
Abstract
Spiders’ ability to spin foraging webs using silk from specialized glands represents a remarkable evolutionary innovation, yet the molecular evolution of silk glands remains unclear. Here, we investigated the evolution of silk glands in Araneoidea spiders through genomic and transcriptomic mining. After the divergence of the Araneoidea clade, numerous new genes with simplified structures and constrained expression emerged, expressed predominantly in silk glands and coexpressed with ancient genes to drive the evolution of silk glands. Among these, SpiCE-DS8, a newly evolved small secretory peptide unique to the Nephilinae subfamily, interacted with the N-terminal of MaSp1b and was incorporated into dragline silk, potentially aiding in silk solidification. In vitro wet-spinning experiments demonstrated that SpiCE-DS8 enhanced fiber properties, likely enabling spiders to arm their foraging webs for more efficient prey capture. These findings highlight the pivotal role of lineage-specific genes in silk gland evolution and provide insights for synthetic silk development.
Similar content being viewed by others


Introduction
The ability of spiders to spin foraging webs using silk secreted by specialized glands is a remarkable evolutionary innovation, considered as impressive as bird feathers and insect wings1,2. The acquisition of the ability to spin foraging webs has allowed spiders to increase their evolutionary diversification while providing a substrate on which they can explore various survival strategies3,4. Among spiders, the superfamily Araneoidea, representing one-quarter of the spider tree of life, serves as an exemplary model of this evolutionary achievement5. These spiders, which spin webs that vary in shape and size, are distributed across a wide range of ecosystems6. Through the synergistic use of robust web frameworks and adhesive silk, they exhibit a unique ability to capture prey efficiently.
The mechanical properties of spider webs are largely attributed to the diverse silk types produced by different glands. Major ampullate (Ma) silk forms the primary framework of the web, providing exceptional tensile strength and toughness for structural support7. Minor ampullate (Mi) silk, which is used for auxiliary support lines or spiral construction, enhances web stability8,9. Moreover, the capture spirals, which are composed of elastic and adhesive silks10,11, endow the web with unparalleled prey capture efficiency. The high toughness and adhesiveness of spider silk directly influence web size, load-bearing capacity, and prey capture success12,13. Notably, the exceptional characteristics of spider silk allow the construction of webs across substrates ranging from delicate grasses and shrubs to the vast branches of towering trees6,14, highlighting the extraordinary versatility and strength of silk in various environments. However, the mechanisms underlying the evolutionary innovation of high-performance silk in Araneoidea spiders, along with the molecular evolution of their silk-producing glands, remain poorly understood.
New genes are considered key drivers of phenotypic evolutionary innovation15,16,17. In both animals and plants, the phylostratigraphic approach for transcriptomic analysis provides new insights into species-specific tissue evolution18. For example, studies have elucidated the single-insertion origin of metazoan larvae19, the chordate origins of the vertebrate brain20, and the rapid growth of bamboo shoots17. By assigning all genes within a target genome to different phylogenetic strata (PS) based on their evolutionary lineage, this approach enables the chronological characterization of gene age within a phylogenetic framework. Integrating gene expression profiles further facilitates the investigation of tissue evolution17,18. Additionally, the phylostratigraphic approach can be used to construct coexpression networks of ancient and new genes, facilitating inference of the potential functions of new genes17,21. Motivated by this framework, we are interested in using several Araneoidea spiders as models to investigate the evolutionary process of silk glands through a phylostratigraphic approach, shedding light on the genetic basis of their remarkable functional adaptations.
Significant progress has been made in understanding the mechanical properties of spider silk14,22,23, which are attributed primarily to the repetitive sequences in spider spidroins and their highly conserved C- and N-terminal domains24,25. Recent studies have demonstrated that Araneoidea-specific components, such as the cysteine-rich protein in Latrodectus (black widow spiders) and the spider silk composition element (SpiCE) in Trichonephila clavipes (golden orb-weavers), substantially enhance the performance of recombinant Ma silk26,27,28. These components are likely encoded by genes unique to Araneoidea spiders and are absent in other spiders, suggesting that the evolution of new genes in this lineage has conferred spiders with superior silk properties. Comparative transcriptomic studies of silk glands in Argiope argentata and cobweb-weaving spiders have revealed lineage-specific gene expression patterns, including silk gland-specific transcripts and divergent expression profiles between fiber- and glue-producing glands, highlighting the crucial role of gland-specific expression components in silk synthesis and evolution29,30. However, it remains uncertain whether these genes are truly unique to Araneoidea spiders or have evolved from preexisting genetic frameworks. Additionally, their systematic identification and functional significance in the evolution of silk glands and silk production have largely remained unexplored. The recent explosion of spider-related data6,23,31 provides a significant opportunity for advancing research in this field.
In this study, we employed progressive whole-genome alignment and stringent homology-based methods to investigate the genes and transcripts of several Araneoidea spiders, tracing a substantial number of genes that emerged after the divergence of the Araneoidea ancestor. Expression analysis revealed that these genes are highly expressed specifically in silk glands and are closely involved in the core interaction networks of spidroin genes. Surprisingly, proteomics analysis revealed that one new gene, SpiCE-DS8, might be associated with the mechanical properties of spider dragline silk. Through in vitro interaction assays and spinning experiments, we validated the functional role of SpiCE-DS8 in enhancing the mechanical performance of silk. These findings provide new insights into the evolution and innovation of spider silk, highlighting the important role of new genes in the functional evolution of silk glands in Araneoidea spiders and inspiring further breakthroughs in material innovation.
Results
Phylogenetic origins and features of Araneoidea spider genes
To identify new genes in Araneoidea spiders accurately, we collected protein sequence data spanning the entire phylogenetic hierarchy of this group. This dataset includes (1) the Nr protein dataset downloaded from the NCBI database; (2) publicly available genome data for 30 Arachnida species (Fig. 1a, Supplementary Data 1); and (3) transcript assemblies for 1116 species, including 1107 spider species, based on 1166 transcriptomes6,23,31 (Supplementary Data 1). In total, 235,689,197 protein sequences were considered. We then assigned the phylogenetic relationships of Trichonephila clavata within different PS levels according to NCBI-Taxonomy. However, we noted discrepancies in the results of NCBI-Taxonomy and those of previous studies32,33. For example, NCBI-Taxonomy classified horseshoe crabs (Limulidae) as nonarachnids6,32,34, whereas phylogenetic analyses have previously placed them within Arachnida. Additionally, NCBI-Taxonomy placed Trichonephila (formerly Nephila) in the Nephilidae family within Araneoidea, while prior studies have placed it in the Araneidae family6,35. To resolve these issues, we reconstructed the phylogeny of 50 Arachnida species, supporting the placement of horseshoe crabs within Arachnida and confirming that Trichonephila should be classified in the Araneidae family of Araneoidea (Supplementary Fig. 2a–c).

a Gene classification into different PS levels on the basis of an organized workflow. b The 16 PS levels of T. clavata, spanning from the genes of the most ancient cellular organisms to the most recent species-specific genes. RAGs refer to relatively ancient genes, while RYGs refer to relatively young genes. c Gene number distribution across the 16 PS levels in T. clavata. The Venn diagram in the lower-right corner shows that genes from PS13 to PS16 are annotated as genes of unknown function. d–f Box plots showing the transition of genes across the 16 PS levels from ancient to young genes, with decreases in the exon length and protein sequence length, and an increase in the Tau index. The Tau index measures the specificity or breadth of gene or transcript expression across analyzed tissues. The gene sets analyzed for each PS level (PS1–PS16) in d–f are identical to those quantified in c. Box plots display the median (center line), the 25th and 75th percentiles (box limits), and the minimum and maximum values within 1.5× the interquartile range (whiskers).
We followed previously established methods to assign the genes of T. clavata to 16 PS levels, ranging from the oldest (PS1: cellular biology) to the most recent (PS16: T. clavata-specific) (Fig. 1b), and included A. argentata and Latrodectus hesperus based on transcript assemblies from non-reference transcriptomes (Supplementary Fig. 3a, b). Here, we focus on the evolution of silk glands. The Araneoidea superfamily is known for having evolved seven silk glands6, so we classify genes found in the ancestral lineage of Araneoidea, before its divergence, as relatively ancient genes (RAGs, PS1–PS12), whereas those that evolved after this divergence are considered relatively young genes (RYGs, PS13–PS16). Among the 37,599 genes in T. clavata, 30,371 are RAGs, and 6868 are RYGs, with 1932 genes being exclusive to T. clavata (Fig. 1c). Furthermore, functional annotation via eggNOG-mapper36 revealed that these postdivergence genes lacked functional annotations (potentially due to lack of homology or incomplete database coverage) (Fig. 1c, Supplementary Fig. 3c), suggesting that these RYGs, which originated after the divergence of the Araneoidea superfamily approximately 170 million years ago31, may possess novel functions. Furthermore, we performed sequence and expression analyses of genes across the 16 PS levels. Compared with ancient genes, young genes tend to have fewer exons and shorter protein sequences (Fig. 1d, e), with higher expression specificity (Fig. 1f). Notably, at PS8–PS16, genes exhibited a progressive reduction in the number of exons, averaging two exons per gene. The protein sequence lengths decreased to an average of 106–143 amino acids (PS13–PS16), while tissue-specific expression gradually increased, with average tau values exceeding 0.8. These results reveal that new gene structures evolve toward simplification and acquire constrained tissue-specific expression patterns, so younger genes tend to have more specialized functions.
