
Abstract
The regulation of polymerisation and translocation of biomolecules is fundamental. Wzc, an integral cytoplasmic membrane tyrosine autokinase protein serves as the master regulator of the biosynthesis and export of many bacterial capsular polysaccharides and exopolysaccharides. Such polysaccharides play essential roles in infection, defence, and some are important industrial products. Wzc comprises a large periplasmic domain, two transmembrane helices and a C-terminal cytoplasmic kinase domain with a tyrosine-rich tail. Wzc regulates polymerisation functions through cycling the formation and dissociation of an octameric complex, driven by changes in the phosphorylation status of the tyrosine-rich tail. E. coli Wzc serves a model for a wider family of polysaccharide co-polymerases. Here, we determine structures of intermediate states with different extents of phosphorylation. Structural and computational data reveal the pre-ordering of the tyrosine-rich tail, the molecular basis underlying the unidirectionality of phosphorylation events, and the underlying structural dynamics on how phosphorylation status is transmitted.
Similar content being viewed by others
Introduction
Many bacterial capsular and secreted extracellular polysaccharides (CPS and EPS) are assembled by a “Wzy-dependent” strategy (reviewed in ref. 1; Fig. 1a, b). These polymers are vital to bacteria as they are involved in immune evasion, stress response and antibiotic resistance. Consequently, the synthesis and export of CPS and EPS represents a potential target for antibacterial intervention1,2. In Wzy-dependent pathways, Wzx is a flippase belonging to the multi-antimicrobial extrusion protein family, which translocates undecaprenol diphosphate-linked polysaccharide repeat units produced at the cytoplasm-membrane interface across the cytoplasmic membrane3. Once available at the periplasmic face of the membrane, the lipid intermediates provide substrates for the Wzy polymerase, a glycosyltransferase possessing a GT-C fold4 which extends the nascent polysaccharide chain by adding one repeat unit at a time. The polymerisation reaction performed by Wzy is regulated by a polysaccharide co-polymerase protein (PCP). There are two classes of PCP, exemplified by Wzz (PCP-1, regulating lipopolysaccharide O-antigen polysaccharide polymerization) and Wzc (PCP-2, regulating polymerization of CPS and EPS)1,5. The Wzy and Wzz proteins from Pectobacterium atrosepticum have been shown to form a complex of one Wzy with eight Wzz proteins6 and it is expected that Wzy and their cognate PCP form a complex.

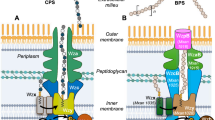
a Schematic model for the assembly and export of capsular polysaccharide (CPS) in E. coli K30, the prototype for Wzy-dependent CPS biogenesis. The repeat unit of E.coli K30 CPS50 is synthesized on an undecaprenol diphosphate carrier lipid at the cytoplasmic side of the inner membrane (IM) by a phosphoglycosyltransferase (PGT; WbaP) and glycosyltransferases (GT(s); WbaZ, WcaO and WcaN), and flipped to the periplasmic face of the membrane by Wzx. The repeat units are polymerized by Wzy, and the polymers are exported by Wza1. Polymerization and export are regulated by Wzc (a PCP-2a family autokinase), which cycles between a phosphorylated state and dephosphorylated state due the activity of the Wzb phosphatase9. Wzi is involved in the surface attachment of CPS1. b The gene cluster for the synthesis and export of the CPS of E. coli K30 (GenBank Acc. AF104912.3). c A cartoon of a Wzc monomer, comprising the periplasmic domain containing motif 1–3, two transmembrane helices, the BY-kinase domain, and the C-terminal tyrosine-rich tail (Y-tail) (adapted from9).
Wzc PCP-2 proteins found in CPS and EPS biosynthetic pathways are integral membrane proteins with two transmembrane helices (TM) that flank a large periplasmic domain7,8,9,10 (Fig. 1c). The second of the two helices connects to a cytoplasmic bacterial tyrosine (BY-) kinase catalytic domain (containing characteristic Walker A and B nucleotide binding motifs), followed by a C-terminal tyrosine-rich tail7,8,9,10. The kinase domain distinguishes PCP-2 proteins from PCP-1 proteins; the kinase mechanism is the focus of this study. Tyrosine kinases were originally thought to be confined to eukaryotes, but are now known to be prevalent among bacteria where they play a variety of roles11. BY-kinases are distinct in both structure and sequence from eukaryotic kinases8, enhancing their potential as antibacterial targets. In Gram-positive bacteria, the transmembrane and cytoplasmic BY-kinase domains of PCP-2 proteins exist as two polypeptides1,12,13 that form a complex which appears to function in the same manner as the single chain Wzc found in Gram-negative bacteria. The BY-kinase domain is highly conserved and Wzc proteins (and their Gram-positive paralogues) almost all possess a C-terminal tyrosine-rich tail7. Both the phosphorylation and dephosphorylation of the Wzc C-terminal tyrosine-rich tail were shown to be required for polymerisation and export, presumably by regulation of the polymerase Wzy and possibly other proteins9,14. Dephosphorylation of Wzc is catalysed by a cognate phosphatase (Wzb a protein tyrosine phosphatase (PTP) family member) in Gram-negative bacteria, and by a protein histidinol phosphatase (PHP) in Gram-positive bacteria15. Mutations eliminating phosphatase function result in drastically reduced CPS production, phenocopying kinase-null mutations14. Mutagenesis of the tyrosine-rich tail established that the regulatory function of Wzc in E. coli CPS biosynthesis is determined by the extent of phosphorylation rather than any individual tyrosine residue9,14,16,17.
PCP proteins form oligomers and we previously reported the cryo-EM structures of octamers of the dephosphorylated form of Wzc from E. coli serotype K30 (the CPS assembly prototype) by using an inactive (kinase) mutant, K540M9. A large central cavity in the octamer is formed by the transmembrane helices (Fig. 1)9. The periplasmic domain has three motifs (numbered 1, 2 and 3), with motif 3 proposed to interact with the outer membrane CPS translocon formed by an octamer of protein Wza9,18. Motif 3 varies in its structural arrangement between monomers (breaking the eight-fold symmetry), and in some monomers motif 3 is significantly disordered9. The C-terminal tail has a tyrosine-rich portion (Y705 to K721) which inserts into the active site of the neighbouring protomer9. A similar insertion of the tyrosine tail that stabilises an octameric ring also seen in the isolated BY-kinases from Staphylococcus aureus13 and E.coli K-1219, indicating a conserved feature. Mass spectrometry data demonstrated that tyrosine phosphorylation of Wzc from E. coli started at either Y718 (not conserved) or Y717 (highly conserved) and progressed sequentially to further tyrosine residues towards the N-terminus. Sequential phosphorylation destabilised the oligomers and WzcK540M4YE (K540M, Y718E, Y717E, Y715E and Y713E) was engineered to resemble the phosphorylated state at the observed stability “tipping-point”. Introduction of an additional mutation (Y708E) resulted in disassembly of the octamer9. An NMR study20 implicated residues 508–514 in recognition by the cognate phosphatase, Wzb15. Since the cryoEM structures indicated these residues are not fully accessible even in WzcK540M4YE mutant (i.e. the least stable octamer)9, we hypothesize that the phosphatase may only be able to access its substrate once the octamer has disassembled. We proposed a model in which the assembly and disassembly of the octamer around the Wzy polymerase was at the heart of the molecular basis of the regulation of Wzc9.
