
Abstract
Highly conserved homeobox genes are closely related to bone formation during embryogenesis, while their role in adult bone resorption remains unclear. In this study, we found that the homeobox gene MSX2 actively participates bone metabolism. Myeloid-specific Msx2 deficiency safeguards bone mass under physiological and pathological conditions. Loss of Msx2 acts as a “brake” in the fusion fate of osteoclasts, resulting in a larger population of pre-osteoclasts. Pre-osteoclasts secrete platelet-derived growth factor-BB (PDGF-BB), which promotes angiogenesis-mediated bone formation. Mechanistically, MSX2 directly binds to the vital osteoclastogenic transcription factor PU.1 and protects it from FBXW7-mediated ubiquitination degradation. Msx2 and Fbxw7 double knockout mitigated the protective effect of MSX2 deficiency on bone mass. Finally, we identified a natural compound, morusinol, that specifically destroys the combination of MSX2 and PU.1, promoting PU.1 degradation and attenuating ovariectomy-induced bone loss. Overall, our results demonstrate that targeting Msx2 is a promising anabolic therapy for osteoporosis.
Similar content being viewed by others


Introduction
Bone is the primary organ that provides support and protection. Disturbed bone homeostasis impairs bone health, leading to osteoporosis and fragility fracture1,2. Bone homeostasis is generally believed to be governed by osteoblast-mediated bone formation and osteoclast-mediated bone resorption2. However, in recent years, these views have broadened because multiple cell types, including endothelial cells, neurons, immune cells, and adipocytes, exert various effects on bone cells within the local environment3,4,5,6. These findings enrich our understanding of bone metabolism regulation and inspire us to explore novel strategies for osteoporosis treatment.
Post-menopausal osteoporosis (PMOP) is the most common type of osteoporosis. In the bone environment of PMOP, osteoclasts are over-activated and mediate significantly enhanced bone resorption, leading to severe bone loss7. Osteoclast is the only cell type in the body capable of absorbing the bone matrix. Induced by macrophage colony-stimulating factor (M-CSF) and receptor activator of nuclear factor-κB ligand (RANKL), bone marrow monocytes/macrophages (BMMs) could proliferate, fuse, and eventually differentiate into multi-nuclear osteoclasts8. Mechanistically, M-CSF and RANKL bind to their receptors CSF1R and RANK, respectively, and stimulate downstream signaling pathways such as MAPKs and NF-κB, leading to the activation of master transcription factors including PU.1 and NFATc1, initiating osteoclastogenesis9,10. Osteoclasts actively participate in bone metabolism and perform endocrine and paracrine functions. Tartrate-resistant acid phosphatase-positive (TRAP+) mono-nuclear preosteoclasts secrete platelet-derived growth factor-BB (PDGF-BB), which enhances H-type vessel (EMCNhi CD31hi) formation and eventually stimulates osteogenesis11. Therefore, restraining osteoclast fusion and maintaining them in the preosteoclast stage could suppress bone resorption while coupling angiogenesis and osteogenesis, exhibiting great potential for osteoporosis treatment.
Characterized by a particular 180 bp homeobox sequence, homeobox genes encode a group of transcription factors containing homeodomain12. The structure of these genes has been highly conserved during evolution, and they generally play critical roles in embryogenesis, cell proliferation, and differentiation13,14,15. Multiple homeobox genes have been implicated in the regulation of bone mass and osteogenesis16,17,18,19. Nevertheless, the current understanding of the role of homeobox genes in PMOP and osteoclastogenesis remains superficial and scattered, and the hub homeobox genes that regulate PMOP remain unclear. Here, we found that msh homeobox 2 (MSX2) plays a central role in PMOP. MSX2 belongs to the Msx family, Antennapedia (ANTP) class of human homeobox genes, and has been reported to be involved in bone development, including skull and tooth formation20,21. Functional haploinsufficiency of MSX2 causes parietal foramina (PFM), and whole-body Msx2 knockout mice exhibit pleiotropic defects in bone mass and ectodermal organ formation22,23. Although Msx2 is generally considered an important regulator of osteogenesis, Msx2 null mutant mice show an osteopetrosis phenotype in a RANK-dependent manner in skeleton24, suggesting that Msx2 might also participate in osteoclastogenesis, and its regulatory effect in the bone environment could be complex. However, the direct role of Msx2 in osteoclast remains unclear.
In this study, we showed that the loss of Msx2 in myeloid cells leads to increased bone mass. Msx2 deficiency impairs osteoclast fusion and exerts the role of “brake” in the progression of osteoclast cell fate, which subsequently maintains osteoclasts at the pre-fusion stage. The augmented numbers of pre-osteoclast secrete PDGF-BB, enhancing H-type vessel formation and further coupling osteogenesis. Mechanistically, MSX2 directly binds to PU.1 and protects it from FBXW7-mediated ubiquitination and degradation. Msx2 and Fbxw7 double conditional knockout mice exhibit recovery of bone resorption and osteoclastogenesis. Finally, we screened a natural compound library and found that morusinol, a flavonoid isolated from Morus alba root bark, can bind to MSX2 and release PU.1, promoting PU.1 degradation. The in vivo analysis confirmed the therapeutic effects of morusinol in an ovariectomy (OVX)-induced osteoporosis model. Our results suggest that MSX2 plays a critical role in PMOP pathogenesis and bone metabolism, coupling osteoclastogenesis with angiogenesis and osteogenesis, and may be a potential target for the treatment of PMOP.
Results
Msx2 is upregulated in post-menopausal osteoporosis and osteoclastogenesis
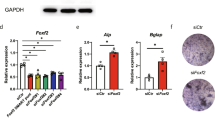
To identify important homeobox genes in PMOP and osteoclastogenesis, we crossed differentially expressed genes from GSE230665 (RNA-Seq datasets compares PMOP bones with postmenopausal healthy bones, |logFC| > 0.8, p < 0.05)25 and osteoporosis-related genes from GeneCards database (score > 1) with 298 homeobox genes and found 11 potential target genes (Supplementary Fig. 1a). PPI network analysis suggested that MSX2 and IRX3 are hub genes (Supplementary Fig. 1b). According to previous studies, global Msx2 knockout impairs osteoclast formation, whereas loss of Irx3 has no effect on osteoclasts26,27. Nevertheless, the direct role of Msx2 in osteoclastogenesis lacks solid evidence, and the underlying mechanism remains unclear; therefore, we focused on Msx2 in this study. To confirm the increased Msx2 expression level in PMOP, we constructed a mouse OVX model and double-labeled MSX2 with the osteoclastic marker Cathepsin K (CTSK) on bone sections from the distal femur. As shown in Supplementary Fig. 1c, d, MSX2+ CTSK+ cell number was significantly higher in the OVX group than in the sham group. These results prompted us to explore the expression pattern Msx2 during osteoclastogenesis. Therefore, we stimulated BMMs with M-CSF and RANKL in vitro and investigated the mRNA and protein levels of Msx2 during osteoclast formation. The mRNA levels were upregulated on day 3 of osteoclastogenesis and decreased on day 6 (Supplementary Fig. 1e). The protein levels of Msx2 also peaked on day 3 and remained constant on day 6 (Supplementary Fig. 1f, g). Together, these findings suggest that Msx2 is upregulated in PMOP and osteoclastogenesis, and may be a potential regulator of osteoclast formation.
Msx2 deficiency in myeloid cells increases bone mass by inhibiting pre-osteoclast fusion
To better understand the role of Msx2 in bone metabolism and osteoclasts, we generated conditional Msx2 knockout mice by crossing Msx2f/f mice with LysM-Cre mice (Msx2 cKO), in which Msx2 was deficient in myeloid lineage cells. The bone mass of 3-month-old female Msx2 cKO mice was higher than that of Msx2f/f controls (Fig. 1a). Micro-computed tomography (Micro-CT) analysis showed that bone volume per tissue volume (BV/TV), trabecular bone number (Tb.N), and cortical bone thickness (Ct.Th) increased, while trabecular bone separation (Tb.Sp) decreased in Msx2 cKO mice; trabecular bone thickness (Tb.Th) remained unchanged (Fig. 1b). Similar to that in females, Msx2 deficiency resulted in elevated BV/TV, Tb.N, Ct.Th, and reduced Tb.Sp in males (Supplementary Fig. 2). To precisely describe the changes in bone resorption in Msx2 cKO mice, we analyzed bone sections of the distal femurs. Although TRAP staining showed comparable numbers of total TRAP-positive cells between Msx2 cKO mice and controls, the number of multi-nuclear TRAP-positive cells was lower, and that of mononuclear TRAP-positive cells was higher in the Msx2 cKO group (Fig. 1c, d). Serum CTX-1 levels were also downregulated in Msx2 cKO mice (Fig. 1e). As more pre-osteoclasts were detected in Msx2 cKO mice, it was proposed that the fusion stage was impaired by Msx2 deficiency. Therefore, we performed in vitro osteoclastogenesis assays by inducing BMMs with M-CSF and RANKL. TRAP staining showed that there were comparable numbers of TRAP+ cells in Msx2 cKO and control groups after 3-day stimulation, and these cells were mainly mononuclear. However, cells from Msx2 cKO mice did not develop mature multi-nuclear TRAP+ osteoclasts on day 6, suggesting that the fusion stage of osteoclastogenesis was impaired (Fig. 1f, g). Accordingly, this impairment resulted in the suppression of resorption activity. As shown in Fig. 1h–j, Msx2 deficient cells generated reduced resorption pit areas on bone slices and produced fewer resorptive enzymes, such as CTSK. To directly compare the fusion ability of cells from Msx2 cKO and control mice, we performed a double fluorescence labeling assay in which fused cells were defined as double-labeled with both DiI and Hoechst (N ≥ 1), whereas unfused cells received only a single label (Fig. 1k). Expectedly, Msx2 deficient cells showed more single labeled cells than control cells when stimulated with RANKL (Fig. 1l). To more accurately evaluate the fusion efficiency of Msx2 cKO cells, we stained F-actin and nuclei using phallodin and DAPI, respectively. We calculated normalized number of fusion events as previously reported, equally considering fusions between two mononucleated cells, one mononucleated cell and one multinucleated cell, and two multinucleated cells. As shown in Fig. 1m, n, Msx2 cKO cells exhibited significantly reduced fusion events. Moreover, we examined genes related to osteoclast fusion and found Msx2 cKO cells expressed lower levels of Cd9, Cd44, Cd47, Dcstamp, and Ocstamp, which was consistent with their impaired fusion ability (Fig. 1o). Taken together, these results indicate that Msx2 deficiency improves bone mass by inhibiting pre-osteoclast fusion.

