1,2-migration be controlled? scope and limitations. J. Am. Chem. Soc. 129, 6931–6942 (2007).
Watson, D. A. et al. Formation of ArF from LPdAr(F): catalytic conversion of aryl triflates to aryl fluorides. Science 325, 1661–1664 (2009).
Lindhardt, A. T., Gøgsig, T. M. & Skrydstrup, T. Studies on the 1,2-migrations in Pd-catalyzed negishi couplings with josiPhos ligands. J. Org. Chem. 74, 135–143 (2009).
Filatova, E. A., Gulevskaya, A. V., Pozharskii, A. F. & Ozeryanskii, V. A. Synthesis and some properties of alkynyl derivatives of 1,3-dialkylperimidones. An example of the 1,2-palladium migration in the Sonogashira reaction. Tetrahedron 72, 1547–1557 (2016).
Sekiguchi, Y., Onnuch, P., Li, Y. & Liu, R. Migratory aryl cross-coupling. J. Am. Chem. Soc. 147, 1224–1230 (2025).
Chowdhury, M. G. et al. A comprehensive account of synthesis and biological activities of α-lidene-benzocycloalkanones and benzoheterocycles. ChemistrySelect 7, e202201468 (2022).
Abbas, A. A., Farghaly, T. A. & Dawood, K. M. Recent advances on anticancer and antimicrobial activities of directly-fluorinated five-membered heterocycles and their benzo-fused systems. RSC Adv. 14, 19752–19779 (2024).
Hissler, M., Dyer, P. W. & Réau, R. Linear organic π-conjugated systems featuring the heavy group 14 and 15 elements. Coord. Chem. Rev. 244, 1–44 (2003).
Baumgartner, T. & Réau, R. Organophosphorus π-conjugated materials. Chem. Rev. 106, 4681–4727 (2006).
Matano, Y. & Imahori, H. Design and synthesis of phosphole-based π systems for novel organic materials. Org. Biomol. Chem. 7, 1258–1271 (2009).
Ren, Y. & Baumgartner, T. Combining form with function–the dawn of phosphole-based functional materials. Dalton Trans. 41, 7792–7800 (2012).
Baumgartner, T. Insights on the design and electron-acceptor properties of conjugated organophosphorus materials. Acc. Chem. Res. 47, 1613–1622 (2014).
Shameem, M. A. & Orthaber, A. Organophosphorus compounds in organic electronics. Chem. Eur. J. 22, 10718–10735 (2016).
Allen, M. C. et al. Renin inhibitors. synthesis of transition-state analog inhibitors containing phosphorus acid derivatives at the scissile bond. J. Med. Chem. 32, 1652–1661 (1989).
Smith, W. W. & Bartlett, P. A. Macrocyclic inhibitors of penicillopepsin. 3. design, synthesis, and evaluation of an inhibitor bridged between P2 and P1‘. J. Am. Chem. Soc. 120, 4622–4628 (1998).
Mucha, A., Kafarski, P. & Berlicki, Ł. Remarkable potential of the α-aminophosphonate/phosphinate structural motif in medicinal chemistry. J. Med. Chem. 54, 5955–5980 (2011).
Clarion, L. et al. C-glycoside mimetics inhibit glioma stem cell proliferation, migration, and invasion. J. Med. Chem. 57, 8293–8306 (2014).
Iwamoto, N. et al. Control of phosphorothioate stereochemistry substantially increases the efficacy of antisense oligonucleotides. Nat. Biotechnol. 35, 845–851 (2017).
Knouse, K. W. et al. Unlocking P(V): reagents for chiral phosphorothioate synthesis. Science 361, 1234–1238 (2018).
Tang, W. J. & Zhang, X. New chiral phosphorus ligands for enantioselective hydrogenation. Chem. Rev. 103, 3029–3069 (2003).
Dutartre, M., Bayardon, J. & Jugé, S. Applications and stereoselective syntheses of P-chirogenic phosphorus compounds. Chem. Soc. Rev. 45, 5771–5794 (2016).
Ni, H. Z., Chan, W.-L. & Lu, Y. X. Phosphine-catalyzed asymmetric organic reactions. Chem. Rev. 118, 9344–9411 (2018).
