
Abstract
Diffuse intrinsic pontine glioma (DIPG) is a fatal brainstem tumor desperately in need of better treatments. Chimeric antigen receptor (CAR) T cell therapies for DIPG have demonstrated clinical tolerability and bioactivity, but not universal benefit. A major obstacle is insufficient CAR T cell trafficking to the tumor. As our recent clinical trials have demonstrated locoregional elevation of CXCL10, a ligand of the chemokine receptor CXCR3, here we aim to leverage this CXCL10 upregulation to enhance cell trafficking by engineering our B7-H3-targeting CAR T cells to overexpress CXCR3 variants. We demonstrate that, compared to unmodified B7-H3 CAR T cells, CXCR3-A-modified CAR T cells migrate more efficiently toward CXCR3 ligands in vitro, and when delivered intracerebroventricularly in orthotopic DIPG mouse models, CXCR3-A-modified CAR T cells show enhanced trafficking into the tumor and improved therapeutic efficacy. Overall, our data support the potential for engineering CXCR3-A expression to enhance CAR T cell trafficking and efficacy against DIPG.
Similar content being viewed by others

Introduction
Central nervous system (CNS) tumors have surpassed leukemia as the leading cause of death among children with cancer1. Among the pediatric CNS tumors, diffuse intrinsic pontine glioma (DIPG) is particularly devastating, affecting approximately 400 children per year in the US at a median age of 72,3. DIPG is an aggressive and universally fatal brainstem tumor, for which surgical excision is not feasible, and chemotherapy is largely ineffective. Focal radiation is the only standard treatment option, yet it solely provides a palliative benefit, as most patients die within a year. Therefore, more effective treatments against DIPG are desperately needed.
Chimeric antigen receptor (CAR) T cell therapies targeting various antigens have been developed to treat pediatric CNS tumors, including DIPG4,5. However, despite evidence of feasibility, tolerability, and some CAR T cell bioactivity in phase 1 clinical trials, the overall efficacy remains limited. Among the challenges6,7, insufficient CAR T cell trafficking to the tumor has been a major obstacle to successful CAR T cell treatment against solid tumors such as DIPG. Therefore, to address this issue, we engineered our B7-H3-targeting CAR T cells (B7-H3 CAR T cells), which have been clinically studied and well tolerated in our phase 1 trial (NCT04185038)8,9, to overexpress the chemokine receptor CXCR3 to promote CAR T cell trafficking and improve their efficacy against DIPG.
CXCR3 is a chemokine receptor that interacts with the chemokines CXCL10, CXCL9, and CXCL11 and directs cell migration to ascend the concentration gradient of these ligands10,11,12,13,14,15. Given that CXCL10 is produced by activated T cells16 and that CXCL10 is upregulated in the cerebrospinal fluid (CSF) of pediatric CNS tumor patients following intracranial CAR T cell treatment8,9, we hypothesized that the interaction between CXCL10 and CXCR3 could be leveraged to enhance CAR T cell trafficking to the tumor by overexpressing CXCR3 in CAR T cells. In addition, with more CXCR3-modified CAR T cell trafficking to the tumor, more CXCL10 would be produced, further amplifying CAR T cell recruitment through positive feedback. Therefore, this iteration of CAR T cell technology could be more effective in treating children with DIPG.
Many efforts to promote CAR T cell trafficking to solid tumors have been devoted to capitalizing on the chemokines upregulated by the tumor types of interest. For example, it was shown that glioblastoma models express CXCL8 post-irradiation, and CAR T cells engineered to express the receptors for CXCL8 can migrate more effectively to the irradiated tumor in the mouse brain by intravenous (IV) injection17. Similar approaches were studied by other groups in various tumor models, in which CAR T cells were modified to express a selection of chemokine receptors based on the chemokine profiles of the tumor contexts18,19,20,21,22,23,24,25. In contrast, our strategy aims to take advantage of the chemokines expressed by antigen-stimulated CAR T cells, such as CXCL10, given the clinical evidence of CXCL10 upregulation following CAR T cell doses8,9,26. While endogenous CXCR3 is expressed on activated T cells10, here we investigate whether overexpressing CXCR3 could further sensitize CAR T cells to migrate toward CXCL10. Particularly, we investigate the trafficking of CXCR3-modified CAR T cells via intracerebroventricular (ICV) delivery in our mouse models, which is clinically significant for CNS tumors.
In this study, we demonstrate that B7-H3 CAR T cells engineered to overexpress the CXCR3 isoform CXCR3-A can migrate more efficiently toward the CXCR3 ligands, such as CXCL10 in vitro, in comparison to the standard B7-H3 CAR T cells. In vivo, CXCR3-A-modified CAR T cells show enhanced trafficking to the tumor sites post ICV delivery and result in improved efficacy in treating orthotopic DIPG xenograft mouse models. Overall, our data support the potential for engineering CXCR3-A overexpression to enhance CAR T cell trafficking to the tumor and improve the efficacy of CAR T cell therapy for DIPG.
Results
B7-H3 CAR T cells are cytotoxic against patient-derived DIPG cells and are able to produce CXCL10
Several CAR T cell therapies with various antigen specificities have been developed8,9,26,27 and clinically investigated in a series of first-in-human phase 1 trials at Seattle Children’s for pediatric CNS tumors using intracranial delivery28,29. We focused our study on the B7-H3 CAR T cells, which have demonstrated clinical tolerability after ICV delivery to children with DIPG8,9. B7-H3 (CD276) is a member of the B7 family of immunoregulatory proteins and is highly expressed on DIPG tumor cells30,31, including our treatment-naïve biopsy-derived DIPG cells32 (PBT-22FH, PBT-27FH, and PBT-29FH) and an autopsy-derived DIPG model (SU-DIPG-13) used in this study (Fig. 1a; additional information on these models in the Methods). We validated that the B7-H3 CAR T cells could kill the primary DIPG cells when co-cultured in vitro (Fig. 1b, Supplementary Fig. 1a) and produce CXCL9 and CXCL10 post-stimulation, although CXCL11 was not significantly released (Fig. 1c, Supplementary Fig. 1b).

a B7-H3 expression on primary DIPG cells (blue) by flow cytometry with isotype controls (red), representative of n = 2 independent experiments. b Relative cell viability of PBT-22FH or PBT-29FH cells after co-cultured with non-transduced (NTD) or CAR T cells at the indicated E:T ratios for 24 hours. c Concentrations of CXCL9, CXCL10, and CXCL11 by ELISA in co-cultures of NTD T or CAR T cells with PBT-22FH or PBT-29FH target cells at E:T of 1:1 for 24 hours. Cell viability was normalized to the vehicle control without T cells in (b). p values were determined by two-way ANOVA with Sidak’s multiple comparisons test (b, n = 3 independent experiments) or one-way ANOVA with Dunnett’s multiple comparisons test (c, n = 3 independent experiments). Significance was compared with NTD T cells at each E:T ratio in (b). Results are presented as means ± SD in (b, c). Source data are provided as a Source Data file.
Manufactured CAR T cells heterogeneously express CXCR3 and can migrate toward CXCL10
CXCR3 is upregulated by activated T cells10, and since T cell activation is the first step to generate CAR T cells among widely used protocols, we expected manufactured CAR T cells to display some endogenous expression of CXCR3. To confirm this, we isolated T cells from healthy donors’ peripheral blood mononuclear cells (PBMC), activated them with clinically used anti-CD3/CD28 beads, and generated CAR T cells, which did result in a heterogeneous increase of CXCR3 surface expression (Fig. 2a). To test CAR T cell migration efficiency in vitro for this study, we first used our standard B7-H3 CAR T cells to establish a trans-well chemotaxis assay. CAR T cells were seeded in the insert wells that allow them to migrate through a membrane with 5-μm pores into the bottom wells with chemoattractant, and the number of CAR T cells in the bottom wells was counted and normalized to the seeded cell number. As the CAR T cells had endogenous CXCR3 expression, they were able to migrate toward CXCL10 in a dose-dependent manner within 2 hours (Fig. 2b). We further established a trans-well killing assay, in which CAR T cells were permitted to migrate for 2 hours then co-cultured overnight with DIPG cells seeded in the bottom wells. As CXCL10 could induce CAR T cell migration, significantly more DIPG cells were killed when seeded with CXCL10 (Fig. 2c). Given that CXCL10 neither directly inhibited DIPG tumor viability (Fig. 2d) nor affected CAR T cell cytotoxicity against DIPG cells (Fig. 2e) in the control experiments, the reduced tumor viability in the cultures with CXCL10 in Fig. 2c reflects CXCL10-induced CAR T cell migration.

