
Abstract
Ferroptosis is a type of iron- and lipid peroxidation–dependent programmed cell death that is involved in various diseases. Some pathogens manipulate host ferroptosis for pathogenesis; however, the potential mechanisms of action remain unclear. Pseudomonas aeruginosa is an opportunistic pathogen that relies on iron for its virulence, biofilm formation, and survival. Here, we report that P. aeruginosa employs the quorum-sensing metabolite, Pseudomonas quinolone signal (PQS), to induce ferroptosis in macrophages through a carnosine-N-methyltransferase (CNMT)-transferrin receptor 1 (TFR1) methylation pathway. Specifically, PQS promotes iron-dependent lipid peroxidation to induce ferroptosis in macrophages. Using high-resolution mass spectrometry–based cellular thermal shift assay (MS-CETSA)/thermal proteome profiling, we identify CNMT as the direct intracellular receptor of PQS in macrophages. Mechanistically, PQS binding increases the histidine methyltransferase (His MTase) activity of CNMT, catalysing methylation of TFR1 at His35. This methylation increases TFR1 protein production, resulting in amplified iron acquisition for ferroptosis. Crucially, the PQS–CNMT–TFR1 axis is distinct from canonical bacterial pathogens that exploit host cell death pathways, revealing the unique strategy of P. aeruginosa to exploit host epigenetic machinery.
Similar content being viewed by others

Introduction
The opportunistic pathogen Pseudomonas aeruginosa causes life-threatening nosocomial infections in the lung, gut, skin, and blood, and is responsible for millions of infections annually, with mortality rates exceeding 40% in immunocompromised patients1. It coordinates virulence through the chemical-based communication mechanism called quorum-sensing (QS)2. QS molecules are released into the extracellular environment and accumulate with increasing bacterial population density. After a threshold, QS molecules regulate bacterial pathogenicity. One of the key QS molecules, Pseudomonas quinolone signal (PQS, 2-heptyl-3-hydroxy-4 (1H)-quinolone), is regulated by the transcriptional activator PqsR (pqs QS system regulator)3. In P. aeruginosa, PQS orchestrates the secretion of diverse virulence factors, such as metabolites, proteins, and membrane vesicles4, that contribute to bacterial pathogenesis, as well as mediates iron acquisition5 and generates reactive oxygen species (ROS)6 for biofilm formation and bacterial survival. From a host–pathogen interaction perspective, PQS modulates host inflammation7, cellular function8, and exhibits direct cytotoxic effects, leading to host cell death through various modalities. PQS promotes ROS production in human lung epithelial cells and inhibits haeme oxygenase-1 (HO-1) and nuclear factor erythroid 2-related factor 2 (NFR2), thus inducing apoptosis9. Moreover, PQS stimulates the production of IL-6 cytokines and induces necroptosis in bone marrow mesenchymal stem cells10. Further, PQS forms a red-coloured complex with Fe3+, resulting in the “red death” of Caenorhabditis elegans11. Although PQS-mediated programmed cell death (PCD) of host cells has been reported, the molecular mechanisms remain unclear.
Ferroptosis is a type of PCD that is triggered by the toxic build-up of lipid peroxides on membranes via the accumulation of iron-dependent ROS and suppression of the antioxidant defence system. These lipid peroxides react with polyunsaturated fatty acids (PUFAs), thereby disrupting cell membrane integrity. This phenomenon is involved in various physiological processes and development of diseases, including infectious diseases12,13,14. Fe3+ mainly enters cells via endocytosis induced by the transferrin receptor (TFR1, CD71), and excess ferrous ion accumulation leads to Fenton reaction and lipid peroxidation, resulting in ferroptosis15,16,17. Studies reveal that secretory proteins, metabolites, and small molecule derivatives from pathogens hijack host ferroptosis18,19. The P. aeruginosa-secreted 15-lipoxygenase (pLoxA) mediates host arachidonic acid–phosphatidylethanolamine (AA–PE) oxidation for lipid peroxidation and degradation of the glutathione peroxidase 4 (GPX4) anti-ferroptosis system to induce ferroptosis20,21,22. Structural analogues of PQS, including the quinazolinone and quinoline derivatives, such as Erastin (ERA)23, DFIQ24, and salsolinol25, are potent inducers of ferroptosis. Although PQS is a key quinolone metabolite secreted by P. aeruginosa, its association with host ferroptosis has not been reported.
Here, we reveal a host–pathogen interaction mechanism through which the P. aeruginosa-derived metabolite PQS manipulates carnosine N-methyltransferase (CNMT, CARNMT1, C9orf41)—a histidine methyltransferase (His MTase)26,27 with uncharacterised roles in iron metabolism—to drive macrophage ferroptosis. Using mass spectrometry–coupled cellular thermal shift assay (MS-CETSA, a.k.a. thermal proteome profiling)28,29, we identified CNMT as the direct intracellular target of PQS. Biochemical validation demonstrated that PQS enhances CNMT-mediated H35 methylation of TFR1, amplifying iron influx and lipid peroxidation cascades. Our findings establish CNMT as the methyltransferase coordinating bacterial QS signalling interacting with host ferroptosis, and suggest a potential PQS-host CNMT/TFR1 interface-based anti-P. aeruginosa treatment.
Results
P. aeruginosa PQS induces macrophage cytotoxicity
First, we compared the cytotoxic effects of P. aeruginosa PAO1 wild-type (PAO1), PQS-deficient mutant (ΔpqsR), and complemented (ΔpqsR + , ΔpqsR + pMiniCTX1-pqsR) strains on RAW264.7 murine macrophages. PAO1, ΔpqsR, and ΔpqsR+ strains had similar growth rates (Fig. 1A). RAW264.7 macrophages were co-incubated with PAO1, ΔpqsR, ΔpqsR + bacteria cultures (Bac) and cell-free supernatants (Sup). The release of cytosolic lactate dehydrogenase (LDH) was used as an indicator of cytotoxicity. Macrophages co-incubated with Bac or Sup of ΔpqsR showed lower levels of cytotoxicity than those incubated with Bac or Sup of PAO1 and ΔpqsR + (Fig. 1B). As the ΔpqsR strain lacks the ability to produce PQS, which is mostly extracellularly secreted by P. aeruginosa, we quantified PQS levels in PAO1, ΔpqsR, and ΔpqsR + Sup using ELISA. ΔpqsR Sup (0.1 μg/mL) had significantly lower PQS than PAO1 Sup (1.4 μg/mL) and ΔpqsR + Sup (1.2 μg/mL) (Fig. 1C). Thus, PQS is likely a key cause of cytotoxicity observed in macrophages co-cultured with P. aeruginosa.

A Growth curves of PAO1, ΔpqsR, and ΔpqsR + strains. B Lactate dehydrogenase (LDH) assay for cell death in RAW264.7 macrophages infected with PAO1, ΔpqsR, and ΔpqsR + (ΔpqsR mutant expressing pqsR from a rescue construct pMiniCTX1-pqsR) Bac (bacteria cultures) or Sup (cell-free supernatants). Multiplicity of infection (MOI) = 100. Data represent the means ± SD (n = 3, two-way ANOVA with Tukey’s multiple comparisons test). C ELISA quantification of PQS concentrations measured in PAO1, ΔpqsR, and ΔpqsR+ supernatants. Data represent the means ± SD (n = 3, one-way ANOVA with Tukey’s multiple comparisons test). D LDH assay for cell death in RAW264.7 macrophages treated with different concentrations of PQS for 24 h, untreated macrophages (Ctrl), and macrophages treated with DMSO as the negative control. Data represent the means ± SD (n = 3, one-way ANOVA with Tukey’s multiple comparisons test). E CCK-8 assay for cell viability in RAW264.7 macrophages treated with different concentrations of PQS for 24 h; IC50 (PQS) = 14.98 μg/mL. Data represent the means ± SD (n = 3). F Microscopic observation of the effect of concentration of IC50 (PQS) exposure for 24 h on RAW264.7 macrophage morphology. Scale bar = 100 µm. G Transcription electron microscopy (TEM) observation of the effect of 10 μg/mL PQS exposure on RAW264.7 macrophage morphology. Nu: cell nucleus, Au-L: autolysosome. 15/40KK: × 15000/ 40,000 magnifications. Scale bar = 2/0.5 μm. H CCK-8 assay for cell viability in BMDM, THP-1, U937, A549, and BEAS-2B cells treated with 10 μg/mL PQS for 24 h. Data represent the means ± SD (n = 3). Source data are provided as a Source Data file.
To assess the direct roles of PQS on macrophage cytotoxicity, we treated RAW264.7 macrophages with purified PQS (Sigma-Aldrich). LDH assay and microscopy observations showed dose-dependent and time-dependent effects of PQS on macrophage cytotoxicity, respectively (Fig. 1D and Supplementary Fig. S1A). Similar results were obtained using the Alamar blue assay (Supplementary Fig. S1B). Using the cell counting kit-8 (CCK-8) assay, the half maximal inhibitory concentration (IC50) of PQS on RAW264.7 macrophages was identified as 14.98 μg/mL (Fig. 1E), which is within the range that is naturally secreted in liquid cultures by P. aeruginosa (approximately 1–20 μg/mL)30. Chlorpromazine (CPZ) is known to preferentially get inserted into the inner leaflet of cell membranes, thereby inhibiting PQS-mediated outer leaflet expansion31. We used CPZ to block PQS entry into cells, and observed that CPZ reduced PQS-induced macrophage cell death (Supplementary Fig. S1C).
Wide-field light microscopy of the morphology of RAW264.7 macrophages treated at IC50 of PQS observed decreased cell numbers compared with untreated RAW264.7 macrophages (Fig. 1F). The trypan blue assay revealed that cell numbers dropped dramatically when RAW264.7 macrophages were treated with PQS (1.4 × 107) compared with control (3.3 × 107) (Supplementary Fig. S1D). Transmission electron microscopy was used to evaluate PQS-induced cell damage at the ultrastructure level, revealing that cells exposed to PQS exhibited distinct changes with many cytoplasmic vacuoles and autolysosomes compared with the control group (Fig. 1G). Scanning electron microscopy results demonstrated that many holes were formed on the membranes of cells treated with PQS (Supplementary Fig. S2A). We tested the effect of PQS treatment on several immortal cells, such as human THP-1 monocytes, U937 monocytes, A549 human lung epithelial cells, and BEAS-2B human bronchial epithelial cells. We also isolated primary bone marrow (BM)-derived macrophages (BMDM), using flow cytometry with the macrophage marker antibodies F4/80 and CD11b (Supplementary Fig. S2B). CCK-8 results were similar to those observed with RAW264.7 macrophages, with cell death observed in all five cell types (BMDM, THP-1, U937, A549, and BEAS-2B), with a much higher death rate of epithelial cells (A549 and BEAS-2B) (Fig. 1H). We chose A549 to observe the morphologies of dead cells using light microscopy. The morphology of PQS-treated A549 cells revealed classical cell death characteristics compared with untreated A549 cells (Supplementary Fig. S2C, D). Then, we assessed whether PQS also causes cell death in the mice lung in an in vivo trachea intubation model using TdT-mediated dUTP nick end-labelled (TUNEL) staining. PQS-treated mice had greater cell death in the lungs than untreated mice (Supplementary Fig. S2E). These results revealed that PQS induces cytotoxicity in many types of host cells as well as in the mice lung in vivo.
