
Abstract
Feeding increases plasma bile acid levels while the nucleus of the solitary tract (NTS) and area postrema (AP) of the brain detect changes in hormones to regulate feeding. However, whether an increase in bile acids activates Takeda G protein-coupled receptor 5 (TGR5) in the NTS and/or AP to lower feeding through a negative feedback pathway is unknown. Here, we discover that infusion of TGR5 agonist CCDC in the NTS of male rats lowered food intake without causing conditional taste avoidance in short-term high fat (HF) fed male rats in association with HF-induced increase in TGR5 expression in the NTS. In contrast, CCDC infusion into the AP failed to lower food intake in HF rats with a reduction in TGR5 expression in the AP. CCDC infusion in the NTS activates TGR5 to reverse HF-induced leptin resistance by enhancing a leptin-leptin receptor-STAT3 signaling axis selectively in the NTS to lower feeding. Finally, metabolomic analysis indicated that HF impaired a refeeding-induced rise of endogenous TGR5 ligand deoxycholic acid in the plasma and subsequently in the NTS in association with hyperphagia, while direct infusion of deoxycholic acid in the NTS of HF rats activated TGR5 to lower feeding and enhanced leptin-STAT3 signaling, thereby altogether demonstrating physiological and pharmacological activation of TGR5 in the NTS regulates food intake. In summary, we discover that an activation of TGR5 in the NTS enhances leptin-STAT3 signaling to lower food intake. Our findings highlight the potential of targeting TGR5 to reverse leptin resistance in the NTS.
Similar content being viewed by others


Introduction
Primary bile acids are synthesized from cholesterol in the liver and undergo various reactions using enzymes of the cytochrome P450 family1. Bile acids created under both the classical and acidic pathway converge at phase II conjugation passing through enzymes such as bile acid-CoA synthase and bile acid-CoA-amino acid N-acetyltransferase leading to glycine/taurine conjugation1. Within the intestine, the gut microbiota will convert primary bile acids into secondary bile acids like deoxycholic acid (DCA)2,3. Bile acids exert three primary physiological functions: facilitating fat digestion4, regulating cholesterol metabolism5 and exerting metabolic effects by signaling through Farnesoid X Receptor (FXR)6 and Takeda G-protein Coupled Receptor (TGR)57.
TGR5 is a bile acid G protein-coupled receptor which activates cAMP-Protein Kinase A (PKA) signaling and is expressed in adipose, liver, and whole brain tissues7,8,9. Although TGR5 activation in adipose tissue and the liver increases energy expenditure and insulin sensitivity, respectively10,11, its documented food intake-lowering effect has so far only been ascribed to the hypothalamus12,13. Interestingly, selective hypothalamic TGR5 knockdown does not disrupt the feeding-lowering effect in rodents receiving dietary supplementation of bile acid12, indicating that extrahypothalamic TGR5 also regulates feeding. Indeed, the nucleus of solitary tract (NTS) and area postrema (AP) have been implicated to detect changes in Glucagon-like-peptide-1 (GLP1) and Growth Differentiation Factor 15 (GDF15) to regulate feeding14,15,16,17, but the effect of TGR5 in the NTS and/or AP is unknown. Together with the fact that bile acid levels in the blood increase shortly after a meal in healthy human subjects18 and that plasma level of bile acids positively correlated with the respective levels in the whole brain19, TGR5 expressed in the brain may have physiological relevance in detecting an increase in circulating bile acid levels to lower food intake and maintain energy homeostasis via a negative feedback loop.
Obesity is a global epidemic20,21 that is characterized by leptin resistance due in part to the inability of leptin to activate signal transducer and activator of transcription 3 (STAT3) via leptin receptor (LepR) b in the hypothalamus to lower feeding22. Interestingly, leptin action in the NTS, but not AP, lowers food intake as well23,24, but whether leptin resistance is acquired by HF in the NTS and the associated signaling pathway involved remains unknown. Given the adverse side effects of nausea associated with weight loss medications such as GLP1R agonists25,26, thus there is clearly a pressing need for novel and effective therapeutics to lower weight in obesity. Herein we sought to explore the pharmacological and physiological relevance of TGR5 in the NTS and/or AP for feeding regulation and its interaction with leptin signaling, in male rats.
Results
TGR5 agonist CCDC, but not leptin, infusion into the NTS lowers food intake in HF rats with impaired leptin-STAT3 signaling in the NTS
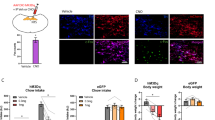
To investigate the sufficient role of TGR5 in the NTS and/or AP in feeding regulation, we found that TGR5 was expressed in the NTS and AP of chow and HF fed male rats as assessed by qPCR (Fig. 1a), like in the hypothalamus12,13. TGR5 expression was detected in both NTS and AP of HF rats with fluorescence in situ hybridization (FISH) indicated as an orange fluorescence (Supplementary Fig. 1a) as well. Next, we infused synthetic TGR5-specific agonist 3-(2-chlorophenyl)-N-(4-chlorophenyl)-N,5-dimethyl-4-isoxazolecarboxamide (CCDC, 100 μM) or sodium carboxymethyl cellulose (CMC, vehicle) into the mediobasal hypothalamus (MBH) of rats targeting the arcuate nucleus as we first wanted to verify whether the CCDC dose we used is functional as hypothalamic TGR5 activation is documented to lower feeding12,13 (Supplementary Fig. 1b). CCDC vs. CMC infusion into the MBH (0.04 μL/min for 5 mins) of chow rats with comparable pre-experimental food intake and body weight indeed lowered food intake 24 h after refeeding (Supplementary Fig. 1c–e). To verify the location of the delivery, we infused a dye (Chicago Sky Blue) at 0.04 μL/min for 5 min into the MBH targeting the arcuate nucleus of chow rats and the dye was found selectively in the arcuate nucleus as detected by bright field microscopy (Supplementary Fig. 1f).

a Relative Tgr5 mRNA expression in the NTS and AP of chow (NTS n = 9; AP n = 7) or HF (NTS n = 6; AP n = 6) rats. b Cumulative food intake of NTS CMC (n = 12) or CCDC (n = 10) HF rats 24 h after food was given back. c Percentage saccharin preference in HF rats following IP saline (n = 5) vs. IP LiCl (n = 5) and NTS CMC (n = 5) vs. NTS CCDC (n = 5) infusion. d Relative PKA activity in the NTS CMC (n = 5) or CCDC (n = 5) HF rats 3 h after infusion. e Cumulative food intake of AP CMC (n = 10) or CCDC (n = 10) HF rats 24 h after food was given back. f Relative LepRb mRNA expression in the NTS (n = 9) and AP (n = 7) chow or NTS (n = 6) and AP (n = 5) HF rats. g Cumulative food intake in NTS CMC (n = 11), STAT3-PI (n = 5), leptin (n = 11), leptin+STAT3-PI (n = 15) chow rats 24 h after food was given back. h Cumulative food intake of NTS CMC (n = 6) or leptin (n = 10) HF rats 24 h after food was given back. i Cumulative food intake of AP CMC (n = 6) or leptin (n = 10) chow rats 24 h after food was given back. j Representative western blot images of p-STAT3, t-STAT3, and GAPDH protein levels in the NTS of chow rats 30 min after CMC, leptin, or leptin+STAT3-PI 5 min acute infusion. k Schematic representation of brainstem tissue section for immunohistological and FISH images. l Representative images (scale bar: 100 μm) of immunohistological staining for p-STAT3 with DAPI (top) and cFOS (bottom) in the NTS of chow rats 30 min after CMC or leptin 5 min acute infusion. m Representative western blot images of p-STAT3, t-STAT3, and GAPDH protein levels in the NTS of HF rats 30 min after CMC or leptin 5 min acute infusion. n Representative images (scale bar: 100 μm) of immunohistological staining for p-STAT3 with DAPI (top) and cFOS (bottom) in the NTS of HF rats 30 min after CMC or leptin 5 min acute infusion. a–f, h, i P values were calculated by two-tailed t tests. g P values were calculated by one-way ANOVA with Tukey’s multiple comparison. Data presented as mean ± SEM. IP intraperitoneal.
We infused equimolar CCDC vs CMC into the NTS of chow and 3 d HF fed rats that were hyperphagic but with similar body weight vs chow fed rats prior to brain injections (Supplementary Fig. 1g, h). We found that CCDC vs. CMC infusion into the NTS lowered food intake in chow rats 24 h after refeeding (Supplementary Fig. 1i). In HF rats, CCDC vs CMC infusion into the NTS was as potent in lowering food intake, did not cause conditioned avoidance and activated PKA (i.e., marker of TGR5 activity9) in the NTS (Fig. 1b–d), while the location of the NTS reagent delivery was confirmed with a dye found selectively in the NTS of HF rats (Supplementary Fig. 1j). Interestingly, HF vs chow increased TGR5 expression in the NTS but in direct contrast decreased TGR5 expression in the AP (Fig. 1a). Consistently, CCDC vs CMC infusion into the AP only lowered food intake in chow (Supplementary Fig. 1k–m) but not in HF rats (Fig. 1e), while the location of the AP delivery was confirmed with a dye found selectively in the AP of HF rats that received AP dye infusion (Supplementary Fig. 1n). Thus, selectively infusing TGR5 agonist CCDC into the NTS, but not AP, lowers food intake in HF male rats.
