
Abstract
Individuals with progressive liver failure risk dying without liver transplantation. However, our understanding of why regenerative responses are disrupted in failing livers is limited. Here, we perform multiomic profiling of healthy and diseased human livers using bulk and single-nucleus RNA- and ATAC-seq. We report that in alcohol-associated liver disease, alterations in the hepatic immune milieu prevent hepatocytes from transitioning to proliferative progenitors. We also find differences in RNA binding protein expression, particularly of the ESRP, PTBP, and SR families, leading to misregulation of developmentally controlled RNA splicing. Our data pinpoint ESRP2 as a disease-sensitive splicing factor and support a causal role for its deficiency in the pathogenesis of severe alcoholic hepatitis. Notably, splicing defects in Tcf4 and Slk, two ESPR2 targets, alter their nuclear localization and activities, disrupting WNT and Hippo signaling pathways that are critical for normal liver regeneration. We further demonstrate that changes in stromal cell populations enrich failing livers with TGF-β, which suppresses the ESRP2-driven epithelial splicing program and replaces functional parenchyma with quasi-progenitor-like cells lacking liver-specific functions. Taken together, these findings indicate that misspliced RNAs are effective biomarkers for alcohol-associated liver disease, and targeting them could improve recovery in affected individuals.
Similar content being viewed by others


Introduction
Alcohol-associated liver disease (ALD) stands as the predominant cause of chronic liver disease, with its incidence and prevalence rising in the US and globally1. Abstinence remains the sole proven cure for ALD, as both the risk and severity of the disease correlate directly with the dose and duration of alcohol exposure. Similar to other drug-induced liver injuries, liver dysfunction in ALD is most severe shortly after toxin exposure and typically normalizes after the noxious agent is eliminated. However, individuals who develop severe alcohol-associated hepatitis (SAH) or alcohol-associated cirrhosis (AC) are at elevated mortality risk from progressive liver failure despite discontinuing alcohol consumption2.
Currently, the only life-saving intervention for ALD patients with acute or chronic liver failure is liver transplantation. Developing alternative treatment options requires understanding the mechanisms that derail normal regenerative responses in livers with SAH or AC. Published work demonstrates a robust correlation between the severity of liver dysfunction and the accumulation of progenitor-like, immature hepatocytes in SAH3,4, and senescent hepatocytes in AC5 and some SAH patients6. There is growing consensus that liver epithelial cells (hepatocytes and cholangiocytes) function as facultative stem/progenitor-like cells to regenerate hepatic parenchyma after adult liver injury7,8,9. Thus, liver failure in ALD likely results from dysregulation of mechanisms that orchestrate state transitions in liver epithelial cells.
Hepatocytes and cholangiocytes in healthy adult livers exhibit high phenotypic plasticity and can shed and regain metabolic and proliferative activities7,9,10,11. Although many studies focus on mechanisms that mediate state transitions by changing gene transcription12, post-transcriptional mechanisms that regulate RNA processing, stability, and translation, as well as modulate protein sequence and activity, have proven to be equally critical13,14,15,16,17,18,19,20. Post-transcriptional gene regulation is crucial for implementing rapid responses during reversible state changes that liver cells require to survive and reconstitute healthy parenchyma after injury21,22,23,24,25,26,27,28. However, the post-transcriptional landscapes of alcohol-associated human liver diseases remain unexplored.
Herein, we leveraged next-generation deep sequencing of liver RNA and multiomics platforms (snRNA-seq and snATAC-seq) to identify mechanisms that drive phenotypic differences in liver epithelial cell populations between healthy human livers and explanted human livers with SAH or AC. Putative mediators were then manipulated in cultured cells and mouse ALD models to determine their pathogenic significance. Our results reveal that RNA processing is globally dysregulated in hepatocytes of livers with decompensated SAH or AC, demonstrating why this occurs and how it promotes liver failure. These discoveries have important clinical implications as they identify prognostic biomarkers and therapeutic targets for ALD.
Results
Altered immune milieu in ALD associates with a quasi-progenitor state of hepatocytes
Recent single-cell resolution studies have significantly advanced our understanding of liver diseases and regenerative processes11,29,30,31,32,33. Particularly for ALD, these have been hindered by the logistics of tissue preservation and cell isolation for library preparation. To circumvent these challenges, we utilized a 10× multiomics platform to obtain matched snRNA-seq and snATAC-seq profiles from fresh frozen livers of unaffected individuals as well as SAH and AC patients (Fig. 1a). While this successfully profiled nuclear RNA, several disadvantages of this approach compared to single-cell and other sequencing approaches include loss of cytoplasmic RNA information (cytoplasmic mRNAs as well as mitochondrial RNAs), lower detected genes and UMI per cell, and a lack of transcript-wide coverage34,35,36. Further, because snRNA-seq only profiles nuclear RNAs, it enriches for un- or partially spliced transcripts. A lengthy nuclear isolation/processing protocol could potentially produce biases in cell types identified compared to single-cell methods34,35,36. Considering these challenges, we performed Illumina-based bulk RNA-seq analyses in parallel to 10× multiomics from matched samples to accurately profile lowly expressed mRNAs and their post-transcriptional processing. After quality control and analysis (CellRanger ARC, Seurat v4, and Signac, see “Methods”), multiomic profiling provided 27,000 single nuclei profiles with an average of 1476 genes detected per nucleus. Within this, ~7000–8000 nuclei were captured from SAH and AC patient livers each, and ~11,900 nuclei from unaffected livers. All samples were integrated and corrected for batch effects using Harmony. To group and identify clusters, WNN analysis was performed, considering both gene expression and chromatin accessibility (Fig. 1b). Cell types were annotated based on differential gene expression and published references for cell-type-specific markers37. Hepatocytes and non-parenchymal cells (NPCs) clusters were annotated based on the expression levels and accessibility of HNF4α and Vimentin (VIM) genes, respectively (Supplementary Fig. 1a). Within the NPC cluster, we identified most of the major human non-hepatocyte liver cell populations, excluding cholangiocytes (Fig. 1b).

a Schematic summarizing single nuclear isolation from human liver samples and transcriptomics using multiomic and deep sequencing approaches. (Created in BioRender. Das, D. (2025) https://BioRender.com/mskjuhf). b rnaUMAP showing distinct cell populations identified from multiomic profiling at single-cell resolution from over 27,000 liver nuclei from 15 individuals (n = 5 each). c Distribution of various liver cells in unaffected and alcohol-associated liver disease (ALD) patients. AC and SAH showed an increased number of non-hepatocyte (NPC) cells. Non-directional Chi-squared test was performed cell numbers showed a p-value < 0.0001, indicating statistically significant change in cell populations depending on conditions. d Distribution of porto-central coordinate \({\eta }_{i}\) values in unaffected liver, AC, and SAH conditions. The histogram shows the frequency of cells at different \({\eta }_{i}\) values for each condition, followed by p-value from using two-sided Kolmogorov–Smirnov (K–S) test, which shows significant differences (p < 0.05) between unaffected and diseased distributions. e Cell type-specific changes in gene expression associated with ALD (examples shown in dot plot, left) and corresponding gene ontology analysis demonstrating affected processes (right). f Average Transcript per million (TPM) values of key pro-inflammatory cytokines as quantified from bulk RNA-seq. Correspoding Average TPM values are shown within each box of the heatmap. SAH severe alcohol-associated hepatitis, AC alcohol-associated cirrhosis.
Increased inflammation, induced by several underlying factors, is a fundamental process associated with the progression of ALDs. Metabolites, including acetaldehyde, acetate, and reactive oxygen species, produced during the hepatic breakdown of alcohol can induce inflammatory responses. These can be worsened by gut leakiness, a condition commonly seen among ALD patients, which leads to the infiltration of LPS and other microbial products into the liver. Furthermore, ALDs sensitize non-immune cells, including hepatocytes, to immune signals that affect their normal function. We observed a predominant increase in the fraction of NPCs in diseased samples, led by a 4.5-fold increase in T-cell numbers, indicating widespread inflammation (Fig. 1c). While liver macrophages exhibited a disease-related increase, we observed a significant shift from a Kupffer cell-dominant population to monocyte-derived macrophages in SAH (Supplementary Fig. 1b). Furthermore, the distribution of hepatocytes along the porto-central axis (see “Methods”) was affected in both SAH and AC conditions (Fig. 1d and Supplementary Fig. 2a–e). Specifically, the transcripts expressed in the Portal (zone 1), Midlobular (zone 2), and Central (zone 3) zones exhibited significantly more heterogeneity for SAH and AC than control, as measured by pairwise Euclidean distance in high-dimensional PC space (Supplementary Fig. 2c, e).
We investigated gene expression programs in SAH and AC patients across various liver cell populations (Fig. 1e, Source data). We noticed that differentially expressed genes were enriched in mediators of inflammation-associated signaling pathways such as IL-1, TNF, and NF-kB (Fig. 1e, Source data). Our analysis indicated that Liver Mϕ are the major source of IL1-β, which increased significantly in SAH patients compared to unaffected individuals (Supplementary Fig. 3). Strikingly, the highest expression levels were observed in Liver Mϕ from Cirrhotic livers. In line with previous observations that activated T cells are the primary source of immunomodulatory cytokine IFNɣ38, we noticed that SAH livers had higher IFNɣ secretion from T cells in parallel to a decrease in lymphocyte-derived TNF-α (Supplementary Fig. 3). Comparing our findings to bulk RNA-seq data showed that, at the transcript level, the average expression of TNF-α and IL1-β undergoes a modest increase. At the same time, TGF-β displayed the most striking upregulation in SAH patients (Fig. 1f, Source data). Elevated levels of inflammatory cytokines and the accumulation of immune cells in SAH, including lymphocytes, polymorphonuclear leukocytes, and monocyte-derived macrophages, reflect the heightened inflammatory milieu characteristic of advanced disease39,40. For instance, although neutrophil numbers in both the circulation and liver increase, their antimicrobial functions (chemotaxis, phagocytosis, oxidative burst, NETosis) may be altered in SAH41,42,43,44.
Increased levels of inflammatory cytokines in SAH livers are known to suppress the activities of transcription factors (TFs) that inhibit the proliferation of mature hepatocytes and promote hepatocyte differentiation39,45. This leads to the accumulation of progenitor-like hepatocytes, which could be detrimental to SAH patient survival3,46. Hence, we evaluated the activities of TFs that function to establish hepatocyte identity early in liver development and compared them to changes in TFs that maintain adult hepatocyte identity and function. We first used our matched ATAC-seq dataset from hepatocytes to investigate the activity of “adult-specific” TFs by estimating the enrichment of Tn5 transposase insertions around known motifs corresponding to these TFs (Fig. 2a, c, d and Supplementary Fig. 4a, c). We observed a metagene level decrease in the chromatin accessibility around the adult-stage TF binding sites in AC and SAH patient hepatocytes relative to healthy hepatocytes. In contrast, despite an increase in their expression, we did not observe a complementary change in the activities of TFs (e.g., SOX9) that are active in developing livers (Fig. 2b, c, d and Supplementary Fig. 4b, d).

