
Abstract
Nucleotide excision repair (NER) represents one of the major molecular machineries that control chromosome stability in all living species. In Eubacteria, the initial stages of the repair process are carried out by the UvrABC excinuclease complex. Despite the wealth of structural data available, some crucial details of the pathway remain elusive. In this study, we present a structural investigation of the Mycobacterium tuberculosis UvrAUvrB complex and of the UvrA dimer, both in complex with damaged DNA. Our analyses yield insights into the DNA binding mode of UvrA, showing an unexplored conformation of Insertion Domains (IDs), underlying the essential role of these domains in DNA coordination. Furthermore, we observe an interplay between the ID and the UvrB Binding Domain (UBD): after the recognition of the damage, the IDs repositions with the concomitant reorganization of UBD, allowing the formation of the complex between UvrA and UvrB. These events are detected along the formation of the uncharacterized UvrA2UvrB1-DNA and the UvrA2UvrB2-DNA complexes which we interpret as hierarchical steps initiating the DNA repair cascade in the NER pathway, resulting in the formation of the pre-incision complex.
Similar content being viewed by others


Introduction
Nucleotide excision repair (NER) is a versatile and highly conserved DNA repair pathway responsible for removing bulky DNA-distorting adducts1. In bacteria, two proteins, namely UvrA and UvrB, serve as the initial NER damage sensors1,2,3,4,5. UvrA is an ATPase in the ATP-binding cassette (ABC) superfamily that functions as a dimer. The catalytic core of each protomer of the functional UvrA2 homodimer consists of two composite nucleotide-binding domains (NBD-I and NBD-II) and each moiety contain an ATP/ADP binding site (ATP-I and ATP-II, belonging to NBD-I and NBD-II, respectively) and a Signature domain (SIG-I and SIG-II belonging to NBD-I and NBD-II, respectively). Two distinctive extra domains sprout from the NBD-I of UvrA: the UvrB Binding Domain (UBD) and the Insertion Domain (ID)6,7,8,9,10 (Fig. 1a). UvrB is annotated as a member of the SF2 helicase family for the presence of conserved helicase motifs (I-VI); it contains the classical RecA-like domains (domains 1a and 3), in addition to three auxiliary domains (domains 1b, 2, and 4) and the β-hairpin motif that projects from domain 1a11,12,13,14,15.

a Crystal structure of MtUvrA2 (PDB: 3ZQJ) in two orientations. UvrA domains were colored according to the schematic diagram (the terms ATP-I and ATP-II are used to indicate the proximal and distal ATP binding site, respectively. Similarly, SIG-I and SIG-II are indicative of signature domains, while UBD stands for UvrB binding domain and ID for Insertion Domain). b Schematic representation of the different steps of prokaryotic NER: damage search process (lesion is depicted with a red star) performed by either UvrA2 or UvrA-UvrB complex (1); damage identification and consequent recruitment of UvrB molecules (2); pre-incision complex formation and UvrA detachment (3); UvrC recruitment and damaged DNA incision (4); restoration of the native dsDNA, operated by UvrD, DNA Polymerase I and ligase (5 and 6). Created in BioRender. Ferraris, D. (2024) BioRender.com/f96q610.
There is consensus on the distinct roles of the two proteins in the early damage detection process: UvrA acts as the primary sensor of DNA double-helix deformation13,16, with the capacity to simultaneously bind damaged DNA and UvrB, behaving as a molecular matchmaker that enables the precise delivery of UvrB to the lesioned site13,17. The discrimination of the lesion and subsequent stable binding represents the first step of the UvrA activity; the binding of UvrB is then the functional prerequisite for the complete pathway to proceed to the pre-incision complex formation, with one UvrB molecule bound to the lesion. Subsequently, UvrC nuclease cleaves the DNA on both sides of the damaged site, the UvrD helicase removes the excised oligonucleotide, and DNA polymerase I fills the gap. Finally, DNA ligase I seals the nick (Fig. 1b).
Despite the amount of available structural information5,6,7,8,10,11,18,19, some critical molecular and structural aspects of the initial steps of bacterial NER remain unclear. In particular, although atomic structures of UvrA bound to damaged DNA7 and UvrA bound to UvrB5 have been reported, a description of the configuration of the UvrA-UvrB complex bound to DNA is still lacking.
Specifically, the conformational changes occurring in UvrA during genome scanning and the overall architecture of the lesion-sensing complex remain elusive9. On the one hand, it has been proposed that UvrA can locate and verify the DNA lesion independently of UvrB16,20,21. On the other hand, forming an UvrA-UvrB complex has been suggested as the bona fide DNA damage-detecting step, initiating the NER pathway5,9,18,22. The precise oligomerization state of the UvrA-UvrB complex remains unclear since the in vitro stoichiometry has been alternatively estimated as UvrA2UvrB2 or UvrA2UvrB15,13,20,21,22. Indeed, it is possible that both forms could populate the NER pathway. UvrA is essential for damage identification, sensing DNA helix distortion and participating in DNA melting and unwinding23,24. However, whether it is UvrA alone or in complex with UvrB the one responsible for the damage recognition mechanism remains to be established. Moreover, two other outstanding questions regarding the transition toward the pre-incision complex remain to be addressed. First, the only available UvrA2UvrB2 structural model5 is symmetric, with the two UvrB molecules capable of binding complementary strands. However, the pre-incision complex is asymmetric and contains a single copy of UvrB25. It is, therefore, unclear how the lesion-containing strand is identified. Second, the UvrA2UvrB2 places UvrB ~80 Å from the damage site. Two mechanisms have been hypothesized to explain the localization of UvrB to the lesion: the “recruitment model” model, which suggests a direct delivery of UvrB on the lesion through UvrA2UvrB2 complex reconfiguration, and the “translocation model”, which implies that UvrA dissociates before the translocation of UvrB to the damage site; UvrB translocates using its intrinsic ATPase activity5,12.
Our study focuses on the NER pathway in Mycobacterium tuberculosis (MTB), which is essential to maintaining infectivity and survival4,26,27. Using cryogenic electron microscopy (cryo-EM), we determined three distinct macromolecular complexes in the MTB NER pathway: UvrA2 bound to DNA (MtUvrA2-DNA), the UvrA2-UvrB2 complex bound to DNA (MtUvrA2UvrB2-DNA), and a previously undescribed UvrA2-UvrB1 complex bound to DNA (MtUvrA2UvrB1-DNA). Our structural, genetic, and biochemical study came to several conclusions. UvrA in the MtUvrA2-DNA complex was found in a distinct configuration compared to a prior structure. Notably, the two IDs are observed clamping on DNA as they mediate the melting and unwinding of the duplex. We suggest a division of labor between the two UvrA protomers. In the MtUvrA2UvrB1-DNA complex, we establish a link between UvrB binding to a particular UvrA monomer and the concomitant release of this monomer’s ID from contact with DNA. The structure of the MtUvrA2UvrB2-DNA complex reveals how the repositioning of UvrA’s IDs may enable the transfer of the lesion to UvrB. We propose that the three structures (MtUvrA2-DNA, MtUvrA2UvrB1-DNA, and MtUvrA2UvrB2-DNA) operate sequentially in the NER pathway.
Results
MtUvrA dimer copurifies with endogenous E. coli dsDNA containing oxidative damage
The MtUvrA-MtUvrB complex (prepared as previously described6,11) was vitrified and cryo-EM data were collected using a Talos Arctica (Suppplementary Table 1). The 2D average analysis revealed the presence of particles consistent with the UvrA dimer, but not compatible with the complex (Supplementary fig. 1). Surprisingly, during the 3D reconstruction step, two distinct sets of particles were identified, corresponding to the DNA-free MtUvrA2 and to MtUvrA2 in complex with dsDNA; these were subsequently processed to achieve a final resolution of 4.9 Å and 4.2 Å, respectively (Supplementary fig. 1, Supplementary Table 1 and Supplementary fig. 2a, b). The structure of MtUvrA2-DNA was reconstructed at higher resolution and will be discussed in a later section, while the structural analysis of MtUvrA2 without DNA is part of the supplementary information.
A posteriori analyses revealed contaminant nucleic acids in the MtUvrA sample used in cryo-EM (Supplementary fig. 3a, b). Given the nature of UvrA as DNA damage detector, we sought to identify the type of lesion possibly present in the copurified DNA. To this end, we conducted a dot blot-based analysis using antibodies specific to prevalent types of DNA lesion, that confirmed the presence of oxidized nucleobases (i.e. 8-oxoguanine), while common UV-induced adducts (i.e. CPDs and 6-4PP) were absent (Supplementary fig. 3c). Taking into account that recombinant MtUvrA was purified in the absence of any intentional external source of DNA damage, we speculate that the exposure of the sample to a pro-oxidative environment resulting from cell lysis, and/or the prolonged binding to a nickel-based matrix, could have induced oxidation of E. coli genomic DNA, which then served as the substrate for UvrA28, overall explaining the abundance of the MtUvrA2-DNA complexes in our sample.
