
Abstract
Pathogenic variants in ATP-dependent chromatin remodeling proteins are a recurrent cause of neurodevelopmental disorders (NDDs). The NURF complex consists of BPTF and either the SMARCA5 or SMARCA1 ISWI-chromatin remodeling enzyme. Pathogenic variants in BPTF and SMARCA5 have been previously implicated in NDDs. Here, we describe 35 individuals from 26 families with de novo or maternally inherited variants in the X-linked SMARCA1 gene. This SMARCA1-related NDD is associated with a spectrum of involvement, including mild to severe ID/DD, delayed or regressive speech development, ASD features, facial dysmorphisms, and other variable features. Individuals carrying SMARCA1 truncating variants exhibit a mildly unique genome-wide DNA methylation profile and a high penetrance of macrocephaly. Genetic dissection of the NURF complex using Smarca1, Smarca5, and Bptf single and double mouse knockouts reveals the importance of NURF composition and dosage for proper forebrain development. We propose that genetic alterations affecting different NURF components, including SMARCA1, result in a NDD with a broad clinical spectrum.
Similar content being viewed by others

Introduction
A frequent cause of neurodevelopmental disorders (NDDs) associated with intellectual disability/developmental delay (ID/DD) and/or autism spectrum disorders (ASD) arise from pathogenic variants in genes encoding proteins that regulate or modify chromatin architecture1,2. Prominent amongst these are genes encoding the catalytic subunits of chromatin remodeling complexes that utilize ATP hydrolysis to reposition nucleosomes. These ATP-dependent chromatin remodeling proteins are critical for DNA replication, transcription, and DNA repair3 and they can be subdivided into the SWI/SNF, ISWI, CHD, and INO80 sub-families. Multiple members from each group have been implicated in distinct NDDs4,5,6,7. The best characterized is the SWI/SNF family, comprising BRG1 and BRM, which are exchangeable subunits within a large 12–15 protein complex known primarily as the BRG1-associated factor (BAF) complex. Pathogenic variants in the genes encoding BRG1 (SMARCA4) and BRM (SMARCA2) are the cause of Coffin-Siris 4 (CS4, MIM 614609) and Nicolaides-Baraitser (MIM 601358) syndromes, respectively8,9. Additionally, pathogenic variants in the genes encoding most other BAF subunits cause similar clinical features, supporting the notion that a defective BAF complex drives a broad neurodevelopmental spectrum with autistic features at the mild end, classic Coffin-Siris syndrome in the middle, and Nicolaides-Baraitser syndrome representing the most severe disease4.
The ISWI family, consisting of the SMARCA1 and SMARCA5 genes are highly conserved orthologs. of the Drosophila ISWI protein that contains an ATPase (or SNF2-) and a HAND-SANT-SLIDE (HSS) domain that interacts with histone H4 tails and linker DNA10. The ISWI complexes are largely heterodimeric, interchangeably pairing one of the ATPase subunits with either a member of the ~180-kDa bromodomain adjacent to zinc finger (BAZ) transcription factor family (BAZ1A, BAZ1B, BAZ2A, BAZ2B), or a larger protein with related chromatin domains (BPTF, CECR2, or RSF1), suggesting the existence of 16 distinct ISWI complexes. While many seem to be mammalian-specific, three ISWI complexes, ACF (BAZ1A), CHRAC (BAZ1A, CHRAC15, CHRAC17), and NURF (BPTF, RbAp 46/48) are conserved from mammals to Drosophila, indicating their functional importance.
Studies associating ISWI genes with NDDs has progressed more slowly than for other chromatin remodelers. The earliest reported connection was the finding that BAZ1B is one of 28 genes within the 1.5 Mb region on 7q11.23 that is commonly deleted in Williams-Beuren syndrome (WBS; MIM 194050), although the contribution of BAZ1B to the WBS phenotype remains inconclusive11,12. The NURF complex was implicated in the Neurodevelopmental Disorder with Dysmorphic Facies and distal Limb anomalies (NEDDFL) syndrome (MIM 617755) through the identification of pathogenic variants in the BPTF gene13,14. Later, germline pathogenic variants in the SMARCA5 gene were described in 12 individuals who presented with developmental delay, short stature, and microcephaly15. While these NDDs are presumed to be unique, there is significant clinical overlap between the syndromes. Consistent with this idea, forebrain-specific inactivation of the murine Bptf gene or the Smarca5 gene both cause severe defects in progenitor proliferation resulting in forebrain hypoplasia that is most severe in the Bptf conditional knockout16,17.
The NURF complex has demonstrated roles in target gene expression during cell differentiation through interactions with a wide range of transcription factors and the H3K4me3 histone mark18,19,20,21,22,23,24,25. In Drosophila, ISWI genome-wide binding data demonstrated peak binding 300 bp downstream of the TSS where it helps position the first two nucleosomes26. Both Smarca5 and Bptf cKO mice show dysregulation of many transcription factors involved in neurogenic fate specification further strengthening a role for NURF in neural development16,17,27.
Interestingly, the initial purification of the mammalian NURF complex showed that it was primarily associated with SMARCA1. Initial expression studies indicated broad tissue distribution for both human and mouse ISWI genes albeit with temporal differences28. In the developing mouse brain, ISWI transcript and protein levels show a consistent pattern with Smarca5 enriched earlier when progenitor cells are abundant, and Smarca1 elevated later and predominantly in terminally differentiated neurons27,29,30. These studies suggest that there are distinct roles for NURF during forebrain development depending on whether it associates with SMARCA5 or SMARCA1. Indeed, in contrast to Smarca5 and Bptf cKO mice, progenitor differentiation is delayed in Smarca1ex6del mutant mice leading to increased neuronal production and mice with larger brains31.
The phenotypic similarities between individuals with BPTF or SMARCA5 pathogenic germline variants combined with NURF subunit exchange, and a distinct phenotype in Smarca1ex6del mutant mice, parallels findings with the BAF complex and the broad phenotypic spectrum caused by different BAF subunit gene mutations. As such, we sought to utilize mouse and human genetic studies to further explore the relationship between NURF subunits and identify a NDD associated with SMARCA1 variants. While several whole genome sequencing screens of large NDD cohorts identified SMARCA1 variants in three individuals, a definitive phenotype or syndrome has not emerged32,33,34. Here, we report the identification of 35 individuals from 26 families with de novo or inherited germline variants within the SMARCA1 gene associated with variable degrees of developmental delay and speech regression and a high penetrance of macrocephaly. In addition, we demonstrate the importance of the NURF complex in murine neocortical development and propose that altered composition and dosage of this complex is a major contributor to variable neurodevelopmental disorders.
Results
Identification of SMARCA1 genetic variants
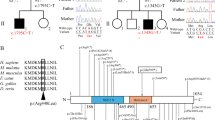
Our initial characterization began with performing exome sequencing on a large six-generation family (Fig. 1A) containing 4 individuals with mild to severe ID/DD and brain overgrowth. This study identified a hemizygous nonsense variant (c.271 C > T [p.Arg91*]) in the SMARCA1 gene (NM_003069.4) in the proband (vi1), his brother (vi2) and two maternal cousins (vi8 and vi9). Both mothers (v2 and v10) of the affected individuals were heterozygous for the variant while the paternal grandfather (iv1) and maternal uncle (v5) were negative. An additional 25 families were ascertained in part through the MatchMaker Exchange network35 for a total of 35 individuals with apparent non-mosaic hemizygous or heterozygous SMARCA1 variants. A total of 25 unique variants were identified in addition to a second unrelated family with the same variant as the index family (Fig. 1B, Table 1). Protein truncating and putative loss-of-function (LOF) variants included seven nonsense variants (c.271 C > T [p.Arg91*], c.565 C > T [p.Arg189*], c.685 C > T [p.Arg229*], c.757 C > T [p.Arg253*], c.916 C > T [p.Arg306*], c.1070 T > G [p.Leu357*], c.1971dup [p.Asn658*]), and three frameshifting insertions/deletions (indels) affecting coding exons (c.543dupG [p.Pro182Alafs*18], c.1071dupA [p.Leu358Ilefs*3], c.3103-3106del [p.Arg1035Glnfs*13]). Fifteen missense variants (c.353 C > T [p.Thr118Ile], c.407 A > G [p.Gln136Arg], c.566 G > A [p.Arg189Gln], c.775 C > G [p.Arg259Gly], c.1295 T > C [p.Met432Thr], c.1514 T > C [p.Val505Ala], c.1680 T > A [p.Phe560Leu], c.1940T>C [p.Ile647Thr], c.2161 G > A [p.Asp721Asn], c.2252 G > A [p.Arg751Gln], c.2311 G > A [p.Glu771Lys], c.2471 C > T [p.Pro824Leu], c.2681 A > T [p.Glu894Val], c.3007 T > C [p.Phe1003Leu], c.3152 C > T [p.Ser1039Leu]) were also identified. The c.7 C > T [p.Gln3*] and c.2897 G > T [p.Gly966Val] variants were previously reported in isolated cases with neurodevelopmental disorders, but absent in our cohort33,34.

A Six generation pedigree of the index family carrying the c.271 C > T [p.Arg91*] variant within SMARCA1. Genetic testing also included the maternal uncle (v5) and grandfather (iv1), who were both negative for the variant. B Schematic diagram highlighting the position of the pathogenic variants identified and the functional domains encoded by SMARCA1. Blue text denotes variants with macrocephaly, purple text refers to previously described variants that were not identified in this study. Numbers refer to amino acid position. Numbers in brackets identify recurring mutations. The motifs comprising the SNF2 ATPase/helicase domain and the HSS domain are highlighted in blue and green, respectively.
Within the 26 index cases, 17 had a maternally inherited variant, four variants occurred de novo, and for five cases the segregation was not determined, although four of these were presumed maternally inherited. Comprehensive genomic sequencing data from all 26 families identified four individuals with other variants of uncertain significance (VUS) that were deemed non-causative while the remainder either had no notable molecular findings or did not provide additional information (SupplementaryTable 1). Importantly, all 25 SMARCA1 variants were predicted as deleterious using multiple bioinformatic algorithms (SIFT, Polyphen2-, MutationTaster, and CADD; see SupplementaryTable 2). We also examined allele counts for all variants using gnomAD v4.1.0. This analysis identified several single reports (allele frequency ≤8.303 × 10−7) while the c.1295 T > C,p.Met432Thr variant was present in four alleles but with an equally low allele frequency (≤3.4 × 10−6; see Supplementary Table 2). While none of our cohort had copy number abnormalities involving SMARCA1, we predicted that copy number variations (CNVs) in the SMARCA1 gene would also be deleterious so we examined the DECIPHER database for CNVs encompassing the SMARCA1 gene. Publicly available data identified 72 CNVs with an almost equal-split between deletions (44%) and duplications (56%). While many individuals with these deletions/duplications had phenotypes that included global developmental delay, intellectual disability, or delayed speech, it was challenging to assign the phenotype to SMARCA1 gain/loss as most CNVs were >10 Mb in size (76% duplications and 97% deletions) and included multiple genes.