a (top) Genome-wide Tn5 insertion enrichment plots around motifs for critical transcription factors regulating mature hepatocyte states—HNF4α, CEBPα, and FOXA2—showing their decreased activity in SAH and AC patients. (bottom) Genome accessibility around their respective chromatin loci is shown below each plot. b Genome-wide Tn5 insertion enrichment plots around motifs for critical transcription factors regulating fetal-like hepatocyte states—SOX9, RELA, and E2F1—showing their unchanged activity in ALD patients. Chromatin accessibility from snATAC-seq was assessed through Seurat and Signac pipelines (see “Methods”). c Feature plots showing expression levels of FOXA2 and SOX9 TFs from snRNA-seq. d Western blot images (above) and corresponding quantitations (below) showing expression levels of TF proteins from human liver lysates (n = 3 liver samples per condition). In (d, BOTTOM), individual data points report biological replicates, and centers and error bars represent mean and Standard deviation, respectively. Blots in (d, TOP) are a representative image of 3 individual western blot trials. Two-way ANOVA statistical test used in (f). * 0.01< p < 0.1, ** 0.001< p < 0.01. SAH severe alcohol-associated hepatitis, AC alcohol-associated cirrhosis.
Additionally, other fetal TFs whose activity is re-established during successful regeneration in mice (e.g., E2F1, RELA)11, did not display robust activation in ALD hepatocytes (Fig. 2b and Supplementary Fig. 4b). Likewise, when we compared the gene expression profiles of disease-state hepatocytes with those of different developmental stages of human hepatocytes47, we noticed a decreased correlation between the gene expression profiles of SAH and adult-stage hepatocytes. However, there was only a modest correlation between the SAH transcriptome and the late-stage fetal hepatocytes, and the correlation with early fetal or hepatoblast stages was even weaker (Supplementary Fig. 4e). Furthermore, when we focused on the direct E2F1 gene targets, we detected only a handful of genes that showed significant changes in expression in SAH hepatocytes, indicating limited E2F1 transcriptional activation in ALD livers48 (Supplementary Fig. 4f). Hence, we propose that the progression of ALDs is associated with a loss of adult hepatocyte identity, yet these livers fail to mount a successful regenerative response due to an incomplete transition to a productive progenitor-like state, which generally aids hepatocyte proliferation in response to liver injury11.
Dysregulated RNA binding protein expression and alternative splicing changes in ALDs
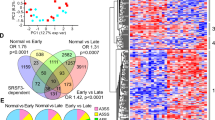
We noticed a significant fraction of transcripts whose expression levels did not align with the chromatin accessibilities at their loci (Fig. 3a–c). Such discordance between gene expression and chromatin accessibility is widespread in genomes49, and several established examples exist where TFs promote gene expression without altering chromatin accessibility50,51,52. However, alternate mechanisms that operate downstream of transcription could also be driving some gene expression changes in diseased liver cells. RNA Binding Proteins (RBPs) orchestrate coordinated changes in groups of RNAs that encode functionally related proteins when cells undergo state transitions53,54,55 and, thus, are attractive candidates as regulators of the dynamic cellular reprogramming that occurs in alcohol-injured livers. Therefore, we used our bulk RNA-seq data to profile human ALD-related differences in RBP expression. We found striking variations in mRNA levels of ~50 RBPs in diseased versus healthy livers (Fig. 3d and Supplementary Fig. 5a).

Scatter plot showing a correlation between gene expression and gene accessibility in a SAH vs unaffected hepatocytes and b Hepatocytes vs Kupffer cells, indicating that gene accessibility changes strikingly correlate with cell type-specific gene expression changes but not with pathogenic cell-state transitions. c Examples of concordance and discordance between gene accessibility and expression between SAH and unaffected hepatocytes. d Heatmap showing expression of top enriched RBPs across unaffected, SAH, and AC patients. e Western blot images and f corresponding quantification demonstrating protein expression levels of various developmentally regulated RBPs from human livers (n = 4 biological replicates). g Venn diagram showing the intersection of misregulated alternative splicing events in SAH and AC. h Distribution of alternative splicing events in SAH and AC patients. i Distribution of RNA binding motifs for developmentally regulated alternative splicing factors around AS events. In (f), individual data points report biological replicates, and centers and error bars represent mean and Standard deviation, respectively. Blots in (e) are a representative image of 3 individual western blot trials. Two-way ANOVA statistical test used in (f). In (i), Fisher’s exact test was used with correction for False discovery rate and a cutoff of q < 0.05. SAH severe alcohol-associated hepatitis, AC alcohol-associated cirrhosis.
Of note, many of the differentially expressed RBPs are critical regulators of RNA processing and metabolism. Using snRNA-seq-based evaluation of the cell type-specificity, we found that each cell type had a unique RBP expression profile and displayed distinct changes in their expression patterns in SAH and AC (Supplementary Fig. 5b). For instance, RBPs such as ESRP2 and A1CF were hepatocyte-specific and downregulated in SAH, whereas CELF2 was primarily expressed in T cells, and its expression was upregulated in SAH (Supplementary Fig. 5b). Similarly, PTBP2, which serves critical splicing functions during neuronal maturation, was upregulated in the liver sinusoidal endothelial cells but downregulated in the plasma cells of SAH patients (Supplementary Fig. 5b). Notably, the changes in mRNA abundance of RBPs were strongly recapitulated at steady-state protein levels in SAH (Fig. 3e, f). Notably, we observed a significant reduction in the levels of ESRP2 and RBM47, which are typically highly expressed in adult livers. In contrast, CELF2 and SRSF1—which show low expression in the adult stage—were upregulated in SAH livers.
The adult RNA splicing factor, ESRP2, is one of the most downregulated RBPs in the SAH livers45. ESRP2 is known to regulate alternative splicing (AS) of ~20% hepatocyte mRNAs and serves essential roles in the genesis and maintenance of the adult transcriptome necessary for supporting the mature phenotype of hepatocytes18,23,25,56. AS adds to the complexity and function of the liver proteome during development, regeneration, and disease20,57,58. Furthermore, regulated changes in splicing are crucial for establishing the identities of several cell types, including hepatocytes18,23. However, their impact on the function and progression of ALDs has not been systematically investigated. We found that both SAH and AC livers undergo widespread changes in splicing (Fig. 3g, Source data) and detected 893 and 993 AS events from SAH and AC patient livers, respectively, with 260 events common to both conditions. Most of the AS events belonged to the skipped exon (SE) and mutually exclusive exon (MXE) categories (Fig. 3h). Inspection of RNA sequences surrounding misspliced exons in SAH and AC revealed strong enrichment/de-enrichment of binding motifs for many developmentally regulated RBPs, including ESRP2 (Fig. 3i). This indicated that altered expression of RBPs could drive some of the splicing changes in SAH by directly binding to the nearby cis-regulatory motifs on target transcripts.
Alternative splicing induces widespread functional aberrations in the SAH proteome
Next, we focused on how alternative splicing contributes to disease progression in SAH. We observed that exons misregulated in SAH were distributed across the open-reading frame (ORF), 5′ and 3′ untranslated regions, and many overlapped with start and stop codons25 (Fig. 4a). A large number of skipped exons were found in the ORF, implying that exon inclusion/exclusion could generate protein isoforms with altered amino acid sequences and functions (Fig. 4a). The inclusion/skipping of exons with lengths as multiples of three maintains the reading frame during translation. While most of these events in SAH maintained the ORF frame, there was a significant fraction (~ 40%) of exons whose inclusion altered the reading frame (Fig. 4b). This presents a major change in protein isoforms encoded when the misregulated exon is closer to the start codon. We also identified a number of missplicing events that led to nonsense-mediated decay, reducing the steady-state transcript levels25 (Fig. 4b).

a Distribution of exons misregulated in SAH patients across gene body and their breakup into transcript regions. b Effect of misregulated exons on ORF of encoded transcript. c Pie chart showing the fraction of alternative spliced genes that encode proteins with known secreted isoforms. d Venn diagram showing the intersection of alternative spliced genes in SAH patients with known secreted and membrane-bound isoforms. e Heatmap showing numbers of predicted alternative splicing induced functional defects in protein products from SAH and AC patients. f RT-PCR based splice assays validating alternative splicing changes in SAH patients. RT-PCR assays for alternative exons were repeated atleast twice and representative results are presented. SAH severe alcohol-associated hepatitis, AC alcohol-associated cirrhosis.
Investigating how the inclusion/exclusion of misregulated exons might affect the sequence and function of the encoded proteins, we noted that many misspliced transcripts encoded for secreted proteins. To identify disease-associated alternative protein isoforms among secreted factors, we cross-referenced the misspliced gene set in SAH to human genes with at least one secreted protein product and found 35 genes dysregulated in SAH (Fig. 4c). Of the 39 misspliced exons identified in these genes, 34 had a homology pair in the mouse genome. Notably, we found that the 151 bp exon in FGFR3 and 69 bp exon in PDGFA, which are targets of ESRP2 in mice livers, are also misregulated in SAH livers. Further, we discovered that of the 714 alternatively spliced genes in SAH, over 20% had membrane-bound protein isoforms, and these included secreted factors FGFR3, CD44, and IL1R1 (Fig. 4d).
We next explored other functional domains encoded by skipped exons in SAH and AC patients using Exon ontology analysis59. This revealed (i) structural features like intrinsically unstructured domains, primary and secondary structure domains, membrane structures, etc. (ii) post-translational modification sites for phosphorylation at serine and threonine residues, alkylation, etc. (iii) intracellular localization signals to nuclei, ER, lysosome, etc. (iv) binding domains for interaction with other proteins, organic cyclic compounds, heterocyclic compounds, etc. and (v) domains critical for the catalytic activity of enzymes such as hydrolases, transferases, etc. (Fig. 4e). We verified many of these individual splicing events using gel-based RT-PCR assays, and the results demonstrated excellent correlation with RNA-seq (Fig. 4f). Despite having fewer misregulated exons than AC, SAH livers showed higher enrichment in specific categories, especially those encoding intrinsically unstructured protein regions, phosphorylation targets, protein binding interactions, and nuclear localization signals.
Decreased inclusion of NLS-containing exons disrupts nuclear localization of SLK and WNT effector TCF4 in SAH livers
We were particularly intrigued by misregulated events that disrupted nuclear localization and nuclear export signals (Fig. 4e), especially for signaling and TFs that are critical to hepatocyte functions. We predicted that excluding a nuclear localization signal (NLS) of a predominantly nuclear protein would lead to its cytoplasmic mislocalization and negatively influence its function. Using RT-PCR-based assays, we evaluated two of such predicted events—93 bp exon in SLK (SLK_93) and the 73 bp exon of TCF4 (TCF4_73)—and confirmed that SLK_93 and TCF4_73 inclusion in SAH livers decreased by ~40% and ~25%, respectively (Fig. 5a, b). “TCF4” is a widely used symbol/alias for the TCF7L2 gene known as “T-cell-specific transcription factor 4” or “transcription factor 7 like 2” (not to be confused with the official symbol of another gene, “transcription factor 4”). Immunohistochemical analysis confirmed that both SLK and TCF4 proteins are predominantly nuclear localized in healthy, unaffected livers, whereas their localization becomes primarily cytoplasmic in SAH livers (Fig. 5c, d).