We next assessed the feeding regulatory impact of leptin signaling in the NTS and/or AP (Supplementary Fig. 1b). First, we confirmed expression of LepRb in the NTS and AP of chow and HF rats using qPCR (Fig. 1f) as documented23,27,28. The expression of LepRb (the green fluorescence) in the NTS and AP of HF rats was alternatively confirmed using FISH (Supplementary Fig 1a). Interestingly, HF vs chow decreased LepRb expression in the NTS but not in the AP (Fig. 1f), which contrasts the TGR5 expression pattern (Fig. 1a). With comparable pre-experimental body weight but higher food intake in HF vs chow rats (Supplementary Fig. 1o, p), leptin vs. CMC infusion into the NTS lowered feeding at 24 h in chow but not in HF rats (Fig. 1g, h). Leptin vs CMC infusion into the AP of chow rats (Supplementary Fig 1q, r) did not lower food intake despite its LepRb expression (Fig. 1i)27.
In parallel to the hypothalamus, we first found that leptin vs CMC infusion into the NTS increased STAT3 phosphorylation (p-STAT3) without affecting total STAT3 (t-STAT3) protein levels in the NTS tissue obtained after 30 min administration (Fig. 1j, Supplementary Fig. 1s). To validate the activation of STAT3 by leptin is selective in the NTS but not in the immediate nearby regions, we performed immunofluorescence staining of p-STAT3 in brainstem tissue section (−13.6 mm to −14.1 mm posterior to the bregma, containing both AP and NTS) obtained at 30 min after NTS leptin administration (Fig. 1k). The immunohistological images were taken from the left side of the brainstem region that contain NTS, AP and the central canal (CC). Leptin vs. CMC administration into the NTS of chow rats increased p-STAT3 positive (p-STAT3+) cell numbers in the NTS but not in the AP in parallel to increased c-FOS positive (c-FOS+) cells (Fig. 1l, Supplementary Fig. 1t, u), indicating that the activation of STAT3 by leptin infused into the NTS is selective to the NTS.
We inhibited STAT3 with STAT3 peptide inhibitor (STAT3-PI) to address its necessary role (Supplementary Fig. 1b) by co-infusing STAT3-PI with leptin into the NTS of chow rats, which negated the ability of leptin to stimulate STAT3 (Fig. 1j, Supplementary Fig. 1s) and lower feeding (Fig. 1g), while STAT3-PI alone vs CMC had no effect (Fig. 1g). Importantly, leptin vs CMC administration into the NTS of HF rats failed to activate STAT3, increase p-STAT3+ cells and c-FOS+ cells (Fig. 1m, n, Supplementary Fig 1v–x). These findings collectively indicate that HF induces leptin resistance and dysregulates feeding by impairing the ability of leptin to activate STAT3 in the NTS.
Activation of TGR5 by CCDC enhances leptin-LepR-STAT3 signaling in the NTS to lower food intake
To investigate whether TGR5 interacts with leptin in the NTS, we co-infused TGR5 agonist CCDC with leptin into the NTS of HF-induced hyperphagic and leptin resistant male rats (Fig. 1h, m, n, Supplementary Fig. 1o) that had comparable pre-experimental feeding and weight (Supplementary Fig. 2a–c). CCDC and leptin infusion into the NTS of HF rats inhibited feeding at 24 h as compared to CCDC alone (Fig. 2a), while the food intake-lowering NTS effect of CCDC + leptin or CCDC was stronger compared to leptin or CMC treatment alone in HF rats, respectively (Fig. 2a). These findings indicate that CCDC in the NTS lowers food intake through NTS leptin-dependent and independent pathways in HF rats. To zoom in on and characterize the leptin-dependent effect of CCDC, we compared the effect of CCDC + leptin vs CCDC alone (independent of leptin) by analyzing the effect of leptin on STAT3 signaling 3 h after CCDC administration at a point when CCDC was determined to activate PKA activity in the NTS (Fig. 1d). We found that CCDC + leptin vs CCDC infusion increased p-STAT3 levels (Fig. 2b, Supplementary Fig. 2d), p-STAT3+ and c-FOS+ cell (Fig. 2c, Supplementary Fig. 2e, f) in the NTS of HF rats. Of note, the effect of NTS CCDC alone on pSTAT3 levels, p-STAT3+ and c-FOS+ cells is minimal to none as compared to the effect of NTS CMC or leptin alone in HF rats (Fig. 1m, n, Supplementary Fig. 1v–x), while the effect of CCDC + leptin vs CCDC alone on pSTAT3, p-STAT3+ and c-FOS+ cell was comparable to the effect of NTS leptin vs CMC in chow rats (Fig. 1j, l, Supplementary Fig. 1s–u). Taken together, our findings demonstrate that CCDC enhances leptin-STAT3 signaling selectively in the NTS.

a Cumulative food intake of NTS CMC (n = 15; data replotted from Fig. 1b, h), leptin (n = 10; data replotted from Fig. 1h), CCDC (n = 10; data replotted from Fig. 1b) or CCDC+leptin (n = 11) HF rats 24 h after food was given back. b Representative western blot images of p-STAT3, t-STAT3, and GAPDH protein levels in NTS CCDC 5 min acute infusion, followed by a 3 h wait, then NTS CMC or leptin 5 min acute infusion, and a subsequent 30 min waiting time in HF rats. c Representative images (scale bar: 100 μm) of immunohistological staining for p-STAT3 with DAPI (top) and cFOS (bottom) in NTS CCDC 5 min acute infusion, followed by a 3 h wait, then NTS CMC or leptin 5 min acute infusion, and a subsequent 30 min waiting time in HF rats. d Relative NTS Tgr5 mRNA expression of NTS shMM (n = 6) or shTgr5 (n = 6) rats. e Relative NTS PKA activity of NTS CMC shMM (n = 6), CCDC shMM (n = 6), CMC shTgr5 (n = 6), or CCDC shTgr5 (n = 6) HF rats, measured 3 h after 5 min acute infusions. f Cumulative food intake of NTS CMC shMM (n = 5), CCDC+leptin shMM (n = 10), CMC shTgr5 (n = 5), or CCDC+leptin shTgr5 (n = 8) HF rats 24 h after food was given back. g Relative NTS LepRb mRNA expression of NTS shMM (n = 6) or shLepR (n = 6) rats. h Cumulative food intake of NTS CMC shMM (n = 7), leptin shMM (n = 7), CMC shLepR (n = 7), leptin shLepR (n = 7) chow rats 24 h after food was given back. i Representative western blot images of p-STAT3, t-STAT3, and GAPDH protein levels in the NTS of NTS shMM or shLepR HF rats receiving a NTS CCDC 5 min acute infusion, followed by a 3 h wait, then NTS leptin 5 min acute infusion, and a subsequent 30 min waiting time. j Cumulative food intake of NTS CCDC+leptin shMM (n = 8) or CCDC+leptin shLepR (n = 8) HF rats 24 h after food was given back. k Cumulative food intake of NTS CMC (n = 15, data replotted from Fig. 1b), STAT3-PI (n = 7), CCDC + STAT3-PI (n = 11), CCDC+leptin+STAT3-PI (n = 6), or CCDC+leptin (n = 11, data replotted from Fig. 2a) HF rats 24 h after food was given back. a, e, f, h, k P values were calculated by one-way ANOVA with Tukey’s multiple comparison. d, g, j P values were calculated by two-tailed t tests. Data presented as mean ± SEM.
To study the necessity of TGR5 in CCDC interaction with leptin, we knocked down TGR5 expression in the NTS of HF rats by injecting a lentivirus expressing the short hairpin RNA (shRNA) of rat TGR5 (shTgr5) or mismatch shRNA (shMM) selectively into the NTS via a bilateral cannulae (3 μL/site in 10 min) (Supplementary Fig. 2a) as described29. We first confirmed that direct injection (3 μL/site in 10 min) of a lentivirus expressing GFP into the NTS selectively infected (i.e., GFP expressing cells) the NTS and not the nearby AP as compared to control (Supplementary Fig. 2g). Importantly, shTgr5 vs shMM injection into the NTS knocked down TGR5 expression by ~60% selectively in the NTS but not the AP tissues of HF rats as well (Fig. 2d, Supplementary Fig. 2h). NTS CCDC vs CMC infusion in shMM HF rats increased PKA activity in the NTS (Fig. 2e) as in non-viral infected HF rats (Fig. 1d), and this effect of NTS CCDC vs CMC on PKA was blocked in shTgr5 HF rats, while shTgr5 vs shMM alone did not alter PKA activity in NTS CMC HF rats (Fig. 2e), indicating that Tgr5 is required for CCDC to activate PKA in the NTS of HF rats. Importantly, NTS CCDC + leptin vs cmc infusion lowered food intake in shMM but not in shTGR5 rats, while NTS shTgr5 vs shMM alone had no effects on feeding (Fig. 2f, Supplementary Fig. 2j, k). Of note, CCDC + leptin in shTgr5 HF rats had comparable food intake as in shMM or shTgr5 vehicle infused HF rats indicating that the full (i.e., leptin-dependent and -independent) effects of CCDC is mediated by Tgr5 in the NTS. Furthermore, shTgr5 and shMM HF rats had similar food intake effects compared to NTS CMC non-viral rats (Fig. 1b) that illustrate viral infection per se did not alter food intake. Thus, these findings demonstrate that Tgr5 in the NTS is required for CCDC infusion into the NTS to lower food intake via leptin-dependent and -independent pathways.