In parallel, we also identified an additional 7 families with SMARCA1 variants, which were deemed either likely non-causative or were not explanatory (SupplementaryTable 3). This cohort included three families with the same variant (c.1150 G > A,p.Val384Met) that has previously been reported in a pedigree of familial schizophrenia and co-segregated with a variant in the AVEN gene, drawing into question the causality of this variant32. Moreover, this variant was present in gnomAD in 16/1195462 alleles in presumably healthy individuals, including 7 hemizygotes (allele frequency 0.00001338; see SupplementaryTable 2). Two other families had an identical variant (c.1610 C > T, p.Pro537Leu) that was similarly present with a high allele frequency in gnomAD. An additional family with the variant (c.113 G > T, p.Gly38Val) was identified in combination with a de novo duplication of 5p15.2 (arr[hg19] 5p15.2p15.1 (13,678,440-16,880,410)x3), which had a high probability of contributing to the phenotype.
The highly conserved ATPase domain (comprising SNF2-N and Helicase-C motifs) and the HSS histone interaction module of SMARCA1 are critical for nucleosome remodeling activity36. Sixteen of the 25 unique variants identified resided within these two functional domains (12 in ATPase, 4 in HSS) while a previously reported case (p.Gly966Val) is also located in the HSS domain34. The remaining identified variants were spread throughout the protein-coding sequence (Fig. 1B).
Clinical findings of individuals with SMARCA1 variants
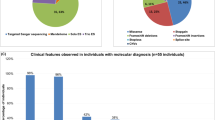
Affected individuals included 26 males and 9 females, ranging from 2 to 49 years at their last clinical assessment. The cohort showed a broad range of clinical features that are summarized in Table 1. Most individuals presented with ID/DD (77%; 27/35) and speech delay and/or speech regression (83%; 29/35). Notably, increased head circumference (OFC \(\ge+2 \) SD) and/or reported macrocephaly/brain overgrowth was reported in many individuals (49%; 17/35) and was identified in the index family and reported in eight individuals with truncating variants (Fig. 1B; Table 1). However, three individuals with truncating variants without brain overgrowth and four individuals with missense variants with brain overgrowth suggest that this feature is more complex and not restricted to individuals with putative LOF variants. Similarly, individuals with CNV deletions encompassing the SMARCA1 gene were not associated with macrocephaly. Increased height (\(\ge+\)2 SD; 29%) at the time of the last exam was reported slightly less frequently arguing against a generalized overgrowth syndrome (Table 1; Supplementary Fig. 1). Other commonly reported clinical features included motor delay (20/34; 59%), seizures/epilepsy (23%; 8/35), and hypotonia (42%; 15/35). Over half the individuals (19/35; 54%) were diagnosed with behavioral difficulties (ASD, ADHD, aggression) and in some instances (9/25) this was linked to regression in speech and mental development. Almost one-third of patients (31%; 11/35) received an ASD diagnosis. Dysmorphic facial features (58%; 19/35) were also commonly reported although clinical descriptions were variable (Table 1). Photographs from seven affected individuals show that frontal bossing, a long face, slanted palpebral fissures, a flat nasal root with a short bulbous nose, anteverted nares, and a thin upper lip were relatively common features of the cohort (Fig. 2).

Facial photographs of seven patients (A–G) highlighting the common features that include frontal bossing, a long face, slanted palpebral fissures, a flat nasal root with a short bulbous nose, anteverted nares, and a thin upper lip. The age at which the photograph was taken is listed, with the variant identified shown above each photograph. Multiple photographs for two individuals (E, G) show that the features remain constant with age. p.Arg259Gln (E) or the (G).
Genomic DNA methylation profiles associated with SMARCA1 variants
Given the broad phenotypic features of individuals with SMARCA1 variants, we explored whether they exhibit a distinguishing methylation profile in their peripheral blood DNA. Genome-wide DNA methylation analysis was performed on DNA derived from peripheral blood from 19 individuals and further stratified by gender. The SMARCA1-M sub-cohort consisted of 12 males with SMARCA1 variants, two of whom had lymphoblast cell line (LB-line) replicate samples which were also used for investigating profile clustering (SMARCA1-M-rep). An additional male sample was also used for test clustering. On the other hand, the SMARCA1-F sub-cohort consisted of seven female individuals carrying SMARCA1 variants with or without phenotypic presentation, three of which have LB-line replicate samples (SMARCA1-F-rep) which were only used for unsupervised clustering.
Initial analysis of all 19 individuals compared to 57 controls matched by age, sex and array type revealed a predominantly hypomethylated profile defined by 232 probes differentiating cases from controls (Fig. 3A). LB replicates also exhibited the same methylation pattern, while the additional test SMARCA1-M sample exhibited a pattern more like controls (Fig. 4A). Gender-specific methylation profiles were also observed. SMARCA1-M samples matched to 60 unaffected controls showed a generally hypomethylated pattern for 212 probes, and the SMARCA1-F samples matched to 56 unaffected controls also displayed lower methylation levels for 221 distinct probes. The two probe sets have absolute methylation difference ranges 5–13% and 5–16%, respectively, and distinguish the SMARCA1 cases from the matched controls (Fig. 3B, C; SupplementaryFig. 2). While the initial global methylation profile does not separate the individual gender sub-cohorts, profiles for each gender were distinguishable up to a moderate effect size. The SMARCA1-F samples and replicates mostly clustered with SMARCA1-M cases but in a separate sub-cluster with a subset of the controls in the heatmap, and the majority of the SMARCA1-M samples and replicates also matched the methylation pattern of controls evident in the heatmaps (Fig. 4B and Fig. 4C, respectively). However, multidimensional scaling (MDS) plots reveal moderate methylation differences between the male samples, female samples, and unaffected samples.

Volcano plots (left) show the mean methylation difference of cases and controls plotted against the -log10(p) values and highlight the selected differential probes for the distinct profiles of A SMARCA1 cohort, B male individuals carrying SMARCA1 variants (SMARCA1-M), and C female individuals with SMARCA1 variants (SMARCA1-F). MDS plots (center) and heatmaps (right) with Euclidean clustering show clustering of cases (red for males, orange for females) and unaffected controls (blue). Columns of the heatmap correspond to the samples and rows correspond to the selected probes.

Other SMARCA1 samples not used in SMARCA1 profile discovery were investigated using unsupervised analysis and examined using the distinct profiles for A SMARCA1 cohort, B SMARCA1-M cohort, and C SMARCA1-F cohort. As expected, replicates cluster with respective similar cases for each profile.
SMARCA1 variants generate stable proteins that localize to the nucleus
We previously reported that an in-frame deletion of exon 6 in the murine Smarca1 gene (Smarca1ex6del mice) resulted in animals with an enlarged brain phenotype31. The Smarca1ex6del mice generate an internally truncated SMARCA1 protein that was unable to hydrolyze ATP and remodel nucleosomes but retained the ability to bind its interaction partners, thereby suggesting that it functions as a dominant negative protein37. A subset of the SMARCA1 cohort in this study presented with macrocephaly, and many of these were caused by truncating variants in the SNF2-N domain or missense variants located in the HSS domain. This raised the question of whether truncating variants produce a stable truncated protein potentially with dominant negative functions, or an unstable protein suggestive of a LOF allele. To examine how different variants affect protein stability, we generated a full-length SMARCA1 expression plasmid with a FLAG-tag at the C-terminal end to distinguish it from the endogenous protein. We observed a stable protein of the proper size after transfection into U2OS cells (Fig. 5A). We next used site-directed mutagenesis to create 4 different variants identified in patients. First, we chose the R253* nonsense variant associated with macrocephaly and the nearby variant R259G that was not associated with macrocephaly. Similarly, we chose two variants within or adjacent to the HSS domain, namely R751Q and E771K, the latter variant associated with macrocephaly. Each of the missense variants contained the FLAG-tag on the C-terminus, while the R253* construct contained an N-terminal FLAG-tag. Following transfection, we observed that all 4 variants produced a stable protein, including the R253* variant which generated a protein of 35 kDa in size, albeit at a somewhat lower protein level than the other variants (Fig. 5A). In addition, we plated cells on cover slips for immunostaining, observing that the WT and all four variant proteins were targeted to the nucleus, as expected. It was also noted that the 35 kDa truncated protein produced by the R253* variant was not restricted to the nucleus and was also present in the cytoplasm (Fig. 5B). Taken together, these data suggest that the stability of the SMARCA1 protein is maintained in the presence of variants located within both the SNF2-N and HSS domains including a nonsense variant that generates a protein one-third the size of full-length SMARCA1.

A Immunoblots for SMARCA1 and FLAG protein following transfection of constructs expressing WT or four different SMARCA1 variants (R253*, R259G, R751Q, and E771K) into U2OS cells. Similar results were obtained from two independent transfections. B Transfected U2OS cells were also analyzed by immunofluorescence for FLAG (green) and SMARCA1 (red) expression. Nuclei were counterstained with DAPI. Scale bars = 20 μm. Images are representative of two independent transfections with >50 transfected cells examined per experiment. C Adult cortex and cerebellum protein lysates isolated from WT and Smarca1 KO mice were immunoblotted for SMARCA1 protein. Vinculin served as a protein loading control. Genotypes of the animals are shown at the bottom of each lane. Gt, Gene trap allele; +, WT allele; Y, Y chromosome; Cre-, absence of Cre recombinase; Cre+, presence of Cre recombinase. D Bar graph (top) depicting the neuroanatomical size differences observed as a percentage of the WT mice. Each numbered bar identifies the brain region analyzed (as indicated in the legend on the right) for area and length differences between Smarca1 cKO (n = 7) and control littermates (n = 5). Schematic illustration of a mouse sagittal brain section showing an overview of the observed changes (bottom). The light and dark yellow shading highlights the statistically significant differences (p < 0.05 and p < 0.01, respectively) while the grey shading denotes regions not analyzed for this study. Source data are provided as a Source Data file.
