
Abstract
Development of the DNA affinity purification and sequencing (DAP-seq) technique has allowed genome-scale studies of transcription factor (TF)-binding sites with high reproducibility. Here, we apply this technique to the human opportunistic pathogen Rhizopus microsporus, a mucoralean fungus belonging to the understudied group of early-diverging fungi. We characterize genome-wide binding sites of 58 TFs encoded by genes regulated through adenine methylation and representing major TF families. This analysis reveals their binding profiles and recognized sequences, expanding and diversifying the catalog of known fungal motifs. By integrating this data with DNA 6-methyladenine profiling, we uncover the extensive direct and indirect impact of this epigenetic modification on the regulation of gene expression. Furthermore, we use the generated data to identify TFs involved in biologically relevant processes such as zinc metabolism and light response. Our work enhances our understanding of regulatory mechanisms in R. microsporus and provides broader insights into gene regulation across the fungal kingdom.
Similar content being viewed by others


Introduction
Transcription factors (TFs) play a central role in gene expression regulation by binding to specific DNA sequences. They can either activate or repress the transcription of target genes, thereby modulating cellular processes in response to various environmental and developmental signals. The complete set of genomic binding sites for a given TF, often referred to as its cistrome1, provides a comprehensive view of the complex transcriptional regulatory networks, which are also influenced by epigenetic modifications to ensure precise control of the transcription2,3,4.
Chromatin immunoprecipitation followed by sequencing (ChIP-seq) has been widely employed to determine TF binding sites (TFBS)5,6,7. However, scaling up this approach for genome-wide cistrome characterization is challenging due to its heavy reliance on antibody quality and availability, as well as its technical complexity when applying to proteins with low expression levels8. These limitations explain why TFBS are only available for specific proteins and are often limited to a few model organisms6. To overcome these issues, in vitro approaches that allow large-scale analyses of TFBS, such as protein binding microarrays (PBM)9 and systematic evolution of ligands by exponential enrichment (SELEX)10, were developed11,12. These methodologies, however, use synthetic short DNA fragments to identify the TF binding motif, which is then used to scan the genome for potential TFBS. The development of DNA affinity purification sequencing (DAP-seq) combines the use of native genomic DNA of ChIP-seq and the scalability of in vitro approaches13,14. With the caveat of lacking post-translational modifications (PTMs) in the expressed protein, this method has enabled broad and comprehensive characterization of a considerable number of TFs from both prokaryotic and eukaryotic representatives13,15,16. Remarkably, this high-throughput methodology relies on the in vitro synthesis of the TFs of interest, followed by pulldown and next-generation sequencing of native or amplified genomic DNA, enabling cistrome characterization in a wide range of non-model organisms15.
Despite the development of these in vitro approaches, a significant gap remains in the study of TFBS in fungi. Fungi are an incredibly diverse kingdom of organisms with substantial medical, ecological, and industrial relevance17,18,19. Yet, compared to other eukaryotes, genome-wide studies of TF binding in fungi are very limited20. Most research has focused on characterizing specific TFs or DNA-binding proteins, predominantly in Dikarya fungi21,22,23, with only a few studies conducting broader analyses of TFBS across different TF families24,25. Noteworthy, since DAP-seq has made TFBS studies possible independent of species and the availability of molecular tools, it has been widely applied to plants13,26,27,28 and bacteria15,16,29, but only to some specific TFs in a smaller-scale study in fungi. In particular, this technique has allowed the elucidation of the fungal gene regulatory networks involved in nitrogen metabolism30 and carbon utilization31.
Beyond the Dikarya, the basal fungal subkingdom has been traditionally neglected. Combined with the limited availability of molecular tools, this has left many aspects of their biology, including gene expression regulation, largely unexplored. Furthermore, comprehensive studies on transcriptional regulators that respond to environmental signals, which are well-characterized in Dikarya fungi30,31,32,33, are notably lacking in early-diverging fungi (EDF). This group, however, encompasses the majority of known fungal phyla and presents a series of unique characteristics that make them of significant scientific interest34,35. Within this group, Rhizopus microsporus (Mucoromycota) stands out due to its high agricultural, industrial, and clinical relevance because of its phytopathogenic potential through symbiotic interaction with endobacteria36,37, the use of specific strains in the production of fermented products38, and its high frequency as a causal agent of mucormycosis, a lethal infection classified by the WHO as high-priority concern39,40,41. Notably, this fungus relies on the use of N6-methyladenine (6mA) as its main DNA epigenetic modification42, a unique characteristic shared with other EDF, ciliates, and green algae43,44,45. This modification is associated with gene expression in all these eukaryotic groups and its study in R. microsporus has revealed that it is essential42, underscoring its crucial role in gene regulation.