a CXCR3 expression on unstimulated T cells, anti-CD3/CD28 beads-stimulated T cells, and generated CAR T cells by flow cytometry, representative of n = 2 independent experiments. b Percentage of CAR T cells that migrated from insert wells through 5-µm pores to bottom wells with CXCL10 at the indicated time points. c Relative cell viability of PBT-29FH cells after co-cultured with CXCL10-induced migrated CAR T cells for 24 hours. CAR T cells were seeded on insert wells to migrate through 5-µm pores to bottom wells with CXCL10 for 2 hours before removal of the insert wells. d Relative cell viability of PBT-29FH cells after cultured with CXCL10 for 24 hours. e Relative cell viability of PBT-29FH cells after directly co-cultured with CAR T cells at E:T of 1:1 in the presence of CXCL10 for 24 hours. Cell viability was normalized to the vehicle control without T cells in (c–e). p values were determined by one-way ANOVA with Dunnett’s multiple comparisons test (b, e, n = 3, and c, d, n = 4 independent experiments). Results are presented as means ± SD in (b–e). ns, not significant. Source data are provided as a Source Data file. Illustrations in (b, c) were created in BioRender. Song, E. (2025) https://BioRender.com/bv050hn.
CXCR3-A-modified CAR T cells show enhanced migration toward CXCL10
To test our hypothesis that engineering CAR T cells to overexpress CXCR3 can further sensitize CAR T cells to migrate toward CXCL10, we developed genetic constructs that allow co-expression of the B7-H3-specific CAR, a truncated epidermal growth factor receptor (EGFRt) transduction marker, and a CXCR3 isoform (Fig. 3a). CXCR3 has three different isoforms in human due to alternative splicing14, namely CXCR3-A, CXCR3-B, and CXCR3-alt, and we generated B7-H3 CAR T cells overexpressing each of these isoforms. While the CAR and EGFRt marker genes were consistently transduced and expressed on the surface across the CAR T cell groups (Fig. 3b), the commercially available antibodies for CXCR3 could only detect elevated CXCR3 expression on the CXCR3-A-modified CAR T cells (CAR-3A) (Fig. 3c), but not on the CAR T cells engineered to overexpress CXCR3-B (CAR-3B) or CXCR3-alt (CAR-3alt) in comparison to the unmodified CAR T cells (CAR-only). Therefore, we performed RT-qPCR to evaluate the relative RNA expression of each CXCR3 isoform. Our data suggest that CXCR3-A was the dominant isoform in the CAR-only T cells (Fig. 3d), and CXCR3-A expression was exponentially increased in the CAR-3A T cells, while CXCR3-B and CXCR3-alt were specifically overexpressed in the CAR-3B and CAR-3alt T cells respectively (Fig. 3e). Of note, the primer pairs to detect CXCR3-A had unavoidable overlaps with the CXCR3-B and CXCR3-alt sequences, which accounted for the artifacts of elevated CXCR3-A expression in the CAR-3B and CAR-3alt T cells.

a Illustration of constructs to generate B7-H3 CAR T cells that overexpress various isoforms of CXCR3. b, c CAR, EGFRt transduction marker, and CXCR3 expression on unmodified and CXCR3-modifed CAR T cells by flow cytometry, representative of n = 2 independent experiments. gMFI, geometric mean fluorescent intensity. d, e Relative expression of CXCR3 isoforms in unmodified or CXCR3-modifed CAR T cells by RT-qPCR. The data were normalized to ACTB (β-actin) control then nominally multiplied by 108 for data display, representative of n = 2 independent experiments. f, g Percentage of CAR T cells that migrated from insert wells to bottom wells with CXCL10 or CXCL11 after 2 hours. p values were determined by two-way ANOVA with Dunnett’s multiple comparisons test (f, n = 3, and g, n = 5 independent experiments). Results are presented as means ± SD in (f, g). Source data are provided as a Source Data file. Illustration in (a) was created in BioRender. Song, E. (2025) https://BioRender.com/07rm22h.
To compare the migration efficiency of all the CXCR3-modified CAR T cells, we first performed the trans-well chemotaxis assay with CXCL10. Compared to the CAR-only T cells, the CAR-3A T cells showed significantly enhanced migration toward CXCL10; however, this was not observed with the CAR-3B or CAR-3alt T cells (Fig. 3f). Based on the literature, it is unsettled whether CXCR3-B promotes cell migration, and CXCR3-alt has been described to primarily interact with CXCL11 among the CXCR3 ligands13,14. To test the activity of the overexpressed CXCR3-alt in the CAR-3alt T cells, we then compared the CAR T cell migration toward CXCL11, and we demonstrated that both the CAR-3A and CAR-3alt T cell groups migrated more efficiently when induced by CXCL11 (Fig. 3g). Although the CAR-3alt T cells may warrant further investigation, given the lack of production of CXCL11 by the CAR T cells (Fig. 1c, Supplementary Fig. 1b), we focused our study on the CAR-3A T cells for further development.
CXCR3-A-modified CAR T cells show enhanced migration and efficacy against DIPG across multiple T cell donors in response to CXCR3 ligands
To validate our study results across multiple T cell donors, we generated the CXCR3-A-modified and unmodified B7-H3 CAR T cells with isolated T cells from three different PBMC donors. Despite donor variability, there was no significant difference between the CAR-3A and CAR-only T cells in terms of the fold expansion during production (Supplementary Fig. 2a), the CAR transduction rate (Supplementary Fig. 2b), the proportion of CD4 and CD8 T cells (Supplementary Fig. 2d), the distribution of T cell phenotypes (Supplementary Fig. 2e), as well as the expression of T cell exhaustion markers including PD1, TIM3, and LAG3 (Supplementary Fig. 2f), while the CXCR3 expression was elevated on the CAR-3A T cells (Supplementary Fig. 2c) across the three donors. Furthermore, we demonstrated that the CAR-3A and CAR-only T cells had similar cytotoxicity and cytokine production when they were directly co-cultured with the patient-derived DIPG cells in vitro (Fig. 4a, b).

a Relative cell viability of PBT-22FH or PBT-29FH cells after co-cultured with CAR T cells at the indicated E:T ratios for 24 hours. b Concentrations of IL-2, IFN-γ, and TNF by ELISA in co-cultures of CAR T cells with PBT-22FH or PBT-29FH target cells at E:T of 10:1 for 24 hours. Cell viability was normalized to the vehicle control without T cells in (a). p values were determined by randomized block (matching the data for each donor) two-way ANOVA with Tukey’s multiple comparisons test (a, n = 3 different T cell donors), randomized block one-way ANOVA with Dunnett’s multiple comparisons test (b, n = 3 different T cell donors). Results are presented as means ± SD. ns, not significant. Source data are provided as a Source Data file.
Next, we compared the migration efficiency of the CAR-3A and CAR-only T cells from the three T cell donors toward CXCR3 ligands, including CXCL9, CXCL10, and CXCL11. In trans-well chemotaxis assays, the CAR-3A T cells showed enhanced migration toward all three ligands of CXCR3 (Fig. 5a). We then performed the trans-well killing assay using DIPG cells cultured with CXCL9, CXCL10, or CXCL11. As shown in Fig. 5b, despite some variations among the donors, when the migration was induced by CXCL9, CXCL10, or CXCL11, the CAR-3A T cells consistently resulted in more DIPG tumor death, when compared to the CAR-only T cells. This enhanced efficacy was corroborated by the increased cytokines produced by the migrated CAR-3A T cells (Fig. 5c). As our data showed that the presence of CXCL9, CXCL10, or CXCL11 would not specifically enhance the cytotoxicity or cytokine production of the CAR-3A T cells when they were directly co-cultured with the DIPG cells in the control experiments (Fig. 5d, e), we confirmed that the enhanced efficacy by the CAR-3A T cells in the trans-well killing assays (Fig. 5b, c) should be attributed to their more efficient migration toward the CXCR3 ligands.