To address the necessary role of leptin receptor and STAT3 in the effect of TGR5 on leptin sensitivity, we first knocked down leptin receptor by injecting lentivirus expressing shRNA targeting rat LepR (shLepR) vs shMM into the NTS (Supplementary Fig. 2a). We first confirmed that LepRb expression was reduced by 40% in the NTS (Fig. 2g) but not in the AP (Supplementary Fig. 2i) in shLepR vs shMM rats, and that leptin vs CMC infusion into the NTS of chow rats lowered food intake in shMM rats (Fig. 2h) as in non-viral infected rats (Fig. 1g). NTS leptin vs CMC infusion instead failed to lower food intake in shLepR chow rats with comparable pre-experimental food intake and body weight (Supplementary Fig. 2l, m). Of note NTS shLepR vs shMM in CMC-infused rats had no effect on feeding (Fig. 2h), altogether demonstrating that leptin receptor in the NTS is required for leptin to lower feeding. Importantly, shLepR vs shMM decreased NTS STAT3 activation (Fig. 2i, Supplementary Fig. 2n) and increased food intake in HF rats that received NTS CCDC + leptin infusion, independent of changes in pre-experimental feeding and weight (Fig. 2j, Supplementary Fig. 2j, k). In parallel, we infused STAT3-PI to inhibit STAT3 signaling in the NTS (Supplementary Fig. 2a). With comparable pre-experimental feeding and weight (Supplementary Fig. 2b, c), we first found that STAT3-PI vs CMC alone did not affect food intake in HF rats (Fig. 2k) as in chow rats (Fig. 1g) nor STAT3-PI altered the feeding-lowering effect of NTS CCDC infusion as NTS CCDC + STAT3-PI still lowered food intake as compared to NTS STAT3-PI alone (Fig. 2k). However, co-administration of STAT3-PI with CCDC + leptin into the NTS of HF rats negated the anorectic effect of CCDC + leptin vs CMC alone (Fig. 2k) that led to comparable food intake in HF rats with NTS CCDC infusion alone (Supplementary Fig. 2o). Taken together, leptin receptor and STAT3 signaling in the NTS are required for the leptin-dependent effect of CCDC.
In summary, pharmacological activation of TGR5 by CCDC reverses HF-induced leptin resistance by enhancing leptin-leptin receptor-STAT3 signaling axis in the NTS.
Activation of TGR5 decreases inflammation and leptin signaling suppressor in the NTS
HF feeding within 1–3 days induces microglial (i.e., the resident macrophages within the central nervous system) inflammation and the associated cytokines interleukin 6 (IL6) and tumor necrosis factor α (TNFα) release in the hypothalamus30,31,32. Microglial inflammation is sufficient and necessary for HF to induce hypothalamic leptin resistance partly via increased expression of neuronal leptin-LepRb-STAT3 signaling suppressor such as tyrosine phosphatase 1B (PTP1B) and suppressor of cytokine signaling 3 (SOCS3)32,33,34,35,36,37, thereby potentially highlighting a paracrine microglia-to-neuron pathway that induces hypothalamic leptin resistance32. Given that TGR5 activation decreases macrophage/microglial inflammation38,39 and inhibits macrophage inflammation-associated atherosclerosis and insulin resistance40,41, we reason that TGR5 activation may alter inflammation to regulate leptin sensitivity in the NTS.
Using NTS tissues that were obtained after the refeeding studies, HF rats that received NTS CMC infusion had increased Cd68 (i.e., a myeloid cell-specific marker for activated microglia) and Il6 expression in the NTS as compared to chow rats (Fig. 3a, b). NTS infusion of CCDC into the NTS of HF rats lowered Cd68 and Il6 expression in the NTS to levels seen in NTS CMC chow rats, while NTS CCDC vs CMC infusion in chow rats had minimal effects (Fig. 3a, b). Tnfa expression was comparable among groups (Fig. 3c). NTS CMC HF rats vs NTS CMC chow rats lowered LepRb expression in the NTS (Fig. 3d), confirming the effect of HF in non-CMC infused rats (Fig. 1f). CCDC vs CMC had no effect on LepRb expression in the NTS of HF nor chow rats (Fig. 3d). However, CCDC vs CMC reduced the expression of Ptp1b in the NTS of HF and chow rats (Fig. 3e) and more importantly prevented HF vs chow-induced increase in Ptp1b expression in CMC rats (Fig. 3e). Socs3 expression remained comparable among groups (Fig. 3f). These findings collectively suggest (and clearly remains to be tested) that the anti-inflammatory effect of TGR5 activation in the NTS may potentially reverse HF-induced leptin resistance by decreasing Ptp1b in the NTS.

Relative NTS Cd68 (a), Il6 (b), Tnfa (c), LepRb (d), Ptp1b (e), and Socs3 (f) mRNA expression of NTS CMC (n = 8) or CCDC (n = 6) chow rats and NTS CMC (n = 8 for a–d; n = 7 for e, f) or CCDC (n = 8 for a–d; n = 7 for e, f) HF rats, measured 24 h after food was given back following 5 min acute infusion. g Representative images (scale bar: 100 μm; 25 μm for the digital zoomed-in images) showing FISH for Tgr5, Aif1, and DAPI or LepR, Aif1, and DAPI in the NTS of HF rats 30 min after CMC 5 min acute infusion. Digital zooms of the boxed regions are shown to the left. Arrowheads indicate cells co-expressing Tgr5 or LepR and Aif1. h Left: Proportion of Aif1+ microglia in the NTS that are Tgr5+ or Tgr5- and LepR+ or LepR- (pie chart). Right: Percentage of Aif1+ microglia expressing Tgr5 or LepR in the NTS (n = 3 per group). i Representative images (scale bar: 100 μm; 25 μm for the digital zoomed-in images) showing FISH for Tgr5, Rbfox3, and DAPI or LepR, Rbfox3, and DAPI in the NTS of HF rats 30 min after CMC 5 min acute infusion. Digital zooms of the boxed regions are shown to the left. Arrowheads indicate cells co-expressing Tgr5 or LepR and Rbfox3. j Left: Proportion of Rbfox3+ neurons of NTS that are Tgr5+ or Tgr5- and Lepr+ or Lepr- (pie chart). Right: Percentage of Rbfox3+ neurons expressing Tgr5 or LepR of NTS (n = 4 per group). k Representative images (scale bar: 100 μm; 25 μm for the digital zoomed-in images) showing FISH for Tgr5, LepR, and DAPI in the NTS of HF rats 30 min after CMC 5 min acute infusion. Digital zooms of the boxed regions are shown to the left. Arrowheads indicate Tgr5 expressing cells that are either LepR+ (top) or LepR- (bottom). l Left: Proportion of Tgr5+ cells in the NTS that are Lepr+ or Lepr- (pie chart). Right: Percentage of Tgr5+ cells that are Lepr- or Lepr+ in the NTS (n = 4). a–f P values were calculated by one-way ANOVA with Tukey’s multiple comparison; h, j P values were calculated by two-tailed t tests; l P values were calculated by two-tailed paired t tests. Data presented as mean ± SEM.
To explore the localization of TGR5 and LepR on microglia and neurons, we used fluorescence in situ hybridization (FISH) to co-stain (i) TGR5 or LepR and microglia, (ii) TGR5 or LepR and neurons, and (iii) TGR5 and LepR. We counted the TGR5 and LepR mRNA expressing microglia and neuron cells in the same NTS region (Fig. 3g–l) that we detected STAT3 activation by leptin (Fig. 1l). To define a positive cell count, we ensured that a FISH probe was within a single cell containing DAPI (Supplementary Fig. 3a–c). For example, in Supplementary Fig. 3a, we show a single cell with an orange and green fluorescence illustrating a Tgr5+ Aif1+ cell (Tgr5+ microglial cell). Accordingly, we first found that among all the Aif1+ microglia cells (total 259 + /− 23 cells) within a NTS section, 51% (131 + /−14 cells) of the microglia cells expressed Tgr5 (Tgr5+) and that was significantly higher than 22% (48 + /−9 cells) of LepR+ microglia cells (Fig. 3g, h). The percentage of LepR+ neurons (i.e., Rbfox3+ cells) was 43% (133 LepR+Rbfox3+ cells / 310 total number of Rbfox3+cells) and that was significantly higher than 31% Tgr5+ neurons (101 Tgr5+Rbfox3+ / 328 total number of Rbfox3+cells) (Fig. 3i, j). Representative images of a Tgr5+ or LepR+ Rbfox3+ (neuron) cell were depicted in Supplementary Fig. 3b. Lastly, 74% of Tgr5+ cells did not express LepR (LepR– 198 / Tgr5+ 270 number of cells) in the NTS and the number of LepR- Tgr5+ cells was significantly higher than LepR+Tgr5+ cells 27% (LepR+ 71 / Tgr5+ 270 number of cells) (Fig. 3k, l). Representative images of a LepR+ or LepR- Tgr5+ cell were depicted in Supplementary Fig. 3c. Of note, there was an average of 249 + /−11 Tgr5+ cells out of a total of 700 + /− 12 cells (DAPI) within the NTS region we counted in all our slides (n = 11). Other potential cell types expressing Tgr5 within the NTS include astrocytes42, although the percentage of them should be relatively low as there were 131 Tgr5+ microglial cells (Fig. 3h) and 101 Tgr5+ neurons (Fig. 3j) as compared to an average of 249 Tgr5+ cells within the NTS. In summary, we found that in the NTS, (i) approximately half of microglia cells express Tgr5 but only ~22% express LepR, (ii) over 70% of Tgr5+ cells do not co-express LepR, and (iii) HF induced-Il6 and Ptp1b expression were negated by CCDC. Thus, we put forward a working hypothesis (and clearly remains to be tested) that activation of microglial Tgr5 in the NTS of HF rats reduces inflammatory cytokine IL-6 release and neuronal Ptp1b expression to enhance leptin-STAT3 signaling and lower food intake via a microglia-neuronal paracrine axis.