While most large-scale projects on cistrome analyses have focused on higher eukaryotes, such as mice and humans, fungal TF landscapes have received far less attention20,46,47. Here, we address this gap by presenting a comprehensive analysis of the R. microsporus cistrome. Using DAP-seq, we identified the binding sites of TFs regulated by 6mA, providing the most detailed gene regulatory network described in filamentous fungi to date. Moreover, a thorough investigation of TF families highly conserved across fungi, both phylogenetically close and distant relatives, offers valuable insights into the evolution of TFs and the mechanisms underlying gene regulation within the fungal kingdom.
Results
The fungal TF landscape
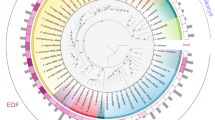
We curated a list of 51 TF families, including the fungal-specific families (APSES, MAT, and Copper fist) based on previous studies46,47 and scanned the genomes of 62 fungal species, 43 of which are classified as EDF (Supplementary Data 1). In addition to the Zn cluster family expansion in Dikarya, particularly in Ascomycota20 (Fig. 1, Supplementary Fig. 1), other remarkable differences were found when comparing EDF representatives with Dikarya and among themselves. Contrary to the Zn cluster family, the HTH/Homeodomain-like family was more expanded in EDF compared to Ascomycota (p < 0.0001, Chi-square test) (Fig. 1, Supplementary Fig. 1), as is the C2H2/CCHC/CH/C5HC2 family in Monoblepharomycota, Neocallimastigomycota, and Chytridiomycota (p < 0.0001, Chi-square test). Some TF families, such as E2F and Tubby, are absent in Dikarya47 (Fig. 1, Supplementary Fig. 1), while others, such as RFX and STAT, are good examples of TF families that are present in Dikarya but are absent or drastically less abundant in EDF.

The abundance of each TF family, relative to the total number of TFs, is indicated for each species. Gray boxes indicate the absence of a given TF family. The bottom part displays the proportion of TF-coding genes relative to the total number of annotated genes, the total number of annotated genes, as well as the genome size, for each species. Information on the genomes analyzed can be found in Supplementary Data 1. Source data are provided as a Source Data file.
We also detected other interesting differences, such as the expansion of the HMG-box family in Glomeromycota, as well as the Helix-loop-helix (HLH) and GATA Zn finger families in Mucoromycota, Mortierellomycota, and some representatives of Chytridiomycota. Additionally, the Zn cluster TFs were particularly abundant in Entomopthoromycota (Fig. 1, Supplementary Fig. 1). This dynamic nature of TF families explains the notable expansion of TF coding genes, which is clearly noticeable in Ascomycota. Nevertheless, TF-coding genes also constitute a notable proportion of total protein-coding genes in Mucoromycota, Glomeromycota, and Entomopthoromycota (Fig. 1). Noteworthy, Mucoromycota has the higher TF landscape diversity based on the Shannon index47, with some TF families, such as WRKY and E2F, being almost exclusively found in this phylum (Fig. 1).