Young genes exhibit expression divergence in silk glands
To better understand the gene expression relationships across different PS levels, we calculated the expression correlations across various tissues to explore the interactions between ancient and young genes. The results revealed widespread correlations for older genes (PS1–PS6) (Fig. 2a, Supplementary Fig. 5a, b), particularly in all silk glands and tissues such as the hemolymph (Hem), pedipalp (Ped), legs (Leg), epidermis (Epi), and venom gland (Ven), whereas the correlations for the fat body (Fat) and ovary (Ova) were weaker. In younger genes, strong correlations persisted in the silk glands between PS7–PS14, but correlations across most tissues were weakened in PS15–PS16. Notably, strong expression correlations were observed between Ped, Leg, and Epi, as well as between Ma and Mi, suggesting potential functional or developmental linkages. For instance, Ma and Mi glands share similar morphological features and collaborate in prey capture through their combined secretions, which may underlie their coordinated gene expression patterns6,24.

a Heatmap of gene expression correlations across the 16 PS levels, calculated using Pearson correlation, revealing a modular expression pattern. Tissue order in each PS heatmap (top-to-bottom, left-to-right) follows the arrow-indicated order in the legend. Correlation strength is shown by the blue-to-red gradient. b Distribution of silk gland-expressed genes and tissue-specific silk gland genes across the 16 PS levels. c Coexpression modules significantly correlated with Ma, Ag, Tu, and Py&Ac. Node size represents normalized expression (log2(TPM + 1)); bold borders indicate silk gland–specific spidroin genes. Pearson correlation coefficients (r) with two-tailed P-values are annotated at the top of the figure. Genes from PS1–PS12 and PS13–PS16 are distinguished by light and dark shading, respectively; tissues are colored as indicated in the legend. Ma major ampullate gland, Mi minor ampullate gland, Tu tubuliform gland, Fl flagelliform gland, Ag aggregate gland, Ac&Py mixed aciniform and pyriform gland, Hem hemolymph, Ped pedipalp, Leg legs, Epi epidermis, Ven venom gland, Fat fat body, Ova ovary.
We examined the expression patterns of genes associated with different PS levels in the silk glands (Fig. 2b). From PS1 to PS11, the number of expressed genes (TPM > 1) in the silk glands consistently exceeded the number of tissue-specific genes (Tau >0.75). However, from PS12 to PS16, tissue-specific genes outnumbered the total expressed genes, suggesting that younger genes play an increasingly important role in silk gland function. To further investigate the relationships between younger genes and silk gland functions, we conducted a weighted gene coexpression network analysis and identified four significant modules associated with silk glands (Fig. 2c, Supplementary Fig. 6). These modules are associated with Ma (r = 0.77, P-value = 7e−09), including 273 RAGs and 86 RYGs; Ag (r = 0.96, P-value = 2e−22), including 543 RAGs and 84 RYGs; Tu (r = 0.98, P-value = 8e−28), including 440 RAGs and 122 RYGs; and Py&Ac (r = 0.95, P-value = 2e−20), including 233 RAGs and 74 RYGs. Spidroin genes were present in the modules associated with Ma, Ag, and Tu, forming complex coexpression networks that integrated both ancient and young genes. Notably, while spidroin genes are core-expressed genes in the silk gland, some RYGs exhibit expression levels that surpass or are comparable to those of spidroins. These findings suggest that younger genes may play critical roles in spidroin-related functions.
Newly evolved genes preferentially expressed in silk glands
Following previously described research strategies37, we constructed a species tree using the genome sequences of 16 spiders (13 Araneoidea spiders and 3 non-Araneoidea spiders) to identify and differentiate newly evolved genes in Araneoidea spiders (Fig. 3a). The protein sequences of the RYGs were aligned to the genomes of these species, including both coding and noncoding regions (for detailed methods, see the Materials and Methods section). A total of 232 genes were identified as having evolved after the divergence of the Araneoidea ancestor, including de novo genes, duplicate genes, and orphan genes (Fig. 3b). The specific classification of genes from PS13 to PS16 is shown in Fig. 3c. For PS13, the numbers of de novo, duplicate, and orphan genes are 2, 30, and 17, respectively; for PS14, the numbers are 2, 10, and 12; for PS15, the numbers are 3, 26, and 41; and for PS16, the numbers are 5, 47, and 37. These results indicate that the number of de novo genes is relatively low in Araneoidea spiders, whereas the accumulation of duplicate and orphan genes may reflect unique mechanisms of genome adaptation during the evolutionary process.

a ML phylogenetic tree of 16 spiders based on concatenated protein sequences of 144 single-copy genes. b Schematic diagram of genes classified as de novo, duplicate, or orphan genes. c Distribution of de novo, duplicate, and orphan genes in PS13–PS16 on the basis of genomic alignment. d Expression patterns of 232 Araneoidea-specific genes across 13 tissues of T. clavata. Trichonephila inaurata ma. refers to Trichonephila inaurata madagascariensis.
The multi-tissue expression profiles of the 232 newly identified genes showed that these genes are predominantly expressed in the six silk glands, with varying degrees of specificity and activity (Fig. 3d, Supplementary Fig. 7). In contrast, their expression levels are considerably lower in tissues such as Hem, Ped, and Leg, with both the number and abundance of transcripts being significantly reduced. Among the silk glands, Ma, Mi, Ag, and Fl exhibited the greatest number of highly expressed genes. Notably, the expression profiles of Ma and Mi were more similar to each other, whereas Ag and Fl exhibited more distinct and specific gene expression patterns. Tu and Py&Ac also showed distinct and specific expression patterns. These findings suggest that the newly evolved genes in Araneoidea spiders play significant roles in silk glands, with their functions becoming increasingly specialized, particularly with possible functional cooperation between the Ma and Mi glands (Figs. 2a–c and 3d).
Two new genes potentially linked to dragline silk function
To investigate the functions of these RYGs in the silk gland, we employed the Ma gland as a model, as described in our previous work31, and divided it into the following three regions: Tail, Sac, and Duct. Proteomic analyses were subsequently performed on each of these regions. Our analysis identified 14 new genes, all of which were supported by proteomic data (Fig. 4a). Among these genes, four were uniquely expressed in the Sac, whereas three were expressed in both the Sac and Duct. Additionally, two genes were expressed in all three regions: Tail, Sac, and Duct. Moreover, the Tail and Duct each contained one gene, highlighting distinct yet interconnected expression patterns across these regions (Fig. 4b). Notably, two of the genes that were common to both the Sac and Duct (SpiCE-DS5 and SpiCE-DS8) were also detected in the dragline silk proteome. Furthermore, transcriptomic data from the three MA gland regions corroborated the expression of these two genes in the Sac and Duct (Fig. 4b). These findings suggest that SpiCE-DS5 and SpiCE-DS8 may play important roles in the function of dragline silk.

a Upset plot of genes identified in dragline silk and the tail, sac, and duct proteomes of the Ma tissue. The numbers highlighted in red in the plot represent new genes identified in the three segments of the Ma gland or dragline silk. The genes SpiCE-DS5 and SpiCE-DS8, indicated by arrows, were also identified in dragline silk. b Transcriptomic and proteomic profiles of 14 new genes in the Ma gland and dragline silk. c Coexpression network of the SpiCE-DS5, SpiCE-DS8, and MaSp genes on the basis of expression profiles from 13 tissues. The RAGs and RYGs are represented in different colors, with the node size reflecting the normalized TPM expression values, which are calculated as log2(TPM + 1). d Gene coexpression network based on single-cell expression levels from the Ma gland. The top five genes of each module are displayed.
The primary components of dragline silk include proteins, inorganic salts, and water38,39. The main protein components are spidroins, along with various auxiliary proteins, whose abundances in the dragline silk are comparable to that of MaSp31. On this basis, we hypothesize that SpiCE-DS5 and SpiCE-DS8 may be involved in the mechanical properties of dragline silk. To further test this hypothesis, we constructed a transcriptomic coexpression network for the SpiCE-DS5, SpiCE-DS8, and MaSp genes by using multitissue data (Fig. 4c). The results revealed that these two genes participate in both direct and indirect interactions with other MaSp family members. For example, SpiCE-DS5 interacts with MaSp-like and MaSp2e, whereas SpiCE-DS8 interacts not only with these two spidroins but also with MaSp1b. Overall, SpiCE-DS5 and SpiCE-DS8 form a complex coexpression network with eight distinct spidroins, encompassing both new and previously characterized genes. These findings support the hypothesis that SpiCE-DS5 and SpiCE-DS8 are functionally related to spidroins in the synthesis and function of dragline silk.
