
Abstract
Current Aβ-targeting therapeutics for Alzheimer’s disease (AD) only slow cognitive decline due to poor understanding of AD pathogenesis. Here we describe a mechanism of AD pathogenesis in which the histone methyltransferase G9a noncanonically regulates translation of hippocampal proteins associated with AD pathology. Correspondingly, we developed a brain-penetrant inhibitor of G9a, MS1262, which restored both age-related learning & memory and noncognitive functions in multiple AD mouse models. Further, comparison of AD pathology-correlated mouse proteomes with those of AD patients found G9a regulates pathological pathways that promote Aβ and neurofibrillary tangles. This mouse-to-human overlap of G9a regulated AD-associated pathologic proteins supports at the molecular level the efficacy of targeting G9a translational mechanism for treating AD patients. Additionally, MS1262 treatment reversed the AD-characteristic expression or phosphorylation of multiple clinically validated biomarkers of AD that have the potential to be used for early-stage AD diagnosis and companion diagnosis of individualized drug effects.
Similar content being viewed by others


Introduction
Alzheimer’s disease (AD), the most common form of dementia in older adults, is a neurodegenerative disorder characterized by progressive decline in cognition, memory, and emotional states1,2. The heterogeneity and multifaceted nature of AD prevents clear mechanistic understanding of disease pathogenesis, which hinders development of effective therapy. The amyloid-beta (Aβ) hypothesis has guided current approaches to AD therapeutics3. However, Aβ-targeting drugs showed limited specificity and efficacy toward AD in the clinic because (i) the primary drug effect is slowing cognitive decline at the earliest AD stages, but there are no biomarkers to conclusively diagnose early-stage AD, (ii) drug effects were determined primarily based on dose-dependent reductions in plaques measured by positron emission tomography (PET), yet large amounts of amyloid plaques are apparently present in healthy, non-demented individuals4, and (iii) no biomarkers other than Aβ plaque size are available to measure effects of these drugs on AD progression. Meanwhile, multiple pathological brain alterations other than formation of Aβ plaque and neurofibrillary tangles (NFTs) occur during AD pathogenesis, most of which are not readily measurable due to lack of corresponding biomarkers5. Thus, new mechanism-based drugs are urgently needed for both effective medication of AD and for derivation of new biomarkers to precisely assess drug effects.
Despite the identification of a few AD-related genetic risk factors6, the exact etiology of AD pathogenesis remains obscure. The histone methyltransferases G9a (EHMT2) and G9a-like protein GLP (hereafter G9a will represent both proteins in their functional dimerized form)7,8 were among the histone-modifying enzymes associated with behavioral abnormalities9. Elevated activity of G9a exists in post-mortem brain tissues from AD patients and familial AD (5xFAD) mice7, which implicated G9a activity in AD pathogenesis. In AD, G9a functioned as an epigenetic (transcriptional) suppressor by catalyzing the dimethylation of lysine 9 of histone 3 at specific genes associated with synaptic transmission such as glutamate receptor genes7. Recently, Johnson et al. revealed that AD has a proteopathic nature or is a proteomic disease10, i.e., AD pathology and cognitive decline strongly correlated with altered expression of a broad spectrum of proteins. However, the canonical transcriptional silencing function of G9a did not explain how expression or post-translational modification of specific proteins is regulated in AD in which G9a was constitutively active. That is, the G9a-regulated mechanism responsible for AD-related proteomic alterations (‘proteopathology”) has yet to be established.
Using our chromatin activity-based chemoproteomics (ChaC)-mass spectrometry (MS) approach11 with UNC0965, a biotinylated version of a G9a inhibitor12, to dissect G9a pathways in AD, we revealed a noncanonical G9a-regulated translation mechanism that contributes to AD proteopathology. Particularly, we detected increased interaction between G9a and several regulators of N6-methyladenosine (m6A) modification, including METTL3, an RNA methyltransferase that catalyzes m6A modification of select mRNAs in AD13,14,15. Based on this G9a-regulated mechanism of proteopathology a new AD therapeutics was developed with a brain-penetrant inhibitor of G9a on multiple AD mouse models. In parallel, we conducted an AD pathology-correlated multiomics analysis to discover a MS1262-reversed, AD mouse-to-patient conserved proteome where multiple clinically validated biomarkers of AD were also identified. Our mechanistic findings show that G9a regulates translation or post-translational modifications (phosphorylation) of a broad range of proteins associated with AD pathology, which putatively suggests that targeting G9a can provide an effective therapeutic for AD patients in the clinic.
Results
In our overall experimental design (Fig. 1A), in vivo ChaC-MS dissection of AD brain tissues revealed a G9a-mediated translational mechanism that limits or reverses AD progression and pathogenesis. Using three AD mouse models, including 5.5-month-old 5xFAD, 5.5-month-old APPNLGF KI16, and 12.25-month-old 3xTg mice that recapitulate age-induced memory deficits17,18 we linked MS1262 treatment-induced improvements in behavior/synaptic function to multiomics changes in the hippocampus m6A epitranscriptome, proteome, and phosphoproteome. Further, we compared these AD mouse data with human AD patient data to identify mouse-to-human conserved changes reversed by MS1262. These results enabled us to ascertain the mechanistic role of G9a activity in AD pathogenesis. Consequently, new protein biomarkers are mechanistically derived to evaluate the drug effects on individual patients for precision medication of AD.

A The overall design for deciphering G9a-mediated AD pathogenesis and the mechanism of G9a-target drug action. B ChaC-MS revealed a noncanonical function of G9a in the translational regulation of AD pathogenesis. The networks shared by 139 G9a (EHMT2) interactors that were identified in at least 3 of 4 ChaC experiments from 5xFAD mouse hippocampus and AD organoids (human). (See also Figures. S1-S3 & S6; Data S1A–C).
Constitutively active G9a regulates a translation mechanism of AD pathogenesis
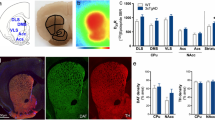
We used the biotin-tagged G9a inhibitor UNC0965 with label-free quantitation (LFQ)19,20 ChaC-MS12 to identify G9a-interacting proteins in the hippocampus of the 5xFAD mice and cerebral organoids derived from an AD iPSC line F033K with the amyloid precursor protein (APP) V717I mutation21. The samples from 5xFAD and WT mice were collected at 3.5 months and 5.5 months. The organoid groups at 88 and 102 days represented early AD (MCI) pathology. Based on LFQ ratios (log2 ≥ 1, t-test: p value ≤ 0.05) that are proportional to the relative binding of individual proteins to G9a, UNC0965 ChaC identified 100 proteins that had enhanced interaction with G9a in the hippocampus of 5xFAD mice and 182 in MCI/AD organoids. Principal component analysis identified a few clusters of G9a interactors. The clusters of G9a interactors from healthy human organoids compared to AD organoids were distinctly separated. The pulldown products from WT mice using UNC0965 formed tighter clusters than those from 5xFAD or 5xFAD MS1262-treated mice, likely due to varying levels of AD progression in 5xFAD mice and their responses to MS1262 (Figure. S1, Supplementary Data S1A, B). We identified heightened H3K9me2 in the dentate gyrus (DG) of both 5xFAD and APPNLGF mice (Figure. S2A–F). In parallel MS1262 treatment reduced Aβ plaque sizes, although this reduction was not highly statistically significant (Figure. S2G–I). These results indicated the high specificity of UNC0965 ChaC-MS to dissect AD heterogeneity with mixed cell types, i.e., UNC0965 captured G9a interactors specifically from AD-related cells that had aberrant G9a activity and associated Aβ plaques.
The predominant functional networks (mapped by STRING)11 overrepresented by 65 conserved G9a interactors in both 5xFAD hippocampus and the MCI/AD organoids were primarily associated with major translational or post-translational processes such as alternative splicing, RNA modification and processing, translation initiation and elongation, ribosome biogenesis, and protein degradation (proteostasis22) (Fig. 1B, Supplementary Data S1C). Particularly, UNC0965 ChaC-MS identified most known cofactors of METTL3, including HNRNPA2B1, which showed enhanced interaction with G9a in AD-related samples. Jiang et al. reported that progression of tauopathy was mediated by interaction of tau with HNRNPA2B1 and m6A RNA23. Similarly, the ChaC-identified interactions of G9a with the METTL3-HNRNPA2B1 translation machinery24 implicated a function of G9a in translation associated with tauopathy or AD pathology. More broadly, the combined ChaC-MS data predicted that, via AD-phenotypic interactions with key regulators of m6A translation such as HNRNPA2B1 and other regulators of ribosomal biogenesis, G9a has a noncanonical (nonepigenetic) function in translational and post-translational regulation of AD pathogenesis.
Also, to determine the clinicopathological relevance of these ChaC findings of the 65 G9a interactors in mouse AD and human AD organoids (listed in Supplementary Data S1A), we used our multiomics approach12 to retrospectively analyze the mRNA expression of the G9a interactors in blood samples from MCI and AD patients compared to control samples from the AddneuroMed Cohort (batch 1) study (GEO Accession: GSE63060)25. We identified 10 and 7 G9a interactor mRNAs that had interaction-correlated overexpression patterns26 in the blood of 80 MCI and 145 AD patients, respectively, compared with a healthy population (n = 104) (Fig. S3, panels A & B). These results indicated select mRNAs although showing small changes in expression are translated into functional proteins as the MCI/AD-characteristic G9a interactors.
MS1262 is a highly potent, selective, brain-penetrant inhibitor of G9a and GLP
We previously discovered potent and selective G9a/GLP inhibitors UNC0642 and UNC147927. We and others also reported that the replacement of the quinazoline core with the quinoline core led to higher inhibitory potency toward G9a and GLP, likely due to higher basicity of N-1 of the quinoline core that is crucial for G9a binding28,29,30. Based on these findings, we designed and synthesized MS1262, a novel G9a/GLP inhibitor that contained the quinoline core substituted with a morpholino group at the 2-position (Fig. 2A). In biochemical assays, MS1262 (G9a: IC50 = 19 ± 14 nM; GLP: IC50 = 6 ± 1 nM) showed 23-fold and 37-fold higher potency than UNC0642 (G9a: IC50 = 433 ± 6 nM; GLP: IC50 = 223 ± 46 nM) for G9a and GLP, respectively (Fig. 2B). Using isothermal titration calorimetry, we confirmed that MS1262 had high binding affinities to G9a (Kd = 74 ± 10 nM) and GLP (Kd = 19 ± 5 nM) (Fig. 2C); these affinities were approximately 3-fold higher than the binding affinities of UNC0642 (G9a: Kd = 230 ± 17 nM, GLP: Kd = 62 ± 16 nM)30. Moreover, MS1262 displayed excellent selectivity for G9a and GLP, lacking significant inhibition ( < 20% at 1 µM) for any of the 21 other methyltransferases tested (Fig. 2D). Similarly, MS1262 effectively reduced histone H3 lysine 9 dimethylation in cells in a concentration-dependent manner, indicating the strong intracellular inhibitory activity of MS1262 (Fig. 2E). Importantly, MS1262 showed good brain penetration in an in vivo mouse pharmacokinetic study. Following a single 5 mg/kg intraperitoneal (i.p.) injection of MS1262 in C57BL/6 mice, MS1262 displayed brain/plasma ratios of 0.64 and 0.73 at 2 and 4 hours post-injection (Fig. 2F). Taken together, these results indicate that MS1262 is a highly potent, selective, brain penetrant G9a/GLP inhibitor suitable for in vivo efficacy studies in AD mouse models.

A Discovery of MS1262. B Concentration-dependent inhibition of G9a (left) and GLP (right) by MS1262 in G9a and GLP enzymatic assays. UNC0642 was used as a control. Data shown are the mean ± SD from four independent experiments. C ITC titrations of MS1262 into G9a (top) and GLP (bottom). The calculated values represent the means ± SD from two independent experiments. D Activity of MS1262 against 21 other methyltransferases at 1 µM. Data are the means ± SD from two duplicate experiments. E Concentration-dependent reduction of the H3K9me2 level by MS1262 in K562 cells. K562 cells were treated with MS1262 at the indicated concentrations for 48 h. Western blot results are representative from at least two independent experiments. F Plasma and brain concentrations of MS1262 over 4 h following a single 5 mg/kg intraperitoneal injection of MS1262 in mice. Data shown are the mean ± SD from three tested mice per time point.