Single-particle cryo-EM analysis of the MtUvrA-MtUvrB-DNA complex reveals three assembly states with a hierarchical reconfiguration
The optimal superposition of MtUvrA2-DNA reconstruction to the BstUvrA2-UvrB2 complex5 (PDB: 3UWX) revealed that both BstUvrB monomers would collide with the dsDNA molecule present in our model (Supplementary fig. 4a), suggesting that critical structural rearrangements should take place to allow the simultaneous binding of UvrB and dsDNA to MtUvrA2. Therefore, we attempted the long-sought structural characterization of the UvrA-UvrB-DNA complex through the use of a synthetic dsDNA molecule (DNA*) with a 10-nucleotide single-stranded 5’ overhang ends with high affinity for UvrB15 and two internal fluorescein-modified nucleobases separated by four base pairs that mimic bulky lesions recognized by UvrA19 (Supplementary Table 3, Supplementary fig. 4b).
We assembled and purified the MtUvrA-MtUvrB complex and subsequently mixed it with the DNA*. The sample was vitrified, and cryo-EM data were collected (Supplementary figs. 5, 6 and Supplementary Table 1). Following 3D reconstruction, three kinds of oligomeric assemblies (namely MtUvrA2-DNA, MtUvrA2UvrB1-DNA and MtUvrA2UvrB2-DNA) were identified and independently processed to a resolution of 3.2 Å, 4.3 Å and 3.7 Å, respectively (Fig. 2, Supplementary fig. 5 and Supplementary fig. 6a).

a The indicated cryo-EM reconstructions are color coded and labeled according to the models shown in b; in the MtUvrA2-DNA and MtUvrA2UvrB1-DNA* reconstructions, the yellow and the red arrowhead points out the lack of signal corresponding to a 7-nt long segment of one DNA strand, and to one MtUvrA protomer ID, respectively. b Cartoon representation of the MtUvrA2-DNA, MtUvrA2UvrB1-DNA* and MtUvrA2UvrB2-DNA* models in two orientations; in the side views it is possible to appreciate the initial bending of the DNA in MtUvrA2-DNA and MtUvrA2UvrB1-DNA* and the final position in the groove of MtUvrA2UvrB2-DNA* structure. The unmodeled DNA strand segment on MtUvrA2-DNA is represented with a dashed line.
The MtUvrA2-DNA reconstruction showed a signal for the dsDNA longer than the one expected for DNA* (Supplementary fig. 7); moreover, the map superposed well (CC = 0.92) with the previously obtained MtUvrA2-DNA map, thus we concluded that it represents the structure of MtUvrA2 associated to the co-purified E. coli DNA. Considering the final resolution of the two reconstructions, we decided to focus our analyses on the latter one.
The MtUvrA2UvrB2-DNA and MtUvrA2UvrB1-DNA reconstructions exhibited anisotropic behavior due to significant preferential orientation, resulting in a lower map quality than expected for their calculated FSC resolution (Supplementary fig. 6b, c). Nevertheless, the quality of both reconstructions enabled the building of a model through rigid-body fitting of the different domains of UvrA and UvrB (Fig. 2b). Both reconstructions did not exhibit signal for dsDNA since the density is compatible with ssDNA overhangs bound to UvrB, exactly as in DNA* (Supplementary fig. 8). Accordingly, the experimental procedure that we designed allowed us to stabilize two complexes between MtUvrA, MtUvrB and DNA* with two distinct stoichiometries, which, in conjunction with the MtUvrA2-DNA, represent three structures of macromolecular assemblies acting in the initial stages of mycobacterial NER.
Remarkably, in the three different protein-DNA complexes it is possible to distinguish two different DNA configurations: indeed, although the central DNA portion in MtUvrA2-DNA is characterized by lower resolution (Supplementary fig. 6b), it is evident that the oligonucleotide is bent ( ~ 28°) and locally melted, as in MtUvrA2UvrB1-DNA* complex, while the DNA in MtUvrA2UvrB2-DNA* complex has been observed in a linear configuration, lying in the groove of UvrA dimer with the same positioning of TmUvrA2-DNA7 (Fig. 2b). In all the complexes, the protein-DNA interactions result in a local alteration of the base pair parameters, among which rise and twist angle values diverge from the expected patterns for a canonical B-DNA structure29,30,31.
The MtUvrA IDs play a central role in DNA unwinding, melting and the flipping of a nucleobase into a potential lesion recognition state
The structure of MtUvrA2-DNA shows the same organization of the core catalytic domains observed in the crystallographic structure of the TmUvrA2-DNA (PDB: 3PIH)7, with slight changes in the Signature-I and the C-terminal Zn-finger β-hairpins761-775 (Supplementary fig. 9a, b). Still, the C-terminal Zn-finger β-hairpins761-775 adopt the same open conformation that was suggested to be correlated to DNA binding. Protein-DNA contacts along the Signature-II domain are mostly conserved, with a minor variation between the loop704-709 and the phosphate backbone due to the deformation of the DNA (Supplementary fig. 9c). A striking difference is observable at the level of the IDs: while in the TmUvrA2-DNA structure they do not contact the DNA molecule, in our MtUvrA2-DNA reconstruction they clamp both sides of the flat segment in the middle of the DNA double helix, which displays a 28° bending and different degrees of unwinding: when compared to the canonical B-DNA, the flat central portion of the DNA displays -16° of unwinding and the nucleobases flanking this region (A/T16 and A/T27) 9° of unwinding (calculated with w3DNA 2.0 software31). As a result, the major groove-corresponding surface of the unwound dsDNA region faces the catalytic core of the MtUvrA dimer, while the widened minor groove faces upwards (Fig. 3a). Each ID holds in place one of the complementary DNA strands, by using the α-hairpin381-405 and the α-hairpin313-335, with the α-helix320-335 deeply inserted into the DNA minor groove (Fig. 3a and Supplementary fig. 10a).

a Closeup of the MtUvrA dimer IDs, showing the structural motifs/elements that are engaged in clamping and deforming the DNA molecule (in orange); in both panels, the region of one DNA strand that is not defined in the model is represented by a dashed line and β-hairpins363-377 were not modeled. b Detailed view of the flipped-out nucleotide; functionally relevant residues of the recognition-ID hydrophobic pocket are displayed as sticks and labeled, as well as Tyr323. c Flipped-out nucleotide in UvrB-DNA complex (PDB: 6O8F)12, accommodating into the hydrophobic pocket between domains 1a and 1b. d SPR sensorgrams of MtUvrA proteins (wild type and mutated variants) binding to the damaged-DNA probe immobilized on a sensor chip. e Calculated Kon, Koff and KD values are shown for each investigated variant. Source data are provided as a Source Data file.
The path of the DNA sugar-phosphate backbone is a key attribute particularly well-defined in our reconstruction, except for a 7 nucleotide-long region that contacts one of the two IDs (Figs. 2 and 3a). The poor signal may indicate DNA flexibility, as a probable result of the base-pairing loss due to the presence of the damage; this base-pairing loss as well as base-stacking alteration in the vicinity of the lesion could be the hallmark that UvrA senses to recognize the presence of the damage site. A more detailed analysis of the ID-DNA interface, complementary to the disordered strand, revealed a notable signal oriented backward with respect to the position of the nucleobases (Supplementary fig. 10b). The better definition of the DNA reconstruction with respect to the ID part, together with the fact that nucleobases scatter electrons particularly well, lead us to interpret this signal as a flipped-out nucleobase (Fig. 3b). Interestingly, the reoriented nucleobase is hosted in a hydrophobic pocket within the ID, adopting an overall conformation that matches well the one observed in the structure of UvrB-DNA complex, in which an extruded cytosine occupies an equivalent hydrophobic pocket between domains 1a and 1b12 (Fig. 3c). It is noteworthy that the side-chain of the tyrosine residue at position 323 of the α-helix320-335 (Y323) appears properly positioned to potentially replace the missing flipped-out base within the DNA π-stacking. As a matter of fact, sequence alignment showed that the Y323 residue is highly conserved among UvrA of different species (Supplementary fig. 11), supporting a key role in the clamping.
A disordered DNA strand and an extruded nucleobase are structural hallmarks found in other helicases32,33,34 and in the lesion recognition process of other DNA repair enzymes35,36,37. Notably, in our structure these two features are exclusively observable at the level of the ID that is engaged in a potential damage recognition conformation, indicating a division of labor between the two IDs. Consequently, we will refer to the ID that hosts the flipped nucleobase as the “recognition-ID”, and we will indicate the other one as the “support-ID” (Supplementary fig. 10a).