While the reported structures offered important insight into the function of Wzc, there remained significant gaps in our molecular understanding, including structural changes that occur during phosphorylation, the determinants of phosphorylation directionality, and regulatory activity.
Here, we report the characterisation of the structures WzcK540M2YE (Y718E, Y717E), WzcK540M3YE (Y718E, Y717E and Y715E) and WzcK540M3YE_N711Y (an additional N711Y mutation) corresponding to different phosphorylation states. These structures are used to develop a molecular dynamics model that rationalises the directionality of the phosphorylation, the dynamics of the C-terminal tail, and transmission of information on the phosphorylation state.
Results
Experimental structural snapshots
In Wzc from E. coli K30, tyrosine residues 708, 713, 715, 717 and 718 are targets for phosphorylation; residue Y718 is neither conserved in Gram-negative PCP-2 proteins, nor is it essential for function in CPS assembly or downstream phosphorylation9. We used mutation of tyrosine to glutamate to mimic phosphorylation and the inactive K540M mutant was used for structural studies as previously described9. The WzcK540M2YE (Y718E and Y717E) mutant corresponds to a doubly phosphorylated form but includes the first conserved and functional phosphorylation event (denoted 1pY). At a global level, the structure (periplasmic, transmembrane and cytoplasmic domains) (Fig. 2a, Supplementary Figs. 1, 4) is very similar to the octameric WzcK540M structure. Briefly, portions of the periplasmic domains do not follow the 8-fold symmetry that is seen in the membrane spanning and cytoplasmic domains. In three subunits, four helices are folded into a bundle that sits on motif 1 from the periplasmic domain. In four subunits, these four helices are mostly disordered, but the ordered portions are oriented parallel to the membrane normal. In one subunit, these helices are entirely disordered (Fig. 2a, Supplementary Figs. 1, 4). Each of these configurations and their arrangement in the octamer are identical to those seen in WzcK540M structure.

a WzcK540M2YE. b WzcK540M3YE. c WzcK540M3YE_N711Y. In each panel, on the left is shown the coulombic map of the entire molecule viewed from the side. The middle image shows the coulombic map of kinase domains only, viewed from the periplasm. The coulombic map of helix α23 and the tyrosine tail as they insert into the active site of the neighbouring kinase is shown on the right.
The remainder of the periplasmic domain is arranged around the central 8-fold axis, creating a large central cavity (Fig. 2a, Supplementary Figs. 1, 4). The transmembrane region has eight pairs of helices arranged as a circle enclosing a central void. The transmembrane helices are not close packed, leaving large portals open to the membrane bilayer. The cytoplasmic BY-kinase domains form a circular arrangement with the C-terminal tyrosine-rich tail reaching into the neighbouring protomer. The presence of ADP and MgCl2 improved the quality of the coulombic map for WzcK540M2YE, permitting unambiguous identification of Y715 at the (neighbouring) kinase active site (Fig. 2a, Supplementary Fig. 1). As a consequence, the interaction between the two protomers is different than previously observed9. In order for Y715 to bind at the catalytic site (as opposed to Y717 seen in WzcK540M structure9) the loop provided by residues 710–714 has changed conformation to become a strand. Y713 sits in the same pocket that contains Y715 in the published WzcK540Mstructure9. This “tyrosine pocket” is formed by loops A534-P536, P645-L647 and residues E675 and S679 (Supplementary Fig. 5d). The arrangement of the residues that form the pocket are identical in the two structures (one with Y715 and the other with Y713 in the pocket) (Supplementary Fig. 5d). The positions of the catalytic site residues and the kinase domain residues that interact with the tail do not however show (at the resolution of our studies) any change in position or conformation.
The WzcK540M3YE (Y718E, Y717E and Y715E) mutant was designed to mimic the conserved doubly phosphorylated species (2pY) and we therefore expected Y713 at the catalytic site. Although a tyrosine was clearly at the kinase active site, poor quality coulombic density unfortunately precluded its unambiguous identification (Fig. 2b, Supplementary Fig. 2). The disorder seen in the C-terminal tail, beginning at S704, was not observed in any other structure (WzcK540M, WzcK540M4YE9, WzcK540M2YE (this study)). No residue was visible in the tyrosine pocket, if Y713 is at the catalytic site then N711 would be expected to be at the pocket. We hypothesised that the pocket does not bind an asparagine side chain, and that leads to the observed disorder. To test this, we further mutated WzcK540M3YE to introduce N711Y (WzcK540M3YE_N711Y). The resulting structure confirmed our hypothesis; it possessed an ordered tail that unambiguously identified Y713 at the catalytic site with N711Y in the tyrosine pocket (Fig. 2c, Supplementary Fig. 3). The relative positions of N711Y and the pocket are different than that observed in WzcK540M and WzcK540M2YE but are almost identical to that previously described in WzcK540M 4YE (Fig. 2c, Supplementary Fig. 5d). The resolution of the EM structures and the flexibility of this region precludes definitive analysis of specific molecular interactions. As a result, it was not possible to convincingly rationalise the preference of the pocket for tyrosine over asparagine, except to note the aromatic ring of tyrosine would make interactions with the hydrophobic ring of Pro 536 that are not possible for asparagine. To accommodate Y713 at the catalytic site and N711Y in the pocket, helix α23 has partially unwound to a strand at its C-terminus (Y705). In the WzcK540M3YE structure, this region of the helix is disordered. Excluding the disorder/order of the C-terminus, the structure of WzcK540M3YE_N711Y was essentially unchanged relative to WzcK540M3YE (Supplementary Fig. 6). The four helices in the periplasmic domain show the same pattern of structures that was observed in WzcK540M and WzcK540M2YE. More broadly, the overall structure of the catalytic site and that of the kinase domain are identical in all the experimental structures.
