
Abstract
Human coronaviruses (CoV) cause respiratory infections that range from mild to severe. CoVs are a large family of viruses with considerable genetic heterogeneity and a multitude of viral types, making preventing and treating these viruses difficult. Comprehensive treatments that inhibit CoV infections fulfill a pressing medical need and may be immensely valuable in managing emerging and endemic CoV infections. As the main protease (Mpro) is highly conserved across many CoVs, this protease has been identified as a route for broad CoV inhibition. We utilize the advanced generative chemistry platform Chemistry42 for de novo molecular design and obtained novel small-molecule, non-peptide-like inhibitors targeting the SARS-CoV-2 Mpro. ISM3312 is identified as an irreversible, covalent Mpro inhibitor from extensive virtual screening and structure-based optimization efforts. ISM3312 exhibits low off-target risk and outstanding antiviral activity against multiple human coronaviruses, including SARS-CoV-2, MERS-CoV, 229E, OC43, NL63, and HKU1 independent of P-glycoprotein (P-gp) inhibition. Furthermore, ISM3312 shows significant inhibitory effects against Nirmatrelvir-resistant Mpro mutants, suggesting ISM3312 may contribute to reduced viral escape in these settings. Incorporating ISM3312 and Nirmatrelvir into antiviral strategy could improve preparedness and reinforce defenses against future coronavirus threats.
Similar content being viewed by others

Introduction
CoVs infections from diverse sources have become globally prevalent, resulting in multiple respiratory diseases with a spectrum of severity. Seven human-infecting CoVs have been recognized: HCoV-229E, HCoV-OC43, HCoV-NL63, HCoV-HKU1, Severe acute respiratory syndrome CoV (SARS-CoV), Middle East respiratory syndrome CoV (MERS-CoV), and SARS-CoV-2. Notably, SARS-CoV and MERS-CoV instigated outbreaks in 2002 and 2012, respectively. Since December 2019, the CoV disease 2019 (COVID-19) caused by SARS-CoV-2 has led to a global pandemic and public health emergency of international concern. The SARS-CoV-2 pandemic has caused over 777 million confirmed cases worldwide with more than 7 million deaths reported by the World Health Organization (WHO) as of April 6, 20251. Vaccines2,3,4,5,6 and neutralizing antibodies7,8,9, mainly targeting spike protein of SARS-CoV-2, have been developed and some have been approved for clinical use. However, most of these interventions have diminished efficacy due to treatment-resistant viral evolution and an immune escape mechanism caused by the high frequency of spike protein mutations in some Omicron subvariants. Conventional vaccines and neutralizing antibodies7 are delivered via intramuscular and intravenous injections with strict low-temperature storage requirements that pose logistical hurdles. Oral antiviral delivery is easily accessible and stable which is expected to alter the trajectory of SARS-CoV-2’s wide and rapid spread. This efficiency will enable patients to self-administer treatment while alleviating a heavy burden on the public health system.
The SARS-CoV-2 main protease (Mpro) or 3C-like protease (3CLpro) is a cysteine protease pivotal for viral replication. Mpro catalyzes the proteolytic cleavage of nascent viral polyprotein 1a and 1ab (pp1a and pp1ab) at conserved sites and directly mediates the maturation of non-structural proteins10,11. Mpro possesses unique substrate selectivity and is highly conserved within the coronavirus family, making it an attractive antiviral drug target12,13. Several Mpro inhibitors are currently clinically approved or in clinical testing against COVID-19. These inhibitors may be classified into two groups according to their chemical properties: i. peptidomimetics like Pfizer’s Nirmatrelvir14, which contains a cyano group that functions as a reversible covalent Mpro inhibitor ii. non-peptidomimetics like Shionogi’s non-covalent Mpro inhibitor Ensitrelvir15, which contains a triazine core. Notably, both drug classes possess unique limitations in their usage. To maintain a clinical response and sufficient drug plasma concentration, Nirmatrelvir must be combined with Ritonavir, a CYP3A and P-glycoprotein (P-gp) inhibitor. Combined usage of Ritonavir is contraindicated with a wide range of drugs and may result in drug-drug interaction (DDI) risk. A high plasma concentration of Ensitrelvir was needed to satisfy its in vivo antiviral efficacy15.
SARS-CoV-2 infection is particularly significant in immunocompromised individuals or those prone to chronic infections, as these conditions can facilitate more frequent viral mutations16,17. Although real-world surveillance has reported few cases of resistant viruses following the use of nirmatrelvir, the potential emergence of resistant strains remains a significant concern18,19,20,21,22. Thus, there is a clinical need to develop Mpro inhibitors targeting new sites that can overcome existing drug-resistant SARS-CoV-2 while mitigating the risk of future evolutionary mutations that may arise following continued treatment.
We employed a state-of-the-art generative chemistry platform, Chemistry42, to carry out the de novo molecular design of novel small-molecule, non-peptide-like inhibitors of SARS-CoV-2 Mpro. This large-scale virtual screening and structure-based optimization pipeline identified ISM3312, an irreversible covalent Mpro inhibitor. In this study, we present the discovery pipeline utilized to identify and develop ISM3312 and test its anti-viral efficacy as an orally delivered treatment for COVID-19. We show that ISM3312 possesses potent antiviral efficacy against multiple human coronaviruses, including current SARS-CoV-2 variants of concern, independent of P-gp inhibition. In vitro serial passaging experiments revealed no mutations within the Mpro binding region of ISM3312, suggesting its binding site may be less prone to developing resistance mutations. Furthermore, ISM3312 inhibited Nirmatrelvir-resistant viruses, which we term Mpro mutants. Finally, the sequential administration of ISM3312 with Nirmatrelvir may effectively mitigates the likelihood of viral resistance mutations of Mpro, thus positioning it as a promising broad-spectrum coronavirus inhibitor and strategic reserve candidate for potential future coronavirus infections.
Results
Discovery of a novel Mpro inhibitor
With the structural characterization of SARS-CoV-2 Mpro, it has become an attractive target for drug development, and numerous compounds have been identified through high-throughput screening12,13. AI-based drug design has been a critical tool in early-stage drug discovery efforts. During the COVID-19 pandemic, several attempts using AI/ML-based approaches generated structures that showed potential activity against SARS-CoV-2 Mpro23,24,25. Our group utilized our generative chemistry platform Chemistry4226,27,28,29, which integrates many generative AI models, virtual screening, and molecular annotation modules to carry out the de novo molecular design of small molecules with desired properties (Fig. 1a). In the utilized version of Chemistry42, there were over 40 generative models, including generative autoencoders27,30, generative adversarial networks31,32,33, flow-based approaches34, evolutionary algorithms35, language models36 and others. These models employ different molecular representations string-based, graph-based, and 3D-based. The model performance is computed based on the scores that molecular structures get from the reward modules such as Pharmacophore module, Pocket module (docking), ReRSA module (synthetic accessibility), Shape module and others22 and based on the structural diversity of the output provided by the model. The higher reward scores accumulated from diverse reward modules22 and the more diverse generated structures in terms of the cosine metric – the better model performance.

a Workflow of Chemistry 42 platform to generate SARS-CoV-2 main proteinase inhibitors. Performance of the design rounds is provided on the right side as bar charts, where SD- and SD+ are the number of inactives and actives in the single-dose experiments respectively, Tier I shows the number of compounds in the IC50 range of >10 µM, Tier II: between 10 µM and 1 µM, Tier III: 1 µM–500 nM, Tier IV: 500 nM–100 nM, Tier V: below 100 nM. The IC50 values of the most active isomers were considered only, less active stereoisomers were not considered in these statistics. b The structures of AI based Mpro inhibitors and IC50 (measurement after 90 min incubation, mean ± SD values shown, n = 3) and EC50 values (mean values shown; n = 2) of each compound. c In vitro ADME and pharmacokinetics data of AI-based Mpro inhibitors. Source data are provided as a Source Data file.
To identify novel Mpro inhibitors, we exploited the published X-ray structures of SARS-CoV-2 Mpro and homologous SARS-CoV Mpro available at the early days of global pandemics with the focus on co-crystals. We prioritized 4C-Ugi reaction-based scaffold (PDB:6W63 and 3V3M) over the peptidomimetic protease inhibitor N3 (PDB: 6LU7) due to its synthetic tractability and superior drug-likeness for potential oral administration. This scaffold gained prominence as Mpro inhibitors following the SARS-CoV outbreak in 2002, with seminal work including the identification of ML188 (PDB: 3V3M)37 and a close analog X77 (PDB:6W63) as early hit compounds. Subsequent work by Mesecar’s group37 then systematically explored this chemotype’s anti-coronavirus potential, establishing preliminary structure-activity relationships (SAR). However, their work only focused on non-covalent inhibition mechanisms. We initially focused on expedited delivery of Mpro inhibitors and synthetic accessibility factor played crucial role for the campaign and getting a molecule via 1-step synthesis route looked attractive for further optimization. Chemistry42 platform has been utilized to search for a good primary hit among the pre-filtered vendor stock (Fig. 1a, Round 1). The structure-based drug design experiment used 4 pharmacophore points from 6W63 ligand, pocket features (mandatory H-bond interaction to His67 and PLI Score), SOM (self-organized map) trained on cysteine protease inhibitors set derived from ChEMBL and stricter synthetic feasibility estimate (in terms of ReRSA) as the key driving force of the reward function. It should be noted that shape and 3D descriptors of the 6W63 ligand were not considered during this experiment to avoid structural bias at this round. The small set of compounds had been prioritized after the in silico experiment. Few of them then showed good in vitro potency against SARS-CoV-2 Mpro, including the set of a-chloroacetamides, known as an appropriate “fast track to hit”38 warhead as this covalent moiety binds via an irreversible interaction with the C145 residue. The first round of the compounds exhibited sub-micromolar potency against Mpro.
Then the generative chemistry engine of Chemistry42 has been utilized to perform versatile hit expansion and optimization (Fig. 1a Round 2). It should be noticed that the generative model pipeline did not require any specific training procedure on the reported chemical space of protease inhibitors (including Mpro inhibitors) and SOM module was disabled for this and the following rounds of molecular design. The docked pose of the best α-chloroacetamide hit (0.98 μM) from Round 1 was used as the template for Round 2. Its pharmacophore features (4 points) and shape features were used for the reward function value optimization. The peptide-like core with the α-chloroacetamide warhead was fixed using Anchor points module as 3D constraints and this constraint was considered by the generative models directly that allowed for more focused structural output. Metabolic Stability Enhancer performed replacements to mitigate soft spots, while ReRSA module forced generation of synthetically accessible Ugi-products. However, the stability of the chloroacetamide warhead was a concern due to its high thiol reactivity39. The CFA warhead has previously been reported by Ojida group to lower the covalent reactivity of EGFR inhibitors instead of acrylamide and chloroacetamide40. Thus, α-chlorofluoroacetamide (CFA) was used instead of chloroacetamide40 as a less reactive covalent warhead to optimize stability in plasma (both human and mice) (Fig. 1a Round 3). The setup of the following iterative molecular design experiments (Round 3) was organized similarly to Round 2 with the exception of the covalent warhead, since it has been changed to CFA. Among the CFA compounds designed during Round 3, lead compound 1A (Fig. 1b) exhibited the most potency against Mpro, with a half-maximal inhibitory concentration (IC50) value of 61 nM. However, compound 1A also inhibited CYP isoforms 1A2, 2C9, 2C19, 2D6, and 3A4 with IC50s of 24.3, 15.6, 9.5, 6.5, 1.45 μM, respectively. The A to B value of 1A in Caco-2 cells was 0.50 × 10−6 cm/s (Efflux Ratio (ER) = 48), which is relatively low permeability, explaining its weak cellular potency in VeroE6 cell lines (concentration for 50% of maximal effect (EC50) = 2.15 μM) and poor bioavailability (F = 9%) in mouse PK (Fig. 1c). Conversely, compound 1B (also designed during Round 3) exhibited an improved ADME profile with better translation of in vitro enzymatic potency to in vitro primary antiviral cell-based assay efficacy than compound 1A. Both compounds 1A and 1B exhibited moderate in vitro liver microsome stability.
Next, we sought to optimize the drug-likeness of lead compound 1B by improving Caco-2 permeability, reducing CYP inhibition risk, and improving oral exposure in mouse PK. During the lead optimization stage, introducing a methyl group (compound 2 and compound 3) substantially contributed to enzymatic potency towards Mpro (Fig. 1b), with IC50 values of 16 nM and 14 nM, respectively. Besides, Compound 2’s CYP inhibition was altered as it inhibited CYPs 1A2, 2C9, 2C19, 2D6, and 3A4 with IC50 values > 50, >50, >50, 32.8, 15.6 μM, respectively. Compound 3 inhibited CYP isoforms 1A2, 2C9, 2C19, 2D6, and 3A4 with all IC50 values > 50 μM, indicating lower DDI risk than compound 2. Furthermore, compounds 2 and 3 exhibited moderate permeability in Caco-2 assays with 1.61 × 10−6 cm/s and 1.34 × 10−6 cm/s, respectively. Consequently, compounds 2 and 3 exhibited improved cellular potency in live virus infected VeroE6 cell lines with EC50 = 68 nM and 71 nM, respectively. Despite decreased in vitro liver microsome stability was observed, we assessed the covalent reactivity of these two compounds by performing the glutathione (GSH) trapping assay at a concentration of 10 μM. Compounds 2 and 3 presented acceptable half-life values (t1/2) of 361 min and 1127 min, respectively (Fig. 1c), which were comparable to the clinically available Bruton’s tyrosine kinase (BTK) inhibitor, ibrutinib.