In addition, we previously analyzed the single-cell transcriptome of the Ma gland31, revealing the presence of multiple cell types. To further explore whether SpiCE-DS5 and SpiCE-DS8 interact with spidroins at the cellular level, we conducted a cell-specific coexpression network analysis by using the Ma gland single-cell expression data. As shown in Fig. 4d, the single-cell coexpression network was divided into eight modules (Supplementary Fig. 8); the top five highly expressed genes in each module are displayed. The analysis revealed that MaSp1a, MaSp1b, MaSp1c, MaSp2e, and SpiCE-DS8 were all present in the network. Notably, the MaSp genes were predominantly expressed in the Tail and Sac, whereas SpiCE-DS8 was expressed mainly in the Sac and Duct (Supplementary Fig. 9a). Based on the process of spidroin synthesis, secretion, and fiber formation in the silk gland31, as well as the convergent silk fiber formation mechanism in silkworm silk gland (Supplementary Fig. 9b). We hypothesize that the MaSp proteins are synthesized in the Tail and Sac, and then flow through the Sac and Duct where they interact with SpiCE-DS8. Given that the Sac and Duct are key regions for the transition of spidroin solutions into fibers, we speculate that SpiCE-DS8 may play a critical role in the fiberization process of MaSp spidroins, possibly aiding in the transition from a liquid silk solution to solid silk fibers.
Evolutionary origin and functional role of SpiCE-DS8
We used SpiCE-DS8 as a seed for homology analysis across all spider transcriptomes collected and assembled in this study, as well as for annotating homologous genes in the genomes of 13 Araneoidea spiders, and addressed potential gaps in annotations from previous studies. The results revealed homologous genes in Nephila pilipes and T. inaurata madagascariensis, whereas an unannotated homologous gene was identified in T. antipodina (Fig. 5a). Notably, all three species belong to the subfamily Nephilinae40,41, suggesting that SpiCE-DS8 is a new orphan gene specific to the Nephilinae lineage. Multiple sequence alignment revealed that the protein sequences encoded by SpiCE-DS8 genes contain a conserved signal peptide region (Fig. 5a), along with an additional 42 amino acid sequence, classifying it as a small secretory peptide. Research has shown that small secretory peptides play critical physiological roles in animals42,43,44 and are often secreted into the extracellular space to perform their functions. This finding aligns with the hypothesis that SpiCE-DS8 may be secreted from the sac and duct into the silk gland lumen, where it could interact with MaSp (Supplementary Fig. 9b), further supporting its potential role in silk fiber formation. We identified two conserved sequence regions within SpiCE-DS8, namely, SpiCE-DS8-L (42 aa) and SpiCE-DS8-S (24 aa), with SpiCE-DS8-S exhibiting greater conservation across the four species.

a Multiple sequence alignment of SpiCE-DS8 across four spiders. b Phylogenetic relationships of SpiCE-DS8 genes and their flanking genes in 16 spiders. The phylogenetic tree was constructed via the maximum likelihood (ML) method on the basis of the sequences of 144 single-copy genes. c Prediction of the 3D structure and interaction model of MaSp1b and SpiCE-DS8-L/SpiCE-DS8-S. d Interaction heatmap showing the N-terminal interaction of SpiCE-DS8-L/SpiCE-DS8-S with MaSp1b. e SpiCE-DS8 interacts with MaSp1b and its N-terminal domain (MaSp1b-N). The input group represents total protein, while the IgG and IP groups represent proteins after immunoprecipitation. The prey protein signal was detected in the IP group but not in the IgG group, indicating an interaction between the two proteins. The bait protein signal was also detected in the IP group, confirming successful immunoprecipitation. These results were reproduced in three independent biological replicates with similar outcomes. Source data are provided as a Source data file.
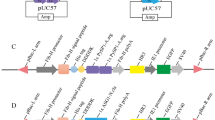
To explore the origin and evolutionary pattern of SpiCE-DS8, four adjacent genes flanking SpiCE-DS8 were included in the analysis. Homology searches and structural annotations of these genes were conducted across the genomes of 15 additional spiders. As shown in Fig. 5b, these four genes are ancient genes that diverged before the common ancestor of Araneoidea. Interestingly, these genes were lost in more ancient branches of the Araneoidea clade (e.g., Theridiida, Tetragnathidae, and Linyphiidae) but were reacquired in the Araneidae family. The SpiCE-DS8 gene itself evolved only in the Nephilinae subfamily and is a unique orphan gene specific to this subfamily (Fig. 3d). This new gene is embedded within the intergenic regions of ancient genes, and similar evolutionary patterns have been reported in previous studies45,46, suggesting that this strategy may be an important mechanism for the emergence of new genes with functional roles.
To further explore the functional role of SpiCE-DS8, we used AlphaFold347 to predict the potential interaction interface between SpiCE-DS8 and MaSp1b (N-terminus, two repeat regions, and the C-terminus) (Fig. 5c). When the 3D structure of MaSp1b alone was predicted, we observed that its N- and C-terminal regions formed ordered helical structures, whereas the repetitive region formed five parallel β-sheet structures. Upon introduction of the SpiCE-DS8-L/SpiCE-DS8-S sequences, the repetitive region of MaSp1b presented an increased number of β-sheet structures, with as many as 9 (SpiCE-DS8-S) or 10 (SpiCE-DS8-L) β-sheets. The ordered folding of the repetitive region is known to facilitate the fiberization process of spidroins48,49, with the conserved SpiCE-DS8-S potentially representing the core functional region of SpiCE-DS8. The interaction heatmap (Fig. 5d) also indicates that SpiCE-DS8-L or SpiCE-DS8-S interacts with the N-terminal region of MaSp1b, with SpiCE-DS8-S showing a notably stronger interaction. Given that previous studies have shown that the N-terminus of MaSp plays a critical role in the transition of spidroins from a soluble state to a solid fiber state50,51, we speculate that SpiCE-DS8, by interacting with the N-terminus of MaSp, promotes the ordered folding of its repetitive regions, thereby accelerating the solidification of silk protein solutions (Supplementary Fig. 11a, b). Co-immunoprecipitation experiments confirmed the interaction between SpiCE-DS8 and MaSp1b (N-terminal region, one repeat region, and the C-terminal region) (Fig. 5e). Immunoprecipitation targeting only the N-terminus of SpiCE-DS8 and MaSp1b still revealed an interaction (Fig. 5e), suggesting that the N-terminus of MaSp1b interacts with SpiCE-DS8.
In vitro spinning shows SpiCE-DS8 enhances silk properties
Enhancing the mechanical properties of dragline silk is crucial for spider prey capture, as the performance of dragline silk, which serves as the framework of the web, directly determines its ability to support larger or heavier prey12,13. To evaluate whether SpiCE-DS8 can improve the performance of dragline silk, we conducted in vitro spinning of fibroin and spider silk analogs. We adapted the wet spinning process proposed by Michael Wöltje et al.52 for this study, modifying it to suit the silk protein spinning workflow (Fig. 6a, Supplementary Movie 1). Fibers were spun from a solution containing 4% fibroin protein derived from both silkworm cocoons (Chuan Shan × Shu Shui) and cocoons of the spidroin-expressing silkworm (SP2-KI: 80% MaSp)53, with the addition of SpiCE-DS8 protein during the process. When a multi-functional variable-speed spinneret was used, the wet-spun fibers exhibited smooth surfaces (Fig. 6b). Remarkably, fibers that were spun with SpiCE-DS8-L and SpiCE-DS8-S showed even tighter and smoother surfaces, indicating that these two proteins exerted a significant impact on the fiber surface structure.

a Schematic of the in vitro spinning process for a silk fibroin and SpiCE-DS8 mixed solution. Image created with Adobe Illustrator 2020. b Scanning electron microscopy (SEM) image of spun silk fibers. This experiment was performed in three independent biological replicates, with similar results observed. c Determination of the secondary structure content of spun silk fibers using FTIR spectroscopy. d Stress‒strain curve of spun silk fibers. Fibroin represents the silk fibroin of silkworm cocoons, and SP2-KI represents cocoons of the spidroin-expressing silkworm. Data reflect measurements from 30 independent biological replicates (n = 30). Source data are provided as a Source data file.
