
Abstract
Prematurity, defined as birth before 37 weeks of gestation, is the leading cause of mortality in children under five, affecting ~11% of live births worldwide (≈15 million annually). Despite advances in neonatal care, preterm infants remain at high risk of complications. In neonatal intensive care units, gastric residuals (GRs) are routinely monitored to guide enteral feeding, yet their microbial composition remains poorly understood. We performed metagenomic sequencing of 199 stool and 69 GR samples from 39 preterm infants during hospitalization to characterize stomach and gut microbiomes. To our knowledge, this is the first metagenomic sequencing of the GR in premature infants. We identified 11 GR microbial clusters, commonly dominated by Staphylococcus, Streptococcus, and Klebsiella, with microbial diversity correlating with aspiration frequency. Colonization was dynamic: early GR samples were enriched with Staphylococcus epidermidis and Bradyrhizobium, while later samples featured Escherichia coli, Staphylococcus hominis, and Streptococcus thermophilus. Stool samples formed eight microbial clusters, frequently enriched with Enterobacteriaceae. S. epidermidis was linked to higher gestational age and lower richness, whereas Bifidobacterium breve, a beneficial commensal, appeared later. Comparative analysis showed overlap between gut and gastric microbiota, with GR samples more dynamic and less subject-specific. Strain-level analysis revealed both individual-specific and widely shared taxa, including a pathogenic Klebsiella aerogenes strain associated with bacteremia, detectable a week before clinical isolation. These findings provide new insights into microbial colonization dynamics of preterm infants.
Similar content being viewed by others


Introduction
The gastrointestinal tract is heavily colonized by trillions of microorganisms known as the gut microbiota, which critically support host physiology, healthy development, and function1,2,3,4. Inoculation of the gut microbiome of newborns begins at birth, and the neonatal gut environment and maturity impact its composition5. Establishment of the gut barrier leads to a shift in microbial composition from the facultative anaerobes (such as Enterobactericea and Enterococci) to strict anaerobes (such as Bacteroides, Bifidobacterium, and Clostridium), reflecting an adaptation to the decreasing oxygen levels within the gut lumen6,7. Changes in the composition of the gut microbiota (dysbiosis) have been associated with a wide array of diseases, including inflammatory, autoimmune, neurological, and metabolic disorders8,9,10,11,12,13. Intriguingly, gut microbial dysbiosis during neonatal development is linked to disease development later in life, such as allergy, asthma, and autoimmune disorders14,15,16. Furthermore, bacteria residing in an infant’s gut are pivotal in allowing the infant to maximize absorption from their diet17. One of the bacteria that has been shown to benefit infants and be associated with a healthy infant gut microbial environment is Bifidobacterium18,19,20,21,22,23,24. Infants hospitalized in the neonatal intensive care unit (NICU) are not provided with various milk formulations25 and probiotics19,21, containing Bifidobacterium, despite its considerable impact on shaping the gut microbiome, due to health ministry policy19,26,27.
Preterm birth affects approximately 15 million infants annually, representing approximately 11% of live births worldwide, with incidence rates continuing to rise28. Although advances in neonatology have enhanced survival rates among the most premature and critically ill infants, these populations remain at increased risk for a broad spectrum of long-term adverse health outcomes28. Neonatal morbidities of preterm infants include respiratory morbidities such as respiratory distress syndrome, later leading to bronchopulmonary dysplasia, neurological morbidity including intraventricular hemorrhage and periventricular leukomalacia, retinopathy of prematurity, early and late onset sepsis, necrotizing enterocolitis, and more29.
Necrotizing enterocolitis (NEC) is the leading gastrointestinal cause of morbidity and mortality in neonatal intensive care units (NICUs), primarily affecting very-low-birth-weight (VLBW) premature infants (birth weight <1500 g), with a prevalence of 5%–10% and a mortality rate of 25%–30%30. NEC is a result of a multifactorial process that includes reduced blood flow to an immature gut (including disruptions to early immunological maturation)31,32. This may lead to increased gut permeability33,34. Changes to the gut wall that follow bacterial dysbiosis allow for the translocation of bacteria from the gut lumen into the bloodstream, potentially causing bacteremia and sepsis35,36,37. Prevention of life-threatening conditions in preterm infants often incorporates multiple clinical interventions, including prolonged intensive care monitoring and antibiotic administration, which can alter the developing microbiome.
Repetitive examination of gastric residuals (GR), collected by aspirating the gastric content of preterm infants, is a common practice performed in the NICU of Hadassah Medical Center that helps assess infant health status and guides the initiation and progression of enteral feeding in these infants38,39. Gastric residue monitoring aims to use abnormalities or increases in gastric residual volumes as early predictors of feeding intolerance, potentially reflecting NEC40,41,42. However, recent studies suggest that in the absence of clinical signs of feeding intolerance, avoiding pre-feeding monitoring of gastric residues is safe, and does not increase the risk for NEC. Furthermore, there have been reports that routine monitoring of GR does not improve clinical outcomes and may delay advancement of enteral nutrition and weight gain43,44,45,46.
Anatomically, the stomach and gut are interconnected segments within the seamless continuum of the gastrointestinal tract, presenting different settings for microbial colonization owing to the contrasting environmental conditions, such as gastric acidity and motility. Bacteria may overcome these environmental dissimilarities and utilize the common tract connecting the two organs to translocate from one niche to another. A study using 16S sequencing to profile the gastric microbiome reported the presence of Bacteroides spp. and Bifidobacterium spp. within this niche in premature infants47. To date, the gastric microbiome of premature infants remains largely unexplored, particularly in terms of comprehensive characterization using metagenomic sequencing approaches.
To expand the knowledge of the GR and stool microbiome development in premature infants, we performed metagenomic sequencing of 69 GR samples and 199 samples from 39 subjects and profiled their microbial composition throughout their postnatal hospitalization. We identified composition-derived clusters within the samples and carried out comparisons involving both microbial and clinical metrics. Our results support and highlight some of the gut microbiome developmental changes occurring throughout its maturation process, such as the transition from Staphylococcus epidermidis-dominated communities and the chronological emergence of Bifidobacterium-dominated bacterial populations. Among the gastric microbiome, we found co-occurrence of bacteria also present across stool samples, including members of the Klebsiella and Staphylococcus genera. Lastly, using sequenced data, we identified a strain of Klebsiella aerogenes, ultimately associated with bacteremia a week before its initial detection within a blood sample, highlighting the potential of sequencing methods to provide early detection of the presence of pathogenic strains, preceding future systemic infections.