Considering both compounds’ favorable in vitro ADMET characteristics, we evaluated the in vivo pharmacokinetic (PK) profiles of compounds 2 and 3 (see the Supplementary Table 1 and Supplementary Fig. 1 for detailed PK result). Both 2 and 3 displayed similar, good oral exposure and bioavailability in mice, with F values of 75.8% and 72.0%, respectively (Fig. 1c). Compound 3 possessed reasonable bioavailability (F = 49.0%) in dogs and showed superior protease-selective inhibition towards SARS-CoV-2 Mpro in a 76-protease profiling panel (Fig. 1c, Supplementary Table 2). The maximum tolerated dose of compound 3 in vivo was up to 1000 mpk, BID in mice and 600 mpk, BID in dogs. Furthermore, compound 3 was well tolerated in a 14-day GLP tox study. Based on these findings, compound 3 (ISM3312) was selected as a preclinical candidate for further evaluation in the treatment of COVID-19 and currently Phase I study in healthy volunteers has been completed in China.
ISM3312 irreversibly inhibits Mpro
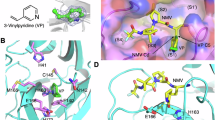
To determine ISM3312’s mechanism of action, we obtained the structure of ISM3312 bound to SARS-CoV-2 Mpro (PDB: 8WTS) at 1.56 Å resolution using X-ray diffraction (Fig. 2a and Supplementary Table 3). The crystal structure revealed that ISM3312 is covalently attached to Mpro via a carbon-sulfur bond formed between the sulfur atom of the catalytic C145 and the carbon atom of ISM3312’s CFA warhead (Fig. 2b and Supplementary Fig. 2). The CFA warhead can interact with the active site through multiple potential hydrogen bonds: i. the fluorine group of the CFA accepts a weak hydrogen bond from the backbone of C145 (NH, 3.2 Å) ii. the warhead amide oxygen accepts a hydrogen bond from the backbone of G143 (NH, 2.9 Å) iii. the pyrimidine group of ISM3312 forms one hydrogen bond with sidechain of H163 (2.9 Å). The tetrahydropyran (THP) group is extended to the solvent area and the attached amine oxygen accepts one hydrogen bond with the backbone of E166 (NH, 2.9 Å). Moreover, the benzene ring is deep within the M49-proximal hydrophobic sub-pocket, forming a π-π stacking interaction with H41. To profile the irreversible binding nature of ISM3312, we performed an enzymatic dilution assay against Mpro and compared ISM3312 to existing reversible Mpro inhibitors Nirmatrelvir, GC376, and Ensitrelvir. ISM3312 maintained its irreversible inhibitory activity towards Mpro for the entire four-hour incubation period. (Fig. 2c). In contrast to ISM3312, Nirmatrelvir, Ensitrelvir, and GC376 inhibitory activity diminished over time and was below 50% at the terminal four-hour timepoint (Fig. 2c), highlighting their reversible inhibitory nature, what’s more, the kinact (0.034 min−1) and KI (0.010 μM) values were also determined. Mpro across coronaviruses share the same catalytic mechanism and key activity residues, including C145 and H41. As predicted from the binding model of ISM3312 for Mpro, ISM3312 effectively binds to a variety of CoV catalytic pockets without collision (Fig. 2d). To test whether ISM3312 could effectively inhibit Mpro across multiple coronavirus subtypes, we performed an enzymatic inhibition assay against Mpro proteins from all coronaviruses known to infect humans. ISM3312 exhibited a broad and potent inhibition against Mpro derived from all coronaviruses tested (Fig. 2e). Nirmatrelvir exhibited higher IC50 values for all Mpro proteins other than SARS-CoV-2 variants Mpro assayed relative to ISM3312. At the same time, Ensitrelvir was ineffective against three of the eight coronavirus Mpro proteins (Fig. 2e).

a Crystal structure of ISM3312 in complex with SARS-CoV-2 Mpro. Cartoon representation of dimeric Mpro bound with ISM3312. Mpro is in grey and ISM3312 is shown as magenta ball-and-stick models in the active sites of Mpro. b ISM3312 binds to the pocket of the SARS-CoV-2 Mpro active site and interacts with the amino acid residues surrounding it by forming hydrogen bonds (shown in dashed line). The residues involved are visualized as sticks. c Enzymatic dilution assay against SARS-CoV-2 Mpro highlights the irreversible binding mechanism of ISM3312. d The predicted binding model of ISM3312 with Mpro from human coronavirus. e ISM3312 showed broad and potent inhibition against Mpro proteins from the listed coronavirus types. Measurement after 90 min incubation, mean values are shown; n = 3. Source data are provided as a Source Data file.
ISM3312 inhibits various CoVs in vitro
We evaluated the antiviral activity of ISM3312 and multiple existing Mpro inhibitors based on their ability to inhibit SARS-CoV-2 after infection of VeroE6 cells. The cytotoxicity of the compounds was measured in VeroE6 cells using the CellTiter-Glo® system in which the 50% cytotoxic concentration (CC50) of ISM3312 was more than 86.8 μM (Supplementary Table 4). ISM3312 displayed comparable, potent antiviral efficacy against a spectrum of tested SARS-CoV-2 variants including the ancestral, Delta, BA.2.3, BA.5, XBB.1, and EG.5 (Fig. 3a). The EC50 values against the different SARS-CoV-2 variants for ISM3312, Nirmatrelvir, and Ensitrelvir are summarized in Fig. 3b. Unlike Nirmatrelvir, ISM3312 inhibitory function was largely independent of the presence of a P-gp inhibitor, Elacridar (Fig. 3a). P-gp, or multidrug resistance protein 1 (MDR1), diminishes drug efficacy by removing the drug from the target tissues. P-gp is extensively distributed and expressed in the liver, pancreas, kidney, and colon. Since Nirmatrelvir, Ensitrelvir, and Remdesivir are substrates for P-gp24, their clinical usage, especially of Nirmatrelvir, necessitates treatment with PK boosters regarding dosage and administration. EC50 value of ISM3312 was significantly lower than the Nirmatrelvir group without Elacridar, and there was no significant difference between ISM3312 and the Nirmatrelvir group containing Elacridar (Fig. 3a). In the absence of Elacridar, the EC50 of Nirmatrelvir against different SARS-CoV-2 strains increased by 49 to 81 times, Ensitrelvir increased by 2 to 13 times, and ISM3312 increased by 1 to 2 times. This demonstrated that the antiviral effect of ISM3312 is independent of P-gp inhibitor Elacridar (Fig. 3b). P-gp inhibitors CP-100356 and Elacridar exhibit comparable effects (Supplementary Fig. 3). We characterized the caco-2 permeability data of Nirmatrelvir (Papp A to B < 0.1 × 10−6 cm/s, efflux ratio (ER) 1522) and Ensitrelvir (Papp A to B 0.42 × 10−6 cm/s, ER 68.8). Evidently, the cellular potency of Nirmatrelvir and Ensitrelvir is more influenced by their permeability and efflux ratio compared to ISM3312. For different strains of SARS-CoV-2, the selectivity index (SI = CC50/EC50) of ISM3312 was significantly greater than 100 (Supplementary Table 5).

a. Compound inhibition and EC50 values are calculated according to SARS-CoV-2’s CPE rates. Three independent experiments were performed with eight concentration gradients, each with quadruplicate wells. b EC50 values and fold-change relative to EC50 of different drugs with Elacridar (in parentheses) of different SARS-CoV-2 variants by VeroE6 CPE-based assay. c The inhibition of compounds and the value of EC50 is calculated according to MERS-CoV inhibition rate by indirect immunofluorescence assay (IFA). Inhibition Rate = [1 - (Infection Rate of the Test Compound - Cell Control) / Infection Rate of the Virus Control] × 100%. Following the analysis of inhibition rates, the EC50 is determined using a four-parameter fitting process. Representative images are shown on the right at 0.19 μM. Scale bar=100 μm. n = 3 biological replicates. d EC50 values of respective compounds against infection with 229E (Alphacoronavirus), NL63 (Alphacoronavirus), OC43 (Betacoronavirus) using IFA. Representative images are shown on the right at 0.19 μM (229E), 0.78 μM (NL63), and 0.19 μM (OC43), respectively. n = 3 biological replicates. In the bar plots, data are presented as the mean±s.e.m. Data were analyzed by using two-way ANOVA (a, c) or a one-way ANOVA (d) followed by Tukey’s multiple comparisons test. Exact P values are reported in the figure. Source data are provided as a Source Data file.
Next, the antiviral efficacy of ISM3312 was assessed against other human CoVs at the cellular level using an indirect immunofluorescence assay (IFA). The cytotoxicity of ISM3312 and multiple reference compounds were evaluated on Huh7 cells with the CellTiter-Glo® system (Supplementary Table 4). Viral antigens were labeled, and inhibition rates were measured at the indicated drug dilutions. ISM3312 demonstrated a lower EC50 against the two MERS-CoV variants (EMC-2012, 40 nM, and the mouse-adapted virus MA30, 50 nM) (Fig. 3c) relative to Nirmatrelvir and Ensitrelvir. The representative images shown on the right at a concentration of 0.19 μM indicated that ISM3312 inhibited MERS-CoV replication more potently than the other drugs at the same concentration (Fig. 3c). ISM3312 exhibited lower EC50s against the low pathogenicity coronavirus strains 229E, NL63, and OC43 than Nirmatrelvir and Ensitrelvir with EC50 values of 19, 167, and 56 nM, respectively (Fig. 3d). The representative fluorescent images on the right showed that, at the same drug concentrations, ISM3312 exhibits superior inhibitory activity against 229E (0.19 μM), NL63 (0.78 μM), and OC43 (0.19 μM) compared to Nirmatrelvir and Ensitrelvir (Fig. 3d). ISM3312 showed comparable or stronger antiviral activity than Nirmatrelvir and Ensitrelvir against CoVs in vitro, independent of P-gp inhibitors.
ISM3312 inhibits CoVs in human organoids
Next, we tested ISM3312 in an ex vivo human proximal airway organoid model as a more clinically relevant experimental system. Human proximal airway organoids are comprised of the major types of human airway epithelial cells and are widely used tools for understanding human respiratory viral pathogenesis41,42,43. ISM3312, Nirmatrelvir, and Ensitrelvir significantly reduced BA.2.3 virus loads and protein expression in both apical and basolateral viral release of 2D airway organoids (Fig. 4a, b). ISM3312 (200 nM) inhibited the production of virus particles by more than 100-fold relative to untreated controls (Fig. 4a). Additionally, IFA staining revealed a pronounced antiviral effect of ISM3312 against SARS-CoV-2 (Fig. 4b).

Comparison of the antiviral efficacy of ISM3312, Nimatrelvir, and Ensitrelvir against SARS-CoV-2 BA.2.3 was evaluated in 2D human proximal airway organoids. Viral RNA of supernatants from the apical and basolateral chambers of the 2D proximal airway organoids was quantified by real-time PCR. Representative data from independent experiments utilizing organoids from three different donors were displayed (a), n = 3 biological replicates. Organoids were stained with anti-NP (green) and DAPI (blue). Representative immunofluorescence images of SARS-CoV-2 infected organoids at 72 h post-infection (b), with a scale bar indicating 50 μm. Anti-MERS-CoV activity measurement of ISM3312 in 2D Airway Organoids. Viral RNA in the apical and basolateral chamber supernatants were quantified by qRT–PCR (c), n = 3 biological replicates. Representative immunofluorescence images of MERS-CoV infected organoids at 72 h post-infection (d), with a scale bar indicating 50 μm. e–g Viral RNA gene copy number were detected to evaluate antiviral efficacy of ISM3312 against 229E, NL63, OC43, and HKU1 in 2D airway organoids at two concentrations (20 nM, 200 nM). 229E, NL63, and HKU1 were performed with three biological replicates, while OC43 was performed with three biological samples and three technical replicates. Representative immunofluorescence images were shown in f and g. Scale bar = 50 μm. Data were analyzed by using two-way ANOVA followed by Tukey’s multiple comparisons test (a, c, e). Data in Fig.4a, c, e are presented as mean values ± SEM. Exact P values are reported in the figure. Source data are provided as a Source Data file.
Next, we tested whether ISM3312 similarly inhibits other human CoVs replication in human airway organoids. We selected beta CoVs (MERS-CoV, OC43, HKU1) and alpha CoVs (229E and NL63) as model viruses in the organoid system. Interestingly, ISM3312 exhibited broad antiviral activity against various CoVs at 20 or 200 nM concentration, including MERS-CoV (Fig. 4c, d), low-pathogenicity CoVs 229E, NL63, OC43 and HKU1 (Fig. 4e–g). Furthermore, it exhibited dose dependency against 229E, NL63, and OC43 infections, and at a concentration of 200 nM, ISM3312 demonstrated markedly increased antiviral potency (Fig. 4e–g). These data demonstrated that ISM3312 inhibited CoV infection at nanomolar concentrations in a physiologically relevant organoid system.
ISM3312 inhibits a spectrum of CoVs in vivo
To examine whether the results in vitro of ISM3312’s antiviral activity could be translated into in vivo model systems that mimic human CoV infection. We utilized the H11-K18-hACE2 C57BL/6 mice44,45,46, which are highly susceptible to SARS-CoV-2 infection and the gold standard model system for testing therapeutic interventions in this field. Mice were infected with ancestral SARS-CoV-2 strain (Fig. 5a), Omicron BA.2.3 variant (Fig. 5b), and XBB.1 variant (Fig. 5c). The mice were administered intragastrically with vehicle, Nirmatrelvir (300 mpk) or ISM3312 (75, 150, 300 mpk) starting at 2 h post infection, and twice daily.