Refeeding increases endogenous TGR5 agonist DCA in the NTS of chow but not HF rats, while direct DCA infusion into the NTS of HF rats activates TGR5 to lower feeding
Having detected a sufficient role of TGR5 in the NTS for feeding regulation and its potential link(s) to leptin sensitivity (Figs. 1–3), we finally explore whether TGR5 in the NTS could be regulated by endogenous bile acids to impact feeding. First, we collected plasma samples and NTS tissues from chow and HF-induced hyperphagic rats with comparable pre-experimental weight that underwent fasting and refeeding conditions (Supplementary Fig. 4a, b). Unconjugated, taurine-conjugated, and glycine-conjugated bile acid levels were measured from the NTS and plasma samples using ultra-performance liquid chromatography-coupled time-of-flight mass spectrometry (UPLC-MS) metabolite profiling. Chow rats refed for 0.5 h vs. fasting increased total plasma bile acid levels (Fig. 4a) as reported18,43,44. Interestingly, we found that this refed-induced rise of endogenous plasma bile acid levels was in parallel to an elevation of plasma TGR5 ligands deoxycholic acid (DCA) and hyodeoxycholic acid (HDCA) as well as other bile acids such as chenodeoxycholic acid (CDCA), and cholic acid (CA), but not the rest of unconjugated and taurine/glycine-conjugated bile acids including TGR5 ligands taurodeoxycholic acid (TDCA), taurohyodeoxycholic acid (THDCA), glycodeoxycholic acid (GDCA), and glycolithocholic acid (GLCA)7,45 (Fig. 4b, Supplementary Fig. 4c, d). In HF-induced hyperphagic rats, refeeding for 0.5 h vs fasting increased total plasma bile acid levels (Fig. 4c) and HDCA, CA, ursodeoxycholic acid (UDCA), and taurochenodeoxycholic acid (TCDCA) but not DCA and CDCA (Fig. 4d, Supplementary Fig. 4e, f). Of note, DCA is the only bile acid TGR5 ligand whose plasma levels were significantly increased in chow but not in HF-induced hyperphagic rats after 0.5 h of refeeding (Fig. 4e).

a Total bile acid levels in the plasma of chow rats during fasting (n = 5) or at 0.5 h after refed (n = 5). b Unconjugated bile acid levels in the plasma of chow rats during fasting (n = 5) or at 0.5 h after refed (n = 5). c Total bile acid levels in the plasma of HF rats during fasting (n = 6) or at 0.5 h after refed (n = 5). d Unconjugated bile acid levels in the plasma of HF rats during fasting (n = 6) or at 0.5 h after refed (n = 5). e DCA levels in the plasma of chow and HF rats during fasting (chow n = 5; HF n = 6) or at 0.5 h after refed (chow n = 5; HF n = 5). f Unconjugated bile acid levels in the NTS (pmol/g of tissue) of chow rats at 0.5 h (n = 6) or 2 h (n = 5) after refed. g Total bile acid levels in the NTS (pmol/g of tissue) of chow rats at 0.5 h (n = 6) or 2 h (n = 5) after refed. h Unconjugated bile acid levels in the NTS (pmol/g of tissue) of HF rats at 0.5 h (n = 5) or 2 h (n = 6) after refed. i Total bile acid levels in the NTS (pmol/g of tissue) of HF rats at 0.5 h (n = 5) or 2 h (n = 6) after refed. j DCA levels in the NTS (pmol/g of tissue) of chow and HF rats at 0.5 h (chow n = 6; HF n = 5) or 2 h (chow n = 5; HF n = 6) after refed. k Cumulative food intake of NTS saline shMM (n = 8), DCA shMM (n = 8), saline shTgr5 (n = 7), DCA shTgr5 (n = 8) HF rats 24 h after food was given back. l Representative western blot images of p-STAT3, t-STAT3, and GAPDH protein levels in the NTS of HF rats receiving a NTS DCA 5 min acute infusion, followed by a 3 h wait, then NTS CMC or leptin 5 min acute infusion, and a subsequent 30 min waiting time. m Illustration of TGR5 and leptin-STAT3 signaling in feeding regulation and associated experimental approaches. a–d, f–i P values were calculated by two-tailed t tests; e, j, k P values were calculated by one-way ANOVA with Tukey’s multiple comparison. Data presented as mean ± SEM. DCA deoxycholic acid, HDCA hyodeoxycholic acid, CDCA chenodeoxycholic acid, CA cholic acid, UDCA ursodeoxycholic acid, α-MCA α-muricholic acid, β-MCA β-muricholic acid.
If changes in plasma bile acid levels reflect a progressive change in bile acid levels in the NTS that impact feeding regulation, the bile acids in the plasma that are increased by refeeding at 0.5 h would require time to cross the blood-brain-barrier and accumulate in specific brain regions such as the NTS. Given that refeeding for 2 h decreases DCA levels in the hypothalamus of obese vs chow mice as well12, we decided to measure the level of bile acids in the NTS (pmol/g of tissue) at 2 h vs 0.5 h after refeeding in chow and HF-induced hyperphagic rats with comparable body weight (Supplementary Fig. 4a, b) to evaluate for potential progressive changes of bile acid levels in the NTS incurred by refeeding. First, we found that refeeding for 2 h vs. 0.5 h indeed increased DCA and HDCA levels in the NTS, while CA and α-muricholic acid (α-MCA) levels were decreased (Fig. 4f, Supplementary Fig. 4g, h), resulting in comparable total bile acid levels (Fig. 4g). In direct contrast, refeeding for 2 h vs 0.5 h in HF rats failed to increase DCA but was able to increase HDCA levels in the NTS as well as inhibiting CDCA but with no effects on CA, and α-MCA levels in the NTS (Fig. 4h, Supplementary Fig. 4i, j). Total bile acid levels remained comparable as well (Fig. 4i). Similar to the effect on plasma DCA levels by 0.5 h of refeeding (Fig. 4e), DCA levels in the NTS were increased by ~2-fold progressively from 0.5 to 2 h of refeeding in chow but not in HF rats (Fig. 4j). Thus, refeeding in chow rats progressively increases the level of DCA in the plasma and the NTS, and the rise of endogenous DCA levels is abolished in HF rats. These findings suggest that (i) a physiological rise of DCA in the plasma and then the NTS trigger a negative feedback pathway via the TGR5 to lower feeding, and (ii) HF feeding disrupts the rise of DCA levels in the NTS and subsequently feeding regulation.
To address whether the physiological TGR5 ligand DCA regulates feeding, we infused DCA (7.7 pmol/g, 0.2 ul/site over 5 min) into the NTS of HF rats to mimic the ~2.2-fold rise of NTS DCA levels induced by 2 h vs. 0.5 h refeeding in chow rats that was disrupted by HF (Fig. 4j), and repeated the feeding studies in HF rats that received shTgr5 vs. shMM lentiviral administration into the NTS to knockdown TGR5 expression in the NTS (Supplementary Fig. 4k, Fig. 2d). With comparable pre-experimental food intake and weight in the NTS shTgr5 vs. shMM lentiviral infected HF rats (Supplementary Fig. 4l, m), DCA vs saline infusion into the NTS lowered food intake at 24 h in shMM but not in shTgr5 HF rats (Fig. 4k). Of note, NTS shTgr5 vs shMM in saline infused rats had comparable food intake after 24 h refeeding (Fig. 4k). Lastly, DCA + leptin infusion into the NTS of HF rats as compared to DCA infusion alone activated STAT3 in the NTS tissues obtained after 30 min of acute bolus leptin infusion (Fig. 4l, Supplementary Fig. 4n), to a similar level seen in NTS leptin vs saline-infused chow rats (Fig. 1j, Supplementary Fig. 1s). Together with the fact that NTS leptin vs saline infusion failed to activate STAT3 in HF rats (Fig. 1m, n), these findings demonstrate that DCA enhances the ability of leptin to activate leptin-STAT3 signaling in the NTS (Fig. 4l). Collectively, our findings indicate that a physiological activation of TGR5 by an endogenous TGR5 ligand DCA activates TGR5 in the NTS of HF rats to lower feeding and enhance leptin-STAT3 signaling.
Discussion
In this study, we report that activation of STAT3 in the NTS is required for leptin to lower feeding in healthy conditions, while high fat feeding impairs the ability of leptin to activate STAT3 in the NTS and lower feeding in parallel to inducing hyperphagia, thereby highlighting that the NTS is a site of leptin resistance. More importantly, we discover that knockdown of TGR5 or leptin receptor selectively in the NTS negates the ability of TGR5 agonist to enhance leptin-STAT3 signaling in the NTS and lower feeding in HF rats with leptin resistance in the NTS (Fig. 4m), thereby unveiling activation of TGR5 in the NTS is sufficient to reverse leptin resistance to lower food intake.
TGR5 agonist infused into the AP of HF rats did not lower feeding. This contrasting effect of TGR5 action in the NTS vs. AP is of great interest as GLP1R activation in both the NTS and AP lower food intake while the GLP1 effect in the AP induces aversion14. GDF15 action in the AP lowers feeding in association with food aversion as well, but leptin fails to lower food intake in the AP23 and fails to induce aversive effects46,47. Consistently, CCDC infusion into the AP did not lower food intake in HF rats, while the food lowering effect of CCDC infusion into the NTS did not induce conditional taste avoidance suggesting it is independent of nausea. The opposing effect of HF diet on Tgr5 expression in the NTS vs AP may have led to the differential effect of CCDC in NTS vs AP. Very little is known of how TGR5 expression is regulated. However, one study documented that exercise increases TGR5 expression in the skeletal muscle by the unfolded protein response-activated transcriptional regulator Atf6α48. Given that the activated hypothalamic unfolded protein response induces hypothalamic leptin resistance in obese mice49, while HF feeding increases Tgr5 expression in the NTS as currently described, a possibility remains (and clearly remains to be investigated) that HF activates unfolded protein response in the NTS to induce TGR5 expression to enhance leptin signaling in HF and/or obese conditions. It is also important to compare the relative contribution of the NTS, AP and arcuate nucleus in lowering food intake by systemically administering CCDC in HF rats. Additionally, future studies should assess the potential sex-dependent central interaction of TGR5 with leptin.