The inspection of MtUvrA2-DNA reconstruction revealed a continuous extra density above the minor groove corresponding to the flat face of the dsDNA; the signal is large enough to host the two unmodeled ID β-hairpins363-377 (Supplementary fig. 10c), which, in this position, would directly contact the DNA. In general, β-hairpins are conserved motifs in helicases involved in DNA melting24. The MtUvrA ID β-hairpin363-377 is particularly enriched with conserved arginine and tyrosine residues (Supplementary fig. 11), that have the potential to interact with DNA sugar-phosphate backbone and to establish π-stacking contacts with nucleobases, thereby interfering with base pairing. Considering the conformation and the amino acid composition of the MtUvrA ID β-hairpin363-377, we propose that it could play a prominent role in DNA melting. A sequence comparison analysis highlighted a high degree of conservation of this element among species, except for the hyperthermophiles Thermotoga maritima and Aquifex aeolicus. In such cases, the high temperatures of the ecological niche of these organisms may facilitate spontaneous dsDNA local melting, even in the absence of a β-hairpin, equivalent to the MtUvrA ID β-hairpin363-377 (Supplementary fig. 11).
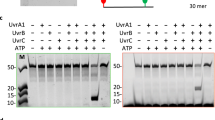
Although previous works highlighted the role of IDs in stabilizing the complex with damaged DNA through DNA locking and melting8, to date there is no structural model describing the molecular details of this phenomena38,39. Although we were unable to unambiguously establish the identity of the dsDNA component fortuitously captured in the MtUvrA2-DNA complex, the overall molecular architecture revealed by our cryo-EM structure is consistent with a genuine lesion recognition state. To verify the hypothesized contribution of the ID β-hairpin363-377 and Y323 to the MtUvrA lesion-sensing function, we challenged the damaged-DNA binding activity of two protein variants: MtUvrA-Δβ-hairpin, lacking the entire ID β-hairpin363-377, and MtUvrA-Y323A, in which Y323 is substituted by an alanine. We performed two pilot experiments (Electrophoretic Mobility Shift Assay - EMSA - and Microscale Thermophoresis - MST) using a DNA fragment with the same nucleotide sequence as DNA*, containing 2 fluorescein-dT (FLUdT) mimicking a bulky nucleobase adducts (probe) (Supplementary Table 3); these analyses indicated that the damaged-DNA binding activity of the two variants is severely compromised, compared to the one of wild type MtUvrA (Supplementary fig. 12 a,b). To obtain further detailed information, we characterized the binding of MtUvrA and mutated variants with the damaged DNA with surface plasmon resonance (SPR): the use of a sensor chip functionalized with the same probe revealed that not only MtUvrA, but also its two protein variants can bind damaged-DNA, although displaying different kinetics (Fig. 3d). In particular, UvrA-∆β-hairpin and UvrA-Y323A mutants displayed a dissociation phase sensibly faster than those characterizing the wild-type protein (Fig. 3e), which explains why with non-real-time assays (EMSA, MST) we detected no binding: the dissociation of the mutants is too fast, leading to a non-stabel interactions with the damaged DNA. Furthermore, the binding of the MtUvrA variants to the DNA ligand resulted in a lower response, as evidenced by the reduced resonance units count (Fig. 3d). Therefore, the biochemical data support our structural observations, indicating that the β-hairpin363-377 and Y323 represent crucial elements for binding damaged DNA by increasing the retention time on the lesion.
Overall, the collective evidence lends support to a model in which the MtUvrA2-DNA structure represents a state wherein UvrA clamps, unwinds and melts dsDNA through the IDs. The recognition-ID is responsible for nucleobase flipping, while β-hairpin363-377 may facilitate melting by interacting with the minor groove face of the DNA. We suggest that this model represents the end point of the UvrA-promoted lesion recognition process.
Concerning the ATP/ADP binding state, we are able to observe the presence of ADP in the distal sites of the dimer (Supplementary Fig. 13), while the proximal sites are empty. This agrees with previous observations that suggest that UvrA dimer is often more stable in the presence of a mixed nucleotide-bound/free species, in which the proximal site is empty and the distal site may be occupied by ATP or ADP19,22,40. In addition, this correlates well with what observed and proposed previously41: the genome scanning activity and the discrimination between native and damaged DNA require ATP hydrolysis at the distal sites, which are demonstrated to consume ATP in a fast way, releasing phosphate; the proximal sites, instead, have been implicated in interactions between UvrA and UvrB, playing an important role in recruiting UvrB to the damaged site40,42. In our model, we are able to see ADP molecules fulfilling the distal sites, supporting the idea that MtUvrA2-DNA structure describes the moment in which UvrA recognizes the lesion, while the proximal sites are empty, coherently with the fact that no UvrB molecule is present.
The binding of one MtUvrB molecule to one MtUvrA protomer triggers the concomitant repositioning of the support-ID with consequent DNA release
The cryo-EM MtUvrA2-DNA structure provides a molecular snapshot of an early stage in the NER pathway, in which MtUvrA stably binds a potentially damaged DNA. To investigate the structural changes required for the pathway to proceed, we focused on the MtUvrA2UvrB1-DNA* cryo-EM structure that gave us the opportunity to evaluate the impact of the binding of one UvrB molecule on the overall architecture of the protein-DNA complex.
The reconstruction revealed a loss of signal for the ID belonging to the MtUvrB-bound MtUvrA protomer (Fig. 2a), suggesting that MtUvrB destabilized the MtUvrA ID conformation. The Signature-I domain of the catalytic core of the same MtUvrA protomer underwent a significant repositioning with respect to MtUvrA2-DNA structure (Fig. 4) subsequent to the C-terminal Zn-finger β-hairpins761-775 repositioning, adopting a conformation that closely resemble the one observed in BstUvrA2UvrB2 structure, the only available structure of UvrA in complex with UvrB5. The Signature-I domain is connected to both the ID and UBD and properly positions the UBD for UvrB binding. The structural arrangement of the UBD upon MtUvrB binding and the MtUvrA ID-DNA interaction appear mutually exclusive, due to steric hindrance, and consequently ID undergoes a conformational change releasing the DNA (Fig. 4 and Supplementary Movie 1). Moreover, the binding of MtUvrB affects the nearby C-terminal Zn-finger β-hairpin761-775 of the adjacent UvrA protomer that switches from the ID in the MtUvrA2-DNA structure to the Signature-I domain in the MtUvrA2UvrB1-DNA* complex (Fig. 4 and Supplementary Movie 1). This observation led us to propose that the C-terminal Zn-finger β-hairpin761-775 functions as a scaffolding element, alternatively supporting the Signature-I domain or the ID, along the structural rearrangements that are functional to the protein-DNA complexes remodeling.

Composite graphical representation of MtUvrA2-DNA (left) and MtUvrA2UvrB1-DNA* (right) structures. In both panels, the MtUvrA domains that undergo a substantial repositioning upon MtUvrB binding (ID, UBD, Signature-I and the β-hairpin761-775) and the DNA are represented as cartoon. The missing domains in each structure (UBD in MtUvrA2-DNA and ID in MtUvrA2UvrB1-DNA*) are represented as plane-colored shapes. The curved arrow in the panel on the right indicates the approx. 40 ° rotation of the Signature-I domain (as evaluated by comparing the position of the α-helix463-488 in the two models). In each image, the red arrow points to the contacts established between either the Zn-module (left) or the Signature-I domain (right) of one MtUvrA protomer and the C-terminal Zn finger β-hairpin761-775 of the opposite one in the dimer. For clarity, the UvrB molecule has been omitted.
The hydrophobic pocket of the MtUvrA recognition-ID hosts the lesion independently of UvrB
Despite the lower resolution of the MtUvrA2UvrB1-DNA* cryo-EM map, we could clearly discern the junction between the single-stranded and the double stranded segments (Fig. 5a and Supplementary fig. 8). This permitted the assignment of the nucleotide sequence of our DNA*. One of the lesion-mimicking fluorescein-modified thymine (FLUdT) of DNA* aligned with the recognition-ID, that is still defined in our structure (Fig. 5b). This evidence suggests that the first MtUvrB binding event induces the release of the support-ID only, while the recognition-ID remains stably associated to the lesioned-strand via the clamping α-hairpins. Moreover, in the MtUvrA2-DNA complex, the flipped-out nucleotide binds at the hydrophobic pocket of the recognition-ID by adopting a pose that perfectly matches the one displayed by the genuine modified-nucleobase (i.e. FLUdT) in the MtUvrA2UvrB1-DNA* complex (Fig. 5b).

a Cartoon representation of the MtUvrA2UvrB1-DNA* with the fluorescein dT highlighted as green spheres. The cryo-EM reconstruction is shown as semitransparent surface. The unpaired-to-paired DNA transition was marked with an arrow. b Superposition of MtUvrA2UvrB1-DNA* and MtUvrA2-DNA structures. The fluorescein dT, the DNA and the recognition-ID in MtUvrA2UvrB1-DNA* appear in green, orange and pink, respectively. The flipped-out nucleobase, the DNA and the recognition-ID in MtUvrA2-DNA are colored in cyan, white and red, respectively. The missing segment of one DNA strand in MtUvrA2-DNA structure is represented with a dashed line.