Characterization of Smarca1 conditional knockout mice
To model a SMARCA1 LOF allele in mice, we obtained Smarca1 targeted ES cells (ESCs) from the UC Davis Knockout Mouse Project (KOMP) repository (MMRRC:062622-UCD; Smarca1tm1a(KOMP)Wtsi) that were selected using a gene-trap insertion technique. The resulting Smarca1 allele contained loxP sites flanking exon 12 and the reporter cassette (En2 SA-IRES-βGEO-pA) within intron 11. The premise is that recognition of the En2 splice acceptor site generates a fusion transcript that encodes a truncated SMARCA1 protein and the β-Gal reporter. In addition, the loxP sites flanking exon 12 allow for Cre excision to generate a frame-shifted null allele. To determine whether the Smarca1tm1a(KOMP)Wtsi mice were constitutively null for SMARCA1 or would require Cre-recombination, we first generated WT, Smarca1tm1a(KOMP)Wtsi mice, and Smarca1tm1a(KOMP)Wtsi mice bred to the Nestin-Cre driver. Protein lysates were prepared from dissected brain regions (cortex and cerebellum) of young adult mice to assess SMARCA1 protein expression by Western blot. We observed that WT male mice (+/Y) produced a SMARCA1 protein of 135 kDA as expected, while SMARCA1 was not detected in lysates obtained from Smarca1tm1a(KOMP)Wtsi (Gt/Y Cre-) or Smarca1tm1a(KOMP)Wtsi with Cre (Gt/Y Cre+) mice (Fig. 5C). The lack of SMARCA1 protein in the Smarca1tm1a(KOMP)Wtsi mice indicates that the gene-trap model generates an unstable transcript and a constitutive null allele that does not require Cre excision. We hereafter refer to these animals as Smarca1 KO mice to distinguish them from the Smarca1ex6del animals.
Smarca1 KO mice were viable and born in normal Mendelian ratios with no obvious gross developmental defects, suggesting that SMARCA1 protein is not essential during embryogenesis or for postnatal survival. Brains dissected from 12 wild-type (WT) and 12 Smarca1 KO adult mice were collected and examined for neuroanatomical differences in 22 distinct brain regions38. The Smarca1 KO brains showed little difference from WT littermates although slight size reductions were observed in total brain width, corpus callosum length, and the area of the granule cell layer of the dentate gyrus (Fig. 5D). Within the E15.5 developing neocortex we observed no differences in the proportion of radial glial (Pax6 + ) or intermediate (Tbr2+) neural progenitor cells (Fig. 6A). Embryonic brains were pulsed for one hour with EdU to label progenitor cells in S-phase, which also showed no deficit in the proliferating proportion of the two progenitor populations. Staining with phospho-histone H3 (PH3), a marker of mitotic cells revealed an increase in the total number of PH3+ cells along the length of the lateral ventricle in the Smarca1 KO animals, yet this had no impact on overall cell number in the developing cortex (Fig. 6B). Cortical lamination was examined by immunostaining postnatal day 0 (P0) brain sections for deep layer (Tbr1, Ctip2) and upper layer (Satb2) neuronal markers. Smarca1 KO and WT brain sections at P0 showed no deficits in cortical lamination (Fig. 6C). Taken together, we conclude that the Smarca1 KO mice do not present with a brain overgrowth phenotype or with major deficits in forebrain development, which is in stark contrast to the phenotype of the Smarca1ex6del mice and many individuals of the SMARCA1 cohort with truncating variants.

A Control (WT) and Smarca1 KO (KO) coronal brain sections isolated at E15.5 were IF-stained to detect radial glia progenitors (PAX6), intermediate progenitors (TBR2), or cells in S-phase marked by their incorporation of EdU (green; 1 hour pulse). An EdU-click chemistry reaction was used to identify progenitor cells in S-phase. Cell nuclei were counter-stained with DAPI (blue). Images were taken at the dorsomedial region of the developing cortex. Graphs below the images depict the proportion of marker+/DAPI+ cells in WT and Smarca1 KO animals (n = 3 mice/genotype). Data are presented as mean values +/− SEM. B Control (WT) and Smarca1 KO (KO) coronal brain sections isolated at E15.5 were IF-stained to detect mitotic progenitor cells (PH3). The graph underneath the image is the proportion of PH3-positive neural progenitor cells along a 500 μm region of the lateral ventricle from WT and Smarca1 KO animals (n = 3 mice/genotype). Data are presented as mean values +/− SEM. Statistical significance was determined using an unpaired t test (two-tailed). *, p-value = 0.0307. C Control (WT) and Smarca1 KO (KO) coronal brain sections isolated at P0 and imaged at the dorsomedial region of the cerebral cortex. Sections were IF-stained for the deep-layer neuronal markers TBR1 (red; layer VI) and CTIP2 (white; layer V), and the upper layer neuronal marker SATB2 (green, layers II-V). The merged images are shown in the left panels. Cell nuclei were counter-stained with HOECHST dye (blue; right panels). Upper graph depicts total nuclei counts (DAPI+) within the P0 cortex of WT and Smarca1 KO mice (n = 3 mice/genotype). The lower graph shows the percentage of marker+ cells to the total nuclei count of WT and Smarca1 KO mice (n = 3 mice/genotype). Data are presented as mean values +/− SEM. For all graphs, gray bars represent WT samples while red bars represent Smarca1 KO samples. Scale bars = 100 μm for PH3 and 20 μm for all other panels. Source data are provided as a Source Data file.
Behavioral analysis of Smarca1 conditional knockout mice
To determine if these animals had altered behavior like other mouse NDD models, we performed a battery of tests to assess locomotor function, anxiety, social interaction, learning and memory. Initially, Smarca1 KO mice were tested for ambulatory movement, circadian activity, and sensorimotor coordination using the beam-break test and the rotorod test. In the beam-break test the mice are housed in a cage containing an array of photobeams and the ambulatory activity of the animals are recorded over a 48-hour period by tabulating the frequency of light beam interruptions (ambulatory activity count; Fig. 7A). Both WT and Smarca1 KO mice were more active during the dark cycle compared to the light cycle (WT, p = 0.0003; KO, p < 0.0001), while the Smarca1 KO mice also showed more activity than the WT mice in the dark (p = 0.0097) but equivalent activity (p = 0.567) during the light cycle (Fig. 7B). As a second measure of locomotor activity we utilized the rotorod test in which the mouse needs to utilize its coordination and endurance to stay on a rotating rod. The latency to fall is measured with each mouse tested 4 times a day for 2 consecutive days. We observed no significant difference in performance between WT and mutant mice (Fig. 7C).

A Plot of hourly ambulatory activity of WT and Smarca1 KO mice (Mutant) in a photobeam-break test. Grey shading depicts evening hours when mice are more active. B Quantification of total beam breaks during the light and dark cycle. Asterisk denotes a multiple comparison 2-way ANOVA test demonstrating a significant difference between WT and mutants in beam breaks during evening hours (dark), p = 0.0097. C The rotorod test measures the ability of the mice to demonstrate coordination and endurance to remain on a rotating rod. Both WT and Smarca1 KO mice (Mutant) showed improvement during consecutive trials (left graph) and similar performance as measured by the latency time (sec) to fall off the rotating rod (right graph). D In the social interaction test the WT and Smarca1 KO mice (Mutant) both spend more time interacting with the stranger mouse (compare social vs. habituation) and showed no difference in the time spent in the interaction zone with the stranger mouse. E In the open field test the WT and Smarca1 KO mice (Mutant) travel similar distances (i) and with a comparable velocity (ii). They also spend an equal amount of time in the brightly lit center of the arena (iii) and in the corners (iv). F In the Morris water maze the WT and Smarca1 KO mice (Mutant) learned to find the platform quicker each day during the trials (left graph). On the test day, they each spent equivalent time in the back right (BR) quadrant where the platform had been located. G In the cue and contextual fear conditioning test, the WT and Smarca1 KO mice (Mutant) showed a similar freezing response when presented with the tone. For all behavior tests a cohort of WT (n = 14) and Smarca1 KO (n = 13) male mice (P60-80) was used. The data are presented as mean values +/− SEM. For all graphs, gray bars represent WT samples while red bars represent Smarca1 KO samples. Source data are provided as a Source Data file.
The next test was the social interaction test in which the mouse is first allowed to become accustomed to a chamber that contains an empty cage. The mouse is then re-introduced to the chamber, but a stranger mouse is present in the cage. Normal behavior is to spend more time in the interaction zone exploring the stranger mouse. During both the habituation phase and when the stranger mouse was present, the Smarca1 KO and WT mice spent an equal amount of time within the interaction zone (Fig. 7D).
To assess anxiety, we then performed the open field test. Placement of the animal in a large open arena with bright illumination in the center of the chamber challenges the mouse’s inclination to explore a new environment over its natural aversion to brightly lit spaces. Mice are normally light aversive and spend less time in the center and more time in the darker corners of the arena. We observed that both WT and Smarca1 KO mice traveled similar distances at comparable velocities with no significant differences in the time spent in the center of the arena or in the corners (Fig. 7E).
To assess learning and memory, we performed two different tests, the Morris water maze, which tests their spatial memory to find a hidden platform, and the fear conditioning test, which tests contextual memory by associating a tone with an aversive foot shock. In the Morris water maze test, mice were trained over a 7-day period to use visual cues to find the platform below the surface of the water. On the probe day (day 8), the platform is removed, and the time spent in the quadrant where the platform was located versus the time spent in the other quadrants is a measure of the mouse’s ability to remember. We observed no significant difference in performance between the WT and Smarca1 KO mice during the training period, where the time to find the platform decreased each day (Fig. 7F, left panel). Similarly, on probe day, the WT and Smarca1 KO mice spent an increased amount of time in the target quadrant suggesting they remembered the location (Fig. 7F, right panel).
In the cue and contextual fear conditioning test, the mouse is placed in a neutral environment during training and given a foot shock shortly after hearing a tone. Fear is measured during training by freezing time, and this increases as they learn to associate the environment and the tone with the aversive stimulus. In subsequent days, the mouse is placed in the same context or given the same cue but does not receive the shock. Both WT and Smarca1 KO mice showed increased freezing time during training and in the contextual or cued memory portion of the test (Fig. 7G; SupplementaryFig. 3). Overall, the Smarca1 KO mice were more active in the dark but otherwise, this battery of behavior tests did not identify any statistically significant differences between the Smarca1 KO mice and their littermate controls that would be considered consistent with the learning deficits identified in individuals with SMARCA1 variants.