Further analysis through synchrotron infrared spectroscopy was conducted to quantify the secondary structure of the fibers in the amide I region (1600–1700 cm−1). The results showed that the addition of SpiCE-DS8 significantly increased the β-sheet content (Fig. 6c). In Fibroin + SpiCE-DS8-L, the β-sheet content rose from 29.35 to 38.14% (Supplementary Data 2), and in Fibroin + SpiCE-DS8-S, it increased to 39.32%. Similarly, among SP2-KI-containing systems, the β-sheet content increased from 28.46.45 to 37.09% in SP2-KI + SpiCE-DS8-L and 38.08% in SP2-KI + SpiCE-DS8-S. These findings suggest that SpiCE-DS8 promoted the formation of β-sheets and enhanced the ordering of the protein structure.
Next, we measured the mechanical properties of the spun silk fibers. The incorporation of SpiCE-DS8-L resulted in a substantial enhancement in the strain, which increased by approximately 67.7% compared with that of pure fibroin (Fig. 6d, Supplementary Data 3). Similarly, the toughness modulus exhibited a remarkable increase of approximately 70.4%, indicating notable improvements in both ductility and toughness. When SpiCE-DS8-S was incorporated into the fibroin matrix, the performance improvements were even more pronounced. Specifically, the strain increased by approximately 82.1%, the strength increased by 20.6%, and the toughness modulus nearly doubled, reflecting significant gains in the ductility, strength, and toughness. The conserved domain of SpiCE-DS8-S played a critical role in enhancing these mechanical properties. Moreover, in systems containing SP2-KI, the SP2-KI + SpiCE-DS8-S composite exhibited exceptional performance, with the strength and toughness moduli improving by approximately 12.3% and 19.2%, respectively. The Young’s modulus for all the samples ranged from 2500 to 4000 MPa, indicating high rigidity capable of resisting deformation. Although the introduction of SpiCE-DS8 slightly reduced the Young’s modulus, it substantially improved the ductility and toughness of the material, thus enhancing its overall mechanical performance.
In conclusion, the incorporation of SpiCE-DS8 significantly improved the mechanical properties of silk, with particularly pronounced effects observed for the conserved SpiCE-DS8-S region. For spiders, this enhancement aids in the capture of larger and heavier prey, whereas for humans, it offers substantial potential for the artificial synthesis of silkworm and spider silk and the development of high-performance materials, highlighting its dual value in biological and engineering applications.
Discussion
The evolution of new genes plays a pivotal role in driving phenotypic diversification16,17,46. While silk production is a universal trait among spiders6, considerable variation exists in how different species utilize silk functionally. Araneoidea spiders possess up to seven distinct silk glands, each of which is responsible for the secretion of specialized silks—such as dragline silk7, which supports the structural integrity of the web, and glue silk10,11, which is crucial for prey capture. These silk specialization mechanisms enhance both predation efficiency and ecological adaptability. However, the evolutionary processes that underlie the functional diversification of silk glands and the performance optimization of dragline silk remain poorly understood. To address this gap, in this study, we integrated comprehensive genomic and transcriptomic data with robust phylogenetic analysis and gene expression profiling. By doing so, we aimed to elucidate the molecular mechanisms driving the evolutionary divergence of silk gland gene expression, thereby providing new insights into the molecular basis for silk performance optimization in Araneoidea spiders.
Through progressive alignment and rigorous ortholog tracing, we identified new genes that originated after the divergence of the Araneoidea ancestor in T. clavata, A. argentata, and L. hesperus (Fig. 1b, c, Supplementary Fig. 3). Intriguingly, these genes were uniformly annotated as unknown function, consistent with their status as novel genetic elements that potentially encode new biological functions. Sequence and structural analyses further supported this observation, revealing simplified architectures and tissue-specific expression patterns of these genes. The average length of these recently evolved genes was less than 143 amino acids, suggesting that their compact sizes may facilitate rapid evolutionary adaptations. Although functional characterization of these new genes remains challenging, expression profiling demonstrated predominant expression in silk glands. Notably, Araneoidea spiders, which experienced only the ancestral arthropod whole-genome duplication event54, have evolved remarkable web diversity and exceptional dragline silk properties6,14. Strikingly, more than half of the new genes were expressed in silk glands (Fig. 1c), with a significant proportion integrated into the spidroin expression network, even surpassing spidroin expression levels in some cases (Fig. 2c). Our findings elucidated an underexplored aspect of spider genome evolution, highliting a potential connection between the origin of these new genes and the specialization of silk glands and underscoring their critical roles in silk production and function.
Comparative genomic analyses revealed that the evolution of new genes in spiders is primarily driven by gene duplication events and the accumulation of orphan genes (Fig. 3c). In the Ma and Mi glands, new genes exhibited highly conserved expression patterns (Fig. 3d), which may reflect their functional synergy and their expression correlation is tightly conserved across the entire phylogenetic hierarchy (Fig. 2a), suggesting functional coordination between these silk-producing glands8. Ma-derived dragline silk demonstrates exceptional tensile strength and toughness, serving as the structural framework and safety line55,56, whereas Mi silk exhibits a higher elastic modulus to withstand dynamic loading57. In both Ag and Fl glands, distinct yet overlapping expression profiles of the new genes are observed. The silk produced by the Fl glands, which is involved in prey capture, and the wet adhesive produced by the Ag glands, which attaches to the Fl silk, complement each other. The Ag glands are unique to Araneoidea spiders, imparting wet adhesive properties to their webs22. These tissue-specific expression patterns suggest that the new genes may facilitate the functional specialization of silk glands by modulating the composition and material properties of silk proteins. This modulation is crucial for tailoring the mechanical properties of each silk type to meet specific ecological demands, thereby enhancing the adaptive versatility of spider webs.
Proteomic analyses have identified two RAGs, SpiCE-DS5 and SpiCE-DS8, which are expressed in both the sac and duct regions of the Ma gland and are secreted into spider silk, suggesting their potential involvement in silk performance (Fig. 4a, b). The expression of these genes in multiple regions of the gland indicates potential dual functions, contributing to both silk protein synthesis and postsecretion optimization31. Coexpression network analysis revealed interactions among SpiCE-DS5, SpiCE-DS8, and MaSp (Fig. 4c). Single-cell coexpression module analysis further revealed that SpiCE-DS8 functioned as a hub gene (Fig. 4d), playing a central role in the MaSp network through direct and indirect interactions. Interestingly, evolutionary analyses revealed that SpiCE-DS8 was an orphan gene that was exclusive to the Nephilinae subfamily, existing as a single-copy gene and nested within ancient genes (Fig. 5b). This pattern of gene nesting and reacquisition following the loss of conserved genes suggests that the evolution of SpiCE-DS8 is likely driven by functional demands45,46. SpiCE-DS8 encodes a small secretory peptide with extracellular secretion activity that interacts with MaSp within the silk gland lumen (Fig. 5a). Structural predictions and interaction assays confirmed that SpiCE-DS8 interacts with the N-terminal domain of MaSp (Fig. 5c, d). This interaction likely facilitates the orderly formation of β-sheets in repetitive regions, a key step in the transformation of liquid silk protein into solid fibers48,49. This finding underscores the molecular intricacy of silk production and suggests that SpiCE-DS8 may play a pivotal role in influencing silk fiber quality and mechanical properties by guiding protein folding and assembly within the lumen.
The Ma gland of the spider and the silk gland of the silkworm share morphologically similar spinning apparatuses and convergent silk-secretion characteristics (Supplementary Fig. 9)31. Notably, the mechanical properties of silkworm cocoon silk and spider dragline silk are primarily attributed to fibroin heavy chain (FibH, the dominant component of silkworm cocoon silk) and MaSp proteins14,22,23, respectively. Although FibH and MaSp exhibit almost no sequence homology, they share a common architectural organization: a conserved N-terminal domain, a highly variable repeat region, and a conserved C-terminal domain (Supplementary Fig. 12a). Crucially, while their N-terminal sequences differ significantly (Supplementary Fig. 12b), AlphaFold3 predictions revealed that the N-terminal domain of FibH (FibH-N) can also interact with SpiCE-DS8 (Supplementary Fig. 12c, e). This finding strongly suggests that SpiCE-DS8 may similarly enhance the mechanical properties of silkworm silk. Based on this evidence, we further validated the functional role of SpiCE-DS8 using both native silkworm silk and transgenic MaSp-expressing silkworm silk.