The capacity of the recombinant MtUvrA to bind and copurify with endogenous E. coli oxidized DNA, the low dissociation rate showed through SPR data and the extensive interactions shown in the MtUvrA2-DNA structure underpin that MtUvrA is able to lock the lesioned-DNA in a highly stable complex as previously described13,16. On the other hand, in MtUvrA2UvrB1-DNA* reconstruction, we observed the release of one ID (support-ID) with the concomitant binding of one UvrB molecule to the same UvrA protomer. This poses the question on how UvrB recognizes the UBD associated to the support-ID; we speculate that the driving force for the first UvrB binding event relies on a thermodynamic principle: it is likely that the binding energy of each ID is different, because of the presence of the lesion on one side of the DNA; the binding affinity of the recognition-ID is expected to be higher than one of the support-ID. This results in a low free-energy gain during the release of the support-ID and therefore the binding, with the associated conformational changes, is favored in the support-ID-containing UvrA protomer. The absence of a signal for MtUvrB or the UBD in the MtUvrA2-DNA reconstruction suggests that MtUvrB is dispensable to reach such a “locked-in-damage” stage. Moreover, the MtUvrA2UvrB1-DNA* points out that MtUvrB triggers a dramatic structural remodeling of the IDs in MtUvrA2-DNA complex. Our findings support previous works that established that UvrA is responsible for the first damage identification event, exhibiting discrimination between damaged and undamaged DNA based on the residence time on the DNA, subsequently addressing UvrB to the lesion16,40.
The binding of two UvrB molecules destabilizes the UvrA dimer interface and set the ground for the lesion hand-off between UvrA and UvrB
The overall “Venetian gondola” assembly of MtUvrA2UvrB2-DNA* resembled the one observed in the BstUvrA2UvrB2 structure (PDB: 3UWX)5. In both models, each UvrB interacts with a UBD and a Signature-II domain of opposite protomers (Fig. 2b). Both structures exhibited the same conformation for the catalytic core, including the interplay between the Signature-I domain and the C-terminal Zn-finger β-hairpin761-775, as observed in the MtUvrA2UvrB1-DNA* complex. Additionally, the disengagement of both IDs makes the DNA* free to position inside the MtUvrA2 central groove; the resolution in the central region of the DNA is very low, which is probably caused by the DNA being disordered in this part: indeed, we decided not to model it; nevertheless, a B-DNA cannot fit in the density in that region.
This conformation in which both IDs are open and two UvrB molecules are bound further confirms the association between MtUvrB binding and ID displacement.
A remarkable similarity between MtUvrA2UvrB2-DNA* and BstUvrA2UvrB2 structures is that the IDs share a similar orientation (Fig. 6a). In both cases, each ID opens wide to approach the ATP-binding domain II. Nevertheless, in BstUvrA2UvrB2 structure the ID repositioning is less pronounced, as a possible consequence of the interactions established between the symmetry mates in the crystal lattice. Given that the same structural configuration was observed in the presence as well as in the absence of DNA, we postulate that UvrB binding not only displaces the IDs, but it also drives the proper IDs repositioning towards the ATP-binding domains II.

a The image on the left represents the models of both the UvrA-UvrB heterodimers observed in the MtUvrA2UvrB2-DNA* structure; the UBDs were not represented for a better visualization. The image on the right corresponds to an equivalent heterodimeric component within the BstUvrA2UvrB2 structure. The ID, the ATP-binding domain II (ATP-II) and UvrB are differently colored and labeled. The distances between ID and ATP-II, and the tip of α-hairpin381-405 and Asn117 of UvrB (Ans116 in BstUvrB) are indicated as double arrows. b Structure-based hypothetical model for the lesion hand-off (two 180° rotated images are shown). Upon optimal superposition, the ID of MtUvrA2UvrB2-DNA* structure was superposed and replaced with the ID and the DNA of MtUvrA2-DNA structure; the DNA and the UvrB β-hairpin98-116 were shifted to avoid clashes and marked with black arrows. Important elements in the model are highlighted and labeled. The missing region in one DNA strand is represented with a dashed line. The green arrow indicates the direction of the short-range translocation of UvrB on the DNA.
Remarkably, the structural rearrangement induced by MtUvrB brings each ID at a 26 Å distance to the MtUvrB molecule bound to the opposite MtUvrA protomer (Fig. 6a). Given that the recognition-ID represents the primary damage recognition element in MtUvrA and that the support-ID disengages DNA upon the first MtUvrB binding event, we are tempted to speculate that the recognition-ID repositioning could move the lesion in close proximity to the already bound MtUvrB (Fig. 6b). Although this hypothesis would require further demonstration, the proposed mechanism satisfies four important requirements for the formation of the pre-incision complex: i) the UvrB β-hairpin inserts through the DNA major groove43,44, whereas the recognition-ID of UvrA interacts with DNA through the minor groove; therefore, the repositioning of the recognition-ID, together with the lesion held in place, would make the major groove accessible for the insertion of UvrB β-hairpin. ii) The damaged nucleobase recognized by the recognition-ID locates in the 3’-to-5’ DNA strand; consequently, the recognition-ID holds the lesion and repositions towards a pre-bound UvrB, which is capable of 5’-3’-directed translocation45. iii) The melted DNA region matches the proposed 6nt-long bubble necessary for UvrB interaction23. Finally, iv) the flipped-out lesion in UvrA agrees with the proposed UvrB lesion recognition mechanism through a base extruded from the DNA helix12,14.
In our cryo-EM single particle analysis of MtUvrA2UvrB2-DNA*, we detect flexibility between two halves. Each half contains one MtUvrB molecule, a DNA fragment and all domains within one MtUvrA protomer except for the UBD that belongs to the second protomer (Supplementary fig. 5). So, the MtUvrA2UvrB2-DNA* complex is flexible and behaves as two rigid body halves that hinge through the MtUvrA dimerization interface. This is an unprecedented characteristic with respect to previous UvrA structures that showed an extensive interaction between UvrA protomers of 2849 Å2. In contrast, in MtUvrA2UvrB2-DNA* the UvrA dimer interface is 1189 Å2, a decrease of 60% (Supplementary Table 4). Moreover, this observation agrees with the BstUvrA2UvrB2 structure, which shows a comparable decrease. Therefore, both structures align with the idea that the structural rearrangement produced by the second UvrB molecule binding triggers UvrA interprotomer destabilization; analyzing these three structures in succession (MtUvrA2-DNA, MtUvrA2UvrB1-DNA* and MtUvrA2UvrB2-DNA*), it is possible to observe that the binding of the DNA in MtUvrA2-DNA triggers the opening of the C-terminal Zn-finger β-hairpins761-775 with respect to MtUvrA2, which causes the repositioning of the SIG-1 domain, leading to the opening of the ID and to the binding of UvrB on the same protomer that exposed the ID; the same events happen for the other protomer, leading to the configuration of MtUvrA2UvrB2-DNA*, in which the dimerization interface is destabilized. It is plausible, therefore, that the destabilization event is caused by a series of conformational changes, triggered by the damage recognition. Since UvrA requires dimerization to bind DNA46, the UvrA dimer destabilization and potential disassembling could culminate in DNA release. DNA release from UvrA is strictly required during the lesion hand-off to UvrB. Therefore, these two key features of MtUvrA2UvrB2-DNA* structure (ID repositioning and UvrA dimer disassembly) support a model in which the recognition-ID repositions to transfer the damage to UvrB.
Discussion
The structures and biochemical analysis presented here elucidate some of the unanswered questions regarding the damage detection mechanism of UvrA and the transfer of the lesion to UvrB. In particular, we observe three conformations that we interpret as representative of sequential events in the initial stages of NER. This observation leads us to propose a model regarding the process that bridges the damage recognition by UvrA and the formation of the pre-incision complex. As showed in Fig. 7, UvrA first dimerizes to properly bind DNA7 (Fig. 7, step 1 and 2); then, unknown conformational changes, probably triggered by ATP/ADP binding and hydrolysis, lead to UvrA in a locked-in-damage state represented by our MtUvrA2-DNA structure (Fig. 7, step 3), in which it clamps, bends and unwinds DNA through the IDs. In this context, several experimental evidences show that DNA bending and melting is functional for the NER pathway for a proper incision operation47,48,49. Moreover, it has been proposed that UvrA, also independently of UvrB, is able to induce a slight bending of DNA that gradually becomes more severe along the pathway to a point of DNA kinking and wrapping in the pre-incision complex49,50,51. Our observations are in good agreement with single-molecule studies; indeed, Stracy et al. recorded two different modes of UvrA’s interactions with DNA during its searching for lesions16: a slowly moving state in which UvrA makes transient interaction with the DNA ( < < 15 ms) and a more stably bound state. The authors concluded that these long DNA-bound state represents the damage verification process in which UvrA takes time to sample DNA conformations to verify the presence of a lesion. In this sense we can suggest that what we observed in our MtUvrA2-DNA model is not related to the transient binding mechanism while it can be associated to the verification step, the long-bound state, in which UvrA tests the presence of the lesions through conformational changes leading to active distortions of the DNA molecule. Subsequently to the damage recognition, the binding of the first UvrB molecule displaces the support-ID releasing one DNA strand (Fig. 7, step 4 and Supplementary Movie 2). However, in the MtUvrA2UvrB1-DNA* reconstruction, UvrB binds a ssDNA, which deviates from the natural situation where the dsDNA is expected to be present. In such a position, UvrB would clash with an unprocessed dsDNA (Supplementary fig. 4), while it does not when we modeled UvrB as observed in the 6O8E pdb, namely in complex with a bent, unwound DNA showing a flipped-out nucleotide (Supplementary fig. 8). The second UvrB binding event seems to lead to UvrA dimer disassembly, while the recognition-ID repositioning could facilitate UvrB binding to the bubble around the lesion (Fig. 7, step 5 and Supplementary Movie 2). The decreased stability of the UvrA dimer and the ID displacement potentially preparing the lesion hand-off are consistent with the previously reported disassembly of UvrA during the lesion transfer to UvrB25. Therefore, it is plausible that, once primed by the second UvrB binding event, the two phenomena simultaneously occur.