With these additional structures, a complete set of snapshots now exists for different (phosphorylation) states of the octamer: WzcK540M (0pY), WzcK540M2YE (1pY), WzcK540M3YE (2pY), and WzcK540M4YE (3pY) (Fig. 3a, Supplementary Fig. 4), WzcK540M5YE (4pY) does not form a stable octamer9. Comparison of the structures reveals that the differences within the tail and kinase active site are accompanied by a progressive rotation of the transmembrane and periplasmic domains by 3° (relative to 0pY) for 1pY, 5° for 2pY and 12° for 3pY (Fig. 3b). The interaction between N-terminal helix α1 and helix α23 (which directly connects to the moving C-terminal tyrosine tail) changes as Wzc progresses through the states (Fig. 3b, c). Relative to the remainder of the structure, the position and structure of these two helices undergo only very small changes between the 0pY and 1pY states (Fig. 3b, c). In 2pY, helix α23 has partially unwound, retaining only one helical turn, and consequently has a diminished interface with α1 (Figs. 2b, c, 3b, c). In 3pY, α23 is completely unwound and retains no interaction with α19. Throughout these changes, the helical conformation of α1 is preserved, with only minor positional shifts observed (Fig. 3b, c). A rotation of the transmembrane and periplasmic regions, relative to the kinase ring, is observed in comparison of the octamer structures (Fig. 4).

a The structures of the octamers reveal shifts in periplasmic and transmembrane domains relative to WzcK540M. b Superposition using the kinase domain of the monomer from each snapshot shows the movement in the transmembrane helices and periplasmic domain. Zoomed insets highlight conformational changes involving helices α1 and α23. c Structure of the C-terminal region beginning at residue 700 and helix α1 is highlighted, with the kinase domain of neighbouring monomer coloured grey.

a Side view, b top view comparing the structures of WzcK540M (PDB ID: 7NHR) and WzcK540M4YE (PDB ID: 7NIB). WzcK540M (PDB ID: 7NHR) is coloured gray, and WzcK540M4YE (PDB ID: 7NIB) is coloured by chain. c Structure comparison of the four states. The octameric cytoplasmic kinase domain is used for the structural alignment.
Molecular dynamics analysis of phosphorylation
To gain insight into transitions between phosphorylation states, we performed a series of MD simulations. Studying the entire molecule was too computationally expensive to be feasible, so we used the soluble cytoplasmic catalytic domains and the tyrosine-rich tails of two adjacent Wzc subunits (Fig. 5a). We modelled four phosphorylation states (0pY, 1pY, 2pY and 3pY) (Supplementary Table 2) and compared these simulations with the experimental structures from the Tyr-to-Glu mutants. The distance between the Cβ of the tyrosine and the Cα of D564 from the adjacent subunit was used as metric for structural changes (Fig. 5a). The simulation reveals that stability of the dimer is inversely correlated with the number of phosphorylated tyrosines. In essence, phosphotyrosines positioned C-terminal to the residue at the active site are progressively destabilising. Repeating the MD calculations with ADP at the active site, we observed the complete displacement of the phosphotyrosine from the active site in each model (Fig. 5b, Supplementary Fig. 7). When Mg2+ was introduced into the model, it stabilised the position of ADP by coordinating to both ADP and phosphotyrosine. We investigated the Tyr-to-Glu mutants in MD and observed that the mutated residue quickly leaves the active site in all cases (Fig. 5b, Supplementary Fig. 8). This validates our assumption that the substitution is a good mimic for the natural phosphorylation of the tyrosine. After leaving the active site, the negatively charged residue (phosphotyrosine or glutamate) was not observed to bind elsewhere on the structure. The model showed that in order for Y708 to enter the active site, the C-terminal tail pulls helix α23 away from helix α1, resembling closely the structure of 3pY (WzcK540M4YE9) (Fig. 5c).

a The portion of the structure used in the MD simulation and the metric used in comparison. The metric was defined as the distance between the carbon beta of the tyrosine (or glutamic acid) and the carbon alpha of D564, located in the active site of the adjacent subunit. b Boxplots of this metric for each structure. The boxplots represent the variation of the distance over time and the replicates (n = 3000 MD simulations frames. The box bounds the interquartile range divided by the median, with the whiskers extending to a maximum of 1.5 times the interquartile range beyond the box). See Supplementary Table 2 for the summary of the MD simulations, Supplementary Fig. 7 for the time trace. c Comparison between the initial and final frames of the 3YE (2pY) showing the displacement of E713 from the active site and the binding of Y708. Helix \({{{\rm{\alpha }}}}1\) is shown for reference but was not included in the simulations. Source data are provided as a Source Data file.
Mechanism for transmembrane signal transduction
To assess the functional role of the helix \(\alpha 1\) and \(\alpha 23\) interactions, we performed MD simulation of two neighbouring protomers embedded in a phospholipid bilayer. For these simulations, we selected 0pY (where the interaction is strongest) and 3pY (where the α23 and α1 interactions are absent). To measure the conformational changes, we calculated the angles between each of the helices TMH1 (transmembrane helix 1), TMH2, \(\alpha 1\) and the axis perpendicular to the membrane plane, as well as the relative angle between TMH1 and TMH2 (Fig. 6a, b). In the absence of phosphorylation with Y717 at the active site (Fig. 5b), the C-terminal tail around the active site exhibits low flexibility (Fig. 6c, d). In the absence of ADP, pY708 remained at the 3pY active site, resulting in limited flexibility and preservation of a D28-K701 contact that links helices \(\alpha 1\) and \(\alpha 23\) (Fig. 6b–e). In the presence of ADP pY708 was displaced, leading to increased C-terminal flexibility and disruption of the D28-K701 contact which, in turn, mobilised the α1 helix (Fig. 6b–e). Similar results were observed when simulating three neighbouring protomers in a phospholipid bilayer (Supplementary Figs. 9, 10). Superposition of the initial and final frames of the simulation revealed a structural rearrangement in the protein driven by rotation of the helices \(\alpha 1\) and TMH1, which is transmitted to the periplasmic domain (similar observation were made with WzcK540M4YE) (Fig. 6b–d). MD analysis shows that phosphorylation of the tail induces conformational changes in transmembrane and periplasmic domains via direct coupling between helices α23 and α1. To probe the D28-K701 contact, mutants were created (D28A, K710A and D28A/K701A) and all mutants resulted in a drastic reduction in CPS production (Fig. 6f) confirming the importance of this contact.