RT-PCR-based splicing assay demonstrating decreased inclusion of a 93 bp exon of SLK and b 73 bp exon of TCF4 in SAH patients, both of which encode NLS sequence. IHC images from human patient samples demonstrating nuclear localization of c SLK, and d TCF4 is disrupted in SAH patient hepatocytes, immunostaining results are representative from 4 biological replicates in each condition. e Western blot analysis from human liver lysates demonstrating increased phosphorylation of Polo-like kinase (PLK) in SAH patients (n = 4 biological replicates). f RT-PCR-based splicing assay (top) demonstrating exclusion of the 93 bp exon in SLK transcript upon ASO treatment and western blotting (bottom) showing increased phosphorylation of PLK1 in AML12 cells after ASO treatment. Presented data is representative of 3 independent trials, but individual lanes represent technical replicates from a given trial. g Immunofluorescence staining showing decreased nuclear localization of SLK protein in AML12 cells upon ASO-induced exclusion of 93 bp exon in SLK transcript. h quantitation from MTT assay showing increased proliferation in AML12 cells treated with ASO against 93 bp exon of SLK. Data points reflect technical replicates (n = 4) for each condition, centers and error bars represent mean and Standard deviation, respectively. Presented data is representative of 3 independent trials. Student t-test was used to assess significance. ***p < 0.001. i Cumulative plot showing decreased expression of TCF4 target genes in AH patients. Kolmogorov–Smirnov (K–S) test was used to assess statistical significance, with p-value presented in top left for each comparison. j Chromvar plots showing decreased TCF4 activity in SAH hepatocytes in our scATAC-seq dataset. k RT-PCR-based splicing assay showing increased exclusion of the 73 bp exon in Tcf4 transcript upon ASO treatment in AML12 cells. Differences in PSI values are shown as mean ± SD below image. l Immunofluorescence staining showing decreased nuclear localization of TCF4 protein in AML12 cells upon ASO-induced exclusion of 73 bp exon in TCF4 transcript. Presented images are representative of 3 independent trials. m Heatmap showing qRT-PCR-based expression levels of WNT downstream target genes in AML12 cells treated with control ASO, CHIR99021 and/or Tcf4_73-targeting ASO. n Schematic demonstrating how missplicing in TCF4 and SLK leads to altered WNT signaling and proliferation program in SAH patients. Note that the inclusion of the 73 bp exon of TCF4 changes the reading frame for the downstream exon. (Created in BioRender. Das, D. (2025) https://BioRender.com/q3o6yv5). All RT-PCR assays for alternative exons were repeated atleast twice and representative results are presented. Scale bar in images for (g) and (l) reflect 5 μm. ASO anti-sense oligonucleotide.
We next evaluated how these disease-induced splicing events lead to functional disruption of SLK and TCF4. Ste20-like kinase (SLK) regulates cytoskeletal remodeling and cell migration60,61 and, when in the cytoplasm, it can phosphorylate and activate the multi-stage regulator of mitosis, PLK162. We hypothesized that AS-mediated removal of the NLS in SLK protein would lead to its cytoplasmic mislocalization, resulting in PLK activation. Western blot (WB) analysis of SLK, PLK, and phosphorylated PLK proteins revealed a significant increase in both the levels of PLK and the fraction of phosphorylated PLK in SAH patients. In contrast, the SLK levels were relatively unchanged (Fig. 5e and Supplementary Fig. 6a).
To further test whether the AS switch in SLK is sufficient to alter its localization/function, cultured AML12 cells at ~60–70% confluency were serum-starved and exposed to 1% serum, along with Slk_93-specific or scrambled control antisense oligonucleotides (ASOs). The Slk_93 targeting ASO reduced the inclusion levels to ~11%, compared to ~70% inclusion in the control ASO treatment (Fig. 5f). As predicted, immunofluorescence (IF) analysis showed a predominant absence of SLK in the cell nucleus upon ASO treatment (Fig. 5g and Supplementary Fig. 7a, c, d), and WB for PLK and phosphorylated PLK revealed significantly increased levels of PLK phosphorylation (Fig. 5f). Additionally, colorimetric MTT assay showed a significant increase in absorbance at 550 nm upon treatment with Slk_93-targeting ASO, indicating increased proliferation of hepatocytes (Fig. 5h). These data are consistent with the hypothesis that expressing cytoplasmic SLK isoform in SAH due to AS promotes PLK phosphorylation and cellular proliferation (Fig. 5n).
Next, we evaluated the effect of the NLS-containing exon in TCF4 (also known as TCF7L2), a major co-effector of WNT signaling in the liver. The WNT pathway is a critical signaling cascade that regulates development, homeostasis, regeneration, and disease progression in the liver, controlling various aspects of hepatocytes, including cell polarity, proliferation, differentiation, zonation, metabolism, and survival63. Canonical WNT signaling has β-Catenin as its chief effector, which normally forms a degradation complex with axin, adenomatous polyposis coli, and diversin, facilitating its phosphorylation and degradation. Activation of WNT signaling requires the hypo-phosphorylation and release of β-Catenin, enabling its nuclear translocation, where it partners with T-cell factor/lymphoid-enhancing factor (TCF/LEF) family of TFs to activate expression of target genes63. The TCF4 function is key to regulating virtually every aspect of liver function, especially glucose production and other metabolic processes64,65. We hypothesized that the cytoplasmic mislocalization caused by decreased inclusion of TCF4_73 would disrupt the WNT signaling pathway in ALD livers. A cumulative plot of TCF4 target gene expression revealed a significant decrease in their expression in SAH and AC patients, as indicated by the leftward shift of these curves compared to those of normal individuals (Fig. 5i). ChromVAR66 analysis of snATAC-seq to assess the TCF4 motif activities revealed that, unlike normal and AC hepatocytes, SAH hepatocytes had a significant reduction in the chromVAR scores for TCF4 motifs, indicating reduced TCF4 activities (Fig. 5j). We also found a marked decrease in portal and central localized transcripts in SAH compared to the normal livers, suggesting a general loss of zonal identity, which is consistent with the role of the WNT pathway in establishing the porto-central zonation of many hepatic genes63 (Supplementary Fig. 6b). We next investigated if splicing misregulation of TCF4_73 is sufficient to alter TCF4 protein localization/function. Targeted ASO reduced the inclusion of NLS-containing TCF4 exon to 27% (Fig. 5k), led to a decrease in nuclear-localized TCF4 protein (Fig. 5l and Supplementary Fig. 7b, e, f), and abrogated the WNT activation by GSK-3β inhibitor, relative to controls (Fig. 5m and Supplementary Fig. 7g). These data suggest that in SAH, disease-induced missplicing causes aberrant localization of TCF4, decreasing its transcriptional activity, especially in zone 3 hepatocytes (Fig. 5n).
Loss of ESRP2 exacerbates pathologies associated with an extended chronic+binge alcohol diet in mice
To identify potential RBPs that regulate the splicing of SLK and TCF4 NLS-containing exons, we inspected the genome sequence around Slk_93 and Tcf4_73. We identified several occurrences of the 5-mer sequence UUGGG, the consensus binding motif for the epithelial-specific splicing factor ESRP2. Importantly, ESRP2-deficient mice livers (ESRP2 KO) showed a marked reduction in the inclusion of both exons (Fig. 6a, b). Using in vivo eCLIP-seq (enhanced-Crosslinking Immunoprecipitation-seq) of flag-tagged ESRP218, we observed a predominant enrichment of ESRP2 eCLIP Tags (direct binding events) in the upstream intronic region of both the 93 bp Slk exon and 73 bp Tcf4 exon (Fig. 6a, b), indicating direct regulation by ESRP2. Hence, we propose that in SAH livers, decreased ESRP2 expression/activity inhibits the inclusion of NLS-containing exons in SLK and TCF4 transcripts, leading to the cytoplasmic relocalization of both proteins.

The inclusion of the NLS-containing a 93 bp Slk exon and b 73 bp Tcf4 exon decreases significantly in ESRP2 KO mice. The green and blue tracks represent RNA-sequencing coverages from WT C57BL/6J and ESRP2 KO mouse hepatocytes on the UCSC genome browser, respectively. eCLIP tags for ESRP2 are shown in black in the panel above RNA-seq tracks to demonstrate ESRP2 binding to the upstream introns. c Schematic showing extended NIAAA diet schedule on mice (20D + 2B). Mice were fed ad libitum Lieber-DeCarli ethanol liquid diet for 20 days along with two oral gavages of ethanol (5 g kg−1)—one each at day 10 and day 20. d ALT levels and qRT-PCR-based quantitation of TNF-α, Esrp2, CD133, and Hnf4a genes. Data points reflect biological replicates, centers and error bars represent mean and Standard deviation, respectively. Student t-test, and 2-way ANOVA was used to assess significance for ALT and gene expression, respectively. *p < 0.05. e Histological characterization of WT and ESRP2 KO mice after 20D + 2B diet. (n = 5 biological replicate). f Gel-based RT-PCR assay showing ESRP2 depletion leads to SAH-like transitions in alternative splicing profiles in 20D + 2B mice liver (n = 3 biological replicate). Differences in PSI values are shown as mean ± SD below each image. Scale bar in images for (e) reflect 100 μm.
As discussed above, the consensus motif for ESRP2 was one of the most enriched sequence motifs around exons that are misspliced in SAH livers, primarily in the upstream/downstream intronic regions (Supplementary Fig. 8 and Fig. 3i). We, therefore, characterized the transcriptome-wide splicing defects associated with alcohol feeding in mice and tested whether ESRP2 mediates the maintenance of adult hepatocyte characteristics in this context. To recapitulate the early stages of alcohol-associated hepatitis, we devised an extended version of the NIAAA model for our studies (20D + 2B) where mice were fed ad libitum Lieber-DeCarli ethanol liquid diet for 20 days and administered two oral gavages of ethanol (5 g/kg−1)—one each at day 10 and day 20 (Fig. 6c). Livers were collected 24 h after the second binge to profile transcriptomic changes. We found that 20D + 2B fed mice recapitulated over 20% of the gene expression defects in SAH livers. Importantly, mice lacking ESRP2 exhibited exacerbated alcohol-associated phenotypes on the 20D + 2B diet, compared to WT mice (Supplementary Fig. 9a, b), consistent with what was previously observed in the 10D + 1B NIAAA model45. Specifically, Esrp2 KO mice displayed severe hepatocyte injury, elevated levels of inflammatory cytokines, and decreased expression of adult TFs (Fig. 6d). Significant increase in steatosis (as seen from H&E and Oil red O staining), inflammation (as seen from MPO and F480 staining), cellular proliferation (as seen from Cyclin D1 staining) were noted, and portal fibrosis was worsened in the Esrp2 KO mice (Fig. 6e).
While both WT and ESRP2-deficient livers exhibited splicing changes in several genes upon 20D + 2B alcohol feeding, Esrp2 KOs recapitulated splicing defects in a larger proportion of genes misregulated in SAH livers (Supplementary Fig. 9c, Source data). Several missplicing events in genes, including Slk, Arhgef10L, and the key regulators of the Hippo signaling pathway, such as Nf2, Tead1, and Yap, were validated by RT-PCR (Fig. 6f). While alcohol feeding in WT mice only produced modest changes, alcohol-fed Esrp2 KOs showed a much stronger misregulation in splicing. This was particularly the case for SLK_93 NLS-containing exon, which closely resembled levels observed in human SAH livers. Further, in line with our predictions about adult-to-fetal reprogramming, Esrp2 KO mice exhibited increased proliferation on ethanol diet, which could be partially explained by the expression of the cytoplasmic isoforms of SLK and TCF4. Notably, the cumulative plot of TCF4 target gene expression revealed a significant decrease in their expression in ESRP2-deficient livers compared to other control groups, indicating reduced hepatic activity of TCF4 in Esrp2 KO relative to WT mice on an ethanol diet (Supplementary Fig. 9d).
Increased TGF-β signaling drives ESRP2 suppression in SAH
To pinpoint upstream signaling events most critical to suppressing the adult identity of hepatocytes in SAH, we implemented the NicheNet pipeline67. This approach integrates mRNA abundance data with the signaling and gene regulatory network information to identify which ligands influenced gene expression in hepatocytes and predict intermediate signaling factors. First, to model genes that potentially predict hepatocyte maturity, we utilized a gene list of interest (Source data) that had been previously benchmarked for predicting the successful differentiation of hepatocytes from iPSCs68. We then performed a data-driven prioritization of ligands expressed in NPCs based on how well they predict the expression of the gene list of interest (Fig. 7a panel-1). Among the top identified ligands, TGF-β showed increased expression in most NPC populations in SAH livers (Fig. 7a panel-3), with the highest expression levels in T lymphocytes (Fig. 7a panel-2, 7b). Furthermore, TGF-β displayed strong regulatory potential on several target genes, including PPARg, SOX9, CTNNB1, and HIF1A (Fig. 7a panel-4). We also identified HGF as another ligand that could regulate hepatocyte identity by potentially regulating the expression of MET.