a Percentage of CAR T cells that migrated from insert wells to bottom wells with vehicle, 300 ng/mL CXCL9, 100 ng/mL CXCL10, or 300 ng/mL CXCL11 after 2 hours. b Relative cell viability of PBT-29FH cells after co-cultured with migrated CAR T cells for 24 hours. CAR T cells were seeded on insert wells to migrate to bottom wells with vehicle, 300 ng/mL CXCL9, 100 ng/mL CXCL10, or 300 ng/mL CXCL11 for 2 hours before removal of the insert wells. c Concentrations of IL-2, IFN-γ, and TNF by ELISA in cultures from (b) after 24 hours. d Relative cell viability of PBT-29FH cells after directly co-cultured with CAR T cells at E:T of 1:1 in the presence of vehicle, 300 ng/mL CXCL9, 100 ng/mL CXCL10, or 300 ng/mL CXCL11 for 24 hours. e Concentrations of IL-2, IFN-γ, and TNF by ELISA in cultures from (d) after 24 hours. Cell viability was normalized to the vehicle control without T cells in (b, d). p values were determined by randomized block (matching the data for each donor) two-way ANOVA with Sidak’s multiple comparisons test (a–e, n = 3 different T cell donors). Results are presented as means ± SD. ns, not significant. Source data are provided as a Source Data file.
Paracrine chemokines can lead to enhanced efficacy by CXCR3-A-modified CAR T cells against DIPG
As antigen-stimulated CAR T cells would produce CXCL9 and CXCL10 (Fig. 1c, Supplementary Fig. 1b), we investigated if the chemokines produced by CAR T cells from co-culturing with DIPG cells would be sufficient to induce CAR T cell migration in trans-well killing assays. We first prepared CAR T cell-conditioned medium (CM) by stimulating the B7-H3 CAR T cells with DIPG cells and harvesting the supernatant after 24 hours, then we applied the CM in a trans-well killing assay. As shown in Fig. 6a, while the CAR T cell CM alone had some direct inhibitory effect on the DIPG cells, the CM induced the CAR-3A T cells to migrate and kill DIPG cells, as evidenced by the significantly more reduced tumor viability compared to the CM-only group. The tumor viability with the migrated CAR-3A T cells was also substantially lower (though not statistically significant) in comparison to the CAR-only T cell group, when attracted by the CM (Fig. 6a).

a Relative cell viability of PBT-29FH cells after co-cultured with migrated CAR T cells for 24 hours. CAR T cells were seeded on insert wells to migrate for 2 hours to bottom wells with conditioned medium (CM) from antigen-stimulated CAR T cell culture, before removal of the insert wells. b Relative cell viability of PBT-29FH cells after co-cultured with continuously migrating CAR T cells for 24 hours. CAR T cells were seeded on insert wells and allowed to migrate for 24 hours to the bottom wells without chemoattractant. c Concentrations of CXCL10 by ELISA at the indicated time points in cultures of DIPG cells with added IFN-γ at the indicated doses. p values were determined by randomized block (matching the data for each donor) one-way ANOVA with Dunnett’s multiple comparisons test (a, n = 3 different T cell donors) or paired one-tailed t test (b, n = 3 different T cell donors), or lognormal one-way ANOVA with Dunnett’s multiple comparisons test (c, n = 4 different DIPG models). Results are presented as means ± SD in (a, b). Source data are provided as a Source Data file. Illustration in (b) was created in BioRender. Song, E. (2025) https://BioRender.com/bv050hn.
Next, we tested whether the CAR-3A T cells would demonstrate enhanced efficacy in a long-term trans-well killing assay without exogenously added chemoattractant, in which seeded CAR T cells were given 24 hours to continuously migrate from the insert wells to the bottom wells with only DIPG cells (Fig. 6b). Since some CAR T cells would pass through the insert pores in the absence of exogenous chemoattractant (Fig. 2b), they would be stimulated by the DIPG cells and release chemokines including CXCL9 and CXCL10 in the bottom wells, which should attract more CAR T cells to actively migrate and in turn lead to more chemokine production as positive feedback in the following hours. Given that the CAR-3A T cells were more efficient migrating toward CXCL9 and CXCL10 than the CAR-only T cells, we hypothesized that more DIPG cells would be killed by the migrated CAR-3A T cells via the amplified positive feedback loop. Indeed, the CAR-3A T cells consistently outperformed the CAR-only T cells in this assay, either in repeated experiments using CAR T cells of the same donor (Supplementary Fig. 3) or across all three T cell donors (Fig. 6b), supporting our hypothesis.
Moreover, we investigated whether DIPG tumors would contribute to the paracrine CXCL10, as CXCL10 is inducible by IFN-γ33, a major cytokine produced by stimulated CAR T cells (Fig. 4b). As demonstrated in Fig. 6c, while our primary DIPG cells would not make CXCL10 in the absence of IFN-γ, a substantial amount of CXCL10 was produced by the DIPG cells when cultured with IFN-γ. Therefore, the production of CXCL10 can be reinforced by DIPG tumors in response to IFN-γ when treated by CAR T cells.
CXCR3-A-modified CAR T cells show enhanced trafficking and therapeutic efficacy when delivered intracerebroventricularly in orthotopic DIPG models
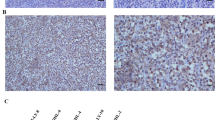
Based on our encouraging in vitro data, we investigated the effect of engineering CXCR3-A in CAR T cells against orthotopic DIPG mouse models. We first intracranially implanted luciferase-labeled SU-DIPG-13 cells into NSG mice, which is an aggressive and well-studied DIPG in vivo model34. As illustrated in Fig. 7a, after 7 days, the mice were treated with the CAR-3A, CAR-only, or non-transduced (NTD) T cells of the same donor via either IV or ICV injection, as both routes have been used in clinical studies for pediatric CNS tumor patients. To compare CAR T cell trafficking from the periphery via IV or from the CSF via ICV to the tumor, we sacrificed mice from each treatment group at the 24-hour and 48-hour time points to harvest the mouse brains. The IHC staining for human CD3, CD4, and CD8 qualitatively indicated increased presence of the CAR-3A T cells around the tumor sites, compared with the CAR-only or NTD T cells, at both the time points post-delivery to the mouse CSF through ICV injection (representative images shown in Fig. 7b; additional images shown in Supplementary Fig. 4). In contrast, there was lack of T cell trafficking to the brain through IV administration during the first 48 hours (Supplementary Fig. 5), although we could not rule out the possibility that IV injected CAR T cells may arrive at the tumor sites at a later time point. The tumor areas were defined based on H&E staining (Fig. 7b, Supplementary Figs. 4, 5) and confirmed by IHC staining for human Ki67 and B7-H3 (Supplementary Fig. 6).

a Illustration of the experimental plan (details described in the Methods section). b Representative images from n = 2 mice in each group at each time point of H&E staining for the tumors and IHC staining for human CD3 around the corresponding tumor areas from the mouse brains collected on day 1 and day 2 (additional images shown in Supplementary Fig. 4). General areas of the SU-DIPG-13 tumors are indicated by black arrows. Scale bars in the images represent 300 µm. Illustration in (a) was created in BioRender. Song, E. (2025) https://BioRender.com/5ikgow3.
Next, we investigated whether the production of CXCL10 at the tumor sites correlated with the increased presence of the CAR-3A T cells post-ICV injection. In areas corresponding to the previously displayed tumor sections (Fig. 7b, Supplementary Figs. 4, 6a), the IHC staining for human CXCL10 suggested limited CXCL10 at the tumor sites where few CAR-only T cells had arrived at 24 hours post ICV injection (Fig. 8), and the CXCL10 qualitatively increased at the 48-hour time point, corresponding to additional CAR-only T cell arrival (Fig. 8). In comparison, there appeared to be greater abundance of CXCL10 at the tumor sites where the ICV delivered CAR-3A T cells had efficiently trafficked to within 24 hours (Fig. 8). Of note, the CAR-3A T cell-infiltrated tumor areas showed qualitatively less CXCL10 at the 48-hour time point (Fig. 8), which was likely related to the substantial tumor regression at this time point in this group (Supplementary Fig. 6), reducing antigen stimulation for the CAR-3A T cells to release CXCL10 and decreasing CXCL10 secretion by the tumor cells in response to IFN-γ. Overall, the ICV delivered CAR-3A T cells not only demonstrated enhanced trafficking to the tumor, but also resulted in elevated CXCL10 at the tumor sites.