a Weight loss and survival curve of Nirmatrelvir and ISM3312 against ancestral SARS-CoV-2 using H11-K18-hACE2 C57BL/6 mice (Mock, n = 4 mice; other groups, n = 8 mice). Virus titers in ancestral SARS-CoV-2 infected lungs (dpi 1) and brains (dpi 3) were quantified by FFA (Mock, n = 4 mice; other groups, n = 8 mice). b Weight loss and survival curve of Nirmatrelvir and ISM3312 against SARS-CoV-2 BA.2.3 using H11-K18-hACE2 C57BL/6 mice (n = 8 mice). Virus titers in the SARS-CoV-2 BA.2.3 infected lungs (dpi 1) and brains (dpi 4) were quantified by FFA (Mock, n = 4 mice; other groups, n = 8 mice). c Virus titers in the SARS-CoV-2 XBB.1 infected lungs (dpi 1) and brains (dpi 4) were quantified by FFA (n = 5 mice). d. Weight loss and survival curve of ISM3312 against MERS-CoV mouse adapted virus MA30 using hDPP4 knockin C57BL/6 mice (n = 6 mice). Viral titers in MERS-CoV MA30 infected lungs were quantified by FFA (n = 4–5 mice). e Viral gene copy number in 229E infected lungs from hAPN KI BALB/c mice were quantified by qRT–PCR (n = 5 mice). f Viral gene copy number in NL63 infected lungs from Ad5-hACE2 sensitized IFNARKO BALB/c mice were quantified by qRT–PCR (n = 4–5 mice). g. Weight loss and survival curve of ISM3312 against OC43 using WT C57BL/6 mice (n = 5–6 mice). Viral gene copy numbers in the OC43 infected brains and spinal cords at 5 dpi were quantified by qRT–PCR (n = 4 mice). Data were analyzed by using one-way ANOVA (a, c) or two-way ANOVA followed by Tukey’s multiple comparisons test (d, g). Two-sided unpaired t-tests were performed in e and f with Welch’s correction in e (data of Dpi 3). Data are presented as mean values ± SEM. Exact P values are reported in the figure. Source data are provided as a Source Data file.
ISM3312 exhibited 100% protection against ancestral SARS-CoV-2 strain, mirroring the efficacy of Nirmatrelvir at 300 mg/kg (mpk) (Fig. 5a). Notably, viral titers in the lung at day 1 post-infection (dpi) and brain at 3 dpi. (Fig. 5a) exhibited a significant reduction at the dosage of 75 mpk BID, with ISM3312 displaying a dose-dependent response. Additionally, ISM3312 demonstrated significant protective effects against the BA.2.3 variant, achieving viral inhibition in the lungs and brain at a dose as low as 150 mpk, which was comparable to Nirmatrelvir at 300 mpk (Fig. 5b). ISM3312 treatment attenuated the formation of pulmonary lesions (including inflammation, edema, and hemorrhage) (Supplementary Fig. 4b). Furthermore, ISM3312 effectively inhibited XBB.1 variant infection in lung and brain tissue (Fig. 5c). These data demonstrate that ISM3312 possesses strong antiviral activity and protection against a spectrum of SARS-CoV-2 variants.
Unlike reversible covalent or non-covalent inhibitors, which require sustained plasma concentrations above the unbound in vitro EC50 over a 24 h period, the irreversible covalent mechanism of ISM3312 may allow for distinct PK/PD requirements. To evaluate this, we conducted a pharmacokinetic study in C57BL/6 J mice under a 75 mg/kg BID for 4 days (Supplementary Fig. 5). The unbound plasma concentration of ISM3312 exceeded EC50 for WT SARS-CoV-2 for 4 h and for the BA.2.3 variant for 6 h post-dose. This transient but potent target coverage aligns with ISM3312’s irreversible mechanism and in vitro kinetics data—where covalent binding ensures prolonged inhibition even after drug clearance, thereby enabling robust in vivo efficacy despite shorter half-life compared to reversible covalent and non-covalent inhibitors.
After obtaining promising in vivo efficacy data with SARS-CoV-2, we hypothesized that ISM3312 may be an effective antiviral against other human CoVs. Thus, we utilized the hDPP4 knock-in (KI) C57BL/6 mice infected with mouse-adapted MERS-CoV MA30 to mimic MERS-CoV infection47. This model causes a fatal pulmonary disease phenotype associated with diffuse alveolar damage that recapitulates important pathological features found in patients47. Remarkably, ISM3312 was highly effective in protecting against MERS-CoV infection and significantly reduced viral load (>4 logs) at 75 mpk after two treatments on 1 dpi. ISM3312 dose-dependently inhibited MERS-CoV virus replication (Fig. 5d). Furthermore, ISM3312 treatment reduced alveolar wall thickening and edema at 4 dpi (Supplementary Fig. 4c).
To assess the antiviral effects of ISM3312 against low-pathogenicity CoVs, we utilized hAPN KI BALB/c to infect with 229E, and Ad5-hACE2 adenoviruses transduced IFNARKO BALB/c mice to infect with NL63 as described previously48. ISM3312 significantly reduced viral RNA copy number in 229E-infected mouse lungs (Fig. 5e) and caused a slight decrease in NL63 infection (2–4 folds) (Fig. 5j). Additionally, we used a mouse-adapted OC43 virus (VR759) developed from a suckling mouse model, which causes acute encephalitis, to evaluate the antiviral activity of ISM3312. ISM3312 protected against viral infection, which resulted in a significant reduction in viral RNA copy number in mouse brain and spinal cord tissues (Fig. 5g). These collective findings argue in favor of ISM3312 as a broad-spectrum antiviral agent against an array of human CoVs.
ISM3312 compensates for Nirmatrelvir-resistant viruses
Treatment with antiviral agents may select for treatment-resistant viral variants and subsequent therapeutic failure. For SARS-CoV-2, the selection of Mpro-resistant isolates induced by Nirmatrelvir has been reported by many groups with key resistant mutations, including S144A, E166A, E166V, and L167F reported and validated in vitro20,49. T98I/R/del (n = 4), E166V (n = 3), and W207L/R/del (n = 4) SARS-CoV-2 Mpro mutations were observed in PAXLOVID-treated subjects from clinical trial50. We utilized serial viral passages to evaluate the risk of drug-resistant mutations following ISM3312 treatment and measured ISM3312’s efficacy against Nirmatrelvir-resistant mutants. Specifically, we passaged Omicron BA.2.3 strain using VeroE6 cells for eighteen consecutive times with increasing concentrations of ISM3312 and Nirmatrelvir. We refer to the eighteen consecutive passaged groups as ISM3312 P18 and Nirmatrelvir P18 which showed resistance towards ISM3312 and Nirmatrelvir compared to the control P18, respectively (Fig. 6a, b). ISM3312 exhibited moderate-level resistance (about 15-fold increase in EC50) against the ISM3312 P18 virus compared to the Ctrl virus and low-level resistance (about 8-fold increase) against the Nirmatrelvir P18 virus. Strikingly, Nirmatrelvir displayed high-level resistance (about 153-fold increase) against the Nirmatrelvir P18 virus and no resistance (<threefold) against the ISM3312 P18 virus, relative to their respective the Ctrl viruses (Fig. 6c). Thus, sequential treatment with ISM3312 and Nirmatrelvir may enhance virus clearance and slow the acquired resistance rate.

SARS-CoV-2 BA.2.3 was serially passaged 18 times in VeroE6 cells with escalating concentrations of ISM3312 and Nirmatrelvir and P-gp inhibitor Elacridar (n = 3 biological replicates) (Method). Inhibition of passage eighteen viruses by ISM3312 (a) and Nirmatrelvir (b). c EC50 of ISM3312 and Nirmatrelvir against passage 18 resistant SARS-CoV-2. Fold change of EC50 mean values relative to inhibition of Ctrl strain from three biologically independent experiments. Color coding for variant frequency: white, no resistance ( < threefold); light blue, low-level resistance (3- to 10-fold); Medium blue, moderate-level resistance (10- to 50-fold); dark blue, high-level resistance ( > 50-fold). d Mutations in Mpro of ISM3312 and Nirmatrelvir resistance from the indicated passages. Dots indicate Ctrl at that residue. Mutations are shaded according to frequency. e IC50 (Compounds are compared for IC50 values under identical incubation time and conditions in each mutant assay) of Nirmatrelvir, Ensitrelvir, and ISM3312 against a panel of Mpro mutants induced by Omicron (P132H) and Nirmatrelvir (Y54A, N133H, F140A, E166A, E166V, L167F, H172Y) those previous reported and ISM3312 (T21I, L50L, P252L were bold labeled) using biochemical FRET assay. Colorimetric mapping of the dAffinity value (kcal/mol) for ISM3312-Mpro complex (f) and Nirmatrelvir-Mpro complex (g) by virtual alanine scanning. Residues around the binding sites are shown. Colors range from blue (negative values, indicating increased protein-ligand affinity) to red (positive values, indicating decreased protein-ligand affinity). ISM3312 is shown in pink, and Nirmatrelvir is shown in orange. h The affinity values for four mutations, including F140A, E166A, E166V and L167F. dAffinity values of E166A and E166V mutations in the Nirmatrelvir-bound system (bold labeled) were significantly higher than those in the ISM3312-bound system. Colorimetric mapping of the dStability value (kcal/mol) for ISM3312-Mpro comple x (i) and Nirmatrelvir-Mpro complex ( j) by virtual alanine scanning. Colors range from blue (negative values, indicating increased protein stability) to red (positive values, indicating decreased protein stability). k The dStability values for 4 mutations, including F140A, E166A, E166V and L167F. Data are presented as mean values ± SEM. Source data are provided as a Source Data file.
Viruses from early (7) and terminal (18) passages were sequenced via next generation sequencing (NGS) through which we identified three dominant mutations (T21I, L50F, 50^NP and P252L) in Mpro from ISM3312 P18 (Fig. 6d). These residues are distal to the drug-binding sites and in silico mutations analysis suggests that these mutations are unlikely to directly modulate protein-ligand interactions and hence have limited impact on binding affinity (Supplementary Fig. 6a–d). We then conducted comprehensive mutation assays to investigate potential allosteric or compensatory mechanisms that might influence drug activity. The enzymatic inhibition assay showed that the IC50 values of ISM3312 against the single T21I, L50F, and P252L mutations and combined mutations (bold labeled) in Mpro were comparable to the WT enzyme, with no significant resistance observed (<threefold), except 50^NP + P252L mutant with low level resistance (4.738-fold) (Fig. 6e). Previous reports indicated the mutations of T21I, L50F and P252L are predicted to increase protein stability and reduction in susceptibility of SARS-CoV-2 replication to improve viral fitness20,49,51,52,53. In addition, the P252L mutation is precisely located at the dimer-dimer interface of Mpro, and this residue may affect protein-protein interactions within the enzyme-substrate interactions complex54, thereby restoring the fitness of viral replication, this may partially explain resistance to ISM3312. We also observed mutations at L50F, S144A, E166A/V, A191V, F294K, and T304I in the Nirmatrelvir P18 virus (Fig. 6d). Several of these mutations were reported in previous works as contributors of resistance to Nirmatrelvir20,53.
To investigate efficacy of ISM3312 against Nirmatrelvir resistant mutants, we employed an enzymatic inhibition assay to test the inhibitory activities against a panel of Mpro mutants induced by Omicron (P132H) and Nirmatrelvir induced mutations (Y54A, N133H, F140A, E166A, E166V, L167F, H172Y) reported in the literature19,20,49,50,51,53. The IC50 values of ISM3312, Ensitrelvir, and Nirmatrelvir against P132H mutant compared to WT enzyme were comparable, and no significant resistance was observed (<threefold) (Fig. 6e). This finding aligns with previous observations of the inhibitory effects of these drugs against Omicron strains. Nirmatrelvir displayed significant resistance, ranging from 50- to 3187-fold, against N133H, E166A, E166V, and the combination of L50F + E166A + L167F mutants. Additionally, it exhibited moderate resistance to H172Y and low-level resistance to Y54A, F140A, and L167F mutants (Fig. 6e). Ensitrelvir displayed moderate- to high-levels resistance against all tested mutations (Fig. 6e). However, ISM3312 only showed moderate-level resistance (20- to 22-fold) against N133H and H172Y mutants, while presenting low-level resistance against the other tested variants (Fig. 6e).
In silico alanine screening has been used to evaluate the impact of single amino acid mutations on protein structure. To elucidate the structural mechanism underlying drug resistance, we applied the models to provide plausible explanations for the structural basis through which ISM3312 may overcome mutational resistance caused by F140A, E166A, E166V, and L167F induced by Nirmatrelvir. As shown in Fig. 6f–j, interaction sites critical for decreasing protein-ligand affinity and protein stability are marked in warm colors. In the context of the Mpro-ISM3312 complex, in silico alanine mutations of H41, G143, H163, and H164 are predicted to decrease affinity (Fig. 6f). In contrast, mutations of T26, L27, H41, Y54, F140, H163, H164, L167, H172, V186, R188, and Q192 are predicted to decrease protein stability (Fig. 6i). In Mpro- Nirmatrelvir complex, alanine mutation of residues L27, M49, F140, L141, S144, H163, E166, R188 and T190 (Fig. 6g) are predicted to decrease affinity whereas mutations of L27, H41, M49, P52, F140, S144, H163, H164, M165, L167, H172, V186, R188 and Q192 may reduce protein stability (Fig. 6j). These results indicate that the Nirmatrelvir-Mpro complex is more susceptible to losses of binding affinity or protein stability as mutations at a greater number of residues are predicted to impact this complex negatively. One explanation for this differential loss of affinity may be due to the different covalent reactivity of tested ligands. Irreversible ISM3312 has a stronger covalent reactivity and a slower off rate than the reversible Nirmatrelvir, meaning Mpro mutations may be less likely to affect its binding affinity. Differences in the respective protein-ligand interactions, binding site occupancies, and structure integrity may also contribute to the predicted differences in alanine mutation-induced protein stability.
Based on the calculated dAffinity (Fig. 6h) and dStability (Fig. 6k) values of F140A, E166A, E166V, and L167F, we concluded that dAffinity values of E166A and E166V mutations in the Nirmatrelvir-bound system (bold labeled) were significantly higher than those in the ISM3312-bound system. This finding suggested that the differential fold-change values of E166A and E166V in Nirmatrelvir relative to ISM3312 may be attributed to changes in their binding affinities. Residue E166 is located at the S1 pocket, and a set of hydrogen bonds are established between ligands (ISM3312 or Nirmatrelvir) and the side chain of F140, H163, E166, and H172. Thus, the intermolecular interaction involving E166 may be important in stabilizing the hydrogen bond network. The NH moiety of lactam in Nirmatrelvir forms a strong hydrogen bond (3.20 Å, Supplementary Fig. 6f) with the sidechain of E166. In comparison, the CH of pyrimidine in ISM3312 forms a weak hydrogen bond (3.26 Å, Supplementary Fig. 6e). Thus, when E166 is replaced with alanine or valine, these network-stabilizing interactions are no longer observed, which is reflected in the reduced binding affinity of Nirmatrelvir. Therefore, greater affinity losses are observed in the Mpro-Nirmatrelvir complex than in the Mpro-ISM3312 complex. The virtual mutations yield results that agree with experimental data, indicating that this modeling approach is valuable in understanding the structural basis of ISM3312’s broad inhibitory activity towards different Mpro mutants.