Analysis of compound NURF mutants
Previous biochemical studies have suggested that the NURF complex can associate with either SMARCA1 or SMARCA5 protein39. The severe microcephaly observed in Smarca5 and Bptf conditional knockouts further suggests that Smarca1 and Smarca5 may regulate brain growth via different mechanisms. To determine how specific NURF complexes relate to cortical growth, we examined brain size and cortical area from age-matched mice containing single or double gene knockouts of the Smarca1, Smarca5, or Bptf genes that comprise the NURF complex. The comparison showed that for the single gene knockouts, the Bptf knockout animals (Bptf cKO) had the most severe cortical phenotype (Fig. 8A), followed by the Smarca5 mutants, which matched previous reports16,17. The Smarca1ex6del mice were subjected to the same neuroanatomical analysis as the Smarca1 KO mice. This analysis showed that the total brain area of the Smarca1ex6del mice was 10% larger than controls (SupplementaryFig. 4), which was not fully reflected in the cortical area measurements, where we observed a 1% increase. Moreover, all combinations of double mutants (Smarca1/Smarca5 dKO, Smarca5/Smarca1ex6del, and Smarca5/Bptf dKO) resulted in a phenotype no more severe than the Bptf mutants alone (Fig. 8A). Surprisingly, the enlarged brain of the Smarca1ex6del mice was reduced to the same extent as the other NURF mutants when combined with Smarca5 ablation. Taken together, these data reveal that proper cortical development requires NURF, and that Smarca5 fully compensates when Smarca1 is completely ablated (no difference in brain size in Smarca1 KO) but not when some SMARCA1 protein is produced (e.g. Smarca1ex6del mice).

A Image showing size differences of brains (P20) dissected from different NURF mutant genotypes or their control littermates (WT). Numbers correspond to the cortical area ratio between the mutant and WT animals (n = 2). Ex6DEL refers to the Smarca1ex6del line. cKO, conditional knockout; dKO, double conditional knockout. B P10 cerebellar extracts isolated from control (WT) or Smarca1 KO animals immunoblotted for SMARCA1, SMARCA5, and CECR2. C) Co-immunoprecipitation assays performed from WT (upper panels) or Smarca1 KO (lower panels) cerebellar extracts with antibodies to SMARCA5 or rabbit IgG. 5% of the total IP lysate was loaded as input. Experiments in B and C were repeated two additional times using lysates prepared from different animals. Source data are provided as a Source Data file. D Model of NURF complex subunit switching. (i) The NURF complex is expressed in proliferating neuroprogenitors (NPCs) and in differentiated neurons during murine corticogenesis. (ii) During normal cortical development (WT), the catalytic subunit of NURF is SMARCA5 during growth which is switched to SMARCA1 during differentiation. (iii) Ablation of the Bptf gene (Bptf cKO) prevents formation of the NURF complex and results in minimal growth and a severely hypoplastic cortex. Individuals with BPTF pathogenic variants also present with microcephaly. (iv) When Smarca5 is inactivated (Smarca5 cKO), there is some compensation by SMARCA1 during growth that results in reduced cortical volume but a slightly larger cortex than the Bptf cKO mice. Individuals with SMARCA5 variants also present with microcephaly. (v) Smarca1 KO mice have a normal sized cortex which results from near complete compensation by the SMARCA5 protein. (vi) In the Smarca1ex6del mice, subunit switching occurs normally but the mutant protein lacks remodeling activity creating a dominant negative effect that delays differentiation and results in a larger brain size. Individuals with SMARCA1 variants and macrocephaly are predicted to lack remodeling activity and function in a dominant negative fashion, while variants not associated with macrocephaly are predicted to be functional hypomorphs.
The less severe hypoplastic neocortex of the Smarca5 cKO mice compared to the Bptf cKO animals likely results from the reported up-regulation of Smarca116,27. To further explore compensation by SMARCA5 in the Smarca1 KO, we examined SMARCA5 protein levels and the formation and composition of the CERF complex40 within the cerebellum of WT and Smarca1 KO mice. The switch to the cerebellum and analysis of the CERF complex was chosen for three reasons: (1) the lack of commercially available antibodies specific to BPTF for analysis of the NURF complex; (2) the Smarca5 cKO and Smarca1ex6del mutant animals show similar differential effects on cerebellar size to those observed in the neocortex; and (3) the CERF complex is enriched in the cerebellum27,37. We observed that SMARCA5 and CECR2 proteins were not upregulated but expressed at comparable levels in the WT and Smarca1 KO cerebellar extracts isolated from P21 mice (Fig. 8B). Immunoblots also confirmed that SMARCA1 protein was absent in the Smarca1 KO samples (Fig. 8B). Using SMARCA5 antibodies for immunoprecipitation (IP) assays, CECR2 was not co-purified in the WT samples (Fig. 8C, top panel) suggesting that the CERF complex comprised only SMARCA1 at P21, consistent with previous findings37,40. However, in Smarca1 KO lysates, the SMARCA5 antibody was able to co-IP the CECR2 protein, suggesting that it functionally compensates for the loss of SMARCA1 protein (Fig. 8C, lower panel).
Discussion
We describe 35 individuals with a variable neurodevelopmental syndrome caused by both truncating and missense variants in the X-linked SMARCA1 gene. Including the index family, a total of 10 different nonsense or frameshifting variants were identified that provide strong evidence of disease causation for the SMARCA1 gene. Three quarters of the affected individuals described were hemizygous males, most with maternally inherited variants. Heterozygous females comprised the remaining 25% of affected cases, and these individuals often showed a milder phenotype. The most prevalent clinical findings were ID/DD (77%) and speech delay and/or speech regression (83%), while numerous other organ systems were more variably involved. One third of individuals received an ASD diagnosis suggesting that SMARCA1 may also be identified in individuals with ASD. Given that many genes encoding chromatin remodeling proteins result in syndromic NDDs2,7,9,14,15, our data further show that SMARCA1-related neurodevelopmental disorder is a broad and variable clinical entity.
Attempts to stratify variant type with phenotypic features highlight the complexity of the genetic mechanism(s) underlying this distinct SMARCA1-related NDD. The index family carried a nonsense variant (p.Arg91*) that resulted in a brain overgrowth, a feature common to half the cohort. However, amongst individuals with truncating variants, the penetrance of macrocephaly was 70% suggesting that a straight LOF mechanism does not result in macrocephaly. It is noteworthy that macrocephaly was not detected in any of the CNV cases, suggesting that LOF alleles may function via a separate genetic mechanism. The normal brain size and lack of a phenotype observed in the Smarca1 KO mice suggest that LOF alleles may also be subject to functional compensation by SMARCA5. The exogenous expression of the p.Arg253* truncating variant in U2OS cells generated a truncated 35 kDa SMARCA1 protein that was stable and partially mislocalized to the cytoplasm, thereby suggesting that some truncating variants may generate truncated proteins with hypomorphic or dominant-negative activity. Collectively, these findings indicate that SMARCA1 truncating variants do not simply equate to LOF alleles and that further work is required to determine which truncating variants generate stable truncated proteins, and which ones are LOF alleles due to nonsense-mediated decay.
A dominant-negative genetic mechanism is supported by data from the Smarca1ex6del mouse model. These animals produce an internally truncated protein that can incorporate into complexes that lack remodeling activity and result in enhanced chromatin accessibility and a macrocephalic phenotype31,37,41. Similarly, SMARCA1 truncating variants capable of producing a stable truncated protein and associated with macrocephaly would be predicted to function in a dominant-negative fashion. Three of four missense variants associated with macrocephaly are in the HSS domain, which may prevent remodeling activity and could lead to dominant-negative effects on gene expression, similar to the Smarca1ex6del mice. Equally plausible is that the shared phenotypes between SMARCA1 truncating and missense variants are indicative that SMARCA1 variants function as hypomorphic alleles. Regardless, the genetic mechanism(s) causing the SMARCA1-related NDD is complex and may be variant dependent, yet we predict that they all impact NURF composition and NURF function in the developing brain. Some examination of SMARCA5 and/or BPTF protein levels in individuals with SMARCA1 pathogenic variants may also prove interesting in defining how these factors modulate phenotypic variability.
Another approach used to stratify the SMARCA1 phenotype was to generate genome-wide methylation profiles. These have been described in a growing number of genetic conditions, including NDDs caused by variants in other chromatin remodeling complexes, and they can be used to develop sensitive and specific molecular biomarkers referred to as epi-signatures42,43. While we demonstrated evidence of a mild global DNA methylation profile shared by patients with truncating variants and overgrowth, these changes were not sufficiently robust to be considered an epi-signature at this time and are further reflective of the phenotypic complexity underlying the SMARCA1-related NDD. Our ongoing work in expanding the DNA methylation data set may enable the future development of a sensitive biomarker that can be used for diagnostic purposes.
One other genotype-phenotype consideration remains noteworthy. An individual with a p.Ile647Thr variant developed psychotic symptoms that were notable because of a previously reported family with schizophrenia in four affected siblings with a p.Val384Met variant32. The p.Val384Met variant was recurrent as we identified three additional families, including one with similar phenotypic features (Supplementary Table 3). Although the pathogenicity of the p.Val384Met variant remains questionable as causative for the SMARCA1-related NDD (see Supplementary Table 2), future studies exploring how SMARCA1 variants contribute to psychoses and/or schizophrenia seem warranted.
Nine 9 affected females were identified in our cohort, including 7 with truncating variants. Females with truncating variants were evenly split between those with macrocephaly (3) and those without (4). Macrocephaly was also discordant in two sets of mother-daughter pairs, one family with the daughter presenting as macrocephalic, while in the other family, the mother was macrocephalic. Overall, there was some indication that the learning disabilities were milder in the female patients. For example, within the index family, individuals V2 (44 yrs) and V10 (49 yrs) had no reported learning disability, and although head circumference was mildly increased (+1.0 and +1.5 SD, respectively), macrocephaly and overgrowth were not significant. Individual VI8 (14 yrs; daughter of V10) was macrocephalic (+4.9 SD) with a mild learning disability. However, there was sparse clinical data available from mothers of affected individuals that precluded a full assessment of whether obligate carriers regularly present with milder features of the disorder. Many of these effects could be attributed to differences in X-inactivation skewing, but such studies were only available from three females, and they were discordant with phenotype. As such, further investigation is required to fully assess whether X-inactivation skewing is a key modulator of the phenotype in affected females.