Representative images from n = 2 mice in each group at each time point of IHC staining for human CXCL10 around the corresponding tumor areas from the mouse brains collected on day 1 and day 2, as illustrated in Fig. 5a. General areas of the SU-DIPG-13 tumors are indicated by black arrows. Scale bars in the images represent 300 µm.
To compare the therapeutic efficacy of the CAR-3A and CAR-only T cells, we included additional mice in each treatment group (n = 5 or 6) as depicted in Fig. 7a for weekly bioluminescent imaging (BLI) to measure the signals from the DIPG tumors. As SU-DIPG-13 cells could metastasize to the spinal cord (Fig. 9a), we quantified the BLI signals from the whole body of the mice in this experiment. When the CAR T cells were locoregionally delivered via ICV injection, the CAR-3A T cells reduced the tumor signals to the background level within 7 days (Fig. 9c) and maintained complete tumor regression in this study (Fig. 9d). In contrast, although the ICV administered CAR-only T cells significantly inhibited the tumor growth compared to the NTD T cells (Fig. 9c), rapid rebound of tumor signals from either the brain or the spinal cord was observed in 4 out of 5 mice from this group (Fig. 9a, d), which led to their fatality based on protocol-defined euthanasia criteria (Fig. 9e, Supplementary Table 1).

a BLI images of mice at the indicated imaging time points. b, c Log10 of bioluminescent total flux from SU-DIPG-13 tumor cells in each mouse on day 7. d Bioluminescent total flux from SU-DIPG-13 tumor cells at the indicated time points. e Kaplan-Meier plot for mouse survival. f Mouse weight at the indicated time points. n = 5 mice in the IV NTD T, IV CAR-only T, ICV NTD T, and ICV CAR-only T groups, and n = 6 mice in the IV CAR-3A T and ICV CAR-3A T groups (a–f). Results are presented as means ± SD in (b, c). p-values were determined by one-way ANOVA with Tukey’s multiple comparisons test (b, c) or log-rank (Mantel–Cox) test (e). ns, not significant. Source data are provided as a Source Data file.
To be noted, prior to the end of this study, one mouse treated with ICV injected CAR-3A T cells met the euthanasia criteria without an apparent tumor signal from previous BLI images (Fig. 9a). H&E and IHC staining for the mouse brain showed the presence of xenograft tumor (human Ki67+) yet with low expression of human B7-H3 (Supplementary Fig. 7), suggesting tumor antigen escape as a resistance mechanism under the selective pressure by the CAR-3A T cells. The fact that this coincided with loss of luciferase expression indicated that the tumor relapse was derived from a limited number of double-negative tumor cells of this aggressive model. To address the issue of tumor antigen escape from CAR T cells, a variety of strategies have been preclinically and clinically developed5,35,36,37,38,39,40,41,42,43,44, including Seattle Children’s ongoing phase 1 trial to investigate CAR T cells with multiple specificities against DIPG (NCT05768880).
On the other hand, neither CAR T cell group demonstrated efficacy when delivered systemically through IV injection (Fig. 9a, b, d, e, Supplementary Table 1), which was most likely due to the insufficient trafficking of CAR T cells from the periphery to the brain (Supplementary Fig. 5) and the aggressiveness of this in vivo model.
Mouse weight loss and neurologic symptoms were correlated with tumor progression (Fig. 9f), and there was no evident toxicity by the CAR-3A T cells in comparison with the CAR-only T cells.
To validate these findings, we also evaluated the safety and efficacy of the CAR-3A T cells in a second DIPG in vivo model and used T cells from another donor. In brief, luciferase-labeled PBT-27FH cells were intracranially engrafted in NSG mice for 35 days before T cell administration through ICV injection (Supplementary Fig. 8a). The BLI signals from the mouse brain suggested a substantial decrease in tumor burden by the CAR-3A T cells within 7 days of treatment (Supplementary Fig. 8b), indicating efficient tumor engagement by the CAR-3A T cells, while the tumor signals remained above baseline in most mice treated with the CAR-only T cells. Despite the extended time for the tumor to progress in this model (Supplementary Figs. 8c, 9), the mice treated with the CAR-3A T cells had a survival advantage compared to the mice in the NTD or CAR-only T cell groups (Supplementary Fig. 8d, Supplementary Table 2), though the comparison to the latter did not reach statistical significance. To be noted, the statistical power of this study was unexpectedly reduced, because two mice in the CAR-3A T cell group without tumor BLI signal (Supplementary Fig. 9) were euthanized per requests by the veterinary staff due to non-tumor-related causes (vaginal prolapse, day 182; bacterial infection, day 193). IHC staining for human Ki67 and B7-H3 confirmed absence of tumor in these brains (Supplementary Fig. 10). There was no weight loss (Supplementary Fig. 8e) or sign of illness such as lethargy, hunching posture at rest, or hair loss exhibited by the mice in relation to the treatment of the CAR-3A T cells, supporting the safety of this CAR T cell engineering approach.
Discussion
As DIPG is universally fatal and responsible for nearly 25,000 years of lost potential life each year in the US alone2,3, more effective treatments are desperately needed. Here, we developed a new iteration of CAR T cell therapy by engineering B7-H3 CAR T cells to overexpress CXCR3-A, in order to improve CAR T cell trafficking to the tumor. We demonstrate that, compared to the standard B7-H3 CAR T cell therapy, our CXCR3-A-modified CAR T cells migrate more efficiently toward the CXCR3 ligands (e.g., CXCL10) in vitro and show enhanced trafficking and therapeutic efficacy when delivered intracerebroventricularly in our orthotopic DIPG mouse models. Because CXCL10 is produced by activated CAR T cells as well as by DIPG cells in response to IFN-γ from activated CAR T cells, this CXCL10 could attract more CXCR3-A-modified CAR T cells to migrate to the tumor, which would result in a positive feedback loop by amplifying the CXCL10 production and further recruitment of CAR T cells, thus leading to improved therapeutic efficacy against DIPG (Fig. 10).

a, b Some of the ICV delivered CAR T cells encounter tumor and get stimulated by the tumor antigen, leading to some tumor cell death. c, d The stimulated CAR T cells produce chemokines such as CXCL10, which can recruit additional CAR T cells that endogenously express CXCR3. DIPG tumor cells also express CXCL10 in response to IFN-γ released by stimulated CAR T cells. e By engineering the CAR T cells to overexpress CXCR3-A, more CAR T cells respond and migrate to the tumor site. f This effect in turn results in more CXCL10 production, followed by more CAR T cell recruitment through positive feedback, which leads to more tumor cell death, thus improving the trafficking and efficacy of CAR T cell therapies for DIPG. Created in BioRender. Song, E. (2025) https://BioRender.com/dv8wt3z.
It is worth noting that while the tumor immune microenvironment (TIME) of DIPG has been described as immunologically inert45,46, other immune cells could be attracted to the tumor via chemotaxis following CAR T cell administration and may in turn contribute to the chemokine and cytokine production in the TIME. For example, microglia, the CNS resident immune cells, express CXCR3 and thus could be recruited by the CXCL10 from the activated CAR T cells47, and microglia can also produce CXCL10 and IFN-γ among other cytokines upon activation48,49. These interactions among CAR T cells and other immune cell types may reinforce a pro-inflammatory environment in the tumor and further recruit other anti-tumor immune cells, provided that CAR T cells can efficiently migrate to the tumor to initiate this feedforward reaction. To study these interactions and how CXCR3-A-modified CAR T cells may enhance these interactions, immunocompetent syngeneic DIPG mouse models and murine CAR T cells would be required, and while these models would have their own limitations, we are establishing these models in our lab for future investigations.
Our clinical data with the standard B7-H3 CAR T cells indicated some CAR T cell activity and elevated CXCL10 in the CSF of patients with DIPG8,9, which may be sufficient to initiate the positive feedback loop for recruiting the CXCR3-A-modified CAR T cells. Nevertheless, our approach could further synergize with strategies that promote the production of CXCR3 ligands from the tumor cells prior to CAR T cell administration, to increase the initial tumor engagement by the CXCR3-A-modified CAR T cells. For example, oncolytic virus encoding a CXCL11 gene has been preclinically developed50, which could target tumor cells to express CXCL11 and attract more CXCR3-A-modified CAR T cells immediately after the injection. In addition, since DIPG cells would make CXCL10 in response to IFN-γ, oncolytic virus encoding an IFN-γ gene may provide a similar effect for enhancing the trafficking of the CXCR3-A-modified CAR T cells as a combination therapy for DIPG.