Results
Study design
Premature infants, born before completing 37 weeks of gestation, are initially colonized with microbes within the first days of life48,49,50,51,52. These acquired microbial populations, consisting of bacteria, fungi, and viruses, play a pivotal role in the infant’s development and have been associated with various health conditions53,54,55. During their postnatal hospitalization, infants frequently undergo a wide spectrum of medical interventions and tests that have the potential to alter the microbial profile, such as antibiotic treatment56,57,58. Observing the establishment and dynamics of the microbiome in preterm infants, in correlation with their respective clinical status, has the potential to help understand the relationship between the host microbiome and health. An interesting clinical practice carried out in some neonatal intensive care units (NICUs) is the monitoring of gastric residual (GR) samples, which consists of aspiration of stomach content before feeding, as well as in cases of clinical indications40. To the best of our knowledge, this is the first study to characterize the microbial communities of the gastric residuals in premature infants using metagenomic sequencing methods.
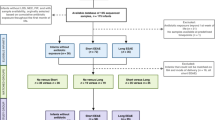
To pursue these challenges, we collaborated with the NICU of Hadassah Ein Kerem Medical Center, and collected samples from premature infants during their post-delivery hospitalization (Fig. 1). A total of 199 stool and 69 GR samples were collected from 39 premature infants (median gestational age 29 weeks, mean gestational age 29 weeks + 1 day) and sequenced using metagenomic sequencing (6,273,587 mean read coverage). Subjects were randomized into a high or low frequency gastric residuals collection group consisting of 19 and 20 participants, respectively (Methods). While all subjects underwent gastric residuals aspiration upon clinical indication, gastric residuals were routinely aspirated prior to feeding only in infants within the high-frequency group. Necrotizing enterocolitis (NEC) occurred in five infants (3 and 2 subjects from the low and high-frequency groups, respectively). Five premature infants, some of whom developed NEC, died during their hospitalization period (also, 3 and 2 cases from the low and high-frequency study groups, respectively, Fig. 1). Out of five cases of NEC, two (40%) were diagnosed before ten days of life, and in both incidences, a positive blood culture was obtained a day prior to the diagnosis, suggesting a NEC-associated sepsis rate comparable to those reported elsewhere in premature infants59,60,61. Averages of clinical metrics across the study groups were similar, including length of hospitalization (60.2 days vs. 50.2 days; p value = 0.28), birth weight z-score (0.049 vs. −0.548; p value = 0.059), delta z-score of discharge weight (1.44 vs. 1.15; p value = 0.24), gestational age (28w + 5 d vs. 29w + 4 d; p value = 0.35), and birthweight (1170 grams vs. 1130 grams; p value = 0.76; Table 1). Antibiotic exposure was common across premature infants throughout their hospitalization in the NICU. Ampicillin, Amikacin, and Vancomycin were the most commonly administered antibiotics within our cohort, across both study groups. Antibiotic treatment rates were high in both the high and low-frequency study groups (73.68% vs. 90%, respectively, p value = 0.363 Chi-Squared test). Mean length of antibiotic courses was similar between the high and low-frequency study groups (3.06 days vs. 3.31 days; p value = 0.54 two-sided t-test).

Subjects (y-axis) were divided into two groups (high- and low-frequency residual collection, top and bottom, respectively), lines represent the hospitalization period, across gestational age (x-axis), and dots represent sequenced stool (brown) and gastric-residual (green) samples. The line type represents the clinical outcome of each infant.
Gastric residuals microbiome
To examine the various patterns of the gastric residual (GR) microbiome of preterm infants, we performed metagenomic sequencing on 69 samples from 30 preterm infants. In general, the GR samples often had low-diversity microbial communities, dominated by a single/few bacterial species (Fig. 2A), enabling a clean clustering into 11 distinct clusters (Fig. 2A, B; using partitioning around means (PAM) algorithm, with Bray-Curtis dissimilarity; Methods). Each of the 11 clusters can be characterized by its dominant representative bacteria, commonly from the Klebsiella, Streptococcus, and Staphylococcus genera (Fig. 2A), known inhabitants of the gut and skin microbial populations62,63,64,65,66,67,68,69,70. The cluster assignment was done irrespective of the GR collection frequency, and we found most clusters to contain samples from both frequencies (Fig. 2C).

A Microbial profiles of all GR samples. Each bar represents a single sample, and samples are grouped according to their assigned cluster. Bars are colored based on the common taxa (Others appear in gray). GR collection frequency for the sample subject is represented in a dot under bars. B, C Principal coordinate analysis (PCoA) of GR samples. Each sample is represented as a dot on the plot and is colored according to its assigned cluster (B) or the GR sampling frequency of the infant (C). D, E Comparison of community evenness (Shannon index) of GR samples across clusters and GR collection frequency, respectively. Boxes show median and represent the 25% and 75% quantiles, whiskers extend to 1.5 times the interquartile range from box edges. p-value was calculated using a two-sided t-test. Each dot represents a sample, and the number of samples within each cluster is noted under each box. F Dynamics of early life GR stability. Lines connecting between the first (left) and last (right) sample represent the trajectory of GR microbial composition of subjects throughout hospitalization. The width of the lines connecting the columns is proportional to the number of infants sharing this trajectory (coloring as in (A)). Clusters appear as the y-axis and are colored according to the representative bacteria.
Next, we explored the bacterial richness of GR samples, and as the stomach is known for its acidity, we a priori expected to find bacteria that thrive in low-pH conditions, such as Lactobacilli or Helicobacter, which are commonly found in acidic niches such as the vagina, and mature gastric microbiome, respectively71,72,73,74,75. In theory, the acidic environment in the stomach could present challenging conditions for bacterial colonization, permitting only a few shared colonization-eligible bacteria, thus resulting in a likely low bacterial richness. To our surprise, Lactobacillus species were not abundant in any of the GR samples (Fig. 2A). Most GR samples had low-diversity communities, as reflected by Shannon index (Fig. 2D). When considering individual clusters, we found that clusters RC4, RC5, and RC7, which were dominated by Streptococcus mitis, Staphylococcus hominis, and Klebsiella aerogenes, respectively, had the richest communities (Fig. 2D). With respect to GR collection frequency, we found that higher collection frequency tended to have a richer GR community, potentially due to constant perturbation of the biological niche, yet this association was not statistically significant (Fig. 2E).
Finally, we assessed the stability of the GR microbiome during the hospitalization period of the infants in the NICU. To understand the intra-subject variance of this developing microbial niche, we selected the first and last GR samples collected per infant and compared the assigned microbial cluster at the beginning and end of hospitalization. We found that clusters RC2, RC5, and RC8, which were rich in Escherichia coli, Staphylococcus hominis, and Streptococcus thermophilus, respectively, were completely missing in the first GR samples (Fig. 2E). In contrast, clusters RC1 and RC3, which were rich in Bradyrhizobium and Staphylococcus epidermidis, respectively, were completely missing in the last GR samples (Fig. 2E). Taken together, these dynamics could suggest an age-dependent development in the GR environment where some early colonizers do not sustain in the developing stomach. Additionally, some gastric bacteria may lack the ability to efficiently thrive in a young GR niche while retaining the capacity to colonize it later on in early life.