Cistrome characterization of R. microsporus using DAP-seq
The diversity of TF landscapes found in Mucoromycota prompted us to investigate the cistrome of R. microsporus. For that purpose, we performed DAP-seq on a curated list of 88 TFs from major TF families (Supplementary Table 1) encoded by genes harboring 6mA concentrated in regions of high methylation density known as Methylated Adenine Clusters (MACs) under the same growth conditions used to generate DAP-seq samples42. MACs usually appear downstream of the transcription start site (TSS) of transcriptionally active genes of EDF42,43,44. We processed the datasets generated for each TF and retained only those with > 5% of the reads aligning to the predicted peaks for the respective TF (FRIP, Fragments of Reads in Peaks)13,15 (Fig. 2a and Supplementary Table 1). Of the initial 88 TFs, 41 passed this filter, yielding a success ratio of 46.6%. To assess the data robustness and reproducibility, we conducted a second replicate of the experiment using the same TF candidates. In this replicate, 57 TFs passed the filter (64.7% success ratio), including 40 of the 41 TFs from the first attempt. In total, 58 TFs produced reliable results in at least one replicate and were considered for cistrome analysis. These TFs belonged to 11 different families, including the broadly distributed bZip, GATA Zn finger, HLH, Homeobox, and Heat-shock (HSF) families (Fig. 2b). Among these, TFs from the HLH, Homeobox, GATA, and HSF performed particularly well, with success rates of 89%, 92%, 82%, and 87%, respectively (Fig. 2b), whereas all TFs from CBF, Tubby, and PHD-type Zn finger failed to produce reliable data (Fig. 2b). These results highlighted the influence of family-specific properties on success rates of DAP-seq experiments, as previously described13. Western blot analysis revealed that some proteins failed to yield results due to low or absent expression, which may be attributed to the absence of a required cofactor or other issues related to their in vitro expression (Supplementary Fig. 2)

a Filtering results for DAP-seq experiments indicating FRIP and the percentage of aligned reads. Each dot represents one TF. Only TFs with FRIP > 5% were considered. The left and the right panel are for the first and second replicate, respectively. b Proportion of TFs that passed filtering for each family. Gray color indicates FRIP < 5%. c TF motif clustering based on motif similarity using the R package motifStack. The outer circle indicates the TF family. d Comparison of R. microsporus DAP-seq motifs against JASPAR motif database. The best hit for each R. microsporus TF motif was compared against all new motifs identified. The color code represents the score (-log10 P-value) for each motif comparison. e Peak frequency distribution over protein-coding genes for bZip, HLH, HSF, GATA, and Homeobox families. TSS was extended 1 kb upstream and downstream and split into 25 equally sized bins. f Proportion of peaks in R. microsporus genomic features. The same TF families as in (e) were considered. Source data are provided as a Source Data file.
A total of 50,597 peaks were identified after filtering, with the number of peaks per TF ranging from 20 to several thousand (Supplementary Fig. 3). Subsequently, we determined the motif bound by each TF, observing a high degree of similarity between experimental replicates (Supplementary Fig. 4), even from those with FRIP > 5% in one replicate, supporting the reliability of the cistrome characterization. Clustering the identified motifs based on their similarity revealed that motifs from all HLH, GATA, Homeobox, HSF, and bZip were grouped together with TFs of the same family, indicating well-conserved target sequences within these families (Fig. 2c).
Currently, only 307 motifs from fungal species are deposited in the public repository JASPAR48, with 292 derived from Saccharomyces cerevisiae and none from any EDF representative48. We queried our motif catalog against the JASPAR curated database and retained the closest matches for each of the 58 TFs based on motif similarity. Sequences recognized by bZip TFs displayed some similarity to motifs bound by S. cerevisiae CRE (cAMP response element) factors Cst6/Aca2 and Sko1 (Fig. 2d)49,50,51. HLH TFs bound to similar motifs to those recognized by Tye7 and Cbf1 (Fig. 2d), which are also HLH TFs in S. cerevisiae52,53. Motifs from homeobox KN TFs also showed moderate similarity to the motif bound by S. cerevisiae Tos8, a TF involved in meiosis and cell damage response54. Interestingly, motifs bound by HSF TFs also displayed some similarity to those recognized by Mga1 and Hsf1 (Fig. 2d), which are heat-shock factors involved in pseudohyphal growth and the essential regulation of heat-response genes in S. cerevisiae55,56,57. The similarities observed in this comparative analysis underscore the robustness of our dataset. In addition to identifying motifs, we analyzed TF binding relative to genomic features. Peaks tended to accumulate 0–250 bp upstream the TSS of protein-coding genes, with a large proportion located in promoter regions or the first exon (Fig. 2e, f). Noteworthy, HSF TF peaks accumulated closer to and downstream of the TSS (Fig. 2e), suggesting distinct regulatory mechanisms among TF families. A genome-wide coverage analysis also revealed differences between TF families. While all GATA TFs were bound to similar regions (computing the sequencing coverage in 20 kb bins) notable differences were observed between the HSF and HLH TFs (Supplementary Fig. 5), highlighting the diversity between all these TF families.
The different TF families show target gene and functional diversification
To gain deeper insight into the role of each TF in gene expression regulation, we identified their putative targets. The 58 analyzed TFs displayed a wide range in the number of targets, from just six in the case of CP2-TF1 to several hundred for GATA Znf6, HOX2, or HLH Myc1 (Supplementary Data 2). Using this information, we generated a gene network including the 58 TFs studied and their respective predicted targets (Fig. 3a), which can be explored at https://mycocosm.jgi.doe.gov/Rhimi59_2/dapseq/network;WfDeCM?organism=Rhimi59_2. This network revealed well-differentiated groups of genes regulated by specific TF families. Notably, two distinct sets of genes regulated by GATA, HLH, Homeobox, and Fork-head TFs could be identified. In contrast, HSF and bZip TFs, with the sole exception of bZip5, were found to regulate a unique set of target genes (Fig. 3a). Interestingly, the HMG-box TFs showed a remarkable heterogeneity in their target gene set, with a high degree of variability among the targets of the three TFs from this family that were analyzed (Fig. 3a).