PQS triggers iron-dependent ferroptosis in host cells
Various cell death types, such as apoptosis, necroptosis, pyroptosis, ferroptosis, and autophagic cell death, have been noted after bacterial infections32. To identify the cell death type induced by PQS, we used several cell death inhibitors, including ferrostatin-1 (Fer-1, a ferroptosis inhibitor), Necrostatin-1 (NEC, a necroptosis inhibitor), Z-VAD (VAD, a pan-caspases inhibitor to block apoptosis), Disulfiram (DIS, a pyroptosis inhibitor), and 3-methyladenine (3-ME, an autophagy inhibitor), before PQS treatment followed by a CCK-8 assay. Only Fer-1 (which removes ferrous ions produced by lipid hydrogen peroxide33) rescued the cell death effect caused by PQS, whereas minimal effects were observed for the other four cell death blockers (NEC, VAD, DIS, and 3-ME) (Fig. 2A). This result suggests ferroptosis is the predominant death modality.

A CCK-8 assay for cell viability in RAW264.7 macrophages pretreated with different cell death inhibitors—Fer-1, NEC, VAD, DIS, and 3-ME—for 3 h and then treated with 10 μg/mL PQS for 24 h. Data represent the means ± SD (n = 3, one-way ANOVA with Tukey’s multiple comparisons test). B Quantification of total iron in RAW264.7 macrophages treated with PQS or PQS + Fer-1; macrophages treated with Ferric ammonium (FA) and DMSO served as the positive and negative controls, respectively. Data represent the means ± SD (n = 3, one-way ANOVA with Tukey’s multiple comparisons test). Quantification of total iron (C) and CCK-8 assay for cell viability (D) in RAW264.7 macrophages treated with or without PQS in RPMI1640 + FBS medium (normal iron supplemented by FBS) and iron-free RPMI1640 medium without FBS. Data represent the means ± SD (n = 3, two-way ANOVA with Tukey’s multiple comparisons test). E Representative images of Phen Green fluorescence staining to label labile free iron in RAW264.7 macrophages treated with PQS or PQS + Fer-1; macrophages treated with FA served as the positive control. Phen Green: green; Hoechst nucleus: blue. Scale bar = 20 µm. F Representative images of C11-BODIPY fluorescence staining to label lipid ROS in RAW264.7 macrophages treated with PQS or PQS+Fer-1; macrophages treated with ERA served as the positive control. C11-BODIPY: green; Hoechst nucleus: blue. Scale bar = 20 µm. G Flow cytometry for measurement of lipid ROS with 5 μM C11-BODIPY lipid probe in RAW264.7 macrophages treated with PQS or PQS + Fer-1; macrophages treated with ERA and DMSO served as the positive and negative control, respectively. H Flow cytometry for measurement of intracellular ROS with 10 μM DCF probe in RAW264.7 macrophages treated with PQS or PQS+Fer-1; macrophages treated with ERA and DMSO served as the positive and negative control, respectively. I CCK-8 assay for cell viability in RAW264.7 macrophages pretreated with different ferroptosis inhibitors, Lip-1, Trolox, and VK2, for 3 h and then treated with IC50 (PQS) for 24 h; macrophages treated with DMSO served as the negative control. Data represent the means ± SD (n = 3, one-way ANOVA with Tukey’s multiple comparisons test). J Flow cytometry for measurement of mitochondrial ROS with MitoSOX probe in RAW264.7 macrophages treated with PQS or PQS + Fer-1; macrophages treated with ERA and DMSO served as the positive and negative controls, respectively. Source data are provided as a Source Data file.
The total labile free iron concentration was 3.9-fold higher in PQS-treated RAW264.7 macrophages than in control cells, whereas no accumulation was observed in PQS + Fer-1-treated macrophages (Fig. 2B). Moreover, both ferric and ferrous iron were increased in PQS-treated RAW264.7 macrophages than in controls (Supplementary Fig. S3A). To examine the source of elevated levels of free iron, we replaced RPMI1640 + FBS medium (iron supplemented by foetal bovine serum [FBS]) with an iron-free RPMI1640 medium without FBS when cells had reached 80% confluence, along with PQS addition. Only minimal levels of iron were detected in cells grown in iron-free RPMI1640 medium without FBS (Fig. 2C), indicating that PQS-induced elevated cellular free iron is a result of extracellular uptake from the RPMI1640 + FBS medium. Intriguingly, we also observed that PQS induced lower cytotoxicity in cells cultured in medium without iron supplementation, which confirmed iron dependence of PQS-dependent cytotoxicity (Fig. 2D). Phen Green fluorescence staining to label labile free iron in PQS-treated RAW264.7 macrophages revealed accumulation of labile free iron, whereas no accumulation was detected in PQS+Fer-1-treated cells (Fig. 2E), indicating that PQS elevated labile iron pools (LIP) in macrophages.
As lipid peroxidation (ROS) is the hallmark of ferroptosis, we also evaluated lipid ROS levels by staining cells with C11-BODIPY. Confocal microscopy showed greater lipid ROS accumulation in PQS-treated cells than that in the control group and comparable with that in ERA-treated cells, whereas no accumulation was observed in PQS+Fer-1-treated cells (Fig. 2F). ERA (system xc- inhibitor) is a ferroptosis inducer that was used as a positive control in this study. Flow cytometry and lipid ROS quantification showed that PQS efficiently increased lipid ROS to levels comparable to those induced by ERA (Fig. 2G and Supplementary Fig. S3B, C). Ferroptosis may also increase intracellular ROS. DCFH-DA (DCF) fluorescence and flow cytometry results confirmed that PQS increased intracellular ROS in macrophages (Fig. 2H and Supplementary Fig. S3D, E). Further, we used other ferroptosis inhibitors, including liproxstatin-1 (Lip-1), Trolox, and Vitamin K2 (VK2)—that can inhibit ROS-dependent lipid peroxidation—to confirm whether they can inhibit PQS-induced cell death. CCK-8 assay revealed that these ferroptosis inhibitors also attenuated PQS-induced cell death, consistent with the results obtained using Fer-1, confirming that PQS induced iron- and ROS-lipid peroxidation–mediated ferroptosis (Fig. 2I).
Ferroptosis is often accompanied by mitochondrial dysfunction34. MitoFerroGreen and mitoSOX fluorescence analyses indicated that PQS increased mitochondrial labile free iron and ROS in macrophages (Supplementary Fig. S3F and Fig. 2J). Moreover, the mitochondrial membrane potential (MMP, ΔΨm) decreases in ferroptosis35. Tetramethylrhodamine ethyl ester perchlorate (TMRE) fluorescence and flow cytometry results showed that PQS decreases MMP (Supplementary Fig. S3G, H, I). Mitochondria is the main site for ATP production, and ATP levels increase during ferroptosis36. Measurement of ATP showed that PQS increases ATP levels in macrophages (Supplementary Fig. S3J). Further, liperfluo fluorescence results showed greater accumulation of intracellular lipid peroxides in PQS-treated cells than in the control group (Supplementary Fig. S4A, B). Moreover, intracellular malondialdehyde (MDA) and 4-Hydroxynonenal (HNE) levels in PQS-treated RAW264.7 macrophages were higher than in non-treated cells (Supplementary Fig. S4C, D).
Next, studies using A549 human lung epithelial cells revealed that lipid ROS levels within PQS-treated cells was higher than those in control cells, indicating higher ferroptosis levels (Supplementary Fig. S4E). C11-BODIPY fluorescence staining showed lipid ROS accumulation in PQS-treated cells, whereas no accumulation was noted in non-treated cells (Supplementary Fig. S4F, G), confirming pan-cell toxicity of PQS. These results indicated that PQS induces host cell ferroptosis by increasing labile iron concentration, lipid ROS production, intracellular ROS production, lipid peroxides, MDA, and 4-HNE.
CNMT serves as an intracellular effector of PQS
Next, we explored the intracellular targets and the mechanism of action of PQS in host cells, especially within host immune cells. The cellular thermal shift assay in combination with MS-CETSA was used to identify plausible intracellular protein targets of PQS (Fig. 3A). The MS-CETSA technique exploits altered thermal stability and solubility of ligand-bound proteins, which are then quantified at proteome-wide level using protein mass spectrometry to identify putative targets. After processing the data using the T-test statistic methods, CNMT26 was identified as the top potential intracellular target protein of PQS (Fig. 3B). CNMT belongs to the S-adenosyl-L-methionine (SAM)-dependent methyltransferase superfamily in which the binding methyltransferases utilise the ubiquitous methyl donor SAM as a cofactor to methylate proteins37. Then, CETSA western blotting showed that cell lysates treated with PQS (Tm = 58 °C) had a Tm shift of 12 °C relative to lysates treated with DMSO (Tm = 46 °C) (Fig. 3C), revealing a 12 °C thermal stabilisation of CNMT by PQS.

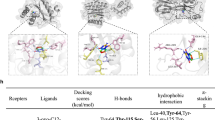
A MS-CETSA experimental procedure to identify the potential direct target of PQS. Schematic diagrams were created using BioRender.com. B Volcano plot of T-test screening of MS-CETSA assay results to identify CNMT as the potential direct target of PQS. Data are presented as the log2 fold change in protein abundance (x-axis) versus the -log10 p-value (y-axis) for each protein, derived from triplicate experiments. Two-sided moderated t test p-values were calculated. C Representative western blot analysis of CETSA experiments of RAW264.7 cell lysate in the presence of PQS or DMSO under 37, 46, 49, 52, 55, and 58 °C. Thermally stable Superoxide Dismutase 1 (SOD1) served as a loading control. D The interaction model of the CNMT protein with PQS visualised using Chimera. E Interaction map between residues of the CNMT protein with PQS. F Quantifying the binding affinity of CNMT protein with PQS using MST assay (n = 3). G Steady-state kinetics of CNMT and CNMT + PQS for carnosine. Kinetic measurements were performed using a bioluminescence methyltransferase assay, MTase-Glo (Promega). Data are presented as mean ± SD (n = 3, two-way ANOVA with Tukey’s multiple comparisons test). Source data are provided as a Source Data file.