MS1262 inhibition of G9a activity rescues AD-related deficits in behavior and synaptic function
The 5xFAD and APPNLGF mice experience memory and affective behavioral deficits that progressively worsen with age31,32,33,34. Previously, we showed that 5xFAD mice exhibited a series of hippocampus-dependent memory and affective deficits, including impaired spatial memory in the novel place recognition (NPR) test, elevated innate anxiety in the open field and zero maze tests, and increased depression-like behavior in the forced swim test35. Therefore, we assessed these behaviors in response to long-term, intermittent G9a inhibition by MS1262 (Fig. 3A and B). Locomotion in an open field was unaltered between wild-type (WT) controls and either 5xFAD or APPNLGF mice in response to MS1262 or vehicle treatment (Fig. 3C and H), indicating that G9a inhibition did not affect locomotion. For the NPR test to assess spatial memory, animals treated with MS1262 showed a significant preference for the object in the novel location measured by the discrimination ratio (5xFAD: p < 0.0001, APPNLGF: p < 0.001), indicative of improved spatial memory (Fig. 3D and I). Notably, the discrimination ratio of MS1262-treated mice was rescued to the level of age-matched WT animals for both AD mouse models (Fig. 3D and I). 5xFAD mice were also tested in the Barnes maze as an additional spatial memory measure (Figure. S4A, B). 5xFAD animals treated with MS1262 displayed significantly faster escape latencies than 5xFAD animals that received vehicle treatment, further demonstrating improved spatial memory (Figure. S4C). Notably, 5xFAD—MS1262 mouse performance in the Barnes maze were similar to those of WT control animals (Figure. S4C). For open field and zero maze tests to assess anxiety-like behavior, MS1262 treatment showed no effects on the amount of time 5xFAD mice spent in the center of an open field (Fig. 3E) but MS1262 treatment increased the time APPNLGF mice spent in the center of the open field as compared with both wildtype and vehicle-treated controls (Fig. 3J). MS1262 treatment further increased the time 5xFAD and APPNLGF mice spent in open arms in the zero maze, bringing them closer to wild-type control animals (Fig. 3F and K). These results suggest G9a inhibition has anti-anxiolytic effects in multiple AD mouse models. MS1262-treated animals also spent less time immobile in the forced swim test compared with vehicle-treated animals, suggestive of less depression-like behavior (Fig. 3G and L). Together, these results suggest that G9a inhibition effectively rescued both learning and memory as well as affective deficits in these AD mouse models.

A Experimental timeline for drug administration and novel place recognition test. B Depiction of the paradigm used to test memory and affective related behavior. C Locomotion in an open field was unaffected under wild-type, 5xFAD, vehicle, and chronic MS1262 treatment. D Preference for the novel-located object during retrieval was significantly reduced in 5xFAD mice compared with wild-type controls and was completely rescued by chronic MS1262 treatment, as measured by discrimination ratio (see “Methods” for calculations). E Time spent in the center of an open field was unaffected under wild-type, 5xFAD, vehicle, and chronic MS1262. F MS1262 administration rescued anxiety-like behavior in 5xFAD mice back to wild-type levels demonstrated by increased time spent in the open arms of a zero maze. G Depressive-like behavioral deficits in 5xFAD mice were rescued to wildtype levels after chronic MS1262 treatment as measured by immobile time during the forced swimming paradigm. H Locomotion in an open field was unaffected under wildtype, APPNLGF, vehicle, and chronic MS1262 treatment. I Preference for the novel-located object during retrieval was significantly reduced in APPNLGF mice compared to wildtype controls and was rescued by chronic MS1262 treatment as measured by discrimination ratio. J Time spent in the center of an open field was unaffected under wildtype, APPNLGF, while chronic MS1262 treatment led to an increase in time spent in the center. K MS1262 administration rescued anxiety-like behavior in APPNLGF mice back to wildtype levels demonstrated by increased time spent in the open arms of a zero maze. L Depressive-like behavioral deficits in APPNLGF mice were rescued to wildtype levels after chronic MS1262 treatment as measured by immobile time during the forced swimming paradigm. Data are visualized as mean +/- SEM with each individual displayed as a point. Wildtype (C57BL/6 J) (n = 7), 5xFAD (n = 10), 5xFAD vehicle (n = 14), 5xFAD MS1262 (n = 12) and Wildtype (C57BL/6J-Aw-J/J) (n = 7), APPNLGF vehicle (n = 10), APPNLGF MS1262 (n = 10), mice were utilized for behavioral studies. Significance was assessed by ANOVA and two-sided Tukey’s posthoc test between each condition. ns = not significant, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. (See also Figures. S4 & S5 & Data S3).
Since 5xFAD and APPNLGF mice are accelerated Aβ deposition models of AD, we also utilized 12.25-month-old 3xTg mice, which are a slower onset model of AD that is characterized by both Aβ plaque deposition and tau tangle formation and better accounts for the natural physiological aging process17,18. 3xTg mice also display behavioral deficits associated with memory loss and affective behavior dysfunction. We assessed the behavior of 12.25-month-old 3xTg mice that received 6-week intermittent G9a inhibition by MS1262 in the open field, NPR, Barnes maze, elevated zero maze, and forced swim test (Figure. S5A, B–H and I). Locomotion in an open field was unaltered between 3xTg mice in response to MS1262 or vehicle treatment (Figure. S5C), indicating that G9a inhibition did not affect locomotion. For the NPR test assessing spatial memory, 3xTg animals treated with MS1262 showed a significant preference for the novelly-located object measured by the discrimination ratio compared to vehicle treated controls (p < 0.01), indicative of improved spatial memory (Figure. S5D). 3xTg mice were also tested in the Barnes maze as an additional spatial memory measure. 3xTg animals treated with MS1262 displayed significantly faster escape latency than 3xTg animals that received vehicle treatment, further demonstrating improved spatial memory (Figure. S5J). For open field to assess anxiety-like behavior, MS1262 treatment showed no effects on the amount of time 3xTg mice spent in the center of an open field (Figure. S5E). However, the MS1262-treated 3xTg mice spent significantly more time in open arms of the zero-maze compared with vehicle-treated control animals. (Figure. S5F). These results suggest the anti-anxiolytic effects by G9a inhibition in AD mice. For the forced swim test to assess depression-like behavior, MS1262-treated 3xTg animals spent less time immobile compared with vehicle-treated animals, suggestive of less depression-like behavior (Figure. S5G). Together, these results suggest that G9a inhibition effectively rescued learning, memory, and affective deficits in the 3xTg AD mouse model that displays both plaque and tau tangle pathology, further implying broad applicability of effective AD treatment by MS1262.
Additionally, to assess the effect of MS1262 treatment on synaptic transmission and intrinsic properties of hippocampal cells, we recorded evoked action potential and pharmacologically isolated spontaneous excitatory postsynaptic currents (sEPSCs) of hippocampal dentate granule cells (GCs) from vehicle- and MS1262-treated 5xFAD mice. The rationale of selecting dentate gyrus GCs for recording was based upon our previous findings showing that the hippocampus-dependent behaviors mentioned above were dentate gyrus dependent36. No significant differences were observed between vehicle and MS1262-treated animals in membrane capacity, input resistance, resting membrane potential, and intrinsic excitability (Fig. 4A-D). Interestingly, while sEPSC amplitude was unaffected, the frequency of sEPSCs recorded from dentate GCs in vehicle-treated 5xFAD mice was significantly decreased compared to WT control mice, and was reversed by MS1262 treatment (Fig. 4F-I). These results suggested that long-term G9a inhibition by MS1262 in 5xFAD mice increased excitatory glutamatergic synaptic transmission onto the dentate GCs. With the improvement in learning and memory, as well as increased GC sEPSC frequency, we thought to interrogate hippocampal long-term potentiation (LTP), which has been shown to be essential for the consolidation of spatial memory37,38,39. LTP in the CA1 of the hippocampus, an electrophysiological measure of synaptic functioning for learning and memory was lost in 5xFAD mice compared to wildtype animals, and completely restored after intermittent MS1262 treatment (Fig. 4J-K). These results suggested that long-term G9a inhibition by MS1262 in 5xFAD mice increased excitatory glutamatergic synaptic transmission onto the dentate GCs and restored healthy LTP, which results in rescued spatial memory function after MS1262 treatment.

A Quantification of membrane capacity between WT mice and 5xFAD mice treated with vehicle and MS1262 (n = 15/21/22 cells for WT/Vehicle/MS1262) ANOVA followed by Tukey’s test visualized as mean ± SEM. B Quantification of input resistance between WT mice and 5xFAD mice treated with vehicle and MS1262 (n = 15/21/22 cells for WT/Vehicle/MS1262) ANOVA followed by Tukey’s test visualized as mean ± SEM. C Quantification of resting membrane potential between WT mice and 5xFAD mice treated with vehicle and MS1262 (n = 15/21/22 cells for WT/Vehicle/MS1262) ANOVA followed by Tukey’s test visualized as mean ± SEM. D Mean ( ± SEM) number of action potential elicited in response to increasing step current from dentate granule cells of WT mice and 5xFAD mice injected with vehicle (black) or MS1262 (blue). (n = 15/21/22 cells for WT/Vehicle/ MS1262. ANOVA followed by Tukey’s test. E Representative traces of sEPSCs recorded from dentate granule cells derived from WT mice and 5xFAD mice injected chronically with vehicle (left) or MS1262 (right). F, G Cumulative probability (f) and average amplitude distribution (g) of sEPSCs (WT, n = 15 cells from 3 mice, 5xFAD-Vehicle, n = 16 cells from 3 mice; 5xFAD-MS1262, n = 15 cells from 3 mice) ANOVA followed by Tukey’s test visualized as mean ± SEM. H, I Cumulative probability distribution (h) and average frequency (i) of sEPSCs (WT, n = 15 cells from 3 mice, 5xFAD-Vehicle, n = 16 cells from 3 mice; 5xFAD-MS1262, n = 15 cells from 3 mice. P = 0.0004, Kruskal-Wallis test, Dunn’s test for multiple comparisons p < 0.0001 and p = .0453) visualized as mean ± SEM. J, K fEPSP slope (J) as measured as a percent of baseline and LTP visualized in a bar graph (k). (WT, n = 8 slices from 3 mice, 5xFAD-vehicle, n = 4 slices from 3 mice; 5xFAD-MS1262, n = 9 slices from 4 mice. ANOVA followed by Tukey’s test *p < 0.05, ****p < 0.0001 Data are visualized as mean ± SEM. (See also Data S3).
G9a activity regulates translational machinery, including m6A-mediated translation, to contribute to AD-related protein networks
To dissect a G9a-mediated mechanism of AD pathogenesis and the corresponding action mechanism of MS1262 reversal of AD symptoms, we performed multiomics experiments, including ChaC-MS, m6A RNA immunoprecipitation (MeRIP)-seq, and tandem mass tag (TMT)-based quantitative proteomics/phosphoproteomics19,20 using microdissected hippocampal samples from the same mouse cohorts used for the aforementioned behavioral studies (Fig. 3 and Fig. 4). These samples included AD mouse lines (5xFAD or APPNLGF KI) and age-matched wild-type mice with or without MS1262 treatment. Because the hippocampus is the primary brain area affected by AD, the MS1262-induced changes in G9a binding or changes in the hippocampus m6A epitranscriptome or proteome/phosphoproteome revealed AD-related, G9a-associated pathways and reflected the inhibitor effects on AD pathogenesis at the molecular level.