Xu, G., Senanayake, C. H. & Tang, W. J. P-chiral phosphorus ligands based on a 2,3-dihydrobenzo[d][1,3]oxaphosphole motif for asymmetric catalysis. Acc. Chem. Res. 52, 1101–1112 (2019).
Zeni, G. & Larock, R. C. Synthesis of heterocycles via palladium-catalyzed oxidative addition. Chem. Rev. 106, 4644–4680 (2006).
Platon, M., Amardeil, R., Djakovitch, L. & Hierso, J.-C. Progress in palladium-based catalytic systems for the sustainable synthesis of annulated heterocycles: a focus on indole backbones. Chem. Soc. Rev. 41, 3929–3968 (2012).
Wu, X.-F., Neumann, H. & Beller, M. Synthesis of heterocycles via palladium-catalyzed carbonylations. Chem. Rev. 113, 1–35 (2013).
Kaur, N. Palladium-catalyzed approach to the synthesis of S-heterocycles. Catal. Rev. 57, 478–564 (2015).
Kaur, N. Palladium catalysts: synthesis of five-membered N-heterocycles fused with other heterocycles. Catal. Rev. 57, 1–78 (2015).
Braun, M. Stereoselective domino heck‐suzuki reactions. Eur. J. Org. Chem. 26, e202201282 (2023).
Shintani, R., Okamoto, K. & Hayashi, T. Rhodium-catalyzed isomerization of α-arylpropargyl alcohols to indanones: involvement of an unexpected reaction cascade. J. Am. Chem. Soc. 127, 2872–2873 (2005).
Shintani, R. & Hayashi, T. Rhodium-catalyzed addition of arylzinc reagents to aryl alkynyl ketones: synthesis of β,β-disubstituted indanones. Org. Lett. 7, 2071–2073 (2005).
Shintani, R. et al. Rhodium-catalyzed asymmetric synthesis of indanones: development of a new “axially chiral” bisphosphine ligand. J. Am. Chem. Soc. 128, 2772–2773 (2006).
Alberico, D., Scott, M. E. & Lautens, M. Aryl−aryl bond formation by transition-metal-catalyzed direct arylation. Chem. Rev. 107, 174–238 (2007).
Liu, C., Zhang, H., Shi, W. & Lei, A. Bond formations between two nucleophiles: transition metal catalyzed oxidative cross-coupling reactions. Chem. Rev. 111, 1780–1824 (2011).
Le Bras, J. & Muzart, J. Intermolecular dehydrogenative heck reactions. Chem. Rev. 111, 1170–1214 (2011).
Yeung, C. S. & Dong, V. M. Catalytic dehydrogenative cross-coupling: forming carbon−carbon bonds by oxidizing two carbon−hydrogen bonds. Chem. Rev. 111, 1215–1292 (2011).
Liu, C. et al. Oxidative coupling between two hydrocarbons: an update of recent C–H functionalizations. Chem. Rev. 115, 12138–12204 (2015).
Yang, Y., Lan, J. & You, J. Oxidative C–H/C–H coupling reactions between two (hetero)arenes. Chem. Rev. 117, 8787–8863 (2017).
Gandeepan, P. et al. 3d transition metals for C–H activation. Chem. Rev. 119, 2192–2452 (2019).
Liang, Y.-F. et al. Carbon–carbon bond cleavage for late-stage functionalization. Chem. Rev. 123, 12313–12370 (2023).
Zhao, J. & Larock, R. C. Synthesis of substituted carbazoles, indoles, and dibenzofurans by vinylic to aryl palladium migration. J. Org. Chem. 71, 5340–5348 (2006).
Hu, T.-J. et al. Highly stereoselective synthesis of 1,3‐dienes through an aryl to vinyl 1,4‐palladium migration/heck sequence. Angew. Chem. Int. Ed. 57, 5871–5875 (2018).
Wei, D., Hu, T.-J., Feng, C.-G. & Lin, G.-Q. Synthesis of substituted naphthalenes by 1,4‐palladium migration involved annulation with internal alkynes. Chin. J. Chem. 36, 743–748 (2018).