Overview of the gut microbiome of preterm infants
First, to get an overview of the premature infant gut microbiome and its development across the hospitalization period (mean 55.3 days, median 55 days), we performed metagenomic sequencing and profiled the microbiome of 132 stool samples from 39 subjects (Methods; Fig. 3A). Overall, the premature infants’ gut microbiome maintained low bacterial diversity, naturally falling into eight distinct clusters (generated using partitioning around medioids (PAM) analysis with Bray-Curtis dissimilarity; Methods, Fig. 3B). Each cluster was characterized by a dominant representative bacteria, mainly from the Enterobacteriaceae family, such as Escherichia coli and Klebsiella spp (Fig. 3A). As expected, some of the cluster-representative taxa were previously reported as popular colonizers, and potentially pathobionts, of the premature infant gut, such as members of the Staphylococcus, Klebsiella, Enterococcus and Escherichia genera52,76,77,78,79,80,81. We found the clustering classification to be independent of the mode of delivery, age at sampling, and the frequency of gastric residuals sampling (Supplementary Fig. 1A, B, C). Interestingly, while probiotics are not administered in this NICU, we found Bifidobacterium species in 17 premature infants, which is considered a measure of overall health in early life18,21,22,24 (Supplementary Fig. 2).

A Microbial profiles of 132 stool samples. Each bar represents a single sample, and samples are grouped according to their assigned cluster. Bars are colored based on the taxa. B Principal coordinate analysis (PCoA) of stool samples. Each sample is represented as a dot on the plot and is colored according to its assigned cluster. C Comparison of the community evenness (Shannon index) of stool samples between clusters. Boxes show the median and represent the 25% and 75% quantiles, whiskers extend to 1.5 times the interquartile range from box edges. Each dot represents a sample, and the number of samples within each cluster is noted under each box. p-value was calculated using a two-sided Kruskal–Wallis test. D, E Comparison of the birthweight and gestational age at birth, respectively, between clusters of subjects’ first samples. For each infant, the first sample was used to categorize the infant to a specific cluster. Box statistics as in (C) F Dynamics of early life stool microbiome stability. Lines connecting between the first (left) and last (right) samples represent the trajectory of stool microbial composition of subjects throughout hospitalization. The width of the lines connecting the columns is proportional to the number of infants sharing this trajectory. Clusters appear as the y-axis and are colored according to the representative bacteria.
Our observation that various bacteria can excel and thrive in the premature infant’s gut raises the question of whether different dominant taxa allow for different microbial community dynamics to develop. To compare the different microbial clusters in terms of community evenness and richness, we calculated the Shannon and the Chao1 indices, respectively. We found that while most of the clusters had similar in richness and evennes metrics, SC6, dominated by Staphylococcus epidermidis, maintained a markedly decrease in both evenness and species richness (Fig. 3C and Supplementary Fig. 3). These findings suggest a colonization of the immature gut environment by S.epidermidis resulting in an uneven and low diversity microbial community. Additionally, these findings may hint that S. epidermidis, when dominating a niche, challenges other bacteria seeking to thrive alongside it.
Earliest microbiome profile and its clinical associations
Next, we sought to understand if the assigned microbial cluster can provide insightful information regarding the infant’s health status. To evaluate a possible association between the microbial profile and the subject’s anthropometric measurements, we decided to focus on the earliest time point for each infant, collected within the first two weeks of life. We then examined the association of the assigned cluster of the first stool sample with birthweight and gestational age at birth (Fig. 3D, E). We found that infants in SC6, dominated by Staphylococcus epidermidis, had an overall advanced gestational age at birth with a relatively low birthweight (small for gestational age - SGA). In contrast, infants in SC1, dominated by Escherichia coli, had the highest median gestational age and weight at birth. Our results highlight a possible association between S. epidermidis-dominated stool samples in early life and birth weight that is inappropriate for their age. To check whether mode of delivery was a confounding factor in our results, we validated that birthweight and gestational age at birth were not associated with mode of delivery (Supplementary Fig. 4).
Microbiome dynamics during hospitalizations
Premature infants are often hospitalized for a considerable period following birth, allowing us to examine the dynamics of their microbiome during hospitalization. To examine whether the gut microbiome converges towards a consensus community, we compared the microbial composition of the first and last sample of each infant, highlighting shifts in microbial cluster assignments throughout the hospitalization period (Fig. 3F). Surprisingly, we found that none of the infants had their last stool sample assigned the S. epidermidis-rich SC6 despite it being a relatively prevalent cluster among the first stool samples. Notably we found SC3, a cluster rich in Bifidobacterium breve, to be represented across the last stool samples despite being absent in the first samples (Supplementary Fig. 2). Overall, when examining additional time points throughout the hospitalization, we observed that samples annotated as SC6 tended to be collected at earlier time points, whereas samples assigned to SC3 were collected at later stages (Supplementary Fig. 5).
Shared microbial strains colonize both gastric and gut environments
First, we compared the overall microbial composition in the two environments to assess the impact of the different niche settings on their residing microbiome. Using a principal coordinate analysis with the Bray-Curtis dissimilarity metric, we found no significant separation between the two sample types, implying the presence of similar bacteria across the two niches (Fig. 4A). Specifically, we identified Staphylococcus epidermidis, Escherichia coli, and Klebsiella pneumoniae to maintain a pivotal role in explaining the variability across these samples (Fig. 4B–D).

Principal coordinate analysis (PCoA) of stool and GR samples, colored by sample type (A) and S.epidermidis (B), K.pneumoniae (C), and E.coli (D) relative abundance. Each sample is represented as a dot on the plot. E Comparison by sample type of average microbial dissimilarity across two consecutive samples. Each dot represents the estimated subject-specific average daily Bray-Curtis dissimilarity derived from pairs of consecutive samples, divided by the number of days between them. Measurements (dots) from the same infant are connected with a dashed line. p-value was calculated using a two-sided paired Wilcoxon test (n = 15 sample pairs). Boxes show the median and represent the 25% and 75% quantiles, whiskers extend to 1.5 times the interquartile range from box edges. F, G, H Strain-level analysis of key taxa across all stool (n = 199) and GR (n = 69) samples. Phylogenetic trees of S.epidermidis (F), K.pneumoniae (G), and E.coli (H). Each leaf (dots) represents a single sample. The phylogenetic distance between dominant strains of two samples is the sum of the distances on the x-axis (branches of the tree). Samples are colored by the subject from which the sample was taken, and shaped according to the sample type.