First, ChaC-MS analysis revealed that MS1262 treatment reversed G9a binding to most regulators of translation, RNA processing, and ribosome biogenesis (correlation coefficient = -0.483, Figure. S6 and Supplementary Data S1C). Meanwhile, MS1262 reversed AD-related binding of G9a to major translation regulators, which confirmed the dependence of G9a translational function on its activity in AD. Specifically, most of ChaC-identified G9a-interacting m6A regulators with translation functions were implicated in AD or neurodegenerative diseases. For example, increased m6A writer (METTL3, RBM15) and decreased m6A eraser (FTO, ALKBH5) expression were reported along with an overall increase in m6A modification with age/AD-progression in the hippocampus and cortex of various mouse models (APP/PS1, APPNL-G-F/MAPTP301S) and AD patients13,15,23,40. Similarly, oligomeric tau was shown to complex with m6A-modified transcripts through HNRNPA2B1, a m6A reader, to regulate translation and promote neurodegeneration23. In addition, MS1262 inhibition also reversed AD-characteristic G9a interactions with proteins involved in postsynaptic neurotransmitter receptor internalization such as Rabphilin‐3 A (Rph3A), a synaptic vesicle protein41, Mecp242,43, and clathrin-dependent endocytosis44. Thus, combined ChaC results revealed that G9a activity associates m6A translation regulatory complexes with AD-dysregulated synaptic processes.
Because of our recent finding that G9a is the upstream regulator of METTL3/m6A-mediated translation axis in chronic inflammatory conditions45 we utilized MeRIP-seq to investigate whether/how inhibition of G9a activity by MS1262 treatment affects the genome-wide, AD-characteristic m6A distribution. As expected, we observed a global increase in m6A modification in the hippocampus of 5XFAD mouse line, compared to age-matched wild-type controls (Fig. 5A, Supplementary Data S2A–B). Specifically, increased m6A was observed around the 5’-UTR and coding sequence (CDS) of transcripts with concomitant reduction around the 3’-UTR region (Figure. S7A). Notably, G9a inhibition reversed AD-driven changes to m6A methylome, as evidenced by negative overall correlation of -0.59 (Fig. 5B), with MS1262 treatment reducing m6A around the 5’-UTR and CDS of AD-dysregulated transcripts with concomitant increase around the 3’-UTR (Figure. S7A). Overall, these AD-dysregulated, MS1262-reversed m6A-tagged transcripts are primarily involved in synaptic signaling and brain development (Fig. 5C, S7B & Supplementary Data S2C) and are also in various central/peripheral nervous system disorders (Figure. S7C). Importantly, these MeRIP-seq results indicated that AD-activated G9a is an upstream regulator of the m6A-mediated translation of AD-related neuropathologic processes.

A Violin plot showing an increase in m6A level in the hippocampus of 5x-FAD mice compared to age-matched wildtype controls (AD vs. WT), which is reversed upon MS1262 treatment (AD-tr vs. AD). Hippocampus tissue from four wild type (WT) mice, four 5x-FAD (AD) mice, and four 5x-FAD mice treated with MS1262 (AD-tr) was used for m6A sequencing. A two-tailed paired t-test (****p < 0.0001) was used, with the number of transcripts (n = 3172) for which the m6A modification ratio could be calculated for both comparisons indicated at the top. B A three-way comparison showing that MS1262 treatment reverses AD-mediated changes in the m6A methylome of hippocampus. Hippocampus tissue from four wild type (WT) mice, four 5x-FAD (AD) mice, and four 5x-FAD mice treated with MS1262 (AD-tr) was used for m6A sequencing. Weighted fold-change, from all m6A peaks mapping to a gene, is shown for AD vs WT (x-axis) and AD-tr vs AD (y-axis) mice. Select transcripts with higher (red, n = 461) or lower (blue, n = 57) m6A-modification in the hippocampus of AD mice, compared to age matched wildtype controls, that was reversed following MS1262 treatment are highlighted. Linear regression showing overall trend (blue line with 95% CI highlighted by grey shading on either side), Pearson correlation (\(R\)) and associated P-value are shown. (C) Enrichment analysis for AD-dysregulated, MS1262 reversed, m6A modified transcripts highlighted in (B). Top 20 terms with significant over-representation are shown, with grey cells indicating a lack of significant enrichment. P-values were calculated using cumulative hypergeometric distribution followed by Benjamini-Hochberg correction to account for multiple testing. (See also Figure. S7 & Data S2).
In parallel, we performed AD-correlated proteomic and phosphoproteomic experiments using micro-dissected hippocampus tissue from a large cohort of 5xFAD mice exhibiting mid/late-stage AD (C57BL/6 J; n = 4) with or without MS1262 treatment, along with age matched wild type controls (C57BL/6 J; n = 3), resulting in identification of 7899 proteins and 14,788 phosphorylated sites (in 4262 protein groups) in total. Similar experiments in a second mouse model of AD, i.e., wild type (C57BL/6J-Aw-J/J; n = 3) mice versus APPNLGF KI (C57BL/6J-Aw-J/J; n = 4) mice with or without MS1262 treatment identified 6576 proteins and 13,6687 phosphorylated sites (in 3854 protein groups) across all hippocampal samples (Fig. 6A and Supplementary Data S1D). Principal component analysis showed not only good separation of wild-type, AD (5xFAD/APP-NLFG), and MS1262-treated AD (5xFAD/APP-NLFG) samples but also showed high quantitative reproducibility between biological replicates (Figure. S8A).

A Venn diagrams summarizing total number of proteins and phospho-sites identified in 5x-FAD and APP-NLGF mouse models in this study. Briefly, global/phospho-proteomic experiments were performed using proteins extracted from hippocampus of age matched wild-type controls (WT; n = 3), 5x-FAD/APPNLGF mice at mid/late-stage Alzheimer’s (AD; n = 4), and AD mice treated with MS1262 (AD-tr; n = 4). Samples were TMT-labeled and run in quadruplicate. B Bar charts summarizing number of proteins (global proteomics) and phospho-sites (phosphor-proteomics) showing statistically significantly (log2(FC) ± 0.2; p < 0.05; two-tailed unpaired Student’s t test) changes in comparison of AD vs WT and AD-MS1262 treated vs AD mice. C Heatmap summarizing pathway activity z-scores, calculated by Ingenuity Pathway Analysis (IPA) using differentially regulated proteins and phospho-sites shown in (B) for indicated comparisons (AD vs WT & AD-MS1262 treated vs AD) in 5x-FAD and APP-NLFG mice. MS1262 treatment reverses/ameliorates pathway activity changes seen in both mouse models of AD, when compared to age matched wild-type (WT) controls. D Heatmap summarizing MS1262-affected diseases and functions based on dysregulated proteins/phosphosites shown in (B). Z-scores were calculated using Ingenuity Pathway Analysis (IPA). E The network of indicated signaling pathways (see also Figures. S8-S9 & Data S1D–E).
In line with AD-characteristic, MS1262-reversed m6A distribution (Fig. 5B), we observed increased expression of m6A writers (e.g., METTL3), reduction in m6A erasers (e.g., FTO) and dysregulation of several m6A readers (e.g., HNRNPA2B1) in the hippocampus of 5xFAD and/or APP-NLGF mice, compared to age matched controls; with MS1262 treatment reversing AD-dysregulated expression of several m6A regulators (Figure. S8B). In addition, approximately 47% (5xFAD) and 42% (APP-NLGF) of proteins that showed AD-characteristic dysregulation that was reversed following MS1262 treatment, classified as ‘AD/G9a-coregulated’ proteins, were translated from m6A-tagged RNAs (Figure. S8C) further confirming the function of constitutively active G9a in regulating translation of AD-related proteins.
Overall, 9764 entities (global = 1346; phosphosites = 8418; log2FC\(\pm\) 0.2 and P < 0.05) were differentially regulated (Fig. 6B), with two major clusters of proteins whose expression or phosphorylation levels were either up- or downregulated in 5xFAD and/or APP-NLGF mice compared to age matched controls; a pattern that was reversed following MS1262 treatment (Figure. S8E and Supplementary Data S1D). Because MS1262 specifically inhibited AD-activated G9a, proteins with MS1262-reversed expression or phosphorylation were classified as ‘AD/G9a-coregulated’. Notably, these AD/G9a-coregulated proteins showed more pronounced phosphorylation changes than their expression changes, which confirmed that G9a activity broadly regulates AD pathogenesis at the translational or post-translational (i.e., phosphorylation) levels. Further, we estimated pathway activity scores to find AD-dysregulated (i.e., AD versus wild type) and MS1262-reversed (i.e., treated AD versus AD) biological processes/pathways in the hippocampus of 5xFAD and APPNLGF mice (Fig. 6C, S9 & Supplementary Data S1E). Notably, most AD/G9a coregulated proteins that were conserved in both 5xFAD and APPNLGF KI mice overrepresented the pathways closely associated with dysregulated synaptic functions (Fig. 6D, S9B–E & Supplementary Data S1D). As examples, MS1262 suppressed AD-characteristic expression of proteins associated with synaptic structure and function, such as NRP146,47, tenascin-c48, and SHANK349 (Figure. S8E). Also, two of these AD/G9a-coregulated proteins were microtubule-associated protein products of MAPT genes (Figure. S8D)50,51. We observed that MS1262 treatment suppressed Tau peptide expression that is characteristically increased in AD brains. Because NFTs composed of abnormally hyperphosphorylated Tau are a hallmark of AD52, this result demonstrated the specificity of MS1262 in targeting AD tauopathy.
G9a-associated pathways mechanistically contribute to both cognitive and non-cognitive symptoms of AD
Broadly on the basis of MS1262-induced phosphorylation changes, we observed that MS1262 reactivated the interactive pathways involved in calcium signaling, CREB signaling in neurons, and synaptogenesis signaling (Fig. 6E), which are suppressed during AD pathogenesis53,54,55. Specifically, SHANK3 is a scaffolding protein in the postsynaptic density; phosphorylated forms of SHANK3 have different synaptic properties56,57. We found that MS1262 reversed CaMKII-mediated phosphorylation of SHANK357 and another synaptic protein GluN2A-subunit-containing NMDAR58 (Figure. S9C) in correlation with improved cognition and rescued synaptic function of MS1262-treated AD mice (Figs. 3 and 4). In addition, other AD-suppressed, MS1262-reactivated signaling pathways (Fig. 6C) included i) the CDK5 pathway that has critical functions in synaptic functions and memory consolidation59 (Figure. S9D), ii) Dopamine- and cAMP-regulated phosphoprotein (DARPP-32) that robustly integrates dopamine and glutamate signals involved in synaptic plasticity and long-term memory60 (Fig. 6C & S9D), iii) Opioid signaling whose activation limited the production of Aβ61,62 (Figure. S9D), iv) actin-based motility signaling that is associated with synaptic loss63,64, and v) the signaling involving the SNARE complex, oxytocin in spinal neurons, and endocannabinoid system that contributes to neurodegeneration and associated impairments in learning, memory, and cognition65 (Figs. 6C, 6E & S9).
In correlation with the observation that injection of oxytocin reversed some damages caused by amyloid plaques66, MS1262 treatment reactivated oxytocin in spinal neurons or brain by reversing the phosphorylation states of proteins related to synaptic plasticity and memory formation67. Also, MS1262 treatment upregulated protein components of brain pyrimidine biosynthesis and pyrimidine salvage pathways that are essential for synapses production68 (Figs. 6C, 6E & S9). Other proteins abnormally expressed in AD and restored to normal levels by MS1262 were gonadotropin releasing hormone, a neuropeptide central regulator of neurogenic and neuroprotective functions and an activator of cAMP-mediated signaling69,70, and G-protein coupled receptors involved in numerous key neurotransmitter systems in the brain that were disrupted in AD71,72 (Fig. 6C & S9). Similarly, MS1262 reversed activation states of numerous AD-associated signaling pathways (Fig. 6C, S9A & S9D), including (i) HIF1a signaling related to microglia dysfunction in AD73, (ii) Granulocyte-macrophage colony-stimulating factor contributing to AD-related neuroinflammation74, (iii) Triacylglycerol biosynthesis75, (iv) RHOGDI that regulates the actin cytoskeleton and spine dynamics76, (v) Impaired PI3K-AKT-GSK-3beta-mTOR pathway in AD-associated microglia77, and (vi) Mitochondrial dysfunction78,79 (Figure. S9D-E).