Molecular docking was performed using the ligand PQS against the ligand-binding domain of CNMT to predict the interaction sites. Overall, nine CNMT amino acids—L32 (leucine, position 32 from the initial amino acid), S34 (serine), A35 (alanine), A36 (alanine), E49 (glutamic acid), L52 (leucine), E53 (glutamic acid), H56 (histidine), and W221 (tryptophan)—appeared to bind to PQS (Fig. 3D). Mapping the interactions between PQS and the residues within the ligand-binding site on CNMT using the LIGPLOT v. 4.5.338 software showed that PQS binds to the CNMT protein through a hydrogen bond with A35 and hydrophobic forces with residues L32, S34, A36, L52, E53, H56, and W221 (Fig. 3E)—almost similar to analysis obtained with molecular docking (Fig. 3D). Next, a microscale thermophoresis (MST) assay revealed that the dissociation constant (Kd) value of CNMT with its ligand PQS was 79.68 ± 15.34 nM (Fig. 3F), confirming that PQS directly binds to CNMT as a ligand.
The reported function of the transmethylase CNMT is to catalyse the transfer of a methyl group of SAM on carnosine (CAR, N-β-alanyl-L-histidine) to form anserine (ANS, β-alanyl-Npi-methyl-histidine) (Supplementary Fig. S5)37. Thus, we further determined the effects of PQS on the enzymatic activity of CNMT by quantifying the production of the by-product SAH using a bioluminescence assay. CNMT + PQS resulted in faster carnosine methylation (Km = 2.601 μM, kcat = 7.189 min−1, kcat/Km = 2.764 min−1 μM−1) than CNMT alone (Km = 4.631 μM, kcat = 7.091 min−1, kcat/Km = 1.531 min−1 μM−1) (Fig. 3G), indicating that PQS enhances CNMT catalytic efficiency (kcat/Km) on carnosine. These results showed that PQS directly binds the intracellular receptor CNMT to promote its enzymatic activity.
CNMT is responsible for PQS-triggered ferroptosis in macrophages
Next, we verified whether CNMT activity is related to ferroptosis. CNMT knockdown (shCNMT) macrophages were constructed via lentiviral transduction (Supplementary Fig. S6A, S6B and Supplementary Table S1). RT-qPCR confirmed that CNMT mRNA levels were 4.3-fold lower in shCNMT macrophages than in RAW264.7 macrophages transfected with the scramble control, confirming CNMT knockdown (Supplementary Fig. S6C). shCNMT+ complemented macrophages were generated by transiently transfecting shCNMT macrophages with the pCMV-CNMT plasmid. shCNMT+complemented macrophages were used to ascertain the role of CNMT in PQS-induced cell death. Then, a CCK-8 assay was performed to evaluate the role of CNMT in PQS-induced cell death. PQS-treated shCNMT macrophages (71.2%) showed increased cell viability than PQS-treated normal RAW264.7 macrophages (49.1%) and PQS-treated shCNMT+complemented macrophages (53.2%) (Fig. 4A), indicating that CNMT contributes to PQS-induced cell death.

A CCK-8 assay for cell viability in RAW264.7, shCNMT, and shCNMT + (shCNMT expressing CNMT from a rescue construct pCMV-CNMT) macrophages treated with PQS for 24 h. Data are presented as mean ± SD (n = 3, two-way ANOVA with Tukey’s multiple comparisons test). B Representative images of Phen Green fluorescence staining of label labile free iron in shCNMT macrophages treated with or without PQS. Phen Green: green; Hoechst (nucleus): blue. Scale bar = 20 µm. C Quantification of labile free irons in shCNMT macrophages treated in (B). Data are presented as mean ± SD (n = 3, one-way ANOVA with Tukey’s multiple comparisons test). D Representative images of C11-BODIPY fluorescence staining to label lipid ROS in shCNMT macrophages treated with or without PQS; ERA as the positive control. C11-BODIPY: green; Hoechst: blue. Scale bar = 20 µm. E Quantification of lipid ROS in shCNMT macrophages treated in (D). Data are presented as mean ± SD (n = 3, one-way ANOVA with Tukey’s multiple comparisons test). Source data are provided as a Source Data file.
Phen Green fluorescence staining revealed slightly greater accumulation of labile free iron in PQS-treated shCNMT macrophages than in shCNMT macrophages (Fig. 4B, C); however, these levels were lower than those in PQS-treated normal RAW264.7 macrophages (second panel of Fig. 4B vs. second panel of Fig. 2E), indicating that CNMT is crucial for PQS-induced iron influx in macrophages. C11-BODIPY fluorescence staining showed that lipid peroxidation was slightly higher in PQS-treated shCNMT macrophages than in shCNMT macrophages (Fig. 4D, E); however, this was lower than that in PQS-treated normal RAW264.7 macrophages (second panel of Fig. 4D vs. second panel of Fig. 2F), indicating that CNMT is also essential for PQS-induced lipid peroxidation in macrophages. DCF fluorescence staining demonstrated a slightly greater intracellular ROS accumulation in PQS-treated shCNMT macrophages than in shCNMT macrophages (Supplementary Fig. S6D, E); however, this was lower than that in PQS-treated normal RAW264.7 macrophages (second panel of Supplementary Fig. S6D vs. second panel of Supplementary Fig. S3C), indicating that CNMT is crucial for PQS-induced cellular ROS in macrophages. Further, Liperfluo fluorescence staining to label intracellular lipid peroxides showed slightly higher levels in PQS-treated shCNMT macrophages than in shCNMT macrophages (Supplementary Fig. S6F, G); however, this was lower than that in PQS-treated normal RAW264.7 macrophages (second panel of Supplementary Fig. S6F vs. second panel of Supplementary Fig. S4A), indicating that CNMT is also essential for PQS-induced lipid peroxide generation in macrophages. These results demonstrate that PQS promotes ferroptosis-associated biomarkers (labile free iron, lipid ROS, cellular ROS, and lipid peroxides) in a CNMT-dependent manner.
PQS-activated CNMT–TFR1 axis mediates iron overload to induce ferroptosis in macrophages
To explore how CNMT could affect ferroptosis, we performed proteomics analysis to discover potential ferroptosis-related proteins affected by PQS-enhanced CNMT. The expression levels of global proteins in shCNMT RAW264.7 macrophages were compared to those in normal RAW264.7 macrophages after PQS treatment for 24 h (Fig. 5A). Proteomic profiling demonstrated that 122 proteins were downregulated in PQS-treated shCNMT RAW264.7 macrophages compared with PQS-treated RAW264.7 macrophages (Fig. 5B, C and Supplementary Data 1). Subcellular localisation analysis showed that 20.87% of differentially expressed genes encoded plasma membrane proteins. Further, Gene Set Enrichment Analysis (GSEA) demonstrated that the transmembrane transport biological process was enriched (Fig. S7A, S7B). Among the 122 proteins downregulated in PQS-treated shCNMT RAW264.7 macrophages, the iron uptake–associated single-pass membrane receptor TFR1 was directly related to ferroptosis as the critical downstream effector of CNMT15. TFR1 is the main entrance through which cells absorb iron ions. TFR1 exists as a homodimer, with each subunit bound to a molecule of transferrin (TF), which can bind to two ferric ions each, making a total of four ferric ions. The complex is internalised through clathrin-dependent endocytosis, followed by the release of ferrous ions into LIP. Excessive levels of ferrous ions lead to the Fenton reaction and lipid peroxidation, ultimately resulting in ferroptosis (Fig. 5D).

A Proteome analysis to discover the potential direct ferroptosis-related target of PQS-enhanced CNMT. Schematic diagrams were created using BioRender.com. B Heatmap of significantly differentially expressed proteins (DEPs) in the shCNMT RAW264.7 macrophages+PQS compared with RAW264.7 macrophages based on proteome data. C A volcano plot of the DEPs between the RAW264.7 macrophages + PQS and shCNMT RAW264.7 macrophages+PQS based on proteome data was analysed. D Model of TFR1-dependent iron accumulation leading to lipid peroxidation and ferroptosis. Schematic diagrams were created using BioRender.com. E CO-IP assay of CNMT-Flag from the lysates of HEK293T cells co-transfected with TFR1-HA (n = 3). F Interaction model of the CNMT protein with the 67-amino acid TFR1 cytoplasmic region visualised using Chimera. G Interaction assay between CNMT and TFR1 using the Y2H system. The transformants were plated onto low-stringency Leu and Trp defect (left) and high-stringency Leu, Trp, His, and Ade defect (right) selection Synthetic Dextrose (SD) plates, respectively. CNMT* = CNMTK157G+E354G double mutation. H CCK-8 assay for cell viability in RAW264.7, shCNMT, shCNMT + siRNA-TFR1 double knockdown, and siTFR1 macrophages treated with PQS for 24 h. Data are presented as mean ± SD (n = 3, one-way ANOVA with Tukey’s multiple comparisons test). I Representative images of Phen Green fluorescence staining to label labile free iron in shCNMT + siTFR1 macrophages treated with or without PQS. Phen Green: green; Hoechst nucleus: blue. Scale bar = 20 µm. J Representative images of Phen Green fluorescence staining to label labile free iron in siTFR1 macrophages treated with or without PQS. Phen Green: green; Hoechst: blue. Scale bar = 20 µm. Source data are provided as a Source Data file.
Next, CNMT–TFR1 protein interactions were assessed using co-immunoprecipitation (CO-IP). Western blotting results confirmed that CNMT-Flag directly interacted with TFR1-HA in HEK293T cell lysates upon Flag magnet beads pull-down assay (Fig. 5E). TFR1 has 61 amino acids in the N-terminal cytoplasmic region39, with one side always facing the cytoplasm, whereas CNMT is an intracellular protein37; thus, CNMT only can bind to the cytoplasmic region of TFR1. Therefore, molecular docking was performed using CNMT against the cytoplasmic region of TFR1 to predict the exact interaction sites. Two amino acids, K157 (lysine) and E354 (glutamic acid), appeared to bind to two TFR1 amino acids, N11 (asparagine) and L12 (leucine) (Fig. 5F). A yeast two-hybrid assay (Y2H) was performed to confirm the interaction between CNMT and TFR1, with the T + p53 group as a positive control (Fig. 5G). To evaluate whether the CNMT residues K157 and E354 are required for binding to TFR1, we repeated the Y2H using a mutated CNMT (CNMT*) (K157G and E354G). No interactions were observed between the CNMT* protein and TFR1 (Fig. 5G), which showed that CNMT binds to the TFR1 protein via key residues K157 and E354.