Discussion
Developing a more effective broad-spectrum anti-CoV drug remains a high unmet need to overcome current drug resistance and mitigate future evolution. Leveraging a generative AI-driven drug discovery platform, we produced ISM3312, an irreversible covalent Mpro inhitor upon 4C-Ugi reaction-based scaffold with the optimization of its α-chlorofluoroacetamide warhead and physicochemical properties. The uniqueness of this scaffold also gained parallel interest from other groups. For example, Wang’s group reported noncovalent inhibitor 23 R (PDB: 7KX5)55 and covalent inhibitor Jun9-62-2R (PDB: 7RN1) based on ML188. The dihaloacetamide covalent analogue Jun9-62-2R displayed moderate potency against Mpro with IC50 = 0.43 μM56. Yang’s group incorporated methyl diketone as the covalent warhead achieving a potent inhibitor Y180 (PDB: 7FAZ) with good in vitro antiviral activity with EC50 = 11.4 nM57. Ojida’s group later also introduced their CFA-group as covalent warhead afterwards in 4C-Ugi reaction-based scaffold58 resulting in compound 18 with IC50 56 nM. Furthermore, they introduced CFA warhead in peptidomimetic Mpro inhibitor59. Compared with these reported 4C-Ugi reaction-based Mpro inhibitors, a magic methyl group was introduced in the quaternary carbon of ISM3312, which formed a unique binding conformation, conferring superior enzymatic potency (IC50 = 14 nM) and cellular antiviral activity (EC50 = 71 nM). Besdies, this molecule also demonstrated much improved in vitro /in vivo pharmacokinetics properties, and a desired safety profile with low DDI risk. Most importantly, ISM3312 has a novel unique irreversible binding kinetics compared to Nirmatrelvir and Ensitrelvir, facilitating its ability to inhibit multiple CoVs and variants. ISM3312 is the first clinical Ugi-like scaffold Mpro inhibitor55,56,57,58,59 and first clinical compound with the novel α-chlorofluoroacetamide covalent warhead.
ISM3312 demonstrated excellent off-target selectivity and broad-spectrum activity against Mpro proteins derived from various human coronaviruses 229E, OC43, HKU1, NL63, MERS-CoV, SARS-CoV, and SARS-CoV-2. Furthermore, ISM3312 exhibited robust inhibition of various coronaviruses and clinically relevant SARS-CoV-2 variants in the human proximal airway organoid model and multiple murine CoV models. Importantly, ISM3312 possesses superior activity to Nirmatrelvir in vitro against SARS-CoV-2, MERS-CoV, 229E, NL63, OC43, and HKU1 with lower EC50 values and comparable in vivo activity against SARS-CoV-2. Additionally, the efficacy of ISM3312 is not affected by the P-gp transporter at the cellular level. The broad-spectrum antiviral efficacy may enable its use in preventing and controlling emerging viral infectious diseases.
Drug-resistant mutations have been well-documented in treating viral infections caused by HIV60, hepatitis B virus (HBV)61, hepatitis C virus (HCV)62, herpesviruses63,64, and influenza viruses65. For SARS-CoV-2, Mpro inhibitor-resistant mutants have been reported by several groups wherein key resistant mutations identified are S144A, E166A, E166V, and L167F based on the original strain20,49,50,66. To investigate resistant mutants against ISM3312, we employed in vitro serial passaging of the clinically prevalent Omicron BA.2.3 strain with ISM3312 or Nirmatrelvir. The sequencing results indicated that the mutations in the Mpro derived from BA.2.3, which confers resistance to Nirmatrelvir, are consistent with those previously reported in the literature20,49,50,66. Moreover, ISM3312 inhibited Nirmatrelvir-resistant viruses while Nirmatrelvir retained some inhibition towards ISM3312-resistant viruses. Sequential treatment with ISM3312 and Nirmatrelvir may combat drug-resistant strains, although further work is needed to confirm this hypothesis. In agreement with this hypothesis, the biochemical enzymatic assay revealed that ISM3312 exhibited higher inhibitory activity against mutants that were Nirmatrelvir-resistant (Fig. 6e). Furthermore, the mutation sites generated from the serial passaging virus treated with ISM3312 were not located within the enzymatic activity binding domain. This finding suggests that ISM3312 may be less likely to induce resistance mutations at drug-binding sites, which will be the focus of future studies. Thus, ISM3312 has the potential to overcome resistance to currently available COVID treatments, which may aid in the treatment of resistant strains that arise from the widespread use of drugs with the same mechanism of action.
Based on these positive and encouraging drug profile, ISM3312 is being evaluated in a phase 1 clinical trial (CTR20230768). Notably, ISM3312 was successfully synthesized from bench scale to the kilogram scale in only two steps from 4 commercially available starting materials in high step economy and atom efficiency, providing an overall yield of 15% with excellent chemical (98.1%) and optical purity ( > 98% de) (Supplementary Fig. 7). This yield was achieved without chromatographic purification and chiral separation. Thus, a critical advantage of ISM3312 is that its large-scale development cost would be considerably lower than currently available clinical Mpro inhibitors like Nirmatrelvir.
In summary, the novel Mpro inhibitor ISM3312 generated by a generative AI engine has demonstrated its broad-spectrum activity against human coronaviruses, including SARS-CoV-2, MERS-CoV, and certain common cold-causing coronaviruses. Its ability to specifically target the Mpro protein, crucial for viral replication, confers a strategic advantage in combating coronavirus outbreaks. The incorporation of this potent compound into our armamentarium will enhance preparedness efforts and fortify defenses against unforeseen challenges posed by emerging coronaviruses.
Methods
Ethics statement
In this study, the protocol and any amendment(s) or procedures involving the care and use of animals in this study will be reviewed and approved by the Preclinical R&D Unit of Medicilon Institutional Animal Care and Use Committee (IACUC) and WuXi AppTec (Shanghai) Co., Ltd IACUC. The infectious animal study was reviewed and approved by Institutional Animal Care and Use Committees of the First Affiliated Hospital of Guangzhou Medical University (2022276) and Guangzhou Customs Inspection and Quarantine Technology Center (IQTC202314). Human proximal airway organoids were derived from biopsied normal lung tissues collected from patients who underwent thoracic surgery at the First Affiliated Hospital of Guangzhou Medical University. Written informed consent was obtained from all participants prior to sample collection. The study protocol was reviewed and approved by the Ethics Review Committee of the First Affiliated Hospital of Guangzhou Medical University (approval number: ES-2023-193-02). All tissue samples were fully anonymized prior to analysis, and no identifiable personal information was accessed or used at any stage of the research.
Synthesis of compounds 1A, 1B, 2 & 3

To a solution of ethyl 2-chloro-2-fluoroacetate (1 g, 7.12 mmol, 826.45 μL, 1 eq) in THF (4 mL), MeOH (4 mL) and H2O (2 mL) was added NaOH (426.92 mg, 10.67 mmol, 1.5 eq). The reaction mixture was stirred at 25 °C for 12 hrs. TLC (PE: EA = 5:1) showed one new spot formed. The reaction mixture was concentrated under vacuum, adjusted pH=2 by 1 N HCl solution and extracted with EtOAc (50 mL×3). The combined organic phases were dried over anhydrous Na2SO4 and concentrated under vacuum. The reaction mixture was used in the next step directly without further purification. 2-chloro-2-fluoroacetic acid (0.6 g, crude) was obtained as a colorless oil. To a solution of 4-(thiazol-5-yl) aniline (492.8 mg, 2.8 mmol, 1 eq) and 1,1-difluoro-4-isocyanocyclohexane (406.0 mg, 2.8 mmol, 1 eq) in CF3CH2OH (4 mL) was added 2-chloro-2-fluoroacetic acid (313.6 mg, 2.8 mmol, 1 eq) and pyrimidine-5-carbaldehyde (302.4 mg, 2.8 mmol, 1 eq). The reaction mixture was stirred at 25 °C for 1 hr. LC-MS showed reactant was consumed and one peak of desired mass was detected. The reaction was concentrated under vacuum. The crude product was triturated with MTBE (20 mL) and filtered. Crude product (500.0 mg, 910.3 μmol, 32.75% yield) as off-white solid was obtained. Then it was separated by chiral SFC (column: Daicel ChiralPak IG (250*30 mm, 10 μm); mobile phase: [Neu-MeOH]; B%: 40%-40%,5.6;90 min) and concentrated under vacuum. The third peak is compound 1 A (33.89 mg, 60.34 μmol, 7.90% yield) was obtained as a yellow solid.
1H NMR (400 MHz, DMSO-d6): δ = 9.09 (s, 1H), 8.96 (s, 1H), 8.51 (s, 2H), 8.38 (d, J = 7.6 Hz, 1H), 8.33 (s, 1H), 7.74–7.55 (m, 3H), 7.48–7.11 (m, 1H), 6.59–6.38 (m, 1H), 6.09 (s, 1H), 3.85 (d, J = 6.4 Hz, 1H), 2.04–1.73 (m, 6H), 1.62–1.44 (m, 1H), 1.42–1.25 (m, 1H). 13C NMR (101 MHz, DMSO-d6) δ 166.44, 162.77 (d, J = 26.26 Hz), 158.02, 157.84, 154.44, 140.40, 137.21, 135.83, 131.95, 131.40, 128.29, 127.07, 123.51 (t, J = 241.39 Hz), 90.81 (d, J = 246.44 Hz), 60.26, 45.56, 31.20 (t, J = 24.24 Hz), 31.09 (t, J = 24.24 Hz), 27.62, 27.58. HRMS (ESI+): calcd for C23H22N5O2F3ClS [M + H]+ 524.1129, found 524.1126.

To a solution of 4-(trifluoromethoxy)aniline (10 g, 56.46 mmol, 7.63 mL) and pyrimidine-5-carbaldehyde (9.15 g, 84.69 mmol) in DCM (300 mL) was added tetrachlorotitanium (5.35 g, 28.23 mmol, 5.35 mL) and Et3N (17.14 g, 169.37 mmol, 23.57 mL) at 0 °C under N2. The reaction mixture was stirred at room temperature for 2 h. The reaction mixture was poured into ice water (200 mL) and extracted with DCM (2 × 50 mL). The water phase was adjusted to pH = 8 with sat. NaHCO3. The mixture was filtered and the filter cake was washed with DCM (2 × 50 mL). The filtrate was allowed to stand for layer separation. The water phase was extracted with DCM (3 × 50 mL). The several batches of organic phase were combined, washed with brine (60 mL), dried over Na2SO4, filtered and concentrated under vacuum to give a residue. The residue was purified by flash silica gel chromatography (Eluent of 0~100% ethyl acetate/Petroleum ether gradient @ 60 mL/min) to give the intermediate. To a solution of the intermediate (4.5 g, 16.84 mmol) in CF3CH2OH (100 mL) was added (2 R)-2-chloro-2-fluoro-acetic acid (2.27 g, 20.21 mmol) and 1,1-difluoro-4-isocyano-cyclohexane (2.44 g, 16.84 mmol). The mixture was stirred at 20 °C for 23 h. The reaction mixture was concentrated under reduced pressure to give a crude product. The crude product was purified by reversed-phase HPLC and chiral SFC (DAICEL CHIRALCEL OD (250 mm*50 mm, 10 µm); Mobile phase: A: Supercritical CO2, B: Neu-IPA; Isocratic: A:B = 55:45; Flow rate: 200 mL/min), concentrated under vacuum to afford 1B (0.895 g, 37.28% yield, fraction A, SFC: Retention time: 2.922 min).
1H NMR (400 MHz, DMSO-d6): δ 9.03 (s, 1H), 8.54 (s, 2H), 8.43 (d, J = 7.4 Hz, 1H), 7.36 (br s, 4H), 6.69–6.46 (m, 1H), 6.26–6.02 (m, 1H), 3.91 (br d, J = 6.9 Hz, 1H), 2.25–1.74 (m, 6H), 1.66–1.50 (m, 1H), 1.46–1.26 (m, 1H). 13C NMR (101 MHz, DMSO-d6) δ 166.38, 162.69 (d, J = 26.26 Hz), 157.97, 157.80, 148.24, 135.04, 133.32, 128.21, 123.47 (t, J = 241.39 Hz), 122.25 (q, J = 242.4 Hz),121.51, 90.81 (d, J = 246.44 Hz), 60.23, 45.57, 31.18 (t, J = 24.24 Hz), 31.08 (t, J = 24.24 Hz), 27.60, 27.54. HRMS (ESI+): calcd for C21H20N4O3F6Cl [M + H]+ 525.1123, found 525.1117.

A mixture of 4-(trifluoromethoxy)aniline (2 g, 11.29 mmol), 1-(pyrimidin-5-yl)ethan-1-one (1.66 g, 13.56 mmol), AcOH (4.20 g, 69.94 mmol, 4.00 mL) in toluene (30 mL) was degassed and purged with N2 for 3 times, and then the mixture was heated to reflux (120 °C) for 48 hrs with removal of water by Dean‐Stark trap under N2 atmosphere. The reaction mixture was filtered and the filtrate was concentrated under reduced pressure to give curde 1-(pyrimidin-5-yl)-N-(4-(trifluoromethoxy)phenyl)ethan-1-imine, which was used for next step without further purification. To a solution of imine (1 g, 3.56 mmol) in CF3CH2OH (10 mL) was added (2 R)-2-chloro-2-fluoro-acetic acid (667 mg, 3.56 mmol) and 4,4-difluorocyclohexanecarbonitrile (517 mg, 3.56 mmol). The mixture was stirred at 25 °C for 16 h. The reaction mixture was concentrated under reduced pressure to give a residue. The residue was purified by flash silica gel chromatography (Eluent of 0 ~ 70% Ethyl acetate/Petroleum ether gradient) to get product. The product was purified by prep-HPLC. The pure fractions were collected, and the volatile solvent was removed by evaporation. The racemate was further separated by SFC (DAICEL CHIRALPAK AD (250 mm*30 mm, 10 µm)); Mobile phase: A: Supercritical CO2, B: Neu-MeOH; Isocratic: A:B = 90:10; Flow rate: 50 mL/min) to afford two fractions. Compound 2 (28 mg, 30.94% yield, fraction A, SFC: Retention time: 1.816 min) was obtained.