In vitro spinning experiments confirmed that the inclusion of SpiCE-DS8 significantly enhances the mechanical properties of spun silk fibers. The core function of SpiCE-DS8 is to increase the β-sheet content of the protein and optimize the structural order of the fibers (Fig. 6c). This increase in the β-sheet content is directly associated with improvements in fiber toughness and strength58,59, particularly following the introduction of the conserved SpiCE-DS8-S region. Both the fibroin and SP2-KI fibers exhibited increased strength and fracture toughness (Fig. 6d), suggesting that the conserved functional domain of SpiCE-DS8 is crucial for promoting fiber formation and optimizing its structural properties. Integrating these findings with the previously observed interaction between SpiCE-DS8 and MaSp, we demonstrate that SpiCE-DS8 primarily facilitates the ordered folding of the MaSp repetitive regions through its interaction with the N-terminus of silk proteins, thereby promoting the self-organization of the silk fibers. Although previous studies have shown that SpiCE-DS1 (Tc11G063480, also named SpiCE-NMa1)60, a core protein of dragline silk, enhances silk toughness in recombinant spider silk experiments, this gene, with a sequence length of 254 aa, originated from the ancient PS10 and is not specific to the Araneoidea. In contrast, SpiCE-DS8, which was identified in this study, is a smaller protein (67 aa) and an orphan gene unique to the Nephilinae subfamily, with a key 24-aa core sequence. This mechanism underscores the critical roles of new genes in functional adaptation and offers a compelling example of how spiders optimize silk performance by incorporating additional small molecular components.
Generally, the toughness of the foraging web prevents larger or heavier insects from breaking through it, while its stickiness keeps the insects from escaping, thereby effectively capturing prey. Our findings indicate that a significant number of novel genes emerged after the divergence of the Araneoidea ancestors, and these genes are closely associated with silk gland functions, suggesting that these new genes may be a core drivers of the evolution of Araneoidea spiders. Assuming that the population size and foraging web performance of Araneoidea spiders are not disturbed by other external factors and that their size has not changed, changes in the web performance and species diversity can signify their exposure to external factors. Herein, we combine climate change and ecological factors61 to explain the changes in spider webs. As shown in Fig. 7, we propose a food chain relationships in which the radiation of angiosperms prompted the radiation of insects62,63,64,65, and subsequently, the radiation of insects drove the radiation of Araneoidea spiders66. To meet ecological demands, the evolution of new genes contributed to the diversification of Araneoidea spiders, including the emergence of wet glue silk and the enhancement of silk properties, both of which are key to prey capture.

The curve at the bottom of the figure shows approximately 225 million years of global temperature changes, corresponding to temperatures on the right thermometer; this curve was drawn according to the study by Voosen61. The divergence time of Araneoidea was ~182 Mya according to TimeTree98. The timeline of angiosperms, insects, and spiders at the top of the figure relates to geological origins. The red line in the spider web represents the dragline silk, and the thin to thick lines represent the silk toughness enhancements. The green solid circle represents the aggregate glue. Image created with Adobe Illustrator 2020.
Overall, our study demonstrates that the enhancement of silk properties in Araneoidea spiders is driven primarily by new genes, offering new insights into the evolutionary mechanisms associated with foraging webs. From an evolutionary perspective, the improved mechanical properties of dragline silk significantly enhance the ability of spiders to capture larger prey, providing a competitive advantage in terms of survival56,67. From the perspective of materials science, the introduction of SpiCE-DS8 provides new routes for the artificial synthesis of high-performance silk materials60,68. In summary, this study not only offers natural inspiration for the development of advanced materials but also illustrates how new genes transcend species boundaries, highlighting their broad functional potential. This study represents a classic example of the reciprocal influence between natural science and materials science, providing valuable insights into the profound connections between biological evolution and applied science.
Methods
Data acquisition
We compiled publicly available data for 1144 chelicerate species, including 1121 spiders, six scorpions, six ticks, six mites, four horseshoe crabs, and one sea spider (Supplementary Data 1). Annotated genomes were available for 30 of these species. For the remaining 1114 species, we assembled de novo transcript sets from transcriptome data through a multistep process: (1) Raw sequencing data were processed using fastp v0.21 to remove adapter sequences69, filter out low-quality reads, and trim bases; (2) the cleaned data were used for de novo transcriptome assembly with Trinity v2.1170; (3) a custom Python script was employed to extract the longest transcripts from the assembled data; (4) CD-HIT v4.8.171 was used to remove redundant sequences with the parameters -c 0.8, -aS 0.8, -d 0, and -T 40; and 5) TransDecoder v5.5.0 was applied to predict coding regions within the assembled transcripts70.
Orthology assignment and phylogenetic analyses
To screen out sequences set for phylogenetic construction, we used the sea spider (N. striatum) as the query, and the putative one-to-one ortholog groups between the sea spider and 50 chelicerates were exposed by all-vs.-all comparisons of the protein sequences in each pair with the Diamond v0.9.27 (E value < 1e−5) program72. Protein sequences were aligned using MUSCLE v3.8.3173 and reverse translated to codon sequences by using Pal2nal v1474. These codon sequences were concatenated for each species to construct the phylogenetic tree. The maximum likelihood phylogenetic tree was inferred using IQ-TREE v2.1.475, RAxML v8.2.12 (with the parameters -x 12345 -# 1000 -m GTRGAMMA)76 and MrBayes v3.2.7 (with the parameters mcmc ngen: 1,000,000; nst = 6; standard deviation <0.01)77 under the optimal model found by ModelFinder78. Resulting trees were visualized with FigTree v1.4.4 (http://tree.bio.ed.ac.uk/software/figtree/).
Gene PS assignment
We first downloaded the NCBI-Nr database (https://ftp.ncbi.nlm.nih.gov/blast/db/FASTA/nr.gz) (data downloaded on August 10, 2021) and integrated it with the gene sets collected and de novo assembled in this study (Supplementary Data 1) to construct a comprehensive local database. The target genome was subsequently categorized into different PS levels based on the NCBI-Taxonomy database (https://www.ncbi.nlm.nih.gov/taxonomy) and phylogenetic profiles. When discrepancies were observed between the NCBI-Taxonomy results and published phylogenetic relationships6,32,34, we reconstructed the phylogenetic relationships to provide a more reliable basis for gene PS assignment. Following the construction of the local database and the determination of PS levels, genes from the target genome were first assigned to PS1 (cellular organism) using the Diamond BLASTP program (E value < 1e−3) for alignment against the local database, following methodologies described in previous research19,20. The genes were assigned using a progressive alignment strategy. Genes aligning at the first PS level were classified as PS1. Then, genes from PS1 were excluded, and the remaining genes were assigned to PS2 (Eukaryota), continuing progressively until reaching the final PS level (species-specific).
New gene classification and detection
Sixteen spiders with available genomic data were selected to determine phylogenetic relationships, followed by screening for new genes specific to the Araneoidea family (PS13–PS16) based on the gene PS assignments. For Araneoidea-specific genes, the classification into three categories was based on the study by Zhang et al.17,37. (1) The first category was de novo genes, referring to noncoding homologous fragments present in the non-Araneoidea genomes that do not form complete gene sequences79,80. The BLAT v3.5 tool81 was used to scan the genomes of non-Araneoidea spiders (minIdentity = 80, coverage > 20%) to identify these genes. (2) The second category was duplicate genes, which are homologous genes or noncoding homologous fragments that are present in two or more copies in the Araneoidea genomes but absent in non-Araneoidea genomes. The duplicate genes were screened for via DupGen_Finder v1.017,82 with an E-value < 1e−5. (3) The third category was orphan genes; although previous studies have categorized species-specific genes as orphan genes83, they are defined here as genes having neither ancestral homologous segments nor homologous genes specific to the Araneoidea family. Additionally, we used the GMAP tool84 for gene structure annotation in other spider genomes.
RNA-seq data quantification
The quantification of the nonreference transcriptome was performed via the following process (Supplementary Fig. 4): (1) the previous steps (de novo assembly of transcripts, as described in Data acquisition) were used to obtain nonredundant transcripts; (2) HISAT v2.2.185 was used to map the clean reads to the transcripts; and (3) StringTie v2.1.586 was employed to calculate the TPM values of each longest open reading frame on the basis of the gff file (generated by TransDecoder70). The TPM values of T. clavata were calculated directly by using the second and third steps. The transcriptomic data for the T. clavata spider were obtained from our recently developed SpiderDB (https://spider.bioinfotoolkits.net/) database31. The Tau index was calculated by using the R package tispec (https://github.com/roonysgalbi/tispec).
Proteomics
T. clavata spiders (Supplementary Fig. 1) were captured in Dali, Yunnan, China, and transported to the laboratory for Ma gland dissection by the following steps: (1) Ma glands were segmented with scissors as described in our previous study31. Approximately 20 mg per sample (~4–6 female spiders) was taken for protein component identification, and three independent biological replicates were established. (2) After homogenization, each sample was dissolved in 500 μL of 9 M LiSCN solution, centrifuged at 10,000 rpm for 10 min, and the supernatant was collected. (3) Then, the protein concentration was measured via the Bradford assay (Beyotime, China). (4) Following protein concentration determination, the sample was diluted 10-fold with 8 M urea solution and mixed with 5 × SDS–PAGE buffer solution. (5) Separation was performed by NuPAGE and using a 4–12% Bis‒Tris protein gel (Thermo Fisher Scientific, USA). (6) The samples were trypsin-digested, and the peptides were identified via LC‒MS/MS using a Q Exactive HF‒X Column (Thermo Fisher Scientific, USA). (7) The identified peptides were quantified using MaxQuant v1.3.0.587 against a custom protein sequence database comprising 37,607 entries from T. clavata. The search parameters included a minimum peptide length of 7 aa and allowed a maximum of two missed cleavages. Both peptide and protein identifications were filtered at a 1% false discovery rate, with protein identification requiring at least one unique peptide. All reverse database matches and potential contaminants were systematically excluded from the final dataset to ensure data quality. A protein had to be identified in three biological replicates for the identification to be considered valid, and the iBAQ (intensity-based absolute-protein-quantification) value was used to assess protein expression levels.