a NicheNet-based cell interaction analysis summarizing (from left to right) 1. Key ligands that regulate hepatocyte identities in SAH livers, 2. Expression levels of ligands in different NPC cell types (Ligand producing “senders”) in SAH livers, 3. Log fold changes (LFC) in their expression levels in SAH livers compared to unaffected livers, and 4.NicheNet-based prediction of a ligand’s potential to regulate key hepatocyte target genes. TGF-β1 and HGF ligands showed the highest correlation to the gene set defining hepatocyte maturity. b Feature plot illustrating increased expression levels of TGF-β1 specifically in the NPC populations from SAH patients. c Western blot and RT-qPCR analyses showing ESRP2 reduction upon TGF-β1 treatment (5 ng/mL in DMEM/F-12 complete media for 36 h) and restoration upon addition of TGF-βR I/II inhibitor LY2109761 (50 μM for 48 h). TGF-β1 treatment increases steady-state mRNA levels of EMT-transcription factors Snai1 and Cdh2, which return to baseline after inhibitor addition. Phospho-Smad3 expression confirms TGF-β pathway activation, and its subsequent reduction upon inhibitor treatment confirms inhibitor efficacy. Data points reflect technical replicates (n = 3) for each condition, centers and error bars represent mean and Standard deviation, respectively. Presented data is representative of 3 independent trials. Pairwise Welch t-test was used to assess significance. *p < 0.05, **p < 0.01, ****p < 0.0001. d RT-PCR analysis of ESRP2 splicing targets (Slk, Slain2, and Tcf4) demonstrates significant exon skipping (93, 78, and 73 bp, respectively) following TGF-β treatment. These levels return to baseline upon the addition of the TGF-β inhibitor. Exon lengths are shown beside the gene name, and the differences in PSI values are shown as mean ± standard deviation below each image. e Schematic showing a proposed model of the combinatory roles of cytokines and Wnt signaling in regulating cellular transitions in hepatocytes after injury, which is severely misregulated in SAH due to misspliced downstream Wnt mediators. (Created in BioRender. Das, D. (2025) https://BioRender.com/3lchxkt).
TGF-β signaling is critical for initiating and maintaining epithelial-to-mesenchymal transition (EMT) by upregulating key EMT regulators, including Snail, δEF1/SIP1, and Id proteins. Importantly, δEF1/SIP1, Snail, and Twist bind to the promoter regions of ESRPs to suppress their expression and downstream splicing activities69,70. To test the direct contribution of TGF-β in SAH-related downregulation of ESRP2, we chose to treat AML12 cells with TGF-β for 36 h. AML12 hepatocyte cultures are a valuable tool for studying liver disease, drug metabolism, and toxicology; however, they have certain limitations, including altered insulin responsiveness, limited capability to model gluconeogenesis, and their inability to replicate the complex cellular interactions present in a whole liver71,72. Nonetheless, their immortalized epithelial morphology with stable hepatocyte-like features and ease of genetic/pharmacological manipulation led us to test their suitability for modeling SAH-related changes in ESRP2 and its splicing program. AML12 cells exposed to TGF-β showed significant upregulation of SNAI1 mRNA and increased phosphorylation of SMAD3 protein (Fig. 7c). Additionally, TGF-β treatment resulted in increased N-cadherin (Cdh2) mRNA abundance and a pronounced reduction in ESRP2 and E-Cadherin protein levels (Fig. 7c). Further, we noted similar changes in the splicing patterns of Slk and Tcf4 mRNAs upon TGF-β treatment (Fig. 7d), which were initially detected in the SAH livers.
To explore the potential recovery of ESRP2 and its splicing targets in ALD, we introduced a TGF-β receptor type I/II inhibitor (LY2109761), which suppresses basal and TGF-β-stimulated cell migration. Treatment of TGF-β-exposed cells with the inhibitor reduced the pSmad3 levels, indicating effective blockade of the TGF-β signaling pathway (Fig. 7c). Importantly, treating AML12 hepatocyte cultures with the inhibitor reinstated ESRP2 expression and restored the splicing of its target transcripts (Slk, Tcf4, and Slain2) (Fig. 7c, d). Together, these findings highlight the therapeutic potential of TGF-β inhibitors in mitigating ALD-associated spliceopathy, hepatocyte dedifferentiation, and impairment of liver regeneration.
Discussion
SAH and AC can cause liver failure and, thus, are the most lethal outcomes of ALD. It is important to emphasize that although both SAH and AC develop in the context of chronic alcohol consumption, the conditions have very different trajectories. Patients with SAH present abruptly with overt manifestations of profound liver dysfunction, including jaundice, coagulopathy, and multiple organ failure. In contrast, patients with AC typically live for years without any overt symptoms or signs of liver disease and only gradually develop clinical evidence of progressive liver dysfunction after decades of excessive drinking. Nonetheless, SAH can be superimposed upon AC, resulting in “acute on chronic” liver failure and an abrupt increase in mortality. Continued drinking accelerates the rate of progression to liver failure in individuals with either SAH or AC. Conversely, once alcohol consumption is terminated, both conditions typically improve with time. Despite the discontinuation of alcohol consumption, however, liver function progressively deteriorates in a subpopulation of patients with these conditions, necessitating liver transplantation for survival. The mechanisms that underlie the differences in liver resiliency between patients with SAH or AC who recover from ALD versus those who cannot are poorly understood. By the time of liver transplantation, liver dysfunction is similarly severe in SAH and AC patients despite their differing rates of progression to liver failure, presumably because surviving liver epithelial cells in patients with SAH and decompensated AC are dysfunctional and unable to regenerate sufficient healthy parenchyma to avert liver failure3,4.
The opportunity to study explanted livers with advanced alcohol-related disease provides a unique opportunity to delineate mechanisms underlying progressively defective liver repair, as currently available mouse models of ALD cause relatively mild disease that is most reminiscent of early-stage, clinically mild ALD. Previous studies have shown that the transcriptomes of ALD patient livers undergo significant reprogramming, and expression of TFs controlling hepatocyte maturation is deregulated in ALD73. In line with these findings, we found that the abundance, nuclear localization, and activities of TFs that maintain the mature hepatocyte phenotype (e.g., HNF4α, C/EBPα, FOXA2) are reduced in SAH and AC, although transcription of these TFs themselves remains similarly active in diseased and healthy livers. Furthermore, the activities of TFs that enable hepatocytes to transition to a less mature, more proliferative phenotype (e.g., SOX9, RELA, E2F1) are not increased despite the disease-related induction of these TFs. These findings suggest that mechanisms operating downstream of transcription play a critical role in regulating the phenotypic transitions necessary for the recovery of functional parenchyma in livers injured by alcohol. Dysregulation of these processes may trap hepatocytes in an intermediate state, where they are neither fully mature nor functional progenitors (Fig. 7e).
We analyzed explanted livers enriched with these dysfunctional cells to identify mechanisms that maintain their persistence long after the toxin that induced the liver damage is removed. Our data show that global changes in the hepatic expression of RBPs significantly contribute to this pathology. RBPs interact with microRNAs and noncoding RNAs to control the stability, splicing, and translation of suites of mRNAs that must collaborate to orchestrate complex phenotypic switches18,74. Previous studies have demonstrated the importance of the microRNA network in modulating susceptibility to alcohol-related hepatotoxicity75,76. Our work presents evidence about various post-transcriptional mechanisms that dictate ALD outcomes in multiple ways. The results: (i) demonstrate disease-related differences in the mRNA expression of a large number of RBPs; (ii) prove that the differences in transcript abundance are paralleled by differences in protein expression of these RBPs, as well as (iii) changes in their mRNA splicing targets that (iv) result in functional changes in the proteins encoded by those mRNAs, and (v) lead to changes in signaling that broadly control reparative and regenerative responses in injured livers. Together, these foundational findings provide a platform for studies to identify specific RBPs, which function as the hubs that tether this interactome, as these RBPs (or some of their targets) could be manipulated to improve recovery from ALD.
We focused on ESRP2 as a pivotal disease-sensitive RBP because it is absent in the early stages of liver development but (i) upregulated dramatically in the final stages of hepatocyte differentiation, (ii) expressed specifically in hepatocytes of healthy adult livers, and (iii) is known to regulate the adult splicing program of ~20% of the mature hepatocyte transcriptome23. Further, the current analysis confirmed that hepatocyte expression of ESRP2 is virtually undetectable in livers with clinically decompensated ALD45. Thus, ESRP2 is one of the most severely downregulated RBPs in the transcriptome and proteome of hepatocytes in livers with SAH, and its expression is also suppressed in AC. ESRP2 prevents hepatocytes from undergoing EMT and thereby maintains hepatic epithelial integrity56. Epithelial integrity is crucial for executing vital liver-specific functions. After liver injury, cell-to-cell connections in surviving hepatocytes must be relaxed but then re-established efficiently for liver regeneration to reconstitute functional hepatic parenchyma. Consistent with this notion, our earlier work demonstrated that injury-related cytokines that initiate liver regeneration (e.g., TNF-α, IL1-β) suppress ESRP245. Downregulation of ESRP2 inhibits Hippo kinase signaling, enabling the activation of YAP, a factor that promotes hepatocyte EMT23,25. In this study, we demonstrated that decreasing ESRP2 activates Polo-like kinase 1, which is known to expedite the transition through later stages of the cell cycle and facilitate hepatocyte proliferation77. After partial hepatectomy, TNF-α and IL1-β are transiently induced, ESRP2 is transiently repressed, and effective liver regeneration ensues.
However, when ESRP2 suppression is sustained in hepatocytes of injured livers, regeneration is defective, and maladaptive inflammatory and fibrogenic repair responses predominate. Our mouse model of ALD supports a causal role for ESRP2 depletion in this liver growth dysregulation by showing that Esrp2 KO mice develop significantly worse liver inflammation, fibrosis, and spliceopathy than alcohol-fed controls. Our single nuclei analysis of human SAH and AC livers (which “naturally” accumulate ESRP2-deficient hepatocytes) demonstrates significant qualitative and quantitative changes in immune and hepatic stellate cell populations. These changes in stromal cell populations enrich the liver microenvironment with factors that promote inflammation (e.g., TNF-α, IL1-β) and fibrosis (e.g., TGF-β), all of which also suppress ESRP2. Hence, the hepatic microenvironment in ALD favors the sustained downregulation of ESRP2, leading to the persistence of a quasi-progenitor-like state in hepatocytes.