UvrA dimerizes (1) to bind DNA (2); the damaged DNA recognition event triggers unknown conformational changes, leading to the bending, unwinding and melting of the DNA (3), allowing it to assess the presence of the lesion. After damage identification, one UvrB molecule binds and displaces the support-ID, releasing one DNA strand (4); a second UvrB molecule binds, causing the repositioning of the recognition-ID, which holds the DNA and transfers it towards UvrB (5), together with the UvrA dimer disassembly. This conformational state represents a transition state, that allows UvrB to access the lesion bubble and to replace UvrA (5). UvrB translocates the DNA through its ATPase activity to the pre-incision complex final conformation (6). The main events leading to the following conformation were indicated in blue.
Surprisingly, instead of lesion transfer to MtUvrB and complete MtUvrA disassembling, in our MtUvrA2UvrB2-DNA* structure we observed the DNA* deeply inserted in the central groove of the complex with poor signal for the lesion site and only MtUvrA dimer interface destabilization. In this context, our MtUvrA2UvrB2-DNA* structure represents a transition state in which UvrA is unable to transfer the lesion due to the unnatural overhang ends of the DNA*. Therefore, we are tempted to suggest that the DNA bubble containing the lesion would move towards UvrB, dragged by the IDs. The process culminates the pre-incision complex formation through the short-range UvrB translocation to properly position the damage and ready to recruit UvrC (Fig. 7, step 6). It has to be said that this model is speculative: an alternative model may involve the release of DNA by the IDs of UvrA, after the binding of UvrB; this would allow the DNA to slide towards one of the two UvrB molecules positioned on both ends of the complex.
Interestingly, the damage sensing mechanism described here for UvrA is perfectly in line with the available structures of DNA repair proteins that includes modified bases flipping out, DNA bending and, in some cases, unwinding. Particularly remarkable is the agreement with UV-DDB that participates in pyrimidine dimers (CPD and 6-4PP) recognition in eukaryotic NER. UV-DDB bends DNA by 40° allowing a β-hairpin insertion into the double helix and flipping out the modified bases in a lesion-binding pocket52. A second meaningful example is the functional equivalent, but structurally unrelated, yeast protein Rad4 and its human ortholog XPC. As UV-DDB, Rad4 bends DNA by ~40° and inserts a β-hairpin, but in this case, Rad4 flips out the two bases opposite to the lesion53,54.
The present results offer a structural model of the first steps in NER pathway from UvrA in a locked-in-damage recognition state to the lesion hand-off to UvrB. However, the mechanistic events leading to such a UvrA conformation and the role of ATP hydrolysis remained unanswered. Another important missing piece of information is represented by the description of the molecular bases of the capability of UvrA to discriminate between damaged and undamaged DNA, since an experimental model of UvrA bound to unmodified dsDNA is still lacking.
Methods
Purification of MtUvrAUvrB complex
M. tuberculosis UvrA was over-expressed in E. coli BL21 (DE3) strain harboring the plasmid pMtHisUvrA6. M. tuberculosis UvrB gene was provided by gene synthesis (GenScript) and was over-expressed in E. coli BL21 (DE3) strain harboring the plasmid pMtHisUvrB. The two proteins were purified as described in Supplementary information. To obtain the samples used in the cryo-EM experiments, pure MtUvrA- and MtUvrB-containing protein solutions were pooled together in a 1.2 molar excess of MtUvrB and 1 mM ATP and 10 mM MgCl2 were added as stabilizing elements of the complex formation18,32,33. The sample mixture was concentrated to 4.3 mg/mL and loaded onto a Superdex 200 Increase 10/300 GL size exclusion chromatography column with a buffer containing 20 mM Tris HCl pH 8.0, 300 mM NaCl. The chromatographic fractions that, on the basis of chromatogram analysis and subsequent SDS-PAGE, contained hetero-tetrameric complexes, were then pooled and concentrated up to 0.5 mg/mL, obtaining the MtUvrA-MtUvrB sample. The MtUvrA-MtUvrB-DNA* was prepared by incubating the MtUvrA-MtUvrB sample with the FLUdT-containing dsDNA (DNA*, Supplementary Table 3), in a 1.2 molar excess of DNA*. The sample was flash-frozen in liquid nitrogen and stored at -80 °C.
The MtUvrA-∆β-hairpin and MtUvrA-Y323A variants used in the EMSA-, MST- and SPR-based analyses were produced adopting the same procedure used to express and purify the wild type MtUvrA protein, followed by an additional Size Exclusion Chromatographic step using a Superdex 200 Increase 10/300 GL column with a buffer containing 20 mM Tris HCl pH 8.0, 500 mM NaCl, 5% glycerol as the mobile phase.
MtUvrA-MtUvrB DNA complex cryo-EM sample preparation, data collection and data processing
Vitrification of MtUvrA-MtUvrB and MtUvrA-MtUvrB-DNA* samples at 1.75 µM was carried out with a Mark IV Vitrobot (Thermo Fisher Scientific). 3 μl of each sample were applied to a Quantifoil R 1.2/1.3 Cu 300-mesh grid previously glow-discharged at 30 mA for 30” in a GloQube (Quorum Technologies). Immediately after sample application, the grids were blotted in a chamber at 4 °C and 100% humidity, and then plunge-frozen into liquid ethane.
MtUvrA-MtUvrB sample was transferred to a Talos Arctica (Thermo Fisher Scientific) operated at 200 kV equipped with a Falcon III operating in electron counting mode. 3’642 movies were acquired at a nominal magnification of 120’000x corresponding to a pixel size of 0.889 Å/pixel. The total dose was 40 e-/Å2 equally distributed on 40 frames. Data collection statistics are summarized in Supplementary Table 1.
Movies were preprocessed with RELION 3.155 (Supplementary fig. 1). All movies were subjected to motion-correction and dose-weighting using MOTIONCOR256. Estimation of the contrast transfer function (CTF) was performed on aligned dose-weighted sum of power spectra every 4 e-/A2 using CTFFIND 4.1.1057. Motion-corrected micrographs were imported to WARP and particle picking was preformed using the deep convolutional neural network BoxNet2_20180602 using a particle diameter of 60 Å resulting in a total of 240’304 picked particles. Particles were imported in RELION 3.1 and extracted with a box size of 400×400 pixels and a pixel size of 0.889 Å/pixel, inverted, normalized and imported into CRYOSPARC 4.2.158 for further processing. After a first round of 2D classification, particles averages above 60 Å were selected yielding a 148’931 set of particles. The set of particles was further split in two groups depending on their composition through an ab initio job using two models. The two ab initio reconstructed volumes corresponded to MtUvrA2 (68’915 particles) and MtUvrA2-DNA (80’016 particles) and were further cleaned through 3D classification yielding a final number of 22’766 particles and 27’801 particles, respectively. The final resolution of the two structures was 4.9 Å for MtUvrA2 and 4.2 Å for MtUvrA2-DNA, according to an FSC of 0.143 (Supplementary fig. 1). Data processing statistics are summarized in Supplementary Table 1 and a schematic representation of the workflow used is in Supplementary fig. 1.
Vitrified grids of MtUvrA-UvrB-DNA* sample were sent to the CM01 beamline at the ESRF59,60) for a data collection. Movies were recorded using a Titan Krios (Thermo Fisher Scientific) operated at 300 kV equipped with a K3 detector operating in electron counting mode. 21’232 movies were acquired from two twin grids at a nominal magnification of 105’000x in superresolution mode bin2 corresponding to a pixel size of 0.84 Å/pixel. The dose rate during movie acquisition was 18.8 e-/pix/s and the total dose was 40 e-/Å2 equally distributed on 40 frames. Data collection statistics are summarized in Supplementary Table 1.