Dual knockdown shCNMT+siRNA-TFR1 (shCNMT + siTFR1) macrophages were constructed by transfecting shCNMT macrophages with TFR1-targeted siRNA to evaluate the role of the CNMT–TFR1 pathway in PQS-induced cell death (Supplementary Fig. S7C). CCK-8 assays after PQS treatment of macrophages showed that shCNMT+siTFR1 abolished PQS-induced toxicity. shCNMT + siTFR1 macrophages showed a viability of 92.7%, whereas normal macrophages and shCNMT RAW264.7 macrophages exhibited a viability of 49.1% and 78.5%, respectively. siTFR1 macrophages, which served as a positive control, exhibited a viability of 95.2% (Fig. 5H). This result suggested that the CNMT–TFR1 pathway is the major contributor to PQS-induced cell death. Phen Green fluorescence staining of PQS-treated shCNMT + siTFR1 and siTFR1 macrophages showed that PQS treatment did not have a significant effect on labile free iron accumulation in shCNMT + siTFR1 and siTFR1 macrophages (Fig. 5I, J). However, labile free iron accumulation in PQS-treated shCNMT + siTFR1 and siTFR1 macrophages was significantly lower than that in PQS-treated RAW264.7 and PQS-treated shCNMT macrophages (second panels of Fig. 5I, J vs. second panels of Figs. 2E, 4B), indicating that the CNMT–TFR1 pathway contributes to iron overload that results in PQS-induced ferroptosis in macrophages.
Further RNA-seq and proteomics analyses were performed to examine the potential genes and pathways that are affected in RAW264.7 macrophages by PQS treatment. Proteomics data demonstrated that 92 proteins were upregulated in PQS-treated RAW264.7 macrophages compared with non-treated RAW264.7 macrophages (Supplementary Fig. S7F and Supplementary Data 2). RNA-sequencing data analysis revealed 1404 genes upregulated in PQS-treated RAW264.7 macrophages compared with non-treated RAW264.7 macrophages (Supplementary Fig. S7G and Supplementary Data 3). Comparison of RNA-seq and proteomics data revealed nine shared transcripts upregulated by PQS treatment—TFR1, high mobility group protein HMGI-C2 (Hmga2), U6 snRNA-associated Sm-like protein3 (Lsm3), U6 snRNA-associated Sm-like protein5 (Lsm5), non-histone chromosomal protein HMG-17 (Hmgn2), prefoldin subunit 1 (Pfdn1), high mobility group protein HMG-I/HMG-Y (Hmga1), SAFB-like transcription modulator (Sltm), and mesencephalic astrocyte-derived neurotrophic factor (Manf) (Supplementary Fig. S7H)—which confirmed the essential roles of TFR1 in PQS-induced cell death. These results in this section indicate that PQS-enhanced CNMT interacts with TFR1 to enhance ferroptosis via K157 and E354.
PQS binding increases His MTase activity of CNMT, enabling TFR1 methylation at H35
Although we had shown that PQS promotes CNMT–TFR1 pathway–dependent ferroptosis, the molecular mechanism remained unclear. Therefore, we explored the potential regulatory model. TF and Ferritin (Fth1, encoding ferritin heavy chain) are TFR1-related ferroptosis marker genes. Extracellular TF-bound iron interacts with TFR1 membrane protein for cell iron uptake, while ferritin is a protein that stores iron inside cells, and decreased expression of ferritin releases free iron, leading to ferroptosis40. Western blotting studies revealed that TFR1 and TF were upregulated, whereas Fth1 was downregulated in PQS-treated RAW264.7 macrophages (Fig. 6A). Further, TFR1 protein levels were 1.97-fold higher in PQS-treated RAW264.7 macrophages than in RAW264.7 macrophages while remaining unchanged in shCNMT RAW264.7 macrophages (Fig. 6B), which confirmed that PQS promotes ferroptosis-related protein production.

A Western blotting quantification of the expression of TFR1, TF, and Fth1 protein in RAW264.7 macrophages treated with different concentrations of PQS. β-tublin was the internal control. Protein size (kDa) is indicated on the right side. Densitometric analyses of the bands was performed using the ImageJ software. B Western blotting quantification of the TFR1 expression in RAW264.7 macrophages and shCNMT RAW264.7 macrophages treated with or without PQS. β-actin served as the internal control. Protein size (kDa) is indicated on the right side. Densitometric analyses of the bands was performed using the ImageJ software. C Western blotting quantification of the global histidine methylation level in RAW264.7 macrophages treated with different concentrations of PQS using a pan-Met-His antibody. β-tublin served as the internal control. Protein size (kDa) is indicated on the right side. D Western blotting quantification of the TFR1 histidine methylation level within the in vitro methylation reactions on TFR1 with CNMT and PQS using a pan-Met-His antibody. SAM is the methyl donor. Input CNMT and TFR1 served as the internal control. Protein size (kDa) is indicated on the right side. E Western blotting quantification of the TFR1 histidine methylation level within the in vitro methylation reactions on TFR1WT (wide type) or TFR1H35G with CNMT and PQS using a pan-Met-His antibody. SAM is the methyl donor. Input CNMT and TFR1 as served the internal control. Protein size (kDa) is indicated on the right side. F Proposed molecular mechanism model of P. aeruginosa small molecule PQS promoting CNMT enzymatic activity to methylate TFR1 H35, which leads to ferroptosis in macrophages. Schematic diagrams were created using BioRender.com. All data are presented as means ± SD (n = 3). Source data are provided as a Source Data file.
Owing to the His MTase nature of CNMT, we also explored the extent of contribution of PQS to global histidine methylation levels in whole RAW264.7 macrophages by using a pan-methyl-histidine (Met-His) antibody. Intriguingly, western blotting results showed that PQS increased global histidine methylation levels in whole RAW264.7 macrophages (Fig. 6C). Then, via the in vitro methyltransferase assay, we explored whether CNMT can methylate TFR1. The in vitro methylation assay results demonstrated that CNMT directly methylates TFR1, and PQS enhances the methylation level of TFR1 mediated by CNMT (Fig. 6D). These results indicate that CNMT regulates TFR1 at the post-translational level. TFR1 has only one histidine H35 in the 61-amino acid N-terminal cytoplasmic region. Therefore, we hypothesised that CNMT methylates TFR1 H35. Next, we constructed a mutated TFR1 (TFR1H35G, histidine was mutated to glycine) (Supplementary Fig. S8A). The in vitro methyltransferase assay was repeated using TFR1H35 and TFR1H35G, with results revealing no methylation within TFR1H35G even with the addition of CNMT and PQS (Fig. 6E), which confirmed that CNMT methylates TFR1 H35. We also synthesised one TFR1 peptide containing H35 (VDGDNSHVEMKLA) to perform the in vitro methyltransferase assay with CNMT. LC-MS/MS spectrum verified H35 methylation in TFR1 with a 14.02 mass increase after b7 (Supplementary Fig. S8B).
Based on all the above results, we proposed the following model for CNMT/TFR1-dependent PQS-induced ferroptosis. During P. aeruginosa infection, PQS increases TFR1 methylation levels catalysed by CNMT methyltransferase, leading to greater TFR1 protein production, resulting in elevated iron influx, and ultimately, inducing ferroptosis (Fig. 6F).
Discussion
P. aeruginosa uses different molecular mechanisms, including apoptosis, autophagic cell death, pyroptosis, necroptosis, and ferroptosis, to induce PCD during lung infections41,42. Membrane vesicles secreted by P. aeruginosa can directly enter host cells and target mitochondria, activating Caspase-3/7 and leading to mitochondrial apoptosis43. P. aeruginosa type VI secretes PGAP1-like effector that induces host autophagy by activating endoplasmic reticulum stress44. Our study reveals a paradigm-shifting mechanism: P. aeruginosa QS molecule PQS directly triggers ferroptosis in host cells by manipulating host epigenetic regulation (Fig. 7). We first found that P. aeruginosa PqsR-mediated PQS small molecules trigger cytotoxicity in various host macrophages and epithelial cells (Fig. 1). PQS exploits vulnerabilities in iron metabolism by increasing LIP and promotes iron-dependent lipid peroxidation to induce ferroptosis in macrophages (Fig. 2). MS-CETSA results exhibited that CNMT was the direct intracellular receptor of PQS in macrophages. PQS binds CNMT to promote its enzymatic activities (Fig. 3). CNMT is related to ferroptosis; however it may not directly induce ferroptosis (Fig. 4). Proteomics analysis of shCNMT RAW264.7 macrophages revealed that TFR1 is the downstream protein of CNMT that enhances iron influx for ferroptosis (Fig. 5). PQS binding increases the His MTase activity of CNMT, enabling TFR1 methylation at H35, which upregulates TFR1 protein production, creating a feedforward loop for iron influx and ferroptosis (Fig. 6). These findings highlight a PQS–CNMT–TFR1 pathway adopted by P. aeruginosa to induce ferroptosis during infection of host cells. This pathway is particularly significant given the elevated PQS concentrations detected in clinical infections45,46, which overlap with the PQS IC50 (14.98 μg/mL) measured in macrophages (Fig. 1E).

P. aeruginosa utilises its QS metabolite PQS to hijack the host methyltransferase CNMT. PQS binding activates CNMT, which methylates TFR1 at histidine 35 and increases TFR1 expression, amplifying iron uptake and lipid peroxidation to trigger macrophage ferroptosis. This pathway reveals a unique microbial mechanism of epigenetic manipulation. Schematic diagrams were created using BioRender.com.
Owing to the autoinduction feature of QS metabolites, the concentration of P. aeruginosa PQS might spike during infections and can be clinically detected in the infected lung, urine, and blood, thus inducing various direct effects in host cells by exerting immunomodulatory activities3,8. PQS downregulates nuclear transcription factor (NF)-κB and hypoxia-inducible factor 1 (HIF-1) pathways to block host innate immune systems7. Further, PQS stimulates MAP kinase p38 to enhance the bactericidal activity of polymorphonuclear neutrophils and induce apoptosis47. Moreover, PQS also activates airway epithelial bitter taste receptors (taste family 2 bitter receptor proteins; T2Rs) and stimulates T2R-mediated immune responses48. In this study, we reported that PQS promotes iron-dependent lipid peroxidation to induce ferroptosis in macrophages (Fig. 2), a finding that is consistent with the results of previous PQS-related studies stating that PQS employs multiple mechanisms to control host cell responses. Further, we showed that PQS causes mice lung cell death in vivo (Supplementary Fig. S2E); therefore, the regulatory roles of PQS warrant further investigation in mouse lung models.