Preclinical evidence, including results in this study, has suggested the superiority of locoregional CAR T cell delivery to the CNS over systemic dosing in treating CNS tumor models41,51,52, and CAR T cell clinical trials for CNS tumors have demonstrated the feasibility and tolerability of injecting CAR T cells to the CNS of patients4,5,28. As such, it is important to study CAR T cell trafficking by administering CAR T cells to the CNS space in preclinical in vivo models. Here, we demonstrate that CAR T cell trafficking to the CNS tumor can be enhanced by engineering chemokine receptor expression in the CAR T cells when they are ICV delivered.
While our study focused on enhancing the B7-H3 CAR T cell therapy for DIPG, this concept of CXCR3-A-modified CAR T cells is readily applicable to CAR T cell therapies targeting other types of solid tumors, provided with modifications of the CAR specificities for suitable tumor antigens. In addition, we focused on the CXCR3 isoform CXCR3-A for CAR T cell engineering based on our preliminary data and due to the lack of clinical data regarding the CXCL11 levels in the CAR T cell-treated DIPG patients. Nonetheless, the CXCR3-alt-modified CAR T cells could be beneficial in a tumor context where CXCL11 production is relevant.
Of note, the role of CXCR3-B in cell migration has been conflicting based on the current literature13,14. As CXCR3 only has one isoform in mouse, which is the counterpart for CXCR3-A in human, CXCR3-B as well as CXCR3-alt have not been well studied. Here, our data suggested that CXCR3-B overexpression did not significantly increase T cell migration toward CXCL10 or CXCL11 (Fig. 3f, g). However, the overexpression of CXCR3-B was only confirmed by RT-qPCR (Fig. 3e), and we were not able to validate the surface expression of the transduced CXCR3-B due to the absence of commercially available antibody.
We showed enhanced trafficking and efficacy by the CXCR3-A-modified CAR T cells despite the fact that the unmodified CAR T cells upregulate CXCR3 expression due to the activation step in the commonly adopted production process (Fig. 2a). However, as new CAR T cell production protocols are developed, some of which either omit the activation step to preserve the stemness of the CAR T cells53 or generate CAR T cells through inducible pluripotent stem cells (iPSCs) for potential off-the-shelf applications54, in these cases, engineering the CAR T cells to overexpress CXCR3 may be even more essential to promote CAR T cell trafficking and efficacy, as CAR T cells generated by these methods would have less endogenous CXCR3 expression.
Although the ability to assess the toxicity of a CAR T cell therapy is limited in xenograft mouse models, we did not observe any safety concern derived from the engineering of CXCR3-A in the B7-H3 CAR T cells, and given the significantly enhanced therapeutic efficacy demonstrated in our study, CXCR3-A-modified CAR T cells deserve further preclinical and clinical investigations.
Methods
Inclusion and ethics statement
This research does not involve multi-region collaboration. All contributors are acknowledged with authorship or in the Acknowledgements section. All primary human DIPG cell cultures from Seattle Children's were generated following informed consent and are in compliance with an institutional review board approval (#14449). All human peripheral blood mononuclear cells were collected by Bloodworks Northwest with informed consent obtained from each donor under the approved protocol (#20141589) by the institutional review board. All mouse experiments were conducted in accordance with the IACUC-approved protocol ACUC00669 at Seattle Children’s Research Institute.
Primary human DIPG cell culture
PBT-22FH (H3.3 mutant), PBT-27FH (H3.1 mutant), and PBT-29FH (H3.3 mutant) were generated from treatment-naïve patient-derived biopsy samples obtained at diagnosis. For PBT-22FH, PBT-27FH, and PBT-29FH, the tumor tissues were obtained at Seattle Children’s Hospital, and cell cultures were created at Fred Hutchinson Cancer Center as previously described32. SU-DIPG-13 (H3.3 mutant) was autopsy-derived and generously donated by Dr. Michelle Monje (Stanford University). Cells were modified by lentiviral transduction followed by blasticidin selection (Fisher Scientific, #A1113903) to steadily express firefly luciferase. Cells were maintained in NeuroCult NS-A Basal Medium with NS-A Proliferation Supplement (Stemcell Technologies, #5751), 1x Antibiotic/Antimycotic (ThermoFisher Scientific, #15-240-062), 40 ng/mL epidermal growth factor (PeproTech, #AF-100-15), and 40 ng/mL fibroblast growth factor (PeproTech, #AF-100-18B) and were adherent in flasks coated with laminin (R&D Systems, #3446-005-01). All cell culture models were validated by DNA fingerprinting and routinely tested to be mycoplasma negative.
CAR constructs
The medium-spacer human B7-H3-specific CAR construct has previously been described8. In brief, the CAR is comprised of a scFv derived from the MGA 271 monoclonal antibody, a medium-length spacer of IgG4-hinge-CH3, a CD28 transmembrane domain, a 4-1BB co-stimulatory domain, and a CD3ζ signaling domain, and the CAR sequence was appended to a T2A ribosomal skip sequence followed by a truncated EGFR (EGFRt) marker gene, which was appended to a second T2A sequence followed by the methotrexate-resistant human DHFR mutein gene, the last of which was not utilized in this paper. For the CAR constructs co-expressing a CXCR3 isoform, the human DHFR mutein gene was replaced with the open reading frame sequence of CXCR3-A, CXCR3-B, or CXCR3-alt (synthesized by Genewiz).
Lentiviral vector preparation
Lentiviral vectors were produced by transfecting Lenti-X 293-T cells (Takara, #632180), kindly provided by Dr. Bernasconi (University of Bern). In brief, 293-T cells were seeded in T150 flasks that were pre-coated with collagen (Millipore Sigma, #C3867-1VL) and cultured in DMEM medium (Gibco, #11960044) supplemented with 10% fetal bovine serum (Gibco, #A5256701), 10 mM HEPES (Gibco, #15630080), 100 U/mL penicillin/streptomycin (Gibco, #15140122), and 1x GlutaMax (Gibco, #35050061) to grow to 80% confluence, after which the cells were incubated briefly with a cocktail of 3 mL of Opti-MEM (Gibco, #31985070) with 90 μL Lipofectamine 3000 transfection reagent (Invitrogen, # L3000015) that was gently mixed with 15 μg of the vector plasmid of interest and the packaging plasmids of 18 μg of psPAX2 (Addgene, #12260) and 7 μg of pCMV-VSV-G (Addgene, #8454). Additional culture medium was added thereafter. After culturing the transfected 293-T cells for 24 h and 48 h, supernatants that contained lentiviral vectors were collected and filtered through 0.45-mm syringe filters (Millipore Sigma, #SLHP033RS), then centrifuged at 18,000 x g for 20 h at 4 °C. The pellets of lentiviral vectors were resuspended in 2 mL of culture medium, aliquoted, and stored at -80 °C.
Generation of human CAR T cells
Bulk human T cells were isolated from peripheral blood mononuclear cells of healthy donors (Bloodworks Northwest) by negative selection using EasySep Human T Cell Isolation Kit (StemCell, #17951) and maintained in RPMI 1640 medium (Gibco, #21870076) supplemented with 10% fetal bovine serum (Gibco, #A5256701), 10 mM HEPES (Gibco, #15630080), 100 U/mL penicillin/streptomycin (Gibco, #15140122), GlutaMax (Gibco, #35050061), 5 ng/mL recombinant human IL-7 (Biolegend, # 581904), and 0.5 ng/mL recombinant human IL-15 (Biolegend, # 570304). Following isolation, T cells were stimulated with anti-CD3/CD28 Dynabeads (Gibco, #40203D) in the medium at 1 × 106 cells/mL for 24 h and transduced at a multiplicity of infection of 3, after which the transduced T cells were enumerated by a Vi-CELL BLU counter (Beckman Coulter) every other day and fed with the medium to 0.8 × 106 cells/mL density. After the expansion, the cells were cryopreserved in CryoStor CS10 (StemCell, #07930) until further use. To resuscitate cryopreserved cells, the cells were quickly thawed with pre-warmed medium and pelleted by centrifugation, then cultured in the medium overnight to recover, and the viable cell number was determined by a Vi-CELL BLU counter (Beckman Coulter) before being used for functional assays.