Movies were preprocessed with RELION 3.155 (Supplementary fig. 5). All movies were subjected to motion-correction and dose-weighting using MOTIONCOR256. Estimation of the CTF was performed on aligned dose-weighted sum of power spectra every 4 e-/A2 using CTFFIND 4.1.1057. Motion corrected micrographs were imported to WARP and particle picking was preformed using the deep convolutional neural network BoxNet2_20180602 using a particle diameter of 60 Å resulting in a total of 2’158’399 picked particles. Particles were imported in RELION 3.1 and extracted with a box size of 400×400 pixels and a pixel size of 0.84 Å/pixel, inverted, normalized and imported into CRYOSPARC 4.2.158 for further processing. After a first round of 2D classification, particles averages above 60 Å were selected yielding 666’671 particles. The set of particles was further split in three groups depending on their composition and an ab initio model was generated with each set. The initial set of 666’671 selected particles was heterogeneous refined using three ab initio models as references. Three classes corresponding to MtUvrA2UvrB2-DNA* (143’862 particles), MtUvrA2UvrB1-DNA* (138’164 particles) and MtUvrA2-DNA (242’593 particles) were further cleaned through 2D classification yielding a final number of 110’916, 99’428 and 229’676 particles, respectively. The final resolution for the three structures was 4.0 Å for MtUvrA2UvrB2-DNA*, 4.3 Å for MtUvrA2UvrB1-DNA* and 3.2 Å for MtUvrA2-DNA, according to an FSC of 0.143 (Supplementary fig. 6). MtUvrA2UvrB2-DNA* reconstruction was split in two halves and each applied on the full reconstruction for particle subtraction first, and then the complementary half on the subtracted particles for a local refinement. The half reconstruction of the MtUvrA2UvrB2-DNA* produced a map at 3.6 Å and 3.7 Å of resolution. The two halves were merged in a composite map using chimera61. Data processing statistics are summarized in Supplementary Table 1 and a schematic representation of the workflow used is in Supplementary fig. 5.
Model building and refinement
For MtUvrA2-DNA, a previously published structure of TmUvrA (PDB: 3PIH) was used as initial model. UCSF Chimera61 was used to rigidly fit the model in the density map. The changes in sequence and conformations were made using Coot62. The Insertion Domains of MtUvrA crystal structure (PDB: 3ZQJ) were rigid-body fit into the map, with the exception of the non-modeled β hairpin domains, and the model was then refined against the map using Phenix Real Space Refine63. A random nucleotide sequence of DNA was modeled and a density fit restraining the Watson-Crick base pairs ad the Pi-stacking between nucleobases was performed. Nevertheless, the electron density map signal of one DNA strand fades away in the central portion and for this reason only a single strand was modeled there. Starting from this configuration, we parameterized MtUvrA2-DNA in the AMBER14SB64 force field with OL15 torsional modifications65 for DNA using the tleap program. The zinc ions coordination has been modeled via the ZAFF force field66. We parameterized ADP via the GAFF2 force field and computed its point charges using the AM1-BCC semiempirical approach. Subsequently, we solvated it with the OPC water model67 and neutralized with sodium ions. This system underwent a two-phases energy minimization (steepest descent with a 1000 kJ/mol energy tolerance and conjugate gradient minimization with a 100 kJ/mol energy tolerance) using GROMACS 2022.368, obtaining the final model. Figures were prepared with CHIMERAX69, PYMOL70, INKSCAPE71 and BIORENDER72.
For MtUvrA2UvrB1-DNA*, the MtUvrA2-DNA structure and the chain corresponding to UvrB from a previously published model (PDB: 3UWX) was docked into the map, manually adjusted in Coot62 and refined using Phenix Real Space Refine63.
For the MtUvrA2UvrB2-DNA* structure, the previously published model (PDB: 3UWX) was used and a rigid-body fit was applied in order to get the final model, finally refined using Phenix Real Space Refine63. In analogy with the modeling performed for MtUvrA2-DNA, for MtUvrA2UvrB2-DNA* we parameterized the system with the AMBER14SB-OL15-ZAFF force field and solvated it with OPC water and neutralized with sodium ions. We obtained the final model with the same two-phases minimization algorithm used in MtUvrA2-DNA refinement.
Plasmid vectors for the over-expression of the MtUvrA variants in E. coli cells
We generated the plasmid vector that encodes the MtUvrA-Y323A variant by using the pMtHisUvrA6 construct as the DNA template, the primers pair Y323Afor/Y323Arev (see Supplementary Table 3), and the QuikChange II site-directed mutagenesis kit (Stratagene). The protein-encoding open reading frame in the resulting construct was verified by sequencing (Eurofins MWG Operon).
The expression construct for MtHisUvrA-∆β-hairpin was provided by a custom gene synthesis service (GenScript), encoding the protein variant that lacks the Q361-E381 spanning region, corresponding to the entire ID β-hairpin.
DNA substrates
Mutagenic primers and DNA oligonucleotides used in biochemical assays and structural investigation, synthesized by Eurofins Genomics, are reported in Supplementary Table 3.
Dot Blot Assay
All samples were treated with 0.4 M NaOH, 10 mM EDTA pH8 to denature DNA, boiled for 10 min at 94 °C and finally neutralized with 2 M NH4CH3COO- 1:1 v/v. DNA was then loaded on a nitrocellulose membrane activated with 6X SSC buffer through a Dot Blot vacuum manifold (SRC 96 Schleicher and Schuell), followed by two washes with 2X SSC. The membrane was dried, and DNA/membrane were crosslinked for 2 h at 80 °C.
The membrane was then blocked and hybridized with primary antibodies (anti-6-4PPs clone 64M-2; anti-CPDs clone TDM-2; anti-8-oxo-dG (15A3) sc-66036; anti-ssDNA MAB3034) and secondary antibodies HRP-conjugated. Chemiluminescence reaction was induced using Bio-Rad Clarity ECL and developed with Chemidoc Touch (Bio-Rad). SH-SY5Y (ATCC-CRL-2266) cells were untreated or treated with UVC lamp, and then harvested. Genomic DNA was purified using Macherey-Nagel™ NucleoSpin™ Tissue kit, processed as the UvrA sample and loaded on nitrocellulose with serial dilution (125 ng, 62 ng, 31 ng).
Antibody sources and working concentrations are reported: Anti-CPDs TDM-2 (D194-1): CosmoBio, monoclonal, Mouse, 1:1000. Anti-6-4PP (64M2): CosmoBio, monoclonal, Mouse, 1:500. Anti- ssDNA (MAB3034): Merck-Millipore, monoclonal, Mouse, 1:1000. Anti-8OHdG (15A3), Santa Cruz Biotechnology sc-66036, monoclonal, Mouse 1:3000 in 3% BSA. Goat anti-Mouse IgG (H + L): ThermoFisher Scientific, Secondary Antibody HRP (Cat # 31430), 1:10000. All of them were used diluted in TBS 0.1% Tween-20.
Electrophoretic mobility shift assays (EMSA)
A biotinylated dsDNA fragment, containing 2 fluorescein-modified thymine nucleobases (FLUdT) and 5’-protruding ends, was mixed, at a fixed 0.15 µM concentration, with increasing concentrations (0.0-1.5 μM) of wild type MtUvrA, or MtUvrA-Y323, or MtUvrA-∆β-hairpin, in binding buffer (50 mM Tris HCl pH 8.0, 500 mM NaCl, 5% glycerol; final volume=20 µL). Upon a 5 min incubation at 25 °C, samples were electrophoresed on 8% polyacrylamide gel (in 44.5 mM Tris, 44.5 mM boric acid, 1 mM EDTA, pH 8.3) at 100 V for 30 min. The bands were visualized by direct gel imaging using a green light-emitting diode (LED)/605-nm-band-pass filter excitation/emission parameters, respectively.
Surface Plasmon Resonance analysis
A Biacore X100 (GE Healthcare) instrument was used for DNA binding experiments. The biotinylated dsDNA fragment containing 2 fluorescein molecules was immobilized onto the surface of a SA sensor chip (cat # BR100012, GE Healthcare), functionalized with streptavidin; the DNA was immobilized onto the active flow cell (#2), while the other flow cell was used as a reference. Recombinant UvrA proteins (wild type and mutant versions) were diluted to a concentration of 50 nM in HBS-EP+ buffer (GE Healthcare). Considering molecular weights (MW) of ligand and analytes of about 25 kDa and 109 kDa respectively, the appropriate ligand density (RL) on the chip was calculated according to the following equation: RL = (ligand MW/analyte MW) × Rmax × (1/Sm), where Rmax is the maximum binding signal (200 RUs) and Sm corresponds to the binding stoichiometry DNA:protein that is 1:2. Accordingly, the target capture level of the oligonucleotides was of 20 response units (RUs). Increasing concentrations of UvrA proteins were flowed over the SA sensor chip coated with damages dsDNA at a flow rate of 30 μl/min at 25 °C with an association time of 120 s and a dissociation phase of 600 s. A double regeneration step with 50 mM Glycine pH 1.5 and 0.4% SDS was performed following each analytic cycle.
The affinity constant (KD) and the kinetic parameters were evaluated using the BIAcore evaluation software (GE Healthcare) and the reliability of the kinetic constants calculated by assuming a heterogeneous ligand model supported by the quality assessment indicators values.