Lin, J. et al. Tunable construction of multisubstituted 1,3-dienes and allenes via a 1,4-palladium migration/carbene insertion cascade. J. Org. Chem. 87, 12019–12035 (2022).
Chen, Y.-Z. et al. Highly stereoselective synthesis of 2,2‐disubstituted vinylphosphonates via aryl to vinyl 1,4‐palladium migration. Chin. J. Chem. 40, 2188–2192 (2022).
Chen, Z.-Y. et al. Highly stereoselective synthesis of 1,3‐enynes via aryl to vinyl 1,4‐palladium migration/sonogashira coupling sequence. ChemCatChem 16, e202400478 (2024).
Tsitopoulou, M., Clemenceau, A., Thesmar, P. & Baudoin, O. 1,4-Pd migration-enabled synthesis of fused 4-membered rings. J. Am. Chem. Soc. 146, 18811–18816 (2024).
Matsuda, T., Yamaguchi, Y. & Murakami, M. Synthesis of silole skeletons via metathesis reactions. Synlett 2008, 561–564 (2008).
Carr, D. J. et al. Synthesis of 2,3-dihydro-1-phenylbenzo[b]phosphole (1-Phenylphosphindane) and its use as a mechanistic test in the asymmetric appel reaction: decisive evidence against involvement of pseudorotation in the stereoselecting step. J. Org. Chem. 78, 10500–10505 (2013).
Mansuy, D. et al. Thiophene S-oxides as new reactive metabolites: formation by cytochrome P-450 dependent oxidation and reaction with nucleophiles. J. Am. Chem. Soc. 113, 7825–7826 (1991).
Villar, R. et al. Synthesis and cytotoxic activity of lipophilic sulphonamide derivatives of the benzo[b]thiophene 1,1-dioxide. Bioorg. Med. Chem. 12, 963–968 (2004).
Encío, I. et al. Benzo[b]thiophenesulphonamide 1,1-dioxide derivatives inhibit tNOX activity in a redox state-dependent manner. Br. J. Cancer 92, 690–695 (2005).
Chandrasekera, N. S. et al. Synthesis and anti-tubercular activity of 3-substituted benzo[b]thiophene-1,1-dioxides. Peerj 2, e612 (2014).
Kummari, L. K. et al. Antifungal benzo[b]thiophene 1,1-dioxide IMPDH inhibitors exhibit pan-assay interference (PAINS) profiles. Bioorg. Med. Chem. 26, 5408–5419 (2018).
Qi, X. & Lan, Y. Recent advances in theoretical studies on transition-metal-catalyzed carbene transformations. Acc. Chem. Res. 54, 2905–2915 (2021).
He, X., Zhong, K., Heng, D. & Zeng, Z. Is the metal involved or not? A computational study of Cu(I)-catalyzed [4 + 1] annulation of vinyl indole and carbene precursor. Chin. Chem. Lett. 33, 2031–2035 (2022).
Zhong, K., Liu, S., He, X.& Ni, H. Oxidative cyclopalladation triggers the hydroalkylation of alkynes. Chin. Chem. Lett. 34, 108339 (2023).
Lv, W. et al. Palladium-catalyzed intermolecular trans-selective carbofunctionalization of internal alkynes to highly functionalized alkenes. ACS Catal. 10, 10516–10522 (2020).
Shan, C. et al. Mechanistic insight into anti-carbometalation of an alkyne via η2-vinyl-nickel type Z/E isomerization. Org. Chem. Front. 10, 4243–4249 (2023).
Chen, X., Engle, K. M., Wang, D. H. & Yu, J. Q. Palladium(II)‐catalyzed C‒H activation/C‒C cross‐coupling reactions: versatility and practicality. Angew. Chem. Int. Ed. 48, 5094–5115 (2009).
Lyons, T. W. & Sanford, M. S. Palladium-catalyzed ligand-directed C−H functionalization reactions. Chem. Rev. 110, 1147–1169 (2010).
Davies, H. M. L., Du Bois, J. & Yu, J.-Q. C–H functionalization in organic synthesis. Chem. Soc. Rev. 40, 1855–1856 (2011).