Recent studies have indicated that adult hepatocytes require three processes to replicate—de-differentiative signaling initiated by pro-inflammatory cytokines, proliferative signaling driven by growth factors/Wnts, and inhibition of TGF-β signaling78. Although hepatocytes of ethanol-fed ESRP2 KO mice and humans with SAH or AC switch to a less mature phenotype, they may be growth-arrested, given that net hepatic expression of Wnt target genes is suppressed, and the hepatocyte populations are relatively depleted. This evidence of reduced regenerative activity is paradoxical because liver size is not reduced in ESRP2 KO mice18,23, and herein we show that depleting ESRP2 in healthy hepatocytes activates PLK1. This response is predicted to expedite transition through late (i.e., G2-M) phases of the cell cycle. On the other hand, aberrant activation of PLK1 in injured tissues has been reported to subvert the responses that normally repair damaged DNA, leading to mitotic catastrophe and cell senescence77. Indeed, we observed that ethanol-fed ESRP2 KO mice accumulate cyclin D1-positive hepatocytes in hepatic acinar zone 2, where hepatocytes in pre-replicative phases of the cell cycle accumulate in healthy liver8,11,79. Together, these observations suggest that many surviving ESRP2-deficient hepatocytes in livers with SAH or AC may be arrested in pre-replicative stages of the cell cycle and are, thus, non-regenerative despite exhibiting a progenitor-like phenotype (Fig. 7e). This possibility merits further investigation in light of a recent report indicating that the number of biliary-derived bipotent transitional liver progenitor cells (TLPCs) increases in parallel with the accumulation of senescent hepatocytes in many liver diseases, including ALD. TLPCs must differentiate into hepatocytes before replicating9. ESRP2 expression is hepatocyte-restricted in adult liver25, raising the possibility that proper regulation of ESRP2 targets is required to regenerate healthy liver tissue after injury. Some of the ESRP2-regulated factors may serve as potential therapeutic targets; others (e.g., aberrantly spliced secreted proteins) may simply telegraph dysfunctional repair responses and serve as biomarkers for poor ALD outcomes.
Methods
Ethics statement
All mouse experiments were performed and reported according to the National Institutes of Health (NIH) and Institutional guidelines for the use and care of laboratory animals, and experimental protocols were performed as approved by the Institutional Animal Care and Use Committee at the University of Illinois, Urbana-Champaign, and Duke University. For human patient samples, all oversight was provided by Johns Hopkins Medicine Human Research Protection Program under IRB00107893 and IRB00154881. The study design and conduct complied with regulations regarding the use of human study participants and were conducted in accordance with the criteria set by the Declaration of Helsinki.
Sample collection
Human liver samples were acquired from patient explants (right lobe) and donor resections (caudate lobe). Collected samples were either snap-frozen and stored at −80 °C or formalin-fixed and embedded in paraffin until use. The diagnosis of SAH and AC is based on previously published guidelines80,81, and all patients with SAH had some degree of cirrhosis or advanced fibrosis. Human samples were received from the Department of Surgery at Johns Hopkins Hospital (supported by the NIAAA, R24AA025017, Clinical resources for AH investigators). The study design and conduct complied with regulations regarding the use of human study participants and were conducted in accordance with the criteria set by the Declaration of Helsinki.
Experimental animals
Mice were housed on a standard 12-h-light/dark cycle (18–23 °C ambient temperature; 40–60% humidity) and were allowed ad libitum access to water and a standard chow diet (2918 Envigo Teklad). Genomic DNA was derived from tail biopsies, and genotyping was performed using standard procedures. Male WT C57BL/6J (Jackson Laboratory, Bar Harbor, ME) and ESRP2-KO23 mice were fed an extended version (20 days + 2 binges) of the NIAAA model82. Briefly, WT (n = 8) and Esrp2-KO mice (n = 8) were initially fed the control Lieber-DeCarli diet ad libitum for 5 days to acclimatize them to the liquid diet. Subsequently, ethanol-fed groups were allowed free access to the ethanol diet containing 5% (v/v) ethanol for 20 days, and control groups were pair-fed the isocaloric control diet. At days 10 and 20, ethanol-fed and pair-fed mice received a dose of ethanol (5 g/kg body weight) or isocaloric maltose dextrin, respectively, via gavage in the early morning and were euthanized 24 h after the second binge. Whole liver tissues and hepatocytes were isolated from mice following guidelines for euthanization and/or anesthesia. Paraffin-embedded liver sections were stained with hematoxylin and eosin (H&E) or Sirius Red for pathological evaluation, and optimal cutting temperature-embedded liver sections were stained with Oil Red O to visualize lipid droplets. For immunohistochemistry, liver sections were deparaffinized, hydrated, and then incubated in 3% hydrogen peroxide to block endogenous peroxidase activity. Antigen retrieval was performed by heating in a 10 mM sodium citrate buffer (pH 6.0) for 10 min using a microwave. Specimens were blocked in Dako Protein Block solution (Agilent, Santa Clara, CA) for 30 min at room temperature, followed by incubation with the primary antibody overnight at 4 °C. Other sections were also incubated at 4 °C overnight in non-immune sera. Polymer-horseradish peroxidase (HRP) anti-rabbit (Dako) and polymer-HRP anti-goat (ThermoFisher Scientific, Waltham, MA) were used as secondary antibodies, and 3,3′-diaminobenzidine (DAB) was used as the brown color to visualize the protein. Sections were counterstained with hematoxylin.
Nuclei isolation and multiomics library preparation
Intact nuclei were isolated from human liver samples based on a protocol adapted from 10× genomics CG000375. Briefly, frozen tissue was transferred to a pre-cooled Dounce homogenizer containing 1× Homogenization Buffer, as described in ref. 83 (supplemented with 0.5 U/μL RNase In and a 1:100 dilution of PIC III). The sample was homogenized 15× using a Pellet Pestle on ice and incubated for 6 min on ice. The sample was homogenized again five times using a Pestle on ice and incubated for an additional four minutes on ice. Then, the sample was filtered through a 70 μm strainer and then through a 15 μm strainer, and then centrifuged at 500 rcf for 5 min at 4 °C. Pellet was left in 1 ml PBS + 1% BSA + 1 U/μl RNase Inhibitor for 5 min on ice without mixing, and then resuspended. Finally, the nuclear suspension was centrifuged at 500 × g for 5 min at 4 °C, and the obtained pellet was resuspended in 1 mL PBS + 1% BSA + 1 U/μL RNase Inhibitor and used for library preparation.
Single-nuclei library preparation and sequencing
Following nuclear isolation, single-nuclei sequencing libraries for multiomic profiling using scRNA-seq and scATAC-seq were prepared individually from pools of human liver samples from Normal individuals, AH patients, and AC patients using the 10× Genomics Chromium Single Cell Kit and sequenced with Illumina NovaSeq 6000 on a SP/S4 flow cell to obtain 150 bp paired reads. For each condition, we pooled the nuclei into a batch of 3 individuals and another batch of two individuals during library preparation and targeted 9000 and 6000 single nuclei per batch, respectively.
snRNA-seq/snATAC-seq data analysis
Raw data obtained from sequencing were demultiplexed, filtered, and mapped to the hg38 human genome using the Cell Ranger ARC pipeline. Cell Ranger ARC was also used to perform barcode counting, peak calling, counting of ATAC and GEX modules, and generating feature-barcode matrices. The data were integrated using the Seurat pipeline and batch-corrected using the harmony batch correction protocol84. Finally, Seurat85,86 and Signac87 pipelines were used to perform dimensionality reduction, cluster determination, cell type annotation, and differential analysis on clusters.
Briefly, “Read10×_h5” function was used to read in feature barcode matrices from the 10× CellRanger ARC pipeline into the R analysis pipeline. An object was created using “CreateSeuratObject” first to contain the gene expression matrix, followed by the use of “CreateChromatinAssay” to create an object for the scATAC-seq peaks identified from the Multiome pipeline. For each peak, a min.cell cutoff of 10 was used. In the combined Seurat object with ATAC and RNA matrices, cells were filtered such that the percent.mt reads were below 30%, RNA counts were greater than 500 and less than 25,000, and ATAC counts were greater than 1000 and less than 10,000. Assays from all samples and conditions were combined, followed by RNA matrix normalization and batch correction using the “RunHarmony” function. UMAP projections were calculated using HARMONY output for the first 30 dimensions of variation. Next, doubletFinder v3 was used on the RNA matrix data to identify doublets, using parameters as recommended, namely, pN 0.1, pK 0.15. For the ATAC data, normalization was performed using the “RunTFIDF” function, followed by batch correction with the “RunSVD” and “RunHarmony” functions. Lastly, multimodal reduction was calculated using “FindMultiModalNeighbors” and “RunUMAP” with Harmony dimension reduction from both RNA (dim 1:30) and ATAC (dim 2:30) datasets. Clusters were identified using the “FindClusters” function, and the multimodal reduction was used as the input dataset with algorithm 3 and resolution 0.4. Clusters were annotated for cell types using manual curation of known cell-type-specific markers after identification of differentially expressed cluster genes with “FindAllMarkers” function with MAST test and latent variable to correct for as cell read depth (CDR), and min.pct 0.25.
To perform motif analysis, position frequency matrices were extracted from the JASPAR2020 database (species 9606). The “AddMotifs” function was used to add Homo Sapiens (hg38) motif information to the Seurat object. We then computed GC content for each peak using the “RegionStats” function, followed by linking each peak to the nearest gene using the “LinkPeaks” function with ATAC matrix used for peak_assay and RNA matrix for expression_assay. Top differentially enriched peaks in conditions were identified using the “FindMarkers” function using the “LR” test and the “nCount_ATAC” column from the metadata matrix as the latent variable. Further, the “FindMotifs” function was used to find enriched motifs within differentially expressed peaks. Lastly, using the default recommended parameters, Cicero was applied to identify peak linkage within ATAC data.
RNA and protein analysis
RNAs were extracted using TRIzol reagent according to standard protocols. Real-time PCR reactions were performed in triplicate or duplicate using the SYBR green master mix. To verify the inclusion levels of target exons, RT-PCR assays were performed using primers that spanned the flanking exons, and the products were resolved on a 5% acrylamide gel. Details of the exons investigated and the primers used are provided in the Source Data. For protein analysis, frozen human liver samples were homogenized in RIPA lysis buffer. Protein lysates were cleared by spinning the samples twice at 4 °C. Subsequently, samples were separated on SDS-PAGE and analyzed by western blotting. Antibody information with dilution is provided in Source Data.
Illumina RNA-seq
Frozen tissue was quickly thawed and disrupted on ice with a homogenizer. Total RNA was isolated using a Qiagen RNeasy© RNA isolation kit. After RNA quality had been assessed by capillary electrophoresis (bioanalyzer), cDNA libraries were prepared using TruSeq© RNA Library Prep Kit and analyzed with an Illumina NextSeq500. Base-calling and fastq conversion were performed using RTA (2.4.11) and Bcl2fastq (2.18.0.12), respectively.
RNA-seq analysis
For analysis of RNA-seq data, raw reads were subjected to read length and quality filtering using Trimmomatic V0.3888 and aligned to the mouse genome (mm10) using STAR (version 2.6.1d)89. Cufflinks package90 was used to assess differential gene expression events, among which significant events were identified using a stringent cutoff criterion (FDR(q-value) < 0.05, log2(fold change) > 1). rMATS v4.0.2(turbo)91 was used to study differential splicing and events with FDR < 0.1, junction read counts ≥ 10, PSI ≥ 10% were deemed to be significant.