MicroScale Thermophoresis
DNA-binding studies were also performed using MicroScale Thermophoresis (MST) according to previously described methods72,73. Damaged dsDNA fragment was used as the probe and intrinsic fluorescence of the two fluorescein molecules was used to avoid the labeling. Damaged dsDNA was used at a concentration of 10 nM and the ligands (UvrA wild type and mutated proteins) were diluted, starting from the highest concentration of 1 µM, in 50 mM Tris HCl pH 8, 300 mM NaCl, 5% glycerol; measurements were performed using Premium standard-treated glass capillaries and the instrument Monolith NT.115 (Nano-Temper Technologies). The IR laser-power was set to 40/80% the laser on-and-off times were set at 20 and 5 s, respectively. The amplitude normalized data ΔFnorm of each dataset were averaged and plotted against the concentration of the unlabeled ligand on a logarithmic scale. The binding data were analyzed using MO affinity analysis software, as provided by the manufacturer.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The density maps and atomic coordinates reported in this paper have been deposited in the Electron Microscopy Data Bank (EMDB) and RCSB Protein Data Bank (RCSB PDB), respectively, with accession codes as follows: DNA-free MtUvrA2 dimer structure EMD-51169 and PDB ID 9GA2; MtUvrA2 bound to endogenous E. coli DNA at low resolution EMD-51168; MtUvrA2UvrB1 bound to damaged oligonucleotide EMD-51170 and PDB ID 9GA3; MtUvrA2UvrB2 bound to damaged oligonucleotide (half 1) EMD-51171; MtUvrA2UvrB2 bound to damaged oligonucleotide (half 2) EMD-51172; MtUvrA2UvrB2 bound to damaged oligonucleotide EMD-51173 and PDB ID 9GA4; MtUvrA2 bound to endogenous E. coli DNA EMD-51174 and PDB ID 9GA5. The movies were deposited in EMPIAR74,75 under the ascension code: EMPIAR-12274. Source data are provided with this paper.
Change history
20 May 2025
In the version of this article initially published, due to a processing error, an artifact appeared next to the peak of the green trace in Fig. 3d, which is now amended in the HTML and PDF versions of the article.
References
Kisker, C., Kuper, J. & Van Houten, B. Prokaryotic nucleotide excision repair. Cold Spring Harb. Perspect. Biol. 5, a012591 (2013).
Truglio, J. J., Croteau, D. L., Van Houten, B. & Kisker, C. Prokaryotic Nucleotide Excision Repair: The UvrABC System. Chem. Rev. 106, 233–252 (2006).
Wozniak, K. J., & Simmons, L. A. Bacterial DNA excision repair pathways. Nat. rev. Microbiol. 20, 465–477 (2022).
Warner, D. F. The role of DNA repair in M. tuberculosis pathogenesis, Drug Discov. Today: Disease Mechan. 7, e5–e11 (2010).
Pakotiprapha, D., Samuels, M., Shen, K., Hu, J. H. & Jeruzalmi, D. Structure and mechanism of the UvrA-UvrB DNA damage sensor. Nat. Struct. Mol. Biol. 19, 291–298 (2012).
Rossi, F. et al. The biological and structural characterization of Mycobacterium tuberculosis UvrA provides l insights into its mechanism of action. Nucleic Acids Res 39, 7316–7328 (2011).
Jaciuk, M., Nowak, E., Skowronek, K., Tañska, A. & Nowotny, M. Structure of UvrA nucleotide excision repair protein in complex with modified DNA. Nat. Struct. Mol. Biol. 18, 191–198 (2011).
Timmins, J. et al. Structural and Mutational Analyses of Deinococcus radiodurans UvrA2 Provide Insight into DNA Binding and Damage Recognition by UvrAs. Structure 17, 547–558 (2009).
Kraithong, T., Hartley, S., Jeruzalmi, D. & Pakotiprapha, D. A peek inside the machines of bacterial nucleotide excision repair. Int. J. Mol. Sci. 22, 1–20 (2021).
Pakotiprapha, D. et al. Crystal Structure of Bacillus stearothermophilus UvrA Provides Insight into ATP-Modulated Dimerization, UvrB Interaction, and DNA Binding. Mol. Cell 29, 122–133 (2008).
Lahiri, S., Rizzi, M., Rossi, F. & Miggiano, R. Mycobacterium tuberculosis UvrB forms dimers in solution and interacts with UvrA in the absence of ligands. Proteins: Struct., Funct. Bioinforma. 86, 98–109 (2018).
Lee, S. J., Sung, R. J., & Verdine, G. L. Mechanism of DNA Lesion Homing and Recognition by the Uvr Nucleotide Excision Repair System. Research 5641746 (Washington, D.C., 2019). https://doi.org/10.34133/2019/5641746.
DellaVecchia, M. J. et al. Analyzing the handoff of DNA from UvrA to UvrB utilizing DNA-protein photoaffinity labeling. J. Biol. Chem. 279, 45245–45256 (2004).
Truglio, J. J. et al. Structural basis for DNA recognition and processing by UvrB. Nat. Struct. Mol. Biol. 13, 360–364 (2006).
Thakur, M., Kumar, M. B. J. & Muniyappa, K. Mycobacterium tuberculosis UvrB Is a Robust DNA-Stimulated ATPase That Also Possesses Structure-Specific ATP-Dependent DNA Helicase Activity. Biochemistry 55, 5865–5883 (2016).
Stracy, M. et al. Single-molecule imaging of UvrA and UvrB recruitment to DNA lesions in living Escherichia coli. Nat. Commun. 7, 12568 (2016).
Sancar, A. & Hearst, J. E. Mol. Matchmakers. Sci. (1979) 259, 1415–1420 (1993).
Pakotiprapha, D. & Jeruzalmi, D. Small-angle X-ray scattering reveals architecture and A2B2 stoichiometry of the UvrA-UvrB DNA damage sensor. Proteins: Struct., Funct. Bioinforma. 81, 132–139 (2013).
Jaciuk, M. et al. A combined structural and biochemical approach reveals translocation and stalling of UvrB on the DNA lesion as a mechanism of damage verification in bacterial nucleotide excision repair. DNA Repair (Amst) 85, 102746 (2020).
Kad, N. M., Wang, H., Kennedy, G. G., Warshaw, D. M. & Van Houten, B. Collaborative Dynamic DNA Scanning by Nucleotide Excision Repair Proteins Investigated by Single- Molecule Imaging of Quantum-Dot-Labeled Proteins. Mol. Cell 37, 702–713 (2010).
Ghodke, H., Ho, H. N. & van Oijen, A. M. Single-molecule live-cell imaging visualizes parallel pathways of prokaryotic nucleotide excision repair. Nat. Commun. 11, 1477 (2020).
Pakotiprapha, D., Liu, Y., Verdine, G. L. & Jeruzalmi, D. A structural model for the damage-sensing complex in bacterial nucleotide excision repair. J. Biol. Chem. 284, 12837–12844 (2009).
Zou, Y. & Van Houten, B. Strand Opening by the UvrA 2 B Complex Allows Dynamic Recognition of DNA Damage. EMBO J. 18 (1999).
Verhoeven, E. E. A. Architecture of nucleotide excision repair complexes: DNA is wrapped by UvrB before and after damage recognition. EMBO J. 20, 601–611 (2001).
Orren D. K. & Sancar A. The (A)BC excinuclease of Escherichia coli has only the UvrB and UvrC subunits in the incision complex. Proc Natl Acad Sci USA. 86, 5237–5241 (1989).
Warner D. F., Tønjum T. & Mizrahi V. DNA metabolism in mycobacterial pathogenesis. Curr Top Microbiol Immunol. 374, 27–51 (2013).
Darwin, K. H. & Nathan, C. F. Role for Nucleotide Excision Repair in Virulence of Mycobacterium tuberculosis. Infect. Immun. 73, 4581–4587 (2005).
Kelly, M. C., Whitaker, G., White, B. & Smyth, M. R. Nickel(II)-catalysed oxidative guanine and DNA damage beyond 8-oxoguanine. Free Radic. Biol. Med 42, 1680–1689 (2007).
Sagendorf J. M., Markarian N., Berman H. M. & Rohs R. DNAproDB: an expanded database and web-based tool for structural analysis of DNA-protein complexes. Nucleic Acids Res. 48, D277–D287 (2020).
Sagendorf, J. M., Berman, H. M. & Rohs, R. DNAproDB: an interactive tool for structural analysis of DNA–protein complexes. Nucleic Acids Res 45, W89–W97 (2017).
Li, S., Olson, W. K. & Lu, X.-J. Web 3DNA 2.0 for the analysis, visualization, and modeling of 3D nucleic acid structures. Nucleic Acids Res 47, W26–W34 (2019).
Colizzi, F. et al. Asymmetric base-pair opening drives helicase unwinding dynamics. Proc. Natl Acad. Sci. 116, 22471–22477 (2019).
Voter, A. F., Qiu, Y., Tippana, R., Myong, S. & Keck, J. L. A guanine-flipping and sequestration mechanism for G-quadruplex unwinding by RecQ helicases. Nat. Commun. 9, 4201 (2018).