SMARCA1 joins BPTF and SMARCA5 as members of the ISWI family of remodeling complexes that are causative of NDDs14,15. The latter two syndromes show considerable phenotypic overlap with microcephaly, a similar facial gestalt, and sandal gap toes common to each syndrome. Like the human BPTF and SMARCA5 NDDs, microcephaly is a common feature of the forebrain-specific conditional knockout mice for the Smarca5 and Bptf genes16,17. Both Smarca5 and Bptf mouse mutants show a reduction in intermediate progenitors, suggesting that the NURF complex has a critical role in amplifying this progenitor pool. Indeed, Bptf seems to be the critical determinant since none of the double knockouts (Smarca1/Smarca5, Smarca5/Smarca1ex6del, or Smarca5/Bptf) were any more severe than Bptf cKO alone (Fig. 8B).
Individuals with pathogenic variants in SMARCA1 are phenotypically distinct in comparison to BPTF and SMARCA5 NDDs, as suggested primarily by the high penetrance of macrocephaly. The opposite effect of pathogenic SMARCA1 versus SMARCA5 variants on cortical growth might be explained by the fact that SMARCA5 functions in proliferating progenitors while SMARCA1 drives differentiation. Indeed, data from our genetic epistasis experiments provide in vivo evidence for the phenotypic differences observed in individuals with pathogenic variants in these closely related genes, allowing us to propose the following model (Fig. 8D). During normal corticogenesis the NURF complex containing SMARCA5 is required for optimal progenitor proliferation and upon switching catalytic subunits to SMARCA1, NURF promotes neuronal differentiation (Fig. 8D, i and ii). Loss of Bptf in mice eliminates the NURF complex and generates the most severe cortical dysplasia (Fig. 8D, iii). In humans, variants in one copy of the human BPTF gene generate the NEDDFL syndrome with microcephaly13,14. Deletion of the mouse Smarca5 gene also leads to microcephaly but the severity is reduced compared to the Bptf mutants due to the upregulation and partial compensation by the Smarca1 gene (Fig. 8D, iv)16,27. Pathogenic variants in one copy of SMARCA5 cause the SMARCA5-related NDD that presents with microcephaly and significant phenotypic overlap with the NEDDFL syndrome15. When Smarca1 is ablated, we observed only minor differences in brain size and no significant changes in cortical development (Figs. 5D, and 6A-C,) suggesting that Smarca5 can adequately compensate for Smarca1 loss during corticogenesis in mice (Fig. 8D, v). This finding was further supported by our demonstration that SMARCA5 protein can complex with CECR2 in the absence of SMARCA1 protein but not in control samples where SMARCA1 is abundant (Fig. 8C). In instances when SMARCA1 protein is mutated but stable, like observed in the Smarca1ex6del mice, subunit switching occurs normally, but the phenotype depends on the effect of the mutation (Fig. 8D, vi). The Smarca1ex6del mice produce a protein with dominant-negative effects on NURF function and a macrocephalic phenotype. In the SMARCA1-related NDD cohort, we observe a combination of macrocephaly and individuals with a normally sized brain, which may reflect dominant-negative or hypomorphic functions of the variant, respectively. Indeed, the interplay between SMARCA5, SMARCA1, and BPTF protein levels, their temporal and spatial association within the NURF complex, and how that dictates the activity of its genomic targets are critical factors for defining brain size and, more importantly, disease severity. Given that a clinical continuum is observed in Coffin-Siris and Nicolaides-Baraitser syndromes, depending on which BAF encoding gene is mutated4,42, it is not unrealistic to consider that SMARCA1, SMARCA5, and BPTF variants may also align along a clinical NDD spectrum.
Methods
Patient Cohorts
This study is approved by the institutional review board at Seattle Children’s Hospital. Families were identified across several institutions with data shared via nodes of the MatchMaker Exchange (MME) network, including MyGene2, Gene-Matcher, PhenomeCentral, and by querying investigators with large cohorts of patients with intellectual disability, macrocephaly, overgrowth and/or other neurodevelopmental features35. Patient consents were obtained from all individuals for whom identifiable data are presented, including permission to publish photographs. All available clinical data from affected individuals were reviewed by the investigators. Pathogenic variants were identified in SMARCA1 using a combination of trio-based exome sequencing (ES; 23 families), short read whole genome sequencing (WGS; 3 families) in both clinical diagnostic and research settings. Sex, number, and age of patients are provided in Table 1. Gender was not considered in the study design, and participants were not compensated.
Methylation data analysis
The SMARCA1 cohort for methylation analysis consists of 19 individuals from 9 families: 7 females and 12 males for whom samples were available. Among the cohort, four individuals are unaffected female carriers of SMARCA1 variants, 13 carry truncating variants, and 6 have missense variants (See Table 1). DNA samples derived from peripheral blood were collected and anonymized. Replicate samples were also obtained from 6 individuals, 5 using LB-line and 1 using peripheral blood, and were only used for investigating unsupervised clustering using the identified probes for the methylation profiles.
The DNA obtained from the samples collected was subjected to bisulfite conversion using the Illumina Infinium MethylationEPIC BeadChip kit. The manufacturer’s protocols were followed, and quality control of the generated methylation data was done using the minfi R package44. Standard data preprocessing for Illumina arrays was implemented, including background correction and normalization. Density plots of beta values were checked, as well as recorded and predicted gender and age differences. Analysis was done on the pre-processed methylation data as described in previous research45,46,47. Initial filtering of probes includes removing those with failed detected p-values, are located in the X or Y chromosome, cross-reactive, or known to target CpG sites overlapping SNPs. This resulted in 772557 probes before the main analysis.
For differential methylation analysis, matched unaffected controls were first selected from the London Health Sciences Centre EpiSign Knowledge Database (EKD)45,46,47. Batches in the EKD known to cause batch effects and samples with more than 5% failed probes were excluded. Selection was done by matching age, sex, and array type of the cases using the R MatchIt package48 with case-control ratios ranging from 1:3 to 1:8, depending on the number of cases in the analysis. Batch effects and outliers in the original cohort, as well as the case-control training data used for the discovery of differential profiles, were inspected using principal component analysis (PCA).
Differential analysis of methylation signals between cases and matched controls was implemented for defining methylation profiles. A linear regression model was fitted using the limma R package49, with the methylation M values as predictors and case/control labels as the response. Covariates were also included using the estimated blood cell proportions of the samples. Using the empirical Bayes method, moderated t-statistics and corresponding p-values were computed for each probe, and p-values were adjusted using the Benjamini-Hochberg method to control for false discovery rates. Methylation differences were also estimated using beta values. Different parameter combinations involving the ranks by p-value and variable importance using receiver operating characteristic (ROC) curve analysis, as well as correlation, were considered for probes with a minimum methylation difference of 5% between cases and controls. The separation of the two groups was examined using hierarchical clustering and multidimensional scaling (MDS). A metric was also formulated to assess the quality of clustering by considering the minimum distance between the two groups and their respective standard deviations. The final parameter values and the selected differentially methylated probes defining the methylation profile were determined using the best clustering score.
Mouse generation and animal husbandry
Animals were housed in a facility under SPF (specific pathogen-free) conditions under ambient temperature and humidity, and a 12/12 light: dark cycle with water and food ad libitum. All experiments were performed according to the guidelines set by the University of Ottawa’s Animal Care Ethics Committee, maintaining the standards set by the Canadian Council on Animal Care in the Animal Care and Veterinary Services (ACVS) facility of the University of Ottawa. The generation and characterization of the Smarca1ex6del, Smarca5 cKO (Snf2hexon5fl/exon5fl), Bptf cKO (B6.129S1-Bptftm1.1Cwu/J; JAX strain #009367), and Emx1-Cre (B6.129S2-Emx1tm1(cre)Krj/J; JAX strain #005628) mouse lines are published16,17,27,31,50. All animals were maintained on a C57BL/6 background. Smarca1 engineered mESCs obtained from the KOMP repository (MMRRC:062622-UCD; Smarca1tm1a(KOMP)Wtsi) were passaged for 7 days, then harvested and provided to the University of Ottawa Transgenic Mouse Core Facility for blastocyst injections using a previously described protocol51. Six chimeric mice were identified, four showed germline transmission when bred to C57BL/6 female mice, and these were bred to homozygosity to generate the Smarca1 tm1a(KOMP)Wtsi line. These animals generate a truncated Smarca1 transcript after exon 11, whereby it splices to the LacZ reporter sequence. To ensure that we generated mice with a brain-specific null-allele for (Smarca1), we also bred the Smarca1 tm1a(KOMP)Wtsi females to the Nestin-Cre line52. This resulted in the excision of exon 12 and a frame-shift mutation. Indeed, both the Smarca1tm1a(KOMP)Wtsi and the Smarca1 KO animals did not express detectable SMARCA1 protein and were treated as null alleles. Double knockout mice were generated by interbreeding f/f lines prior to breeding to the Nestin-Cre or Emx1-Cre driver line.
Genotyping of the Smarca1 KO mice was performed under the following PCR conditions: a denaturing cycle at 94 °C for 2 min, 35 PCR cycles (94 °C for 20 sec, 63 °C for 25 sec, 72 °C for 25 sec) and a final cycle at 72 °C for 1 min with the following primer sequences: CSD-loxF (5’- GAGATGGCGCAACGCAATTAATG −3’) and CSD-Smarca1-R (5’-AAGAACACACTGGGTGCTAGGTAGG-3’) to amplify the Smarca1 KO allele (350 bp product); and CSD-Smarca1 (5’-CCCCTCAGAGGACAGTTATGCTAGG-3’) and CSD-Smarca1-ttR (5’- GCAGACATCATGAATCTTGCAGGC −3’) to amplify the Smarca1 WT allele (602 bp product).
Behavior studies
Young adult WT (n = 14) and Smarca1 KO (n = 13) male mice (P60-80) were transferred to the uOttawa Animal Behavior and Physiology Core for behavior testing. Mice were handled once a day for five days prior to behavioral testing by being lifted out of the cage and replaced several times. All behavior testing was conducted starting in the morning. Tests were conducted in the following order:
Beam break
Starting in the morning, test mice were habituated for an hour in the test room and then individually placed into a housing cage containing normal food and water. Activity within the cage was monitored for 48 hours by the Photobeam Activity System (San Diego Instruments, San Diego, CA), with X- and Y-axis beam breaks recorded. The cages were maintained on a 7 AM–7 PM light cycle. At the end of the recording period, the mice were returned to their home cages. Beam breaks were analyzed in one-hour bins.
Open field assay
Mice were habituated for 30 minutes before being placed in the middle of a 45 cm × 45 cm x 45 cm white acrylic plastic arena with a coroplast floor. Light levels were set at 300 lux. Mice were allowed to freely investigate the arena for 10 minutes while being recorded from above by Noldus Ethovision 17.5 software (Noldus Instruments, Wageningen, Netherlands). At the end of the recording period, the mice were returned to their home cage.