a Representative 3D Micro-CT images of distal femurs from 3-month-old female Msx2f/f and Msx2 cKO mice. b Trabecular and cortical bone parameters of Msx2f/f (n = 8) and Msx2 cKO (n = 8) mice. c Representative TRAP staining images of distal femurs from 3-month-old female Msx2f/f and Msx2 cKO mice. Scale bar, 100 μm. d Histomorphometric analysis of Oc.N/BS, Mu.Oc.N/BS, and Mo.Oc.N/BS (n = 5). Mu.Oc.N/BS, multinuclear osteoclast number per bone surface; Mo.Oc.N/BS, mononuclear osteoclast number per bone surface. e Serum CTX-1 level (n = 5). f BMMs were induced by 50 ng/mL RANKL for 3 or 6 days, and representative TRAP staining images were shown. Scale bar, 100 μm. g Quantification of N.TRAP+ cells/well and osteoclast number (nucleiå 5) per well (Oc.N/well) (n = 5). h Representative SEM images of bone slice resorption pits. Scale bar, 50 μm. i Relative resorption pits area (n = 5). j BMMs from Msx2f/f and Msx2 cKO mice were stimulated by 50 ng/mL RANKL, CTSK protein level were detected during osteoclastogenesis. Representative bands of 3 independent repeats were shown. k Schematic showing cell-cell fusion assay. l Representative images of cell-cell fusion assay. Scale bar, 100 μm. m Msx2f/f and Msx2 cKO BMMs were stimulated by 50 ng/mL RANKL for 6 days, the cells were stained with phalloidin and DAPI to recognize syncytium. Scale bar, 100 μm. n Quantification of normalized number of fusion events (n = 5). o Relative mRNA expression level of fusion-related genes of pre-osteoclasts from Msx2f/f and Msx2 cKO mice (n = 6). Data are presented as mean ± SD, and p-values were calculated by unpaired, two-tail student’s t-test (with Welch’s correction when appropriate).
Msx2 deficiency in myeloid cells stimulates osteogenesis and angiogenesis in vivo
The coupling between osteogenesis and H-type vessel formation has been widely reported, and mononuclear pre-osteoclast could secrete PDGF-BB and promote angiogenesis11. Here, Msx2 deficiency in osteoclast progenitors also resulted in changes on the bone formation side, with a higher osteocalcin (OCN) -positive cell number, enhanced mineral apposition rate (MAR), and bone formation rate per bone surface (BFR/BS) in Msx2 cKO mice (Fig. 2a–d). Angiography showed that Msx2 cKO mice exhibited elevated vessel volume and vessel surface in the bones (Fig. 2e, f). The number of H-type vessels (CD31hi EMCNhi) was also increased (Fig. 2g, h). Flow cytometry further supported our finding that more CD31hi EMCNhi endothelial cells were detected in the bone marrow of Msx2 cKO mice (Fig. 2i, j). PDGF-BB+ TRAP+ cells are considered to be the main population of pre-osteoclasts that induce angiogenesis11. Therefore, we performed immunofluorescence staining and found more PDGF-BB+ TRAP+ cells in Msx2 cKO mice (Fig. 2k, l). Collectively, the loss of Msx2 in myeloid cells increases bone mass by impairing osteoclastogenesis and enhancing angiogenesis and osteogenesis in vivo.

a OCN staining of distal femurs from Msx2f/f and Msx2 cKO mice. Scale bar, 50 μm. b Quantification of osteocalcin+ cell numbers per bone surface (N.OCN+ cells/ BS) (n = 5). c Representative images of calcein double labeling of trabecular bones from Msx2f/f and Msx2 cKO mice. Scale bar, 25 μm. d Quantification of mineral apposition rate (MAR) and bone formation rate per bone surface (BFR/BS) (n = 5). e Representative images of femora vessels. f Quantification of vessel volume and vessel surface (n = 5). g EMCN and CD31 immunofluorescence staining of distal femurs from Msx2f/f and Msx2 cKO mice. Scale bar, 50 μm. h Quantification of CD31hi EMCNhi vessel area in trabecular bones (CD31hi EMCNhi.Ar) (n = 5). i Representative flow cytometry plots of CD31hi EMCNhi endothelial cells in whole bone marrow cells. j Percentage of CD31hi EMCNhi endothelial cells (n = 3). k Immunofluorescence staining of PDGF-BB and TRAP in distal femurs from Msx2f/f and Msx2 cKO mice. Scale bar, 50 μm. l Quantification of PDGF-BB+ TRAP+ cell numbers per bone surface (n = 5). Data are presented as mean ± SD, and p-values were calculated by unpaired, two-tail student’s t-test.
Msx2 deficiency promotes vessel formation of endothelial cells in vitro
To further confirm the effect of Msx2 deficiency on vessel formation, we first detected differences in PDGF-BB secretion between samples derived from Msx2 cKO and control pre-osteoclasts. Msx2 cKO cells exhibited higher Pdgfb expression levels and produced more PDGF-BB in the medium (Supplementary Fig. 3a–c). Subsequently, we cultured human umbilical vein endothelial cells (HUVECs) with conditioned media derived from Msx2 cKO and control pre-osteoclasts (Supplementary Fig. 3d). The wound-healing assay showed that HUVECs cultured in Msx2 cKO medium exhibited elevated migration activity (Supplementary Fig. 3e, f). The Matrigel tube formation assay also indicated that Msx2 cKO conditioned medium enhanced HUVECs tube formation (Supplementary Fig. 3g, h). The osteogenic ability of bone marrow mesenchymal stem cells (BMSCs) was not affected by myeloid Msx2 conditional knockout (Supplementary Fig. 3i). Moreover, no significant changes in osteogenesis were observed when wild-type BMSCs were treated with conditioned media from Msx2 cKO and control pre-osteoclasts in vitro (Supplementary Fig. 3j). These results suggest that myeloid Msx2 deficiency enhances vessel formation of endothelial cells and that the observed promoting effect on osteogenesis in vivo is indirect.
MSX2 promotes pre-osteoclast fusion through stabilizing PU.1
Given the increased mRNA levels of fusion-related genes such as Cd9, Cd44, Cd47, Dcstamp, and Ocstamp, we used the Signaling Pathways Project data of Genecards database to identify their common transcription factors (TFs). This analysis revealed 18 TFs (Supplementary Fig. 4a). Notably, MSX2 is not among these 18, although it is a well-known TF. Another important role of MSX2 is to serve as a co-transcriptional regulator and to affect mRNA expression through other TFs. Therefore, we analyzed the CHIP-Seq data of fusion-related genes from Genecards, and PU.1 (Spi1) showed a relatively higher frequency and binding score in all five fusion-related genes, which means PU.1 is the most probable TF regulating fusion-related genes (Supplementary Fig. 4b–f). Thus, MSX2 likely regulates the expression of fusion-related genes via PU.1. Overexpressing of PU.1 indeed rescued the formation of multinucleated osteoclasts and the expression of fusion-related genes (Fig. 3a–c). To further explore the effect of MSX2 on PU.1, we investigated whether Msx2 knockout altered the expression of PU.1. After treating Msx2f/f BMMs with Cre or Msx2 adenoviruses, the protein level of PU.1 was altered, whereas the mRNA level of PU.1 remained unchanged (Fig. 3d–f). Moreover, knocking out Msx2 enhanced PU.1 degradation when treated with cycloheximide (CHX) (Fig. 3g, h), whereas overexpression of MSX2 suppressed PU.1 degradation (Supplementary Fig. 5a, b). To clarify the PU.1 degradation pathway, we used the proteasome inhibitor MG132 or the lysosome inhibitor chloroquine (CQ) to treat Msx2f/f and Msx2 cKO pre-osteoclasts, respectively. Protein degradation of PU.1 was prevented by MG132 treatment, while CQ showed no rescue effect, suggesting that MSX2 regulated PU.1 protein degradation mainly through the proteasome pathway (Fig. 3i, j). Subsequently, we performed ubiquitination assays and found Msx2 cKO pre-osteoclasts exhibited increased PU.1 ubiquitination (Fig. 3k). In contrast, overexpression of MSX2 attenuated PU.1 ubiquitination, similar results were obtained in the 293T cell line (Supplementary Fig. 5c, d). To further confirm the reduced PU.1 level in osteoclast in vivo, we labeled PU.1 with osteoclastic marker, CTSK, in bone sections and found the number of PU.1+ CTSK+ cells was significantly reduced in Msx2 cKO mice (Fig. 3l, m). Because PU.1 is crucial for myeloid differentiation, we examined the expression of F4/80, a marker of mature monocytes and macrophages, in bone sections from Msx2 cKO mice. The expression level of F4/80 was comparable in Msx2f/f and Msx2 cKO mice, indicating that myeloid Msx2 deficiency does not no influence on myeloid differentiation and osteoclast precursor formation (Supplementary Fig. 5e, f). In summary, MSX2 promotes the expression of osteoclast fusion-related genes by preventing the proteasome-mediated degradation of PU.1 without affecting macrophage and osteoclast precursor development.