Another important aspect of our study is that a physiological rise in circulating endogenous TGR5 ligand DCA levels incurred by refeeding reflects an elevation in the NTS of chow rats, and that the elevation of DCA in both the plasma and the NTS is impaired in HF-induced hyperphagic rats (Fig. 4m). We propose HF feeding disrupts feeding regulation by inhibiting the physiological rise of endogenous DCA levels and not TGR5 signaling in the NTS as direct administration of DCA into the NTS of HF rats lowers feeding, thereby highlighting the therapeutic potential and physiological relevance of TGR5 agonism in the NTS to lower feeding (Fig. 4m). In parallel, studies have documented that changes in gut microbiome are sufficient and necessary for metformin to alter bile acid levels in the gut and subsequently the plasma to regulate systemic metabolism as microbes express bile salt hydrolase that catalyzes bile acid metabolism and therefore alter bile acid composition2,50,51,52,53,54. We put forward a working hypothesis (and clearly warrants investigations) that HF diet induces changes in gut microbiome that are sufficient and necessary to alter DCA levels and eventually lead to the inability of refeeding to increase DCA in the gut -> plasma -> NTS and trigger a negative feedback pathway by activating TGR5 in the NTS to maintain energy homeostasis.
Future investigations using cell-specific genetic loss-of-function approaches targeting TGR5 in the microglia and leptin receptor in neurons in vivo are needed to address whether microglial TGR5 activation alters cytokine release to neurons to alter Ptp1b expression and impact neuronal leptin receptor-STAT3 signaling to lower feeding via a paracrine microglia-neuronal axis. Additionally, demonstrating if TGR5 agonism can sensitize LepRb in the NTS of obese rodents to lower food intake would help elucidate the translational and therapeutic potential of targeting central TGR5 to combat obesity.
The mechanisms that underlie the ability of TGR5 and GLP1R activation in the NTS to lower feeding may differ as the TGR5 agonist effect in the NTS enhances leptin signaling as currently reported, while the effect of whole-body or lateral ventricle administration of GLP1R agonist is leptin-independent55,56,57 in rats. The difference of leptin-dependency in TGR5 vs GLP1R brain action raises the possibility that combined activation of TGR5 and GLP1R in the NTS may additively or synergistically reduce feeding and weight in obesity as compared to either TGR5 and GLP1R alone, and that the selective activation of both TGR5 and GLP1R in the NTS could bypass AP-dependent nausea as well. Lastly, it would be important to explore the relative contribution of NTS vs hypothalamic TGR5 and GLP1R on feeding regulation with concurrent targeted selective knockdown of TGR5 and/or GLP1R in the NTS and/or hypothalamus as both hypothalamic TGR5 and GLP1R regulate feeding as well12,13,58,59.
In conclusion, we discover that pharmacological activation of TGR5 in the NTS with TGR5 agonist reverses HF-induced leptin resistance by enhancing leptin-leptin receptor-STAT3 signaling axis in the NTS to lower food intake in male rats. We additionally report that refeeding increases endogenous TGR5 ligand DCA levels in the plasma and then in the NTS in chow but not in HF rats, while direct infusion of DCA into the NTS of HF rats lowers food intake, further strengthening the physiological relevance and therapeutic potential of targeting TGR5 in the NTS to lower food intake in vivo.
Methods
Animals
Male Sprague-Dawley rats (Charles River Laboratories), of age 8–10 weeks weighing 300–320 g, were used in the study. Rats were housed in individual cages at 23 °C, subjected a standard 12/12 h dark/light cycle, lights go on at 6AM and turn off at 6PM, and had ad libitum access to water and a RC diet (Teklad Diet 7012, Envigo). The chow diet containing 17% fat, 25% protein, and 58% carbohydrate content (3.1 kcal/g total metabolizable energy). All animal protocols were reviewed and approved by the Institutional Animal Care and Use Committee at the UHN in accordance with the Canadian Council on Animal Care guidelines.
Sex as a biological variable
Male rats were used in this study because we have a well-established 3 d high-fat diet model of hyperphagia, central leptin and insulin signaling resistance that is independent of weight gain using male Sprague-Dawley rats60,61. It is unknown if the findings of this study are applicable to female rats.
Surgical procedures
After 2 days of acclimation to animal facility, the rats were anaesthetized (ketamine, 60 mg/kg; xylazine, 8 mg/kg) and brain cannulation surgeries were performed. For the NTS-targeted cannulation surgery, a bilateral 26-gauge stainless steel guide cannula (C235G, Plastics One Inc.) was stereotaxically implanted into the NTS via coordinates (0 mm on the occipital crest, 0.4 mm lateral to the midline, 7.9 mm below the cranial surface) as described. For the AP-targeted cannulation surgery, a single 26-gauge stainless steel guide cannula (C315G, Plastics One Inc.) was stereotaxically implanted into the AP via coordinates (0.3 mm posterior the occipital crest, 0.0 mm lateral to the midline, 7.9 mm below the cranial surface) as described. For the mediobasal hypothalamus (MBH)-targeted cannulation surgery, a bilateral 26-gauge stainless steel guide cannula (C235G, Plastics One Inc.) was stereotaxically implanted into the MBH via coordinates (3.1 mm posterior to the bregma, 0.4 mm lateral to the midline, 9.6 mm below the cranial surface) as described62. Post-surgical food intake and weight were monitored for 5 days leading up to the experiments, and rats that did not attain at least 90% of their pre-surgical weight were excluded. Despite including rats that were fully recovered from surgery, future studies await to validate these studies with longer surgical recovery times.
Lentiviral infection
For the NTS lentiviral infection, after the NTS-targeted cannulation surgery and while the rats were still anaesthetized, lentivirus (1.0 × 106 IFU) expressing shTGR5 (sc-270404-V), shLepR (sc-270697-V, Santa Cruz Biotechnology Inc.), or shMM (sc-108080, Santa Cruz Biotechnology Inc.) was injected via the NTS bilateral cannula (3 μl/site over 10 min)29. The injection cannula was left within NTS for additional 5 min to prevent the backflow of the lentivirus, and dummy cannula was inserted and secured with dust caps. To verify targeted viral infection of the NTS we used GFP lentivirus (sc-108084, Santa Cruz Biotechnology Inc.) using the same infectious units (1.0 × 106 IFU) and injection rate (3 μl/site over 10 min) as our shRNA lentivirus knockdown studies.
Short-term HF model
On the 2nd day after brain cannulation surgeries, rats were given HF diet with 10% lard oil (57IR, TestDiet) containing 34% fat, 22% protein, and 44% carbohydrate (3.9 kcal/g total metabolizable energy) for 3 days. Less than 5% of the rats did not develop hyperphagia and were excluded.
Feeding study with brain reagent infusion
On the 5th day after brain cannulation surgeries, food was removed from 8 AM to 4 PM during the daytime and was returned at 4 PM after a one-time infusion (0.04 μL/min, 5 min, 0.2 μL/site) of reagent into the NTS, AP, or MBH via the brain cannula, while the pre-experimental food intake and weight were monitored, respectively, before food removal and brain infusion. The accumulative food intake was measured at 24 h after food was given back. At the end of the experiments, while the rats were anesthetized, the AP and NTS tissue samples were collected separately for further analysis. CCDC (ab142091, Abcam) was dissolved firstly in DMSO and then in 1% CMC. Assuming a 10 times dilution for CCDC when infused into the brain, we originally gave CCDC (10 μM) at the concentration of 10 times EC5063 but found that CCDC at this concentration in the NTS did not lower food intake in HF rats (CMC 111.2 + 1.4 kcal n = 12; CCDC 113.1 + 6.6 n = 8). Given that CCDC administered at 5 μg/μL (equivalent to 13.85 mM) in the hypothalamus lowers feeding in obese mice, we increased the CCDC concentration to 100 times EC50 (100 μM) and found that CCDC infused at 100 μM in the NTS lowered feeding in both chow and HF rats (Fig. 1b, d). Leptin (497-LR, R&D Systems) was dissolved firstly in 0.9% saline and then with/without CCDC (final CCDC concentration 100 μM) in 1% CMC to the concentration of 153 μM (equivalent to 0.5 μg/site) as this amount of leptin infused into the hypothalamus or NTS lowers food intake23,64,65. STAT3-PI (Cell-Permeable; 573096, Sigma-Aldrich) was dissolved firstly in 0.9% saline and then with/without leptin (final leptin concentration 153 μM) in 1% CMC to the concentration of 20 μM (4 pmol/site) which is based on a previous study using STAT3-PI that negated leptin action66. DCA (30960, Sigma-Aldrich) was dissolved firstly in 0.5% DMSO and then in 0.9% saline to a final concentration of 192 nM. The reason we infused DCA into the NTS of HF rats at this concentration at 0.2 ul/site over 5 min was because infusing DCA at this concentration would deliver 7.7 pmol of DCA per gram of NTS tissue and elevate DCA levels in the NTS of HF rats by ~2.2 fold (similar to what is seen after 2 h of refeeding in chow rats Fig. 4j). Chicago Sky Blue 6B (31542, Cayman) was infused into the NTS, AP, or MBH at the same rate as other reagents. 50 μm coronal brain sections from perfused rats were mounted on slides, and selective injection sites were confirmed using the Zeiss Axio Imager in brightfield mode. A potential limitation was that we replotted control groups in Fig. 2a, k and in their respective Supplementary Fig. panels (Supplementary Fig. 2b, c and o).