Adult social interaction assay
Mice were habituated for 30 minutes in 100 lux regular light and in red light in the test room for 30 minutes before testing. Each mouse was placed in a 45 cm × 45 cm x 45 cm white acrylic plastic arena with an empty 5.5 cm × 9.6 cm × 30 cm plastic holder with wire mesh walls. The mouse was allowed to investigate freely for five minutes and then removed to a clean holding cage for an intertrial interval of five minutes. A gender-matched social interactor mouse was placed in the holder. The test mouse was then returned to the arena and allowed to investigate freely for five minutes. Test mice activity and interaction time with the holder were recorded by Noldus Ethovision 17.5 software (Noldus Instruments, Wageningen, Netherlands).
Rotarod Assay
Mice were habituated in the test room in regular light for 30 minutes prior to testing. Mice were assessed using a Ugo Basile 47659 Rota-Rod NG (Ugo Basile, Gemonio, Italy). Mice were placed on a 4 rpm rotating rod facing away from the experimenter. The rod accelerated from 10 rpm to 40 rpm in 30 seconds and was maintained at 40 rpm for a maximum test time of five minutes. Latency to fall from the rod was recorded. Mice completed four trials per day, with an intertrial interval of 10 minutes, for two days.
Morris Water Maze Assay
Mice were habituated in the test room in regular light for 30 minutes before testing. The Morris water maze is a large plastic tank (~132 cm diameter, Maze Engineers, Skokie, IL) filled with tap water maintained at 22 °C and made opaque with white tempera paint. A 10 cm diameter platform was placed in the pool in the BR quadrant at ~10 mm below the water level. A black cross and a black square were placed on two of the walls of the room as spatial cues. The arena was recorded by a ceiling-mounted camera and tracked by Noldus Ethovision 17.5 software (Noldus Instruments, Wageningen, Netherlands); the experimenter monitored each trial while hidden behind a curtain in the room. The mouse was placed in the pool at one of four loading locations and allowed to search for the pool for one minute; if the mouse did not find the platform, it was placed on the platform for ~5 seconds and then returned to its home cage. Each mouse completed four trials per day of training, being placed in the pool at each loading location once per day in a different order each day, with an intertrial interval of 20 minutes. The mice were trained for seven days. Mice were subjected to a probe trial on the 8th day of testing, where the platform was removed and the mouse allowed to swim for 1 minute before being removed from the pool.
Fear conditioning
The fear conditioning test was conducted in a Phenotyper chamber (Noldus Instruments, Wageningen, Netherlands) modified with a shock grid floor (Med Associates, Fairfax, VT). Noldus Ethovision 17.5 was used to record activity and freezing periods (Noldus Instruments, Wageningen, Netherlands). On the training day, mice were brought directly from the housing room and placed in the test chamber. After two minutes, the mice received 2 tone-shock pairings (a 30 second tone co-terminated with a 2 second 0.35 mV electric shock) with 1 minute separating the tone-shock sessions. The mice were then returned to their home cage. 24 hours later, the mice were returned to the chamber and recorded for 6 minutes with no tone or shock to assess contextual memory. 24 hours later, the chamber was modified with triangular walls, a textured floor, vanilla scent, and different lighting conditions. The mice were placed in the chamber with no sound for three minutes and then with the same tone from the training day for three minutes to assess cue memory.
Tissue collection, EdU injection, and neuroanatomical analysis
To obtain embryonic brains, time-mated mice were established and checked daily for a vaginal plug, which was considered embryonic day 0.5 (E0.5) of the gestation period. At E15.5, embryos were quickly dissected, heads cut off, and fixed in 4% paraformaldehyde (PFA) at 4o C overnight. For EdU pulse labeling, timed-mated pregnant females were injected intraperitoneally with 100 μg/g body weight of 5-Ethynyl-20-deoxyuridine (EdU; Sigma-Aldrich, Oakville, ON, Canada) 60 min before sacrifice and tissue collection. The following day, heads were placed in a 30% sucrose solution, and once submerged, they were transferred to a 1:1 solution of 30% sucrose solution and OCT (Tissue-Tek, Sakura Americas, Torrance, CA, United States), then snap-frozen in liquid nitrogen and stored at −80 °C until sectioned. Brains analyzed at postnatal day 0.5 (P0.5) were prepared in the same manner as embryonic brains. P20 mice were transcardially perfused with saline followed by 4% paraformaldehyde (PFA) in 0.1 M PBS, prior to dissecting the brains. Brains were incubated in 4% PFA at 4 oC overnight, and the remainder of the protocol was identical to the embryonic brain protocol described above. The protocol used for 2D-neuroanatomical measurements of Smarca1 KO brains has been described in detail38. In addition, cortical area measurements were determined using Fiji from whole-brain images of P21 single and double knockout mouse mutants and their control littermates. Data was presented as a ratio of the mutant cortical area to the WT cortical area for each line (n = 2).
Immunofluorescence
Frozen murine brains embedded in OCT were sectioned (12 µm) using a Leica CM1850 cryostat and then washed in 1xPBST (0.1 M Phosphate-Buffered Saline, 0.1% Triton X-100) before use. Where antigen retrieval was required, slides were incubated in 10 mM Sodium citrate buffer (pH 6.0) that was heated to 80o C for 30 min. For EdU immunodetection, sections were first washed with 1X PBS prior to incubation with an EdU staining solution (100 mM Tris-HCL, pH 7.2, 2 mM CuSO4, 10 mM fluorescent azide, 50 mM ascorbic acid) for 1 hour at room temperature. Slides were then washed and incubated with Hoechst dye before imaging as described below.
Sections were blocked (10% horse serum/PBST) for 30 min at room temperature. All slides were then incubated overnight at 4 oC in primary antibody solution (0.04% Triton X-100, 3 mg/mL bovine serum albumin in 1X PBS) with the following primary antibodies: rabbit anti-Pax6 (1:500; Cedarlane, PRB-278P-100); rabbit anti-Tbr2 (1:200; Abcam, ab23345); rabbit anti-Tbr1 (1:100; Abcam, ab31940); rat anti-Ctip2 (1:300; Sigma, 06-570); mouse anti-Satb2 (1:300; Abcam, ab51502); and rabbit anti-phospho-Histone H3 (1:500, Abcam, ab12345). All primary antibodies have been previously validated in other studies16,17,41,53. Slides were then washed in 1xPBS and incubated for 1 hour at room temperature with a 1:500 dilution of the appropriate secondary antibody. Secondary antibodies included anti-rabbit-647 Alex Fluor (Invitrogen, a31573), anti-mouse-488 Alex Fluor (Invitrogen, a21202), anti-rat-555 (Invitrogen, a48263), or anti-rabbit-555 (Invitrogen, a31572). Sections were then washed several times with 1xPBS, incubated with Hoechst 33342 dye (1 μg/ml; 20-[4-ethoxyphenyl]−5-[4-methyl-1-piperazinyl]−2,50-bi-1H-benzimidazole trihydrochloride trihydrate; Thermo FisherScientific, #H3570) for 15 min at room temperature. Finally, slides were mounted on coverslips with DAKO fluorescent mounting medium (Agilent Technologies, #S3023) and imaged with a Zeiss Axiovert Observer Z1 epifluorescent/light microscope with an AxioCam cooled color camera (Zeiss). Images were exported to Adobe Photoshop CS5 (Adobe Systems, Inc.) for further processing, cell counting, and figure preparation.
Cell counts were performed from the coronal cortical images acquired, using a representative region demarcated with a 200 μm wide column that comprised the full height of the cortex. Within each column, the mean cell number (any marker-positive cell) and total cell count (Hoechst+ nuclei) was quantified from a minimum of three sections from at least three biological replicates. A Student's t-test was used for statistical comparison between control and Smarca1 KO samples. The resulting bar graph plots denote mean values +/- SEM. An asterisk was used to designate statistical significance (p < 0.05).
Protein isolation, immunoblots, and co-immunoprecipitation
Brain cortices or cerebella were quickly dissected from individual animals and snap frozen in liquid nitrogen. Tissues were manually sheared then incubated at 4 oC, with rocking, in ice-cold lysis buffer (20 mM Tris-HCl pH 8, 137 mM NaCl, 1% NP-40, 2 mM EDTA) supplemented with Halt protease inhibitor cocktail (Thermo Fisher Scientific, 78425) for 10 min. Lysed samples were pre-cleared by centrifugation (12,000 x g for 15 min) and proteins quantified using the Bio-Rad Protein Assay Dye Reagent Concentrate (Bio-Rad Laboratories). Protein samples were resolved on Bis-Tris 4–12% gradient gels (NuPage, Invitrogen, USA) by electrophoresis (90–150 V) using the Bio-Rad Mini-PROTEAN Tetra Cell and then blotted onto PVDF membranes (Immobilon-P; Millipore) by wet transfer at 0.35 A for 90 mins using the Bio-Rad Mini Trans-Blot cell. Membranes were blocked (45 min, room temperature) with 5% skim milk in TBST (Tris-buffered saline containing 0.05% Triton X-100), and incubated (4 °C, overnight) with the following antibodies: rabbit anti-SMARCA5 (1:500; Abcam ab72499); rabbit anti-SMARCA1 (1:500; Abcam ab37003); rabbit anti-CECR2 (1:500; gift from Dr. Heather McDermid, Univ. Alberta); mouse anti-β-actin (1:3000; Sigma, A1978). All antibodies have been previously validated in other studies using tissue lysates from genetic knockouts27,31,37,54,55. After washing (5 × 5 min TBST) the membranes were incubated for 1 hour at room temperature with HRP-conjugated goat anti-rabbit (1:5000, Jackson Immunoresearch, 111-035-003) IgG (H + L) secondary antibodies. Membranes were washed 5 × 5 min in TBST after antibody incubations, and signals were detected using the Pierce Supersignal West Fempto (Pierce, Cat # 34095) chemiluminescence substrate.
For co-immunoprecipitation, each reaction consisted of 500 μg of protein lysate, with 1 μg of SMARCA5 antibody (Abcam, ab72499) or rabbit IgG for negative controls. The final volume was adjusted to 500 μl with protease inhibitor (1/1000) and lysis buffer. Reactions were incubated at 4 oC overnight and then 30 ml of Protein A/G magnetic beads (Bioclone Inc, cat # MA102) were added and the incubation continued for another hour. Beads were washed 5 times (at 4 oC in 5 min intervals) in PBS containing 0.3% triton-X before eluting in 0.1 M glycine (pH 2.5) at room temperature for 10 min, with occasional agitation. The elution step was performed 4 times, and volumes were pooled prior to immunoblotting (described above).