Macrophages are usually located in a more complex microenvironment than ordinary cells and often face microbial infection in the frontline, thus evolving stronger molecular mechanisms for iron homoeostasis49. Macrophages have enhanced expression levels of HO-1 and glutathione peroxidase 4 (GPX4) to inhibit ferroptosis50. This may explain heightened epithelial cell sensitivity (Fig. 1H), given their lower baseline GPX4/HO-1 expression. Nevertheless, pathogenic bacteria generally have a mechanism of iron enrichment, which can destroy the ferroptosis defence system in macrophages upon invasion51. Our previous study showed that PQS not only enhances the infection mediated by PqsR receptor protein in bacteria, but also inhibits the innate immune pathway mediated by aryl hydrocarbon receptor (AhR) in host macrophages52. Further, both PQS and vitamin K can be used as ligands of host innate immune AhR transcriptional regulatory proteins53, and we reported in our previous study that PQS and vitamin K have opposite effects on the expression of AhR pathway downstream of P450 cytochrome enzymes52. Recent studies revealed that Vitamin K2-menadione-4 (MK4) can be reduced to hydroquinone (VKH2) by ferroptosis suppressor protein 1 (FSP1), which is a potent radical-trapping antioxidant and phospholipid peroxidation inhibitor and thus has anti-ferroptosis functions54. Thus, PQS may promote ferroptosis, as our results show that PQS overwhelms macrophage iron homoeostasis (Fig. 2).
Post-translational modifications (PTMs) constitute an efficient mechanism for altering the activity, stability, and subcellular location of structural proteins, transcription regulators, and enzymes within cells to regulate ferroptosis55. Protein methylation is a type of SAM-dependent PTM that occurs on diverse amino acid residues. Protein methylation has potential connections with ferroptosis. PtpA, a tyrosine phosphatase secreted by Mycobacterium tuberculosis (Mtb), enters the host nucleus, interacts with the arginine methyltransferase PRMT6, promotes asymmetric dimethylation of histone H3 arginine 2 (H3R2me2a), and inhibits GPX4 to promote ferroptosis56. In our study, P. aeruginosa PQS-enhanced CNMT His MTase was found to methylate TFR1 H35 (Fig. 6). Our findings, combined with previous studies on Mtb, collectively demonstrate that distinct pathogenic species employ divergent strategies to hijack the host’s epigenetic machinery57. Coactivator-associated arginine methyltransferase 1 (CARM1, also known as PRMT4) enhances cardiomyocyte sensitivity to ferroptosis by interacting with NFE2L2 (also known as NRF2) and promoting its methylation, which limits the transcriptional activity of NFE2L2 on GPX458. SETD7, a lysine monomethylase, directly interacts with ubiquitin thioesterase 1 (OTUB1) to catalyse OTUB1 methylation at Lys 122, disrupting the interaction between OTUB1 and the anti-ferroptosis protein ubiquitin conjugating enzyme E2 N (UBE2N/UBC13) in H1299 lung cancer cells59. MS-CETSA thermal profiling (Fig. 3A, B) revealed CNMT as the first methyltransferase directly co-opted by bacterial QS molecules to promote ferroptosis (Fig. 4).
TFR1, a key cell-surface molecule, regulates iron-bound transferrin uptake by receptor-mediated endocytosis60. Several PTMs, including phosphorylation, acetylation, ubiquitination, succinylation, N/O-glycosylation, palmitoylation, and stearoylation, have been identified earlier in TFR1, which have been reported to play multiple roles in cellular functions. Ubiquitin ligase E3 HUWE1/MULE ubiquitylates TFR1 for degradation and suppresses ferroptosis in acute liver injury61. ZDHHC6-mediated stearoylation of TFR1 inhibits the activation of the JNK pathway and promotes mitochondrial fragmentation62. Although various PTMs have been identified in TFR1, methylation has not been investigated. Our structural model proposes that CNMT-mediated H35 methylation of TFR1 (Fig. 6D, E) stabilises its cytoplasmic domain (Fig. 5F), potentially enhancing transferrin recycling efficiency.
Overall, our study has unveiled a host–microbe interaction-mediated regulatory pathway centred on the QS metabolite PQS–CNMT–TFR1 axis in macrophages. This study will provide a theoretical basis for multiple intervention strategies, such as QS inhibition (targeting PqsR to reduce PQS production) and iron chelation (repurposing ferroptosis inhibitors, including Fer-1, Lip-1, Trolox, and VK2) for acute P. aeruginosa infections.
Methods
Ethics statement
All animal experiments were performed according to the standards set forth by the Guide for the Care and Use of Laboratory Animals. All animal studies were conducted according to protocols approved by the Institutional Animal Care Committee of Southern University of Science and Technology (Shenzhen, China) and were performed under protocol no. SL2021051062.
Bacterial strains
Bacteria were cultured in Luria–Bertani (LB) broth (1% tryptone, 0.5% yeast extract, 0.5% NaCl) and ABTGC (B-medium (0.1% MgCl2, 0.1% CaCl2, 0.1% FeCl3) supplemented with 10% A10, 0.2% glucose, and 0.2% casamino acids). The media was solidified with 1.5% Bacto Agar (Difco). The overnight-grown subcultures were transferred into 96-well plates over 24 h, and their growth curves were measured for cell density (OD600) using a Tecan Spark plate reader (Tecan Group Ltd., Mannedorf, Switzerland).
Cell lines
The RAW264.7 murine macrophages (ATCC®TIB-71TM), THP-1 human monocyte (ATCC®TIB-202TM), U937 human leukaemia (ATCC®CRL-3253TM), A549 human epithelium (ATCC®CCL-185™), and BEAS-2B human bronchial epithelium (ATCC®CRL-3588TM) cell lines were grown in Dulbecco’s Modified Eagle (DMEM) or Roswell Park Memorial Institute (RPMI)-1640 medium supplemented with 10% foetal bovine serum (FBS) and 1% penicillin/streptomycin (PS) (Gibco, Texas, USA), followed by incubation at 37 °C in a 5% CO2 incubator. Cell lines were routinely tested for mycoplasma contamination. Mouse primary bone marrow (BM)-derived macrophages (BMDMs) were prepared and cultured following the previous protocols63.
P. aeruginosa mutant and complement strains construction
P. aeruginosa PAO1 deletion mutant strains were generated by homologous recombination. The mutants were constructed by overlapping PCR to incorporate a gentamicin-resistance cassette. The mutant fragments were cloned into pK18, a suicide vector, to generate gene knockout plasmids. Each knockout plasmid was transformed into E. coli strain RK600 and then conjugally transferred from RK600 to PAO1. The resultant integrants were selected on agar plates containing 60 µg/mL gentamicin (Gm60). To resolve merodiploids, Gm-resistant colonies were streaked for single colonies on LB + Gm60 plates containing 10% sucrose. Potential mutants without gentamicin-resistance cassettes were screened by PCR using corresponding flanking primers and confirmed by Sanger sequencing. The ΔpqsR+ complement strain was constructed using a ΔpqsR mutant expressing pqsR from a rescue construct using the pMiniCTX1-pqsR plasmid’s own promoter.
Cell death and viability assay
The cytotoxicity of P. aeruginosa, bacterial supernatant64, and commercially purified PQS (Sigma-Aldrich, Cat Number#94398, ≥96.0% purity by HPLC), CAS Number#108985-27-9) was assayed by using murine RAW264.7 using various assays, including trypan blue, lactate dehydrogenase (LDH), alamar blue, and cell counting kit-8 (CCK-8) assays. RAW264.7 macrophages were grown in 24-well plates in DMEM. Before infection, confluent RAW264.7 cells were washed twice with sterile PBS and incubated in DMEM medium. For bacteria, mid-log phase bacteria (OD600 = 0.6) grown in LB broth were centrifuged and resuspended in PBS. Then, bacteria and relevant supernatant were used to infect RAW264.7 macrophages grown in DMEM medium supplemented with 10% FBS at an MOI of 100. After centrifugation of 24-well culture plates to promote bacterial contact with the macrophages, phagocytosis was allowed to proceed for 30 min. The cells were then washed three times with sterile PBS, and fresh DMEM medium supplemented with 400 μg/mL gentamicin was added. After 2 h infection, the culture supernatants were collected for LDH activity detection. For PQS, different concentrations of PQS were co-cultured with macrophage cells for 24 h. After the co-culture, the culture supernatants were collected for detecting LDH activities. The LDH activities were detected by using a commercial LDH cytotoxicity kit (YAESEN Bio) according to standard procedure. The viability of the remain cells was measured using alamar blue assay (Thermo), and CCK-8 assay (APExBIO) to determine the IC50 of PQS. For the cell death inhibitor (Fer-1, NEC-1, Z-VAD, DIS, and 3-ME) addition assay, cells were pretreated with each inhibitor (both use 10 μM) for 3 h, then PQS was added.
Microscopy imaging of cell death
To examine cell death morphology, cells were treated as described in the 6-well plates (Nunc products, Thermo Fisher Scientific Inc.) for static image capture or in glass-bottom culture dishes (MatTek Corporation) for live imaging. Static bright-field images and live-cell images were captured using an Olympus IX71 or a Zeiss Pascal Confocal microscope. The images were processed using ImageJ or the LSM Image Examiner programme. All image data shown were representative of at least three randomly selected fields.
Confocal fluorescence microscopy imaging and flow cytometry
Cells were plated on 35 mm glass-bottom confocal plates (Corning). Labile iron ions, lipid ROS, cellular ROS, and lipid peroxides were stained with Phen Green, BODIPY C11, DCF, and Liperfuo, respectively. All nuclear staining was performed with Hoechst 33342 (Life Technologies). For confocal imaging, cells were imaged using a Plan-Apochromat 63 × or 100 ×/1.4 oil objective on a Zeiss LSM900 confocal system equipped with an Airyscan module and controlled by the Zen blue software. The signal intensity per cell was measured with ImageJ software v1.53 (NIH). All image data shown were representative of at least three randomly selected fields. For flow cytometry analysis, cells were analysed using a flow cytometer (CytoFLEX and CytExpert 2.4, Beckman Coulter) with a 488-nm laser paired with a 530/30 nm bandpass filter. Data were analysed using FlowJo Software 10 (Treestar).
Transmission electron microscopy (TEM) and Scanning electron microscopy (SEM)
The electron microscopy methods were performed as previously described65. For TEM, cell samples in vitro were prefixed in a mixture of 2% paraformaldehyde and 2.5% glutaraldehyde for 2 h at RT. The cells were stained with 1% uranyl acetate for 1 h at RT, followed by three washes with ddH2O (15 min each). Samples were dehydrated in a gradient of ethanol solutions ranging from 30% to 100% ethanol (30, 50, 70, 80, and 90% for eight min; 100% for eight min 3 times), followed by propylene oxide (two 10 min washes). Samples were gradually embedded with 3 mixtures of EPON812 and propylene oxide (ratios of 1:1, 2:1, and 3:1) for 2 h at 4 °C, followed by embedding in pure EPON812 overnight. Following, the samples were incubated with fresh EPON812 for 8 h. The cell samples were embedded for 4 h at RT, incubated overnight at 37 °C, and cured in the oven at 60 °C for 24 h. The polymerised cell samples were sectioned, and stained with 1% uranyl acetate for 20 min followed by lead citrate for 5 min. 70 nm-thick ultra-thin sections were prepared, and images in vitro were acquired using a Tecnai Spirit 120 kV TEM (FEI, USA) at an acceleration voltage of 80 kV. For SEM, cells were fixed with 2.5% glutaraldehyde overnight and then rinsed with PBS three times. Samples were dehydrated through a graded series of ethanol (30, 50, 70, 95, and 100%) and dried by the tertiary butanol method. Dried specimens were sputter coated with gold-palladium and imaged with a JEOL JSM 66390LV field emission scanning electron microscope operating at 10 kV.