a BMMs were infected by GFP or PU.1 adenovirus and then stimulated by RANKL. Representative images of TRAP staining were shown. Scale bar, 100 μm. b Quantification of Oc.N/well in (a) (n = 5). c BMMs were infected by GFP or PU.1 adenovirus and then stimulated by RANKL for 3 days. Relative mRNA expressions of fusion related genes were measured (n = 5). d BMMs were infected by GFP, Cre, or MSX2 adenovirus and then stimulated by RANKL, relative PU.1 mRNA expression was measured (n = 5). e BMMs were treated as stated in (d), and representative images of MSX2 and PU.1 protein bands were shown. f Quantification of MSX2 and PU.1 relative grey levels (n = 3). g BMMs from Msx2f/f and Msx2 cKO mice were induced by RANKL for 3 days, then cells were treated by cycloheximide (CHX) for indicated hours, the protein levels of PU.1 and MSX2 were detected. h Quantification of PU.1 relative grey level. i BMMs from Msx2f/f and Msx2 cKO mice were induced by RANKL for 3 days, then cells were treated by DMSO, MG132, or chloroquine (CQ) for 4 h, the protein levels of PU.1 and MSX2 were detected. j Relative PU.1 protein level was measured (n = 3). k BMMs from Msx2f/f and Msx2 cKO mice were induced by RANKL for 3 days. Cells were treated with MG132 for 4 h, and Co-IP was performed to examine the ubiquitination level of PU.1. l PU.1 and CTSK immunofluorescence staining of distal femurs from Msx2f/f and Msx2 cKO mice. Scale bar, 100 μm. m Quantification of PU.1+ CTSK+ cell numbers per bone surface (PU.1+ CTSK+.N/ BS) (n = 5). Data are presented as mean ± SD, and p-values were calculated by unpaired, two-tail student’s t-test (f and m), one-way ANOVA with Tukey’s multiple comparisons (b–d) and two-way ANOVA with Sidak’s multiple comparisons (h and j).
MSX2 binds PU.1 and protects it from FBXW7-mediated ubiquitination
To further explore how MSX2 protects PU.1, we hypothesized that MSX2 directly binds to PU.1. Immunofluorescence labeling revealed that MSX2 and PU.1 are both nuclear (Fig. 4a). The Co-IP assay further confirmed the direct combination of MSX2 and PU.1 in both BMMs and HEK-293T cells (Fig. 4b, c). To investigate whether this combination affected PU.1 degradation, we searched for the potential E3 ubiquitin ligases related to PU.1 using UbiBrowser and found FBXW728. FBXW7 is the only E3 ubiquitination ligase for PU.1 that has been confirmed by literature in UbiBrowser database (Fig. 4d). Expectedly, exogenous MSX2 significantly reduced the ubiquitination level of PU.1, which was rescued by FBXW7 overexpression in HEK-293T cells (Fig. 4e). Intriguingly, the MSX2 binding region in PU.1 was also the critical motif responsible for FBXW7 recognition. Specifically, we analyzed the amino acid sequence of PU.1 and found three loci in accordance with the FBXW7 phosphodegron motifs (S/TxxxS/T), which was consistent with a previous study29 (Fig. 4f). Molecular docking assays revealed that MSX2 sheltered PU.1-Ser41 and Ser142, blocking their recognition by FBXW7 (Fig. 4g). We further mutated PU.1-Ser41 and PU.1-Ser142 (PU.1-MUT: PU.1-S41A, S142A) and found that FBXW7 could promote PU.1 ubiquitination, however, this effect was canceled by PU.1 mutation (Fig. 4h). Overall, MSX2 served as a protector by concealing PU.1-Ser41 and Ser142, thereby preventing PU.1 from being ubiquitinated by FBXW7 (Fig. 4i).

a BMMs were stimulated by RANKL for 3 days, and immunofluorescence staining of MSX2 and PU.1 in BMMs were captured. Scale bar, 20 μm. b BMMs were stimulated by RANKL for 3 days, and Co-IP was performed to show the interaction between MSX2 and PU.1. c HEK-293T cells were transfected by PU.1-FLAG and MSX2-HA, and the interaction between MSX2 and PU.1 were examined. d Prediction of the E3 ubiquitination ligase of PU.1 using Ubibrowser. e HEK-293T cells were transfected as indicated and then treated by MG132 for 6 h. The ubiquitination level of PU.1 was detected by Co-IP. f The FBXW7 CPD consensus motif in mouse PU.1. g Molecular docking of MSX2 and PU.1 complex. h HEK-293T cells were transfected as indicated and then treated by MG132 for 6 h. The PU.1 ubiquitination levels were detected by Co-IP. i Schematic of the protective effect of MSX2 on PU.1 ubiquitination degradation. All experiments were repeated 3 times independently, and the representative images were shown (a–c, e, and h).
Msx2 Fbxw7 double knockout blunts the bone phenotype of Msx2 cKO mice
FBXW7 has been reported to participate in osteoclastogenesis and mice with osteoclast-specific Fbxw7 ablation (Fbxw7f/f Ctsk-Cre) exhibit osteoporotic phenotype30. Here, we found that myeloid Fbxw7 deficiency (Fbxw7f/f LysM-Cre, Fbxw7 cKO) also led to low bone mass, with reduced BV/TV, Tb.Th, Tb.N, and elevated Tb.Sp (Supplementary Fig. 6a, b). Loss of Fbxw7 promotes osteoclastogenesis both in vivo and in vitro (Supplementary Fig. 6c–f). Consistently, PU.1 protein level was upregulated by Fbxw7 knockout (Supplementary Fig. 6g). To better confirm the relationship between MSX2, PU.1, and FBXW7 in vivo, we bred Msx2f/f Fbxw7f/f LysM-Cre mice (dKO), in which both Msx2 and Fbxw7 were knocked out in myeloid cells. As expected, PU.1 protein levels were restored by the double knockout (Supplementary Fig. 7a). The Co-IP assay revealed a lower ubiquitination level of PU.1 by double knockout (Supplementary Fig. 7b). The in vitro osteoclastogenesis assay showed recovery of multi-nuclear TRAP-positive cell numbers (Supplementary Fig. 7c, d). The bone slice resorption assay indicated that the double knockout rescued the bone resorptive pit area (Supplementary Fig. 7e, f). Additionally, the normalized number of cell fusion events was partially recovered by the double knockout (Supplementary Fig. 7g, h). Subsequently, we analyzed the bone phenotypes in vivo. The bone mass of dKO mice was lower than that of Msx2 cKO mice, and bone parameters were altered correspondingly (Fig. 5a–c). Furthermore, bone section staining showed that the number of multi-nuclear TRAP-positive cells was recovered in dKO mice, whereas OCN-positive cells were decreased (Fig. 5d–g). The number of H-type vessels also decreased (Fig. 5h, i). Furthermore, while the number of PDGF-BB and TRAP double-positive cells was increased in Msx2 cKO mice, the double knockout significantly reduced this population (Fig. 5j, k). In conclusion, using an Msx2 Fbxw7 double knockout mouse model, we further confirmed that MSX2 protects PU.1 from FBXW7-mediated ubiquitination.

a Representative 3D Micro-CT images of distal femurs from 3-month-old female Msx2f/f and Msx2 cKO and Msx2 Fbxw7 dKO mice. b Bone morphology parameters of distal femurs from Msx2f/f (n = 8), Msx2 cKO (n = 8), and Msx2 Fbxw7 dKO (n = 8) mice. c Serum CTX-1 level of Msx2f/f, Msx2 cKO, and Msx2 Fbxw7 dKO mice (n = 5). d Representative TRAP staining images of distal femurs from Msx2f/f, Msx2 cKO, and Msx2 Fbxw7 dKO mice. Scale bar, 100 μm. e Quantification of Mu.Oc.N/BS in (d) (n = 5). f OCN immunofluorescence staining of distal femurs. Scale bar, 50 μm. g N.OCN+ cells/BS was measured (n = 5). h Representative images of EMCN and CD31 immunofluorescence staining of distal femurs. Scale bar, 50 μm. i CD31hi EMCNhi.Ar was measured (n = 5). j Representative images of PDGF-BB and TRAP immunofluorescence staining. Scale bar, 50 μm. k Quantification of PDGF-BB+ TRAP+ cell numbers/ BS (n = 5). Data are presented as mean ± SD, and p-values were calculated using one-way ANOVA with Tukey’s multiple comparisons.
Targeting MSX2 by morusinol prevents bone loss in OVX mice
Given that Msx2 deficiency led to elevated bone mass, we hypothesized that targeting MSX2 could be a potential therapy for osteoporosis. To test this hypothesis, we investigated the bone phenotypes of Msx2f/f and Msx2 cKO mice after ovariectomy. As shown in Supplementary Fig. 8a, b, OVX surgery led to a reduction of BV/TV from 8.497 ± 1.412% to 2.508 ± 1.012% (approximately 70.5% loss) in Msx2f/f mice, while the proportion of bone loss in Msx2 cKO mice was 60.3% (from 14.54 ± 1.861% to 5.712 ± 1.224%). Moreover, Msx2 cKO mice exhibited increased Tb.N, Ct.Th, and decreased Tb.Sp after OVX (Supplementary Fig. 8b). Histological analysis revealed that Msx2 deficiency resulted in fewer multi-nuclear osteoclasts, and serum CTX-1 levels were also reduced (Supplementary Fig. 8c–e). On the bone formation side, Msx2 cKO mice showed more OCN-positive cells, higher MAR and BFR/BS (Supplementary Fig. 8f–i). Consistently, angiogenesis was more active in Msx2 cKO mice, with more EMCNhi CD31hi vessels (Supplementary Fig. 8j, k). These results confirmed that the bone protective effects of Msx2 deficiency also exist in the OVX mouse model.