We next examined whether the GR microbiome is less or more stable than the gut microbiome. For each subject, we compared the compositional dissimilarity of consecutive GR and consecutive stool samples, using the Bray-Curtis dissimilarity index. Specifically, we looked at consecutive samples that were collected within 3 weeks of each other. Our comparison was conducted across 36 and 78 pairs of GR and stool samples, respectively. We found consecutive GR samples to be less similar to each other compared to stool samples, indicating that the gut microbiome is more stable than the gastric microbial community (Fig. 4E, p value = 0.048, paired Wilcoxon).
Lastly, we turned to evaluate the strain similarity within and across body niches. We compared the dominant strain of common species across all stool (n = 199) and gastric residual samples, focusing on the eight representative taxa of the stool clusters (Fig. 4F–H and Supplementary Fig. 6A–D). We found that most of these key bacteria, including Escherichia coli, Klebsiella pneumoniae, Enterococcus faecalis, and Veillonella parvula, maintained a relatively constant dominant strain in infants throughout their hospitalization (Fig. 4G–H and Supplementary Fig. 6A–D). In contrast, in some cases, like for Staphylococcus epidermidis, similar strains were found across multiple subjects, thus suggesting an environmental mode of acquisition (Fig. 4F).
Early metagenomic detection of pathogenic Klebsiella aerogenes strain
Finally, we focused on a single premature infant (PRE.021) who developed Klebsiella aerogenes sepsis, ultimately resulting in death. When examining the strains of K. aerogenes, another key bacterium in the developing preterm gut microbiome, we found that although similar strains were shared across several subjects, a completely distinct strain was present in PRE.021 (Fig. 5A).

A Phylogenetic tree of K.aerogenes. Each leaf (dots) represents a single sample. The phylogenetic distance between dominant strains of two samples is the sum of the distances on the x-axis (branches of the tree). Samples are colored by the subject from which the sample was taken, and shaped according to the sample type. B Comparison of the genomic content of the dominant K.aerogenes strain across different samples and clinical isolates. Rows represent the dominant K.aerogenes strain of a single sample or clinical isolate and columns represent Uniref90 entries. Uniref90 entries that are either fully-shared or fully-absent across all samples are not shown.
To explore the differences underlying the phylogenetic separation between the dominant K. aerogenes strain in PRE.021 and the strains present in other individuals, we carried out a genomic content comparison analysis using PanPhlAn82, highlighting 377 and 199 Uniref90 and UniProt entries, respectively, that are found exclusively within PRE.021 and completely missing from strains of other subjects (Fig. 5B). We then compared these entries against 12 different K. aerogenes reference genomes, using BioCyc83, and found 11 genes that were present in the PRE.021 strain and were missing from at least 9 of the 12 reference genomes. These include genes associated with: bacterial replication (such as dnaA, dnaC), SoxR regulatory genes (rsxB), catalyzing enzymes (rihC, rfbA, ligB, hrpB), iron-sulfur complexes (iscX), nitrogen transport (ptsN), and multidrug resistance proteins (mdtB). Specifically, hpaR (transcription regulator) and fabG (fatty acid elongation) were found to be associated with pathways and gene ontology (GO) groups that are significantly enriched in the K.aerogenes strain of PRE.021 compared to reference genomes.
In contrast, we noted 619 and 614, Uniref90 and UniProt entries, respectively, that were consistently missing in the PRE.021 strain, despite being present in all strains of other infants. We found 64 genes to be absent in PRE.021’s K. aerogenes yet present in various reference genomes of the bacterium. Of these, cysS, thrS (both involved in tRNA functionality), and fhuA (iron uptake) were most commonly missing in PRE.021 compared to reference genomes. We did not find a specific pathway or GO to be significantly depleted among K. aerogenes of PRE.021 in comparison to references.
Interestingly, we observed this unique strain of K. aerogenes in stool seven days prior to its first clinical isolation from PRE.021’s blood culture, despite multiple such culturing efforts throughout this period. This early detection of the pathogenic strain in stool underscores the clinical potential of metagenomic sequencing in the detection of pathogens at an early stage.
Discussion
In this study, we provide a comprehensive characterization of the gastric residual and stool microbiome of premature infants, revealing distinct microbial community structures and their temporal dynamics during hospitalization. We were able to cluster over 130 stool samples into eight groups based on their bacterial composition, highlighting the varying nature of preterm gut microbiome development across different infants. Despite often being dominated by members of the Enterobactericeae family, we also found stool samples to host a high abundance of Staphylococcus epidermidis. Samples rich in S. epidermidis were mostly prevalent at early time points in the hospitalization and were associated with a lower birthweight for gestational age. Our results reflect the interaction between the gut developmental status and microbial colonization. We assessed the dynamics of gut microbiome composition across hospitalization and noted that S. epidermidis-rich populations were completely missing towards the end of hospitalization. These findings support S. epidermidis as a transient colonizer of the premature infant gut and reflect the changes of gut maturation, which leads to a less S. epidermidis-friendly niche52. In contrast to the dynamic colonization of S. epidermidis, we find Bifidobacterium-rich communities appearing in late time points of the hospitalization. Higher abundance of Bifidobacterium species has been associated with a healthy infant gut18,19,20,21,24,25, and while these species are commonly found in neonatal probiotic supplements due to their beneficial effect19,21,22,23,25, there was no use of probiotics in our cohort (as instructed by the local ministry of health), thus all colonization reported here was independent of such supplements. We find several Bifidobacterium species, notably B. breve, B. bifidum, and B. longum to be present across multiple infants. Our findings reinforce the notion that preterm infant gut development leads towards a more mature and health-associated microbial profile.
To the best of our knowledge, we performed the first metagenomic analysis of the gastric residual (GR) microbiome of preterm infants. Our results suggest variable bacterial colonization patterns and unstable intra-subject microbial profiles in early life. Specifically, we report high abundances of skin and mucosa-associated bacteria such as Staphylococci and Streptococci62,63,64,70. Additionally, we found a positive correlation between frequent GR aspirations and increased GR microbial diversity. Taken together, our findings suggest a possible translocation of bacteria from the oropharynx to the gastric environment via the nasogastric tube. We hypothesize that such translocation may be enabled due to contact between the nasogastric tube and the oropharynx during insertion of the feeding tube, carrying these oral bacteria past the gastro-esophageal sphincter (which remains partly open due to the feeding tube) and into the gastric environment. Furthermore, our findings challenge our initial hypothesis that the stomach environment applies selective pressures on bacterial colonization, permitting only acid-tolerant bacteria to thrive, yet this hypothesis may still hold in term infants with a mature gastrointestinal tract. Interestingly, we analyze the microbial trajectory of GR in preterm infants and note shared bacteria throughout the hospitalization period.