Animal experiments
In our study, the C57BL/6 mice were divided randomly into two groups (n = 6 per group, 6-8 weeks old female, weight = 20– 25 g): the control group receiving 0.9% NaCl (containing 0.1% DMSO) and the PQS group receiving PQS plus 0.9% NaCl (containing 0.1% DMSO). Mice were induced by instilling intratracheally 50 μL of PQS solution (10 mg/mL) to reach 25 mg/kg body weight. After the treatments for 16 h, the mice in each group were euthanised, and lung tissues were collected and stained via TdT-mediated dUTP nickend label (TUNEL) following the manufacturer’s instructions (Roche Applied Science), followed by Hoechst 33258 DAPI stain, before being mounted and visualised.
Total, ferrous, and ferric iron ion quantification
RAW264.7 macrophages were seeded on 24-well dishes at an appropriate density and allowed to adhere overnight. The next day, cells were treated with 10 μg/mL PQS and ferric ammonium (FA, 100 μM) for 24 h. After incubation, cells were scraped and resuspended in 300 µL of HBSS. The total, ferrous, and ferric iron ions were detected by using a commercially total, ferrous, and ferric iron ion quantification kit (Elabscience) according to standard procedure.
Lipid reactive oxygen species (ROS) analysis by C11-BODIPY staining
RAW264.7 macrophages were seeded on 24-well dishes at an appropriate density and allowed to adhere overnight. The next day, cells were treated with 10 μg/mL PQS for 24 h, followed by co-treatment with RSL-3 (0.5 μM) and ERA (2 μM) for 3 h. After incubation, cells were incubated with 5 µM C11-BODIPY (Thermo Fisher) for 30 min at 37 °C. Subsequently, cells were scraped, resuspended in 300 µl of Hanks’ balanced salt solution (HBSS, Gibco), filtered through a 40 µm cell strainer (Falcon tube with cell strainer CAP), and then analysed using a flow cytometer (CytoFLEX and CytExpert 2.4, Beckman Coulter) with a 488 nm laser paired with a 530/30 nm bandpass filter. Data were analysed using FlowJo Software 10 (Treestar).
Cellular ROS production measurement by DCFH-DA staining
RAW264.7 macrophages were seeded on 24-well dishes at an appropriate density and allowed to adhere overnight. The next day, cells were treated with 10 µg/mL PQS for 24 h. followed by co-treatment with RSL-3 (0.5 μM) and ERA (2 μM) for 3 h. Cellular ROS production level was detected by the Reactive Oxygen Species Assay Kit (Beyotime). Cells were incubated with 2’,7’-dichlorofluorescin diacetate (DCFH-DA) at a final concentration of 10 μM for 20 min at 37 °C. The cells were washed with PBS and harvested. The stained cells were analysed by using a Cytoflex S flow cytometer (Beckman). All samples were assayed with lasers emitting at 488 nm, and the fluorescence was collected by a 530/30 nm bandpass filter. The flow cytometric data were analysed using FlowJo software.
Mitochondrial Fe2+, ROS, and potential analyses
For mitochondrial Fe2+ assay, cells were stained with 5 μM MitoFerroGreen (Dojindo, cat. M489) working solution at 37 °C for 30 min. Cells were incubated for 15 min in the dark with the mitochondrial potential sensor TMRE (10 nM; T3168, Invitrogen). The production of mitochondrial ROS was observed upon loading cells with MitoSOX Red (5 mM, M36008, Invitrogen).
Intracellular ATP, MDA, and 4-HNE assay
Intracellular ATP levels were determined using an Enhanced ATP Assay Kit (Beyotime, S0027). The detecting solution was added to a 96-well plate and incubated at room temperature for 5 min. The supernatants were then added to the wells and mixed fast before determining the luminescence signals using the Infinite M200 Microplate reader (Tecan). Total ATP levels were calculated on the basis of the luminescence signals accordingly. Intracellular MDA levels were determined using an LPO MDA assay kit (Beyotime, S0131S). The detection solution was added to a 96-well plate and incubated at room temperature for 5 min. The supernatants were then added to the wells and mixed thoroughly before determining the signals using the Infinite M200 Microplate Reader (Tecan). Lipid peroxidation (4-HNE) was detected by a commercial assay kit (Abcam, Cat Number#: ab238538).
Thermal proteome profiling
RAW264.7 cells were lysed in the CETSA lysis buffer, followed by the five freeze-thaw cycles and centrifugation to isolate the supernatant. The protein concentration was subsequently determined by a BCA assay kit (Thermo Fisher Scientific, USA). The cell lysate was incubated with 5 μg/mL PQS or DMSO control at 37 °C for 5 min and then incubated at 52 °C for 3 min. Three biological replicates were performed. Then, samples were prepared by SISPROT, a highly integrated device for digestion and Tandem Mass Tag (TMT) labelling, as previously described. All labelled peptide samples were mixed and fractionated by tip-based high-pH reversed-phase fractionation. The peptides were analysed by an Orbitrap Exploris 480 coupled with a U3000 HPLC system (Thermo Fisher Scientific) and a 20 cm × 100 μm i.d. analytical column in-house packed with C18 beads (1.9 μm/120 Å, Dr. Maisch GmbH). Samples were dissolved in buffer A [0.1% (v/v) FA in water]. A 120 min separation gradient was configured as 1 − 4% buffer B [100% (v/v) ACN in buffer B] in 3 min, 4 − 22% buffer B in 93 min, 22 − 36% buffer B in 11 min, and 36 − 80% buffer B in 1 min, followed by 5 min washing with 80% buffer B and 6 min washing with 1% buffer B. MS parameters were as follows: Full MS scans over the m/z range of 350 − 1200 were performed at a resolution of 60,000. The Orbitrap Exploris 480 was operated in a data-dependent mode. Full scan and top 50 MS/MS scans were acquired per cycle with an MS1 scan resolution of 60,000 and an MS/MS scan resolution of 30,000 with the turbo-TMT option. HCD fragmentation was set with a normalised collision energy (NCE) of 36 and a dynamic exclusion of 45 s. The acquired raw files were analysed with Proteome Discoverer software (version 2.4.0) and searched against a database of human proteins, the Mouse FASTA database from UniProt (reviewed database entries, downloaded on April 4, 2022). Precursor mass tolerance was set as 20 ppm, and fragment ions were searched at 20 mm. Peptides were searched using the fully tryptic cleavage constraint, with up to two missed cleavages. Carbamidomethylation (C, + 57.02 Da) and TMT (N, K, + 229.163 Da) were used as fixed modifications. FDR control is set to 1% at the PSM and peptide levels. The isolation threshold for report ion quantification is set to 50%.
Western blotting
To analyse protein levels, cells were lysed with RIPA buffer (Biyuntian, P0013K). For CETSA WB, cell lysate and 25 µg/uL PQS were incubated at 37 °C for 5 min, then heated at 37 °C, 46 °C, 49 °C, 55 °C, and 58 °C for 3 min. After completing cooled down to 4 °C. The concentration of the supernatant was then measured using a BCA protein quantitation kit (Thermo Scientific, USA). The total cellular proteins were separated by 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred onto polyvinylidene difluoride (PVDF) membranes. The membranes were treated with 5% non-fat milk for 1 h and incubated with the primary antibodies raised in the mouse/rabbit against DDDDK (Flag) (Abcam, ab205606), HA (Abcam, ab9110), CARNMT1 (CNMT) (Atlas Antibodies, HPA02675), TFR1 (Abcam, ab214039), TF (Abcam, ab277635), Fth1 (Abcam, ab183781), pan-Met-His (Bioworld, BS74112), SOD1 (Abcam, ab308181), actin protein (Abclonal, AC026), and beta-Tublin (Abcam, ab314069) overnight, followed by washing in tris-buffered saline (TBST). The blots were further incubated with the secondary antibody, goat anti-mouse/rabbit-HRP (Abcam, Cambridge, England) for 1 h. The blots were washed in TBST followed by detection with the enhanced chemiluminescence reagent, and ECL Western Blotting Substrate Kit (Abcam, Cambridge, England). ImageJ software was used to measure band intensities.
Molecular docking
The CNMT PDB structure file was downloaded from the Uniprot database and modified in Chimera (UCSF)66. The PQS molecule structure file was downloaded from the Pubchem database. The docking process between proteins and potential ligands was performed in the Autodock Vina plug-in inside Chimera. The software programme LIGPLOT v.4.5.3 was used to map the interactions between proteins and potential ligands. Open Babel software67 was used to transfer proper file formats.
Protein purification and microscale thermophoresis
The human recombinant CNMT protein with N-terminal His-Tag was purchased from ImmunoClone (New York, USA). For the purification of TFR1H35G protein mutants, TFR1-HA were expressed in HEK293T cells via transient transfection. After 48 h transfection, TFR1H35G were isolated from whole cell extracts using anti-HA magnet beads (Beyotime), according to the instructions of the manufacturer. Proteins were eluted with 3 × HA peptides (Beyotime), respectively. The binding of ligands to the CNMT protein was assessed by microscale thermophoresis (MST) experiments using the Monolith R NT. LabelFree (NanoTemper Technologies GmbH). For analysing the interaction affinity, the dissociation constant Kd for each ligand was determined by analysing the normalised fluorescence (Fnorm [‰]) versus ligand concentration using NanoTemper Affinity Analysis software and Prism Graphpad.
Steady-state kinetic measurements
Different concentrations of carnosine (0, 0.19, 0.39, 0.78, 1.56, 3.13, 6.25, 12.5, 25, 50, and 100 μM) was used as a substrate for CNMT. The reaction mixture contained 20 mM Tris (pH 8.0), 50 mM NaCl, 0.1 mg/mL BSA, 1 mM DTT, and 1 μM CNMT methyltransferase. To determine the Michaelis constant (Km) value of the substrate, the concentration of SAM and PQS was kept constant at 40 μM and 10 μg/mL, respectively. Reactions were carried out at 37 °C for 20 min with a total volume of 20 μL, and terminated by heating at 95 °C for 5 min. The activity of CNMT was measured using a bioluminescence assay (MTase-Glo, Promega) in which the reaction by-product SAH is converted into ATP in a two-step reaction, and ATP can be detected through a luciferase reaction68. In general, 5 μL of reaction mixture was transferred to a low-volume 96-well plate, and the luminescence assay was performed according to the manufacturer’s protocol. A Tecan Spark plate reader (Tecan Group Ltd., Mannedorf, Switzerland) was used to measure luminescence signals. Less than 20% of the substrate was consumed in all reactions. The dependence of the velocity of product formation per enzyme on substrate concentration was analysed according to the Michaelis-Menten equation using nonlinear regression in GraphPad Prism 8.0 (GraphPad, USA).