Natural compounds have shown advantages in terms of safety, effectiveness, and convenience in disease treatment31; therefore, we screened approximately 20,000 natural compounds using high-throughput virtual screening (HTVS) to identify potential compounds that could interrupt the MSX2-PU.1 combination. According to the XP Gscore and molecular mechanics-generalized born surface area (MM-GBSA) dG Bind, we identified 13 molecules that showed a relatively high affinity for MSX2, of which four molecules could form direct interactions with MSX2, including isosilybin, dehydrodiconiferyl alcohol, lincomycin hydrochloride monohydrate, and morusinol (Fig. 6a, b). After determining the concentrations used for in vitro treatment, we incubated BMMs with these drugs under RANKL stimulation (Fig. 6c). TRAP staining revealed that morusinol exerted the strongest suppressive effect on osteoclastogenesis (Fig. 6d, e). A bone resorption assay confirmed the inhibitory effect of morusinol on osteoclast function (Fig. 6f, g). The protein level of CTSK and PU.1 was also reduced by morusinol (Fig. 6h, i). To evaluate whether morusinol affects osteoclast fusion, we performed fusion assay in which the morusinol group exhibited significant decreased number of Hoechst+ DiI+ cells (Fig. 6j). Via F-actin staining, we found that morusinol reduced the number of osteoclast fusion events (Fig. 6k, l). Consistently, osteoclast fusion-related gene expression was also suppressed by morusinol (Fig. 6m). To determine whether these effects of morusinol were exerted through MSX2 and PU.1, we performed molecular docking between morusinol and MSX2, which showed that morusinol forms hydrophobic interactions with Val32 and Leu179, a hydrogen bond interaction with Ser178, and π-cation with Lys33 (Fig. 6n). These amino acid residues are important for the interaction between MSX2 and PU.1; thus, morusinol may inhibit osteoclastogenesis by interrupting the combination of MSX2 and PU.1 and promoting PU.1 degradation. As expected, the Co-IP assay indicated that the MSX2-PU.1 complex was reduced under morusinol treatment, while ubiquitinated PU.1 was upregulated (Fig. 6o, p). To further confirm whether morusinol works through the MSX2-FBXW7 axis, we employed morusinol in Fbxw7 cKO cells and found that Fbxw7 deficiency could rescue morusinol-mediated osteoclast inhibition (Supplementary Fig. 9a, b). Accordingly, the expression of fusion-related genes and the protein level of PU.1 and CTSK were recovered (Supplementary Fig. 9c, d). Since natural products generally exhibits extensive polypharmacology, and morusinol reportedly regulates beta3-integrin, tumor necrosis factor, and interleukin 1 beta involved in osteoclastogenesis, we profiled expression changes of these potential targets under morusinol treatment34,35,36. As shown in Supplementary Fig. 9e, morusinol subtly influenced the expression of representative pro-inflammatory cytokines, including Il-1b, Tnf-a, and Il-6, only slightly enhancing Il1-b. Although morusinol significantly reduced beta3-integrin (Supplementary Fig. 9f), it may also be a downstream effect of PU.1, which has been reported to transactivate beta3-integrin in concert with NFATc137. Although we cannot completely exclude the polypharmacology of murosinol, our results indeed proves that MSX2-FBXW7 axis is important for the actions of morusinol.

a Strategy of high throughput virtual screening. b Information of the most likely MSX2-targeting natural compounds. c BMMs were incubated with indicted concentrations of drugs for 48 h, the cell viability was detected by CCK-8 assay and normalized to control group (n = 3). d BMMs were stimulated by RANKL accompanied with isosilybin (25 μM), DCHA (100 μM), lincomycin hydrochloride monohydrate (6.25 μM), or morusinol (2.5 μM), respectively. Representative images of TRAP staining were shown. Scale bar, 100 μm. e Quantification of Oc.N/well in (d) (n = 5). f Bone resorption assay of morusinol treatment. Scale bar, 50 μm. g Quantification of relative resorption pits area (n = 5). h Protein level of PU.1 and CTSK under morusinol treatment. i Relative PU.1 and CTSK grey level in (h) (n = 3). j Cell-cell fusion under morusinol treatment. Scale bar, 100 μm. k BMMs were stimulated by 50 ng/mL RANKL and 2.5 μM morusinol for 6 days, the cells were stained with phalloidin and DAPI. Scale bar, 100 μm. l Quantification of normalized number of fusion events (n = 5). m Relative mRNA expression of osteoclast fusion-related genes (n = 5). n Three-dimensional image of molecular docking between morusinol and MSX2. l Protein level of PU.1 and CTSK under morusinol treatment. o BMMs were stimulated by RANKL with or without morusinol for 3 days, then cells were treated by MG132 for 4 h. Co-IP assay was performed to detect the combination of MSX2 and PU.1. p BMMs were stimulated by RANKL with or without morusinol for 3 days, then cells were treated by MG132 for 4 h, the ubiquitination level of PU.1 was examined. Data are presented as mean ± SD, and p-value were calculated by unpaired, two-tail student’s t test (g, i, l, and m) or one-way ANOVA with Dunnett’s multiple comparisons (c and e).
The positive in vitro results prompted us to further investigate the potential therapeutic effects of morusinol in an OVX mouse model. As shown in Fig. 7a, mice were injected daily with 30 mg/kg morusinol for 6 weeks after sham surgery or ovariectomy. Micro-CT results confirmed the beneficial effects of morusinol on bone mass, manifested by rescued BV/TV, Tb.Th, Ct.Th, and reduced Tb.Sp (Fig. 7b, c). Regarding bone resorption, TRAP staining showed that OVX mice exhibited increased multi-nuclear osteoclasts and reduced mono-nuclear osteoclasts, whereas morusinol treatment reversed this trend (Fig. 7d, e). Morusinol also reduced serum CTX-1 concentration in OVX mice (Fig. 7f). Moreover, we double-labeled PU.1 and CTSK in OVX and OVX + morusinol groups. The number of PU.1+ CTSK+ cells was significantly reduced by morusinol in OVX models, indicating morusinol promotes PU.1 degradation in osteoclasts in vivo (Fig. 7g, h). Similar to that in Msx2 cKO mice, morusinol treatment did not affect macrophage development (Supplementary Fig. 10). In terms of bone formation, morusinol treatment increased OCN-positive cells and enhanced MAR and BFS/BS (Fig. 7i–l). Regarding angiogenesis, EMCNhi CD31hi vessels were reduced in the OVX group, whereas morusinol treatment rescued vessel formation (Fig. 7m, n). Overall, targeting MSX2 with morusinol inhibits bone resorption, enhances bone and vessel formation, and protects bone mass.

a Schematic diagram for morusinol treatment in OVX mice, created by Biorender. b The 3D Micro-CT images of distal femurs from mice in Sham, OVX, and OVX+morusinol groups. c Bone morphology parameters of distal femurs (n = 10). d Representative TRAP staining images from Sham, OVX, and OVX + morusinol groups. Scale bar, 100 μm. e Quantification of Mu.Oc.N/BS and Mo.Oc.N/BS (n = 5). f Serum CTX-1 levels were detected by ELISA (n = 5). g Immunofluorescence staining of PU.1 and CTSK in distal femurs from OVX and OVX+morusinol groups. Scale bar, 50 μm. h Quantification of PU.1+ CTSK+ cell number/ BS (n = 5). i Calcein labeling images from OVX and OVX+morusinol groups. Scale bar, 25 μm. j Quantification of MAR and BFR/BS (n = 5). k Immunofluorescence staining of OCN in distal femurs from OVX and OVX + morusinol groups. Scale bar, 50 μm. l N.OCN+ cells/BS was measured (n = 5). m Representative images of immunofluorescence staining of EMCNhi CD31hi vessels. Scale bar, 50 μm. n Quantification of CD31hi EMCNhi.Ar (n = 5). Data are presented as mean ± SD, and p-values were calculated by unpaired, two-tail student’s t-test (h, j, and l) and one-way ANOVA with Tukey’s multiple comparisons (c, e, f, and n).
Discussion
In this study, we identified Msx2 as a critical homeobox gene that modulates bone homeostasis. Msx2 expression is upregulated in PMOP bones and during osteoclastogenesis. As shown in Fig. 8, knocking out Msx2 increases bone mass by attenuating osteoclastogenesis and enhancing angiogenesis-mediated bone formation. At the cellular level, Msx2 deficiency inhibits cell fusion and results in a larger population of pre-osteoclasts, leading to the enhanced secretion of PDGF-BB. High levels of PDGF-BB promote H-type vessel formation and subsequently stimulate osteogenesis. Molecularly, MSX2 prevents PU.1, one of the master transcription factors involved in osteoclast formation, from FBXW7-mediated ubiquitination and degradation. MSX2 binding sheltered the key motif of PU.1 from FBXW7 recognition. These findings prompted us to screen natural compounds targeting MSX2 and identify morusinol, which directly disrupts the MSX2-PU.1 complex and serves as an anabolic therapy for osteoporosis.