In parallel with MS1262-reduced noncognitive AD-like neuropsychiatric behaviors such as depression and anxiety (Fig. 3), we identified numerous phosphoproteins with MS1262-reversed phosphorylation involved in Relaxin signaling that contains markers of depression in AD80 (Fig. 6C & S9D). Moreover, MS1262 reinstated gustation pathways whose dysregulation was indicative of impaired sensory systems81,82 because auditory and visual measures were used to detect prodromal AD83 (Figure. S9D).
In sum, our proteomic/phosphoproteomic analysis identified the proteins showing AD-regulated, MS1262-reversed expression or phosphorylation in multiple interconnected pathways that synergistically contribute to synaptic plasticity or/and synaptic transmission and whose deficits in multiple AD mouse models were rescued by MS1262. Simultaneous effects of MS1262 treatment on these pathways demonstrated that G9a is an upstream translation regulator of proteins that contribute to AD pathology and associated AD symptoms.
MS1262 reversed protein expressions or phosphorylation that mark early-stages of AD
To determine the clinical relevance of G9a-translational mechanism that regulates AD proteopathology, and to predict MS1262 efficacy for treating AD patients, we compared our proteomic and phosphoproteomic data from non-treated versus MS1262-treated AD mice with the proteomic data of different cohorts of AD patients. We first examined the proteomic data of brain autopsy samples84 from (i) control individuals with low pathology of plaques and tangles, (ii) controls with high Aβ pathology but no detectable cognitive defects, (iii) MCI persons with Aβ pathology and slight but measurable defect in cognition, and (iv) AD patients. As shown in Fig. 7A, the expression of eighteen proteins including MAPT that mark AD patients was reversed in MS1262-treated AD mice. For example, like the effect of IL-33 injection that reversed cognitive deficits in APP/PS1 mice85, MS1262 inhibition of G9a upregulated IL-33 expression. Notably, multiple MS1262-affected proteins were the known biomarkers of AD, e.g., ADCYAP1 was identified as a diagnostic biomarker of AD with high discriminatory ability (AUC = 0.850) and validated in AD brains (AUC = 0.935). PBXIP1 is a CSF marker of AD. In addition, we found that MS1262 reversed phosphorylation of the protein products of 33 AD-risk genes86 (Figure. S9C).

A Heatmap of select AD-risk markers showing patient-correlated protein expression in the hippocampus of 5x-FAD & APPNLGF mice whose expression was reversed following MS1262 treatment. Global proteomics data from three age-matched wild-type controls (WT), four 5x-FAD/APPNLGF mice at mid-/late-stage Alzheimer’s (AD), and four AD mice treated with MS1262 (AD-tr), with each sample run in quadruplicate. Rows are clustered into three groups based on pattern of inhibitor effect: (1) “down_up” = markers down in AD mice, compared to WT controls, whose expression is back up following MS1262 treatment, (2) “up_down” = markers whose expression increases in AD mice, compared to WT controls, but is back down upon treatment, and (3) “not_affected” = inhibitor did not affect expression of these markers. The left annotation column (mouse_model) shows the proteomics dataset (orange = 5x-FAD, green = APPNLGF) a marker belongs to. Right annotation columns [log2(AD/WT) & log2(AD-tr/AD)] show fold change for indicated comparisons. Rightmost heatmap shows AD-marker expression in 39 patients from the Banner Sun cohort (LPC = 12, HPC = 6, MCI = 6, and AD = 15) that were pooled and measured in duplicate using TMT-LC-MS/MS. LPC = controls with low pathology of plaques and tangles; HPC = controls with high Aβ pathology but no detectable cognitive defects; MCI = mild cognitive impairment with Aβ pathology and a slight but measurable defect in cognition; AD = late-stage AD with high pathology scores of plaques and tangles; FC = fold change. B MS1262 reversed cerebrospinal fluid (CSF) proteome of early-stage AD. Heatmap showing AD markers (identified from CSF of symptomatic/non-symptomatic AD patients) dysregulated in 5xFAD and APPNLFG mice, compared to DMSO treated wild-type controls, whose expression is reversed following MS1262 treatment. The annotation column on right indicates ‘up’ (red) or ‘down’ (blue) regulation of said marker in symptomatic AD patients compared to age matched non-symptomatic patients. Protein names highlighted in red have AD-dysregulated and G9a reversed expression pattern that shows mouse-to-human conservation. (see also Data S1F).
Further, to identify mouse-to-patient conserved biomarkers for companion diagnosis at a stage when MS1262 treatment could be effective, we compared our mouse profiles of G9a/AD-coregulated, MS1262-reversed proteins with CSF protein biomarkers of AD progression87. Notably, MS1262 treatment reversed AD-characteristic expression of multiple CSF biomarkers of early-stage AD (Fig. 7B & Supplementary Data S1F). For example, the level of SMOC1, an Aβ plaque-associated synaptic protein88,89 was found elevated in early-stage AD CSF87. Other CSF markers87 showing MS1262 affected/reversed expression (Fig. 7B) included GMFB, a newly characterized G9a-regulated factor in the regulation of neuronal growth and differentiation90, and SCGC, which is a marker of prodromal AD91. We also noticed that two CSF markers showed AD-upregulated, MS1262-suppressed expression in the hippocampus of AD mice but decreased abundance in patient CSF, e.g., neuronal pentraxin receptor (NPTXR/NPTX2) which was identified as a biomarker of AD progression for CSF-based liquid biopsy91,92. These discrepancies of AD-related expression were probably due to different sample origins from the hippocampus or CSF, respectively. Nevertheless, simultaneous identification of these CSF biomarkers of AD as MS1262/G9a-regulated proteins suggested that these clinically validated biomarkers together can be used to stratify appropriate patients who may have maximum response to MS1262 treatment and to determine individual effects of MS1262 treatment.
MS1262 reversed multiple AD-specific brain pathological processes that mediate Aβ plaque and NFT pathology
Notably, 148 out of 348 of AD/G9a-coregulated, MS1262-affected proteins (Fig. 8A–C and Supplementary Data S1D) showed Aβ peptide (residues 6-28) correlated expression in the brain of AD patients (Fig. 8D & S10A and Supplementary Data S1G). The Aβ-correlated expression or phosphorylation of AD/G9a-coregulated proteins confirmed that G9a activity is closely associated with AD pathology and promotes the formation of Aβ plaques.

A Heatmap depicting AD/G9a co-regulated proteins/phosphoproteins showing dysregulation in 5x-FAD/APP-NLGF mice, compared to age-matched controls, whose expression/phosphorylation pattern is reversed following MS1262 treatment. Right annotation columns depict log2(fold-change) for indicated comparisons (AD/WT & AD-tr/AD). B, C Heatmaps summarizing results of GO/pathway enrichment (B) and disease & function (C) analyses for proteins and phospho-proteins belonging to the two clusters identified in (A). P-values were calculated using cumulative hypergeometric distribution followed by Benjamini-Hochberg correction to account for multiple testing. All genes in the genome were used as enrichment background. D Volcano plot illustrating the relationship between expression levels of various proteins and the Aβ peptides (residues 6–28) in the brain of AD patients (n = 488)10. The plot shows the biweight mid-correlation (bicor) coefficient and the corresponding BH adjusted p-value for each protein. Cluster 1 (blue) and cluster 2 (red) proteins from (A) are highlighted with point shape denoting proteins (circle) and phospho-proteins (triangle). Names of select AD markers are shown. E Network illustrating the relationship between AD/G9a-coregulated proteins/phosphoproteins identified in (A) and various AD-patient correlated modules (‘M’) that are dysregulated in the brains of AD patients10. Modules showing dysregulation at protein level only, without concomitant change at the transcriptional level in AD patients, are highlighted using cyan outlines (see also Figure. S10 and Data S1D, F–H).
Next, by comparing AD/G9a-corregulated proteins identified in 5xFAD/APP-NLGF mice with “modules” (M) of co-expressed proteins showing dysregulation in brains of AD patient10 we identified 204 entities with AD-related, MS1262-reversed expression or phosphorylation changes (Fig. 8E & Supplementary Data S1H). Notably, 15 out of 38 of these AD/G9a-corregulated ‘modules’ were only dysregulated at the protein level without concomitant change in transcriptomic networks in the AD patient samples (Fig. 8E, highlighted in cyan). This finding, along with the observation that nearly half of the AD/G9a-corregulated proteins are encoded by m6A-tagged transcripts (Figure. S4C), confirmed the translational function of active G9a.
In the networks of patient protein modules (Fig. 8E), SMOC1, whose level in CSF or in postmortem brain was associated with AD pathology, was an MS1262-targeted component of the M42 matrisome module strongly correlated with AD neuropathology and cognition. The Tau family microtubule-associated proteins93 and STIM2 responsible for neuronal impairment94 were MS1262-targeted components of module M7 MAPK signaling and metabolism that is highly associated with the rate of cognitive decline. Plasma glial fibrillary acidic protein, an astrocyte reactivity biomarkers for AD95 and nitric oxide synthase 1 for synaptic transmission and neuroplasticity96 were MS1262-targeted components of modules M5 post-synaptic density or M11 cell-ECM interaction, respectively, again, major modules strongly correlated to AD neuropathology and cognition. In addition, MS1262 treatment reversed the phosphorylation of components of modules M29, glycosylation/ER, and M42, matrisome, that were correlated with AD endophenotypes, and elevated tau microtubule-binding domain peptide levels correlate with the other MS1262-affected components of M42 matrisome and M11 cell-ECM modules. Overall, AD/G9a-corregulated co-expression modules were involved in neuronal signaling, RNA metabolism and translation, protein transport, cellular energetics, and immune response.
Further, we examined spatial relationships between MS1262-reversed AD patient proteome and Aβ plaques and NFTs, the hallmarks of AD neuropathologies. In addition to MS1262 reversal of the phosphorylation of SMOC1 in M42 matrisome, MS1262 reversed the phosphorylation of many module components found in Aβ plaques or/and NFTs, including M1 and M4 components enriched in both Aβ plaques and NFTs, M7 components primarily associated with Aβ plaque, and M13 and M29 components uniquely enriched in NFTs.
Discussion
Epigenetic mechanisms involving G9a are thought to regulate AD pathogenesis. However, few pathways strongly correlated with AD pathology were identified by transcriptomic analysis of G9a inhibitor (UNC0642)-treated 5xFAD mice7,97. Using G9a inhibitors as the mechanistic probes in combined approaches including chemical proteomics, animal behavioral analysis, and AD-correlated proteomics, we discovered a noncanonical function of aberrantly activated G9a in the translation-regulatory mechanism that underlies AD proteopathology. Accordingly, we have developed this mechanism-based, brain-penetrant small molecule therapeutic that readily crosses the blood-brain barrier to reverse this G9a-mediated proteome in AD.
While no mouse model of AD perfectly recapitulates all aspects of AD pathology, we addressed this limitation through the utilization of three distinct mouse models at multiple ages to show the broad applicability of our treatment; while 5xFAD mice and APPNLGF KI mice demonstrate accelerated amyloid pathology and behavior deficits as seen in AD patients, 3xTg mice display both amyloid and tau pathologies. Further, taking the natural aging process into account, we used 12.25-month-old 3xTg mice to recapitulate age-induced memory deficits and tau pathology. Notably, the high efficacy of MS1262 treatment on AD-associated behavioral deficits was consistently observed for all three AD mouse models that comprehensively recapitulate major pathological features of AD patients (Figs. 3 and 4, Fig. S4 & S5), strengthening our claim that MS1262 may be useful for the treatment of AD in aging patients.
Our electrophysiological experiments (Fig. 4) clearly demonstrated that MS1262 effectively rescues the early (up to 60 minute) stages of LTP, which indicated that this potentiation may underly the long-term memory benefits observed in the behavioral experiments. However, without conclusive evidence of late-stage LTP enhancement, this conclusion remains predominantly suggestive. Furthermore, we are limited in our understating of whether MS1262 exerts pathways specific enhancement of synaptic plasticity, as that would require additional control fEPSP recordings from the same hippocampal sections during the treatment.