Exon ontology analysis
Exon ontology analysis was performed on the set of alternatively spliced cassette exons identified using rMATS. Mouse(mm10) annotations were converted to human (hg19) annotations using UCSC liftover with a minimum base remap ratio set to 0.8. The Exon ontology pipeline59 was then applied to the lifted exons to perform ontology analysis.
eCLIP library preparation & analysis
ESRP2 eCLIP was performed on IP from two independent samples along with paired size-matched input before the IP washes, in concordance with previously published protocols92. Isolated hepatocytes from N-terminal FLAG-tagged ESRP2 (ESRP2Tg/wt) mice were suspended in 1× PBS and crosslinked with 1 pulse of 400 mJ/cm2 of 254 nm UV radiation to stabilize RBP–RNA interactions. Subsequent immunoprecipitation (IP) of FLAG ESRP2-RNA complexes, RNA isolation, library preparation, and sequencing were performed as described previously18. Briefly, cross-linked cells were lysed, sonicated, and treated with RNase I (Thermo Fisher), then incubated with FLAG M2 antibody (Sigma-Aldrich F1804) precoupled to antimouse IgG Dynabeads (Thermo Fisher 11201D). After saving 2% of the sample as input, high-salt and low-salt washes were performed. RNA was then dephosphorylated using FastAP (Thermo Fisher) and T4 PNK (NEB), and a 3′ RNA adaptor was ligated with T4 RNA ligase (NEB). IP and input samples were run on a PAGE gel, transferred to nitrocellulose membranes, and the region between 78 and 155 kDa was excised. The excised region was then treated with proteinase K (NEB) and concentrated by column purification (Zymo). Input samples were then dephosphorylated, and a 3′ RNA adaptor was ligated with T4 RNA ligase (NEB). Next, reverse transcription was performed using AffinityScript (Agilent), followed by ExoSAP-IT (Affymetrix) treatment, RNA degradation via alkaline hydrolysis, and 3′ DNA adaptor ligation with T4 RNA ligase (NEB). The final library was prepared by PCR amplification using Q5 (NEB) and size-selection by gel electrophoresis. Libraries were sequenced on the NovaSeq6000 platform (Illumina).
To analyze the eCLIP reads, the CLIP toolkit pipeline was used93. Briefly, 3′ adaptors were clipped from reads, followed by collapsing of PCR duplicates and removal of the N10 random barcode sequence. Reads were mapped using the bwa aligner and cleaned up for PCR duplicates and alignments to repetitive and ribosomal non-coding RNA regions. Peak calling was performed on the remaining mapped reads, and crosslinking site analysis was performed using CIMS/CITS.
Cell culture
AML12 cells (ATCC, catalog number CRL-225) were seeded into 6-well plates (at a density of 2.0 × 105 cells per well). Sterile cover glasses were placed in the 24-well plates (at a density of 1.0 × 105 cells per well) and cultured in DMEM/F12 medium supplemented with insulin–transferrin–selenium (cat. no. 41400-045; Invitrogen) and 10% fetal bovine serum (heat-inactivated at 56 °C) at 37 °C in the presence of 95% air and 5% CO2. For immunofluorescence, AML12 cells were seeded on sterile cover glasses placed in the 6-well plates. At 70–80% confluency, growth media was replaced with low serum media (2% FBS) and incubated for 4 h prior to TGF-β treatment. Using a final concentration of 5 ng/mL TGF-β (cat. no. 7666-MB; R&D Systems), cells were incubated for 36 h before harvesting. For recovery experiments, the cells were treated with 15 μM TGF-β inhibitor (LY2109761, cat. no. HY-12075; MedChemExpress) for 36–48 h. For activating beta-catenin, AML12 cells at 70% confluency were treated with 3 μM CHIR99021 (cat. no. 4423; Tocris) for 48 h. To induce the skipping of Slk 93-bp exon, we used an antisense morpholino oligonucleotide (ASO), with sequence 5′-AACCTACAGTACACAAGCACGCGG-3′, which binds to the 3′ splice site (3′ss) upstream of Slk exon at the intron-exon junction. For inducing Tcf4 73-bp exon skipping, an ASO (Tcf4_73 ASO) sequence was used: 5′-ATCTGGGAGAC TATACACA CCTGCA-3′, which binds to the 5′ss downstream of Tcf4 exon 73 pre-mRNA sequence. As a control, a non-target standard control sequence was used: 5′-CCTCTTA CCTCAGTTACAAT TTATA-3′ (Gene Tools, LLC, Philomath, Oregon, USA). Morpholinos were delivered at a 10 μM final concentration using Endoporter reagent. Cells were harvested 48 h later, and total RNA plus protein lysates were prepared for subsequent analyses.
Immunofluorescence staining of cultured cells and analysis
AML12 cells with control and experimental treatments were grown on coverslips and harvested between 24 and 48 h, fixed in 4% paraformaldehyde for 10 min, permeabilized using 0.5% Trition-×100 in 1× PBS for 5 min, blocked in 2% Normal Goat Serum, 1% BSA, 0.1% Triton ×100 in 1× PBS, and incubated with primary antibody (1:1000) overnight at 4 °C in a humidified chamber. On the following day, the cells were incubated with a secondary antibody (1:500) at room temperature for 1–2 h, treated with NucBlue for 20 min, and mounted. Antibody information is provided in Source Data. All images were taken using Zeiss LSM 900 with Airyscan 2 at the core facilities of IGB, UIUC. The images were processed using Airyscan Deconvolution and ImageJ, and > 10 nuclei per sample were randomly selected for analysis across biological replicates. These images were analyzed using our custom CellProfiler pipeline, wherein nuclei were defined and separated from the surrounding non-nuclear region using Otsu’s global thresholding method. The intensity for SLK and TCF4 was separately measured for the nuclear and non-nuclear regions, and the nuclear fraction was calculated as the ratio of nuclear to non-nuclear areas. The data were analyzed in Prism using the unpaired non-parametric Mann–Whitney U Test (obtained p-value < 0.0001).
Histological analysis and immunohistochemistry (IHC) from mice
Liver tissue samples were fixed in formalin, embedded in paraffin, and sectioned for histological analysis. Hematoxylin and eosin (H&E) staining was performed to evaluate general liver morphology. Fibrosis was assessed using Picrosirius Red staining (Sigma-Aldrich, 365548) following standard protocols.
For immunohistochemistry (IHC), tissue sections were dewaxed, rehydrated, and incubated with 3% hydrogen peroxide for 10 min to block endogenous peroxidase activity. Antigen retrieval was carried out by heating the sections in 10 mmol/L sodium citrate buffer (pH 6.0) for 10 min. Sections were then blocked with Dako protein block solution (Agilent Technologies, Santa Clara, CA) for 1 h and incubated overnight at 4 °C with primary antibodies against SLK, TCF4, CyclinD1, F4/80, or Myeloperoxidase. The following day, the samples were incubated in HRP-conjugated secondary antibodies for 1 h at room temperature, and then visualized using the Dako DAB Substrate Chromogen System. Antibody information with dilution is provided in Source Data. Imaging was performed using Leica Microsystems.
Frozen liver tissues were sectioned at 20 μm, fixed in 10% formalin, and stained with Oil Red O (Sigma-Aldrich, O0625) for 10 min to visualize lipid accumulation.
RNA-seq data normalization and dimensionality reduction
We normalized the RNA-seq dataset using the Seurat pipeline28, as previously described94,95. This process involved the following steps: NormalizeData(object, Normalization.method = “LogNormalize”), FindVariableFeatures(object, selection.method = “vst”, nfeatures = 2000, clip.max = “auto”, binning.method = “equal_width”), ScaleData(model.use = “linear”, min.cells.to.block = 3000, block.size = 1000, scale.max = 10), and RunPCA(npcs = 50, weight.by.var = TRUE). The resulting normalized “RNA” assay was used for subsequent analyses.
Porto-central coordinates calculation
To determine the position of each cell along the porto-central axis of the hepatic lobule, we calculated a porto-central coordinate (\({{{\rm{\eta }}}}\)) based on the expression of previously identified portal and central marker genes in mice34. It’s important to note that while these marker genes were identified in mouse samples, we applied them to our human RNA-seq dataset, assuming the conservation of zonation patterns across species.
Portal marker genes (\({pLM}\)) included
APOF, APOM, ASGR2, ASS1, C1S, C8B, CPT2, EEF1B2, ELOVL2, FADS1, FBP1, GC, GNMT, HSD17B13, IFITM3, IGF1, IGFALS, NDUFB10, PIGR, S100A1, SERPIND1, SERPINF1, UQCRH, VTN, PCK1, ARG1, and CPS1.
Central marker genes (cLM) included
ALAD, ALDH1A1, C6, CPOX, CSAD, CYP1A2, CYP2E1, GSTM1, HPD, LECT2, MGST1, OAT, PON1, PRODH, RGN, and SLC16A10.
For each cell, \(i\), we calculated the identified porto-central coordinate \({\eta }_{i}\) using the following equations:
Where \({E}_{g,i}\) represents the expression of gene, \(g\), in cell, \(i\).
The resulting \({\eta }_{i}\) values range from 0 to 1, with \({\eta }_{i}\) = 0 indicating a position closer to the pericentral region and \({\eta }_{i}\) = 1 indicating a position closer to the periportal region of the hepatic lobule. It should be noted that some genes from the original list (CML2, CYB5, CYP2C37, CYP2C50, CYP3A11, ATP5A1, CYP2F2, DAK, SERPINA1C, SERPINA1E, and TRF) were not found in our dataset and were therefore excluded from the analysis.
Zonation and comparative analysis
We divided the hepatic lobule into three distinct zones based on its calculated porto-central coordinate \({\eta }_{i}\). Given that \({\eta }_{i}\) ranges from 0 to 1, we equally partitioned this range into three zones:
Where \({Z}_{i}\) represents the zone assignment and \({\eta }_{i}\) is the porto-central coordinate of each cell, i.
We compared the zonation patterns between different hepatic conditions (Normal, AC, and SAH), we implemented a subsampling and distance calculation approach using R (R Core Team, 2021) and the Seurat package28. We performed two rounds of random subsampling without replacement. The number of cells subsampled was determined by the minimum cell count across conditions in the same zone, ensuring equal representation. The maximum amount of cells we subsampled across conditions is 100 to reduce noise and ensure a proper representation. This process resulted in two independent subsets of cells for each zone-condition combination, allowing for robust comparative analysis.
Using the principal component (PC) embeddings from the Seurat objects, for any two cells \(i\) and \(j\) in the same zone in the subsample, we computed the Euclidean distance \({d}_{{ij}}\) in the n-dimensional PC space (Eq. (4)).
Where \({{PC}}_{{ik}}\) and \({{PC}}_{{jk}}\) represent the kth principal component values for cells \(i\) and \(j\). This calculation provides a quantitative measure of cell-to-cell variability in the reduced dimensional space.
Zonation-based gene expression analysis
We developed a method inspired by the Methods of the Halpern et al. paper to investigate gene expression patterns across hepatic zones in different conditions (Unaffected, AC, and SAH)34. The set of codes created generates three key data structures: the Porto-central Matrix (\(M\in {R}^{{\#cells} ,\# z{ones}}\)), Mean Gene Expression (MGE) matrix and a variance-sorted gene list.
Using the zonation data obtained from the previous section, we constructed a binary matrix called the Porto-central Matrix (\(M \in {R}^{{\#cells} ,\# {zones}}\)), where rows represent cells \(i\) and columns represent zones \({Z}_{i}\). Each cell was assigned a value of 1 in its corresponding zone column and 0 in others. To ensure balanced representation across zones, we subsampled an equal number of cells (n = 28) from each zone, resulting in a subsampled matrix. The matrix is then normalized through a two-step process: first, each row is divided by its sum, and then each column is divided by its sum. This can be expressed mathematically as:
Using this weighted normalized matrix and the gene expression data from our Seurat objects \((E\in {R}^{{\#genes},{\#column}})\), we obtain the MGE matrix \((G\in {R}^{{\#gene},{\#zones}})\) with rows of genes and columns of normalized average expression of each gene across zones (Eq. (6)).
We then calculated the variance of gene expression for each gene in the normalized MGE matrix \(G\) (Eq. (7)).
where \({G}_{{gZ}}\) represents the expression of gene \(g\) in zone \(Z\), and \({\bar{G}}_{g}\) is the mean expression of gene \(g\) across all three zones. We then ranked genes in descending order based on their calculated variances. To focus on identifying new zonation-associated genes, we excluded the porto-central gene markers used in the initial zonation calculation from this ranked list.