Generation and Analysis of Smarca1 Variants
Directional subcloning of human SMARCA1 cDNA (NM_001282874.2) into pBRIT-LoxP-NTAP and pBRIT-LoxP-CTAP retroviral vectors (17520 Addgene, Cambridge, MA, USA) has been previously described27. Patient mutation R253* was introduced to the pBRIT-LoxP-SNF2L-NTAP plasmid and patient mutations R259G, E771K, and R751Q were introduced into the pBRIT-LoxP-SNF2L-CTAP plasmid using the New England Biolabs (NEB) Site Directed Mutagenesis kit (NEB, cat#E0554S). Primer sets are listed in Supplementary Table 4. U2OS cells were cultured at 37 °C in DMEM with 10% FBS and 1% penicillin–streptomycin. Briefly, cells were grown to a confluency of ~80% and transfected using Lipofectamine 2000 (Invitrogen, cat# 11668030).
Cells cultured on coverslips were fixed with 4% paraformaldehyde (PFA) for 10 minutes at room temperature (RT). After fixation, cells were permeabilized with 0.5 % NP40 in Immunofluorescence (IF) solution (0.5% BSA in PBS) for 10 minutes at RT, quenched with 50 mM NH4Cl in PBS for 10 minutes at RT, and then blocked with 10% horse serum in IF solution for 10 minutes at RT. Cells were stained with rabbit anti-SNF2L(1:500; Cell Signaling, cat#12483S), rabbit anti-HA(1:500; Cell Signaling, cat#37245) for an hour at RT, followed by incubation with the corresponding secondary Alexa Fluor antibodies (Invitrogen, cat #A11008, cat#A21244) for an hour at RT. Coverslips were mounted onto slides with mounting medium(Agilent Technologies, cat # S302380-2). Cells were imaged with an Axio Imager M1 microscope (Zeiss) using a 63X objective. Images were prepared with Fiji software. All SNF2L and HA-positive cell counts were performed relative to DAPI-labeled nuclei.
Cells were homogenized in ice-cold lysis buffer supplemented with protease inhibitor cocktail (Sigma, United States; P8340) and phosphatase inhibitor cocktail (Fisher, cat # 78441), and incubated for 10 min at 4 oC with gentle mixing. Lysates were centrifuged for 5 min at 13,000 g at 4 oC to remove cell debris and the supernatant was quantified for protein concentration by the Bradford method (BioRad, cat # 500-00006). The protein was diluted with 4X Laemmli buffer containing 15% 2-mercaptoethanol and boiled for 5 minutes at 95 oC. 20 ug of protein was resolved with 10% SDS-PAGE gels under denaturing conditions for 1 hour at 175 V and transferred onto PVDF membranes (BioRad, cat # 162-0177) by wet transfer for 1.5 hours at 0.2 A. Membranes were blocked for 1 hour at RT with 5% skim milk in Tris-buffered saline with 0.05% Tween-20 (-TBST) and blotted with rabbit anti-SNF2L (1:1000; Cell Signaling, cat#12483S), rabbit anti-HA (1:1000; Cell Signaling, cat#37245), and mouse anti-beta actin (1:10,000; Sigma, A1978). Membranes were incubated for 1 hour at RT in horseradish peroxidase (HRP)-conjugated goat anti-rabbit (1:1000) (Sigma, cat#A4914) or goat anti-mouse (1:10000) (Sigma, cat#A5906) secondary antibodies. Membranes were washed 3 times for 5 minutes in TBST after antibody incubations, and chemiluminescent signal was detected using Clarity Western ECL Substrate (BioRad, cat#1705061).
Statistics & reproducibility
Families with SMARCA1 variants were identified across several institutions with data shared via nodes of the MatchMaker Exchange (MME) network. No statistical method was used to predetermine sample size. A random deadline was set to receive clinical data for inclusion in the initial description of the SMARCA1-related NDD. Families continue to be referred and collected for further studies. No data were excluded from the analyses. All animal studies were performed with a minimum of three biological replicates. Cell counts were performed with the investigator blinded to genotype. Raw counts are included in the Source Data file. The counting data were analyzed for statistical significance using an unpaired t-test (two-tailed). P-values > p = 0.05 were considered significant and were demarcated with an asterisk on the graph, with the specific p-value provided in the figure legend. For the open field behavior tests, an unpaired t-test (two-tailed) was used for analysis. For all other behavior tests, a two-way ANOVA (Multiple comparisons) was used to test for significance across groups. Significant differences were included in the figure legend along with the test used. Non-significant p-values were included in the source data only. Unscanned blots are also provided in the Source Data file.
Ethics
Our research complies with all relevant regulations. The clinical study protocol was approved by the institutional review board at Seattle Children’s Hospital (Approval # 13291), and written consent was obtained for all clinical samples obtained and from all individuals for whom identifiable data are presented, including permission to publish photographs. Animal experiments were conducted under the approval and guidance of the University of Ottawa’s Animal Care Ethics Committee (Protocol numbers: OHRI-3773 and OHRI-3762), maintaining the standards set by the Canadian Council on Animal Care.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Clinical data were obtained under an IRB-approved protocol. De-identified clinical data are available upon request from the corresponding authors. For the epi-signature analyses, datasets used in this study that are available publicly are previously described (Am J Hum Genet. 2020;106(3):356–70). Anonymized data for each subject is described in the study. The individual genomic and epigenomic or any other personally identifiable data for other samples in the EpiSign Knowledge Database (EKD) are prohibited from deposition in publicly accessible databases due to institutional and ethics restrictions. Specifically, these include data and samples submitted from external institutions to London Health Sciences EKD that are subject to Institutional Material and Data Transfer agreements, data submitted to London Health Sciences for episignature assessment under Research Services Agreements, and research study cohorts under Institutional Research Ethics Approval (Western University REB 106302; and REB 116108). Some of the software packages used in this study are publicly available as described in the Materials and Methods. EpiSign™ is a commercial software and is not publicly available. Source data are provided with this paper.
References
Gourisankar, S., Krokhotin, A., Wenderski, W. & Crabtree, G. R. Context-specific functions of chromatin remodellers in development and disease. Nat. Rev. Genet. 25, 340–361 (2024).
Valencia, A. M. et al. Landscape of mSWI/SNF chromatin remodeling complex perturbations in neurodevelopmental disorders. Nat. Genet. 55, 1400–1412 (2023).
Clapier, C. R. & Cairns, B. R. The biology of chromatin remodeling complexes. Annu. Rev. Biochem. 78, 273–304 (2009).
Bogershausen, N. & Wollnik, B. Mutational Landscapes and Phenotypic Spectrum of SWI/SNF-Related Intellectual Disability Disorders. Front. Mol. Neurosci. 11, 252 (2018).
Sokpor, G., Xie, Y., Rosenbusch, J. & Tuoc, T. Chromatin Remodeling BAF (SWI/SNF) Complexes in Neural Development and Disorders. Front. Mol. Neurosci. 10, 243 (2017).
Li, Y. et al. The emerging role of ISWI chromatin remodeling complexes in cancer. J. Exp. Clin. Cancer Res. 40, 346 (2021).
D’Souza, L., Channakkar, A. S. & Muralidharan, B. Chromatin remodelling complexes in cerebral cortex development and neurodevelopmental disorders. Neurochem Int 147, 105055 (2021).
Tsurusaki, Y. et al. Mutations affecting components of the SWI/SNF complex cause Coffin-Siris syndrome. Nat. Genet. 44, 376–378 (2012).
Van Houdt, J. K. et al. Heterozygous missense mutations in SMARCA2 cause Nicolaides-Baraitser syndrome. Nat. Genet. 44, 445–449 (2012). S441.
Goodwin, L. R. & Picketts, D. J. The role of ISWI chromatin remodeling complexes in brain development and neurodevelopmental disorders. Mol. Cell Neurosci. 87, 55–64 (2018).
Allegri L. et al BAZ1B is a candidate gene responsible for hypothyroidism in Williams syndrome. Eur. J. Med. Genet. 103894 (2020).
Fusco, C. et al. Smaller and larger deletions of the Williams Beuren syndrome region implicate genes involved in mild facial phenotype, epilepsy and autistic traits. Eur. J. Hum. Genet. 22, 64–70 (2014).
Glinton, K. E. et al. Phenotypic expansion of the BPTF-related neurodevelopmental disorder with dysmorphic facies and distal limb anomalies. Am. J. Med Genet A 185, 1366–1378 (2021).
Stankiewicz, P. et al. Haploinsufficiency of the Chromatin Remodeler BPTF causes syndromic developmental and speech delay, postnatal microcephaly, and dysmorphic features. Am. J. Hum. Genet 101, 503–515 (2017).
Li, D. et al. Pathogenic variants in SMARCA5, a chromatin remodeler, cause a range of syndromic neurodevelopmental features. Sci. Adv. 7, eabf2066 (2021).
Alvarez-Saavedra, M. et al. Snf2h Drives Chromatin Remodeling to Prime Upper Layer Cortical Neuron Development. Front. Mol. Neurosci. 12, 243 (2019).
Zapata G., Yan K. & Picketts D. J. Generation of a mouse model of the neurodevelopmental disorder with dysmorphic facies and distal limb anomalies (NEDDFL) syndrome. Hum. Mol. Genet. 31, 3405–3421 (2022).
Barak, O. et al. Isolation of human NURF: a regulator of Engrailed gene expression. EMBO J. 22, 6089–6100 (2003).
Koludrovic, D. et al. Chromatin-remodelling Complex NURF is essential for differentiation of adult melanocyte stem cells. PLoS Genet 11, e1005555 (2015).
Kwon, S. Y., Grisan, V., Jang, B., Herbert, J. & Badenhorst, P. Genome-wide mapping targets of the metazoan chromatin remodeling Factor NURF reveals nucleosome remodeling at enhancers, core promoters and gene insulators. PLoS Genet. 12, e1005969 (2016).
Landry, J. W. et al. Chromatin remodeling complex NURF regulates thymocyte maturation. Genes Dev. 25, 275–286 (2011).
Qiu, Z. et al. Functional interactions between NURF and Ctcf regulate gene expression. Mol. Cell Biol. 35, 224–237 (2015).
Wysocka, J. et al. A PHD finger of NURF couples histone H3 lysine 4 trimethylation with chromatin remodelling. Nature 442, 86–90 (2006).