Presently, an absence of clinically validated protein makers for AD diagnosis prevents assessment of drug effects from animal studies. We overcome this challenge by comparing the AD-related, MS1262-affected mouse multiomics data with those from AD patients of statistical significance ( > 1000 biospecimen)10. Firstly, our multiomics results validated that the constitutively active G9a regulates the translation and post-translational modification (phosphorylation) of a broad range of AD-related proteins that were preserved across the mouse and human/patient samples (Fig. 8), e.g., the AD-characteristic expression or phosphorylation of these mouse-to-patient conserved proteins were affected or reversed by MS1262 treatment while the levels of mRNAs encoding these proteins also showed little change in AD patients. Mechanistically, our m6A RNA-to-protein correlation analysis identified 21 G9a/AD-coregulated m6A mRNAs (Fig. 6B) whose protein translation was reversed by MS1262 treatment of AD mice (Fig. 7A & 7E), indicating that G9a activity translates these m6A modified transcripts into AD-related proteins. Secondly, MS1262 showed multifaceted, therapeutic effects on AD as AD-activated G9a broadly and simultaneously regulated major signaling pathways that mechanistically contribute to both cognitive and non-cognitive symptoms of AD. Thus, MS1262 effects are not ‘one target (G9a) at a time’ but, instead, the drug effect (target) is proteome-wide. As supporting evidence, Pao et al. reported98 that targeting CDK5 hyperactivity ameliorated neurodegenerative phenotypes. Accordingly, we identified CDK5 signaling as one of many G9a/AD-coregulated pathways that were simultaneously reversed by MS1262 treatment. This identification of CDK5 confirmed that G9a is a broad regulator of AD proteopathic landscape, hence, MS1262 rescued a range of AD-dysregulated pathways that define multiple AD pathological hallmarks such as Aβ plaque and NFT pathology. In addition, AD mice treated with MS1262 for six weeks did not show any signs of toxicity; general appearance, activity, and behavior were all normal or improved. Outward normality was consistent with MS1262 reversal of AD proteome and phosphoproteome of the hippocampus. On a systems evaluation of MS1262 side effects, our AD pathology-correlated proteomic/phosphoproteomic results demonstrated that MS1262 specifically and effectively inhibited G9a that was aberrantly activated in AD-related cells but not non-AD related cells in diseased brain so that little, if any, off-target toxicity was observed. Thirdly, in parallel with the observation that MS1262 rescued AD-related behavior, MS1262 treatment reversed the proteopathologic landscape in AD patients, which was evidenced by the greater number of phosphoproteins in the protein co-expression modules highly correlated with broad AD pathology and cognitive deficits. These proteomic identifications of AD mouse-to-patient conservation (Fig. 7&8) revealed multiple MS1262-affected, clinically validated biomarkers of AD which can be used to evaluate evidence of downstream disease modification and to assess the relation between biomarker changes and clinical outcomes.
Methods
Experimental model and subject details
Human cerebral organoid cultures
A familial AD patient-derived iPSC line with the APP V717I mutation (F033K; male) and a sex- and age-matched healthy control (C-03; male) iPSC line (provided by Dr. Chadwick Hales’ laboratory at Emory University) were cultured on irradiated MEFs in human iPSC medium composed of D-MEM/F12 (Invitrogen), 20% Knockout Serum Replacement (KSR, Invitrogen), 1X Glutamax (Invitrogen), 1X MEM Non-essential Amino Acids (NEAA, Invitrogen), 100 µM β-mercaptoenthanol (Invitrogen), and 10 ng/ml human basic FGF (bFGF, PeproTech) as described21,99,100. Forebrain-specific organoids were generated as described21,100,101. Briefly, human iPSC colonies were detached from the feeder layer with 1 mg/ml collagenase treatment for 1 hour and suspended in embryonic body medium composed of FGF-2-free iPSC medium supplemented with 2 µM Dorsomorphin and 2 µM A-83 in nontreated polystyrene plates for 4 days with a daily medium change. On days 5-6, half of the medium was replaced with induction medium consisting of DMEM/F12, 1X N2 Supplement (Invitrogen), 10 μg/ml Heparin (Sigma), 1X Penicillin/Streptomycin, 1X Non-essential Amino Acids, 1X Glutamax, 4 ng/ml WNT-3A (R&D Systems), 1 μM CHIR99021 (Tocris), and 1 μM SB-431542 (Tocris). On day 7, organoids were embedded in Matrigel (BD Biosciences) and continued to grow in induction medium for 6 more days. On day 14, embedded organoids were mechanically dissociated from Matrigel by pipetting up and down onto the plate with a 5 ml pipette tip. Typically, 10-20 organoids were transferred to each well of a 12-well spinning bioreactor (SpinΩ) containing differentiation medium, consisting of DMEM/F12, 1X N2 and B27 Supplements (Invitrogen), 1X Penicillin/Streptomycin, 100 µM β-mercaptoenthanol (Invitrogen), 1X MEM NEAA, 2.5 μg/ml Insulin (Sigma). At day 71, differentiation medium was exchanged with maturation medium, consisting of Neurobasal (Gibco), 1X B27 Supplement, 1X Penicillin/Streptomycin, 1X β-mercaptoenthanol, 0.2 mM Ascorbic Acid, 20 ng/ml BDNF (Peprotech), 20 ng/ml GDNF (Peprotech), 1 ng/ml TFGβ (Peprotech), and 0.5 mM cAMP (Sigma). All media were changed every other day.
Experimental animals
All animal procedures were conducted in accordance with the NIH Guide for the Care and Use of Laboratory Animals and with the approval of the Institutional Animal Care and Use Committee at the University of North Carolina at Chapel Hill. The 5xFAD (C57BL/6 J) and wild-type (C57BL/6 J) littermate controls (16-24-weeks-old) were obtained from the Jackson laboratory. All 5xFAD mice were heterozygous. APPNLGF (C57BL/6J-Aw-J/J) and wild-type (C57BL/6J-Aw-J/J) mice were obtained from Dr. Mohanish Deshmukh. All APPNLGF mice were homozygous. Both male and female mice were used, and sex was matched in the various groups. 3xTg mice (129S4) were obtained from Dr. Leon Coleman Jr. Both male and female mice were used, and sex was matched in the various groups. No immune deficiencies or other health problems were observed in these lines, and all animals were experimentally and drug-naïve before use. Animals were group housed and bred in a dedicated husbandry facility with 12/12 hour light-dark cycles with ad libitum food and water. All mice were under veterinary supervision. Behavioral experiments were performed in the light phase. Once drug or vehicle administration began, animals were moved to a satellite housing facility with the same light-dark cycle.
Behavioral tests
Handling
Mice were handled five days a week for 6 weeks for 5 min/day leading up to behavioral testing.
Open field and habituation
On days 1 and 2, mice were placed in an empty open field environment (45 cm square plastic chamber) for 10 minutes. After each test, the chamber was cleaned with 70% ethanol to eliminate scents from previously tested mice. The first 5 minutes of the day-1 test was analyzed by Noldus Ethovision XT to monitor animal position and locomotion. A 25 cm square was used to indicate the center of the chamber when analyzing the recordings. Both total locomotion and time spent in the center of the open field were quantified by the Noldus Ethovision XT program.
Novel place recognition
On day 3 of behavioral testing, also known as the encoding phase, two identical glass cylinders (height 4 cm, base diameter 1.5 cm) were fixed to the chamber floor (to prevent object movement) on the same side of the chamber 20 cm apart from each other. Animals were allowed to freely explore the chamber for 5 minutes and could interact with the objects while being recorded. Most animals showed no preference for a single object and the locations of the objects were randomized across mice. On day 4 of behavioral testing, also known as the retrieval phase, the position of one of the objects was moved to the opposite side of the chamber, and animals were recorded for 5 minutes while freely exploring the chamber and interacting with the objects. Videos were scored manually using a separately trained researcher who was blinded to the treatment group. Time spent with each object in both encoding and retrieval were scored. Any mouse that showed a preference for one object over the other (defined by interacting with one object for more than double the time of interacting with the other) during the encoding phase was not included in the analysis. Furthermore, mice that did not spend at least 2 seconds of total object interaction time (IT) were not included in the analysis. The discrimination ratio (DR) was calculated in the following way:
A DR of 0 indicated no preference for either object, and a DR of 0.33 indicates spending twice as much time with the object in the novel location compared with the object in the familiar location.
Elevated zero maze
The apparatus was an elevated white plastic ring platform (width of ring 6 cm, outer diameter 60 cm). The entire ring was elevated 60 cm off the ground. Each animal was placed in the closed arm to start the trial and was recorded for 5 minutes. Time spent in the open arm sections was scored by a trained, blinded researcher. After each trial, the apparatus was cleaned with 70% ethanol.
Forced swim
The apparatus was an acrylic cylinder (diameter 20 cm, height 30 cm) filled with room temperature (23 ± 1oC) water to a depth of 20 cm. Each mouse was recorded during a 5-minute swimming trial, and the video was later scored manually by a trained, blinded researcher for time spent immobile. Time spent immobile was defined as when all four paws of the mouse remained immobile. After each trial, the apparatus was filled with fresh water.
Barnes maze
The apparatus was white plastic circular platform with 20 even spaced holes near the outer rim (diameter 120 cm, elevated 80 cm of the ground). Only one of the 20 holes had an escape chamber that was made dark and comfortable with bedding. Our protocol was adapted from102. Each mouse participated in a 5 minute habituation trial that began in the escape chamber. The mouse was then moved to the center of the platform and allowed to explore for 5 minutes or until escape. At the end of the 5 minutes if the mouse had not found the chamber, it was placed back in the escape chamber. The next following 4 days were the acquisition phase where the mice were placed in the center of the platform and were recorded during a 3-minute trial, and the video was later scored manually by a trained, blinded researcher for time to escape latency. After each trial, the apparatus was cleaned with 70% ethanol.
Method details
Drug treatment
The G9a inhibitor MS1262 was dissolved in DMSO to 10 mg/mL and aliquoted into single doses and stored at −20 °C. Immediately prior to injection, these aliquots were thawed and diluted in 0.9% saline. The final solution that was injected intraperitoneally consisted of 1% DMSO and equated to a 1 mg MS1262/kg of animal weight. Control animals received 1% DMSO in 0.9% saline at weight matched volumes. Mice were randomly selected for MS1262 or vehicle treatment and received 1 injection every 3.5 days for 6 weeks.
Microdissection of hippocampi
After 6 weeks of intermittent MS1262 or vehicle treatment, animals were anesthetized with a 5% isoflurane in oxygen mixture until the animal was no longer responsive to a toe pinch. Animals were then transcardially perfused with ice-cold PBS and the brain was isolated. The brain was sliced bilaterally across the sagittal midline. Each half was then taken, and the hippocampus was carefully dissected. Both hippocampi were placed in a cryogenic tube and flash frozen in liquid nitrogen and placed at -80oC.
Chromatin activity-based chemoproteomics (ChaC) with UNC0965
Human organoids or mouse hippocampus were lysed in a lysis buffer (50 mM Tris (pH7.5), 150 mM NaCl, 0.5%DOC, 0.5% Igepal CA630, 5% Glycerol) containing protease inhibitor cocktail and 1 mM PMSF, then sonicated three times for 5 seconds each. The lysate was centrifuged and the supernant was incubated with UNC0965-bound NeutrAvidin agarose at 4 °C overnight; NeutrAvidin agarose alone served as the control. The samples were washed three times with lysis buffer and three times with detergent-free buffer. On-bead trypsin digestion followed, and peptides were collected and desalted before MS analysis. The experiment used three normal and two AD human organoids, conducted twice. For 5xFAD mouse hippocampi, two mock-treated and three MS1262-treated mice were used in the first experiment, and three of each in the second.