1H NMR (400 MHz, DMSO-d6): δ 9.06 (s, 1H), 8.84 (s, 2H), 7.94–7.67 (m, 2H), 7.54–7.32 (m, 3H), 6.51–6.21 (m, 1H), 3.97–3.74 (m, 1H), 2.03–1.72 (m, 6H), 1.66 (s, 3H), 1.64–1.46 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 168.13, 162.79 (d, J = 25.25 Hz), 157.02, 156.28, 148.65, 134.88, 133.50, 133.44, 133.28, 123.65 (t, J = 240.38 Hz), 122.38 (q, J = 252.5 Hz), 121.82, 121.42, 91.25 (d, J = 247.45 Hz), 67.07, 46.59, 31.79 (t, J = 24.24 Hz), 31.70 (t, J = 24.24 Hz), 27.55, 27.48, 24.34. HRMS (ESI+): calcd for C22H22N4O3F6Cl [M + H]+ 539.1279, found 539.1278.

To a solution of 4-(chlorodifluoromethoxy) aniline (2.0 g, 10.33 mmol) in toluene (60 mL) was added p-TsOH (210.26 mg, 1.22 mmol) and 1-pyrimidin-5-ylethanone (1.15 g, 9.39 mmol) at Dean-Stark trap, The mixture was heated and stirred at 140 °C for 12 h. The mixture was filtered, and the filtered cake was wash with toluene (20 mL*3), the organic layer was concentrated under reduced pressure to give a crude. The crude was purified by flash silica gel chromatography (ISCO®; 40 g SepaFlash® Silica Flash Column, Eluent of 0 ~ 30% Ethyl acetate/Petroleum ether gradient @ 45 mL/min) to give N-(4-(chlorodifluoromethoxy)phenyl)-1-(pyrimidin-5-yl)ethan-1-imine (1.7 g, 5.71 mmol, 60.80% yield). To a solution of imine (1.35 g, 4.53 mmol) in CF3CH2OH (2 mL) were added H3PO4 (104.57 mg, 906.99 μmol, 62.24 μL), the mixture was stirred at 25 °C for 1 h, then (2 R)-2-chloro-2-fluoro-acetic acid (927.50 mg, 5.44 mmol, 66% purity) and 4-isocyanotetrahydropyran (504.02 mg, 4.53 mmol). The mixture was stirred at 25 °C for 12 h. The mixture was concentrated under reduced pressure to give a crude. The crude was purified by flash silica gel chromatography (ISCO®; 40 g SepaFlash® Silica Flash Column, Eluent of 0 ~ 100% Ethyl acetate/Petroleum ether gradient @ 40 mL/min) to give a crude. The crude was separated by prep-HPLC (column: Boston Green ODS 150*30 mm*5 μm; mobile phase: [water(FA)-ACN];B%: 48–78%,7 min) to give the racemate (57 mg, 2.11% yield). The racemate was separated by chiral SFC column: DAICEL CHIRALCEL OD-H (250 mm × 30 mm,5 μm); mobile phase: [Neu-ETOH]; B%: 20–20%, min; Mobile phase: A: Supercritical CO2, B: Neu-ETOH; Isocratic: A: B = 80:20; Flow rate: 60 mL/min). Compound 3 (7.38 mg, 16.04% yield, fraction A, SFC: Retention time: 1.797 min) was obtained.
1H NMR (400 MHz, CD3OD): δ 9.07 (s, 1H), 8.97–8.87 (m, 2H), 7.73 (d, J = 8.4 Hz, 1H), 7.48–7.29 (m, 3H), 6.32–6.15 (m, 1H), 4.11–3.91 (m, 3H), 3.56–3.45 (m, 2H), 1.91–1.74 (m, 5H), 1.71–1.55 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 167.83, 162.68 (d, J = 25.25 Hz), 156.99, 156.28, 149.67, 135.11, 133.50, 133.35, 133.31, 124.59 (t, J = 288.86 Hz), 122.27, 121.87, 91.26 (d, J = 247.45 Hz), 67.19, 66.17, 66.15, 46.43, 32.01, 31.98, 24.25. HRMS (ESI+): calcd for C21H22N4O4F3Cl2 [M + H]+ 521.0965, found 521.0964.
Gram-scale synthesis of ISM3312

(E)-N-(4-(chlorodifluoromethoxy)phenyl)-1-(pyrimidin-5-yl)ethan-1-imine (A): To a solution of SM1 (20.34 g, 167 mmol, 1.0 eq.) in toluene (200 mL) was added SM2 (35.5 g, 89.9 mmol, 1.1 eq.), 19.8 g MgSO4 and concentrated H2SO4 (1.6 g, 16.0 mmol, 0.1 eq.). The mixture was heated to 115 ± 5 °C and stirred for 24 h then the mixture was cooled to 30 ± 10 °C and charged DIPEA (1.0 g, 7.74 mmol, 0.05 eq.) and 5.0 g silica gel then stirred for 0.5 ~ 1 h, filtered with celite, and the wet cake was washed with 20 mL*2 toluene. The filtrate was concentrated to remove solvents and added MTBE (20 mL). The mixture was heated to 55 ~ 60 °C and stirred for 0.5 ~ 1 h. Then the mixture was cooled to -5 ~ 0 °C and stirred for 0.5 ~ 1 h, filtrated, washed with MTBE (20 mL). The wet cake was dried at 50 ± 5 °C to obtain a pale brown solid (23 g, 77.3 mmol, 99.9% purity, 46.3% yield).

(R)-2-((R)-2-chloro-N-(4-(chlorodifluoromethoxy)phenyl)-2-fluoroacetamido)-2-(pyrimidin-5-yl)-N-(tetrahydro-2H-pyran-4-yl)propenamide: To a solution of A (15.28 g, 51.3 mmol, 1.0 eq.) in CF3CF2OH (30 mL) was added SM4 (6.75 g, 60.7 mmol, 1.18 eq.). The mixture was cooled to −15 °C and carefully treated with a solution of SM3 in MTBE (14.71 g, 45.9% w/w, 60.0 mmol, 1.17 eq.) to keep IT between −20 ~ -5 °C. Then the mixture was heated to 25 ~ 30 °C and stirred for 32 h. The mixture was diluted with THF (15 mL) then stirred for 0.5 ~ 1 h. Then the mixture was added n-heptane (30 mL), cooled to 0 ~ 5 °C, stirred for 2 h, and filtrated. The wet cake was washed with THF/heptane (2.6 mL/5.1 mL) to afford the crude product. The crude product was triturated with THF (15 mL) and n-heptane (30 mL) at 0 ~ 5 °C for 1 ~ 2 h. Then the mixture was heated to 20 ~ 30 °C and stirred at 0 ~ 5 °C for 1.5 h. The mixture was filtered, washed with THF/heptane (2.6 mL/5.1 mL). ISM3312 free base was obtained (98.14% purity, 99.22% chiral purity, 32.7% assay yield).
Synthesis of chiral (2 R)-2-chloro-2-fluoroacetic acid

Step 1, preparation of 2-chloro-2-fluoroacetic acid: To a stirred sample of ethyl 2-chloro-2-fluoroacetate (100.56 g, 715.52 mmol, 1 eq) was added dropwise 5 N NaOH (29.05 g of NaOH was dissolved in 143 mL of H2O, 726.30 mmol, 1.02 eq) at 0 °C. The mixture was stirred at 25 °C for 16 h. 19F-NMR showed the most of ethyl 2-chloro-2-fluoroacetate was consumed and desired product was formed. The reaction mixture was extracted with MTBE (100 mL), the organic phase was discarded, and the water layer was adjusted pH=2 by 12 N HCl (61 mL), then extracted with MTBE (150 mL × 3). And the combined organic layers were washed with brine (80 mL), dried over Na2SO4, filtered and concentrated under reduced pressure to remove most of MTBE below 35 °C. 2-chloro-2-fluoroacetic acid (137.5 g, HNMR showed 58% purity, contained ~31% of MTBE and ~6.3% of EtOH) was obtained as a colorless oil. 19F-NMR (376 MHz, DMSO-d6): −144.138 (s, 1 F).
Step 2, preparation of INT1: To a mixture of 2-chloro-2-fluoroacetic acid (137.5 g, 58% purity, 709 mmol, 1 eq) in EtOAc (400 mL) cooled to −5 °C was added dropwise a solution of (1S)-1-phenylethanamine (86 g, 709.7 mmol, 1 eq) in EtOAc (200 mL). The mixture was stirred at 0 °C for 2 h and then warmed to 25 °C for 16 h during which time the two diastereoisomeric salts were deposited as a white solid (135.7 g), optical rotation = −4.9 (c = 1 in MeOH).
Step 3, preparation of INT2: A solution of INT1 (135.7 g) in acetone (400 mL) was stirred at 60 °C for 2 h then stirred at 25 °C for 16 h. The crude product was triturated twice with acetone to give INT2 (79 g) as a white solid. Optical rotation = −9.755 (c = 1 in MeOH).
Step 4, preparation of (2 R)-2-chloro-2-fluoroacetic acid: To a mixture of INT2 (36 g, 154 mmol, 1 eq) in DCM (100 mL) was added dropwise 12 N HCl (13.2 mL, 1.03 eq) at 0 °C. The mixture was stirred at 25 °C for 1 h. The organic phase was separated, and the water layer was washed with DCM (100 mL × 2), the combined organic layer was washed with brine (50 mL) and dried over MgSO4, filtered and concentrated under vacuum below 35 °C. Then the residue was added MTBE (200 mL) and stirred for 1 h, then filtered and the MTBE layer was concentrated under vacuum below 35 °C, (2 R)-2-chloro-2-fluoroacetic acid (25 g, HNMR showed contained ~50% MTBE) as a colorless liquid. Optical rotation = -36(c = 1.5 in MeOH). 1H-NMR: (400 MHz, DMSO-d6): 6.811 (d, J = 49.6 Hz, 1H). 19F-NMR (376 MHz, DMSO-d6): -144.138 (s, 1 F).
CYP inhibition
Test compounds (at 0.05, 0.15, 0.5, 1.5, 5, 15 and 50 µM) or blank PB were incubated in pooled human liver microsomes (at 0.2 mg/mL) in the presence of a cocktail substrate solution (phenacetin for CYP1A2, diclofenac for CYP2C9, S-mephenytoin for CYP2C19, dextromethorphan for 2D6 and midazolam for CYP3A4, respectively), and additional NADPH (at 10 mM) was added to initiate the reaction, all the incubations were conducted for 10 min at 37 °C in a water bath. After incubation, the reaction was terminated by cold stop solution (200 ng/mL tolbutamide and labetalol in ACN). The formation of the selective metabolite from its substrate was determined by LC-MS/MS and reported as enzyme activities (% VC) of each cytochrome P450 isoform. XL fit was used to plot the enzyme activities vs the test compound concentrations, and for non-linear regression analysis of the data. IC50 values were determined using 3- or 4-parameter logistic equations.
Caco-2 permeability
Caco-2 cells purchased from ATCC were cultured for 21 d in an incubator at 37 °C in a 5% CO2 and 90% relative humidity environment. Test compound (2.00 μM) and digoxin (10.0 μM) were dosed at bi-directionally in duplicate, while nadolol and metoprolol were tested at 2.00 μM in A to B direction in duplicate as well. The plate was incubated for 2 h in the CO2 incubator at 37 ± 1 °C, with 5% CO2 at saturated humidity without shaking. After incubation, samples were collected from both the donor and receiver sides. The Lucifer yellow rejection assay was applied to determine the Caco-2 cell monolayer integrity. The concentration of test and control compounds were measured by the LC-MS/MS method for the samples taken at 0 and 120 min. The apparent permeability coefficient Papp (cm/s) was calculated using the equation: Papp = (dCr/dt) × Vr / (A × C0), where dCr/dt is the rate of permeation of the compound across the cells, C0 is the initial concentration in the donor chamber, and A is s the surface area of the cell monolayer. The efflux ratio (ER) was estimated as Efflux Ratio = Papp (BA) / Papp (AB).
GSH trapping assay
Test compounds at 10 µM were incubated with GSH solution at 37 °C, while control /blank groups were set simultaneously. After incubation for each time point (0, 60, 120, 240, 360, and 1440 min), the stop solution was added to terminate the reaction, and the samples were quickly stored in −80 °C refrigerators until the last timepoint incubation was completed. The samples were analyzed using LC-MS/MS, and the remaining percentage of test compound was calculated, which was used to calculate the half-life (t1/2).
Liver microsome metabolic stability assay
Microsomes (final concentration 0.5 mg/mL), 100 mM phosphate buffer pH 7.4, and compound (final concentration 1 μM) were added to the assay plate and allowed to preincubate for 10 min at 37 °C. The reaction was initiated by the addition of NADPH (final concentration 1 mM), and the plate was constantly shaken at 37 °C. After 0, 5-, 15-, 30-, 45-, and 60 min aliquots were taken, and the reaction was quenched using cold acetonitrile. The samples were shaken for 10 min, then centrifuged at 3220 × g for 20 min at 4 °C and analyzed by LC-MS/MS. The in vitro intrinsic clearance was calculated from the rate of compound disappearance.