Gene coexpression analysis and 3D structure prediction
Gene coexpression network analysis was performed via the WGCNA v1.73 R package88. Single-cell data from the Ma gland were obtained from the SpiderDB database31. Based on the classification into 10 cell types, encompassing 9349 single cells, the R package hbWGCNA89 was used to calculate gene coexpression relationships at the single-cell level. Complex predictions were made by using the AlphaFold3 tool90, and visualization and hydrogen bonding force analysis were performed by using ChimeraX v1.6.191.
Co-immunoprecipitation
For co-immunoprecipitation, 293T cells were cultured, transfected with plasmids, and harvested 48–72 h posttransfection for protein extraction. Total protein was extracted using IP lysis buffer containing a protease inhibitor cocktail and stored at −80 °C. Protein A/G beads were prepared and washed with PBS and then incubated with anti-Flag (MBL, M185-3L) or anti-GFP (Roche, 11814460001) antibodies for 1 h, followed by overnight incubation with total protein lysate. After repeated washes to remove nonspecific binding, the immunoprecipitated proteins were treated with IP lysis buffer (Thermo, 87787) and SDS sample buffer, heated, and stored as the IP and input groups. SDS‒PAGE and wet transfer were performed to transfer proteins onto PVDF membranes, which were then incubated with primary and secondary antibodies. Finally, the membranes were exposed, and imaging analysis was conducted.
Protein synthesis and silk protein acquisition
SpiCE-DS8-S and SpiCE-DS8-L protein synthesis was carried out by Sangon Biotech Co., Ltd., Shanghai, China. After HPLC purification and MALDI-TOF MS detection (Supplementary Fig. 10), protein samples with purities greater than 95% were obtained. Silkworm cocoons were provided by the Biology Research Center of Southwest University (Chongqing, China). Cocoons of spidroin-expressing silkworms (SP2-KI) were gifted by Prof. Anjiang Tan from Jiangsu University of Science and Technology (Jiangsu, China; Ref. 53). Briefly, transgenic expression of the spider gene MaSp2 in silkworms was achieved using the BmFibH-R system. Approximately 30 cocoons were collected, which produced a 160 kDa MaSp2 protein (GenBank accession ID: AF350276). In this system, endogenous BmFibH expression was completely silenced, while MaSp2 expression was driven by the regulatory elements of BmFibH. The repetitive sequence of BmFibH was replaced with that of MaSp2, whereas the N- and C-termini retained the native BmFibH sequence. Homozygous transgenic silkworms were successfully generated, with MaSp2 accounting for 51.02% of the total cocoon silk. For silk degumming, 5 g of silkworm cocoons was cut into small pieces and boiled in a 0.02 M Na₂CO₃ solution with continuous stirring for 30 min, after which the silk fibers were obtained92. The silk fibers were then rinsed with distilled water for 1 min, and excess water was removed by squeezing. After three additional rinse cycles, the degummed fibers were dried overnight. For lithium bromide dissolution, 3 g of silk fibers was added to 300 mL of 9.3 M LiBr solution and heated in a 55 °C water bath for 4 h until completely dissolved92,93. The solution was dialyzed against pure water for 48 h using a dialysis membrane with a molecular weight cutoff of 14,000 Da until the conductivity dropped below 10 µS94.
In vitro protein interaction assay monitored by bright-field microscopy
SpiCE-DS8-L protein was dissolved in ultrapure water, and complete dissolution was confirmed by centrifugation at 12,000 × g for 1.5 min, with no observable precipitation. SP2-KI and fibroin powders were independently dissolved in 9.3 M LiBr and dialyzed (14 kDa MWCO) against deionized water for 48 h (conductivity <10 µS/cm). For the interaction assay, equimolar mixtures of SpiCE-DS8-L with either SP2-KI or fibroin were incubated at 23 °C for 30 min, followed by bright-field microscopic analysis (10 µL aliquots).
Wet spinning raw material preparation
To prepare the spinning dope, silk fibroin was desalted, then frozen at −20 °C, and then freeze-dried using a SCIENTZ-10N/A lyophilizer for 24 h. In the control group, 0.4 g of freeze-dried filipin material was dissolved in 10 mL of 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP) to create a 4% (w/v) spinning solution52. For the experimental group, 0.35 g of freeze-dried filipin material was combined with 0.05 g of SpiCE-DS8 protein in 10 mL of HFIP. The resulting turbid suspension was incubated overnight at 37 °C with constant shaking. After incubation, a pale yellow, viscous, clear liquid was obtained, which was suitable for wet spinning.
Wet spinning
A syringe pump delivered the spinning solution at a constant rate of 50 μL/min through a conical needle with an inner diameter of 200 μm, directing it into a preprepared coagulation bath composed of 80% (v/v) ethanol, 0.6 M ammonium acetate, and 1% (w/v) PEG400052. Upon contact with the coagulation bath, distinct fibers formed instantaneously and were retrieved using tweezers. These wet fibers were subsequently mounted onto a spinning wheel and towed at a uniform speed of 0.06 m/s. The entire process ensured that all the fibers were efficiently collected on the spinning wheel.
Fiber characterization
The diameters of different segments within each fiber sample (n = 3) were measured under a microscope (magnification: 50 × 10) (Leica Microsystems CMS GmbH, Germany), and the cross-sectional areas of adjacent fiber sections were subsequently calculated. For mechanical testing, degummed silk fibers were utilized. Experiments were conducted on a dynamic mechanical analyzer (DMA Q800, TA Instruments, USA) under the following controlled conditions: gauge length of 10 mm, stretching speed of 1 mm/min, ambient temperature of 24 °C, and relative humidity of 60%. Valid data were obtained for approximately 35 strain points per sample95. Experimental stress‒strain data were analyzed using TA Universal Analysis software to extract raw data, which were further processed using ORIGIN 8.0 (OriginLab, Northampton, MA, USA) to calculate key mechanical properties, including elongation, maximum strength, elastic modulus, and toughness.
Attenuated total reflection infrared spectroscopy (ATR-FTIR)
In a controlled dry environment, the ATR attachment was integrated into the optical path of the spectrometer (Thermo Fisher Scientific Nicolet 6700, USA). An air background spectrum was initially recorded. The surface of the bulk or film samples was subsequently firmly pressed against the crystalline surface of the ATR attachment. Infrared spectra were collected with a resolution of 4 cm−1, averaging 32 scans per sample across a wavelength range of 400/600–4000 cm−1.
Scanning electron microscopy (SEM)
The microfiber samples were directly affixed to a conductive adhesive and coated with gold for 45 s via a Quorum SC7620 sputtering coater at 10 mA. Following gold sputtering, the samples were imaged using a ZEISS Sigma 300, ZEISS GeminiSEM 300, or TESCAN MIRA LMS scanning electron microscope for morphological analysis and energy mapping. Imaging for morphology was conducted at an accelerating voltage of 3 kV via an SE2 secondary electron detector, whereas spectral mapping was performed at 15 kV.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The datasets generated in this study, including assembled transcripts, gene expression profiles, Tau values, and phylogenetic trees, are publicly available in the Figshare96 repository (https://doi.org/10.6084/m9.figshare.28237562/30193852 and https://doi.org/10.6084/m9.figshare.30193852). The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium (https://proteomecentral.proteomexchange.org) via the iProX partner repository97 with the dataset identifier PXD067171. All datasets are publicly accessible and can be freely used by readers without restrictions. Source data are provided with this paper.
Code availability
All analysis software parameters and custom scripts used in this study are archived together with the datasets in the Figshare repository (https://doi.org/10.6084/m9.figshare.28237562).
References
Averof, M. & Cohen, S. M. Evolutionary origin of insect wings from ancestral gills. Nature 385, 627–630 (1997).
Prum, R. O. & Brush, A. H. The evolutionary origin and diversification of feathers. Q. Rev. Biol. 77, 261–295 (2002).
Harwood, J. D., Sunderland, K. D. & Symondson, W. O. C. Web-location by linyphiid spiders: prey-specific aggregation and foraging strategies. J. Anim. Ecol. 72, 745–756 (2003).
Uetz, G. W. Foraging strategies of spiders. Trends Ecol. Evol. 7, 155–159 (1992).