One significant finding of our GR microbiome analysis is the notable abundance of a Bradyrhizobium species, which are considered to be environmental bacteria often found in soil84,85,86. We first suspected Bradyrhizobium presence to be a result of contamination during the sample preparation, but our contamination analysis did not support this hypothesis (Methods). Within our data, we found Bradyrhizobium presence in GR samples of 9 different infants (5 of which also had Bradyrhizobium in their stool samples). Combining these results, despite not being able to completely rule out possible contamination originating in the sample collection step or the NICU environment, we conclude that the Bradyrhizobium abundance observed genuinely reflects its presence within the premature infant microbiome.
Clinically, our work promotes the potential for early detection of pathogenic bacteria using sequencing approaches. We were able to identify the presence of a pathogenic Klebsiella aerogenes in the stool, a full week before its first isolation from clinical specimens. Importantly, clinical sampling was performed in an attempt to monitor a possible infection as early as nine days ahead of our stool sample time point on a near-daily basis, yet it was still undetected.
The major limitation of this study is the limited dietary information for the premature infants. Sadly, these variables were not collected regularly across all infants and thus could not have been included in the analyses. Due to the relatively small size of our cohort, a larger study will assist in emphasizing the results reported here. As gastric residual collection is often done only upon clinical indication and is uncomfortable for the newborn, it is challenging to obtain parental consent for larger cohorts.
Our findings offer novel insights into the colonization patterns of the neonatal gastrointestinal tract in two different organs, and importantly, present grounds for hope regarding future diagnostic approaches incorporating the sensitivity advantage of metagenomic sequencing.
Methods
Study population
Our study consisted of 39 premature infants, born before 37 weeks of gestation and hospitalized in Hadassah Ein Karem Medical Center, Jerusalem. Study participants were randomized into 2 study arms varying by the frequency of gastric residual (GR) aspiration. Both study groups were exposed to GR collection upon clinical indication, but additionally, in the high-frequency group, sampling also took place before feeding. Samples and clinical data were collected by caregivers at the neonatal intensive care unit. Parents provided informed consent for subject participation. Our study was approved by Hadassah Medical Center’s ethics committee, IRB number 0107-23-HMO.
Sample collection
Stool and gastric residual samples were collected as part of the cohort from premature infants from birth and throughout their hospitalization. Study subjects were categorized into 2 groups, as described above. Stool samples were collected using eSwab® with 1 ml of liquid Amies medium in order to preserve the bacterial population. GR were stored in sterile tubes. Both sample types were collected by the neonatal intensive care team at Hadassah Ein Kerem and stored at 4 °C for up to 24 h, and then collected to the lab and stored long term at −80 °C.
Metagenomic library construction and sequencing
DNA extraction from stool samples was done using DNeasy PowerSoil Pro Kit (#47014, QIAGEN). We used the Nextera XT DNA Library Preparation kit (FC-131-1096, Illumina) to create Illumina Sequencing libraries, in accordance to the manufacturer’s recommended protocol using half of the volume and the DNA. Using NextSeq 500, we performed single-end 150 bp sequencing of samples.
Metagenomic analysis
Using Bowtie2 (2.4.5-1)87 and an in-house pipeline, we removed host reads that aligned to the human genome. Samples were filtered and trimmed for Nextera adapters using fastq-mcf, ea-utils88. (1.05). Taxonomic profiling was done using MetaPhlAn489,90 with our unique database91, on the background of the mpa_vJun23_CHOCOPhlAnSGB_202307 marker set. We carried out strain-level analysis using StrainPhlAn 489,90 with default parameters and --sample_with_n_markers 70 and --markers_in_n_samples 70. Further analysis was done using an in-house R (4.2.2) script utilizing dplyr92 (1.1.2), tidyr93 (1.3.0) and tidyverse94 (2.0.0). Statistical tests comparing between groups were carried out using the stat_compare_means() function of the ggpubr95 package. Plots were created using ggplot296 (3.4.2), colors were used from RColorBrewer97 (1.1–3) and pals98 (1.7). Heatmaps were created using pheatmap99. Alpha and beta diversity were calculated using “diversity” (Shannon index) and “vegdist” (Bray-Curtis dissimilarity) from the vegan100 (2.6–4) package, and the PCoA was created using the ape101 (5.7–1) package. A phylogenetic tree was produced using ggtree102 (3.6.2) and the sankey plots were created using ggsankey103 (0.0.99999).
Statistical analysis
No statistical method was used to predetermine the sample size. The investigators were not blinded to allocation during experiments and outcome assessment. An independent t-test was performed to test between groups when mentioned using the R function “t-test”. Intra-subject paired t-test was done to compare Bray-Curtis dissimilarities of consecutive stool and gastric residual samples.
Clustering analysis
Separation of samples into clusters was done using the partitioning around medians (PAM) model. First, the Bray-Curtis dissimilarity was calculated for each pair of samples, representing the compositional distance between the microbial profiles. Next, to figure out the number of clusters for each sample type, we used the fpc (2.2–13)package104. Lastly, in order to separate the data into clusters, we applied the pam function within the cluster (2.1.6) package105.
Bradyrhizobium contamination analysis
To check for possible contamination during DNA extraction, library preparation and sequencing resulting in the presence of Bradyrhizobium, we used the R package decontam(1.16.0)106. Analysis was performed when taking into account the sample extraction and library preparation batches. To validate the results, we also checked for possible contaminations by sample type, thus analysing the stool and gastric residuals separately.
Gene enrichment and depletion analysis for pathogenic Klebsiella aerogenes
PanPhlAn82 was used to compare the genomic content of Klebsiella aerogenes across strains found in our cohort, as well as reference genomes. For the enrichment, we selected Uniref90 entries that were consistently present in all data of PRE.021 but completely absent from strains of other cohort subjects. Similarly, for the depletion analysis, we selected all the Uniref90 entries that were missing throughout the data of PRE.021 but were always present in strains of other study participants. Next, we used UniProt107 ID Mapping online tool to convert Uniref entries into genes. Using BioCyc83 we compared these gene lists to 12 reference genomes of K.aerogenes, including EA1509E, NCTC418, FDAARGOS_363, FDAARGOS_513, KCTC2190, NCTC9735, NCTC9997, NCTC9793, NCTC8846, NCTC9652, NCTC9667, and NCTC9668. Finally, enrichment and depletion analysis were carried out searching for enriched and depleted genes for pathways, transcriptional/translational regulators, and GO.