RT-qPCR
The RNA was extracted from cells using RNeasy Plus Mini Kit (QIAGEN, #74134) and converted to cDNA with High-Capacity RNA-to-cDNA kit (Applied Biosystems, #4387406). The cDNA was then set up with TB Green (Takara, #RR42LR) and a primer pair (Integrated DNA Technologies) in four replicate wells for qPCR with Light Cycler 480 II (Roche). The expression of the gene of interest was normalized to the expression of actin, then multiplied by a nominal factor for data display.
The forward primer for CXCR3-A and CXCR3-alt is: ATGGTCCTTGAGGTGAGTGA, and the forward primer for CXCR3-B is: GCCCTCACAGGTGAGTGA. The reverse primer for CXCR3-A and CXCR3-B is: CAGAAAGCCAGCCACCAG, and the reverse primer for CXCR3-alt is: AACTTGACCCCTGTGGGAA. The forward primer for ACTB is: CACCATTGGCAATGAGCGGTTC, and the reverse primer for ACTB is: AGGTCTTTGCGGATGTCCACGT.
Flow cytometry
For flow cytometry, the antibodies used included mouse-anti-human B7-H3-PE (BioLegend, clone MIH42, #351004, 1:100), mouse-anti-human CXCR3-FITC (BioLegend, clone G025H7, #353704, 1:100), mouse-anti-human EGFR-PE (BioLegend, clone AY13, #352904, 1:100), mouse-anti-human CD4-PE (BioLegend, clone OKT4, #317410, 1:100), mouse-anti-human CD8-APC (BioLegend, clone RPA-T8, #301049, 1:100), mouse-anti-human CD45RA-PE (BioLegend, clone HI100, #304108, 1:100), mouse-anti-human CD62L-BV421 (BioLegend, clone DREG-56, #304828, 1:100), mouse-anti-human PD1-APC (BioLegend, clone A17188B, #621610, 1:100), mouse-anti-human TIM3-APC (BioLegend, clone A18087E, #364804, 1:100), mouse-anti-human LAG3-APC (BioLegend, clone 7H2C65, #369212, 1:100), and biotinylated Protein L (GenScript, #M00097) followed by streptavidin-PE (BioLegend, #405204) secondary staining to detect the B7-H3-specific CAR expression. For flow cytometry analysis, cells were sampled and washed with PBS (Gibco, #10010023), incubated with antibodies in PBS on ice in darkness for 30 min, washed again with PBS twice, followed by secondary staining with additional wash steps when necessary, then evaluated on a LSR Fortessa (BD Biosciences) or Novocyte 3000 (Agilent) flow cytometer. The cell-surface expression of the proteins of interest was analyzed by FlowJo (Tree Star). Examples of gating strategies are provided in Supplementary Fig. 11.
Luciferase assay
The primary DIPG models used for luciferase assays were modified to express firefly luciferase. D-Luciferin (Revvity, #122799-5) substrate solution was added to the cell culture at the indicated time points for each assay and incubated at room temperature for 15 min before quantifying the bioluminescent signal from each well with a SpectraMax iD5 (Molecular Devices). All the in vitro viability assays were performed three times or with T cells from three different donors, and the bioluminescent signals were normalized to the vehicle-treated control wells as 100% viability.
ELISA
ELISA assays were performed with DuoSet ELISA kits (R&D Systems) to detect CXCL9 (#DY392), CXCL10 (#DY266), CXCL11 (#DY672), IL-2 (#DY202), TNF (#DY210) and IFN-γ (#DY285B) concentrations in the collected supernatants per the manufacturer’s instructions. Quantified by SpectraMax iD5 (Molecular Devices).
Trans-well chemotaxis assay
Trans-well chemotaxis assays were performed with Transwell 24-well Plate with 5.0 µm Pore Polycarbonate Membrane Insert (Corning, #3421) to assess the migration efficiency of CAR T cells toward chemoattractant. In brief, blank medium (vehicle) or medium with chemoattractant was added to the bottom wells, then the same number of CAR T cells was seeded on the insert wells, which allow active T cell migration through the pores to the bottom wells. After the indicated length of incubation, the number of migrated CAR T cells in each bottom well was counted in duplicates with Vi-CELL BLU counter (Beckman Coulter). All the trans-well chemotaxis assays were performed at least three times or with T cells from three different donors, and percent migration was calculated as the migrated cell number over the seeded cell number.
Trans-well killing assay
Trans-well killing assays were performed with a Transwell 24-well Plate with 5.0 µm Pore Polycarbonate Membrane Insert (Corning, #3421). In brief, luciferase-labeled DIPG tumor cells were seeded in the bottom wells, then blank medium (vehicle) or medium with chemoattractant was added to the bottom wells. The same number of CAR T cells was seeded on the insert wells, which allows active T cell migration through the pores to the bottom wells. After 2-hour incubation, the insert wells were removed, and the plate was further incubated overnight to allow the migrated CAR T cells to kill the seeded DIPG tumor cells. At the 24-hour time point, luciferase assay was performed as described above to quantify the viability of the seeded DIPG tumor cells. All the trans-well killings assays were performed three times or with T cells from three donors and the bioluminescent signals were normalized to the vehicle-treated control wells as 100% viability.
The chemoattractants used in this study include recombinant human CXCL9 (#ab283902), CXCL10 (#ab280332) and CXCL11 (#ab283901) from Abcam, as well as conditioned media harvested from co-cultures of CAR T cells and DIPG tumor cells.
Mouse experiments
All mice were housed in a vivarium under protocol-defined conditions. The number of mice in each treatment group is indicated in the figure legends, and the consideration of sex in the study designs is indicated in the Reporting Summary linked to this article.
For the orthotopic xenograft model with SU-DIPG-13, eight-week-old male NSG mice (Jackson Laboratories) were intracranially injected with 50,000 SU-DIPG-13 cells in 2 µL PBS (Cytiva, #SH30256.LS) using a stereotactic surgical setup with the coordinates of 0.8 mm lateral and 1.5 mm posterior from lambda and 2.5 mm deep. Seven days after tumor implantation, 2 × 106 CAR T cells in 5 µL PBS were injected into the lateral ventricle of brain with the coordinates of 1.2 mm lateral and 0.6 mm posterior from bregma and 2.0 mm deep, or intravenously in 200 µL PBS. Mice were anesthetized by intraperitoneal injected cocktail of Ketamine (Patterson Veterinary Supply, #07-894-8462; 100 mg/kg animal) and Xylazine (Patterson Veterinary Supply, #07-808-1947; 8 mg/kg animal) for the surgeries and given the painkiller of buprenorphine (Wedgewood Connect, #BUPREN-INJ010VC; 1 mg/kg animal, extended release) by the veterinary staff before the surgeries. Mice from each treatment group were euthanized at the 24-hour and 48-hour post-treatment time points to collect their brains, which were used for immunohistochemistry (IHC) analysis to evaluate the migration of T cells toward the tumor site as described below. For the rest of the mice in each treatment group, bioluminescent imaging (BLI) was performed weekly to measure the tumor signals, for which mice were anesthetized with isoflurane (Patterson Veterinary Supply, #07-890-8115), subcutaneously injected with D-luciferin (Revvity, #122799-5) substrate solution, and imaged with IVIS Spectrum Imaging System (Perkin Elmer) after 15 min. Mouse weight was recorded on a weekly basis with routine health checks to monitor any sign of illness, such as weight loss, hair loss, cranial distension, lethargy, and hunching posture at rest. Mice with severe symptoms were euthanized based on protocol-defined criteria, followed by the collection of brains, livers, and spleens for IHC analysis.
For the experiment with PBT-27FH, gender-balanced NSG mice were intracranially injected with 350,000 PBT-27FH cells in 2 µL PBS using a stereotactic surgical setup with the same coordinates as described above. 35 days after tumor implantation, 1 × 105 CAR T cells in 3 µL PBS were injected into the lateral ventricle of the brain with the coordinates as described above. BLI was performed to measure the tumor signals. Mouse weight was recorded with routine health checks to monitor any sign of illness and mice with severe symptoms were euthanized based on protocol-defined criteria, followed by the collection of brains, livers and spleens for IHC analysis if needed.