For any two zones \({Z}_{i}\) and \({Z}_{j}\) in the condition, we computed the Euclidean distance \({d}_{{ij}}\) in the n-dimensional gene space (Eq. (8)). This computation uses only genes with a variance of 0.1 and above for each zone. This analysis pipeline was applied separately to each condition (Normal, AC, and SAH).
Zonation mean gene expression analysis
We extracted normalized gene expression data from Seurat objects for Normal liver and SAH conditions. The data was subset based on a predefined list of zonation genes, including both portal and central markers, compiled from previous liver zonation literature.
For each condition (Normal and SAH) and zone (1, 2, and 3), we calculated two types of MGE for each zonation gene: (1) the mean expression across all cells, including those with zero expression, and (2) the non-zero mean expression, focusing only on cells where the gene is detected. This dual approach accounts for both overall expression levels and the sparse nature of single-cell RNA-seq data. The results were compiled into two separate matrices of MGE, each with rows representing individual zonation genes and columns representing each zone within Normal and SAH conditions (6 columns total). For both matrices, genes not detected in any cells for a particular zone and condition were marked as NA. We included genes in the analysis if they showed expression in at least one cell in each zone and condition.
Statistical analysis
To understand the differences between eta distribution across conditions, we utilized the Kolmogorov–Smirnov (K–S) test, which is sensitive to location, shape, and spread differences between distributions, without knowing the underlying distribution of eta values. We take the maximum vertical distance (D-statistic) between two empirical cumulative distribution functions.
where x spans all possible eta values. This D-statistic represents the largest absolute difference between the two distributions at any point. The null hypothesis (H0) posits that both samples are drawn from the same continuous distribution, in which the D-value would be very small. Statistical significance was assessed at α = 0.05, with p < 0.05 indicating significant differences between distributions of different sample sizes. Two pairwise comparisons were performed: Unaffected vs. AC and Unaffected vs SAH. The test was implemented using R’s ks.test() function, with exact p-values calculated for sample sizes where computationally feasible.
To visualize and further characterize the analysis from the K–S test, we generated a Quantile–Quantile (Q–Q) plots comparing each pair of conditions and another comparison between Normal Half-1 vs Normal Half-2 (Supplementary Fig. 2a). Subsampling Normal into Half1 and Half2 and the points are on the line showing that they are the same distribution. For the Unaffected vs. AC and Unaffected vs SAH comparison, the plot revealed distinct patterns across different ranges of eta values. Correlation (r) was calculated to show how closely the points are to the diagonal line. Mean Squared Error shows the average square distance of the points from the diagonal line. Combined, these two measurements quantify how similar the Q–Q plots are.
We bootstrapped the data of the percentage of cells in each zone per condition for n = 500 iterations (Supplementary Fig. 2d). For each iteration, we subsampled data per condition with replacement and calculated the percentage of cells in each zone for each condition. For each subsample, we performed a Wilcoxon test comparing zone percentages across 500 iterations, where p < 0.05 marks significant differences between Unaffected and diseased (AC and SAH). We also calculate means and 95% confidence interval for each condition in each zone.
We employed two complementary statistical tests: the Wilcoxon rank-sum test and the independent two-sided t-test. Cells in each zone in different conditions are independent of each other; we conducted both tests to compare the significance of differences in the distribution of cells between conditions within zones and in gene expression between zones in different conditions. The Wilcoxon test, being non-parametric, allowed us to compare the distribution of cells within zones and the value differences in gene expression between zones without assuming normality, while the t-test provided insights into differences in the mean distribution of cells and expression levels. We considered p-values < 0.05 as statistically significant for both tests.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Multiomics data is deposited on Gene Expression Omnibus with accession number GSE281574. Bulk RNA sequencing data is available under the accession number GSE143318. Source data are provided with this paper.
Code availability
All code developed for portal-central coordinate calculation, zonation characterization, statistical analysis, variability of gene expression calculation, and plotting can be found on GitHub at https://github.com/GoyalLab/Zonation_Analysis. A permanent record of the version presented in this study is available on Zenodo [https://doi.org/10.5281/zenodo.15778946].
References
Devarbhavi, H. et al. Global burden of liver disease: 2023 update. J. Hepatol. 79, 516–537 (2023).
Patel, P. V. & Flamm, S. L. Alcohol-related liver disease including new developments. Clin. Liver Dis. 27, 157–172 (2023).
Jung, Y. et al. Accumulation of hedgehog-responsive progenitors parallels alcoholic liver disease severity in mice and humans. Gastroenterology 134, 1532–1543 (2008).
Dubuquoy, L. et al. Progenitor cell expansion and impaired hepatocyte regeneration in explanted livers from alcoholic hepatitis. Gut 64, 1949–1960 (2015).
Aravinthan, A. et al. Hepatocyte expression of the senescence marker p21 is linked to fibrosis and an adverse liver-related outcome in alcohol-related liver disease. PLoS ONE 8, e72904 (2013).
Sharma, S. et al. Pre-therapy liver transcriptome landscape in Indian and French patients with severe alcoholic hepatitis and steroid responsiveness. Sci. Rep. 7, 6816 (2017).
Gadd, V. L., Aleksieva, N. & Forbes, S. J. Epithelial plasticity during liver injury and regeneration. Cell Stem Cell 27, 557–573 (2020).
Wei, Y. et al. Liver homeostasis is maintained by midlobular zone 2 hepatocytes. Science 371 https://doi.org/10.1126/science.abb1625 (2021).
Pu, W. et al. Bipotent transitional liver progenitor cells contribute to liver regeneration. Nat. Genet 55, 651–664 (2023).
Lin, Y. H. et al. IGFBP2 expressing midlobular hepatocytes preferentially contribute to liver homeostasis and regeneration. Cell Stem Cell 30, 665–676.e664 (2023).
Chembazhi, U. V., Bangru, S., Hernaez, M. & Kalsotra, A. Cellular plasticity balances the metabolic and proliferation dynamics of a regenerating liver. Genome Res. 31, 576–591 (2021).
Tian, W. & Xu, Y. Decoding liver injury: a regulatory role for histone modifications. Int. J. Biochem. Cell Biol. 67, 188–193 (2015).
Chen, Y. & Verfaillie, C. M. MicroRNAs: the fine modulators of liver development and function. Liver Int. 34, 976–990 (2014).
Liu, M. & Chen, P. Proliferation‑inhibiting pathways in liver regeneration (Review). Mol. Med. Rep. 16, 23–35 (2017).
Wang, S. et al. The emerging importance role of m6A modification in liver disease. Biomed. Pharmacother. 162, 114669 (2023).
Kren, B. T. & Steer, C. J. Posttranscriptional regulation of gene expression in liver regeneration: role of mRNA stability. FASEB J. 10, 559–573 (1996).
Berasain, C., Arechederra, M., Argemi, J., Fernandez-Barrena, M. G. & Avila, M. A. Loss of liver function in chronic liver disease: an identity crisis. J. Hepatol. 78, 401–414 (2023).
Bangru, S. et al. ESRP2-microRNA-122 axis promotes the postnatal onset of liver polyploidization and maturation. Genes Dev. 39, 325–347 (2025).
Arif, W., Datar, G. & Kalsotra, A. Intersections of post-transcriptional gene regulatory mechanisms with intermediary metabolism. Biochim. Biophys. Acta Gene Regul. Mech. 1860, 349–362 (2017).
Adesanya, O., Das, D. & Kalsotra, A. Emerging roles of RNA-binding proteins in fatty liver disease. Wiley Interdiscip. Rev. RNA 15, e1840 (2024).
Sen, S., Jumaa, H. & Webster, N. J. Splicing factor SRSF3 is crucial for hepatocyte differentiation and metabolic function. Nat. Commun. 4, 1336 (2013).
Elizalde, M. et al. Splicing regulator SLU7 is essential for maintaining liver homeostasis. J. Clin. Invest. 124, 2909–2920 (2014).
Bhate, A. et al. ESRP2 controls an adult splicing programme in hepatocytes to support postnatal liver maturation. Nat. Commun. 6, 8768 (2015).
Wang, A. W. et al. TRAP-seq identifies cystine/glutamate antiporter as a driver of recovery from liver injury. J. Clin. Invest. 128, 2297–2309 (2018).
Bangru, S. et al. Alternative splicing rewires Hippo signaling pathway in hepatocytes to promote liver regeneration. Nat. Struct. Mol. Biol. 25, 928–939 (2018).
Garate-Rascon, M. et al. Splicing factor SLU7 prevents oxidative stress-mediated hepatocyte nuclear factor 4alpha degradation, preserving hepatic differentiation and protecting from liver damage. Hepatology 74, 2791–2807 (2021).
Kumar, D. et al. Degradation of splicing factor SRSF3 contributes to progressive liver disease. J. Clin. Invest. 129, 4477–4491 (2019).
Arif, W. et al. Splicing factor SRSF1 deficiency in the liver triggers NASH-like pathology and cell death. Nat. Commun. 14, 551 (2023).
Matchett, K. P. et al. Multimodal decoding of human liver regeneration. Nature 630, 158–165 (2024).
Gribben, C. et al. Acquisition of epithelial plasticity in human chronic liver disease. Nature 630, 166–173 (2024).
Xu, J. et al. A spatiotemporal atlas of mouse liver homeostasis and regeneration. Nat. Genet. 56, 953–969 (2024).
Guilliams, M. et al. Spatial proteogenomics reveals distinct and evolutionarily conserved hepatic macrophage niches. Cell 185, 379–396.e338 (2022).
Andrews, T. S. et al. Single-cell, single-nucleus, and spatial RNA sequencing of the human liver identifies cholangiocyte and mesenchymal heterogeneity. Hepatol. Commun. 6, 821–840 (2022).
Gupta, A. et al. Characterization of transcript enrichment and detection bias in single-nucleus RNA-seq for mapping of distinct human adipocyte lineages. Genome Res. 32, 242–257 (2022).
Fischer, J. & Ayers, T. Single nucleus RNA-sequencing: how it’s done, applications and limitations. Emerg. Top. Life Sci. 5, 687–690 (2021).
Santiago, C. P. et al. Comparative analysis of single-cell and single-nucleus RNA-sequencing in a rabbit model of retinal detachment-related proliferative vitreoretinopathy. Ophthalmol. Sci. 3, 100335 (2023).
MacParland, S. A. et al. Single cell RNA sequencing of human liver reveals distinct intrahepatic macrophage populations. Nat. Commun. 9, 4383 (2018).
Horras, C. J., Lamb, C. L. & Mitchell, K. A. Regulation of hepatocyte fate by interferon-gamma. Cytokine Growth Factor Rev. 22, 35–43 (2011).
Gao, B., Ahmad, M. F., Nagy, L. E. & Tsukamoto, H. Inflammatory pathways in alcoholic steatohepatitis. J. Hepatol. 70, 249–259 (2019).
Ma, J. et al. Distinct histopathological phenotypes of severe alcoholic hepatitis suggest different mechanisms driving liver injury and failure. J. Clin. Invest. 132 https://doi.org/10.1172/JCI157780 (2022).
Khan, R. S., Lalor, P. F., Thursz, M. & Newsome, P. N. The role of neutrophils in alcohol-related hepatitis. J. Hepatol. 79, 1037–1048 (2023).
Cho, Y. et al. Neutrophil extracellular traps contribute to liver damage and increase defective low-density neutrophils in alcohol-associated hepatitis. J. Hepatol. 78, 28–44 (2023).