Stool samples subsampling
Within our sequenced stool samples, subjects PRE.006, PRE.008, PRE.010, and PRE.024 had a greater representation. In order to account for this and avoid biased results stemming we subsampled the number of samples from these infants to be included in the analysis. Briefly, we calculated the floor value of the 95th percentile of the number of samples across other study participants. We then randomly selected this number of samples from the over-represented infants, maintaining the first and last sample collected during the hospitalization.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The sequencing data generated in this study, containing human-filtered metagenomic reads, have been deposited in the SRA database under accession code PRJNA1312123. Source data for all figures are included in the Supplementary files. Source data are provided with this paper.
References
Turnbaugh, P. J. et al. The human microbiome project. Nature 449, 804–810 (2007).
Ley, R. E., Lozupone, C. A., Hamady, M., Knight, R. & Gordon, J. I. Worlds within worlds: evolution of the vertebrate gut microbiota. Nat. Rev. Microbiol. 6, 776–788 (2008).
Garrett, W. S., Gordon, J. I. & Glimcher, L. H. Homeostasis and inflammation in the intestine. Cell 140, 859–870 (2010).
Lozupone, C. A., Stombaugh, J. I., Gordon, J. I., Jansson, J. K. & Knight, R. Diversity, stability and resilience of the human gut microbiota. Nature 489, 220–230 (2012).
Blaser, M. J. et al. Lessons learned from the prenatal microbiome controversy. Microbiome 9, 8 (2021).
Penders, J. et al. Factors influencing the composition of the intestinal microbiota in early infancy. Pediatrics 118, 511–521 (2006).
Rinott, E. & Youngster, I. The early gut microbiome and the risk of chronic disease. Hum. Microbiome Early Life 239–254. https://doi.org/10.1016/B978-0-12-818097-6.00010-9 (2020).
Clemente, J. C., Ursell, L. K., Parfrey, L. W. & Knight, R. The impact of the gut microbiota on human health: an integrative view. Cell 148, 1258–1270 (2012).
Mathis, D. & Benoist, C. Microbiota and autoimmune disease: the hosted self. Cell Host Microbe 10, 297–301 (2011).
Hooper, L. V., Littman, D. R. & Macpherson, A. J. Interactions between the microbiota and the immune system. Science 336, 1268–1273 (2012).
Honda, K. & Littman, D. R. The microbiota in adaptive immune homeostasis and disease. Nature 535, 75–84 (2016).
Thaiss, C. A., Zmora, N., Levy, M. & Elinav, E. The microbiome and innate immunity. Nature 535, 65–74 (2016).
Tilg, H., Zmora, N., Adolph, T. E. & Elinav, E. The intestinal microbiota fuelling metabolic inflammation. Nat. Rev. Immunol. 20, 40–54 (2020).
Gensollen, T., Iyer, S. S., Kasper, D. L. & Blumberg, R. S. How colonization by microbiota in early life shapes the immune system. Science 352, 539–544 (2016).
Renz, H., Brandtzaeg, P. & Hornef, M. The impact of perinatal immune development on mucosal homeostasis and chronic inflammation. Nat. Rev. Immunol. 12, 9–23 (2011).
Brodin, P. Immune-microbe interactions early in life: a determinant of health and disease long term. Science 376, 945–950 (2022).
Guarner, F. & Malagelada, J.-R. Gut flora in health and disease. Lancet 361, 512–519 (2003).
Saturio, S. et al. Role of bifidobacteria on infant health. Microorganisms 9, 2415 (2021).
Beck, L. C. et al. Strain-specific impacts of probiotics are a significant driver of gut microbiome development in very preterm infants. Nat. Microbiol. 7, 1525–1535 (2022).
Stewart, C. J. et al. Longitudinal development of the gut microbiome and metabolome in preterm neonates with late onset sepsis and healthy controls. Microbiome 5, 75 (2017).
Alcon-Giner, C. et al. Microbiota supplementation with Bifidobacterium and Lactobacillus modifies the preterm infant gut Microbiota and metabolome: An observational study. Cell Rep. Med. 1, 100077 (2020).
Neumann, C. J. et al. Clinical NEC prevention practices drive different microbiome profiles and functional responses in the preterm intestine. Nat. Commun. 14, 1349 (2023).
Robertson, C. et al. Incidence of necrotising enterocolitis before and after introducing routine prophylactic Lactobacillus and Bifidobacterium probiotics. Arch. Dis. Child. Fetal Neonatal Ed. 105, 380–386 (2020).
O’Neill, I., Schofield, Z. & Hall, L. J. Exploring the role of the microbiota member Bifidobacterium in modulating immune-linked diseases. Emerg. Top. Life Sci. 30, 333–349 (2017).
Prasanna, P. H. P., Grandison, A. S. & Charalampopoulos, D. Bifidobacteria in milk products: an overview of physiological and biochemical properties, exopolysaccharide production, selection criteria of milk products and health benefits. Food Res. Int. 55, 247–262 (2014).
Samara, J. et al. Supplementation with a probiotic mixture accelerates gut microbiome maturation and reduces intestinal inflammation in extremely preterm infants. Cell Host Microbe 30, 696–711.e5 (2022).
Beck, L. C., Berrington, J. E. & Stewart, C. J. Impact of probiotics on gut microbiome of extremely preterm or extremely low birthweight infants. Pediatr. Res. 97, 493–496 (2025).
Walani, S. R. Global burden of preterm birth. Int. J. Gynaecol. Obstet. 150, 31–33 (2020).
Stoll, B. J. et al. Neonatal outcomes of extremely preterm infants from the NICHD Neonatal Research Network. Pediatrics 126, 443–456 (2010).
Neu, J., Modi, N. & Caplan, M. Necrotizing enterocolitis comes in different forms: Historical perspectives and defining the disease. Semin. Fetal Neonatal Med. 23, 370–373 (2018).
Stras, S. F. et al. Maturation of the human intestinal immune system occurs early in fetal development. Dev. Cell 51, 357–373.e5 (2019).
Li, Y. et al. In utero human intestine harbors unique metabolome, including bacterial metabolites. JCI Insight 5, e138751 (2020).
Ancel, P.-Y. et al. Survival and morbidity of preterm children born at 22 through 34 weeks’ gestation in France in 2011: results of the EPIPAGE-2 cohort study. JAMA Pediatr. 169, 230–238 (2015).
Sevastiadou, S. et al. The impact of oral glutamine supplementation on the intestinal permeability and incidence of necrotizing enterocolitis/septicemia in premature neonates. J. Matern. Fetal Neonatal Med. 24, 1294–1300 (2011).
Taft, D. H. et al. Center variation in intestinal Microbiota prior to late-onset sepsis in preterm infants. PLoS ONE 10, e0130604 (2015).