Proteomics sample preparation
The mouse hippocampus tissues were resuspended in 8 M urea, 50 mM Tris-HCl pH 8.0, reduced with dithiothreitol (5 mM final) for 30 min at room temperature, and alkylated with iodoacetamide (15 mM final) for 45 min in the dark at ambient temperature. Samples were diluted 4-fold with 25 mM Tris-HCl pH 8.0, 1 mM CaCl2 and digested with trypsin at a ratio of 1:100 (w/w, trypsin: protein) overnight at ambient temperature. There were three wild-type (WT) controls, 4 AD, and 4 AD-treated with MS1262. Peptides were cleaned by homemade C18 stage tips and the concentration was determined (Peptide assay, Thermo 23275). One hundred microgram each was used for labeling with isobaric stable tandem mass tags (TMT11, Thermo Fisher Scientific, San Jose, CA) following supplier instruction. The mixture of labeled peptides was desalted on Cep-Pak light C18 cartridge (Waters). Phosphopeptides were enriched with High-Select Fe-NTA Phosphopeptide Enrichment Kit (Thermo Scientific). One hundred microgram of peptides was fractionated into 20 fractions on C18 stage tip with 10 mM trimethylammonium bicarbonate (TMAB), pH 8.5 containing 5–50% acetonitrile.
Mass spectrometry
Dried peptides were dissolved in 0.1% formic acid, 2% acetonitrile. One microgram phosphopeptides or 0.5 μg of peptides from each fraction was analyzed on a Q-Exactive HF-X coupled with an Easy nanoLC 1200 (Thermo Fisher Scientific, San Jose, CA). Peptides were loaded on to a nanoEase MZ HSS T3 Column (100 Å, 1.8 µm, 75 µm × 250 mm, Waters). The phosphopeptides were separated with 240-min gradient: a linear gradient of 5 to 20% buffer B over 140 min, 20 to 31% over 50 min, 31 to 75% over 30 min, followed by a ramp to 100% B in 1 min and 19-min wash with 100% B. Analytical separation of unphosphorylated peptides was achieved with 110-min gradient, a linear gradient of 5 to 10% buffer B over 5 min, 10% to 31% buffer B over 70 min, 31% to 75% buffer B over 15 followed a ramp to 100%B in 1 min and 19-min wash with 100%B, where buffer A was aqueous 0.1% formic acid, and buffer B was 80% acetonitrile and 0.1% formic acid. The flow rate was kept at a 250 nl/min. Mass spectrometry experiments were also conducted in a data-dependent mode with full MS (externally calibrated to a resolution of 60,000 at m/z 200) followed by high energy collision-activated dissociation-MS/MS of the top 10 most intense ions with a resolution of 45,000 at m/z 200. High energy collision-activated dissociation-MS/MS was used to dissociate peptides at a normalized collision energy of 32 eV in the presence of nitrogen bath gas atoms. Dynamic exclusion was 45 seconds.
A brief gradient was used for ChaC-UNC0965 products: a linear increase from 5 to 30% buffer B over 29 minutes, then from 30 to 45% over 6 minutes, followed by a quick rise to 100% B in 1 minute and a 14-minute wash with 100% B. Mass spectrometry was performed in data-dependent mode with full MS (externally calibrated to a resolution of 60,000 at m/z 200), followed by high energy collision-activated dissociation-MS/MS of the 15 most intense ions at a resolution of 15,000 at m/z 200. This technique used a normalized collision energy of 27 eV with nitrogen bath gas, and dynamic exclusion was set to 20 seconds.
Raw proteomics data processing and analysis
Peptide identification and quantification with TMT reporter ions were performed using the MaxQuant software version 2.1.0.0 (Max Planck Institute, Germany). Protein database searches were performed against the UniProt human protein sequence database (UP000005640) or Mus musculus (UP000000589). A false discovery rate for both peptide-spectrum match and protein assignment was set at 1%. Search parameters included up to two missed cleavages at Lys/Arg on the sequence, oxidation of methionine, protein N-terminal acetylation, and phosphorylation of serine, threonine, and tyrosine as dynamic modifications. Carbamidomethylation of cysteine residues was considered as a static modification. Peptide identifications are reported by filtering of reverse and contaminant entries and assigning to their leading razor protein. Data processing and statistical analysis were performed on Perseus (Version 1.6.10.50). Protein quantitation was performed on biological replicates and a two-sample t-test statistics was used with a p-value of 5% to report statistically significant protein or phosphopeptide abundance fold-changes.
Analysis of ChaC MS data. The analysis identified statistically significant candidates with a p-value ≤ 0.05 and log2FC ≥ 1 as potential specific G9a interactors by comparing UNC0965 pulldown to the negative control. For biological analysis, only those conserved in both 5xFAD mouse hippocampi and human organoids across all ChaC-UNC0965 experiments were considered, and this analysis was performed using STRING.
m6A RNA immunoprecipitation sequencing (m6A/MeRIP-Seq) and data analysis
The m6A/MeRIP-Seq was performed as described with some modifications [2]. Briefly, total RNA was extracted from hippocampus of 5x-FAD (AD, n = 4), MS1262 treated 5x-FAD (AD-tr, n = 4), and age matched wildtype (WT, n = 4) controls using TRIzol (Life Technologies) followed by purification using illustraTM RNA spin Mini kit (GE Healthcare, UK). Then, mRNA was isolated from 10 μg total RNA using Dynabeads Oligo (dT)25 (Thermo Fisher; 61006) according to manufacturer’s instructions. 10% of a total of 150 ng mRNA was used as input, while the rest was incubated with 3 ug anti-m6A polyclonal antibody (Synaptic Systems; 202003) that was pre-conjugated to Dynabeads Protein A (Thermo Fisher; 10001D) in 500 uL IP buffer (50 mM Tris, pH 7.4, 150 mM NaCl, 0.1% Igepal CA-630) for 2 hours at 4 °C. After washing the beads twice with IP-buffer and twice with high-salt wash buffer (50 mM Tris pH 7.4, 500 mM NaCl, 0.1% Igepal CA-630) for 5 min each, the m6A-tagged mRNA was eluted using 100 uL IP-buffer containing 6.7 mM N6-Methyladenosine (Sigma-Aldrich; M2780) and 40 U RNase Inhibitor (NEB, M0314S) followed by recovery using RNA Clean and Concentrator-5 spin columns (Zymo; R1015).
The input and m6A-IPed mRNA were subjected to library generation using SMART-seq protocol as described [3]. For first strand cDNA synthesis, the mRNA was mixed with 0.25 µL RNase inhibitor and 1 µL CDS primer (5’-AAGCAGTGGTATCAACGCAGAGTACT30VN-3’) and heated to 70 oC for 2 min. Then the mixture containing 0.5 µL of 100 mM DTT, 0.3 µL of 200 mM MgCl2, 1 µL of 10 mM dNTPs, 0.25 µL RNase inhibitor, 1 µL of 10 µM TSO primer (5’-AAGCAGTGGTATCAACGCAGAGTACATrGrGrG-3’), 2 µL of 5X SMARTScribe RT buffer and 0.5 µL SMARTScribe reverse transcriptase (Takara, 639536) was added to perform reverse transcription. The cDNA was then amplified by Advantage Polymerase Mix (TAKARA, 639201) with IS primer (5’-AAGCAGTGGTATCAACGCAGAGT-3’). After purification with 0.8X AMPure XP beads (Fisher Scientific, A63880), the fragmentation of 100 pg cDNA was performed with EZ Tn5 Transposase (Lucigen, TNP92110). Fragments of cDNA were amplified by KAPA HiFi hotstart readymix (EMSCO/FISHER, KK2601) with the Nextera i7 primer and Nextera i5 primer. The DNA was purified with 0.8X AMPure XP beads and quantified by qPCR with KAPA Library Quantification Kit (Fisher, NC0078468). The DNA from different samples was pooled at equal molar amounts, and the final sequencing library was loaded at concentrations of 2.7 pM and sequenced on a NextSeq 550 (Illumina) for single-end sequencing.
Raw sequencing data were de-multiplexed using bcl2fastq2 v2.17.1.14 (Illumina) and adapters were removed using Trimmomatic-0.32 software. Then, single-end reads from input and m6A IP samples were mapped to the GRCm38/mm10 mouse reference genome using STAR version 2.7.6a [4]. The resulting bam files from the STAR alignment were sorted and indexed using SAMtools version 1.10 [5] and MACS2 version 2.1.2 [6] was used to call m6A peaks using the bam files for each m6A-IP/input pair. The R packages GenomicRanges v1.36.1 and AnnotationHub v.3.4.0 were used to identify genes overlapping the peaks determined by MACS2, and CHIPseeker v.1.30.3 was used to create profile plots. Weighted average fold-change, from all m6A peaks mapping to a gene, was used to identify AD/G9a-coregulated m6A-modified transcripts and enrichment analysis was carried out using metascape.
Immunohistology, image acquisition, and imaging analysis
Mice were perfused with PBS and brain samples were collected following perfusion and fixed with 4% paraformaldehyde (PFA). Briefly, mice were exposed to a lethal dose of isoflurane in an incubation chamber. At cessation of breathing, the chest cavity was exposed, a 27 G needle inserted into the left ventricle and PBS ( ~ 20 ml) first instilled by peristaltic pump. The brain was extracted and incubated in 10 ml of 4% PFA for 48 hours and then switched to 30% sucrose for 72 hours days, until fully submerged. Brains were sectioned on a microtome at a thickness of 40μm and then stored in antifreeze solution at −20 °C until further usage. Free-floating antibody staining was conducted on brain slices. Free-floating antibody staining was conducted on brain slices. Specifically, a 30-minute incubation in 0.3% Triton-X in PBS. A blocking step was then performed using 5% donkey serum in 0.1% PBST. Sections were then transferred to the primary antibody solution in 0.1% PBST and kept at 4 °C for 48 h, with intermittent brief periods of shaking. After primary incubation, sections were subjected to three 30-min wash steps in 0.1% PBST. Sections were then transferred to a secondary antibody solution in 0.1% PBST for 24 h at 4 °C, with intermittent brief periods of shaking. Sections were washed three times for 30 min each in 0.1% PBT. Sections were mounted on charged glass slides using Diamond prolong gold mounting medium (Thermo Fisher Scientific, no. P36961) and no. 1.5 glass coverslips (Electron Microscopy Sciences, no. 72204-02). The following primary antibodies were used at the following concentrations: mouse anti-H3K9me2 1:250 (Sigma Aldrich, 05-1249), a rabbit Aβ antibody (Cell Signaling Technology, #9888) was used at 1:1000. All brain slices were imaged with an Olympus FV3000 microscope. Imaging volumetric analysis was performed using Imaris. Specifically, A region of interest (ROI) surface was drawn around the hippocampus, and their volumes were recorded.
Slice preparation
Acute slices were prepared from 5.25 month-5.5 month male 5xFAD mice treated with MS1262 (1 mg/kg) or vehicle as described previously. Animals were anesthetized with isoflurane (5% in O2) and transcardially perfused with ice-cold oxygenated artificial cerebrospinal fluid (ACSF) containing the following items (in mM): 92 NMDG, 30 NaHCO3, 25 glucose, 20 HEPES, 10 MgSO4, 5 sodium ascorbate, 3 sodium pyruvate, 2.5 KCl, 2 thiourea, 1.25 NaH2PO4, and 0.5 CaCl2 (pH 7.3, 310 mOsm). Brains were rapidly removed, and acute transverse hippocampal slices (280 μm) were cut using a Leica vibratome (VT1200, Germany). Next, slices were warmed to 34.5 °C for 8 minutes. Then, slices were maintained in the holding chamber containing HEPES ACSF (in mM): 92 NaCl, 30 NaHCO3, 25 glucose, 20 HEPES, 5 sodium ascorbate, 3 sodium pyruvate, 2.5 KCl, 2 thiourea, 2 MgSO4, 2 CaCl2, 1.25 NaH2PO4 (pH 7.3, 10 mOsm) at ambient temperature for at least 1 hour before recording. Electrophysiological recordings were made at 32 °C using a heater controller (TC-324C, Warner Instruments) in ACSF containing (in mM): 125 NaCl, 26 NaHCO3, 20 glucose, 2.5 KCl, 2 CaCl2, 1.3 MgSO4, 1.25 NaH2PO4, (pH 7.3, 310 mOsm). The flow rate was 2 ml/min.