a The \({{{{\rm{\theta }}}}}_{{{{\rm{TMH}}}}1},\,{{{{\rm{\theta }}}}}_{{{{\rm{TMH}}}}2},\,{{{{\rm{\theta }}}}}_{{{{\rm{\alpha }}}}1}\) angles are defined as the angles relative to the axis normal to the membrane, and \({{{{\rm{\theta }}}}}_{{{{\rm{TMH}}}}1-{{{\rm{TMH}}}}2}\) is the angle between TMH1 and TMH2. b Boxplot of the metrics for each system. The boxplots represent the variation of the angles over time and the replicates (n = 1500 MD simulations frames. The box bounds the interquartile range divided by the median, with the whiskers extending to a maximum of 1.5 times the interquartile range beyond the box). c Superposition (using the kinase domain) of the initial (white) and final frames (coloured) reveals structural shifts for each state of Wzc. d Time trace of the minimum distance between the residues D28 and K701. e Close up view showing the changes in the interactions between K701 and D28. See Supplementary Table 2 for the summary of the MD simulations. Source data are provided as a Source Data file. f Immunoblot (anti-K30 CPS) of whole cell lysates separated by SDS-PAGE. The wildtype E. coli strain E69 (serotype O9a:H12:K30) was used as a positive control, and E. coli strain CWG285 (Δwzc) was used as a negative control. WT, D28A, K701A, D28A/K701A represent plasmids encoding wild-type Wzc, Wzc with the D28A mutation, Wzc with the K701A mutations, and Wzc with the D28A/K701A mutations respectively. As CPS does not migrate strictly according to size, protein standard is not useful to indicate the molecular weight. These data are representative of three biological replicates.
Discussion
The assembly and disassembly of the Wzc octamer is driven by phosphorylation and dephosphorylation of the C-terminal tyrosine-rich tail9,14 and this cycling underpins the function of Wzc in regulating polymerisation21 (Fig. 1a). The observation that octameric Wzz (a PCP-1 protein) encloses a single Wzy protomer to form a complex6 implies that octameric Wzc might do the same. Given the activity of Wzy is regulated by PCP proteins (Wzz or Wzc), to promote glycan chain extension, the simplest hypothesis is that the stability of the octamer and thus the PCP-Wzy complex determines Wzy activity. Here we report experimental structures of mutants of Wzc that correspond to intermediates in the phosphorylation pathway. Together with earlier work9, these now provide a complete structural description of Wzc octamers, from the most stable (0pY) form to the stability tipping point (3pY). The mutant corresponding to 4pY, “beyond the tipping point”, cannot be isolated as an octamer9. Mass spectrometry had suggested phosphorylation proceeded sequentially from C to N-terminal in direction9. The structural and MD data reported here confirm this hypothesis, showing that the tyrosine most stably positioned at the active site is the one immediately N-terminal to the phosphotyrosine (or the engineered Tyr-to-Glu mutation).
Description of the intermediates provides a molecular description of the structural changes that accompany the progressive phosphorylation of Wzc and reveal the transmembrane signalling mechanism that is essential for CPS production. During the progressive phosphorylation sequence, the kinase domain is a rigid body (Supplementary Fig. 5a). Structural alignment of the kinase domains of monomers of the full-length protein reveals increasing tilting of the transmembrane and periplasmic regions (Fig. 3). When the octamer is viewed from the periplasm to the cytoplasm, progressive phosphorylation results in a clockwise rotation of transmembrane and periplasmic regions relative to the kinase ring (Fig. 4b–c). These rotations are underpinned by changing interactions in the cytoplasmic regions between helix α1 and C-terminal helix α23 (connected to the tyrosine-rich tail). The movement of helix α1 is directly transmitted to transmembrane helix α2 (Figs. 3b, 6c) and then to the periplasmic domain and, in this way, the phosphorylation signal from the cytoplasm is coupled to the periplasm. This general concept of conformational coupling across the membrane is analogous to signaling mechanisms of some transmembrane receptors, such as the histidine kinase of the two-component system in bacteria22,23 and the EGF receptor in eukaryotes24,25. In these proteins, an environmental signal is transmitted into the cell via transmembrane conformational coupling. For Wzc, the information of the cytoplasmic phosphorylation is transmitted to the periplasmic region. In the context of CPS production, the periplasmic region of Wzc is thought to engage with the outer membrane translocon, Wza and genetic26 and preliminary structural data27 support Wza-Wzc interactions. Such interactions could conceivably facilitate feedback from Wza to the biosynthesis machinery. E. coli mutants lacking Wza exhibit drastic reduction in CPS synthesis28, consistent with signal across the periplasm that promotes synthesis of CPS only when subsequent translocation of the polymer is possible. Polymerisation does proceed in the absence of translocation in E. coli with a mutated Wza that forms stable oligomers but is compromised in its proper insertion into the outer membrane29. This is consistent with a hypothesis that it is the formation of Wza-Wzc interface that results in a signal that regulates polymerisation. Gram-positive PCP-2 proteins typically employ two polypeptides that combine to constitute a Wzc equivalent (Supplementary Fig. 11). Analogues to helix α1, helix α23 and ion pair (D28 K701) found in Wzc are conserved in Gram-negative PCP-2, suggesting a common coupling mechanism.
When a phosphotyrosine residue is displaced from the active site the C-terminal peptide becomes mobile. The YxY sequence pattern is highly (but not absolutely) conserved in the BY-kinase family. In Wzc, when Y717 (0pY) is positioned at the active site, Y715 is bound at the tyrosine pocket and is pre-oriented to shift into the active site. When Y715 occupies the active site (1pY), Y713 is bound in the same pocket. When Y708 occupies the active site (3pY, WzcK540M4YE9), Y706 also sits at the pocket but with a different relative position. Once Y708 is phosphorylated (4pY), the octamer becomes unstable, consistent with the lack of any phosphorylation of Y7069. The presence of the negatively charged E675 in the tyrosine pocket we suggest is incompatible with binding a negatively charged phosphorylated tyrosine (or glutamic acid side chain). One intermediate, 2pY which has Y713 at the active site, does not follow the YxY pattern, since position 711 is asparagine. Notably in the WzcK540M3YE structure the stretch of residues connecting the α23 to the active site is entirely disordered and molecular dynamics of 2pY showed this stretch of residues to be highly mobile (Fig. 5b). We show that it is binding of the i-2 tyrosine to the tyrosine pocket (where i is the number of the residue positioned at the kinase active site) orders the tail between the tyrosine at the active site and helix α23. A mutant of 2pY (Wzc3YE_N711Y) stabilises the C-terminal tail was functional in vivo, restoring immunoreactive CPS to levels approximating the wild type (Fig. 7a). This is consistent with a model in which the more stable the Wzc octamer the higher the Wzy activity. We note the reduction of CPS production by destabilising D28A and K701A mutants further support this model. The Wzc3YE_N711Y shows greater phosphorylation relative to Wzc3YE (Fig. 7b) and proteomic analysis of purified WzcYF_713Y/N711Y reveals both N711Y and Y713 are phosphorylated (Supplementary Fig. 12).