a Gene network for the 58 TFs (bigger rounded circles) and their respective targets. TF families and and their targets are color-coded as indicated in the legend. An interactive DAP-seq viewer is available at https://mycocosm.jgi.doe.gov/Rhimi59_2/dapseq/network;WfDeCM?organism=Rhimi59_2. b Functional classification of TF targets. The top 2 most enriched GO Biological Process terms for each TF were considered, and the P-value enrichment (-log10) was plotted for each term and each TF target set. White indicates no significant association was found for that term (P-value > 0.05, Fisher’s exact test, two-tailed). Colored rectangles group clusters of TFs targeting genes with similar functions. The colors correspond to those used for each TF family, as shown in (a) and Fig. 2c. Source data are provided as a Source Data file.
Given that the function of most TFs of R. microsporus is unknown, we performed functional enrichment analysis on the target gene sets for each TF. The targets of HLH TFs showed two well-defined clustering patterns associated with diverse biological functions, including lipid metabolism, redox homeostasis, and GTP metabolism (Fig. 3b, orange squares), with some biological functions shared with Homeobox TF targets. GATA TF targets also showed a clear functional enrichment pattern, regulating genes involved in amino acid biosynthesis and other relevant processes, such as carbohydrate transport and sulfate assimilation (Fig. 3b, purple squares). Since the TFs studied in this work are largely uncharacterized, this analysis also provided insights into their potential biological roles by examining their targets. For example, the TF Copf3 was found to target genes involved in ribosome biogenesis, while HLH Myc6 appeared to play a role in sodium ion transport (Fig. 3b). To further investigate the overlap between target genes of different TFs, we compared all pairwise combinations for each main family based on their predicted gene targets (Supplementary Data 3), which revealed potential coregulatory pairs.
The interplay between 6mA and TFs in gene expression regulation
Symmetric DNA 6mA plays an essential role in R. microsporus, with 35% of its genes containing MACs, including the 58 TFs identified in this study, and thus being directly regulated by 6mA42. Analyzing the targets of these 58 TFs could provide insights into the indirect role of this epigenetic modification in gene expression regulation. Peaks from the 58 TFs predicted that 6494 genes (60.1 % of total genes) were regulated by these TFs, whereas the total number of methylated genes in the same conditions was 3825 (35.7% of total genes). Comparing these two datasets revealed 2222 genes that were both potentially regulated by TFs and 6mA, accounting for more than one-third of TF-regulated genes and almost 60% of all methylated genes (Fig. 4a). This resulted in a total of 8097 genes (75.6% of total genes) directly or indirectly regulated by 6mA. Subsequently, we analyzed the targets of each TF in relation to the presence or absence of 6mA and found both individual and family-specific differences. The targets of HLH TFs, FK1, and Znf1 TFs were more frequently methylated than those of other TFs (Fig. 4b). Further analysis of the binding signal of each TF across all MACs in the R. microsporus genome revealed a strong preference for binding to these regions by HLH Myc2, HLH Myc3, HLH Myc8, HLH Myc12, Znf1, and FK1 TFs (Fig. 4c), which explains the aforementioned results. To assess whether the motif recognized by each TF was enriched in MACs, the number of binding motifs of these TFs across all MACs was compared to those found in an equal number of randomly generated genomic regions of the same average length using a Fisher’s exact test. No significant enrichment was observed for any of the six cases (HLH Myc2 OR = 0.84, p = 0.0502, HLH Myc3 OR = 0.96, p = 0.3800, HLH Myc8 OR = 0.89, p = 0.1471, HLH Myc12 OR = 0.85, p = 0.0801, Znf1 OR = 0.92, p = 0.2150, FK1 OR = 0.84, p = 0.0670) suggesting that the preferential binding of these TFs to MACs is not due to a higher frequency of their binding motifs in these regions.