Xiao, H. et al. Dual functions of largest NURF subunit NURF301 in nucleosome sliding and transcription factor interactions. Mol. Cell 8, 531–543 (2001).
Xu, B. et al. The Chromatin Remodeler BPTF Activates a stemness gene-expression program essential for the maintenance of adult hematopoietic stem cells. Stem Cell Rep. 10, 675–683 (2018).
Sala, A. et al. Genome-wide characterization of chromatin binding and nucleosome spacing activity of the nucleosome remodelling ATPase ISWI. EMBO J. 30, 1766–1777 (2011).
Alvarez-Saavedra, M. et al. Snf2h-mediated chromatin organization and histone H1 dynamics govern cerebellar morphogenesis and neural maturation. Nat. Commun. 5, 4181 (2014).
Uhlen, M. et al. Proteomics. Tissue-based map of the human proteome. Science 347, 1260419 (2015).
Lazzaro, M. A. & Picketts, D. J. Cloning and characterization of the murine Imitation Switch (ISWI) genes: differential expression patterns suggest distinct developmental roles for Snf2h and Snf2l. J. Neurochem 77, 1145–1156 (2001).
Telley, L. et al. Sequential transcriptional waves direct the differentiation of newborn neurons in the mouse neocortex. Science 351, 1443–1446 (2016).
Yip, D. J. et al. Snf2l regulates Foxg1-dependent progenitor cell expansion in the developing brain. Dev. Cell 22, 871–878 (2012).
Homann, O. R. et al. Whole-genome sequencing in multiplex families with psychoses reveals mutations in the SHANK2 and SMARCA1 genes segregating with illness. Mol. Psychiatry 21, 1690–1695 (2016).
Karaca, E. et al. Genes that affect brain structure and function identified by rare variant analyses of mendelian neurologic disease. Neuron 88, 499–513 (2015).
Lopes, F. et al. Identification of novel genetic causes of Rett syndrome-like phenotypes. J. Med. Genet. 53, 190–199 (2016).
Philippakis, A. A. et al. The Matchmaker Exchange: a platform for rare disease gene discovery. Hum. Mutat. 36, 915–921 (2015).
Leonard, J. D. & Narlikar, G. J. A nucleotide-driven switch regulates flanking DNA length sensing by a dimeric chromatin remodeler. Mol. Cell 57, 850–859 (2015).
Goodwin, L. R., Zapata, G., Timpano, S., Marenger, J. & Picketts, D. J. Impaired SNF2L Chromatin remodeling prolongs accessibility at promoters enriched for Fos/Jun binding sites and delays granule neuron differentiation. Front. Mol. Neurosci. 14, 680280 (2021).
Collins, S. C. et al. A method for parasagittal sectioning for neuroanatomical quantification of brain structures in the adult mouse. Curr. Protoc. Mouse Biol. 8, e48 (2018).
Oppikofer, M. et al. Expansion of the ISWI chromatin remodeler family with new active complexes. EMBO Rep. 18, 1697–1706 (2017).
Banting, G. S. et al. CECR2, a protein involved in neurulation, forms a novel chromatin remodeling complex with SNF2L. Hum. Mol. Genet. 14, 513–524 (2005).
Ahmed, R. et al. Transgenic mice with an R342X mutation in Phf6 display clinical features of Borjeson-Forssman-Lehmann Syndrome. Hum. Mol. Genet. 30, 575–594 (2021).
Aref-Eshghi, E. et al. BAFopathies’ DNA methylation epi-signatures demonstrate diagnostic utility and functional continuum of Coffin-Siris and Nicolaides-Baraitser syndromes. Nat. Commun. 9, 4885 (2018).
Levy, M. A. et al. Novel diagnostic DNA methylation episignatures expand and refine the epigenetic landscapes of Mendelian disorders. HGG Adv. 3, 100075 (2022).
Aryee, M. J. et al. Minfi: a flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics 30, 1363–1369 (2014).
Aref-Eshghi, E. et al. Evaluation of DNA methylation episignatures for diagnosis and phenotype correlations in 42 Mendelian neurodevelopmental disorders. Am. J. Hum. Genet. 106, 356–370 (2020).
Sadikovic, B., Levy, M. A. & Aref-Eshghi, E. Functional annotation of genomic variation: DNA methylation episignatures in neurodevelopmental Mendelian disorders. Hum. Mol. Genet. 29, R27–R32 (2020).
Sadikovic, B. et al. Clinical epigenomics: genome-wide DNA methylation analysis for the diagnosis of Mendelian disorders. Genet Med. 23, 1065–1074 (2021).
Ho, D., Imai, K., King, G. & Stuart, E. A. MatchIt: nonparametric preprocessing for parametric causal inference. J. Stat. Softw. 42, 1–28 (2011).
Ritchie, M. E. et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47 (2015).
Gorski, J. A. et al. Cortical excitatory neurons and glia, but not GABAergic neurons, are produced in the Emx1-expressing lineage. J. Neurosci. 22, 6309–6314 (2002).
Longenecker G., Kulkarni A. B. Generation of gene knockout mice by ES cell microinjection. Curr. Protoc. Cell Biol. Chapter 19, Unit 19 14 19 14 11-36 (2009).
Berube, N. G. et al. The chromatin-remodeling protein ATRX is critical for neuronal survival during corticogenesis. J. Clin. Invest. 115, 258–267 (2005).
Huh, M. S. et al. Stalled replication forks within heterochromatin require ATRX for protection. Cell Death Dis. 7, e2220 (2016).
Niri, F., Terpstra, A. N., Lim, K. R. Q. & McDermid, H. E. Chromatin remodeling factor CECR2 forms tissue-specific complexes with CCAR2 and LUZP1. Biochem. Cell Biol. 99, 759–765 (2021).
Norton, K. A. et al. Implantation failure and embryo loss contribute to subfertility in female mice mutant for chromatin remodeler Cecr2. Biol. Reprod. 104, 835–849 (2021).
Acknowledgements
We thank the families and referring physicians for their participation in this study. The authors would like to thank Drs. Pierre Mattar and Rashmi Kothary for critical reading of the manuscript. Special gratitude to Barbara and Eric Mann, whose visionary generosity made this research and discovery possible. We are grateful to all donors to Seattle Children’s Research Institute who invest in breakthrough discoveries that help prevent, treat, and eliminate childhood disease. Prediction of functional impact of variants using computational tools was performed by Lindsay Clark at Research Scientific Computing, Seattle Children’s Research Institute. We would also like to thank Dr. Kerstin Ure and the uOttawa Animal and Behavior Core for performing the behavior studies on the Smarca1 KO mice and for helping with the analyses. This work was supported by operating grants from the Canadian Institutes of Health Research (PJT180577 and MOP142398; DJP); the Qatar National Research Fund (NPRP 5-175-3-051; TB-O); the National Human Genome Research Institute grant R01 HG009141 and National Institute of Mental Health of the National Institutes of Health award U01MH119689 (EO’H), SFARI, United States (704413; EBR) and from the Stanley Center for Psychiatric Research at the Broad Institute (EO’H); the Research Foundation – Flanders (1861419N, G041821N, and G056122N; SW); the Queen Elisabeth Medical Foundation (SW); the Italian Ministry for Education, University and Research (Ministero dell’Istruzione, dell’Università e della Ricerca - MIUR) PRIN2020 code 20203P8C3X (AB); and #NEXTGENERATIONEU (NGEU) funded by the Ministry of University and Research (MUR), National Recovery and Resilience Plan (NRRP), project MNESYS (PE0000006) – A Multiscale integrated approach to the study of the nervous system in health and disease (DN. 1553 11.10.2022) to EG. ML is a full member of the ERN EpiCARE. AMI wishes to thank Drs. Jillian Parboosingh and Morteza Seifi and acknowledge work done under the Care4Rare Canada Consortium funded by Genome Canada and the Ontario Genomics Institute, the Canadian Institutes of Health Research, Ontario Research Fund, Genome Alberta, Genome British Columbia, Genome Quebec, Children’s Hospital of Eastern Ontario Foundation, and the Alberta Children’s Hospital Foundation. The authors would also like to acknowledge the contributions of Christopher A. Walsh, Muna Al-Saffar, Jennifer E. Neil, Haley Gardiner, Ganeshwaran H. Mochida, Lynn Pais, Brigitte Melly, Anne O’Donnell-Luria, Emma Eastman, and Fatima Khan in the ascertainment and assessment of affected individuals. This work is dedicated to the memory of Professor Lionel van Maldergem who has made remarkable contributions to the field of genetics, including significant contributions to this work.
Author information
Authors and Affiliations
Contributions
K.Y. performed all animal husbandry, cortical dissections, and IF analysis. S.T. performed immunoblots and co-IP experiments. M.L. and P.J. performed site-directed mutagenesis and introduced expression constructs into U2OS cells for analysis. E.P., N.O., and G.M.M. compiled clinical data and generated the pedigree and growth figures. R.R. and B.S. performed the differential methylation analysis and corresponding figures. B.Y. and S.C. performed brain morphometric analyses. A.Z., E.B.B., J.P., K.G.M., M.J.G.S., W.B.D., K.L.P., D.M.F.M., A.F.J., P.Ja., M.P.B., F.S.S., A.B., V.A., E.G., M.K., B.I., S.Co., B.C., W.D., K.E.S., K.S., N.S., S.W., R.O., A.M.I., D.C.K., T.B.O., R.C.Y., M.C.K., S.B., A.P., S.M., S.N., S.Ch., S.A.P., K.M., B.B., K.K., E.O., Z.B., K.A.D., T.K., A.G., B.A., K.B., P.M., P.Jo., F.D., E.H., L.P., L.C., D.L., and D.B. contributed patient clinical data, photos, genomic samples and/or DNA sequencing information. K.Y. and D.J.P. prepared all other figures. G.M.M. and D.J.P. designed, supervised, and provided funding for the project. G.M.M., D.B., A.Z., B.S., and D.J.P. contributed to the writing of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
KGM and MJGS are employees of GeneDX, LLC. All remaining authors declare no competing financial interests.
Peer review
Peer review information
Nature Communications thanks Toshiyuki Yamamoto and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Mirzaa, G.M., Yan, K., Relator, R. et al. Pathogenic variants in SMARCA1 cause an X-linked neurodevelopmental disorder modulated by NURF complex composition. Nat Commun 16, 9875 (2025). https://doi.org/10.1038/s41467-025-64838-5
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-64838-5
This article is cited by
-
Chromatin remodelers in heart development and congenital heart defects
The Nucleus (2026)