Leptin signaling study
On the 5th day after NTS cannulation surgeries, food was removed from 8 AM to 4 PM during the daytime. CMC/CCDC (100 μM) pre-treatment (0.04 μL/min, 5 min, 0.2 μL/site) was given via NTS cannula 3 h prior to the leptin infusion at around 1 PM if needed. At 4 PM, CMC/STAT3-PI (20 μM)/leptin (153 μM)/leptin (153 μM) + STAT3-PI (20 μM) was given via the NTS cannula (0.04 μL/min, 5 min, 0.2 μL/site). At 30 min after leptin infusion, the rats were sacrificed for NTS tissue collection or were subjected to brain perfusion for histological analysis.
Tissues collection
At the end of the studies, the rats with brain cannulation were anesthetized (ketamine, 90 mg/kg; xylazine, 10 mg/kg) and injected with 2–3 μL/site bromophenol blue over 20–30 sec through the cannula to verify the correct placement of the cannula. Rats were euthanized by decapitation, and those that showed injection of dye beyond the correct NTS, AP, or MBH location were excluded from the studies. The AP and NTS tissue samples were collected separately, as previously described29,67. Once the whole brain was harvested from the anaesthetized rats via decapitation, the cerebellum was lifted to expose the caudal part of the brain. Then, the whole brain was isolated from the skull, bathed in ice-cold PBS, and placed on a metal plate cooled by dry ice. The dorsal vagal complex was obtained by cutting from the point of the obex to the end of the vagal triangle, and a micro knife was used to isolate the AP and NTS tissues, guided by a rat brain atlas as well as with visual assistance from markers such as the cuneate nucleus and central canal. All tissues were immediately frozen in liquid nitrogen and stored at −80 °C for future analysis.
Histological analysis
The rat brain perfusion was performed on rats that were anesthetized (ketamine, 90 mg/kg; xylazine, 10 mg/kg), followed by transcardial infusion with ice-cold PBS and then 4% paraformaldehyde. After perfusion, the rats were euthanized by decapitation, and the brain was isolated and was immersed in 4% paraformaldehyde overnight at 4 °C. The brain immersions were repeated with 10%, 20%, and 30% sucrose in PBS, and the brain stem containing dorsal vagal complex was cutoff, embedded into the Tissue-Tek O.C.T Compound (4583, Sakura Finetek), frozen in liquid nitrogen, and stored at −80 °C. Brain stem coronal sections (10 μm) were cut with a cryostat (Leica CM1950, Leica Microsystems) starting from the medial NTS (−12.96 mm posterior to the bregma) to the obex (−14.4 mm posterior to the bregma) based on the rat brain stereotaxic coordinates68, and the brainstem tissue sections containing intact AP and NTS were selected from −13.6 mm to −14.1 mm posterior to the bregma to perform immunofluorescent staining and FISH (Fig. 1k).
For pSTAT3 and cFOS immunostaining, tissue sections were sequentially incubated in base (1% NaOH with 1% H2O2), 0.3% glycine, and 0.3% SDS unmasking solution in PBS for 10 min. Sections for pSTAT3 were then incubated in blocking solution (1% Triton X-100 and 5% donkey serum in PBS) for 1 h at room temperature, while cFOS sections were incubated in blocking solution (1% Triton X-100 and 5% BSA in PBS) for 1 h at room temperature. Afterwards, sections were incubated overnight at 4 °C with either anti-pSTAT3 (Tyr705) Ab (9131, Cell Signaling; 1:100) or cFOS mAb (9F6, 2250, Cell Signaling; 1:1000), followed by 3 washes in PBS.
For pSTAT3 detection, sections were incubated for 2 h at room temperature with Alexa Fluor 488-conjugated donkey anti-rabbit IgG secondary antibody (A-21206, Thermo Fisher Scientific) and DAPI, followed by 3 additional PBS washes. For cFOS detection, sections were incubated for 2 h at room temperature with a biotinylated anti-rabbit IgG secondary antibody from the VECTASTAIN ABC peroxidase kit (PK-4001, Vector Laboratories), followed by 30 min in the ABC complex. The antigen was visualized using DAB (3,3’-diaminobenzidine) peroxidase substrate (SK-4105, Vector Laboratories), producing a brown stain. Immunostaining images were acquired using either an Olympus BX51WI microscope equipped with a QImaging Retiga 2000R digital camera or a Zeiss Axio Imager with a CCD camera and ZenPro 3.5 software.
The FISH was performed using the RNAscope Multiplex Fluorescent Kit v2 (UM323100, ACD; Advanced Cell Diagnostics). The sections were pretreated based on the manufacturer’s instructions with the experimental condition optimizations as: boiling slides for 3 min in target retrieval buffer instead of 5 min; using Protease III for digestion at 40 °C for 15 min instead of 30 min. The sections were hybridized with probes detecting rat TGR5 (Rn-Gpbar, 316731-C1, ACD), rat Lepr (Rn-Lepr, 415951-C2, ACD), rat Aif1 (Rn-Aif1, 457731-C3, ACD), and rat Rbfox3 (Rn-Rbfox3, 436351-C3, ACD) mRNAs. Of note, the rat LepR probes we used targeted all the LepR isoforms, as ACD does not provide LepRb specific probes. Negative and positive controls were processed in parallel with the target probes to ensure tissue RNA integrity and optimal assay performance. Following hybridization, probes were labeled with TSA fluorescent dyes (Vivid 650 for Lepr or TGR5, and Vivid 570 for Aif1 or Rbfox3). After DAPI incubation, the slides were coverslipped with ProLong Gold Antifade Mountant (P36930, Thermo Fisher Scientific). The fluorescent images were captured using a Zeiss Axio Imager and ZenPro 3.5 software.
The fluorescent images were captured from the left side of brainstem region containing NTS, AP and central canal at consistent magnification, illumination, and exposure time. Photoshop (Adobe) was used to uniformly adjust brightness and contrast, and the counter tool was used to quantify the cell numbers. The quantification was performed in the consistent region of NTS for all the immunofluorescent staining and FISH images (Fig. 1k). For immunofluorescent staining, the p-STAT3 fluorescent signaling overlay with DAPI was seen as p-STAT3+ cells. The fluorescent puncta of FISH signaling from a gene probe within the DAPI staining were considered as the gene expressing cells.
Calculation of FISH ratios: To compare the proportion of Tgr5+/microglia (Aif1+) to LepR+/microglia (Aif1+) we ensured that in each section there were comparable numbers of microglia (Aif1+) cells. To determine the ratio, we calculated the Tgr5+ microglia or LepR+ microglia cells to the total number of microglia present in these sections, this is reported in Fig. 3h. A similar calculation was done to report neurons expressing Tgr5 or LepR in Fig. 3j. In Fig. 3l we calculated the total number of Tgr5+ cells and then determined how many Tgr5+ cells are LepR+ or LepR- to provide the ratio reported.
Western blot analysis
NTS tissue samples ( ~ 10 mg) were collected from rats that were anesthetized (ketamine, 90 mg/kg; xylazine, 10 mg/kg), then euthanized by decapitation. Tissues were homogenized for 3 min in 200 μL of RIPA lysis buffer (9806, Cell Signaling) with PMSF (1 mM) using a tissue homogenizer and were centrifuged at 12,500 g for 10 min at 4 °C to take the supernatant from the samples. The protein concentration within the sample supernatants was determined using the Pierce BCA protein assay kit (23225, Thermo Fisher Scientific), and 15 μg protein was subject to electrophoresis on SDS-polyacrylamide gel electrophoresis and transferred to polyvinylidene fluoride membranes. The membranes were incubated with blocking buffer (10% bovine serum albumin in TBST) at room temperature for 1 h, followed by incubation in the primary antibodies (as indicated below) diluted in the blocking buffer at 4 °C overnight. The membranes were washed for 4 times with TBST and incubated with the HRP-conjugated anti-rabbit IgG secondary antibody (7074, Cell Signaling; 1:4000) diluted in 5% skim milk at room temperature for 1.5 h. Finally, the membranes were washed with TBST for another 4 times, and the blot signaling was detected using Clarity Western ECL Substrate (1705060, Bio-Rad). Immunoblots were imaged using an image system (ChemiDoc Imaging system, Bio-Rad), and protein levels were quantified by ImageJ 1.53 (National Institute of Health). For the primary antibody incubations, the membranes were firstly incubated with the anti-p-STAT3 (Tyr705) mAb (D3A7, 9145, Cell Signaling; 1:1000) and the anti-GAPDH mAb (14C10, 2118, Cell Signaling; 1:2000). After imaging, the membranes were stripped of the antibodies with Restore Plus western blot stripping buffer (46430, Thermo Fisher Scientific) and were incubated again with the anti-STAT3 mAb (79D7, 4904, Cell Signaling; 1:1000) after re-blocking.
Quantitative PCR analysis
NTS ( ~ 10 mg) and AP ( ~ 1 mg) tissue samples were collected from rats that were anesthetized (ketamine, 90 mg/kg; xylazine, 10 mg/kg), then euthanized by decapitation. Tissues were weighed, added to lysis buffer, and then homogenized. RNA was isolated using the PureLink RNA Mini Kit (12183025, Thermo Fisher Scientific) and was subjected to DNase I digestion. Then, RNA was quantified by measuring the absorbance at 260 and 280 nm (Cytation 5, BioTek Instruments), and cDNA was generated using 2 mg of RNA and the SuperScript Vilo cDNA Synthesis Kit (11754050, Thermo Fisher Scientific). Quantitative PCR was performed using TaqMan Gene Expression master mix (4369016, Thermo Fisher Scientific) and TaqMan primers (Thermo Fisher Scientific) for rat ribosomal protein 18 s (assay identifier Rn01428913_gH), rat Tgr5 (assay identifier Rn01400316_s1), rat Leprb (assay identifier Rn00664624_m1)23,28, rat Cd68 (assay identifier Rn01495634_g1), Il6 (assay identifier Rn01410330_m1), Tnfa (assay identifier Rn99999017_m1), Socs3 (assay identifier Rn00585674_s1), and Ptp1b (assay identifier Rn01423685_m1) using a quantitative PCR machine (QuantStudio 7 Flex; Applied Biosystems). Relative gene expression was normalized to rat ribosomal protein 18 s as the reference gene using the comparative cycle threshold method.