In wild-type myeloid cells, MSX2 binds PU.1 and prevents PU.1 from FBXW7 mediated ubiquitination, which safeguards fusion cell fate of osteoclast and results in bone resorption. Targeting Msx2 promotes PU.1 degradation and acts as a “brake” in the progression of the fusion fate of osteoclast, resulting in a larger population of pre-osteoclasts. Pre-osteoclasts secrete PDGF-BB and enhance angiogenesis-mediated bone formation (upper part of Fig. 8). Since voluminous prior studies have reported that MSX2 is important in regulating Wnt signaling and bone formation in concert with osteogenic transcription factors, myeloid-specific targeting MSX2 is necessary to achieve net anabolic effects (lower part of Fig. 8). The schematic diagram was designed by Figdraw.
Homeobox genes are a large and diverse group of genes characterized by possessing at least one 180 bp homeobox DNA sequence that encodes a highly conserved 60 amino acid homeodomain12. Homeodomain proteins are generally considered transcription factors or co-transcription factors that play vital roles in diverse physiological and pathological processes38. Approximately 15% of all transcription factors within the animal genome are homeodomain proteins. Studies have shown that homeodomain protein families, such as MSX, DLX, and PBX, regulate cranium and limb development during embryogenesis39,40,41. Moreover, it has been reported that homeodomain proteins play an active role in adult bone mass regulation42,43. Nevertheless, most existing studies have focused on the impact of homeodomain proteins on osteogenesis, with little understanding of their roles in osteoclastogenesis. A few studies have revealed that homeobox genes, such as Lhx2, Dlx5, and Pax6, affect osteoclast formation44,45,46; however, these minority studies are fragmented and unsystematic. Here, we used the expression profiles of PMOP bones to identify 11 osteoporosis-related homeobox genes. PPI network analysis indicated MSX2 as a hub gene. Furthermore, in vitro and in vivo experiments confirmed elevated Msx2 expression in osteoporotic bones and osteoclastogenesis. Collectively, these results inspired us to explore the potential role of MSX2 in osteoclast formation and bone resorption.
Voluminous previous studies have reported that MSX2 is closely associated with bone development. MSX2 mutations cause Boston-type craniosynostosis, and haploinsufficiency of human MSX2 causes deficient ossification around the parietal notch23. In mouse models, whole-body Msx2 deficient mice are viable but with a variety of malformations, including PFM, defective cerebellum, tooth, hair follicle, and mammary gland development22. Msx2 deficient P30 mice exhibit an osteopenia phenotype with predominant osteoblast defects. These phenotypes are considered to be associated with the crucial role of Msx2 in osteoblast differentiation. It has been reported that Msx2 promotes primary murine osteoblast differentiation and calcification in concert with typical osteoblastic transcription factors, and ectopic Msx2 rescues the impaired osteoblastogenesis induced by Runx2 deficiency47,48. Mechanistically, Msx2 expression responds to BMP2/4, and further activates canonical Wnt signaling in osteoblast progenitors49,50. Notably, there are also studies suggesting the potentially important role of Msx2 in osteoclastogenesis. In a Msx2−/− transgenic mouse model, Msx2 null mutation leads to periodontal osteopetrosis, with impaired osteoclast generation in alveolar bones26. It has also been reported that Msx2 deficient adult mice show a slight osteopetrosis phenotype in the skull, tibia, and mandible, which could be reversed by RANK overexpression24. Therefore, the role of Msx2 in different types of bone cells is complex, and conditional Msx2 deficient models are warranted. In this study, we found that PMOP bone samples showed increased Msx2 expression in osteoclasts, indicating that Msx2 in osteoclasts also plays an important role in bone metabolism during adulthood. We generated myeloid-specific Msx2 deficiency mice and demonstrated that knocking out Msx2 in osteoclast progenitors improves bone mass, with impaired osteoclastogenesis mainly manifesting as impaired osteoclast fusion rather than reduced osteoclast numbers. Although our results confirmed the direct role of MSX2 in osteoclasts and bone homeostasis, osteoblast-specific Msx2 knockout and inducible gene knockout systems are still needed to characterize the effect of MSX2 on bone mass more precisely.
In recent years, an increasing number of studies have demonstrated that bones exhibit important endocrine and paracrine behaviors. Crosstalk between different cells in the bone environment provides new insights into the regulation of bone metabolism and general health. The most classic and well-known interaction between bone cells is that osteoblast lineage cells could secrete RANKL and induce osteoclastogenesis51. In 2014, Xie et al. first reported that pre-fusion osteoclasts release PDGF-BB, which promotes H-type vessel formation and subsequently stimulates osteogenesis11. Since then, molecules affecting osteoclast fusion and angiogenesis, such as SIGLEC15, L-plastin, and TLR2, have been successively discovered52,53,54. Antibodies targeting these molecules have also been developed52. Moreover, a recent study also demonstrated that adipocytes in bone marrow could couple angiogenesis through estrogen-related receptor α55. These studies emphasize the important role of angiogenesis in mediating bone cell crosstalk and regulating bone mass. Here, we demonstrate that Msx2 deficiency in myeloid cells reduces bone resorption and increases bone mass. Additionally, we observed that angiogenesis-mediated bone formation was enhanced as a secondary effect of osteoclast-specific Msx2 knockout. These findings are attributed to the role of MSX2 in osteoclast fusion. Specifically, Msx2 deficiency impairs osteoclast fusion and exerts the role of “brake” in the progression of osteoclast cell fate, leading to increased pre-osteoclast numbers and PDGF-BB secretion.
Osteoclast fusion is a well-organized process triggered by RANKL, during which mono-nuclear pre-osteoclasts form multi-nuclear mature osteoclasts. The fusion process involves proteins that mediate recognition and initial association between fusing cells, fusogens that drive the actual fusion events, and various accessory proteins56. Classic fusion-related proteins in osteoclastogenesis include CD9, CD44, CD47, OC-STAMP, and DC-STAMP, the most well-known fusogen56. In recent years, novel fusion-related proteins such as SIGLEC15, L-plastin, and TLR2 have been identified52,53,54. In this study, we showed that the expression of classic fusion-related molecules in osteoclastogenesis is downregulated in Msx2 cKO mice. However, PU.1, rather than MSX2, was predicted to be the common transcription factor of fusion-related genes. Considering PU.1 participates in the entire process of osteoclast differentiation and promotes osteoclast commitment and maturity of osteoclast33,57,58, we proposed that the “brake” role of targeting MSX2 acts on PU.1-“clutch”. Indeed, we found that MSX2 could regulate PU.1 protein levels without affecting PU.1 mRNA levels. Mechanistically, MSX2 could directly bind to PU.1 and prevent it from FBXW7-mediated degradation. FBXW7 is known to recognize a conserved S/TxxxS/T motif59,60, and we found that the key phosphorylation loci of PU.1 (Ser41, Ser142) are sheltered by MSX2, leading to reduced ubiquitination and degradation of PU.1. Msx2 and Fbxw7 double knockout rescued PU.1, and subsequently osteoclast fusion, which was also confirmed in vivo. These results demonstrate that MSX2 could prevent PU.1 from FBXW7-mediated degradation through direct binding. Nevertheless, although our results provide sufficient evidence for the indirect role of MSX2 in fusion-related genes through PU.1, it is still difficult to rule out the direct transcriptional effect of MSX2 on these genes.
Anti-resorptive drugs such as bisphosphonates and denosumab are the first-line agents for osteoporosis treatment. However, these drugs cause severe adverse events, poor compliance, and easy recurrence61, which prompt us to identify and develop new drugs. Recently, drugs such as teriparatide and romosozumab have been developed and applied clinically, drawing more attention to anabolic therapeutic strategies for osteoporosis treatment62. In this study, we demonstrated that targeting MSX2 could be an anabolic strategy. Knocking out Msx2 in osteoclast progenitors inhibited bone resorption, enhanced angiogenesis-mediated bone formation, and rescued bone mass in an OVX-induced osteoporosis mouse model. In addition, the expression pattern of Msx2 indicates that targeting MSX2 primarily affects the osteoclast fusion stage, which evades the potential risk of PU.1 deficiency in myeloid development. Natural products are critical sources of drugs. Compared to synthetic compounds and biological agents, natural products have greater molecular rigidity, which makes them more valuable in tackling protein-protein interactions. Moreover, their use in traditional medicine provides helpful references for efficiency and safety31. In this study, we found that morusinol can target MSX2 and disrupt the interaction between MSX2 and PU.1. Morusinol treatment attenuates multi-nuclear osteoclast formation both in vitro and in vivo. Meanwhile, morusinol promoted vessel formation and osteogenesis. Although we could not completely exclude the polypharmacology of murosinol, our results indicated that morusinol exhibits great potential for the treatment of osteoporosis.
In conclusion, this study demonstrates that Msx2 is a critical homeobox gene regulating osteoclastogenesis and bone homeostasis. Targeting MSX2 acts as a “brake” in the progression of osteoclast cell fate by promoting PU.1 degradation, which subsequently enhances angiogenesis-mediated bone formation. Morusinol targets MSX2 and serves as an anabolic therapy for osteoporosis.