Nagesh, P. T. et al. In vivo Bruton’s tyrosine kinase inhibition attenuates alcohol-associated liver disease by regulating CD84-mediated granulopoiesis. Sci. Transl. Med 16, eadg1915 (2024).
Cho, Y. & Szabo, G. Two faces of neutrophils in liver disease development and progression. Hepatology 74, 503–512 (2021).
Hyun, J. et al. Epithelial splicing regulatory protein 2-mediated alternative splicing reprograms hepatocytes in severe alcoholic hepatitis. J. Clin. Invest. 130, 2129–2145 (2020).
Sancho-Bru, P. et al. Liver progenitor cell markers correlate with liver damage and predict short-term mortality in patients with alcoholic hepatitis. Hepatology 55, 1931–1941 (2012).
Wesley, B. T. et al. Single-cell atlas of human liver development reveals pathways directing hepatic cell fates. Nat. Cell Biol. 24, 1487–1498 (2022).
Denechaud, P. D. et al. E2F1 mediates sustained lipogenesis and contributes to hepatic steatosis. J. Clin. Invest. 126, 137–150 (2016).
Kiani, K., Sanford, E. M., Goyal, Y. & Raj, A. Changes in chromatin accessibility are not concordant with transcriptional changes for single-factor perturbations. Mol. Syst. Biol. 18, e10979 (2022).
John, S. et al. Chromatin accessibility pre-determines glucocorticoid receptor binding patterns. Nat. Genet. 43, 264–268 (2011).
Biddie, S. C. et al. Transcription factor AP1 potentiates chromatin accessibility and glucocorticoid receptor binding. Mol. Cell 43, 145–155 (2011).
Samstein, R. M. et al. Foxp3 exploits a pre-existent enhancer landscape for regulatory T cell lineage specification. Cell 151, 153–166 (2012).
Wheeler, J. R. et al. RNA-binding proteins direct myogenic cell fate decisions. Elife 11 https://doi.org/10.7554/eLife.75844 (2022).
Parra, A. S. & Johnston, C. A. Emerging roles of RNA-binding proteins in neurodevelopment. J. Dev. Biol. 10 https://doi.org/10.3390/jdb10020023 (2022).
Tao, Y., Zhang, Q., Wang, H., Yang, X. & Mu, H. Alternative splicing and related RNA binding proteins in human health and disease. Signal Transduct. Target Ther. 9, 26 (2024).
Derham, J. M. & Kalsotra, A. The discovery, function, and regulation of epithelial splicing regulatory proteins (ESRP) 1 and 2. Biochem. Soc. Trans. 51, 1097–1109 (2023).
Bangru, S. & Kalsotra, A. Cellular and molecular basis of liver regeneration. Semin. Cell Dev. Biol. 100, 74–87 (2020).
Dewald, Z. et al. Altered drug metabolism and increased susceptibility to fatty liver disease in a mouse model of myotonic dystrophy. Nat. Commun. 15, 9062 (2024).
Tranchevent, L. C. et al. Identification of protein features encoded by alternative exons using Exon Ontology. Genome Res. 27, 1087–1097 (2017).
Al-Zahrani, K. N., Baron, K. D. & Sabourin, L. A. Ste20-like kinase SLK, at the crossroads: a matter of life and death. Cell Adh Migr. 7, 1–10 (2013).
Garland, B., Delisle, S., Al-Zahrani, K. N., Pryce, B. R. & Sabourin, L. A. The Ste20-like kinase - a Jack of all trades? J. Cell Sci. 134 https://doi.org/10.1242/jcs.258269 (2021).
Qian, Y. W., Erikson, E. & Maller, J. L. Purification and cloning of a protein kinase that phosphorylates and activates the polo-like kinase Plx1. Science 282, 1701–1704 (1998).
Thompson, M. D. & Monga, S. P. WNT/beta-catenin signaling in liver health and disease. Hepatology 45, 1298–1305 (2007).
Boj, S. F. et al. Diabetes risk gene and Wnt effector Tcf7l2/TCF4 controls hepatic response to perinatal and adult metabolic demand. Cell 151, 1595–1607 (2012).
Ip, W., Shao, W., Chiang, Y. T. & Jin, T. The Wnt signaling pathway effector TCF7L2 is upregulated by insulin and represses hepatic gluconeogenesis. Am. J. Physiol. Endocrinol. Metab. 303, E1166–E1176 (2012).
Schep, A. N., Wu, B., Buenrostro, J. D. & Greenleaf, W. J. chromVAR: inferring transcription-factor-associated accessibility from single-cell epigenomic data. Nat. Methods 14, 975–978 (2017).
Browaeys, R., Saelens, W. & Saeys, Y. NicheNet: modeling intercellular communication by linking ligands to target genes. Nat. Methods 17, 159–162 (2020).
Zabulica, M. et al. Guide to the assessment of mature liver gene expression in stem cell-derived hepatocytes. Stem Cells Dev. 28, 907–919 (2019).
Warzecha, C. C., Sato, T. K., Nabet, B., Hogenesch, J. B. & Carstens, R. P. ESRP1 and ESRP2 are epithelial cell-type-specific regulators of FGFR2 splicing. Mol. Cell 33, 591–601 (2009).
Horiguchi, K. et al. TGF-beta drives epithelial-mesenchymal transition through deltaEF1-mediated downregulation of ESRP. Oncogene 31, 3190–3201 (2012).
Wu, J. C., Merlino, G. & Fausto, N. Establishment and characterization of differentiated, nontransformed hepatocyte cell lines derived from mice transgenic for transforming growth factor alpha. Proc. Natl. Acad. Sci. USA 91, 674–678 (1994).
Sefried, S., Haring, H. U., Weigert, C. & Eckstein, S. S. Suitability of hepatocyte cell lines HepG2, AML12 and THLE-2 for investigation of insulin signalling and hepatokine gene expression. Open Biol. 8 https://doi.org/10.1098/rsob.180147 (2018).
Argemi, J. et al. Defective HNF4alpha-dependent gene expression as a driver of hepatocellular failure in alcoholic hepatitis. Nat. Commun. 10, 3126 (2019).
Sommerauer, C. & Kutter, C. Noncoding RNAs and RNA-binding proteins: emerging governors of liver physiology and metabolic diseases. Am. J. Physiol. Cell Physiol. 323, C1003–C1017 (2022).
Wang, X., He, Y., Mackowiak, B. & Gao, B. MicroRNAs as regulators, biomarkers and therapeutic targets in liver diseases. Gut 70, 784–795 (2021).
Jouve, M., Carpentier, R., Kraiem, S., Legrand, N. & Sobolewski, C. MiRNAs in alcohol-related liver diseases and hepatocellular carcinoma: a step toward new therapeutic approaches? Cancers 15 https://doi.org/10.3390/cancers15235557 (2023).
Iliaki, S., Beyaert, R. & Afonina, I. S. Polo-like kinase 1 (PLK1) signaling in cancer and beyond. Biochem. Pharm. 193, 114747 (2021).
Oliva-Vilarnau, N. et al. Wnt/beta-catenin and NFkappaB signaling synergize to trigger growth factor-free regeneration of adult primary human hepatocytes. Hepatology 79, 1337–1351 (2024).
He, L. et al. Proliferation tracing reveals regional hepatocyte generation in liver homeostasis and repair. Science 371 https://doi.org/10.1126/science.abc4346 (2021).
Crabb, D. W., Im, G. Y., Szabo, G., Mellinger, J. L. & Lucey, M. R. Diagnosis and treatment of alcohol-associated liver diseases: 2019 practice guidance from the American Association for the Study of Liver Diseases. Hepatology 71, 306–333 (2020).
Crabb, D. W. et al. Standard definitions and common data elements for clinical trials in patients with alcoholic hepatitis: recommendation from the NIAAA alcoholic hepatitis consortia. Gastroenterology 150, 785–790 (2016).
Bertola, A., Mathews, S., Ki, S. H., Wang, H. & Gao, B. Mouse model of chronic and binge ethanol feeding (the NIAAA model). Nat. Protoc. 8, 627–637 (2013).
Corces, M. R. et al. An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nat. Methods 14, 959–962 (2017).
Korsunsky, I. et al. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Methods 16, 1289–1296 (2019).
Hao, Y. et al. Integrated analysis of multimodal single-cell data. Cell 184, 3573–3587.e3529 (2021).
Stuart, T. et al. Comprehensive integration of single-cell data. Cell 177, 1888–1902.e1821 (2019).
Stuart, T., Srivastava, A., Madad, S., Lareau, C. A. & Satija, R. Single-cell chromatin state analysis with Signac. Nat. Methods 18, 1333–1341 (2021).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
Trapnell, C. et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 7, 562–578 (2012).
Shen, S. et al. rMATS: robust and flexible detection of differential alternative splicing from replicate RNA-Seq data. Proc. Natl. Acad. Sci. USA 111, E5593–E5601 (2014).
Van Nostrand, E. L. et al. Robust transcriptome-wide discovery of RNA-binding protein binding sites with enhanced CLIP (eCLIP). Nat. Methods 13, 508–514 (2016).
Shah, A., Qian, Y., Weyn-Vanhentenryck, S. M. & Zhang, C. CLIP Tool Kit (CTK): a flexible and robust pipeline to analyze CLIP sequencing data. Bioinformatics 33, 566–567 (2017).
Zhang, Z. et al. Synthetic DNA barcodes identify singlets in scRNA-seq datasets and evaluate doublet algorithms. Cell Genom. 4, 100592 (2024).
Goyal, Y. et al. Diverse clonal fates emerge upon drug treatment of homogeneous cancer cells. Nature 620, 651–659 (2023).
Acknowledgements
This work was supported by the NIH grants R01-AA010154, R01-HL126845, and R21-HD104039 (to A.K.); the NIH grants R01-AA010154, 5R01-DK077794, 1R56-DK1343340 (to A.M.D.); and the NIH R24 AA025017 (to Z.S.), the Duke Endowment (to A.M.D.), Chan-Zuckerberg Biohub Chicago Award to (A.K. and Y.G.) and Muscular Dystrophy Association Research Grant MDA1072487 (to A.K.), the Herbert E. Carter fellowship in Biochemistry (to U.V.C.), the Michael A. Recny fellowship in Biochemistry (to S.N.), Charles F. Kade fellowship in Biochemistry (to D.D.) the NIH Tissue microenvironment training program T32-EB019944 and UIUC Scott Dissertation Completion Fellowship (to S.B.). Y.G. also acknowledges support from the CFAR subproject on an NIH award (P30AI117943). We acknowledge support from the Transgenic Mouse Core, High-throughput Sequencing and Genotyping Core, and Histology-Microscopy Core facilities at the University of Illinois, Urbana-Champaign. Schematics were created with BioRender as cited in the figure captions.
Author information
Authors and Affiliations
Contributions
U.V.C. and S.B. designed the research studies, conducted experiments, acquired data, performed computational analysis, analyzed data, and wrote and revised the manuscript; R.D., D.D., S.N., K.T., A.B., and I.P. conducted experiments and acquired data; Z.S. and B.P. provided human liver samples and bulk RNA-seq data; A.L. and Y.G. performed computational analysis; A.K. and A.M.D. designed the research studies, analyzed data, wrote and revised the manuscript, secured funding for the research. All authors discussed the results and edited the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Nicholas Webster and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Chembazhi, U.V., Bangru, S., Dutta, R.K. et al. Dysregulated RNA splicing impairs regeneration in alcohol-associated liver disease. Nat Commun 16, 8049 (2025). https://doi.org/10.1038/s41467-025-63251-2
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-63251-2