a Immunoblot (anti-K30 CPS) of whole cell lysates separated by SDS-PAGE. The wildtype E. coli serotype K30 strain (E69) was used as a positive control, and E. coli CWG285 (Δwzc) was used as a negative control. WT, WT_N711Y, 3YE, 3YE_N711Y represent plasmids encoding wild-type Wzc and derivatives with the N711Y, Y715E/Y717E/Y718E and Y715E/Y717E/Y718E/N711Y mutations, respectively. As CPS does not migrate strictly according to size, protein standard is not useful to indicate the molecular weight. These data are representative of three biological replicates. b Corresponding western immunoblot of purified Wzc proteins probed with anti-pTyr antibody (top) and anti-hexahistidine antibody (bottom). The experiment represents one of three technical replicates. c Western immunoblot of purified Wzc proteins using anti-pTyr antibody (top) and anti-hexahistidine antibody (bottom). The mutations are identified above each lane. The C-terminal sequences of the tyrosine-rich portion of YF_713Y, YF_N711Y and YF_713Y/N711Y are shown below. The data shows one of three technical replicates. d The ratchet mechanism. The YxY motif acts as a clamp stabilizing the octamer but also ordering the C-terminal tail (top). The tyrosine-rich tails in the octamer are phosphorylated and move unidirectionally through the active sites of adjacent monomers (top right). Phosphorylation progressively destabilizes the octamer which, after the fourth phosphorylation, disassembles (top right and bottom). Wzc regulates the polymerization activity of Wzy (purple) and is thought to form a complex with the translocon Wza (green). The cycling of the structural arrangement of the octamer is driven by (auto)phosphorylation and dephosphorylation (catalysed by Wzb) and the cycling is required for the production and export of CPS (bottom).
We propose that the YxY pattern serves as a molecular ratchet, that by selecting a tyrosine over phosphotyrosine, ensures phosphorylations in unidirectional (moving from the C to the N). In addition, the pocket prepositions its bound tyrosine to translate to the active site (Fig. 7d). The tyrosine pocket is found in both E. coli K-12 (PDB ID: 3LA6) (Supplementary Fig. 5b, e) and CapABK55M (PDB ID: 2VED) (Supplementary Fig. 5c, f), but, as yet, phosphorylated intermediates for these species have not been described. The conservation of the YxY motif and tyrosine pocket, suggests the ratchet is also a conserved feature of the PCP-2 system.
The CPS assembly mechanism described in E. coli K30 is also widespread in Klebsiella pneumoniae, with the loci likely being transferred by horizontal gene transfer. In hypervirulent K. pneumoniae (HvKP) isolates, the typically heterogenous CPS chain-lengths in non-HvKp isolate are replaced by homogeneous long chain polymers resulting in a hypermucoviscous phenotype evident in colony morphology and high viscous cultures in broth30. This property is imparted by an additional protein designated RmpD that interacts with Wzc. While the mechanism underlying RmpD function is not yet known, based on our data suggesting stability of the Wzc octamer is key to polymerisation activity, we propose RmpD most likely acts to influence stability of the Wzc Wzy complex.
Control of polymerisation is a feature of the chemistry of living things that is often hard to replicate in the laboratory. The structural snapshots here help advance our understanding of how Wzc regulates CPS polymerisation. CPS (and EPS) are important defence mechanisms against the host immune system for pathogenic bacteria. Since the BY-kinase is conserved among bacteria but is distinct from eukaryotic counterparts, the BY-kinases may be promising targets for antibacterial drug development.
Methods
Analysis of K30 CPS production in vivo
Wild-type Wzc and the 3YE mutant derivatives9 were produced from pBAD2431 derivatives under control of an L-arabinose-inducible promoter. The N711Y mutant derivatives were constructed by site-directed mutagenesis and using Gibson assembly following the manufacture’s protocol (New England Biolabs). E. coli CWG28514 was transformed with the various plasmids encoding Wzc derivatives and the strains were grown at 37 °C in LB broth (with no added L-arabinose) to an O.D.600nm of 0.6–0.8. L-Arabinose serves as the inducer for transcription of the cloned genes from the vector pBAD promoter, but basal levels of transcription are still observed in the absence of inducer. E. coli E69 (O9a:K30) and CWG285 were grown as K30 capsule positive and negative controls, respectively. One OD unit of each culture were collected by centrifugation at 12,000 × g for 2 min and the pellets were lysed in 1× SDS-PAGE loading buffer for 10 min in a boiling water bath. Protein was removed by digestion with 0.5 mg/mL proteinase K at 55 °C for 1 h. Samples (10 µL) were separated by SDS-PAGE (Tris/glycine) on 8% acrylamide gels at 150 V for 70 min using the BioRad Mini-protean system. Capsular polysaccharide was transferred to nitrocellulose (Amersham Protran 0.45 µm NC, Cytiva) at 350 mA for 45 min in 25 mM Tris, 150 mM glycine, 20 % (v/v) methanol, pH 8.3. Immunoblots were blocked in 5% (w/v) skimmed milk in TBST (10 mM Tris-Cl, pH 7.5, 150 mM NaCl, 0.05% (v/v) Tween-20). Capsular polysaccharide was detected with rabbit anti-K30 antiserum32 as the primary antibody at 1:3000 dilution and goat anti-rabbit alkaline phosphatase (AP) conjugated antibody (Cedar Lane, CLCC43008, 1:3000) as the secondary antibody. AP was detected with nitroblue tetrazolium and 5-bromo-4-chloro-3-indolyl phosphate (Roche).
Phosphotyrosine western immunoblot of purified Wzc proteins
The detection of phosphorylated Wzc proteins was performed as described previously9. In total, 8 µl purified Wzc proteins at 0.2 mg/ml were loaded and analysed by western immunoblotting. For the detection of phosphotyrosine, the monoclonal anti-pTyr antibody from mouse (Sigma, P4110, 1:5000 dilution) was used as primary antibody and HRP conjugated anti-mouse IgG antibody (Promega, W4021, 1:5000 dilution) as secondary antibody. The loading amount of Wzc proteins was detected by the anti-polyHistidine-peroxidase antibody (Merck, A7058, 1:5000 dilution). The uncropped blots are shown in Supplementary Fig. 14.