Methods
Mice
The LysM-Cre (#004781) mice and Msx2f/f mice (#025441) were from Jackson Laboratory (USA). Mice carrying Fbxw7 conditional null alleles (Fbxw7f/f, NM-CKO-200327) were from Shanghai Model Organisms (Shanghai, China). Msx2f/f mice were crossed with LysM-Cre mice to generate Msx2f/+, LysM-Cre+ mice. Myeloid-specific Msx2 deletion was obtained by breeding Msx2f/+, LysM-Cre+ mice with Msx2f/f mice, and Msx2f/f littermates were used as control. Three-month-old female and male mice samples were collected for further analysis. Myeloid-specific Fbxw7 deletion was acquired by crossing Fbxw7f/f mice with LysM-Cre mice through a similar strategy. Three-month-old female Fbxw7f/f, LysM-Cre+ mice and their littermate controls were used for phenotype assessment. To obtain Msx2, Fbxw7 dKO mice, Msx2f/f, LysM-Cre+ mice were firstly crossed with Fbxw7f/f mice to generate Msx2f/+, Fbxw7f/+, LysM-Cre+ mice, which were then crossed with Msx2f/f mice to obtain Msx2f/f, Fbxw7f/+, LysM-Cre+ mice. The Msx2f/f, LysM-Cre+ mice and Msx2f/f, Fbxw7f/f, LysM-Cre+ mice were generated by breeding Msx2f/f, Fbxw7f/+, LysM-Cre+ mice with Msx2f/f, Fbxw7f/+ mice, and the Msx2f/f littermates were used as control. Three-month-old female Msx2, Fbxw7 dKO mice were used for further evaluation. Female wild-type C57BL/6 mice were purchased from SLAC Laboratory Animal Company (Shanghai, China). All animal procedures were ethically approved by Sir Run Run Shaw Hospital Committee for Animal Resources, and were conducted in compliance with the approved ethical guidelines. Mice were housed under a 12 h dark/12 h light cycle, 22 °C ambient temperature, and 50% humidity. All mice were routinely genotyped using standard PCR protocols, and the primers used are listed in Supplementary Table 1. All experimental animals in this study were anesthetized by intraperitoneal injection of 1% sodium pentobarbital before surgery or sacrifice to reduce the pain.
Ovariectomy (OVX)-induced osteoporosis mouse model
Eight-week-old female mice were anesthetized and subjected to a sham operation or bilateral ovariectomy. All mice were given 1 week for postoperative recovery. Msx2f/f and Msx2 cKO mice were euthanized 6 weeks after recovery. Femur and blood samples were collected for further analysis. For the morusinol treatment, 30 mice were randomly categorized into three groups: Sham, OVX, and OVX + murosinol, with 10 mice per group. Morusinol was dissolved in dimethyl sulfoxide and diluted in PBS (vehicle). Mice from the Sham and OVX groups received vehicle injections daily, whereas mice in the OVX + morusinol group received morusinol injections (30 mg/kg/day) one week after the surgery. After a 6-week injection period, the mice were sacrificed for further analysis.
Micro-CT
The femurs were scanned using a Skyscan 1275 instrument (Bruker). The scanning parameters were set as follows: 50 kV, 200 mA, and 9 μm voxel size. For the region of interest (ROI), we selected sections from 100 slices below the growth plate to 300 slices below the growth plate for trabecular bones, and from 300 slices below the growth plate to 350 slices below the growth plate for cortical bones. Bone parameters, including BV/TV, Tb.Th, Tb.N, Tb.Sp, and Ct.Th, were analyzed using CTAn software (Bruker). The 3D reconstructed images were obtained by stacking the 2D images from the ROI using the CTVox software (Bruker).
Bone histology and immunofluorescence
The femurs were fixed with 4% paraformaldehyde for 48 h and then decalcified for 2 weeks. The samples were then embedded in paraffin and cut into 4-μm-thick sections. TRAP staining was performed using a commercial kit (Sigma-Aldrich). For the immunofluorescence staining, we incubated bone sections with primary antibodies at 4 °C overnight. The antibodies used for immunofluorescence analysis were OCN (ab93876, Abcam), CTSK (sc-48353, Santa Cruz), MSX2 (DF4126, Affinity), CD31 (ab182981, Abcam), EMCN (sc-65495, Santa Cruz), PDGF-BB (ab23914, Abcam), and TRAP (ab191406, Abcam). The sections were then incubated with fluorescence-conjugated secondary antibodies for 2 h at room temperature. The images were captured using a fluorescence microscope, and we quantified the positively stained cells in five randomly selected fields of the distal metaphysis of femurs using Halo software.
Angiography
Bone blood vessel imaging was conducted using Micro-CT according to previous reports63,64. Mice were anesthetized, the left ventricle was exposed, and a needle was inserted. The vasculature was washed with saline solution 3 times and then fixed with 4% paraformaldehyde. The vessel dye (MV-120, Flow Tech) was then injected into the vasculature. Subsequently, mice were incubated at 4 °C overnight to crosslink the dye, and then femurs were collected and decalcified for 2 weeks. The decalcified bones were subjected to micro-CT scanning, and the vessel volume and vessel surface were measured.
Flow cytometry
Flow cytometry was performed as previously reported53. Briefly, fresh femurs from Msx2f/f and Msx2 cKO mice were collected, and the muscles, periosteum, and epiphysis were removed. The bone marrow was flushed and collected, and the bones were crushed in cold PBS and digested by collagenase together with the flushed bone marrow at 37 °C for 20 min to obtain the whole bone marrow cells. After filtration and washing, the cells were then incubated with an APC-conjugated CD31 antibody (17-0311-82, eBioscience), PE-conjugated EMCN monoclonal antibody (sc-65495, Santa Cruz), BV510 conjugated TER119 antibody (563995, BD Horizon), or FITC-conjugated CD45 antibody (553079, BD Pharmingen). Fluorescence-activated cell sorting (FACS) was performed, and CD31+CD45-TER119- cells were sorted as bone marrow endothelial cells.
Calcein double labeling
Calcein (#C7600) was purchased from Solarbio (Beijing, China). Calcein double labeling experiments were conducted as previously described32. Briefly, mice were intraperitoneally injected with calcein (15 mg/kg) 10 and 3 days before euthanasia. Femurs were collected and sectioned. Calcein labels were captured using a fluorescence microscope, and the MAR was calculated according to a previously reported standard65.
In vitro osteoclastogenesis
The femurs and tibias of 6-week-old mice were collected and used for BMMs extraction. Bone marrow cells were flushed out and cultured in complete α-MEM supplemented by 30 ng/mL M-CSF (416-ML, R&D) for 4 days. Non-adherent cells were removed, and BMMs were seeded into 96-well plates at a density of 8000 cells/well. For osteoclast stimulation, cells were induced with 30 ng/mL M-CSF and 50 ng/mL RANKL (462-TEC, R&D) for 5–6 days until multi-nuclear osteoclasts were generated.
Preparation of conditional medium
Conditioned medium derived from pre-osteoclast was prepared according to previous reports11,53. Briefly, BMMs were stimulated with 30 ng/mL M-CSF and 50 ng/mL RANKL for 3 days to generate pre-osteoclasts. The serum-containing pre-osteoclast-derived conditional medium was collected 2 days after pre-osteoclast formation. To obtain serum-free conditional medium, the cells were then cultured for 1 more day in a serum-free medium supplemented with M-CSF and RANKL, and the medium was collected. All conditional media were centrifuged (1000 × g, 10 min, 4 °C) and stored at −80 °C before use. Serum-containing conditioned medium was used for the tube formation and osteogenesis assays, while serum-free conditioned medium was used for the wound healing assay.
Bone slice resorption assay
The bone resorption assay was conducted as previously reported66. Briefly, BMMs were seeded onto bovine bone slices at a density of 8000 per well and stimulated with 50 ng/mL RANKL. After mature osteoclasts were generated, the cells were treated with RANKL for 5 days. Then, bone slices were collected, and cells were gently removed from the surface of the bone slices. Resorption pits were observed using a scanning electron microscope.
Cell-cell fusion assay
The cell-cell fusion assay was performed as described previously67. Briefly, BMMs were incubated with 30 ng/mL M-CSF and 50 ng/mL RANKL for 3 days. The cells were collected and categorized into two equal duplicates. Each duplicate cells were labeled with either Hoechst (40731ES, Yeason, 5 μg/mL, 37 °C, 5 min) or CM-DiI (40718ES, Yeason, 2.5 μM, 37 °C, 15 min). The labeled cells were washed 3 times with PBS, and then equal cells with a single label (Hoechst or CM-DiI) were cultured together under 30 ng/mL M-CSF and 100 ng/mL RANKL. Fluorescence images were captured 12 h later.
Fusion efficiency calculation
To precisely evaluate the fusion efficiency of osteoclasts, we calculated the normalized number of fusion events according to a previous report. Osteoclasts were fixed with 4% paraformaldehyde for 15 min and then permeabilized by Triton-100 for 5 min. The F-actin of syncytia was stained by phalloidin (CA1610, Solarbio) for 1 h at room temperature, and the nuclei were stained by DAPI (C0060, Solarbio). The fluorescence images were captured using Nikon fluorescence microscopy. The normalized number of fusion events was calculated as Σ(Ni-1)/Ntotal, in which Ni equals the number of nuclei in a syncytium, and Ntotal equals the total number of nuclei in syncytia and unfused cells. Ni-1 could precisely reflect the number of fusion events to form a syncytium with Ni nuclei irrespective of whether the syncytium was generated by fusion between two mono-nuclear cells, one mono-nuclear cell and one multi-nuclear cell or two multi-nuclear cells; Ntotal was used to normalize the subtle discrepancy of cell density among samples.
In vitro osteogenesis
BMSCs were isolated as previously reported55. For in vitro osteogenesis, BMSCs were incubated with a mixed medium: 50% osteogenic medium (10% FBS, 50 μg/mL ascorbic acid, 5 mM β-glycerophosphate, and 100 mM dexamethasone in α-MEM) and 50% serum-containing conditional medium from pre-osteoclast. ALP staining was performed 7 days after stimulation, and ARS staining was performed 21 days after stimulation.