World Spider Catalog. World Spider Catalog. Version 26. Natural History Museum Bern. Available from: http://wsc.nmbe.ch (accessed on 8 November 2024) (2024).
Kallal, R. J. et al. Converging on the orb: denser taxon sampling elucidates spider phylogeny and new analytical methods support repeated evolution of the orb web. Cladistics 37, 298–316 (2021).
Swanson, B. O., Blackledge, T. A., Summers, A. P. & Hayashi, C. Y. Spider dragline silk: correlated and mosaic evolution in high-performance biological materials. Evolution 60, 2539–2551 (2006).
Papadopoulos, P., Ene, R., Weidner, I. & Kremer, F. Similarities in the structural organization of major and minor ampullate spider silk. Macromol. Rapid. Comm. 30, 851–857 (2009).
Greving, I. et al. Structural diversity of native major ampullate, minor ampullate, cylindriform, and flagelliform silk proteins in solution. Biomacromolecules 21, 3387–3393 (2020).
Choresh, O., Bayarmagnai, B. & Lewis, R. V. Spider web glue: two proteins expressed from opposite strands of the same DNA sequence. Biomacromolecules 10, 2852–2856 (2009).
Adrianos, S. L. et al. Nephila clavipes Flagelliform silk-like GGX motifs contribute to extensibility and spacer motifs contribute to strength in synthetic spider silk fibers. Biomacromolecules 14, 1751–1760 (2013).
Agnarsson, I. & Blackledge, T. A. Can a spider web be too sticky? Tensile mechanics constrains the evolution of capture spiral stickiness in orb-weaving spiders. J. Zool. 278, 134–140 (2009).
Sensenig, A. T., Kelly, S. P., Lorentz, K. A., Lesher, B. & Blackledge, T. A. Mechanical performance of spider orb webs is tuned for high-speed prey. J. Exp. Biol. 216, 3388–3394 (2013).
Agnarsson, I., Kuntner, M. & Blackledge, T. A. Bioprospecting finds the toughest biological material: extraordinary silk from a giant riverine orb spider. PLoS ONE 5, e11234 (2010).
Zhang, J.-Y. & Zhou, Q. On the regulatory evolution of new genes throughout their life history. Mol. Biol. Evol. 36, 15–27 (2019).
Cui, X. et al. Young genes out of the male: an insight from evolutionary age analysis of the pollen transcriptome. Mol. Plant 8, 935–945 (2015).
Jin, G. et al. New genes interacted with recent whole-genome duplicates in the fast stem growth of bamboos. Mol. Biol. Evol. 38, 5752–5768 (2021).
Domazet-Lošo, T. & Tautz, D. A phylogenetically based transcriptome age index mirrors ontogenetic divergence patterns. Nature 468, 815–818 (2010).
Wang, J. et al. Evolutionary transcriptomics of metazoan biphasic life cycle supports a single intercalation origin of metazoan larvae. Nat. Ecol. Evol. 4, 725–736 (2020).
Šestak, M. S. & Domazet-Lošo, T. Phylostratigraphic profiles in zebrafish uncover chordate origins of the vertebrate brain. Mol. Biol. Evol. 32, 299–312 (2015).
Ruprecht, C. et al. Phylogenomic analysis of gene co-expression networks reveals the evolution of functional modules. Plant J. 90, 447–465 (2017).
Correa-Garhwal, S. M., Baker, R. H., Clarke, T. H., Ayoub, N. A. & Hayashi, C. Y. The evolutionary history of cribellate orb-weaver capture thread spidroins. BMC Ecol. Evol. 22, 89 (2022).
Arakawa, K. et al. 1000 spider silkomes: Linking sequences to silk physical properties. Sci. Adv. 8, eabo6043 (2022).
Babb, P. L. et al. The Nephila clavipes genome highlights the diversity of spider silk genes and their complex expression. Nat. Genet. 49, 895–903 (2017).
Sanggaard, K. W. et al. Spider genomes provide insight into composition and evolution of venom and silk. Nat. Commun. 5, 3765 (2014).
Pham, T. et al. Dragline silk: a fiber assembled with low-molecular-weight cysteine-rich proteins. Biomacromolecules 15, 4073–4081 (2014).
Larracas, C. et al. Comprehensive proteomic analysis of spider dragline silk from black widows: a recipe to build synthetic silk fibers. Int. J. Mol. Sci. 17, 1537 (2016).
Chaw, R. C., Correa-Garhwal, S. M., Clarke, T. H., Ayoub, N. A. & Hayashi, C. Y. Proteomic evidence for components of spider silk synthesis from black widow silk glands and fibers. J. Proteome Res. 14, 4223–4231 (2015).
Clarke, T. H. et al. Evolutionary shifts in gene expression decoupled from gene duplication across functionally distinct spider silk glands. Sci. Rep. 7, 8393 (2017).
Chaw, R. C., Clarke III, T. H., Arensburger, P., Ayoub, N. A. & Hayashi, C. Y. Gene expression profiling reveals candidate genes for defining spider silk gland types. Insect Biochem. Mol. Biol. 135, 103594 (2021).
Hu, W. et al. A molecular atlas reveals the tri-sectional spinning mechanism of spider dragline silk. Nat. Commun. 14, 837 (2023).
Ballesteros, J. A. & Sharma, P. P. A critical appraisal of the placement of Xiphosura (Chelicerata) with account of known sources of phylogenetic error. Syst. Biol. 68, 896–917 (2019).
Lozano-Fernandez, J. et al. Increasing species sampling in chelicerate genomic-scale datasets provides support for monophyly of Acari and Arachnida. Nat. commun. 10, 2295 (2019).
Fernández, R. et al. Phylogenomics, diversification dynamics, and comparative transcriptomics across the spider tree of life. Curr. Biol. 28, 1489–1497.e5 (2018).
Wheeler, W. C. et al. The spider tree of life: phylogeny of Araneae based on target-gene analyses from an extensive taxon sampling. Cladistics 33, 574–616 (2017).
Cantalapiedra, C. P., Hernández-Plaza, A., Letunic, I., Bork, P. & Huerta-Cepas, J. eggNOG-mapper v2: functional annotation, orthology assignments, and domain prediction at the metagenomic scale. Mol. Biol. Evol. 38, 5825–5829 (2021).
Zhang, L. et al. Rapid evolution of protein diversity by de novo origination in Oryza. Nat. Ecol. Evol. 3, 679–690 (2019).
Sponner, A., Unger, E., Grosse, F. & Weisshart, K. Differential polymerization of the two main protein components of dragline silk during fibre spinning. Nat. Mater. 4, 772–775 (2005).
Sponner, A. et al. Characterization of the protein components of Nephila clavipes dragline silk. Biochemistry 44, 4727–4736 (2005).
Kallal, R. J., Dimitrov, D., Arnedo, M. A., Giribet, G. & Hormiga, G. Monophyly, taxon sampling, and the nature of ranks in the classification of orb-weaving spiders (Araneae: Araneoidea). Syst. Biol. 69, 401–411 (2020).
Dimitrov, D. et al. Rounding up the usual suspects: a standard target-gene approach for resolving the interfamilial phylogenetic relationships of ecribellate orb-weaving spiders with a new family-rank classification (Araneae, Araneoidea). Cladistics 33, 221–250 (2017).
Hancock, R. E. W. & Scott, M. G. The role of antimicrobial peptides in animal defenses. P. Natl. Acad. Sci. USA 97, 8856–8861 (2000).
Maróti, G., Kereszt, A., Kondorosi, E. & Mergaert, P. Natural roles of antimicrobial peptides in microbes, plants and animals. Res. Microbiol. 162, 363–374 (2011).
Titos, I. et al. A gut-secreted peptide suppresses arousability from sleep. Cell 186, 1382–1397.e21 (2023).
Ruiz-Orera, J. et al. Origins of de novo genes in human and chimpanzee. PLoS Genet. 11, e1005721 (2015).
Zhang, W., Landback, P., Gschwend, A. R., Shen, B. & Long, M. New genes drive the evolution of gene interaction networks in the human and mouse genomes. Genome Biol. 16, 202 (2015).
Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021).
Mi, J. et al. High-strength and ultra-tough whole spider silk fibers spun from transgenic silkworms. Matter 6, 3661–3683 (2023).
Li, J. et al. Spider silk-inspired artificial fibers. Adv. Sci. 9, e2103965 (2022).
Gaines, W. A., Sehorn, M. G. & Marcotte, W. R. Spidroin N-terminal domain promotes a pH-dependent association of silk proteins during self-assembly. J. Biol. Chem. 285, 40745–40753 (2010).
Heiby, J. C., Goretzki, B., Johnson, C. M., Hellmich, U. A. & Neuweiler, H. Methionine in a protein hydrophobic core drives tight interactions required for assembly of spider silk. Nat. Commun. 10, 4378 (2019).