Expression and purification of Wzc proteins
Proteins were expressed in E.coli TOP10 cells and purified essentially as previously described9. Cells were grown at 37 °C until the OD600 was around 0.8 before induction with 0.02% arabinose at 20 °C overnight. Cells were harvested by centrifugation and resuspended with lysis buffer (20 mM Na phosphate, pH 7.0, 500 mM NaCl). Constant Systems cell disruptor was used to break the cells, and unbroken cells were removed by centrifugation at 20,000 × g for 1 h at 4 °C. The supernatant from the previous step was collected for further ultracentrifugation at 186,000 × g for 1 h at 4 °C to obtain the cell membranes. The membranes were solubilized with 20 mM Na phosphate, pH 7.0, 500 mM NaCl, 1.0% DDM at 4 °C and undissolved membranes were removed by ultracentrifugation. The supernatant containing Wzc proteins was loaded to ABT nickel resin (Cat. No. 6BCL-NTANi-100). After successive washes with 20 mM Imidazole and 50 mM Imidazole, the protein was eluted with 20 mM Na phosphate, pH 7.0, 500 mM NaCl, 0.003% LMNG and 300 mM Imidazole. Size-exclusion chromatography was performed for further purification using a Superose 6 10/300 increase column (Cytiva) equilibrated with 20 mM HEPES, 150 mM NaCl, 0.001% LMNG, 2 mM tris(2-carboxyethyl)phosphine (TECP), pH 7.3.
Cryo-EM analysis
Purified Wzc proteins (WzcK540M2YE, WzcK540M3YE and WzcK540M3YE_N711Y) at ~2 mg/ml were incubated with 1 mM ADP and 10 mM MgCl2 at 4 °C for 1 h. 3.5 μl sample was applied to glow-discharged Quantifoil gold R1.2/1.3 grids (300-mesh) and the grids were blotted for about 3 sec under 100% humidity and 4 °C before vitrification in liquid ethane using Vitrobot.
The datasets were collected on Krios G4 equipped with cold FEG, Falcon 4 direct electron detector and Selectris energy filter. Data was collected using EPU software at a magnification of ×165,000 with the pixel size of 0.737 Å/pixel. Three datasets for the WzcK540M2YE ADP complex, and two datasets for the WzcK540M3YE ADP complex, were collected and combined for data processing respectively. One dataset for WzcK540M3YE_N711Y ADP complex was collected for data processing. Detailed parameters for data collection were listed in the Supplementary Table 1.
The datasets were processed using cryoSPARC software33 and detailed workflows are described in Supplementary Figs. Briefly, the EM movies were motion-corrected in cryoSPARC and contrast transfer function values were estimated by CTFFIND34. Particles were picked through blob picking, or template picking using the templates generated from a small number of auto-picked particles. Multiple rounds of 2D classification were carried out and good 2D classes were selected for further processing. Multiple rounds of heterogeneous refinement were performed in cryoSPARC. Good classes were selected for further non-uniform refinement35 to get final maps, either without applying symmetry (C1) or with C8 symmetry.
Model building and refinement
Coot was used for the manual buildings and adjustments for the PDBs of WzcK540M2YE, WzcK540M3YE and WzcK540M3YE_N711Y. Ligand restraints were generated by eLBOW within PHENIX36. Structure refinements were carried out using PHENIX. PyMOL (The PyMOL Molecular Graphics System, Version 2.2.0 Schrödinger, LLC), Chimera37 and ChimeraX38,39 were used to prepare the figures of cryo-EM maps and structures.
MD Simulations
The cryo-EM structures for the WT Wzc octamer9 and the Tyr-to-Glu mutants were used to build molecular models for the four states of Wzc. The missing residues, including the missing residues in the C-terminal tail for states 2 to 4 were modelled based on the structures obtained from an AlphaFold2 multimer40. To study the effect of the phosphorylation on the C-terminal tail dynamics, we performed MD simulations of a dimer of the kinase domains (Fig. 5a). For each state, a series of MD simulations with different phosphorylation levels, in the presence or absence of ADP and Mg2+ and for Tyr-to-Glu mutations were performed. The CHARMM-GUI builder41 was used to set up the systems for the MD simulations. The kinase domain dimers were solvated with TIP3P waters and 0.15 M NaCl was added to neutralise the charge of the systems. The systems were equilibrated for 1 ns maintaining the structure of the protein restrained. Three repeats of 500 ns of MD simulation were then performed with position restraints removed for each system.
To study the role of helix \(\alpha 1\) in signal transduction, MD simulations of two adjacent subunits were performed. For these simulations the entire structure of the protomer, excluding motif 3, was used. Four systems were built: 1) phosphorylation 2) triply phosphorylated with Y718 also phosphorylated; 3) triply phosphorylated with Y718 also phosphorylated bound to ADP and 4) Wzc4YE. The CHARMM-GUI membrane builder41 was used to set up the systems for the MD simulations. The systems were inserted in a bilayer containing 25% 1-palmitoyl, 2-oleoyl phosphatidylglycerol and 75% 1-palmitoyl, 2-oleoyl phosphatidylethanolamine and solvated with TIP3P waters and 0.15 M NaCl. The input files for minimization and equilibration provided by CHARMM-GUI were used. The system was first minimized followed by a series of NVT and NPT equilibration steps consisting of gradual removal of the restraints from lipid and protein atoms for a total time of 2 ns. Three repeats of 500 ns of unrestrained atomistic MD simulations, for each configuration of the molecular system were performed.
MD simulations were performed using GROMACS 202142 and CHARMM36m force field43 with a timestep of 2 fs. All simulations were performed at 300 K and 1 bar with protein, lipids and solvent separately coupled to an external bath, using the velocity-rescale thermostat44 and Parrinello-Rahman45 for pressure coupling. All bonds were constrained with the LINCS algorithm46. The long-range electrostatic interactions were computed with the Particle Mesh Ewald method47, while a verlet cut-off method was used to compute the non-bonded interactions. MD simulations were analysed using GROMACS tools, MDAnalysis48 and in-house protocols. Summary of MD simulations were listed in Supplementary Table 2. All images were generated using PyMOL (The PyMOL Molecular Graphics System, Version 2.5.7 Schrödinger, LLC).