Tissue processing, IHC, and histopathologic analysis
Mouse brains were fixed in 10% neutral buffered formalin (Fisher Scientific, #57-35) for 24–72 hours, then transferred into 70% ethanol for long-term storage. Wet tissues were submitted to the Microscopy and Histopathology CoLab at Seattle Children’s Research Institute for paraffin embedding. Formalin-fixed paraffin-embedded blocks were then submitted to the Experimental Histopathology lab at Fred Hutchinson Cancer Center for further processing. Tissue sections were cut 4-5 µm thick for Hematoxylin-Eosin (H&E) stains and IHC. IHC stains included rabbit-anti-human CD3 (Fisher Scientific, clone SP7, #RM9107S, 1:300), rabbit-anti-human CD4 (Millipore Sigma, clone EP204, #104R-26, 1:100), mouse-anti-human CD8 (Agilent, clone C8/144B, #M7103, 1:200), mouse-anti-human CXCL10 (R&D Systems, clone 33036, #MAB266-100, 1:500), rabbit-anti-human B7-H3 (Abcam, clone EPR20115, #ab219648, 1:5000), rabbit-anti-human Ki67 (Cell Signaling, clone D3B5, #12202, 1:1000) and rabbit-anti-albumin (Genetex; polyclonal, #GTX102419, 1:2000) with a hematoxylin counterstain. H&E and IHC images were produced with an Aperio AT slide scanner.
Statistical analysis
The statistical analyses were performed as noted in the figure legends using Prism 10 (GraphPad) to determine the p values. p < 0.05 is considered statistically significant.
Illustrations
The illustrations and schematics in this manuscript were created in https://BioRender.com as noted in the figure legends under a BioRender Academic Publication License with agreement numbers PA28QI59EP, DB28QI59GR, QE28QI67SF, TZ28QI59IN, OU28QI79PR, XD28QI8XIF and ZC28QI79RQ.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Requests for materials newly generated in this study (i.e. the plasmids with the CAR construct co-expressing CXCR3-A, CXCR3-B, or CXCR3-alt) should be addressed to the corresponding author and be will be promptly reviewed by the intellectual property office of Seattle Children’s Research Institute to verify whether the request is subject to any intellectual property or confidentiality obligations. Any materials that can be shared will be released via a Material Transfer Agreement. The remaining data are available within the Article, Supplementary Information or Source Data file. Source data are provided with this paper.
References
Siegel, R. L., Miller, K. D., Wagle, N. S. & Jemal, A. Cancer statistics, 2023. CA Cancer J. Clin. 73, 17–48 (2023).
Cooney, T. et al. Contemporary survival endpoints: an International Diffuse Intrinsic Pontine Glioma Registry study. Neuro Oncol. 19, 1279–1280 (2017).
Vitanza, N. A. & Monje, M. Diffuse intrinsic pontine glioma: from diagnosis to next-generation clinical trials. Curr. Treat. Options Neurol. 21, 37 (2019).
Ronsley, R. et al. CAR T cell therapy for pediatric central nervous system tumors: a review of the literature and current North American trials. Cancer Metastasis Rev https://doi.org/10.1007/s10555-024-10208-4 (2024).
Timpanaro, A. et al. Evolving CAR T-cell therapy to overcome the barriers in treating pediatric central nervous system tumors. Cancer Discov. 15, 890–902 (2025).
Uslu, U. & June, C. H. Beyond the blood: expanding CAR T cell therapy to solid tumors. Nat. Biotechnol. https://doi.org/10.1038/s41587-024-02446-2 (2024).
Ferreras, C. et al. Facing CAR T cell challenges on the deadliest paediatric brain tumours. Cells 10 https://doi.org/10.3390/cells10112940 (2021).
Vitanza, N. A. et al. Intraventricular B7-H3 CAR T cells for diffuse intrinsic pontine glioma: preliminary first-in-human bioactivity and safety. Cancer Discov. 13, 114–131 (2023).
Vitanza, N. A. et al. Intracerebroventricular B7-H3-targeting CAR T cells for diffuse intrinsic pontine glioma: a phase 1 trial. Nat. Med. https://doi.org/10.1038/s41591-024-03451-3 (2025).
Loetscher, M. et al. Chemokine receptor specific for IP10 and mig: structure, function, and expression in activated T-lymphocytes. J. Exp. Med. 184, 963–969 (1996).
Loetscher, M., Loetscher, P., Brass, N., Meese, E. & Moser, B. Lymphocyte-specific chemokine receptor CXCR3: regulation, chemokine binding and gene localization. Eur. J. Immunol. 28, 3696–3705 (1998).
Smit, M. J. et al. CXCR3-mediated chemotaxis of human T cells is regulated by a Gi- and phospholipase C-dependent pathway and not via activation of MEK/p44/p42 MAPK nor Akt/PI-3 kinase. Blood 102, 1959–1965 (2003).
Korniejewska, A., McKnight, A. J., Johnson, Z., Watson, M. L. & Ward, S. G. Expression and agonist responsiveness of CXCR3 variants in human T lymphocytes. Immunology 132, 503–515 (2011).
Reynders, N. et al. The distinct roles of CXCR3 variants and their ligands in the tumor microenvironment. Cells 8 https://doi.org/10.3390/cells8060613 (2019).
Karin, N. CXCR3 ligands in cancer and autoimmunity, chemoattraction of effector T cells, and beyond. Front Immunol. 11, 976 (2020).
Biddison, W. E. et al. Chemokine and matrix metalloproteinase secretion by myelin proteolipid protein-specific CD8+ T cells: potential roles in inflammation. J. Immunol. 158, 3046–3053 (1997).
Jin, L. et al. CXCR1- or CXCR2-modified CAR T cells co-opt IL-8 for maximal antitumor efficacy in solid tumors. Nat. Commun. 10, 4016 (2019).
Craddock, J. A. et al. Enhanced tumor trafficking of GD2 chimeric antigen receptor T cells by expression of the chemokine receptor CCR2b. J. Immunother. 33, 780–788 (2010).
Lesch, S. et al. T cells armed with C-X-C chemokine receptor type 6 enhance adoptive cell therapy for pancreatic tumours. Nat. Biomed. Eng. 5, 1246–1260 (2021).
Li, H. et al. Targeting brain lesions of non-small cell lung cancer by enhancing CCL2-mediated CAR-T cell migration. Nat. Commun. 13, 2154 (2022).
Liu, G. et al. CXCR2-modified CAR-T cells have enhanced trafficking ability that improves treatment of hepatocellular carcinoma. Eur. J. Immunol. 50, 712–724 (2020).
Moon, E. K. et al. Expression of a functional CCR2 receptor enhances tumor localization and tumor eradication by retargeted human T cells expressing a mesothelin-specific chimeric antibody receptor. Clin. Cancer Res. 17, 4719–4730 (2011).
Sun, R. et al. CXCR4-modified CAR-T cells suppresses MDSCs recruitment via STAT3/NF-kappaB/SDF-1alpha axis to enhance efficacy against pancreatic cancer. Mol. Ther. 31, 3193–3209 (2023).
Wang, Y. et al. Chemokine Receptor CCR2b enhanced anti-tumor function of chimeric antigen Receptor T cells targeting mesothelin in a non-small-cell lung carcinoma model. Front Immunol. 12, 628906 (2021).
Whilding, L. M. et al. CAR T-cells targeting the Integrin αvβ6 and Co-Expressing the Chemokine Receptor CXCR2 demonstrate enhanced homing and efficacy against several solid malignancies. Cancers 11 https://doi.org/10.3390/cancers11050674 (2019).
Vitanza, N. A. et al. Locoregional infusion of HER2-specific CAR T cells in children and young adults with recurrent or refractory CNS tumors: an interim analysis. Nat. Med. 27, 1544–1552 (2021).
Ravanpay, A. C. et al. EGFR806-CAR T cells selectively target a tumor-restricted EGFR epitope in glioblastoma. Oncotarget 10, 7080–7095 (2019).
Vitanza, N. A. et al. Locoregional CAR T cells for children with CNS tumors: Clinical procedure and catheter safety. Neoplasia 36, 100870 (2023).
Vitanza, N. A. et al. Locoregional CAR T cells for the treatment of CNS tumors in children: investigational drug service pharmacy activities. J. Hematol. Oncol. Pharm. 14, 148–154 (2024).