Electrophysiology
Slices were visualized on a fixed-stage upright microscope (Olympus BX51WI) equipped with ×4 and ×40 objectives and differential interference contrast optics, infrared illumination, and an infrared-sensitive camera. Whole-cell patch-clamp recordings were performed with glass pipettes with resistance of 4.0–6.0 MΩ when filled with internal solution containing (in mM): 130 K-gluconate, 20 HEPES, 4 MgCl2, 4 Na-ATP, 2 NaCl, 0.5 EGTA, 0.4 Na-GTP (pH 7.2, 290 mOsm). Series resistance (Rs) was monitored throughout all experiments, and cells with Rs changes over 20% were discarded. All recordings were made from granule cells located in the middle or outer layer of the dentate gyrus. For spontaneous excitatory postsynaptic currents (sEPSCs) recordings, dentate granule cells were held at -70 mV in voltage-clamp mode and bicuculine (20 μM; GABAA receptor antagonist; Tocris Bioscience) was added to the ACSF. Intrinsic cellular properties were recorded in current-clamp mode. To test neuronal input resistance, hyperpolarizing current pulses (20 pA, 200 ms) were applied to the neurons. Resting membrane potential was measured as the membrane potential baseline value obtained in current-clamp mode in the absence of current injection. The current-voltage relationship experiments (to evaluate action potential firing rate) consisted of a series of current injections (500-ms duration) between 0 pA and +240 pA in 20 pA delivered in step-wise increments. Data were acquired with a Multiclamp 700B amplifier and digitized with a Digidata 1440 A using pCLAMP 10.7 acquisition software (Molecular Devices). If the data failed a normalcy test, then a nonparametric test was used in its place.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All data needed to evaluate the conclusions in the paper are present in the paper or the Supplementary Materials. The GEO accession to the RNA/m6A-seq datasets is publicly available with GSE278032. The secure token is ifqdamkubraffgf. ProteomeXchange dataset has also been made public via the PRIDE database (ProteomeXchange accession: PXD047045). Raw files and any other data required for reanalysis are available from the corresponding author (X.C.) upon request. Source data are provided with this paper.
References
Bennett, D. A. et al. Epigenomics of Alzheimer’s disease. Transl. Res 165, 200–220 (2015).
Satoh, J., Asahina, N., Kitano, S. & Kino, Y. A comprehensive profile of ChIP-Seq-based PU.1/Spi1 Target Genes in Microglia. Gene Regul. Syst. Bio 8, 127–139 (2014).
Rabinovici, G. D. & La Joie, R. Amyloid-targeting monoclonal antibodies for Alzheimer’s disease. JAMA 330, 507–509 (2023).
Pike, K. E. et al. Beta-amyloid imaging and memory in non-demented individuals: evidence for preclinical Alzheimer’s disease. Brain 130, 2837–2844 (2007).
Jack, C. R. Jr. et al. NIA-AA research framework: toward a biological definition of Alzheimer’s disease. Alzheimers Dement 14, 535–562 (2018).
Luukkainen, L. et al. Mutation analysis of the genes linked to early onset alzheimer’s disease and frontotemporal lobar degeneration. J. Alzheimers Dis. 69, 775–782 (2019).
Zheng, Y. et al. Inhibition of EHMT1/2 rescues synaptic and cognitive functions for Alzheimer’s disease. Brain 142, 787–807 (2019).
Vedadi, M. et al. A chemical probe selectively inhibits G9a and GLP methyltransferase activity in cells. Nat. Chem. Biol. 7, 566–574 (2011).
Schaefer, A. et al. Control of cognition and adaptive behavior by the GLP/G9a epigenetic suppressor complex. Neuron 64, 678–691 (2009).
Johnson, E. C. B. et al. Large-scale deep multi-layer analysis of Alzheimer’s disease brain reveals strong proteomic disease-related changes not observed at the RNA level. Nat. Neurosci. 25, 213–225 (2022).
Liu, C. et al. A chromatin activity-based chemoproteomic approach reveals a transcriptional repressome for gene-specific silencing. Nat. Commun. 5, 5733 (2014).
Wrobel, J. A. et al. Multi-omic dissection of oncogenically active epiproteomes identifies drivers of proliferative and invasive breast tumors. iScience 17, 359–378 (2019).
Huang, H., Camats-Perna, J., Medeiros, R., Anggono, V. & Widagdo, J. Altered expression of the m6A Methyltransferase METTL3 in Alzheimer’s disease. eNeuro. https://doi.org/10.1523/eneuro.0125-20.2020 (2020).
Zhao, F. et al. METTL3-dependent RNA m(6)A dysregulation contributes to neurodegeneration in Alzheimer’s disease through aberrant cell cycle events. Mol. Neurodegener. 16, 70 (2021).
Han, M. et al. Abnormality of m6A mRNA Methylation is involved in Alzheimer’s disease. Front Neurosci. 14, 98 (2020).
Watamura, N. et al. An isogenic panel of App knock-in mouse models: profiling beta-secretase inhibition and endosomal abnormalities. Sci. Adv. 8, eabm6155 (2022).
Oddo, S. et al. Triple-transgenic model of Alzheimer’s disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron 39, 409–421 (2003).
Billings, L. M., Oddo, S., Green, K. N., McGaugh, J. L. & LaFerla, F. M. Intraneuronal Abeta causes the onset of early Alzheimer’s disease-related cognitive deficits in transgenic mice. Neuron 45, 675–688 (2005).
Feltcher, M. E. et al. Label-free quantitative proteomics reveals a role for the mycobacterium tuberculosis SecA2 pathway in exporting solute binding proteins and Mce transporters to the cell wall. Mol. Cell Proteom. 14, 1501–1516 (2015).
Ankney, J. A., Muneer, A. & Chen, X. Relative and absolute quantitation in mass spectrometry-based proteomics. Annu Rev. Anal. Chem. (Palo Alto Calif.) 11, 49–77 (2018).
Kuehner, J. N. et al. 5-hydroxymethylcytosine is dynamically regulated during forebrain organoid development and aberrantly altered in Alzheimer’s disease. Cell Rep. 35, 109042 (2021).
Lopez-Otin, C., Blasco, M. A., Partridge, L., Serrano, M. & Kroemer, G. Hallmarks of aging: An expanding universe. Cell 186, 243–278 (2023).
Jiang, L. et al. Interaction of tau with HNRNPA2B1 and N6-methyladenosine RNA mediates the progression of tauopathy. Mol. Cell 81, 4209–4227.e12 (2021).
Lin, S., Choe, J., Du, P., Triboulet, R. & Gregory, R. I. The m(6)A Methyltransferase METTL3 Promotes Translation in Human Cancer Cells. Mol. Cell 62, 335–345 (2016).
Sood, S. et al. A novel multi-tissue RNA diagnostic of healthy ageing relates to cognitive health status. Genome Biol. 16, 185 (2015).
Wang, L. et al. Novel RNA-affinity proteogenomics dissects tumor heterogeneity for revealing personalized markers in precision prognosis of cancer. Cell Chem. Biol. 25, 619–633 (2018).
Liu, F. et al. Discovery of an in vivo chemical probe of the lysine methyltransferases G9a and GLP. J. Medicinal Chem. 56, 8931–8942 (2013).
Srimongkolpithak, N., Sundriyal, S., Li, F. L., Vedadi, M. & Fuchter, M. J. Identification of 2,4-diamino-6,7-dimethoxyquinoline derivatives as G9a inhibitors. Medchemcomm 5, 1821–1828 (2014).
Xiong, Y. et al. Structure-activity relationship studies of G9a-like protein (GLP) inhibitors. Bioorg. Medicinal Chem. 25, 4414–4423 (2017).
Park, K. S. et al. Discovery of the first-in-class G9a/GLP covalent inhibitors. J. Med Chem. 65, 10506–10522 (2022).
Oakley, H. et al. Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: potential factors in amyloid plaque formation. J. Neurosci. 26, 10129–10140 (2006).
Locci, A. et al. Comparison of memory, affective behavior, and neuropathology in APP(NLGF) knock-in mice to 5xFAD and APP/PS1 mice. Behav. Brain Res 404, 113192 (2021).
Saito, T. et al. Single App knock-in mouse models of Alzheimer’s disease. Nat. Neurosci. 17, 661–663 (2014).
Masuda, A. et al. Cognitive deficits in single App knock-in mouse models. Neurobiol. Learn Mem. 135, 73–82 (2016).
Li, Y. D. et al. Activation of hypothalamic-enhanced adult-born neurons restores cognitive and affective function in Alzheimer’s disease. Cell Stem Cell 30, 415–432 e6 (2023).
Li, Y. D. et al. Hypothalamic modulation of adult hippocampal neurogenesis in mice confers activity-dependent regulation of memory and anxiety-like behavior. Nat. Neurosci. 25, 630–645 (2022).
Whitlock, J. R., Heynen, A. J., Shuler, M. G. & Bear, M. F. Learning induces long-term potentiation in the hippocampus. Science 313, 1093–1097 (2006).
Kemp, A. & Manahan-Vaughan, D. Hippocampal long-term depression: master or minion in declarative memory processes? Trends Neurosci. 30, 111–118 (2007).
Ge, Y. et al. Hippocampal long-term depression is required for the consolidation of spatial memory. Proc. Natl Acad. Sci. USA 107, 16697–16702 (2010).
Jiang, L. et al. β-amyloid accumulation enhances microtubule associated protein tau pathology in an APPNL-GF/MAPTP301S mouse model of Alzheimer’s disease. Front Neurosci. 18, 1372297 (2024)
Tan, M. G. et al. Decreased rabphilin 3A immunoreactivity in Alzheimer’s disease is associated with Abeta burden. Neurochem Int 64, 29–36 (2014).
Xie, A. J. et al. Tau overexpression impairs neuronal endocytosis by decreasing the GTPase dynamin 1 through the miR-132/MeCP2 pathway. Aging Cell 18, e12929 (2019).
Huang, J. L. et al. MeCP2 prevents age-associated cognitive decline via restoring synaptic plasticity in a senescence-accelerated mouse model. Aging Cell 20, e13451 (2021).
Srinivasan, S., Gal, J., Bachstetter, A. & Nelson, P. T. Alpha adaptins show isoform-specific association with neurofibrillary tangles in Alzheimer’s disease. Neuropathol. Appl Neurobiol. 48, e12776 (2022).
Muneer, A. et al. Non-canonical function of histone methyltransferase G9a in the translational regulation of chronic inflammation. Cell Chem. Biol. 30, 1525–1541 e7 (2023).
Lim, K. H., Yang, S., Kim, S. H. & Joo, J. Y. Identifying new COVID-19 receptor neuropilin-1 in severe Alzheimer’s disease patients group brain using genome-wide association study approach. Front Genet 12, 741175 (2021).
Shen, W. B. et al. SARS-CoV-2 invades cognitive centers of the brain and induces Alzheimer’s-like neuropathology. Preprint at bioRxiv https://doi.org/10.1101/2022.01.31.478476 (2022).
Xie, K. et al. Tenascin-C deficiency ameliorates Alzheimer’s disease-related pathology in mice. Neurobiol. Aging 34, 2389–2398 (2013).
Kouser, M. et al. Loss of predominant Shank3 isoforms results in hippocampus-dependent impairments in behavior and synaptic transmission. J. Neurosci. 33, 18448–18468 (2013).
Rademakers, R., Cruts, M. & van Broeckhoven, C. The role of tau (MAPT) in frontotemporal dementia and related tauopathies. Hum. Mutat. 24, 277–295 (2004).
Bouleau, S. & Tricoire, H. Drosophila models of Alzheimer’s disease: advances, limits, and perspectives. J. Alzheimers Dis. 45, 1015–1038 (2015).
Chakrovorty, A., Bhattacharjee, B., Saxena, A., Samadder, A. & Nandi, S. Current naturopathy to combat Alzheimer’s disease. Curr. Neuropharmacol. (2022).
Callens, M., Loncke, J. & Bultynck, G. Dysregulated Ca(2+) homeostasis as a central theme in neurodegeneration: lessons from Alzheimer’s disease and Wolfram syndrome. Cells 11, 1963 (2022).
Sharma, V. K. & Singh, T. G. CREB: a multifaceted target for Alzheimer’s disease. Curr. Alzheimer Res 17, 1280–1293 (2020).
Jackson, J. et al. Targeting the synapse in Alzheimer’s disease. Front Neurosci. 13, 735 (2019).