Proteomics analysis
An SDS-PAGE gel band for purified WzcYF_713Y/N711Y was sliced, destained, reduced with 50 mM TCEP for 10 min at 60 °C, alkylated with 100 mM iodoacetamide (IAA) at room temperature for 30 min in the dark, and incubated with trypsin at 37 °C overnight for proteolytic digestion. Digested peptides were dried using a SpeedVac vacuum concentrator (Thermo Fisher Scientific) and resolubilized with 100% H2O with 1% formic acid for LC-MS/MS analysis. The tryptic peptides were analyzed on a Dionex Ultimate 3000 UHPLC coupled to an Orbitrap Eclipse Tribrid mass spectrometer (Thermo Fisher Scientific). The peptides were firstly loaded onto a 75 μm ×2 cm precolumn and separated on a 75μm × 15 cm Pepmap C18 analytical column (Thermo Fisher Scientific) with a binary buffer system. Buffer A was 0.1% formic acid (FA) in 100% H2O and buffer B was 0.1% FA in 80% acetonitrile with 20% H2O. Briefly a linear gradient of 0–40% buffer B was used to separate the peptide. The Eclipse mass spectrometer was operated in positive ion mode using data-dependent acquisition with a 3 s cycle time. Precursors and products were detected in the Orbitrap analyzer at a resolving power of 60,000 and 30,000 (@ m/z 200), respectively. Precursor signals with an intensity > 1.0 × 104 and charge state between 2 and 7 were isolated with the quadrupole using a 0.7 m/z isolation window (0.5 m/z offset) and subjected to MS/MS fragmentation using higher energy collision-induced dissociation (30% relative fragmentation energy). MS/MS scans were collected at an AGC setting of 1.0 × 104 or a maximum fill time of 100 ms and precursors within 10 ppm were dynamically excluded for 30 s. Raw data files were processed using Proteome Discover 3.0 (Thermo Fisher Scientific) against a sequence database containing sequence of WzcYF_713Y/N711Y (the construct used in this study) as well as a list of common contaminants to identify the phosphorylation sites with the following search parameters: trypsin digestion, fixed modification with carbamidomethyl (C); dynamic modifications with oxidation (M), acetylated protein N terminus, and phosphorylation (Y); up to 2 missed cleavage sites, peptide length from 6 to l44; precursor tolerance (10 ppm); fragment mass tolerance (0.6 Da) The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE49 partner repository with the dataset identifier PXD061798.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
EM maps and models have been deposited in the EMDB and wwPDB under accession codes EMD-50042 and PDB 9EXO (C1 WzcK540M2YE ADP complex); EMD-50043 and PDB 9EXP (C8 WzcK540M2YE ADP complex); EMD-50044 and PDB 9I2Q (C1 WzcK540M3YE ADP complex); EMD-50045 and PDB 9I2R (C8 WzcK540M3YE ADP complex); EMD-50046 and PDB 9EXQ (C1 WzcK540M3YE_711Y ADP complex); EMD-50047 and PDB 9EXR (C8 WzcK540M3YE_711Y ADP complex). All constructs are available from the authors upon request until their deposition and release by ADDGENE (Wzc_N711Y: #217920; Wzc3YE_N711Y: #217919; WzcK540M3YE_N711Y: #217918; WzcK540M3YE: #217917; WzcK540M2YE: #1734519). The MD simulations data generated in this study are available as supplementary dataset on Zenodo (https://doi.org/10.5281/zenodo.15025687). The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD061798. PDB codes of previously published structures used in this study are 3LA6 and 2VED. Source data are provided as a Source Data file. Source data are provided with this paper.
References
Whitfield, C., Wear, S. S. & Sande, C. Assembly of bacterial capsular polysaccharides and exopolysaccharides. Annu. Rev. Microbiol. 74, 521–543 (2020).
Ernst, C. M. et al. Adaptive evolution of virulence and persistence in carbapenem-resistant Klebsiella pneumoniae. Nat. Med. 26, 705–711 (2020).
Le Bas, A. et al. Structure of WzxE the lipid III flippase for enterobacterial common antigen polysaccharide. Open Biol. 15, 240310 (2025).
Meitil, I. K. S., Gippert, G. P., Barrett, K., Hunt, C. J. & Henrissat, B. Diversity of sugar-diphospholipid-utilizing glycosyltransferase families. Commun. Biol. 7, 285 (2024).
Woodward, R. et al. In vitro bacterial polysaccharide biosynthesis: defining the functions of Wzy and Wzz. Nat. Chem. Biol. 6, 418–423 (2010).
Weckener, M. et al. The lipid linked oligosaccharide polymerase Wzy and its regulating co-polymerase, Wzz, from enterobacterial common antigen biosynthesis form a complex. Open Biol. 13, 220373 (2023).
Cuthbertson, L., Mainprize, I. L., Naismith, J. H. & Whitfield, C. Pivotal roles of the outer membrane polysaccharide export and polysaccharide copolymerase protein families in export of extracellular polysaccharides in gram-negative bacteria. Microbiol Mol. Biol. Rev. 73, 155–177 (2009).
Jadeau, F. et al. BYKdb: the Bacterial protein tYrosine Kinase database. Nucleic Acids Res. 40, D321–D324 (2012).
Yang, Y. et al. The molecular basis of regulation of bacterial capsule assembly by Wzc. Nat. Commun. 12, 4349 (2021).
Morona, R., Purins, L., Tocilj, A., Matte, A. & Cygler, M. Sequence-structure relationships in polysaccharide co-polymerase (PCP) proteins. Trends Biochem Sci. 34, 78–84 (2009).
Chao, J. D., Wong, D. & Av-Gay, Y. Microbial protein-tyrosine kinases. J. Biol. Chem. 289, 9463–9472 (2014).
Soulat, D. et al. Staphylococcus aureus operates protein-tyrosine phosphorylation through a specific mechanism. J. Biol. Chem. 281, 14048–14056 (2006).
Olivares-Illana, V. et al. Structural basis for the regulation mechanism of the tyrosine kinase CapB from Staphylococcus aureus. PLoS Biol. 6, e143 (2008).
Wugeditsch, T. et al. Phosphorylation of Wzc, a tyrosine autokinase, is essential for assembly of group 1 capsular polysaccharides in Escherichia coli. J. Biol. Chem. 276, 2361–2371 (2001).
Hagelueken, G., Huang, H., Mainprize, I. L., Whitfield, C. & Naismith, J. H. Crystal structures of Wzb of Escherichia coli and CpsB of Streptococcus pneumoniae, representatives of two families of tyrosine phosphatases that regulate capsule assembly. J. Mol. Biol. 392, 678–688 (2009).
Nadler, C. et al. Cycling of Etk and Etp phosphorylation states is involved in formation of group 4 capsule by Escherichia coli. PLoS One 7, e37984 (2012).
Obadia, B. et al. Influence of tyrosine-kinase Wzc activity on colanic acid production in Escherichia coli K12 cells. J. Mol. Biol. 367, 42–53 (2007).
Dong, C. et al. Wza the translocon for E. coli capsular polysaccharides defines a new class of membrane protein. Nature 444, 226–229 (2006).
Bechet, E. et al. Identification of structural and molecular determinants of the tyrosine-kinase Wzc and implications in capsular polysaccharide export. Mol. Microbiol 77, 1315–1325 (2010).
Alphonse, S., Djemil, I., Piserchio, A. & Ghose, R. Structural basis for the recognition of the bacterial tyrosine kinase Wzc by its cognate tyrosine phosphatase Wzb. Proc. Natl Acad. Sci. 119, e2201800119 (2022).
Whitfield, C. Biosynthesis and assembly of capsular polysaccharides in Escherichia coli. Annu Rev. Biochem 75, 39–68 (2006).
Liu, J. et al. Mutational analysis of dimeric linkers in peri- and cytoplasmic domains of histidine kinase DctB reveals their functional roles in signal transduction. Open Biol. 4, 140023 (2014).