Haydar, D. et al. Cell-surface antigen profiling of pediatric brain tumors: B7-H3 is consistently expressed and can be targeted via local or systemic CAR T-cell delivery. Neuro Oncol. 23, 999–1011 (2021).
Majzner, R. G. et al. CAR T cells targeting B7-H3, a pan-cancer antigen, demonstrate potent preclinical activity against pediatric solid tumors and brain tumors. Clin. Cancer Res 25, 2560–2574 (2019).
Biery, M. C. et al. A Protocol for the generation of treatment-naive biopsy-derived diffuse intrinsic pontine glioma and diffuse midline glioma models. J. Exp. Neurol. 1, 158–167 (2020).
Luster, A. D. & Ravetch, J. V. Biochemical characterization of a gamma interferon-inducible cytokine (IP-10). J. Exp. Med. 166, 1084–1097 (1987).
Mount, C. W. et al. Potent antitumor efficacy of anti-GD2 CAR T cells in H3-K27M(+) diffuse midline gliomas. Nat. Med. 24, 572–579 (2018).
Hegde, M. et al. Tandem CAR T cells targeting HER2 and IL13Ralpha2 mitigate tumor antigen escape. J. Clin. Invest. 126, 3036–3052 (2016).
Schmidts, A. et al. Tandem chimeric antigen receptor (CAR) T cells targeting EGFRvIII and IL-13Ralpha2 are effective against heterogeneous glioblastoma. Neurooncol. Adv. 5, vdac185 (2023).
Muhammad, N. et al. A novel TanCAR targeting IL13Ralpha2 and EphA2 for enhanced glioblastoma therapy. Mol. Ther. Oncol. 24, 729–741 (2022).
Yang, M. et al. Tandem CAR-T cells targeting CD70 and B7-H3 exhibit potent preclinical activity against multiple solid tumors. Theranostics 10, 7622–7634 (2020).
Bagley, S. J. et al. Intrathecal bivalent CAR T cells targeting EGFR and IL13Ralpha2 in recurrent glioblastoma: phase 1 trial interim results. Nat. Med 30, 1320–1329 (2024).
Bielamowicz, K. et al. Trivalent CAR T cells overcome interpatient antigenic variability in glioblastoma. Neuro Oncol. 20, 506–518 (2018).
Donovan, L. K. et al. Locoregional delivery of CAR T cells to the cerebrospinal fluid for treatment of metastatic medulloblastoma and ependymoma. Nat. Med 26, 720–731 (2020).
Choi, B. D. et al. CAR-T cells secreting BiTEs circumvent antigen escape without detectable toxicity. Nat. Biotechnol. 37, 1049–1058 (2019).
Choi, B. D. et al. Intraventricular CARv3-TEAM-E T Cells in Recurrent Glioblastoma. N. Engl. J. Med 390, 1290–1298 (2024).
Song, E. Z. et al. The IAP antagonist birinapant enhances chimeric antigen receptor T cell therapy for glioblastoma by overcoming antigen heterogeneity. Mol. Ther. Oncol. 27, 288–304 (2022).
Lin, G. L. et al. Non-inflammatory tumor microenvironment of diffuse intrinsic pontine glioma. Acta Neuropathol. Commun. 6, 51 (2018).
Lieberman, N. A. P. et al. Characterization of the immune microenvironment of diffuse intrinsic pontine glioma: implications for development of immunotherapy. Neuro Oncol. 21, 83–94 (2019).
Flynn, G., Maru, S., Loughlin, J., Romero, I. A. & Male, D. Regulation of chemokine receptor expression in human microglia and astrocytes. J. Neuroimmunol. 136, 84–93 (2003).
Cheeran, M. C., Hu, S., Sheng, W. S., Peterson, P. K. & Lokensgard, J. R. CXCL10 production from cytomegalovirus-stimulated microglia is regulated by both human and viral interleukin-10. J. Virol. 77, 4502–4515 (2003).
Wang, X. & Suzuki, Y. Microglia produce IFN-gamma independently from T cells during acute toxoplasmosis in the brain. J. Interferon Cytokine Res. 27, 599–605 (2007).
Wang, G. et al. CXCL11-armed oncolytic adenoviruses enhance CAR-T cell therapeutic efficacy and reprogram tumor microenvironment in glioblastoma. Mol. Ther. 31, 134–153 (2023).
Brown, C. E. et al. Optimization of IL13Ralpha2-Targeted Chimeric Antigen Receptor T Cells for Improved Anti-tumor Efficacy against Glioblastoma. Mol. Ther. 26, 31–44 (2018).
Theruvath, J. et al. Locoregionally administered B7-H3-targeted CAR T cells for treatment of atypical teratoid/rhabdoid tumors. Nat. Med 26, 712–719 (2020).
Ghassemi, S. et al. Rapid manufacturing of non-activated potent CAR T cells. Nat. Biomed. Eng. 6, 118–128 (2022).
Mishra, H. K. & Kalyuzhny, A. Revolutionizing cancer treatments through stem cell-derived CAR T Cells for immunotherapy: opening new horizons for the future of oncology. Cells 13 https://doi.org/10.3390/cells13181516 (2024).
Acknowledgements
We thank the services provided by Seattle Children’s administrative staff, Flow Cytometry Core and Office of Animal Care, as well as the Experimental Histopathology Shared Resource (RRID: SCR 022606) of the Fred Hutch/University of Washington/Seattle Children’s Cancer Consortium (P30 CA015704). We thank the funding provided by the Washington Research Foundation (to E.Z.S.), the Invent at Seattle Children’s Postdoctoral Scholars Program (to E.Z.S.), the ChadTough Defeat DIPG Foundation (to A.T.), the American-Italian Cancer Foundation (to A.T.), the Yuvaan Tiwari Foundation (to N.A.V.), the DIPG/DMG Research Funding Alliance (to N.A.V.), the We Love You Connie Foundation (to N.A.V.), the St. Baldrick’s Foundation (to N.A.V.) and the National Cancer Institute of the National Institutes of Health (NIH) (R37CA289981 to N.A.V.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. We also thank the generous support from the Avery Huffman DIPG Foundation, Ezra Blue’s Flight for Cancer, Liv Like a Unicorn, Live Gray’s Way, Love for Lucy, the McKenna Claire Foundation, the Pediatric Brain Tumor Research Fund Guild of Seattle Children’s, the Team Cozzi Foundation, and Unravel Pediatric Cancer.
Author information
Authors and Affiliations
Contributions
Conceptualization was by E.Z.S., M.C.J. and N.A.V.; methodology and experimental design was by E.Z.S., A.T., M.M., L.E-S., L.Z.L., S.J., M.D.D., J.B.F., M.K.E., S.S.P., V.K., S.S., M.C.J., M.C.B. and N.A.V.; investigation was by E.Z.S., M.M., L.E-S., D.S.L., L.I.W.; data analysis was by E.Z.S., L.E-S. and N.A.V.; writing was by E.Z.S. and N.A.V with input from all authors; supervision was by N.A.V.
Corresponding author
Ethics declarations
Competing interests
E.Z.S., M.C.J. and N.A.V. are inventors on pending patents related to this study, which are disclosed in U.S. Provisional Patent Application No. 63/569,626, entitled “CXCR3-ENGINEERED ANTI-B7H3 CAR T CELLS”, U.S. Provisional Patent Application No. 63/653,023, entitled “CXCR3 ISOFORMS TO IMPROVE ANTI-B7-H3 RECOMBINANT RECEPTOR TRAFFICKING”, and International Patent Application No. PCT/US2025/021354, entitled “CXCR3 ISOFORMS TO IMPROVE RECOMBINANT RECEPTOR TRAFFICKING”. M.C.J. holds equity in and is the Chief Scientific Officer of BrainChild Bio, Inc. M.C.J. holds equity in, is a Board Observer for and serves as a member of the Joint Steering Committee of Umoja Biopharma, Inc. N.A.V. holds equity in and serves as the Scientific Advisory Board Chair for BrainChild Bio, Inc. All other authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Laura Donovan, Joshua Bernstock, and Maria Vinci, who co-reviewed with Fabio Scirocchi, for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Song, E.Z., Timpanaro, A., Meechan, M. et al. Engineered CXCR3-A expression enhances B7-H3-targeting CAR T cell migration and efficacy against diffuse intrinsic pontine glioma. Nat Commun 16, 9914 (2025). https://doi.org/10.1038/s41467-025-64861-6
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-64861-6