PKA kinase activity measurement
The NTS tissue samples were weighed ( ~ 10 mg) and were homogenized in lysis buffer (5 μL/mg). The supernatant was collected and further diluted for 1000 times. PKA kinase activity was measured using the PKA Kinase Activity Assay Kit (ab139435, Abcam).
Bile acid analysis
Plasma and NTS tissue samples for bile acid measurement were obtained from the anesthetized rats that had fasted from 8 AM to 4 PM and that had been refed for 0.5 or 2 h after fasting. Plasma was obtained via cardiac puncture, and 50 μL of plasma samples was extracted by adding 310 μL methanol (containing 0.1% ammonium hydroxide, and 5 ng chlorpropamide as an internal standard) and then centrifuged (16,000 g, 10 min) to collect supernatant (200 μL). NTS (~10 mg) tissue samples were accurately weighed and homogenized in 100 μL double-distilled water. After adding 310 μL methanol (containing 0.1% ammonium hydroxide, and 25 ng chlorpropamide as an internal standard), the samples were centrifuged (16,000 g, 10 min) to obtain supernatant (300 μL).
Bile acids were quantified with an Ekspert ultra liquid chromatography 100 coupled with an AB SCIEX Triple TOF 5600 with an ESI source. For liquid chromatography separation, Waters XBridge Peptide BEH C18 column (100 mm × 2.1 mm i.d., 3.5 μm) at 40 °C and a flow rate of 0.4 ml/min was used. The solvent of the mobile phase was a mixture of 0.1% formic acid and 10 mM acetic acid amine in water (A) and 0.1% formic acid in 80% methanol and 20% acetonitrile (B). The gradient flow was first set at 35% B for 0.5 min, linearly increased to 60% B during the next 2.5 min, linearly increased to 80% B during the next 7 min, linearly increased to 90% B during the next 6 min, linearly decreased to 35% B during the next 4.5 min and maintained at this composition for an additional 2.5 min. The injection volume was 5 μL. The m/z range was set at 200–800 Da for TOF MS scans and 50–800 Da to produce ion scans in auto MS/MS acquisition. Raw data was processed using the PeakView 1.2 and MultiQuant 2.1 software based on the m/z value and the sample retention time.
Conditional Taste Avoidance test
NTS-cannulated rats were fed a HFD for 3 d before undergoing a conditional taste avoidance test as described69. Rats were habituated to 1 h daily access to water, with two bottles of unflavored water placed in each cage for 5 d. On day 6, all rats received two bottles containing 0.1% saccharin (Sigma-Aldrich, 109185-250 G) instead of water. Immediately after 1 h saccharin exposure, rats received an NTS infusion of CCDC or CMC at the same dose and infusion rate as feeding studies (0.04 \(\mu\)L/min, for 5 min). Another cohort of rats on this training paradigm received an intraperitoneal injection of LiCl (22 mg/kg) (Fisher Scientific, L121-100) or saline as positive and negative controls, respectively. The following day, rats were given 1 h access to two bottles of water. On the next day, they underwent a two-bottle choice test, where they had 1 h access to both water and 0.1% saccharin solution. The position of the two bottles were switched on the second day of the choice test to counterbalance any side preference. Saccharin and water intake were measured and the percentage of saccharin intake relative to total fluid intake was reported.
Statistical analysis
Statistical analyses were performed using Prism 8.0 (GraphPad Software). Student t test (two-sided with 95% confidence level) was used to compare two groups. One-way ANOVA with Tukey post hoc test was performed for more than two groups with single variable, and two-way ANOVA with Tukey post hoc test was performed when there were more than two groups with two variables. Differences were considered significant at P < 0.05, and all results were presented as mean + SEM.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All the relevant data are available from the authors on request and/or are included within the manuscript and supplemental materials. Source data has been included with this paper. Source data are provided with this paper.
References
Fleishman, J. S. & Kumar, S. Bile acid metabolism and signaling in health and disease: Molecular mechanisms and therapeutic targets. Signal Transduct. Target. Ther. 9, 97 (2024).
Ridlon, J. M., Kang, D. J. & Hylemon, P. B. Bile salt biotransformations by human intestinal bacteria. J. Lipid Res 47, 241–259 (2006).
Ridlon, J. M., Kang, D. J., Hylemon, P. B. & Bajaj, J. S. Bile acids and the gut microbiome. Curr. Opin. Gastroenterol. 30, 332–338 (2014).
Borgström, B., Dahlqvist, A., Lundh, G. & Sjövall, J. Studies of intestinal digestion and absorption in the human. J. Clin. Investig. 36, 1521–1536 (1957).
Pandak, W. M. et al. Effects of CYP7A1 overexpression on cholesterol and bile acid homeostasis. Am. J. Physiol. -Gastrointest. Liver Physiol. 281, G878–G889 (2001).
Wang, H., Chen, J., Hollister, K., Sowers, L. C. & Forman, B. M. Endogenous bile acids are ligands for the nuclear receptor FXR/BAR. Mol. Cell 3, 543–553 (1999).
Kawamata, Y. et al. A G protein-coupled receptor responsive to bile acids. J. Biol. Chem. 278, 9435–9440 (2003).
Maruyama, T. et al. Identification of membrane-type receptor for bile acids (M-BAR). Biochem Biophys. Res Commun. 298, 714–719 (2002).
Watanabe, M. et al. Bile acids induce energy expenditure by promoting intracellular thyroid hormone activation. Nature 439, 484–489 (2006).
Velazquez-Villegas, L. A. et al. TGR5 signalling promotes mitochondrial fission and beige remodelling of white adipose tissue. Nat. Commun. 9, 245 (2018).
Holter, M. M. et al. Compound 18 improves glucose tolerance in a hepatocyte TGR5-dependent manner in mice. Nutrients 12, https://doi.org/10.3390/nu12072124 (2020).
Castellanos-Jankiewicz, A. et al. Hypothalamic bile acid-TGR5 signaling protects from obesity. Cell Metab. 33, 1483–1492 e1410 (2021).
Perino, A. et al. Central anorexigenic actions of bile acids are mediated by TGR5. Nat. Metab. 3, 595–603 (2021).
Huang, K. P. et al. Dissociable hindbrain GLP1R circuits for satiety and aversion. Nature 632, 585–593 (2024).
Zhang, S. Y. et al. Metformin triggers a kidney GDF15-dependent area postrema axis to regulate food intake and body weight. Cell Metab. 35, 875–886.e875 (2023).
Tsai, V. W. et al. The anorectic actions of the TGFβ cytokine MIC-1/GDF15 require an intact brainstem area postrema and nucleus of the solitary tract. PLoS One 9, e100370 (2014).
Hayes, M. R., Skibicka, K. P. & Grill, H. J. Caudal brainstem processing is sufficient for behavioral, sympathetic, and parasympathetic responses driven by peripheral and hindbrain glucagon-like-peptide-1 receptor stimulation. Endocrinology 149, 4059–4068 (2008).
Angelin, B. & Bjorkhem, I. Postprandial serum bile acids in healthy man. Evidence for differences in absorptive pattern between individual bile acids. Gut 18, 606–609 (1977).
Higashi, T. et al. Unconjugated bile acids in rat brain: Analytical method based on LC/ESI-MS/MS with chemical derivatization and estimation of their origin by comparison to serum levels. Steroids 125, 107–113 (2017).
Collaboration, N. C. D. R. F. Worldwide trends in body-mass index, underweight, overweight, and obesity from 1975 to 2016: a pooled analysis of 2416 population-based measurement studies in 128.9 million children, adolescents, and adults. Lancet 390, 2627–2642 (2017).
Sun, H. et al. IDF Diabetes Atlas: Global, regional and country-level diabetes prevalence estimates for 2021 and projections for 2045. Diab Res Clin. Pr. 183, 109119 (2022).
Pan, W. W. & Myers, M. G. Jr. Leptin and the maintenance of elevated body weight. Nat. Rev. Neurosci. 19, 95–105 (2018).
Kanoski, S. E. et al. Endogenous leptin receptor signaling in the medial nucleus tractus solitarius affects meal size and potentiates intestinal satiation signals. Am. J. Physiol. Endocrinol. Metab. 303, E496–E503 (2012).
Cheng, W. et al. Leptin receptor–expressing nucleus tractus solitarius neurons suppress food intake independently of GLP1 in mice. JCI Insight 5, https://doi.org/10.1172/jci.insight.134359 (2020).
Bettge, K., Kahle, M., Abd El Aziz, M. S., Meier, J. J. & Nauck, M. A. Occurrence of nausea, vomiting and diarrhoea reported as adverse events in clinical trials studying glucagon-like peptide-1 receptor agonists: A systematic analysis of published clinical trials. Diab, Obes. Metab. 19, 336–347 (2017).
Weghuber, D. et al. Once-weekly semaglutide in adolescents with obesity. N. Engl. J. Med. 387, 2245–2257 (2022).
Grill, H. J. et al. Evidence that the caudal brainstem is a target for the inhibitory effect of leptin on food intake. Endocrinology 143, 239–246 (2002).
Hayes, M. R. et al. Endogenous leptin signaling in the caudal nucleus tractus solitarius and area postrema is required for energy balance regulation. Cell Metab. 11, 77–83 (2010).