Marians, K. J. Crawling and Wiggling on DNA. Structure 8, R227–R235 (2000).
Tubbs, J. L., Pegg, A. E. & Tainer, J. A. DNA binding, nucleotide flipping, and the helix-turn-helix motif in base repair by O6-alkylguanine-DNA alkyltransferase and its implications for cancer chemotherapy. DNA Repair (Amst.) 6, 1100–1115 (2007).
Hollis, T., Ichikawa, Y. & Ellenberger, T. DNA bending and a flip-out mechanism for base excision by the helix–hairpin–helix DNA glycosylase, Escherichia coli AlkA. EMBO J. 19, 758–766 (2000).
Demple, B. Enzyme Structures: DNA repair flips out. Curr. Biol. 5, 719–721 (1995).
Gade, P. et al. Structural and functional analyses of the echinomycin resistance conferring protein Ecm16 from Streptomyces lasalocidi. Sci. Rep. 13, 7980 (2023).
Wagner, K., Moolenaar, G. F. & Goosen, N. Role of the insertion domain and the zinc-finger motif of Escherichia coli UvrA in damage recognition and ATP hydrolysis. DNA Repair (Amst.) 10, 483–496 (2011).
Charman, R. J. & Kad, N. M. Single molecule iSCAT imaging reveals a fast, energy efficient search mode for the DNA repair protein UvrA. Nanoscale 14, 5174–5184 (2022).
Smith, B. T., Grossman, A. D. & Walker, G. C. Localization of UvrA and Effect of DNA Damage on the Chromosome of Bacillus subtilis. J. Bacteriol. 184, 488–493 (2002).
Kraithong, T. et al. Real-time investigation of the roles of ATP hydrolysis by UvrA and UvrB during DNA damage recognition in nucleotide excision repair. DNA Repair (Amst) 97, 103024 (2021).
Eryilmaz, J. et al. Structural insights into the cryptic DNA-dependent ATPase activity of UvrB. J. Mol. Biol. 357, 62–72 (2006).
Waters, T. R., Eryilmaz, J., Geddes, S. & Barrett, T. E. Damage detection by the UvrABC pathway: Crystal structure of UvrB bound to fluorescein-adducted DNA. FEBS Lett. 580, 6423–6427 (2006).
Grossman, L. & Yeung, A.T. The UvrABC endonuclease of Escherichia coli. Photochemistry and Photobiology, 51, 749–755 (1990).
Mazur, S. J. & Grossman, L. Dimerization of Escherichia coli UvrA and its binding to undamaged and ultraviolet light damaged DNA. Biochemistry 30, 4432–4443 (1991).
Zou, Y. Strand opening by the UvrA2B complex allows dynamic recognition of DNA damage. EMBO J. 18, 4889–4901 (1999).
Zou, Y., Walker, R., Bassett, H., Geacintov, N. E. & Van Houten, B. Formation of DNA Repair Intermediates and Incision by the ATP-dependent UvrB-UvrC Endonuclease. J. Biol. Chem. 272, 4820–4827 (1997).
Gordienko, I. UvrAB activity at a damaged DNA site: is unpaired DNA present? EMBO J. 16, 880–888 (1997).
Shi, Q., Thresher, R., Sancar, A. & Griffith, J. Electron microscopic study of (A)BC excinuclease. J. Mol. Biol. 226, 425–432 (1992).
Wang, H. et al. DNA wrapping is required for DNA damage recognition in the Escherichia coli DNA nucleotide excision repair pathway. Proc. Natl Acad. Sci. USA 106, 12849–12854 (2009).
Scrima, A. et al. Structural Basis of UV DNA-Damage Recognition by the DDB1-DDB2 Complex. Cell 135, 1213–1223 (2008).
Chen, X. et al. Kinetic gating mechanism of DNA damage recognition by Rad4/XPC. Nat. Commun. 6, 5849 (2015).
Paul, D. et al. Tethering-facilitated DNA ‘opening’ and complementary roles of β-hairpin motifs in the Rad4/XPC DNA damage sensor protein. Nucleic Acids Res 48, 12348–12364 (2021).
Zivanov, J., Nakane, T. & Scheres, S. H. W. Estimation of high-order aberrations and anisotropic magnification from cryo-EM data sets in RELION -3.1. IUCrJ 7, 253–267 (2020).
Zheng, X. et al. Mechanism of ligand activation of a eukaryotic cyclic nucleotide−gated channel. Nat. Struct. Mol. Biol. 27, 625–634 (2020).
Rohou, A. & Grigorieff, N. CTFFIND4: Fast and accurate defocus estimation from electron micrographs. J. Struct. Biol. 192, 216–221 (2015).
Punjani, A., Rubinstein, J. L., Fleet, D. J. & Brubaker, M. A. cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296 (2017).
Kandiah, E. et al. CM01: a facility for cryo-electron microscopy at the European Synchrotron. Acta Crystallogr D. Struct. Biol. 75, 528–535 (2019).
Sanjuan, A. C. & Miggiano, R. Italian CryoEM Bag (ICB) [dataset]. European Synchrotron Radiation Facility. https://doi.org/10.15151/ESRF-ES-893681585 (2025).
Pettersen, E. F. et al. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput Chem. 25, 1605–1612 (2004).
Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. Features and development of Coot. Acta Crystallogr D. Biol. Crystallogr 66, 486–501 (2010).
Afonine, P. V. et al. Real-space refinement in PHENIX for cryo-EM and crystallography. Acta Crystallogr D. Struct. Biol. 74, 531–544 (2018).
Maier, J. A. et al. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput 11, 3696–3713 (2015).
Galindo-Murillo, R. et al. Assessing the Current State of Amber Force Field Modifications for DNA. J. Chem. Theory Comput 12, 4114–4127 (2016).
Peters, M. B. et al. Structural Survey of Zinc-Containing Proteins and Development of the Zinc AMBER Force Field (ZAFF). J. Chem. Theory Comput 6, 2935–2947 (2010).
Izadi, S., Anandakrishnan, R. & Onufriev, A. V. Building Water Models: A Different Approach. J. Phys. Chem. Lett. 5, 3863–3871 (2014).
Abraham, M. J. et al. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1–2, 19–25 (2015).
Meng, E. C. et al. UCSF ChimeraX: Tools for structure building and analysis. Protein Sci. 32, e4792 (2023).
Schrödinger, L., & DeLano, W. (2020). PyMOL. Retrieved from http://www.pymol.org/pymol
Inkscape Project. (2020). Inkscape. Retrieved from https://inkscape.org.
Duhr S. & Braun D. Why molecules move along a temperature gradient. Proc Natl Acad Sci USA. 103, 19678–19682 (2006).
Wienken, C. J., Baaske, P., Rothbauer, U., Braun, D. & Duhr, S. Protein-binding assays in biological liquids using microscale thermophoresis. Nat. Commun. 1, 100 (2010).
Correction to ‘EMPIAR: The Electron Microscopy Public Image Archive’. Nucleic Acids Res 51, 3499–3499 (2023).
Iudin, A. et al. EMPIAR: the Electron Microscopy Public Image Archive. Nucleic Acids Res 51, D1503–D1511 (2023).
Acknowledgements
We acknowledge the European Synchrotron Radiation Facility for provision of beam time on CM01 and we would like to thank Dr. Eaazhisai Kandiah for assistance. Part of the cryo-EM experiments were carried out at the NoLimits center of the University of Milan. This research was supported by Fondazione Cariplo (grant n. 2016–0604 received by R.M.) and by the Italian Ministry of University and Research with the call PRIN 2022 PNRR (grant n. P2022P8KMF received by R.M. and A.C.S.). The authors would like to thank all the students of the FREE-MOVER project of the University of Piemonte Orientale for the fruitful discussion of the scientific data included in the manuscript.
Author information
Authors and Affiliations
Contributions
R.M. and A.C.S. and D.J. conceptualized the experiments. M.G. and G.F. purified the recombinants proteins for biochemical assays and cryo-EM experiments. M.G., R.M., A.C.S., F.R. and M.R. performed and analyzed the biochemical experiments including surface plasmon resonance, EMSA and microscale thermophoresis. D.R. and S.S. performed dot blot assay and DNA damage quantification. M.G. and A.C.S. performed cryo-EM sample preparation and validation. M.G. and A.C.S. processed and analyzed the cryo-EM datasets. M.G., A.C.S., M.B. and R.C. performed model building and refinement. M.G., R.M. and A.C.S. wrote the manuscript with input from D.J., F.R. and M.B.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Genta, M., Ferrara, G., Capelli, R. et al. Mechanistic understanding of UvrA damage detection and lesion hand-off to UvrB in Nucleotide Excision Repair. Nat Commun 16, 3416 (2025). https://doi.org/10.1038/s41467-025-58670-0
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-58670-0
This article is cited by
-
Structural snapshots of the mechanism of ATP-dependent DNA damage recognition by UvrA
Nature Communications (2025)