Wu, C. H. et al. A bidirectional switch in the Shank3 phosphorylation state biases synapses toward up- or downscaling. Elife 11, e74277 (2022).
Jeong, J., Li, Y. & Roche, K. W. CaMKII phosphorylation regulates synaptic enrichment of Shank3. eNeuro. https://doi.org/10.1523/eneuro.0481-20.2021 (2021).
Hanson, J. E. et al. GluN2A NMDA receptor enhancement improves brain oscillations, synchrony, and cognitive functions in dravet syndrome and Alzheimer’s disease models. Cell Rep. 30, 381–396 e4 (2020).
Liu, S. L. et al. The role of Cdk5 in Alzheimer’s disease. Mol. Neurobiol. 53, 4328–4342 (2016).
Liang, Z., Liu, F., Grundke-Iqbal, I., Iqbal, K. & Gong, C. X. Down-regulation of cAMP-dependent protein kinase by over-activated calpain in Alzheimer's disease brain. J. Neurochem 103, 2462–2470 (2007).
Ni, Y. et al. Activation of beta2-adrenergic receptor stimulates gamma-secretase activity and accelerates amyloid plaque formation. Nat. Med 12, 1390–1396 (2006).
Sarajarvi, T. et al. Genetic variation in delta-opioid receptor associates with increased beta- and gamma-secretase activity in the late stages of Alzheimer’s disease. J. Alzheimers Dis. 48, 507–516 (2015).
Sabo, S. L., Ikin, A. F., Buxbaum, J. D. & Greengard, P. The amyloid precursor protein and its regulatory protein, FE65, in growth cones and synapses in vitro and in vivo. J. Neurosci. 23, 5407–5415 (2003).
Sabo, S. L., Ikin, A. F., Buxbaum, J. D. & Greengard, P. The Alzheimer amyloid precursor protein (APP) and FE65, an APP-binding protein, regulate cell movement. J. Cell Biol. 153, 1403–1414 (2001).
Margiotta, A. et al. Role of SNAREs in neurodegenerative diseases. Cells 10, 991 (2021).
El-Ganainy, S. O. et al. Intranasal oxytocin attenuates cognitive impairment, beta-amyloid burden and tau deposition in female rats with Alzheimer’s disease: interplay of ERK1/2/GSK3beta/Caspase-3. Neurochem Res 47, 2345–2356 (2022).
Ghosh, A. & Giese, K. P. Calcium/calmodulin-dependent kinase II and Alzheimer’s disease. Mol. Brain 8, 78 (2015).
Pesini, A. et al. Brain pyrimidine nucleotide synthesis and Alzheimer disease. Aging (Albany NY) 11, 8433–8462 (2019).
Hoffmann, H. M. Boosting cognition with a hormone. Science 377, 1042–1043 (2022).
Caceres, A., Vargas, J. E. & Gonzalez, J. R. APOE and MS4A6A interact with GnRH signaling in Alzheimer’s disease: Enrichment of epistatic effects. Alzheimers Dement 13, 493–497 (2017).
Moutinho, M. et al. The niacin receptor HCAR2 modulates microglial response and limits disease progression in a mouse model of Alzheimer’s disease. Sci. Transl. Med 14, eabl7634 (2022).
Huang, Y. et al. G protein-biased GPR3 signaling ameliorates amyloid pathology in a preclinical Alzheimer’s disease mouse model. Proc. Natl Acad. Sci. USA 119, e2204828119 (2022).
Baik, S. H. et al. A breakdown in metabolic reprogramming causes microglia dysfunction in Alzheimer’s disease. Cell Metab. 30, 493–507 e6 (2019).
Ahmed, M. M. et al. Innate immune system activation and neuroinflammation in down syndrome and neurodegeneration: therapeutic targets or partners? Front Aging Neurosci. 13, 718426 (2021).
Banks, W. A. The blood-brain barrier as an endocrine tissue. Nat. Rev. Endocrinol. 15, 444–455 (2019).
Aguilar, B. J., Zhu, Y. & Lu, Q. Rho GTPases as therapeutic targets in Alzheimer’s disease. Alzheimers Res Ther. 9, 97 (2017).
Wang, S. et al. TREM2 drives microglia response to amyloid-beta via SYK-dependent and -independent pathways. Cell 185, 4153–4169 e19 (2022).
Swerdlow, R. H. The Alzheimer’s disease mitochondrial cascade hypothesis: a current overview. J. Alzheimers Dis. 92, 751–768(2023).
Gabrielli, A. P. et al. Mitochondria profoundly influence apolipoprotein E biology. J. Alzheimers Dis. 92, 591–604 (2023).
Lee, J. H. et al. Altered relaxin family receptors RXFP1 and RXFP3 in the neocortex of depressed Alzheimer’s disease patients. Psychopharmacol. (Berl.) 233, 591–598 (2016).
Kouzuki, M. et al. Comparison of olfactory and gustatory disorders in Alzheimer’s disease. Neurol. Sci. 39, 321–328 (2018).
Doorduijn, A. S. et al. Olfactory and gustatory functioning and food preferences of patients with Alzheimer’s disease and mild cognitive impairment compared to controls: the NUDAD project. J. Neurol. 267, 144–152 (2020).
Murphy, C. Olfactory and other sensory impairments in Alzheimer disease. Nat. Rev. Neurol. 15, 11–24 (2019).
Bai, B. et al. Deep multilayer brain proteomics identifies molecular networks in Alzheimer’s disease progression. Neuron 105, 975–991 e7 (2020).
Fu, A. K. et al. IL-33 ameliorates Alzheimer’s disease-like pathology and cognitive decline. Proc. Natl Acad. Sci. USA 113, E2705–E2713 (2016).
Kunkle, B. W. et al. Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Abeta, tau, immunity and lipid processing. Nat. Genet 51, 414–430 (2019).
Johnson, E. C. B. et al. Cerebrospinal fluid proteomics define the natural history of autosomal dominant Alzheimer’s disease. Nat. Med 29, 1979–1988 (2023).
Drummond, E. et al. The amyloid plaque proteome in early onset Alzheimer’s disease and Down syndrome. Acta Neuropathol. Commun. 10, 53 (2022).
Dammer, E. B. et al. Multi-platform proteomic analysis of Alzheimer’s disease cerebrospinal fluid and plasma reveals network biomarkers associated with proteostasis and the matrisome. Alzheimers Res Ther. 14, 174 (2022).
Bellver-Sanchis, A. et al. G9a inhibition promotes neuroprotection through GMFB regulation in Alzheimer’s disease. Aging Dis. 15, 311–337(2023).
Libiger, O. et al. Longitudinal CSF proteomics identifies NPTX2 as a prognostic biomarker of Alzheimer’s disease. Alzheimers Dement 17, 1976–1987 (2021).
Lim, B. et al. Liquid biopsy of cerebrospinal fluid identifies neuronal pentraxin receptor (NPTXR) as a biomarker of progression of Alzheimer’s disease. Clin. Chem. Lab Med 57, 1875–1881 (2019).
Tzioras, M., McGeachan, R. I., Durrant, C. S. & Spires-Jones, T. L. Synaptic degeneration in Alzheimer disease. Nat. Rev. Neurol. 19, 19–38 (2023).
Skobeleva, K. et al. The STIM1/2-regulated calcium homeostasis is impaired in hippocampal neurons of the 5xFAD mouse model of Alzheimer’s disease. Int J. Mol. Sci. 23, 14810 (2022).
Chatterjee, P. et al. Plasma glial fibrillary acidic protein is associated with 18F-SMBT-1 PET: two putative astrocyte reactivity biomarkers for alzheimer’s disease. J. Alzheimers Dis. (2023).
Poon, C. H. et al. Functional roles of neuronal nitric oxide synthase in neurodegenerative diseases and mood disorders. Curr. Alzheimer Res 18, 831–840 (2021).
Grinan-Ferre, C. et al. Pharmacological inhibition of G9a/GLP restores cognition and reduces oxidative stress, neuroinflammation and beta-Amyloid plaques in an early-onset Alzheimer’s disease mouse model. Aging (Albany NY) 11, 11591–11608 (2019).
Pao, P. C. et al. A Cdk5-derived peptide inhibits Cdk5/p25 activity and improves neurodegenerative phenotypes. Proc. Natl Acad. Sci. USA 120, e2217864120 (2023).
Wen, Z. et al. Synaptic dysregulation in a human iPS cell model of mental disorders. Nature 515, 414–418 (2014).
Kang, Y. et al. A human forebrain organoid model of fragile X syndrome exhibits altered neurogenesis and highlights new treatment strategies. Nat. Neurosci. 24,1377–1391 (2021).
Qian, X. et al. Brain-region-specific organoids using mini-bioreactors for modeling ZIKV exposure. Cell 165, 1238–1254 (2016).
Pitts, M. W. et al. Barnes maze procedure for spatial learning and memory in mice. Bio. Protoc. 8, e2744 (2018).
Acknowledgements
This work was supported in part by grants NIH 1R41AG085859-01, R21AG071229, 1R41DK133051-01A1, and R01 GM133107-01, (to X.C.), R01MH111773, R01MH122692, RF1AG058160, R01NS104530 (to J.S.), R01AG084184, R01HD088626 and an endowed professorship from the Icahn School of Medicine at Mount Sinai (to J.J.), NIH T32GM135095 (to R.N.S.). R01AG065611 (to Z. W.). R35NS137480 and Dr. Miriam and Sheldon G. Adelson Medical Research Foundation (to G.-l.M.), R35NS116843 and RF1AG079557 (to H.S.). We used Thermo-Fisher Scientific Q Exactive HF-x that was upgraded with funding from UNC Lineberger Comprehensive Cancer Center. We used the NMR Spectrometer Systems at Mount Sinai, acquired with funding from the NIH’s SIG grants 1S10OD025132 and 1S10OD028504. The indication of using MS1262 for AD therapy is protected by US provisional patent applications jointly filed by UNC-CH and Mount Sinai.
Author information
Authors and Affiliations
Contributions
Xian Chen conceived the project and wrote the manuscript. X.C., J.S., and J.J. supervised cross-species proteomics/multiomics analysis, animal studies, MS1262 synthesis and characterization, respectively. L.X., R.N.S., and Y.X. performed and interpreted the experiments. L.X. conducted all proteomic experiments, analyzed the data, and wrote the report. J.A.W. conducted mouse-to-patient correlated multiomics analysis. F.Z., H.S., and G.M. conducted m6A RIP-seq experiments. A.M. analyzed m6A RIP-seq data, proteomics, and patient data, and wrote the report. R.N.S. and J.S. designed and performed drug treatment, analyzed animal behavior, microdissected mouse hippocampal tissues, prepared the figures, and wrote part of the manuscript. Y.X., K.S.P., J.V., J.L., and J.J. designed, synthesized, and characterized MS1262 and wrote the report. C. X. and Z. W. prepared organoids. M.D. provided APPNLGF mice. L.G.C. Jr. provided 3xTg mice. Y-J.L., B.A, and P.D. conducted the electrophysiology experiments and data analysis. Y-D.L., C.D., L.Q., and Y.L. assisted with drug treatment and mouse brain tissue dissection.
Corresponding authors
Ethics declarations
Competing interests
X.C. is the Founder of Transchromix, LLC. J.J. is a cofounder and equity shareholder in Cullgen, Inc., a scientific cofounder and scientific advisory board member of Onsero Therapeutics, Inc., and a consultant for Cullgen, Inc., EpiCypher, Inc., Accent Therapeutics, Inc, and Tavotek Biotherapeutics, Inc. The Jin laboratory received research funds from Celgene Corporation, Levo Therapeutics, Inc., Cullgen, Inc. and Cullinan Oncology, Inc. The remaining authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Xie, L., Sheehy, R.N., Muneer, A. et al. Development of a brain-penetrant G9a methylase inhibitor to target Alzheimer’s disease-associated proteopathology. Nat Commun 16, 4222 (2025). https://doi.org/10.1038/s41467-025-59128-z
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-59128-z
This article is cited by
-
Dysregulation of post-translational modifications in glioma: advances in pathological mechanisms and clinical targeting strategies
Journal of Translational Medicine (2026)