Engle, K. M., Mei, T.-S., Wasa, M. & Yu, J.-Q. Weak coordination as a powerful means for developing broadly useful C–H functionalization reactions. Acc. Chem. Res. 45, 788–802 (2012).
Diesel, J. & Cramer, N. Generation of heteroatom stereocenters by enantioselective C–H functionalization. ACS Catal. 9, 9164–9177 (2019).
Shao, Q. et al. From Pd(OAc)2 to chiral catalysts: the discovery and development of bifunctional mono-N-protected amino acid ligands for diverse C–H functionalization reactions. Acc. Chem. Res. 53, 833–851 (2020).
Zhang, W., Chi, Y. & Zhang, X. Developing chiral ligands for asymmetric hydrogenation. Acc. Chem. Res. 40, 1278–1290 (2007).
Zhao, Q. et al. Noncovalent interaction-assisted ferrocenyl phosphine ligands in asymmetric catalysis. Acc. Chem. Res. 53, 1905–1921 (2020).
Wan, F. & Tang, W. J. Phosphorus ligands from the zhang lab: design, asymmetric hydrogenation, and industrial applications. Chin. J. Chem. 39, 954–968 (2021).
Cao, Z., He, D. Y. & Tang, W. J. Catalysis and synthesis enabled by P-chiral dihydrobenzooxaphosphole ligands. Org. Process. Res. Dev. 28, 949–977 (2024).
Hayashi, T. & Yamasaki, K. Rhodium-catalyzed asymmetric 1,4-addition and its related asymmetric reactions. Chem. Rev. 103, 2829–2844 (2003).
Tian, P., Dong, H.-Q. & Lin, G.-Q. Rhodium-catalyzed asymmetric arylation. ACS Catal. 2, 95–119 (2011).
Jean, M., Casanova, B., Gnoatto, S. & van de Weghe, P. Strategy of total synthesis based on the use of Rh-catalyzed stereoselective 1,4-addition. Org. Biomol. Chem. 13, 9168–9175 (2015).
Heravi, M. M., Dehghani, M. & Zadsirjan, V. Rh-catalyzed asymmetric 1,4-addition reactions to α,β-unsaturated carbonyl and related compounds: an update. Tetrahedron.: Asymmetry 27, 513–588 (2016).
Burns, A. R., Lam, H. W. & Roy, I. D. Enantioselective, rhodium-catalyzed 1,4-addition of organoboron reagents to electron-deficient alkenes. Org. React. 93, 1–686 (2017).
Huang, Y. & Hayashi, T. Chiral diene ligands in asymmetric catalysis. Chem. Rev. 122, 14346–14404 (2022).
Yin, L. et al. Asymmetric synthesis of P-stereogenic phosphindane oxides via kinetic resolution and their biological activity. Nat. Commun. 15, 2548 (2024).
Li, W.-C., Ming, J. & Chen, S. Kinetic resolution of P-chiral phosphindole oxides through rhodium-catalyzed asymmetric arylation. Org. Lett. 26, 3987–3990 (2024).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (22261039, 22461028, and 21901132), the Program for Innovative Research Team in Universities of Inner Mongolia Autonomous Region (NMGIRT2324), the Science and Technology Leading Talent Team in Inner Mongolia Autonomous Region (2022LJRC0001), and the Inner Mongolia Institute of Science and Technology Industrial Technology Innovation Program (2024RCYJ02005). We also acknowledge the support from Center of Advanced Analysis & Gene Sequencing of Zhengzhou University.
Author information
Authors and Affiliations
Contributions
J.M. conceived the idea and guided the project. W.L., J.Z., S.B., and G.L. performed the experiments and analyzed the data. Y.L. directed the part of computational study. L.Z. carried out the computational study. J.M. and S.C. wrote the manuscript. All authors approved the submission of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Li, WC., Zhang, L., Bai, S. et al. Synthesis of benzoheterocycles by palladium-catalyzed migratory cyclization through an unexpected reaction cascade. Nat Commun 16, 3367 (2025). https://doi.org/10.1038/s41467-025-58633-5
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-58633-5