Wöltje, M., Isenberg, K. L., Cherif, C. & Aibibu, D. Continuous wet spinning of regenerated silk fibers from spinning dopes containing 4% fibroin protein. Int. J. Mol. Sci. 24, 13492 (2023).
Yu, Y. et al. Custom-designed, mass silk production in genetically engineered silkworms. PNAS nexus 3, pgae128 (2024).
Schwager, E. E. et al. The house spider genome reveals an ancient whole-genome duplication during arachnid evolution. BMC Biol. 15, 62 (2017).
Harmer, A. M. T., Blackledge, T. A., Madin, J. S. & Herberstein, M. E. High-performance spider webs: integrating biomechanics, ecology and behaviour. J. R. Soc. Interface 8, 457–471 (2011).
Blackledge, T. A. Spider silk: a brief review and prospectus on research linking biomechanics and ecology in draglines and orb webs. J. Arachnol. 40, 1–12 (2012).
Dicko, C., Knight, D., Kenney, J. M. & Vollrath, F. Secondary structures and conformational changes in flagelliform, cylindrical, major, and minor ampullate silk proteins. Temperature and concentration effects. Biomacromolecules 5, 2105–2115 (2004).
Jin, H.-J. & Kaplan, D. L. Mechanism of silk processing in insects and spiders. Nature 424, 1057–1061 (2003).
Kaplan, D., Adams, W. W., Farmer, B. & Viney, C. Silk: biology, structure, properties, and genetics. ACS Publications 54, 2–16 (1994).
Kono, N. et al. Multicomponent nature underlies the extraordinary mechanical properties of spider dragline silk. P. Natl. Acad. Sci. USA 118, e2107065118 (2021).
Voosen, P. Project traces 500 million years of roller-coaster climate. Science 364, 716–717 (2019).
Gandolfo, M. A., Nixon, K. C. & Crepet, W. L. Cretaceous flowers of Nymphaeaceae and implications for complex insect entrapment pollination mechanisms in early angiosperms. P. Natl. Acad. Sci. USA 101, 8056–8060 (2004).
Bao, T., Wang, B., Li, J. & Dilcher, D. Pollination of Cretaceous flowers. P. Natl. Acad. Sci. USA 116, 24707–24711 (2019).
Leslie, A. B., Simpson, C. & Mander, L. Reproductive innovations and pulsed rise in plant complexity. Science 373, 1368–1372 (2021).
Xiao, L., Labandeira, C., Dilcher, D. & Ren, D. Florivory of early cretaceous flowers by functionally diverse insects: implications for early angiosperm pollination. Proc. Biol. Sci. 288, 20210320 (2021).
Penney, D. & Ortuño, V. M. Oldest true orb-weaving spider (Araneae: Araneidae). Bio. Lett. 2, 447–450 (2006).
Kono, N., Nakamura, H., Mori, M., Tomita, M. & Arakawa, K. Spidroin profiling of cribellate spiders provides insight into the evolution of spider prey capture strategies. Sci. Rep. 10, 15721 (2020).
Xia, X.-X. et al. Native-sized recombinant spider silk protein produced in metabolically engineered Escherichia coli results in a strong fiber. P. Natl. Acad. Sci. USA 107, 14059–14063 (2010).
Chen, S., Zhou, Y., Chen, Y. & Gu, J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890 (2018).
Haas, B. J. et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 8, 1494–1512 (2013).
Fu, L., Niu, B., Zhu, Z., Wu, S. & Li, W. CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics 28, 3150–3152 (2012).
Buchfink, B., Xie, C. & Huson, D. H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 12, 59–60 (2015).
Edgar, R. C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797 (2004).
Suyama, M., Torrents, D. & Bork, P. PAL2NAL: robust conversion of protein sequence alignments into the corresponding codon alignments. Nucleic Acids Res. 34, W609–W612 (2006).
Nguyen, L.-T., Schmidt, H. A., von Haeseler, A. & Minh, B. Q. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274 (2015).
Stamatakis, A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313 (2014).
Ronquist, F. et al. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 61, 539–542 (2012).
Kalyaanamoorthy, S., Minh, B. Q., Wong, T. K. F., von Haeseler, A. & Jermiin, L. S. ModelFinder: fast model selection for accurate phylogenetic estimates. Nat. Methods 14, 587–589 (2017).
Levine, M. T., Jones, C. D., Kern, A. D., Lindfors, H. A. & Begun, D. J. Novel genes derived from noncoding DNA in Drosophila melanogaster are frequently X-linked and exhibit testis-biased expression. P. Natl. Acad. Sci. USA 103, 9935–9939 (2006).
McLysaght, A. & Hurst, L. D. Open questions in the study of de novo genes: what, how and why. Nat. Rev. Genet. 17, 567–578 (2016).
Kent, W. J. BLAT-the BLAST-like alignment tool. Genome Res. 12, 656–664 (2002).
Qiao, X. et al. Gene duplication and evolution in recurring polyploidization-diploidization cycles in plants. Genome Biol. 20, 38 (2019).
Tautz, D. & Domazet-Lošo, T. The evolutionary origin of orphan genes. Nat. Rev. Genet. 12, 692–702 (2011).
Wu, T. D. & Watanabe, C. K. GMAP: a genomic mapping and alignment program for mRNA and EST sequences. Bioinformatics 21, 1859–1875 (2005).
Kim, D., Langmead, B. & Salzberg, S. L. HISAT: a fast spliced aligner with low memory requirements. Nat. Methods 12, 357–360 (2015).
Pertea, M. et al. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 33, 290–295 (2015).
Cox, J. & Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 26, 1367–1372 (2008).
Langfelder, P. & Horvath, S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinforma. 9, 559 (2008).
Morabito, S., Reese, F., Rahimzadeh, N., Miyoshi, E. & Swarup, V. hdWGCNA identifies co-expression networks in high-dimensional transcriptomics data. Cell Rep. Methods 3, 100498 (2023).
Abramson, J. et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature 630, 493–500 (2024).
Pettersen, E. F. et al. UCSF ChimeraX: structure visualization for researchers, educators, and developers. Protein Sci. 30, 70–82 (2021).
Sofia, S., McCarthy, M. B., Gronowicz, G. & Kaplan, D. L. Functionalized silk-based biomaterials for bone formation. J. Biomed. Mater. Res. 54, 139–148 (2001).
Rockwood, D. N. et al. Materials fabrication from Bombyx mori silk fibroin. Nat. Protoc. 6, 1612–1631 (2011).
Zheng, Z. et al. Lithium-free processing of silk fibroin. J. Biomater. Appl. 31, 450–463 (2016).
Peng, Z. et al. Overexpression of bond-forming active protein for efficient production of silk with structural changes and properties enhanced in silkworm. Int. J. Biol. Macromol. 264, 129780 (2024).
Jia, A. et al. A newly evolved small secretory peptide enhances mechanical properties of spider silk. figshare https://doi.org/10.6084/m9.figshare.28237562 (2025).
Chen, T. et al. iProX in 2021: connecting proteomics data sharing with big data. Nucleic Acids Res. 50, D1522–D1527 (2021).
Kumar, S. et al. TimeTree 5: An expanded resource for species divergence times. Mol. Biol. Evol. 39, msac174 (2022).
Acknowledgements
This work was supported by the National Key Research and Development Program of China (2023YFF0713900; Y.W.), Fundamental Research Funds for the Central Universities (Swu-XDJH202309; Z.D.), and Science and Technology Innovation Key R&D Program of Chongqing (CSTB2022TIAD-STX0015; Y.W.). The authors thank Professor AJ Tan from Jiangsu University of Science and Technology for providing the cocoons of the spidroin-expressing silkworm (SP2-KI). The authors thank YR Wang and HC Jiang at Southwest University for providing help in data collection and analysis.
Author information
Authors and Affiliations
Contributions
A.J., Y.M., and Y.W. designed the research, analyzed the data, wrote the manuscript, and prepared the figures and tables. T.Y. conducted new gene data analysis and the preparation of corresponding charts. G.Z. assisted with the data collection and analysis. A.J., Y.M., and S.M. designed the entire experimental process, Z.D. providing fibroin materials, and Y.M. and Q.W. carried out the artificial spinning experiment. S.M., W.H., Z.D., and Z.Z. provided comments and revisions on the manuscript. Y.W. supervised the research, coordinated the production of the data, and supervised the writing of the manuscript. Y.W. finalized the manuscript with input from all the authors. All the authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare that they have no competing interests.
Peer review
Peer review information
Nature Communications thanks Amanda Markee, Mario Palma and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Jia, A., Mao, Y., Yang, T. et al. A newly evolved small secretory peptide enhances mechanical properties of spider silk. Nat Commun 16, 10071 (2025). https://doi.org/10.1038/s41467-025-65026-1
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-65026-1