Li, R. J. W. et al. A glucose-sensing mechanism with glucose transporter 1 and pyruvate kinase in the area postrema regulates hepatic glucose production in rats. J. Biol. Chem. 299, 104633 (2023).
Thaler, J. P. et al. Obesity is associated with hypothalamic injury in rodents and humans. J. Clin. Invest 122, 153–162 (2012).
Nakamura, Y., Si, Q. S. & Kataoka, K. Lipopolysaccharide-induced microglial activation in culture: temporal profiles of morphological change and release of cytokines and nitric oxide. Neurosci. Res 35, 95–100 (1999).
Valdearcos, M. et al. Microglia dictate the impact of saturated fat consumption on hypothalamic inflammation and neuronal function. Cell Rep. 9, 2124–2138 (2014).
Valdearcos, M. et al. Microglial inflammatory signaling orchestrates the hypothalamic immune response to dietary excess and mediates obesity susceptibility. Cell Metab. 26, 185–197.e183 (2017).
Zabolotny, J. M. et al. Protein-tyrosine phosphatase 1B expression is induced by inflammation in vivo. J. Biol. Chem. 283, 14230–14241 (2008).
Zhang, X. et al. Hypothalamic IKKbeta/NF-kappaB and ER stress link overnutrition to energy imbalance and obesity. Cell 135, 61–73 (2008).
Mori, H. et al. Socs3 deficiency in the brain elevates leptin sensitivity and confers resistance to diet-induced obesity. Nat. Med 10, 739–743 (2004).
Bence, K. K. et al. Neuronal PTP1B regulates body weight, adiposity and leptin action. Nat. Med 12, 917–924 (2006).
Yanguas-Casas, N., Barreda-Manso, M. A., Nieto-Sampedro, M. & Romero-Ramirez, L. TUDCA: An agonist of the bile acid receptor GPBAR1/TGR5 with anti-inflammatory effects in microglial cells. J. Cell Physiol. 232, 2231–2245 (2017).
Zhu, H. et al. Hyodeoxycholic acid inhibits lipopolysaccharide-induced microglia inflammatory responses through regulating TGR5/AKT/NF-kappaB signaling pathway. J. Psychopharmacol. 36, 849–859 (2022).
Perino, A. et al. TGR5 reduces macrophage migration through mTOR-induced C/EBPbeta differential translation. J. Clin. Invest 124, 5424–5436 (2014).
Pols, T. W. et al. TGR5 activation inhibits atherosclerosis by reducing macrophage inflammation and lipid loading. Cell Metab. 14, 747–757 (2011).
Keitel, V. et al. The bile acid receptor TGR5 (Gpbar-1) acts as a neurosteroid receptor in brain. Glia 58, 1794–1805 (2010).
LaRusso, N. F., Korman, M. G., Hoffman, N. E. & Hofmann, A. F. Dynamics of the enterohepatic circulation of bile acids. Postprandial serum concentrations of conjugates of cholic acid in health, cholecystectomized patients, and patients with bile acid malabsorption. N. Engl. J. Med 291, 689–692 (1974).
Angelin, B., Bjorkhem, I., Einarsson, K. & Ewerth, S. Hepatic uptake of bile acids in man. Fasting and postprandial concentrations of individual bile acids in portal venous and systemic blood serum. J. Clin. Invest 70, 724–731 (1982).
Zheng, X. et al. Hyocholic acid species improve glucose homeostasis through a distinct TGR5 and FXR signaling mechanism. Cell Metab. 33, 791–803.e797 (2021).
Thiele, T. E. et al. Central infusion of GLP-1, but not leptin, produces conditioned taste aversions in rats. Am. J. Physiol. 272, R726–R730 (1997).
Patel, J. D. & Ebenezer, I. S. The effect of intraperitoneal administration of leptin on short-term food intake in rats. Eur. J. Pharm. 580, 143–152 (2008).
Sasaki, T. et al. The exercise-inducible bile acid receptor Tgr5 improves skeletal muscle function in mice. J. Biol. Chem. 293, 10322–10332 (2018).
Ozcan, L. et al. Endoplasmic reticulum stress plays a central role in development of leptin resistance. Cell Metab. 9, 35–51 (2009).
Zhang, S. Y. et al. FXR in the dorsal vagal complex is sufficient and necessary for upper small intestinal microbiome-mediated changes of TCDCA to alter insulin action in rats. Gut 70, 1675–1683 (2021).
Bauer, P. V. et al. Lactobacillus gasseri in the upper small intestine impacts an ACSL3-dependent fatty acid-sensing pathway regulating whole-body glucose homeostasis. Cell Metab. 27, 572–587.e576 (2018).
Bauer, P. V. et al. Metformin alters upper small intestinal microbiota that impact a glucose-SGLT1-sensing glucoregulatory pathway. Cell Metab. 27, 101–117.e105 (2018).
Sun, L. et al. Gut microbiota and intestinal FXR mediate the clinical benefits of metformin. Nat. Med 24, 1919–1929 (2018).
Sayin, S. I. et al. Gut microbiota regulates bile acid metabolism by reducing the levels of tauro-beta-muricholic acid, a naturally occurring FXR antagonist. Cell Metab. 17, 225–235 (2013).
Young, A. A. et al. Glucose-lowering and insulin-sensitizing actions of exendin-4: studies in obese diabetic (ob/ob, db/db) mice, diabetic fatty Zucker rats, and diabetic rhesus monkeys (Macaca mulatta). Diabetes 48, 1026–1034 (1999).
Rodriquez de Fonseca, F. et al. Peripheral versus central effects of glucagon-like peptide-1 receptor agonists on satiety and body weight loss in Zucker obese rats. Metabolism 49, 709–717 (2000).
Gedulin, B. R. et al. Exenatide (exendin-4) improves insulin sensitivity and beta-cell mass in insulin-resistant obese fa/fa Zucker rats independent of glycemia and body weight. Endocrinology 146, 2069–2076 (2005).
Turton, M. D. et al. A role for glucagon-like peptide-1 in the central regulation of feeding. Nature 379, 69–72 (1996).
Schick, R. R., Zimmermann, J. P., vorm Walde, T. & Schusdziarra, V. Peptides that regulate food intake: glucagon-like peptide 1-(7-36) amide acts at lateral and medial hypothalamic sites to suppress feeding in rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 284, R1427–R1435 (2003).
Pocai, A. et al. Restoration of hypothalamic lipid sensing normalizes energy and glucose homeostasis in overfed rats. J. Clin. Invest 116, 1081–1091 (2006).
Wang, P. Y. T. et al. Upper intestinal lipids trigger a gut–brain–liver axis to regulate glucose production. Nature 452, 1012–1016 (2008).
Abraham, M. A., Rasti, M., Bauer, P. V. & Lam, T. K. T. Leptin enhances hypothalamic lactate dehydrogenase A (LDHA)-dependent glucose sensing to lower glucose production in high-fat-fed rats. J. Biol. Chem. 293, 4159–4166 (2018).
Evans, K. A. et al. Discovery of 3-aryl-4-isoxazolecarboxamides as TGR5 receptor agonists. J. Med Chem. 52, 7962–7965 (2009).
Leinninger, G. M. et al. Leptin acts via leptin receptor-expressing lateral hypothalamic neurons to modulate the mesolimbic dopamine system and suppress feeding. Cell Metab. 10, 89–98 (2009).
Satoh, N. et al. The arcuate nucleus as a primary site of satiety effect of leptin in rats. Neurosci. Lett. 224, 149–152 (1997).
Buettner, C. et al. Critical role of STAT3 in leptin’s metabolic actions. Cell Metab. 4, 49–60 (2006).
Zhang, S. Y., Danaei, Z., Bruce, K., Chiu, J. F. M. & Lam, T. K. T. Acute Activation of GFRAL in the Area Postrema Contributes to Glucose Regulation Independent of Weight. Diabetes 73, 426–433 (2024).
Paxinos, G. & Watson, C. The rat brain in stereotaxic coordinates: hard cover edition. (Elsevier, 2006).
Filippi, B. M. et al. Insulin signals through the dorsal vagal complex to regulate energy balance. Diabetes 63, 892–899 (2014).
Acknowledgements
K.B. is supported by an Ontario Graduate Scholarship and a Banting and Best Diabetes Center graduate studentship. S.-Y.Z. is supported by a Canadian Institutes of Health Research (CIHR) post-doctoral fellowship. A.N.G. is supported by the E. Fidlar Ontario Graduate Scholarship, a Banting and Best Diabetes Center graduate studentship, and the University of Toronto MD/PhD program. A.W.X. is supported by the Joseph and Vera Long Foundation. Z.Y. is supported by a Banting and Best Diabetes Center post-doctoral fellowship. This work is supported by a CIHR grant (PJT-189957) to T.K.T.L. who holds a Tier 1 Canada Research Chair in Diabetes and Obesity at the Toronto General Hospital Research Institute and the University of Toronto.
Author information
Authors and Affiliations
Contributions
K.B. and S.-Y.Z. conducted and designed experiments, performed data analyses, and wrote the manuscript. A.N.G., M.W. and Z.Y. assisted with rat studies. T.P.B. and A.W.X. assisted in histological analysis. P. W. assisted in measuring bile acid levels in the plasma and the NTS tissues. T.K.T.L. supervised the project, designed experiments, and edited the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous, reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Bruce, K., Zhang, SY., Garrido, A.N. et al. Pharmacological and physiological activation of TGR5 in the NTS lowers food intake by enhancing leptin-STAT3 signaling. Nat Commun 16, 4990 (2025). https://doi.org/10.1038/s41467-025-60331-1
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-60331-1