Plasma protein binding assay
The extent of protein binding was determined by equilibrium dialysis with a 96-well device. The test compound was spiked into CD-1 mouse and human plasma and the final concentration was 2 μM. An aliquot of 100 μL of plasma samples were dialyzed against an equal volume of dialysis buffer (Phosphate Buffered Saline, 100 mM, pH 7.4 ± 0.1) for 4 h with 5% CO2 at 37 °C. Triplicate incubations were performed. An aliquot of plasma sample was harvested before the incubation and used as T0 samples for recovery calculation. After incubation, 50 μL of samples were transferred from the plasma side as well as the buffer side into new 96-well plates. Each sample was added with equal volume of opposite blank matrix (buffer or plasma) to reach a final volume of 100 μL with volume ratio of plasma: Dialysis Buffer at 1:1 (v:v) in each well. All samples were further processed by adding 300 μL stop solution (Acetonitrile/Methanol (50:50, v: v)) containing internal standards (tolbutamide at 200 ng/mL). The resulting supernatant was diluted 1:1 with pure water and sealed and shaken for 10 min prior to LC-MS/MS (API 4000, AB Sciex, MA, USA) analysis.
The following equations for calculation were used:
[F] = Concentration of compound in free fraction (from receiver chamber)
[T] = Concentration of compound in total plasma fraction (from donor chamber)
[T0] = Concentration of compound in T0 samples (before dialysis)
Pharmacokinetic profile (mice)
The protocol and any amendment(s) or procedures involving the care and use of animals in this study will be reviewed and approved by the Preclinical R&D Unit of Medicilon Institutional Animal Care and Use Committee (IACUC) and WuXi AppTec (Shanghai) Co., Ltd IACUC. All mice were housed in a specific pathogen-free (SPF) facility under a 12 h light/12 h dark cycle, at a constant temperature of 22 ± 2 °C and relative humidity of 50 ± 10%. Male CD-1 mice (n = 3 per group, age: 7–9 weeks, IACUC no.: PK01-001-2019v1.0 for 1A) were subcutaneously injected with compound 1A at a dose of 20 mg/kg dissolved in 0.1 % Tween 80 and 1 % carboxymethyl cellulose in 98.9% distilled water, (homogenous hazy suspension, 4 mg/mL), and intravenously injected with 5 mg/kg dissolved in 20% HP-β-CD in water:10 mM PBS(pH7.4) with 0.5%Tween80 = 10:90 (clear solution, 1 mg/mL). Male CD-1 mice (n = 3 per group, age: 7–9 weeks, IACUC no.: 09013-21049 for 1B) were subcutaneously injected with compound 1B at a dose of 20 mg/kg dissolved in 20% HP-β-CD in water: 10 mM PBS(pH7.4) with 0.5%Tween80 = 10:90 (homogenous white suspension, 2 mg/mL), and intravenously injected with 1 mg/kg dissolved in 20% HP-β-CD in water: 10 mM PBS (pH7.4) with 0.5%Tween 80 = 10:90 (clear solution, 0.2 mg/mL).
Male CD-1 mice (n = 3 per group, age: 7–9 weeks, IACUC no.: 09013-21161 and 09013-21182 for 2) were orally given with 2 at a dose of 100 mg/kg dissolved in 0.1 % Tween 80 and 1 % carboxymethyl cellulose in 98.9% water (white suspension, 10 mg/mL) and intravenously injected with 1 mg/kg dissolved in 20% HP-β-CD in water: 10 mM PBS(pH7.4) with 0.5%Tween80 = 10:90 (clear solution, 0.2 mg/mL).
Male CD-1 mice (n = 3 per group, age: 7–9 weeks, IACUC no.: 09013-21357 for 3) were orally given with 3 at a dose of 100 mg/kg dissolved in 20% HP-β-CD in water: 10 mM PBS (pH7.4) with 0.5%Tween 80 = 10:90 (hazy solution, 10 mg/mL) and intravenously injected with 1 mg/kg dissolved in 20% HP-β-CD in water: 10 mM PBS (pH7.4) with 0.5%Tween 80 = 10:90 (clear solution, 0.2 mg/mL).
Blood samples (about 0.025 mL per time point) were collected at 0.083, 0.25, 0.5, 1, 2, 4, 8, and 24 h post-dose. All blood samples were transferred into pre-chilled commercial EDTA-K2 tubes and placed on wet-ice until centrifugation. Plasma samples were obtained following the standard procedures and the concentrations of the compound in plasma samples were analyzed by LC-MS/MS. PK parameters were calculated by non-compartmental analysis with the Phoenix WinNonlin software.
Female C57BL/6 J mice (n = 3 per group, age: 7–9 weeks, IACUC no.: 09013-22371 for 3) were orally given with 3 at a dose of 75 mg/kg dissolved in PEG400 30% + Solutol HS15 20% + 100 mM pH 3.0 citrate buffer 50% (clear solution,7.5 mg/mL) BID for 4 days. Blood samples (about 0.025 mL per time point) were collected at 0.25, 0.5, 1, 4, 8, 8.5, 10, 12, 16 and 24 h post-dose. All blood samples were transferred into pre-chilled commercial EDTA-K2 tubes and placed on wet-ice until centrifugation. Plasma samples were obtained following the standard procedures and the concentrations of the compound in plasma samples were analyzed by LC-MS/MS. PK parameters were calculated by non-compartmental analysis with the Phoenix WinNonlin software.
Pharmacokinetic profile (dog)
The protocol and any amendment(s) or procedures involving the care and use of animals in this study will be reviewed and approved by the WuXi AppTec (Nanjing) Co., Ltd Institutional Animal Care and Use Committee (IACUC no.: NJ-20241025-Dogs). Male Beagle dogs (n = 3 per group, non-naïve) and female Beagle dogs (n = 3 per group, non-naïve) (IACUC no.:)were treated with a solution of compound 3 dissolved in 8% (v/v) VE-TPGS + 0.5% (w/v) HPMC E5 in 100 mM pH 4.5 citrate buffer (clear solution, 4 mg/mL) for oral at the administrated dose of 20 mg/kg (PO) and Saline (clear solution, 2 mg/mL) at the intravenous administrated dose of 2 mg/kg (IV). Blood samples (about 0.025 mL per time point) were collected at 0.083, 0.25, 0.5, 1, 2, 4, 8, and 24 h post-dose after administration. All blood samples were transferred into pre-chilled commercial EDTA-K2 tubes and placed on wet-ice until centrifugation. Serum Plasma samples were obtained following the standard procedures and the compound concentrations in the plasma samples supernatant were analyzed by LC-MS/MS. PK parameters were calculated using non-compartmental analysis with the Phoenix WinNonlin software.
Human coronavirus proteases (Mpros) activity inhibition assays
The human coronavirus proteases in reaction buffer (20 mM Tris (pH 7.3), 100 mM, NaCl, 1 mM EDTA, 5 mM TCEP, and 0.1% BSA) were pre-incubated with the test compounds at Room temperature for 30 min, then the reaction was initiated by adding corresponding substrate and incubation at 30 °C for 1 h. Reference compound GC376 was provided by WuXi AppTec and was included in each plate to ensure assay robustness. Test compounds were tested at a single dose or 10 doses of titration in duplicate. Compounds were added to an assay plate (384w format) using ECHO in duplicate wells. The final concentration is 10 μM for the single-dose experiment. For the full dose-response experiment, samples were 3-fold serially diluted starting from 25 μM for ten doses and added to an assay plate in duplicate wells. The final concentration (μM) of each compound was 25, 8.33, 2.778, 0.926, 0.309, 0.103, 0.034, 0.011, 0.0038, and 0.0013. Mpro protein (25 μL) was added to an assay plate containing test compounds using a Multidrop. The test compound and Mpro protein were pre-incubated at RT for 30 min. Then, substrate (5 μL, 150 μM) was added to an assay plate. For 100% inhibition controls (HPE, high percent effect), 10 μM of Nirmatrelvir was added. The same volume of DMSO was added for no inhibition controls (ZPE, zero percent effect). The final DMSO concentration was 1%. Each activity testing point had a relevant background control without the enzyme to remove the fluorescence interference of the compound. After 60 min incubation at 30 oC, the fluorescence signal (RFU) was detected using a microplate reader M2e (SpectraMax) at Ex/Em = 340 nm/490 nm. The inhibition activity was calculated using the formula below; IC50 values were calculated using the Inhibition% data with the GraphPad Prism software using the nonlinear regression model of log(inhibitor) vs. response - variable slope (four parameters):
Inhibition% = ((CPD ‒ BGHPE) ‒ (ZPE ‒ BGZPE)) / ((HPE ‒ BGHPE) ‒ (ZPE ‒ BGZPE)) × 100. The time values were plotted on the X-axis, while the enzyme kinetics data were plotted on the Y-axis. The formula Y = (Vi/Kobs) * (1 - Exp(-Kobs*X)) +C was applied to obtain different Kobs values for various compound concentrations. Kobs against [I] were then plotted and fitted to obtain kinact and KI. Mpro enzymatic assay conditions for human coronaviruses are shown in Supplementary Table 6.
Jump dilution assay for SARS-CoV-2 Mpro
Test compounds and SARS-CoV-2 Mpro enzyme were incubated at a 1:1 molar ratio of 2 μM compound and 2 μM enzyme in assay buffer for 20 min. The mixture was then diluted 50-fold into assay buffer, then 5 μL transferred to wells of a 384-well assay plate. Enzyme activity was monitored at Ex/Em of 340 nm/490 nm after addition of 5 μL of 60 μM peptide substrate. Final reaction conditions were 20 nM enzyme with 20 nM compound and 30 μM peptide substrate. Data will be exhibited as inhibition rate using DMSO controls with and without Mpro enzyme.
Protein expression and purification of SARS-CoV-2 Mpro
Full length SARS-CoV-2 Mpro (NC_045512) were expressed and purified as previously described13. Coronavirus Mpro exists as monomer in the solution. The final purified Mpro was stored in 50 mM Tris, 1 mM EDTA, pH7.5.
Crystallization, data collection and structure determination
SARS-CoV-2 Mpro was incubated with 2.95 mM compound for 1 h and the complex (20 mg/ml) was crystallized by hanging drop vapor diffusion method at 18 °C. Crystals were mounted under the well buffer containing 0.05 M Calcium Chloride, 0.1 M Bis-Tris pH 6.5, 30% v/v PEG Monomethyl ether 550. The cryo-protectant solution contained 20% glycerol. Data was collected at European Synchrotron Radiation Facility with PILATUS3 2 M as detector. Data integration and scaling were carried out using the program XDS. The structure was determined by molecular replacement with the Phaser (CCP4) using PDB: 6LU7 as a search template. The initial model was built and refined via iterative cycles of manual model adjustment and refinement with Coot and Refmac5. Ligands were placed on the omit map and further refined.
Model preparation, virtual alanine, and resistance mutation scanning
The SARS-CoV-2 3CL-ISM3312 complex was obtained from our crystal coordinates (8WTS), and the 3CL-NV complex (7WFS) was retrieved from the PDB database. All the ions and water molecules from the two crystal structures were eliminated. Then, the MOE-Quick prep tool was used to optimize and prepare the ligands and protein targets to modify the breaks or defects of the protein structure.
A virtual evaluation of the impact of SARS-CoV-2 3CL mutations on the protein’s stability and the affinity toward ISM3312 or Nirmatrelvir (NV) was conducted using the “Protein Design” module of MOE using the previously mentioned complex structure. In particular, the “Alanine Scan” and “Resistance Scan” tools were used to perform two virtual mutagenesis experiments. First, we applied the Alanine Scan interface, in which each of the amino acids around the binding site was mutated into an alanine residue, calculating at each given time the energy difference between the mutated protein and the WT form concerning both the potential energy of the protein itself (dStability) and the affinity toward ISM3312 or NV (dAffinity). Then, we used the Resistance Scan interface to investigate the impact of a selected pool of mutations: F140A, E166A, E166V, and L167F. For both types of calculations, the conformational sampling was carried out through LowModeMD using the AMBER10:EHT forced field coupled with the generalized Born implicit solvent model; the dAffinity value was determined through the GBVI/VSA method.
Single-dose duplicate protease profiling
ISM3312 was tested in a single dose duplicate at 10 µM against 76 proteases at Reaction Biology Corporation. Control compounds were tested in a 10-dose IC50 with 3-fold serial dilution starting at 10 µM*. (*Start at different concentrations for some enzymes.) The protease activities were monitored as a time-course measurement of the increase in fluorescence signal from fluorescently labeled peptide substrate, and the initial linear portion of slope (signal/min) was analyzed.
FRET assay for SARS-CoV-2 mutants Mpro
The screening of inhibitors and IC50 measurements were performed using a fluorogenic substrate (dabcyl-KTSAVLQ/SGFRKM-edans) derived from the cleavage sites on SARS- CoV -2 Mpro and was synthesized by GenScript. In the FRET assay, Mpro of SARS-CoV-2 with mutations were incubated with ISM3312 and the edans/dabcyl FRET substrate derived from the cleavage sites. After different incubation times shown below, the enhanced fluorescence due to the cleavage of this substrate catalyzed by the Mpro was monitored at 340 nm with excitation at 490 nm. The IC50 value of the individual sample was measured in a reaction mixture containing 20 mM Tris (pH 7.3), 100 mM NaCl, 1 mM EDTA, 5 mM TCEP, and 0.1% BSA. The experimental conditions in detail are listed in Supplementary Table 7.
Mice, viruses, and cells for virus infection
SARS-CoV-2 infection used specific pathogen-free 6-8-week-old H11-K18-hACE2 and OC43 infection used WT C57BL/6 mice were purchased from GemPharmatech Co., Ltd. (Jiangsu, China) and maintained in the Animal Care Facilities at the Guangzhou Medical University. All mice were housed in a specific pathogen-free (SPF) facility under a 12 h light/12-h dark cycle, at a constant temperature of 22 ± 2 °C and relative humidity of 50 ± 10%. For MERS-CoV infection, hDPP4 KI C57BL/6 mice47 were donated by Professor Stanley Perlman. hAPN KI BALB/c mice were used for 229E infection. Ad5-hACE2 transduced IFNAR-/- BALB/c mice were used for NL63 infection as previous described48, and five days post-transduction, mice were infected with NL63 intranasally in a total volume of 50 μL DMEM48.