In vitro methyltransferase assay of CNMT on TFR1 by western blotting
For the substrate TFR1 protein, the methyltransferase assay was carried out in a 50 µL reaction system containing 1 µg purified CNMT, 2 µg purified TFR1, 0.1 mM SAM in 50 mM Tris-HCl (pH 8.0), 10% glycerol, 20 mM KCl, 5 mM MgCl2, and 1 mM phenylmethylsulfonyl fluoride (PMSF). The reaction was incubated at 30 °C overnight. For western blotting, rabbit anti-pan-methyl-Histidine (Met-His) antibody (Bioworld) was used to detect methylation levels within in vitro methyltransferase assay.
ELISA analysis
The amount of PQS inside cells was analysed by ELISA. After cell collection, the concentration of PQS and anserine were quantified by the PQS ELISA kit (Huabodeyi, HBDY-927791O2, China), following the manufacturer’s instructions. Each experiment was repeated three times.
CNMT knockdown by lentiviral transduction
CNMT knockdowns were achieved using the Ubigene shRNA system. The shRNA lentiviral transduction particles were purchased from Millipore-Sigma to deliver and stably express shRNAs in RAW264.7 cells. Specifically, shRNA Control Transduction Particles and shRNA Lentiviral Transduction Particles (targeting mouse) were used. The cells were transduced with lentiviral particles (2 MOI) for 48 h at 37 °C with polybrene (8 µg/mL), followed by puromycin (3 µg/mL) selection for 48–72 h.
Tandem mass tag-based quantitative proteomic analysis
Total cellular proteins were extracted, and trypsin was digested. The digested peptides were labelled with TMT reagents. Subsequently, TMT labelled peptides were separated by a RIGOL L-3000 dual gradient High-Performance Liquid Chromatography (HPLC) using an Agela Durashell-C18 column. An HPLC-separated peptide was identified by nano UPLC-MS/MS, consisting of a Nanoflow HPLC system (EASY-nLC 1000 system from Thermo Scientific) with an EASY-Spray C18 column and an Orbitrap Fusion Lumos mass spectrometer (Thermo Scientific). Then, the protein was identified using the Uniprot_HUMAN database. The resulting MS/MS data were processed using Proteome Discoverer 2.4. The final results were functionally annotated by Gene Ontology (GO)69, KEGG, and STRING (https://www.string-db.org)70. The data were compared with the Uniprot reference proteome of Mus musculus GRCm38.p6 reference proteome database. Differentially expressed proteins are defined when the adjusted P-values is greater than 0.05 and the fold change is more than 1.5 or less than − 1.5.
RNA isolation and Illumina RNA-seq
Total cellular RNA was extracted using the RNeasy Kit (Qiagen, USA). Then, extracted total RNAs were purified using the Ribo-Zero rRNA Removal Kit (Epicentre Biotechnologies, Madison, USA). The integrity of RNA was determined by TapeStation RNA ScreenTape with RNA size distributions. For Illumina RNA-seq, the libraries of RNA samples were generated using NEBNext R Ultra™ Directional RNA Library Prep Kit for Illumina R (NEB, Massachusetts, USA). The libraries were sequenced using an Illumina HiSeq platform to generate paired-end reads. Artemis was used to show the mapping profiles in RNA-seq. The quality of raw sequence data was assessed using FastQC (Babraham Bioinformatics). Adaptor sequences were removed by the trimming function in CLC. RNA sequence analysis was done using the “RNA-seq analysis’ module in CLC genomics Workbench 20 (CLC Bio, Denmark) using Mus musculus GRCm38.p6 reference genome downloaded from the NCBI database. Differential gene expression was analysed using the DESeq2 package in R software. Differentially expressed genes are defined when the adjusted P-values is less than 0.05 and the (log2[fold change] is greater than 1 or less than −1.
Co-immunoprecipitation analysis
Protein interactions were assessed using co-immunoprecipitation. Briefly, cells were washed once with cold PBS and lysed in cold NP-40 Lysis Buffer (Beyotime, P0013F) containing protease inhibitors. The supernatants of the cell lysates were isolated at 12,500 × g for 30 min at 4 °C and were incubated with anti-Flag magnetic beads overnight at 4 °C, followed by extensive washing with NP-40 Lysis Buffer. Subsequently, western blotting steps were performed as previously described.
Yeast two-hybrid assay
The Matchmaker Two-Hybrid System and CNMT gene, cloned into the bait Gal4-BD vector pGBKT7 were used for the yeast two-hybrid assay as described. To investigate the interaction between CNMT with TFR1, the pGBKT7-CNMT and pGADT7-TFR1 plasmids were separately transformed or co-transformed into Y2HGold competent yeast cells. The transformants were plated onto low-stringency (lacking leucine and tryptophan) and high-stringency (lacking adenine, histidine, leucine, and tryptophan) selection plates for detection.
Real-time (RT)-quantitative (q) PCR
To quantify mRNA levels, total cellular RNA was extracted. RT-qPCR was performed with GAPDH as the internal control using the QuantStudio 1 instrument (Applied Biosystems, California, USA), and three replicates were performed.
Quantification and statistical analysis
All statistical analyses were performed using GraphPad Prism software with the two or one-way ANOVA with Tukey’s multiple comparisons test and Benjamini-Hochberg-corrected P-values. Data represent the means ± SD of three independent experiments unless otherwise indicated. P-values of < 0.05 were considered statistically significant. All false discovery rate controls were performed with the Benjamini-Hochberg procedure, and a false discovery rate of 10% (q < 0.1) was selected as the significant threshold. Statistical details for all tests performed can be found in the Figure legends.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All data are available in the manuscript and supplemental information. The RNA-seq data are uploaded to the NCBI SRA database with the following accession numbers: PRJNA1026331. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) via the iProX partner repository with the dataset identifiers PXD045976 (Proteome data), PXD061292 (MS-CETSA data). All data are publicly available as of the date of publication. Source data are provided in this paper.
References
Rossi, E. et al. Pseudomonas aeruginosa adaptation and evolution in patients with cystic fibrosis. Nat. Rev. Microbiol. 19, 331–342 (2021).
Papenfort, K. & Bassler, B. L. Quorum sensing signal–response systems in Gram-negative bacteria. Nat. Rev. Microbiol. 14, 576–588 (2016).
Lin, J., Cheng, J., Wang, Y. & Shen, X. The Pseudomonas quinolone signal (PQS): not just for quorum sensing anymore. Front. Cell. Infect. Microbiol. 8, 230 (2018).
Jia, T. et al. Hfq-binding small RNA PqsS regulates Pseudomonas aeruginosa pqs quorum sensing system and virulence. NPJ Biofilms Microbiomes 10, 82 (2024).
Lin, J. et al. A Pseudomonas T6SS effector recruits PQS-containing outer membrane vesicles for iron acquisition. Nat. Commun. 8, 14888 (2017).
Häussler, S. & Becker, T. The pseudomonas quinolone signal (PQS) balances life and death in Pseudomonas aeruginosa populations. PLoS Pathog. 4, e1000166 (2008).
Legendre, C. et al. Pseudomonas aeruginosa alkyl quinolones repress hypoxia-inducible factor 1 (HIF-1) signaling through HIF-1α degradation. Infect. Immun. 80, 3985–3992 (2012).
Rieger, B. et al. Pseudomonas Quinolone signal molecule PQS behaves like a B Class inhibitor at the IQ site of mitochondrial complex I. FASEB BioAdv. 2, 188–202 (2020).
Abdalla, M. Y. et al. Pseudomonas quinolone signal induces oxidative stress and inhibits heme oxygenase-1 expression in lung epithelial cells. Infect. immun. 85, (2017).
Holban, A.-M., Bleotu, C., Chifiriuc, M. C., Bezirtzoglou, E. & Lazar, V. Role of Pseudomonas aeruginosa quorum sensing (QS) molecules on the viability and cytokine profile of human mesenchymal stem cells. Virulence 5, 303–310 (2014).
Zaborin, A. et al. Red death in Caenorhabditis elegans caused by Pseudomonas aeruginosa PAO1. Proc. Natl. Acad. Sci. USA 106, 6327–6332 (2009).
Dixon, S. J. et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 149, 1060–1072 (2012).
Stockwell, B. R. Ferroptosis turns 10: Emerging mechanisms, physiological functions, and therapeutic applications. Cell 185, 2401–2421 (2022).
Yuan, J. & Ofengeim, D. A guide to cell death pathways. Nat. Rev. Mol. Cell Biol. 25, 379–395 (2024).
Galy, B., Conrad, M. & Muckenthaler, M. Mechanisms controlling cellular and systemic iron homeostasis. Nat. Rev. Mol. Cell Biol. 25, 133–155 (2024).
Ai, Y., Meng, Y., Yan, B., Zhou, Q. & Wang, X. The biochemical pathways of apoptotic, necroptotic, pyroptotic, and ferroptotic cell death. Mol. Cell 84, 170–179 (2024).
Lei, G., Mao, C., Yan, Y., Zhuang, L. & Gan, B. Ferroptosis, radiotherapy, and combination therapeutic strategies. Protein Cell 12, 836–857 (2021).
Amaral, E. P. et al. A major role for ferroptosis in Mycobacterium tuberculosis–induced cell death and tissue necrosis. J. Exp. Med. 216, 556–570 (2019).
Dubey, N. et al. Mycobacterium tuberculosis peptidyl prolyl isomerase a interacts with host integrin receptor to exacerbate disease progression. J. Infect. Dis. 224, 1383–1393 (2021).
Dar, H. H. et al. Pseudomonas aeruginosa utilizes host polyunsaturated phosphatidylethanolamines to trigger theft-ferroptosis in bronchial epithelium. J. Clin. Investig. 128, 4639–4653 (2019).
Dar, H. H. et al. A new thiol-independent mechanism of epithelial host defense against Pseudomonas aeruginosa: iNOS/NO• sabotage of theft-ferroptosis. Redox Biol. 45, 102045 (2021).
Dar, H. H. et al. P. aeruginosa augments irradiation injury via 15-lipoxygenase–catalyzed generation of 15-HpETE-PE and induction of theft-ferroptosis. JCI Insight 7, e156013 (2022).
Dixon, S. J. et al. Pharmacological inhibition of cystine–glutamate exchange induces endoplasmic reticulum stress and ferroptosis. Elife 3, e02523 (2014).