Lee, C.-C. et al. Gut dysbiosis, bacterial colonization and translocation, and neonatal sepsis in very-low-birth-weight preterm infants. Front. Microbiol. 12, 746111 (2021).
Carl, M. A. et al. Sepsis from the gut: the enteric habitat of bacteria that cause late-onset neonatal bloodstream infections. Clin. Infect. Dis. 58, 1211–1218 (2014).
Gregory, K. E. & Connolly, T. C. Enteral feeding practices in the NICU: results from a 2009 Neonatal Enteral Feeding Survey. Adv. Neonatal Care 12, 46–55 (2012).
Parker, L. et al. Aspiration and evaluation of gastric residuals in the neonatal intensive care unit: state of the science: state of the science. J. Perinat. Neonatal Nurs. 29, 51–59 (2015).
Abiramalatha, T., Thanigainathan, S. & Ninan, B. Routine monitoring of gastric residual for prevention of necrotising enterocolitis in preterm infants. Cochrane Database Syst. Rev. 7, CD012937 (2019).
Giuliani, F. et al. Necrotizing enterocolitis: risk factor analysis and role of gastric residuals in very low birth weight infants. Arch. Dis. Child. 93, w227–pw227 (2008).
Li, Y.-F. et al. Gastric residual evaluation in preterm neonates: a useful monitoring technique or a hindrance?. Pediatr. Neonatol. 55, 335–340 (2014).
Branagan, A. et al. Influence of gastric residual assessment in preterm neonates on time to achieve enteral feeding (the GRASS trial)-Multi-centre, assessor-blinded randomised clinical trial. Eur. J. Pediatr. 183, 2325–2332 (2024).
Parker, L. A. et al. Effect of gastric residual evaluation on enteral intake in extremely preterm infants: a randomized clinical trial: a randomized clinical trial. JAMA Pediatr. 173, 534–543 (2019).
Akar, S. & Turgut, M. Do we control gastric residuals unnecessarily in premature newborns? AGRA study: avoidance of gastric residual aspiration. World J. Pediatr. Surg. 3, e000056 (2020).
Kumar, J. et al. Routine prefeed gastric aspiration in preterm infants: a systematic review and meta-analysis. Eur. J. Pediatr. 180, 2367–2377 (2021).
Patel, K., Konduru, K., Patra, A. K., Chandel, D. S. & Panigrahi, P. Trends and determinants of gastric bacterial colonization of preterm neonates in a NICU setting. PLoS ONE 10, e0114664 (2015).
Stewart, C. J. et al. Temporal development of the gut microbiome in early childhood from the TEDDY study. Nature 562, 583–588 (2018).
Roswall, J. et al. Developmental trajectory of the healthy human gut microbiota during the first 5 years of life. Cell Host Microbe 29, 765–776.e3 (2021).
Ferretti, P. et al. Mother-to-infant microbial transmission from different body sites shapes the developing infant gut microbiome. Cell Host Microbe 24, 133–145.e5 (2018).
Yassour, M. et al. Natural history of the infant gut microbiome and impact of antibiotic treatment on bacterial strain diversity and stability. Sci. Transl. Med. 8, 343ra81 (2016).
Rao, C. et al. Multi-kingdom ecological drivers of microbiota assembly in preterm infants. Nature 591, 633–638 (2021).
Lara-Marquez, M. L. & Laubach, S. Cesarean section and chronic immune disorders. Pediatrics 136, S224–S225 (2015).
Yuan, C. et al. Association between cesarean birth and risk of obesity in offspring in childhood, adolescence, and early adulthood. JAMA Pediatr. 170, e162385 (2016).
Virtanen, S. M. et al. Microbial exposure in infancy and subsequent appearance of type 1 diabetes mellitus-associated autoantibodies: a cohort study. JAMA Pediatr. 168, 755–763 (2014).
Flannery, D. D. et al. Temporal trends and center variation in early antibiotic use among premature infants. JAMA Netw. Open 1, e180164 (2018).
Gibson, M. K., Crofts, T. S. & Dantas, G. Antibiotics and the developing infant gut microbiota and resistome. Curr. Opin. Microbiol. 27, 51–56 (2015).
Gasparrini, A. J. et al. Persistent metagenomic signatures of early-life hospitalization and antibiotic treatment in the infant gut microbiota and resistome. Nat. Microbiol. 4, 2285–2297 (2019).
Garg, P. M. et al. Clinical impact of NEC-associated sepsis on outcomes in preterm infants. Pediatr. Res. 92, 1705–1715 (2022).
Bizzarro, M. J., Ehrenkranz, R. A. & Gallagher, P. G. Concurrent bloodstream infections in infants with necrotizing enterocolitis. J. Pediatr. 164, 61–66 (2014).
Cole, C. R. et al. Bloodstream infections in very low birth weight infants with intestinal failure. J. Pediatr. 160, 54–9.e2 (2012).
Harris-Tryon, T. A. & Grice, E. A. Microbiota and maintenance of skin barrier function. Science 376, 940–945 (2022).
Oh, J. et al. Temporal stability of the human skin microbiome. Cell 165, 854–866 (2016).
Oh, J. et al. Biogeography and individuality shape function in the human skin metagenome. Nature 514, 59–64 (2014).
Podschun, R. & Ullmann, U. Klebsiellaspp. As nosocomial pathogens: epidemiology, taxonomy, typing methods, and pathogenicity factors. Clin. Microbiol. Rev. 11, 589–603 (1998).
Kim, S., Covington, A. & Pamer, E. G. The intestinal microbiota: antibiotics, colonization resistance, and enteric pathogens. Immunol. Rev. 279, 90–105 (2017).
Bray, A. S. et al. Klebsiella pneumoniae employs a type VI secretion system to overcome microbiota-mediated colonization resistance. Nat. Commun. 16, 940 (2025).
Calderon-Gonzalez, R. et al. Modelling the gastrointestinal carriage of Klebsiella pneumoniae infections. MBio 14, e0312122 (2023).
Russo, T. A. & Marr, C. M. Hypervirulent Klebsiella pneumoniae clinical and molecular perspectives. J. Intern Med. 287, 283–300 (2019).
Joglekar, P. et al. Integrated genomic and functional analyses of human skin-associated Staphylococcus reveal extensive inter- and intra-species diversity. Proc. Natl. Acad. Sci. USA 120, e2310585120 (2023).
Lamont, R. F. et al. The vaginal microbiome: new information about genital tract flora using molecular based techniques. BJOG 118, 533–549 (2011).
Ravel, J. et al. Vaginal microbiome of reproductive-age women. Proc. Natl. Acad. Sci. USA 108, 4680–4687 (2011).
Relman, D. A. Microbiology: learning about who we are. Nature 486, 194–195 (2012).