The SARS-CoV-2 strains used in this research were isolated from COVID-19 patients in Guangzhou. The SARS-CoV-2 variants, including WT (GenBank: MT123291), Delta (IQTC-IM2175251), BA.2.3 (IQTC-IM22003633), BA.5 (GDPCC-303), XBB.1 (IQTC-1596943), and EG.5 (IQTC-IM23676) were preserved in Guangzhou Customs Inspection and Quarantine Technology Center (IQTC) BSL-3 Laboratory. These viruses were propagated in Vero E6 cells and stored at −80 °C. MERS-CoV EMC2012 and mouse-adapted MA30 strains were preserved in Biosafety Level 3 (BSL-3) Laboratory. All the infection experiments were performed at BSL-3 in Guangzhou Customs Inspection and Quarantine Technology Center or Korea Centers for Disease Control and Prevention (KCDC). Other low pathogenic coronavirus 229E, NL63, OC43, and HKU1 were propagated and preserved in BSL-2 in Guangzhou Medical University. The mice were administered intragastrically with vehicle, Nirmatrelvir (300 mpk) or ISM3312 (75, 150, 300 mpk) starting at 2 h post infection, and twice daily.
VeroE6 (ATCC, CRL-1586), Vero81 (ATCC, CCL-81), Huh7 (JCRB0403), and Huh7-hACE2 (In-house, constructed from Huh7) cells were grown in Dulbecco’s modified Eagle’s medium (DMEM, GIBCO, Grand Island, NY) supplemented with 10% fetal bovine serum (FBS), 1% penicillin/streptomycin. The cells were cultured at 37 °C in a fully humidified atmosphere containing 5% CO2.
Air-liquid interface culture of human proximal airway organoids
Three lines of human airway organoids were established using biopsied normal human lung tissues from patients who underwent surgical operations at the First Affiliated Hospital of Guangzhou Medical University as described previously43. In brief, human airway organoids were packaged in 3D Matrigel and maintained in an expansion medium for the proliferation of airway epithelial cells. After shearing mechanically into fragments and seeded in transwell inserts till approximately 90% cell confluence, the cells were maintained at the air-liquid interface by removing the expansion medium from the apical chamber, leaving it empty, while the basal differentiation medium was replaced every two days for 14 days till full proximal differentiation was achieved.
Cell viability assay
Cell viability was evaluated using a Cell Titer-Glo Luminescent Cell Viability Assay kit (Promega) according to the manufacturer’s instructions. In brief, VeroE6, Huh7, or human proximal airway organoid cells were seeded into opaque-walled 96-well plates for 24 h, and the cells were incubated with a medium containing the indicated concentration inhibitor at 37 °C for three days. Subsequently, the 100 μL of Cell Titer-Glo reagent was added to each well and following a 5 min shaking and 10 min incubation, luminescence was measured by GloMax 20/20 (TurnerBio Systems). The half-cytotoxic concentration (CC50) was assessed in the absence of viruses.
In vitro antiviral cell-based assay
For Fig. 1b, SARS-CoV-2 was provided by the Korea Centers for Disease Control and Prevention (KCDC). Vero cells were acquired from the ATCC and maintained in DMEM supplemented with 10% FBS and 1% penicillin/streptomycin. DMEM supplemented with 2% FBS and 1% penicillin/streptomycin was used as the assay medium. The main reagents used in this assay are Anti-SARS-CoV-2 N protein antibody, Alexa Fluor 488 goat anti-rabbit IgG (H + L) secondary antibody, and Hoechst 33342. Ten-point dose-response curves (DRC) are generated for each compound. Vero cells are seeded at 1.2 × 104 cells per well in black 384-well, μClear plates (Greiner Bio-One) 24 h before the experiment. SARS-CoV-2 is added at a multiplicity of infection (MOI) of ~0.0125 for viral infection. The cells are fixed at 24 hpi with 4% paraformaldehyde and analyzed by immunofluorescence. The acquired images are analyzed using software to quantify cell numbers and infection ratios, and antiviral activity is normalized to positive (mock) and negative (0.5% DMSO) controls in each assay plate. DRCs are fitted by sigmoidal dose-response models using XLfit 4 Software or Prism with the following equation:
EC50 and CC50 values are calculated. Remdesivir was used as a positive control.
In Fig. 3, for cytopathic effect (CPE): 2 × 104 Vero E6 cells were seeded in a 96-well plate for 24 h. The compounds with different dilution concentrations were mixed with SARS-CoV-2 (MOI = 0.01), and 200 μL mixtures were inoculated onto monolayer Vero E6 cells. Seventy-two hours after inoculation, CPE was scored by Celigo Image Cytometer. The inhibition of compounds and the value of EC50 is calculated from SARS-CoV-2’s CPE rates. Three independent experiments were performed with eight concentration gradients, each with quadruplicate wells, and one representative is shown.
In Fig. 3 and Supplementary Fig. 3, for indirect immunofluorescence assay (cell line): Huh7 cell (for MERS-CoV, 229E, OC43), Huh7-hACE2 cell (for NL63) and VeroE6 (SARS-CoV-2) were seeded into a black 96-well cell culture plates with clear bottom at a cell density of 2 × 104 per well and allowed to adhere until the cells were approximately 90% confluent, followed by treatment with different concentrations of drugs or an equivalent amount of solvent for 1 h. Then, the cells were infected with MERS-CoV, 229E, OC43, NL63 or SARS-CoV-2 at a multiplicity of infection (MOI) of 0.0125, 0.03, 0.25, 0.013, 0.05 at 37 °C for 1 h, followed by changing to fresh medium with the indicated concentrations of drugs. Infected cells were detected 24–48 h later using an immunofluorescence assay (IFA) as described previously. In brief, the infection and replication of the virus were determined by detecting the nucleoprotein (N) of different coronaviruses (Sino Biological, China) and followed by Alexa Fluor488-labelled anti-rabbit secondary antibody (Jackson). All the cells were stained with 4,6-diamidino-2-phenylindole (DAPI, Sigma, USA) for nuclear visualization. Celigo Image Cytometer or Perkin Elmer Operetta High Content Imaging System were used to scan and analyze the average infection ratio. EC50 was subsequently calculated. All SARS-CoV-2 and MERS-CoV infection experiments were performed in a biosafety level 3 laboratory. All primary antibodies used in this study were validated for their specific applications based on manufacturer information from Sino Biological (https://www.sinobiological.com).
In Fig. 4, for human proximal airway organoid infection: Mature airway organoids were inoculated with various coronaviruses, including SARS-CoV-2 BA.2.3, MERS-CoV, 229E, NL63, OC43, and HKU1 from the apical side at indicated MOIs (MOI = 0.5 for SARS-CoV-2, MOI = 1 for MERS-CoV and HKU1, MOI = 0.1 for 229E, NL63 and OC43) for two hours. After removal of the inoculum, the 2D airway organoids were washed with DPBS twice and maintained in a differentiation medium supplemented with ISM3312 or DMSO or other control drugs. At the indicated time points post infection, cell-free medium in the apical or basolateral chambers were harvested for the quantification of viral load, whereas the organoids were in parallel fixed in 4% paraformaldehyde (PFA) for the detection of intracellular virions using immunofluorescence staining. Briefly, total RNA was extracted from tissues or supernatants using RNA Purification Kit (EZB). Real-time PCR was performed using the EZProbe One Step qRT-PCR Probe Kit (ROX2 plus) on QuantStudio 6 (ABI) to measure viral loads (The primers are as shown in Supplementary Table 8). After 72 h post-infection, fix the transwell for virus antigen immunofluorescence staining. Finally, capture images using the Zeiss LSM880 High-Speed Super Resolution Laser Scanning Microscope.
Focus forming assay (FFA)
In some experiments, virus was titered using an FFA. VeroE6 cells were seeded in 96-well plates one day before infection. Virus cultures or lung homogenate were serially diluted and used to inoculate Vero E6 cells at 37 °C for 1 h. Inocula were then removed before adding 100 μl 1.6% carboxymethylcellulose warmed to 37 °C per well. After 24 h, cells were fixed with 4% PFA and permeabilized with 0.2% Triton X-100. Cells were then incubated with a rabbit anti-SARS-CoV-2 nucleocapsid protein polyclonal antibody (Cat. No.: 40143-T62, Sino Biological, Inc. Beijing), followed by an HRP-labelled goat anti-rabbit secondary antibody (Cat. No.: 111-035-144, Jackson ImmunoResearch Laboratories, Inc. West Grove, PA). The foci were visualized by TrueBlue™ Peroxidase Substrate (KPL, Gaithersburg, MD), and counted with an ELISPOT reader (Cellular Technology Ltd. Cleveland, OH). Viral titers were calculated as FFU per ml or per gram tissue. The titration determination method for MERS-CoV is like that of SARS-CoV-2, utilizing Vero 81 cells and staining with corresponding antibodies against MERS-CoV.
Viral passage in the presence of ISM3312 or Nirmatrelvir in VeroE6 Cells
To study drug-resistant mutations against ISM3312 or Nirmatrelvir, SARS-CoV-2-BA.2.3 was cultured in the presence of increasing concentrations of ISM3312 or Nirmatrelvir and efflux inhibitor Elacridar. To initiate passaging, VeroE6 cells were seeded in a 12-well plate at a density of 1.5 × 105 cells per well in complete medium (DMEM + 10% fetal bovine serum + penicillin/streptomycin), and both drug and virus were then added the following day. The drug was prepared in a threefold dilution series based on its original EC50, and the virus was added at 0.05 MOI per well. After 2-3 days post infection, the supernatants were harvest according to the CPE, and the supernatants were passaged to each well in the next culture plate. For each passage, multiple new cultures were initiated with the same concentration, as well as one-third lower and three-times higher concentrations. The passage exhibiting the maximum sustainable compound concentration was chosen for subsequent passaging. The passage culture was set up in triplicate (Rep 1, Rep 2, Rep 3) and passaging was performed independently. As passage control, BA.2.3 virus was passaged without ISM3312 or Nirmatrelvir in three independent wells. Values of EC50 for P18 virus against ISM3312 or Nirmatrelvir were determined based on CPE. Viral RNA was extracted for Next-Generation Sequencing (NGS) to identify the mutations in P7 and P18 derived from ISM3312 or Nirmatrelvir compared to passage control.
Sequencing of SARS-CoV-2 passaged in Vero E6 cells
Virus RNA was extracted using QIAamp Viral RNA Mini Kit (Qiagen, Germany). Viral RNA was reverse-transcribed into cDNA and then was synthesized double-stranded DNA. Next, the DNA libraries were prepared using TruePrepTM DNA Library Prep Kit V2 for Illumina® (Vazyme, TD503) according to the manufacturer’s instructions for NGS. Agilent 2100 was used for quality control of the libraries showing the peaks near 350 bp. The cDNA libraries concentration was measured by Qubit dsDNA HS Assay Kit (Thermo Fisher Scientific Inc.). Then the sequencing was performed on Illumina NovaSeq 6000 system to 150 bp pair-end with 300 bp cycles Reagent Kit. Raw data have been deposited to SRA database (PRJNA1154822). The NGS raw reads were firstly processed by CLC Genomics Workbench (version 22.0.1, Qiagen) for removing low-quality reads, short reads and adapter contamination with default parameters. Further, the remaining sequences were aligned to SARS-CoV-2 isolate WuhanHu-1 reference genome downloaded through NCBI (GenBank accession MN908947.3) and exported the consensus sequence. After initial alignment, differences compared to the reference sequence were identified using low frequency variant detection tool in CLC Genomics Workbench with default parameters (threshold of >1%). Differences in nucleotide sequence that result in amino acid substitutions compared to the control BA.2.3 in this study.
Statistical analysis
ANOVA and Student’s t tests were used to analyze differences in mean values between groups using GraphPad Prism 9. All results are expressed as mean ± standard error of the mean (SEM) or standard error and were corrected for multiple comparisons. P values of <0.05 were considered statistically significant.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The atomic coordinates included in this study have been deposited in the PDB with the accession codes: 8WTS. NGS raw data have been deposited into SRA database (PRJNA1154822). All data supporting the results of this manuscript are available in the article or Supplementary Information. Source data are provided with this paper.
Code availability
Chemistry42 is an industry-grade commercial software platform available at http://chemistry42.com.
References
World Health Organization. Coronavirus disease (COVID-19) pandemic. https://www.who.int/emergencies/diseases/novel-coronavirus-2019 (accessed 28 Apr 2025).
Polack, F. P. et al. Safety and Efficacy of the BNT162b2 mRNA Covid-19 Vaccine. N. Engl. J. Med. 383, 2603–2615 (2020).
Baden, L. R. et al. Efficacy and Safety of the mRNA-1273 SARS-CoV-2 Vaccine. N. Engl. J. Med. 384, 403–416 (2021).
Voysey, M. et al. Safety and efficacy of the ChAdOx1 nCoV-19 vaccine (AZD1222) against SARS-CoV-2: an interim analysis of four randomised controlled trials in Brazil, South Africa, and the UK. Lancet 397, 99–111 (2021).
Sadoff, J. et al. Safety and Efficacy of Single-Dose Ad26.COV2.S Vaccine against Covid-19. N. Engl. J. Med. 384, 2187–2201 (2021).
Zhang, Y. et al. Safety, tolerability, and immunogenicity of an inactivated SARS-CoV-2 vaccine in healthy adults aged 18–59 years: a randomised, double-blind, placebo-controlled, phase 1/2 clinical trial. Lancet Infect. Dis. 21, 181–192 (2021).
Weinreich, D. M. et al. REGN-COV2, a Neutralizing Antibody Cocktail, in Outpatients with Covid-19. N. Engl. J. Med. 384, 238–251 (2021).
Chen, P. et al. SARS-CoV-2 Neutralizing Antibody LY-CoV555 in Outpatients with Covid-19. N. Engl. J. Med. 384, 229–237 (2021).
Gupta, A. et al. Early Treatment for Covid-19 with SARS-CoV-2 Neutralizing Antibody Sotrovimab. N. Engl. J. Med. 385, 1941–1950 (2021).