In vitro tube formation
In vitro tube formation assays were performed using Matrigel (356237; BD Biosciences) and prepared according to the manufacturer’s instructions. HUVECs (CL-0675, Procell) were incubated with a mixed medium: 50% endothelial cell medium and 50% conditioned media derived from Msx2f/f and Msx2 cKO pre-osteoclasts for 12 h. The Matrigel was polymerized in 24-well plates at 37 °C and the cells were seeded on the gel at a density of 150,000 per well. After incubation at 37 °C for another 4 h, the tube formation pictures were captured using a microscope.
Wound healing assay
HUVECs were seeded into a 6-well plate and cultured until they reached confluence. Cells were wounded using a sterile pipette tip and imaged under a microscope. The cells were then treated with serum-free conditional media derived from Msx2f/f and Msx2 cKO pre-osteoclasts. Twelve hours later, the wounds were captured again, and the migration area was calculated according to a previous report68.
RNA extraction and real-time PCR
Total RNA was extracted with an Ultrapure RNA extraction kit purchased from CWBio. RNA was reverse-transcribed using a reverse transcription kit (Accurate Biotechnology). Real-time PCR was performed using 2 × SYBR Green Pro Taq HS Premix (Accurate Biotechnology) according to the manufacturer’s instructions. The relative gene expressions were calculated with the −ΔCT method, and β-actin was used as the internal control. The primers used in this study are listed in Supplementary Table 2.
Plasmid transfection and adenovirus infection
The plasmids used in this study were commercially constructed using the pcDNA3.1 vector by GeneChem, and all the tags were fused on the C terminus. For plasmid transfection, HEK-293T cells (CL-0005, Procell) were transfected with 2 μg plasmid using lipofectamine 3000 and P3000 for 24 h. The medium was then changed, and the cells were cultured for another 48 h before the experiments.
Cre, MSX2, and PU.1 adenovirus were commercially constructed using pAdM-FH-GFP vector by WZBiosciences. For adenovirus infection, BMMs were cultured and infected with adenovirus (MOI = 50) for 6 h, then the media were changed, and BMMs were cultured for another 48 h before the experiments.
Immunoblots
The cells were lysed in RIPA lysis buffer containing protease inhibitors. The extracted proteins were then subjected to immunoblot assays, as previously reported32. The primary antibodies used in this study were anti-NFATc1 (sc-7294, Santa Cruz), anti-MSX2 (ab227720, Abcam), anti-β-actin (66009-1-Ig, Proteintech), anti-CTSK (sc-48353, Santa Cruz), anti-PU.1 (ab227835, Abcam), anti-FLAG (20543-1-AP, Proteintech), anti-HA (51064-2-AP, Proteintech), anti-MYC (16286-1-AP, Proteintech), anti-Ubiquitin (10201-2-AP, Proteintech), and anti-FBXW7 (28424-1-AP, Proteintech). After overnight incubation with primary antibodies at 4 °C, corresponding HRP-conjugated secondary antibodies were used for band detection.
Co-IP
The Co-IP assay was performed as previously reported32. Briefly, cell lysates were immunoprecipitated with anti-PU.1 or anti-FLAG overnight at 4 °C. The mixture was then incubated with protein A/G agarose beads (36417ES; Yeason). Next, the immune complexes were collected and subjected to immunoblotting. Primary antibodies from different sources and specific secondary antibody (AS062, Abclonal and abs20213, Absin) were used to avoid heavy/light chain interference.
Molecular docking and HTVS
The structures of MSX2 and PU.1 were constructed using Alphafold and optimized using PROCHECK69,70. Molecular docking was performed using HDOCK, according to previous reports71,72. For HTVS, we selected the interaction site of MSX2 as a docking pocket and performed HTVS. Standard precision and extra precision (XP) analyses were performed to identify the top hits. The MM-GBSA was calculated to quantitatively determine the binding affinity between the small molecules identified by XP and MSX2. The threshold values were set as follows: XP GScore ≤ 7, MM-GBSA dG Bind < −30 kcal/mol.
Statistical analyses
All data are presented as means ± SD. Differences were determined using GraphPad Prism software (version 8.0). Unpaired two-tailed student’s t-test was used for two-group comparisons. One-way and two-way ANOVAs with post-hoc tests for comparisons of more than two groups are shown in the figure legends. Differences were considered significant at p < 0.05.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Source data are provided with this paper. Genes expression profiles of PMOP in femur were previously published in GSE230665. All supporting data are available for reproducibility purposes from the corresponding author upon request. Source data are provided with this paper.
References
Compston, J. E., McClung, M. R. & Leslie, W. D. Osteoporosis. Lancet 393, 364–376 (2019).
Weivoda, M. M. & Bradley, E. W. Macrophages and bone remodeling. J. Bone Miner. Res. 38, 359–369 (2023).
Liu, X., Zhang, P., Gu, Y., Guo, Q. & Liu, Y. Type H vessels: functions in bone development and diseases. Front. Cell Dev. Biol. 11, 1–10 (2023).
Chen, Y., Guo, B., Ma, G. & Cao, H. Sensory nerve regulation of bone homeostasis: emerging therapeutic opportunities for bone-related diseases. Ageing Res. Rev. 99, 102372 (2024).
Fischer, V. & Haffner-Luntzer, M. Interaction between bone and immune cells: implications for postmenopausal osteoporosis. Semin. Cell Dev. Biol. 123, 14–21 (2022).
Li, J., Chen, X., Lu, L. & Yu, X. The relationship between bone marrow adipose tissue and bone metabolism in postmenopausal osteoporosis. Cytokine Growth Factor Rev. 52, 88–98 (2020).
Song, S., Guo, Y., Yang, Y. & Fu, D. Advances in pathogenesis and therapeutic strategies for osteoporosis. Pharm. Ther. 237, 108168 (2022).
Elson, A., Anuj, A., Barnea-Zohar, M. & Reuven, N. The origins and formation of bone-resorbing osteoclasts. Bone 164, 116538 (2022).
Jiang, T., et al. Role and regulation of transcription factors in osteoclastogenesis. Int. J. Mol. Sci. 24, 16175 (2023).
Takayanagi, H. RANKL as the master regulator of osteoclast differentiation. J. Bone Min. Metab. 39, 13–18 (2021).
Xie, H., et al. PDGF-BB secreted by preosteoclasts induces angiogenesis during coupling with osteogenesis. Nat. Med. 20, 1270–1278 (2014).
Garcia-Fernàndez, J. The genesis and evolution of homeobox gene clusters. Nat. Rev. Genet. 6, 881–892 (2005).
Zhao, J. et al. PDX1 + cell budding morphogenesis in a stem cell-derived islet spheroid system. Nat. Commun. 1, 18 (2024).
Kim, K. H., Rosen, A., Bruneau, B. G., Hui, C. C. & Backx, P. H. Iroquois homeodomain transcription factors in heart development and function. Circ. Res. 110, 1513–1524 (2012).
Kuriki, M., Korb, A., Comai, G. & Tajbakhsh, S. Interplay between Pitx2 and Pax7 temporally governs specification of extraocular muscle stem cells. PLoS Genet. 20, 1–27 (2024).
Yu, X. et al. TGFβ-induced factor homeobox 2 blocks osteoblastic differentiation through targeting pSmad3/HDAC4/H4ac/Runx2 axis. J. Cell Physiol. 234, 21284–21293 (2019).
Yang, H., Liang, Y., Cao, Y., Cao, Y. & Fan, Z. Homeobox C8 inhibited the osteo-/dentinogenic differentiation and migration ability of stem cells of the apical papilla via activating KDM1A. J. Cell Physiol. 235, 8432–8445 (2020).
Heo, J. S., Lee, S. G. & Kim, H. O. Distal-less homeobox 5 is a master regulator of the osteogenesis of human mesenchymal stem cells. Int. J. Mol. Med. 40, 1486–1494 (2017).
Xu, J. et al. Shox2 regulates osteogenic differentiation and pattern formation during hard palate development in mice. J. Biol. Chem. 294, 18294–18305 (2019).
Babajko, S., de La Dure-Molla, M., Jedeon, K. & Berdal, A. MSX2 in ameloblast cell fate and activity. Front. Physiol. 6, 1–10 (2015).
Alappat, S., Zhang, Z. Y. & Chen, Y. P. Msx homeobox gene family and craniofacial development. Cell Res. 13, 429–442 (2003).
Satokata, I. et al. Msx2 deficiency in mice causes pleiotropic defects in bone growth and ectodermal organ formation. Nat. Genet. 24, 391–395 (2000).
Wilkie, A. O. M. et al. Functional haploinsufficiency of the human homeobox gene MSX2 causes defects in skull ossification. Nat. Genet. 24, 387–390 (2000).
Castaneda, B., et al. Role of RANKL (TNFSF11)-dependent osteopetrosis in the dental phenotype of Msx2 null mutant mice. PLoS ONE 8, e80054 (2013).
Xie, L. et al. Comparisons of gene expression between peripheral blood mononuclear cells and bone tissue in osteoporosis. Medicine 102, E33829 (2023).
Aïoub, M. et al. Msx2 -/- transgenic mice develop compound amelogenesis imperfecta, dentinogenesis imperfecta and periodental osteopetrosis. Bone 41, 851–859 (2007).
Tan, Z. et al. IRX3 and IRX5 inhibit adipogenic differentiation of hypertrophic chondrocytes and promote osteogenesis. J. Bone Miner. Res. 35, 2444–2457 (2020).
Wang, X. et al. UbiBrowser 2.0: a comprehensive resource for proteome-wide known and predicted ubiquitin ligase/deubiquitinase-substrate interactions in eukaryotic species. Nucleic Acids Res. 50, D719–D728 (2022).