Serrano, C., Harris, P. R., Smith, P. D. & Bimczok, D. Interactions between H. pylori and the gastric microbiome: impact on gastric homeostasis and disease. Curr. Opin. Physiol. 21, 57–64 (2021).
Reshetnyak, V. I., Burmistrov, A. I. & Maev, I. V. Helicobacter pylori: commensal, symbiont or pathogen?. World J. Gastroenterol. 27, 545–560 (2021).
Gregory, K. E. et al. Influence of maternal breast milk ingestion on acquisition of the intestinal microbiome in preterm infants. Microbiome 4, 68 (2016).
Gibson, M. K. et al. Developmental dynamics of the preterm infant gut microbiota and antibiotic resistome. Nat. Microbiol. 1, 16024 (2016).
DiBartolomeo, M. E. & Claud, E. C. The developing microbiome of the preterm infant. Clin. Ther. 38, 733–739 (2016).
La Rosa, P. S. et al. Patterned progression of bacterial populations in the premature infant gut. Proc. Natl. Acad. Sci. USA 111, 12522–12527 (2014).
Costello, E. K., Carlisle, E. M., Bik, E. M., Morowitz, M. J. & Relman, D. A. Microbiome assembly across multiple body sites in low-birthweight infants. MBio 4, e00782–13 (2013).
Stewart, C. J. et al. Temporal bacterial and metabolic development of the preterm gut reveals specific signatures in health and disease. Microbiome 4, 67 (2016).
Beghini, F. et al. Integrating taxonomic, functional, and strain-level profiling of diverse microbial communities with bioBakery 3. Elife 10, e65088 (2021).
Karp, P. D. et al. The BioCyc collection of microbial genomes and metabolic pathways. Brief. Bioinform. 20, 1085–1093 (2019).
Ormeño-Orrillo, E. & Martínez-Romero, E. A genomotaxonomy view of the Bradyrhizobium genus. Front. Microbiol. 10, 1334 (2019).
VanInsberghe, D. et al. Non-symbiotic Bradyrhizobium ecotypes dominate North American forest soils. ISME J. 9, 2435–2441 (2015).
Jones, F. P. et al. Novel European free-living, non-diazotrophic Bradyrhizobium isolates from contrasting soils that lack nodulation and nitrogen fixation genes - a genome comparison. Sci. Rep. 6, 25858 (2016).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012).
Aronesty, E. Comparison of sequencing utility programs. Open Bioinforma. J. 7, 1–8 (2013).
Blanco-Miguez, A. et al. Extending and improving metagenomic taxonomic profiling with uncharacterized species with MetaPhlAn 4. Nat Biotechnol 41, 1633–1644 (2023).
Truong, D. T., Tett, A., Pasolli, E., Huttenhower, C. & Segata, N. Microbial strain-level population structure and genetic diversity from metagenomes. Genome Res. 27, 626–638 (2017).
Ennis, D., Shmorak, S., Jantscher-Krenn, E. & Yassour, M. Longitudinal quantification of Bifidobacterium longum subsp. Infantis reveals late colonization in the infant gut independent of maternal milk HMO composition. Nat. Commun. 15, 894 (2024).
Dplyr: Dplyr: A Grammar of Data Manipulation. (Github).
tidyr. https://tidyr.tidyverse.org.
Wickham, H. et al. Welcome to the tidyverse. J. Open Source Softw. 4, 1686 (2019).
ggplot2-Based Publication-Ready Plots. https://rpkgs.datanovia.com/ggpubr/.
ggplot2. https://ggplot2.tidyverse.org.
ColorBrewer: Color Advice for Maps. https://colorbrewer2.org/#type=sequential&scheme=BuGn&n=3.
Kovesi, P. Good colour maps: How to design them. arXiv [cs.GR] https://doi.org/10.48550/ARXIV.1509.03700 (2015).
Kolde, R. pheatmap: Pretty Heatmaps. https://rdrr.io/cran/pheatmap/ (2019).
Paradis, E. & Schliep, K. ape 5.0: an environment for modern phylogenetics and evolutionary analyses in R. Bioinformatics 35, 526–528 (2019).
Yu, G., Lam, T. T.-Y., Zhu, H. & Guan, Y. Two methods for mapping and visualizing associated data on phylogeny using ggtree. Mol. Biol. Evol. 35, 3041–3043 (2018).
Sjoberg, D. Ggsankey: Make Sankey, Alluvial and Sankey Bump Plots in Ggplot. (Github).
Akhanli, S. E. & Hennig, C. Comparing clusterings and numbers of clusters by aggregation of calibrated clustering validity indexes. Stat. Comput. 30, 1523–1544 (2020).
Maechler. M., Rousseeuw, P., Struyf, A., Hubert, M. & Hornik, K. cluster: Cluster Analysis Basics and Extensions. https://CRAN.R-project.org/package=cluster. (2023).
Davis, N. M., Proctor, D. M., Holmes, S. P., Relman, D. A. & Callahan, B. J. Simple statistical identification and removal of contaminant sequences in marker-gene and metagenomics data. Microbiome 6, 226 (2018).
UniProt: The universal protein knowledgebase in 2021. Nucleic Acids Res. 49, D480–D489 (2021).
Acknowledgements
We thank the participants and their families for joining the research, and Dr Abed Nasereddin and Dr Idit Shiff from the Genomics Applications Laboratory at the Faculty of Medicine of the Hebrew University for their support with DNA sequencing. Prof Jacob Strahilevitz and Prof Jacob Moran Gilad from the Clinical Microbiology Department at Hadassah Medical Center for providing the clinical strain isolates. This study was funded in part by the Israel Precision Medicine Partnership (IPMP), grant number 3061/22, and by the Azrieli Family Foundation (Faculty fellowship for MY). MY holds the Rosalind, Paul, and Robin Berlin Faculty Development Chair in Perinatal Research.
Author information
Authors and Affiliations
Contributions
M.Y. and N.O.S. conceptualized the study. N.O.S. and S.E.F. recruited study participants and gained informed consent from the parents of study subjects. N.M., E.H., N.R., and S.S. transferred samples from the hospital to the lab storage. N.M., N.R., and S.S. were in charge of sample DNA extraction and library sequencing. N.M performed all computational analysis. L.J. and S.S. cultured and isolated strains used for this work. M.Y., N.M., and N.O.S. wrote the manuscript with contributions from all other authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Eugene Dempsey and Parvesh Garg for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Moriel, N., Jones, L., Harpenas, E. et al. Development of the preterm infant gut and gastric residuals microbiome. Nat Commun 16, 9848 (2025). https://doi.org/10.1038/s41467-025-64819-8
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-64819-8