Gushchin I., et al. Mechanism of transmembrane signaling by sensor histidine kinases. Science 356, https://doi.org/10.1126/science.aah6345 (2017).
Jura, N. et al. Mechanism for activation of the EGF receptor catalytic domain by the juxtamembrane segment. Cell 137, 1293–1307 (2009).
Endres, N. F. et al. Conformational coupling across the plasma membrane in activation of the EGF receptor. Cell 152, 543–556 (2013).
Reid, A. N. & Whitfield, C. functional analysis of conserved gene products involved in assembly of Escherichia coli capsules and exopolysaccharides: evidence for molecular recognition between Wza and Wzc for colanic acid biosynthesis. J. Bacteriol. 187, 5470–5481 (2005).
Collins, R. F. et al. The 3D structure of a periplasm-spanning platform required for assembly of group 1 capsular polysaccharides in Escherichia coli. Proc. Natl Acad. Sci. USA 104, 2390–2395 (2007).
Drummelsmith, J. Whitfield C. Translocation of group 1 capsular polysaccharide to the surface of Escherichia coli requires a multimeric complex in the outer membrane. EMBO J. 19, 57–66 (2000).
Nesper, J. et al. Translocation of group 1 capsular polysaccharide in Escherichia coli serotype K30. Structural and functional analysis of the outer membrane lipoprotein Wza. J. Biol. Chem. 278, 49763–49772 (2003).
Ovchinnikova, O. G. et al. Hypermucoviscosity regulator RmpD interacts with Wzc and controls capsular polysaccharide chain length. mBio 14, e0080023 (2023).
Guzman, L. M., Belin, D., Carson, M. J. & Beckwith, J. Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J. Bacteriol. 177, 4121–4130 (1995).
Dodgson, C., Amor, P. & Whitfield, C. Distribution of the rol gene encoding the regulator of lipopolysaccharide O-chain length in Escherichia coli and its influence on the expression of group I capsular K antigens. J. Bacteriol. 178, 1895–1902 (1996).
Punjani, A., Rubinstein, J. L., Fleet, D. J. & Brubaker, M. A. cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296 (2017).
Rohou, A. & Grigorieff, N. CTFFIND4: fast and accurate defocus estimation from electron micrographs. J. Struct. Biol. 192, 216–221 (2015).
Punjani, A., Zhang, H. & Fleet, D. J. Non-uniform refinement: adaptive regularization improves single-particle cryo-EM reconstruction. Nat. Methods 17, 1214–1221 (2020).
Liebschner, D. et al. Macromolecular structure determination using X-rays, neutrons and electrons: recent developments in Phenix. Acta Crystallogr. Sect. D. 75, 861–877 (2019).
Pettersen, E. F. et al. UCSF Chimera–a visualization system for exploratory research and analysis. J. Comput Chem. 25, 1605–1612 (2004).
Pettersen, E. F. et al. UCSF ChimeraX: structure visualization for researchers, educators, and developers. Protein Sci. 30, 70–82 (2021).
Goddard, T. D. et al. UCSF ChimeraX: Meeting modern challenges in visualization and analysis. Protein Sci. 27, 14–25 (2018).
Richard E., et al. Protein complex prediction with AlphaFold-Multimer. Preprint bioRxiv, https://doi.org/10.1101/2021.10.04.463034 (2022).
Jo, S., Kim, T., Iyer, V. G. & Im, W. CHARMM-GUI: a web-based graphical user interface for CHARMM. J. Comput Chem. 29, 1859–1865 (2008).
Abraham, M. J. et al. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1–2, 19–25 (2015).
Huang, J. & MacKerell, A. D. Jr CHARMM36 all-atom additive protein force field: validation based on comparison to NMR data. J. Comput Chem. 34, 2135–2145 (2013).
Bussi, G., Donadio, D. & Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 126, 014101 (2007).
Parrinello, M. & Rahman, A. Polymorphic transitions in single crystals: a new molecular dynamics method. J. Appl. Phys. 52, 7182–7190 (1981).
Hess, B., Bekker, H., Berendsen, H. J. C. & Fraaije, J. G. E. M. LINCS: a linear constraint solver for molecular simulations. J. Computational Chem. 18, 1463–1472 (1997).
Darden, T., York, D. & Pedersen, L. Particle mesh Ewald: an N⋅log(N) method for Ewald sums in large systems. J. Chem. Phys. 98, 10089–10092 (1993).
Michaud-Agrawal, N., Denning, E. J., Woolf, T. B. & Beckstein, O. MDAnalysis: a toolkit for the analysis of molecular dynamics simulations. J. Computational Chem. 32, 2319–2327 (2011).
Perez-Riverol, Y. et al. The PRIDE database at 20 years: 2025 update. Nucleic Acids Res. 53, D543–d553 (2025).
Chakraborty, A. K., Friebolin, H. & Stirm, S. Primary structure of the Escherichia coli serotype K30 capsular polysaccharide. J. Bacteriol. 141, 971–972 (1980).
Acknowledgements
The Rosalind Franklin Institute is funded by UK Research and Innovation through the Engineering and Physical Sciences Research Council (EPSRC). Experimental funding is provided by the Wellcome Trust through the Electrifying Life Science (220526/Z/20/Z to J.H.N.). J.L., Y.Y. are also supported by the Fundamental Research Funds for the Central Universities. C.W. was supported by a Canada Research Chair and research funding from the Canadian Institutes of Health Research (FDN-2016-148364). Mass spectrometry work is supported by Medical Research Council programme grant (MR/V028839/1) awarded to C.V.R. P.J.S.’s lab was funded by Wellcome (208361/Z/17/Z), MRC, BBSRC, EPSRC, NIH, JPIAMR and the Howard Dalton Centre. This project made use of time on ARCHER2 granted via the UK High-End Computing Consortium for Biomolecular Simulation, HECBioSim (http://www.hecbiosim.ac.uk), supported by EPSRC (grant no. EP/R029407/1). P.J.S. would like to thank the SCRTP at Warwick for use of the computing infrastructure.
Author information
Authors and Affiliations
Contributions
Y.Y. purified the proteins. Y.Y., J.L., and J.H.N. carried out an EM analysis. M.B. and P.J.S. carried out MD simulations. B.R.C., M.R.A., N.M.K. and C.W. carried out in vivo analysis. H.S. and C.V.R. carried out mass spectrometry, with the contribution from A.L.B. on sample handling. All authors contributed to the writing of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Ranajeet Ghose and the other, anonymous, reviewer for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yang, Y., Batista, M., Clarke, B.R. et al. Molecular basis for the phosphorylation of bacterial tyrosine kinase Wzc. Nat Commun 16, 3437 (2025). https://doi.org/10.1038/s41467-025-58693-7
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-58693-7