Mishra, M. et al. FBW7 inhibits myeloid differentiation in acute myeloid Leukemia via GSK3-dependent ubiquitination of PU.1. Mol. Cancer Res. 19, 261–273 (2021).
Fukushima, H. et al. NOTCH2 Hajdu-Cheney mutations escape SCFFBW7-dependent proteolysis to promote osteoporosis. Mol. Cell 68, 645–658.e5 (2017).
Rodrigues, T., Reker, D., Schneider, P. & Schneider, G. Counting on natural products for drug design. Nat. Chem. 8, 531–541 (2016).
Ma, Q. et al. The SFRP1 inhibitor WAY-316606 attenuates osteoclastogenesis through dual modulation of canonical Wnt signaling. J. Bone Miner. Res. 37, 152–166 (2022).
Carey, H. A. et al. Enhancer variants reveal a conserved transcription factor network governed by PU.1 during osteoclast differentiation. Bone Res. 6, 1–12 (2018).
Lee, J. J. et al. Morusinol extracted from Morus alba inhibits arterial thrombosis and modulates platelet activation for the treatment of cardiovascular disease. J. Atheroscler. Thromb. 19, 516–522 (2012).
Kwon, H. W. Pharmacological actions of morusinol on modulation of platelet functions via integrin αIIb/β3 signaling. J. Appl. Biol. Chem. 66, 171–178 (2023).
Zelová, H. et al. Evaluation of anti-inflammatory activity of prenylated substances isolated from Morus alba and Morus nigra. J. Nat. Prod. 77, 1297–1303 (2014).
Crotti, T. N. et al. PU.1 and NFATc1 mediate osteoclastic induction of the mouse β3 integrin promoter. J. Cell Physiol. 215, 636–644 (2008).
Holland, P. W. H., Booth, H. A. F. & Bruford, E. A. Classification and nomenclature of all human homeobox genes. BMC Biol. 5, 1–28 (2007).
Takechi, M., Adachi, N., Hirai, T., Kuratani, S. & Kuraku, S. The Dlx genes as clues to vertebrate genomics and craniofacial evolution. Semin Cell Dev. Biol. 24, 110–118 (2013).
Berdal, A. et al. Differential impact of Msx1 and Msx2 homeogenes on mouse maxillofacial skeleton. Cells Tissues Organs. 189, 126–132 (2008).
Eyal, S. et al. Bone morphology is regulated modularly by global and regional genetic programs. Dev. (Camb.) 146, 1–15 (2019).
Oono, F., Sakamoto, Y., Tachi, Y., Mabashi-Asazuma, H. & Iida, K. Effect of Cdx2 polymorphism on the relationship between dietary calcium intake and peak bone mass in young Japanese women. Nutrients 12, 191 (2020).
Isaac, J. et al. DLX3 regulates bone mass by targeting genes supporting osteoblast differentiation and mineral homeostasis in vivo. Cell Death Differ. 21, 1365–1376 (2014).
Kim, J. H. et al. Lhx2 regulates bone remodeling in mice by modulating RANKL signaling in osteoclasts. Cell Death Differ. 21, 1613–1621 (2014).
Lézot, F. et al. Dlx homeobox gene family expression in osteoclasts. J. Cell Physiol. 223, 779–787 (2010).
Jie, Z. et al. Activating; -catenin/Pax6 axis negatively regulates osteoclastogenesis by selectively inhibiting phosphorylation of p38/MAPK. FASEB J. 33, 4236–4247 (2019).
Mirzayans, F., Lavy, R., Penner-Chea, J. & Berry, F. B. Initiation of early osteoblast differentiation events through the direct transcriptional regulation of Msx2 by FOXC1. PLoS ONE 7, 1–8 (2012).
Ichida, F. et al. Reciprocal roles of Msx2 in regulation of osteoblast and adipocyte differentiation. J. Biol. Chem. 279, 34015–34022 (2004).
Cheng, S. L., Shao, J. S., Cai, J., Sierra, O. L. & Towler, D. A. Msx2 exerts bone anabolism via canonical Wnt signaling. J. Biol. Chem. 283, 20505–20522 (2008).
Sun, J., Ishii, M., Ting, M. C. & Maxson, R. Foxc1 controls the growth of the murine frontal bone rudiment by direct regulation of a Bmp response threshold of Msx2. Development 140, 1034–1044 (2013).
Martin, T. J. & Sims, N. A. RANKL/OPG; Critical role in bone physiology. Rev. Endocr. Metab. Disord. 16, 131–139 (2015).
Zhen, G. et al. An antibody against Siglec-15 promotes bone formation and fracture healing by increasing TRAP+ mononuclear cells and PDGF-BB secretion. Bone Res. 9, 47 (2021).
Li, X. et al. Targeting actin-bundling protein L-plastin as an anabolic therapy for bone loss. Sci. Adv. 6, 1–13 (2020).
Dou, C. et al. Sialylation of TLR2 initiates osteoclast fusion. Bone Res. 10, 1–11 (2022).
Huang, T. et al. Targeting adipocyte ESRRA promotes osteogenesis and vascular formation in adipocyte-rich bone marrow. Nat. Commun. 15, 1–20 (2024).
Kodama, J. & Kaito, T. Osteoclast multinucleation: review of current literature. Int. J. Mol. Sci. 21, 1–35 (2020).
Izawa, N. et al. Cooperation of PU.1 with IRF8 and NFATc1 defines chromatin landscapes during RANKL-induced osteoclastogenesis. J. Bone Miner. Res. 34, 1143–1154 (2019).
de la Rica, L. et al. PU.1 target genes undergo Tet2-coupled demethylation and DNMT3b-mediated methylation in monocyte-to-osteoclast differentiation. Genome Biol. 14, 1–21 (2013).
Doble, B. W. & Woodgett, J. R. GSK-3: tricks of the trade for a multi-tasking kinase. J. Cell Sci. 116, 1175–1186 (2003).
Davis, R. J., Welcker, M. & Clurman, B. E. Tumor suppression by the Fbw7Ubiquitin ligase: mechanisms and opportunities. Cancer Cell 26, 455–464 (2014).
Ramchand, S. K. & Leder, B. Z. Sequential therapy for the long-term treatment of postmenopausal osteoporosis. J. Clin. Endocrinol. Metab. 1–9, https://doi.org/10.1210/clinem/dgad496 (2023).
Langdahl, B. L. et al. Romosozumab (sclerostin monoclonal antibody) versus teriparatide in postmenopausal women with osteoporosis transitioning from oral bisphosphonate therapy: a randomised, open-label, phase 3 trial. Lancet. 390, 1585–1594 (2017).
Wang, Y. et al. The hypoxia-inducible factor α pathway couples angiogenesis to osteogenesis during skeletal development. J. Clin. Investig. 117, 1616–1626 (2007).
Duvall, C. L., Taylor, W. R., Weiss, D. & Guldberg, R. E. Quantitative microcomputed tomography analysis of collateral vessel development after ischemic injury. Am. J. Physiol. Heart Circ. Physiol. 287, 302–310 (2004).
Recker, R. R. et al. Issues in modern bone histomorphometry. Bone 49, 955–964 (2011).
Wang, S. et al. An antioxidant sesquiterpene inhibits osteoclastogenesis via blocking IPMK/TRAF6 and counteracts OVX-induced osteoporosis in mice. J. Bone Miner. Res. 36, 1850–1865 (2021).
Dou, C. et al. Graphene-based microRNA transfection blocks preosteoclast fusion to increase bone formation and vascularization. Adv. Sci. 5, 1–10 (2018).
Huang, J. et al. Harmine enhances type H vessel formation and prevents bone loss in ovariectomized mice. Theranostics 8, 2435–2446 (2018).
Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021).
Hodsdon, M. E., Ponder, J. W. & Cistola, D. P. The NMR solution structure of intestinal fatty acid-binding protein complexed with palmitate: application of a novel distance geometry algorithm. J. Mol. Biol. 264, 585–602 (1996).
Muzzammel, H. et al. Identification of RdRp inhibitors against SARS-CoV-2 through E-pharmacophore-based virtual screening, molecular docking and MD simulations approaches. Int. J. Biol. Macromol. 237, 1–10 (2023).
Sahin, K. In silico identification of angiotensin-1 converting enzyme inhibitors using text mining and virtual screening. J. Biomol. Struct. Dyn. 40, 1152–1162 (2022).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (82002334, 82372440), The Key Project of National Natural Science Foundation of China (82330077), Union Fund Project of National Natural Science Foundation of China (U21A20351), and Natural Science Foundation of Zhejiang Province (Q20H060042, LQ24H090007, LQN25H070001).
Author information
Authors and Affiliations
Contributions
Q.M., S.F., Z.X., and Z.J. conceived the study. S.W., H.X. (Hong Xue), L.N., P.Y., Y.S., and J.Z. performed most of the experiments. Q.M., H.W., and H.X. (Hongwei Xie) performed the bioinformatic analysis. B.Z., Q.W., and C.J provided experimental assistance. Q.M., Z.J,. and S.W. oversaw the collection of results and data interpretation. Q.M. and Z.J. wrote the manuscript. A.Q., S.F., L.N., and Z.X. contributed ideas and reviewed the manuscript. All authors have seen and approved the final version of the paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Ma, Q., Wang, S., Xue, H. et al. Targeting Msx2 as a brake in the fusion fate of osteoclasts and an anabolic therapy in pre-clinical models of osteoporosis. Nat Commun 16, 7228 (2025). https://doi.org/10.1038/s41467-025-61938-0
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-61938-0