V’Kovski, P., Kratzel, A., Steiner, S., Stalder, H. & Thiel, V. Coronavirus biology and replication: implications for SARS-CoV-2. Nat. Rev. Microbiol. 19, 155–170 (2021).
Narwal, M., Armache, J. P., Edwards, T. J. & Murakami, K. S. SARS-CoV-2 polyprotein substrate regulates the stepwise M(pro) cleavage reaction. J. Biol. Chem. 299, 104697 (2023).
Zhang, L. et al. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science 368, 409–412 (2020).
Jin, Z. et al. Structure of M(pro) from SARS-CoV-2 and discovery of its inhibitors. Nature 582, 289–293 (2020).
Najjar-Debbiny, R. et al. Effectiveness of Paxlovid in Reducing Severe Coronavirus Disease 2019 and Mortality in High-Risk Patients. Clin. Infect. Dis. 76, e342–e349 (2023).
Unoh, Y. et al. Discovery of S-217622, a Noncovalent Oral SARS-CoV-2 3CL Protease Inhibitor Clinical Candidate for Treating COVID-19. J. Med. Chem. 65, 6499–6512 (2022).
Harari, S. et al. Drivers of adaptive evolution during chronic SARS-CoV-2 infections. Nat. Med. 28, 1501–1508 (2022).
Corey, L. et al. SARS-CoV-2 variants in patients with immunosuppression. N. Engl. J. Med. 385, 562–566 (2022).
Ip, J. D. et al. Global prevalence of SARS-CoV-2 3CL protease mutations associated with nirmatrelvir or ensitrelvir resistance. EBioMedicine 91, 104559 (2023).
Hu, Y. et al. Naturally Occurring Mutations of SARS-CoV-2 Main Protease Confer Drug Resistance to Nirmatrelvir. ACS Cent. Sci. 9, 1658–1669 (2023).
Iketani, S. et al. Multiple pathways for SARS-CoV-2 resistance to nirmatrelvir. Nature 613, 558–564 (2023).
Zhu, Y. et al. In vitro selection and analysis of SARS-CoV-2 nirmatrelvir resistance mutations contributing to clinical virus resistance surveillance. Sci. Adv. 10, eadl4013 (2024).
Sasi, V. M. et al. Predicting antiviral resistance mutations in SARS-CoV-2 main protease with computational and experimental screening. Biochemistry 61, 2495–2505 (2022).
Elend, L. et al. Design of SARS-CoV-2 Main Protease Inhibitors Using Artificial Intelligence and Molecular Dynamic Simulations. Molecules 27, https://doi.org/10.3390/molecules27134020 (2022).
Glaab, E., Manoharan, G. B. & Abankwa, D. Pharmacophore Model for SARS-CoV-2 3CLpro Small-Molecule Inhibitors and in Vitro Experimental Validation of Computationally Screened Inhibitors. J. Chem. Inf. Model 61, 4082–4096 (2021).
Morris, A. et al. Discovery of SARS-CoV-2 main protease inhibitors using a synthesis-directed de novo design model. Chem. Commun. (Camb.) 57, 5909–5912 (2021).
Ivanenkov, Y. A. et al. Chemistry42: An AI-Driven Platform for Molecular Design and Optimization. J. Chem. Inf. Model 63, 695–701 (2023).
Zhavoronkov, A. et al. Deep learning enables rapid identification of potent DDR1 kinase inhibitors. Nat. Biotechnol. 37, 1038–1040 (2019).
Ren, F. et al. AlphaFold accelerates artificial intelligence powered drug discovery: efficient discovery of a novel CDK20 small molecule inhibitor. Chem. Sci. 14, 1443–1452 (2023).
Ren, F. et al. A small-molecule TNIK inhibitor targets fibrosis in preclinical and clinical models. Nat. Biotechnol. https://doi.org/10.1038/s41587-024-02143-0 (2024).
Polykovskiy, D. et al. Entangled conditional adversarial autoencoder for de novo drug discovery. Mol. Pharmaceutics 15, 4398–4405 (2018).
Khrabrov, K., Zhavoronkov, A., Kadurin, A., Nikolenko, S. & Aliper, A. DruGAN: An Advanced Generative Adversarial Autoencoder Model for de Novo Generation of New Molecules with Desired Molecular Properties in Silico
Kadurin, A. et al. The cornucopia of meaningful leads: Applying deep adversarial autoencoders for new molecule development in oncology. Oncotarget 8, 10883 (2016).
Putin, E. et al. Adversarial threshold neural computer for molecular de novo design. Mol. pharmaceutics 15, 4386–4397 (2018).
Kuznetsov, M. & Polykovskiy, D. In Proceedings of the AAAI Conference on Artificial Intelligence. 8226-8234.
Devi, R. V., Sathya, S. S. & Coumar, M. S. Evolutionary algorithms for de novo drug design–A survey. Appl. Soft Comput. 27, 543–552 (2015).
Segler, M. H., Kogej, T., Tyrchan, C. & Waller, M. P. Generating focused molecule libraries for drug discovery with recurrent neural networks. ACS Cent. Sci. 4, 120–131 (2018).
Karypidou, K. et al. Synthesis, biological evaluation and molecular modeling of a novel series of fused 1, 2, 3-triazoles as potential anti-coronavirus agents. Bioorg. Med. Chem. Lett. 28, 3472–3476 (2018).
Powers, J. C., Asgian, J. L., Ekici, O. D. & James, K. E. Irreversible inhibitors of serine, cysteine, and threonine proteases. Chem. Rev. 102, 4639–4750 (2002).
Huang, F., Han, X., Xiao, X. & Zhou, J. Covalent Warheads Targeting Cysteine Residue: The Promising Approach in Drug Development. Molecules 27, https://doi.org/10.3390/molecules27227728 (2022).
Shindo, N. et al. Selective and reversible modification of kinase cysteines with chlorofluoroacetamides. Nat. Chem. Biol. 15, 250–258 (2019).
Katsura, H. et al. Human Lung Stem Cell-Based Alveolospheres Provide Insights into SARS-CoV-2-Mediated Interferon Responses and Pneumocyte Dysfunction. Cell Stem Cell 27, 890–904.e898 (2020).
Han, Y. et al. Identification of SARS-CoV-2 inhibitors using lung and colonic organoids. Nature 589, 270–275 (2021).
Zhou, J. et al. Differentiated human airway organoids to assess infectivity of emerging influenza virus. Proc. Natl. Acad. Sci. 115, 6822–6827 (2018).
Chen, D.-Y. et al. Spike and nsp6 are key determinants of SARS-CoV-2 Omicron BA.1 attenuation. Nature 615, 143–150 (2023).
Chen, X. et al. Preclinical evaluation of the SARS-CoV-2 Mpro inhibitor RAY1216 shows improved pharmacokinetics compared with nirmatrelvir. Nat. Microbiol. 9, 1075–1088 (2024).
Zhang, L. et al. SARS-CoV-2 crosses the blood-brain barrier accompanied with basement membrane disruption without tight junctions alteration. Signal Transduct. Target. Ther. 6, 337 (2021).
Li, K. et al. Mouse-adapted MERS coronavirus causes lethal lung disease in human DPP4 knockin mice. Proc. Natl. Acad. Sci. 114, E3119–E3128 (2017).
Liu, D. et al. Mouse models susceptible to HCoV-229E and HCoV-NL63 and cross protection from challenge with SARS-CoV-2. Proc. Natl. Acad. Sci. USA 120, e2202820120 (2023).
Jochmans, D. et al. The Substitutions L50F, E166A, and L167F in SARS-CoV-2 3CLpro Are Selected by a Protease Inhibitor In Vitro and Confer Resistance To Nirmatrelvir. mBio 14, e0281522 (2023).
U.S. Food and Drug Administration. Fact Sheet for Healthcare Providers: Emergency Use Authorization (EUA) for Paxlovid™ (nirmatrelvir tablets; ritonavir tablets). https://www.fda.gov/media/155050/download (accessed 28 Apr 2025).
Zhou, Y. et al. Nirmatrelvir-resistant SARS-CoV-2 variants with high fitness in an infectious cell culture system. Sci. Adv. 8, eadd7197 (2022).
Moghadasi, S. A. et al. Transmissible SARS-CoV-2 variants with resistance to clinical protease inhibitors. Sci. Adv. 9, eade8778 (2023).
Moghadasi, S. A., Biswas, R. G., Harki, D. A. & Harris, R. S. Rapid resistance profiling of SARS-CoV-2 protease inhibitors. Res Sq. https://doi.org/10.21203/rs.3.rs-2627723/v1 (2023).
Lewandowski, E. M. et al. Distal protein-protein interactions contribute to nirmatrelvir resistance. Nat. Commun. 16, 1266 (2025).
Kitamura, N. et al. Expedited approach toward the rational design of noncovalent SARS-CoV-2 main protease inhibitors. J. medicinal Chem. 65, 2848–2865 (2021).
Ma, C. et al. Discovery of di-and trihaloacetamides as covalent SARS-CoV-2 main protease inhibitors with high target specificity. J. Am. Chem. Soc. 143, 20697–20709 (2021).
Quan, B.-X. et al. An orally available Mpro inhibitor is effective against wild-type SARS-CoV-2 and variants including Omicron. Nat. Microbiol 7, 716–725 (2022).
Yamane, D. et al. Selective covalent targeting of SARS-CoV-2 main protease by enantiopure chlorofluoroacetamide. Chem. Sci. 13, 3027–3034 (2022).
Hirose, Y. et al. Discovery of chlorofluoroacetamide-based covalent inhibitors for severe acute respiratory syndrome coronavirus 2 3CL protease. J. Medicinal Chem. 65, 13852–13865 (2022).
Simen, B. B. et al. Low-abundance drug-resistant viral variants in chronically HIV-infected, antiretroviral treatment-naive patients significantly impact treatment outcomes. J. Infect. Dis. 199, 693–701 (2009).
Locarnini, S. Hepatitis B viral resistance: mechanisms and diagnosis. J. Hepatol. 39, S124–S132 (2003).
Wyles, D. L. & Luetkemeyer, A. F. Understanding Hepatitis C Virus Drug Resistance: Clinical Implications for Current and Future Regimens. Top. Antivir. Med. 25, 103–109 (2017).
Reusser, P. Herpesvirus resistance to antiviral drugs: a review of the mechanisms, clinical importance and therapeutic options. J. Hosp. Infect. 33, 235–248 (1996).
Piret, J. & Boivin, G. Resistance of herpes simplex viruses to nucleoside analogues: mechanisms, prevalence, and management. Antimicrob. Agents Chemother. 55, 459–472 (2011).
Smyk, J. M., Szydlowska, N., Szulc, W. & Majewska, A. Evolution of Influenza Viruses-Drug Resistance, Treatment Options, and Prospects. Int. J. Mol. Sci. 23, https://doi.org/10.3390/ijms232012244 (2022).
Huang, C. et al. A new generation M(pro) inhibitor with potent activity against SARS-CoV-2 Omicron variants. Signal Transduct. Target Ther. 8, 128 (2023).
Acknowledgements
We would like to thank WuXi synthetic, biological and WIND teams for valuable assistance and relevant materials. We sincerely appreciate the contribution from Junwen Qiao, Congcong Zhu, Yushu Yin, Bei Zhang, Jue Wang, Xiaoxia Lin, Jing Shang, Yichen Liu, Ju Wang and Lihong Zhuang to the project progress. We sincerely appreciate Fadi Pulous’ valuable suggestions for polishing the article. We would also like to thank Intel Inc. for their generous financial support of the current project and Dr. Stephanie M. Cope for assistance with the support organization. We also would like to acknowledge the HPC COVID-19 Consortium and Amazon Web Services for providing computing power support for our molecular generations targeting SARS-CoV-2 main protease. A. A.-G. acknowledges support from Dr. Anders G. Frøseth and the Canada 150 Research Chairs Program. We thank Guangzhou Customs District Technology Center for providing the BSL-3 Laboratory to complete the authentic SARS-CoV-2 and MERS-CoV experiments. This work was supported by Ministry of Science and Technology of China (MoST) (2022YFC2303700, AR.Z., 2022YFC2604100 J.S), Major Project of Guangzhou National Laboratory (No. SRPG22-012 J.S), National Natural Science Foundation of China (82495200 JC.Z, 82495203 JC.Z, 82025001 JC.Z, 82471787 J.S), Guangdong Basic and Applied Basic Research Foundation (2022B1515020059 J.S, 2021B1515130005 JX.Z), Basic and Applied Basic Research Projects of Guangzhou Basic Research Program (2023A04J0161 Q.Y), ZHONGNANSHAN MEDICAIFOUNDATION OF GUANGDONG PROVINCE (No. ZNSA-2020013 JC.Z), the 111 Project (D18010 JC.Z).
Author information
Authors and Affiliations
Contributions
J.S, D.S, Q.Y, D.W, J.P, XY.D, Z.C and managed the project, designed, planned experiments and wrote the manuscript. Y.A.I., B.A.Z., S.L, D.S.B., V.A.T., D.A.P., A.S.M., M.N.M. and Q.Z developed the proposed generative platform and drove molecular generations. Y.F and B.Y helped with the crystallization part of the project. L.W., T.L., X.L., A.A-G., A.A., X.D., X.C. and M.Z. helped to write the manuscript and provided general advice. JX.Z., N.Z, F.R., XW.C., A.Z. and JC.Z. led the project and helped to revise the manuscript. J.S, D.W, Q.Y, H.G, Z.C, Y.H, Z.L, R.L, X.H, J.Z, J.Y, X.G, J.T, AR.Z carried out all the mice and in vitro experiments associated virus infection in BSL-2 and BSL-3 laboratories in China. JY.S, M.Y, Z.X, G.P helped to collect the patient samples and human airway organoid culture. P.W, helped to analyze the NGS data.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Daniel Wacker, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Sun, J., Sun, D., Yang, Q. et al. A novel, covalent broad-spectrum inhibitor targeting human coronavirus Mpro. Nat Commun 16, 4546 (2025). https://doi.org/10.1038/s41467-025-59870-4
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-59870-4