Bow, Y.-D. et al. A novel quinoline derivative, DFIQ, sensitizes NSCLC cells to ferroptosis by promoting oxidative stress accompanied by autophagic dysfunction and mitochondrial damage. Cancer Cell Int. 23, 171 (2023).
Wang, H. et al. Acteoside alleviates salsolinol-induced Parkinson’s disease by inhibiting ferroptosis via activating Nrf2/SLC7A11/GPX4 pathway. Exp. Neurol. 385, 115084 (2025).
Wang, K. et al. Metabolic labeling based methylome profiling enables functional dissection of histidine methylation in C3H1 zinc fingers. Nat. Commun. 15, 7459 (2024).
Shimazu, T. et al. Histidine N1-position-specific methyltransferase CARNMT1 targets C3H zinc finger proteins and modulates RNA metabolism. Genes Dev. 37, 724–742 (2023).
Dai, L. et al. Horizontal cell biology: monitoring global changes of protein interaction states with the proteome-wide cellular thermal shift assay (CETSA). Annu. Rev. Biochem. 88, 383–408 (2019).
Huber, K. V. et al. Proteome-wide drug and metabolite interaction mapping by thermal-stability profiling. Nat. Methods 12, 1055–1057 (2015).
Florez, C., Raab, J. E., Cooke, A. C. & Schertzer, J. W. Membrane distribution of thePseudomonas quinolone signal modulates outer membrane vesicle production in Pseudomonas aeruginosa. MBio 8, (2017).
Schertzer, J. W. & Whiteley, M. A bilayer-couple model of bacterial outer membrane vesicle biogenesis. MBio 3, (2012).
Bertheloot, D., Latz, E. & Franklin, B. S. Necroptosis, pyroptosis and apoptosis: an intricate game of cell death. Cell. Mol. Immunol. 18, 1106–1121 (2021).
Miotto, G. et al. Insight into the mechanism of ferroptosis inhibition by ferrostatin-1. Redox Biol. 28, 101328 (2020).
Otasevic, V., Vucetic, M., Grigorov, I., Martinovic, V. & Stancic, A. Ferroptosis in different pathological contexts seen through the eyes of mitochondria. Oxid. Med. Cell. Longev. 2021, 5537330 (2021).
Gao, M. et al. Role of mitochondria in ferroptosis. Mol. Cell 73, 354–363 (2019).
Lee, H. et al. Energy-stress-mediated AMPK activation inhibits ferroptosis. Nat. Cell Biol. 22, 225–234 (2020).
Boldyrev, A. A., Aldini, G. & Derave, W. Physiology and pathophysiology of carnosine.Physiol. Rev.93, 1803–1845 (2013).
Wallace, A. C., Laskowski, R. A. & Thornton, J. M. LIGPLOT: a program to generate schematic diagrams of protein-ligand interactions. Protein Eng. Des. Selection 8, 127–134 (1995).
Candelaria, P. V., Leoh, L. S., Penichet, M. L. & Daniels-Wells, T. R. Antibodies targeting the transferrin receptor 1 (TfR1) as direct anti-cancer agents. Front. Immunol. 12, 607692 (2021).
Chen, X., Yu, C., Kang, R., Kroemer, G. & Tang, D. Cellular degradation systems in ferroptosis. Cell Death Differ. 28, 1135–1148 (2021).
Zheng, Y. et al. Ferroptosis, pyroptosis and necroptosis in acute respiratory distress syndrome. Cell Death Discov. 9, 91 (2023).
Deshpande, R. & Zou, C. Pseudomonas aeruginosa induced cell death in acute lung injury and acute respiratory distress syndrome. Int. J. Mol. Sci. 21, 5356 (2020).
Deo, P. et al. Mitochondrial dysfunction caused by outer membrane vesicles from Gram-negative bacteria activates intrinsic apoptosis and inflammation. Nat. Microbiol. 5, 1418–1427 (2020).
Jiang, F. et al. The Pseudomonas aeruginosa type VI secretion PGAP1-like effector induces host autophagy by activating endoplasmic reticulum stress. Cell Rep. 16, 1502–1509 (2016).
Di Menna, L. et al. The bacterial quorum sensing molecule, 2-heptyl-3-hydroxy-4-quinolone (PQS), inhibits signal transduction mechanisms in brain tissue and is behaviorally active in mice. Pharmacol. Res. 170, 105691 (2021).
Barr, H. L. et al. Diagnostic and prognostic significance of systemic alkyl quinolones for P. aeruginosa in cystic fibrosis: a longitudinal study; response to comments. J. Cyst. Fibros. 16, e21 (2017).
Hänsch, G. M., Prior, B., Brenner-Weiss, G., Obst, U. & Overhage, J. The Pseudomonas quinolone signal (PQS) stimulates chemotaxis of polymorphonuclear neutrophils. J. Appl. Biomater. Funct. Mater. 12, 21–26 (2014).
Freund, J. R. et al. Activation of airway epithelial bitter taste receptors by Pseudomonas aeruginosa quinolones modulates calcium, cyclic-AMP, and nitric oxide signaling. J. Biol. Chem. 293, 9824–9840 (2018).
Mayneris-Perxachs, J., Moreno-Navarrete, J. M. & Fernández-Real, J. M. The role of iron in host–microbiota crosstalk and its effects on systemic glucose metabolism. Nat. Rev. Endocrinol. 18, 683–698 (2022).
Fuhrmann, D. C., Mondorf, A., Beifuß, J., Jung, M. & Brüne, B. Hypoxia inhibits ferritinophagy, increases mitochondrial ferritin, and protects from ferroptosis. Redox Biol. 36, 101670 (2020).
Wang, P. & Lu, Y.-Q. Ferroptosis: a critical moderator in the life cycle of immune cells. Front. Immunol. 13, 877634 (2022).
Jia, T. et al. The AhR ligand phthiocol and vitamin K analogs as Pseudomonas aeruginosa quorum sensing inhibitors. Front. Microbiol. 13, 896687 (2022).
Moura-Alves, P. et al. Host monitoring of quorum sensing during Pseudomonas aeruginosa infection. Science 366, eaaw1629 (2019).
Mishima, E. et al. A non-canonical vitamin K cycle is a potent ferroptosis suppressor. Nature 608, 778–783 (2022).
Wang, Y., Yan, D., Liu, J., Tang, D. & Chen, X. Protein modification and degradation in ferroptosis.Redox Biol. 75, 103259 (2024).
Qiang, L. et al. A mycobacterial effector promotes ferroptosis-dependent pathogenicity and dissemination. Nat. Commun. 14, 1430 (2023).
Zhang, Q. & Cao, X. Epigenetic regulation of the innate immune response to infection. Nat. Rev. Immunol. 19, 417–432 (2019).
Wang, Y. et al. PRMT4 promotes ferroptosis to aggravate doxorubicin-induced cardiomyopathy via inhibition of the Nrf2/GPX4 pathway. Cell Death Differ. 29, 1982–1995 (2022).
Deng, H. et al. SET7 methylates the deubiquitinase OTUB1 at Lys 122 to impair its binding to E2 enzyme UBC13 and relieve its suppressive role on ferroptosis. J. Biol. Chem. 299, (2023).
Cheng, Y., Zak, O., Aisen, P., Harrison, S. C. & Walz, T. Structure of the human transferrin receptor-transferrin complex. Cell 116, 565–576 (2004).
Wu, Y. et al. Ubiquitin ligase E3 HUWE1/MULE targets transferrin receptor for degradation and suppresses ferroptosis in acute liver injury. Cell Death Differ. 29, 1705–1718 (2022).
Senyilmaz, D. et al. Regulation of mitochondrial morphology and function by stearoylation of TFR1. Nature 525, 124–128 (2015).
Ying, W., Cheruku, P.S., Bazer, F.W., Safe, S.H. & Zhou, B. Investigation of macrophage polarization using bone marrow derived macrophages.J. Vis. Exp. 76, 50323 (2013).
Xu, Q. et al. Cytotoxic rhamnolipid micelles drive acute virulence in Pseudomonas aeruginosa. Infect. Immun. 92, e00407–e00423 (2024).
Zhang, X. et al. Ischemia-induced upregulation of autophagy preludes dysfunctional lysosomal storage and associated synaptic impairments in neurons. Autophagy 17, 1519–1542 (2021).
Pettersen, E. F. et al. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 (2004).
O’Boyle, N. M. et al. Open babel: An open chemical toolbox. J. Cheminform. 3, 1–14 (2011).
Hsiao, K., Zegzouti, H. & Goueli, S. A. Methyltransferase-Glo: a universal, bioluminescent and homogenous assay for monitoring all classes of methyltransferases. Epigenomics 8, 321–339 (2016).
Zhang, X. et al. TMT based proteomic analysis of human follicular fluid from overweight/obese and normal-weight patients with polycystic ovary syndrome. Front. Endocrinol. 10, 821 (2019).
Franceschini, A. et al. STRING v9. 1: protein-protein interaction networks, with increased coverage and integration. Nucleic Acids Res. 41, D808–D815 (2012).
Acknowledgements
The authors would like to thank Prof. Yijin Wang from the Southern University of Science and Technology for discussing the possibility of mitochondrial assays. This work was supported by the National Key Research and Development Programme of China (2022YFC2304700 to L.Z., and 2022YFC2304401 to L.Y.); National Natural Science Foundation of China (32300068 to T.J., 91951204 to L.Y., 32270196 to L.Y., 32200155 to L.Y., 32200053 to L.Y., 32300060 to L.Y., and 32400154 to X.D.); Guangdong Basic and Applied Basic Research Foundation (2025A1515010592 to T.J., 2020A1515010316 to L.Y., and 2023A1515110104 to X.D.); Shenzhen Science and Technology Programme KQTD20200909113758004 to L.Y.; Key University Laboratory of Metabolism and Health of Guangdong (2022KSYS007 to L.Y.), and Southern University of Science and Technology high level of special funds-Cell Homoeostasis and Major Diseases (G03117K001 to L.Y.).
Author information
Authors and Affiliations
Contributions
Conceptualisation, L.Y., C.T., and T.J.; methodology, T.J., F.L., T.L.; investigation, A.R., P.L., Y.L.L., Y.Z., X.D.; writing – original draft, T.J., T.L.; writing – review & editing, L.Y., C.T., Y.L., Z.Z.; funding acquisition, L.Y., C.T., T.J., L.Z.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Kayla Simanek, Liangbin Hu, and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Jia, T., Li, F., Li, T. et al. A Pseudomonas aeruginosa quorum-sensing metabolite manipulates macrophage ferroptosis through a methylation pathway. Nat Commun 16, 9992 (2025). https://doi.org/10.1038/s41467-025-65142-y
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-65142-y