a Venn diagram indicating TF target genes and methylated (6mA genes) in the R. microsporus genome. b Proportion of methylated (garnet) and unmethylated (light pink) genes for each TF-target set. c TF profile coverage over MACs. DAP coverage signal for each TF was plotted over R. microsporus MACs. All MACs were sized to 500 bp and extended 500 bp upstream and downstream. An empty backbone plasmid was used as a control (no protein expressed). Vertical dotted lines indicate MAC start and end. d Differential expression (log2FC) of genes demethylated when mta1 expression was reduced. e Volcano plots for target genes of Znf1, HLH2, GATA Znf3 and GATA Znf10 TFs. log2FC and P-values are indicated for each gene. Only genes with log2FC|1| and P-value < 0.05 were considered differentially expressed. f Genome view of genes whose promoter was bound by R. microsporus Znf1 and their expression was regulated by zinc. Scale bar = 500 bp. g Relative expression (RT-qPCR) of zinc-regulated genes for the wild-type and Znf1 mutants growing in zinc-rich media (purple) and zinc-depleted media (gray). Significant induction in the Znf1 mutant in the absence of zinc was tested using one-way ANOVA (P-value cut-off <0.05). Those not induced were predicted as Znf1 targets and are indicated with an orange line. Three replicates were conducted for each condition. Data is represented as mean ± SD. Source data are provided as a Source Data file.
Given that all genes encoding the selected TFs were methylated, we investigated which lost methylation when Mta1 expression, the main methyltransferase involved in 6mA deposition in this fungus, was reduced42. Five of the studied TFs lost methylation (Znf1, HLH2, HLH Myc5, GATA Znf3 and GATA Znf10), leading to downregulation in four of them (Fig. 4d). A total of 20 to 33% of their target genes were also differentially expressed (Supplementary Table 2). Moreover, we analyzed the effects on their respective target genes and found a significantly higher proportion of downregulated genes among the differentially expressed (DE) target genes of Znf1 and GATA Znf10 (Supplementary Table 2 and Fig. 4e), indicating that the loss of methylation also has an indirect effect on the targets of transcription factors regulated by this epigenetic modification.
DAP-seq data provides insights into the regulatory mechanisms underlying important biological processes in EDF
The large number of analyzed TFs and the high degree of representativity of our dataset allowed us to explore the regulatory mechanisms underlying specific processes in R. microsporus, beyond the 6mA epigenetic regulation. As a proof of concept, we investigated the regulation of gene expression by zinc. Recently, we developed an expression system to control gene expression in Mucorales based on the use of the promoter of the zrt1 gene, which encodes a zinc permease whose expression is dependent on zinc availability42. However, the TFs that regulate the expression of this and other genes related to zinc metabolism in EDF were unknown. Following the same approach previously used to analyze MACs, we scanned the promoter of the zrt1 gene using the coverage information from all zinc finger TFs analyzed in this study, which could be potential regulators of this gene58. We found a binding peak for the TF Znf1 upstream of the zrt1 TSS (Supplementary Fig. 6a). In S. cerevisiae, the ortholog of the zrt1 gene is regulated by Zap1, a TF of C2H2/CCHC/CH/C5HC2 zinc finger family59. Znf1 is the only representative of this TF family in our dataset (Supplementary Data 1). Interestingly, we observed that several of the R. microsporus orthologs of Zap1-regulated genes involved in zinc metabolism60,61 (Supplementary Fig. 6b) showed strong Znf1 binding peaks in their promoters and were regulated by zinc availability in the medium (Fig. 4f and Supplementary Fig. 6b), suggesting that they are Znf1 targets. To further investigate the role of znf1 in R. microsporus, we generated a knockout mutant (Supplementary Fig. 6c). Expression analysis by RT-qPCR of eight potential Znf1 targets in both the wild-type strain and the znf1 mutant under zinc-depleted and zinc-replete conditions revealed that the zrt1, zrt3, and zrg17 orthologs were not induced in the mutant under zinc-depleted conditions, suggesting a direct regulatory role of Znf1 in their expression (Fig. 4g). The remaining potential Znf1 targets displayed expression patterns similar to those in the wild-type strain, suggesting functional redundancy with other TFs contributing to their regulation in R. microsporus (Fig. 4g). Disruption of znf1 resulted in reduced growth under zinc-depleted conditions compared to the wild-type strain (Supplementary Fig. 6d), while the sporulation rate remained unaffected (Supplementary Fig. 6e).
Another aspect that strongly captured our interest during the analysis of the R. microsporus cistrome was the regulation of sexual reproduction. Zygomycete fungi are characterized by having two distinct mating types, distinguished by the presence of the sexP (+) or sexM (-) genes in their mating locus. These genes encode HMG-box TFs that regulate mating-specific genes in each type62,63. While some target genes of sexP and sexM are known62, the TFs that bind to their promoters and regulate their expression remain unidentified. Using the data from all the TFs analyzed, we scanned the promoter region of the sexP gene in R. microsporus and found a robust binding peak for bZip5 (Supplementary Fig. 6). This TF, which binds to regions and regulates genes different from the other bZip factors analyzed (Fig. 3b and Supplementary Fig. 7), represents a promising candidate for studying gene regulation in the sexual reproduction of zygomycete fungi. Unfortunately, the R. microsporus strain used in this study (ATCC 11559) has not successfully undergone mating under laboratory conditions, so further investigations using other strains or species will be necessary to elucidate the involvement of bZip5 in the regulation of reproduction.
DAP-seq data unveils white-collar control of light-regulated genes in R. microsporus
The set of TFs analyzed by DAP-seq included a White Collar-2 (WC-2) protein of the WC-2C/D type, here referred to as GATA Znf10 or WC-2C (Supplementary Fig. 8a), making possible the identification of blue light-responsive genes regulated by the white-collar photoreceptor composed of WC-2C and an uncharacterized white collar-1 protein64. While the genetic regulation of light responses has been extensively studied in Mucorales65,66,67,68,69,70 and the biological role of white -collar proteins is well-documented64,71,72, the genes directly or indirectly regulated by these photoreceptors in this fungal group remain unknown. We detected strong binding signals of WC-2C to 1394 potential target genes (Supplementary Data 2), with particular enrichment in the promoter region (Fig. 5a). The binding motif was identified as GATC (Fig. 5b), which is consistent with the consensus binding sequence described for white collar proteins in fungi21,73,74.

a WC-2C (GATA Znf10) and control (empty backbone plasmid) DAP coverage over predicted WC-2C targets. Genes were sized to 1 kb and coverage was computed over genes and 2 kb upstream and downstream. b Motif bound by WC-2C in R. microsporus obtained using the 100 most significant peaks, as scored by signal value in column 7 of the narrowPeak file from WC-2C. c Ligh-induced and repressed genes regulated by WC-2C. A volcano plot shows all differentially expressed genes (log2FC|1| and P-value > 0.05) in yellow and those with a WC-2C peak in their promoter rounded with a purple circle. d Genomic snapshot showing the WC-2C coverage and RNA-seq expression in light and dark growth conditions for the 1888704 gene. Scale bar = 500 bp. e Relative expression of zinc-regulated genes (RT-qPCR) for the wild-type and wc-2c mutants growing in the dark (dark blue) and under light exposure (yellow). Significant induction in the wc-2c mutant in the presence of light was tested using one-way ANOVA (P-value cut-off <0.05). Those not induced were predicted as WC-2C targets and are indicated with a purple line. Three replicates were conducted for each condition. Data is represented as mean ± SD. f Percentage of orthologous promoters with the WC-2C binding motifs. Promoter (600 bp) was extracted from each ortholog and scanned with FIMO using the R. microsporus WC-2C binding motif. Source data are provided as a Source Data file.
To identify the light-responsive genes regulated by the blue-light photoreceptor containing WC-2C, we compared its predicted targets with the genes regulated by light in R. microsporus42. Among the 2871 genes regulated by light, 427 genes (14.8%) showed a WC-2C binding peak in their promoter regions. Of these, the majority—344 genes—were induced, while 83 genes were repressed (Fig. 5c), indicating that WC-2C predominantly functions as a transcriptional activator in this fungus. To validate the involvement of WC-2C in regulating these genes, we generated a mutant strain with a disrupted wc-2c gene (Supplementary Fig. 8b) and analyzed the expression of eight light-responsive genes in both the wild-type and mutant strains under light and dark conditions. Six of these genes failed to be upregulated in response to light in the mutant (Fig. 5e), confirming that WC-2C is critical for the light-responsive expression of most genes it binds. Notably, the wc-2c mutation resulted in increased sporulation under dark growth conditions compared to the wild-type strain (Supplementary Fig. 8c, d). In addition, the mutant exhibited reduced aerial mycelium development and an increased accumulation of sporangiophores at the base of the mycelium (Supplementary Fig. 8e). This phenotype is consistent with the loss-of-function of the light-inducible crgA gene (ID 1913569) in R. microsporus, one of the WC-2C targets that failed to increase expression in the wc-2c knockout mutant (Fig. 5e), which causes a loss of aerial mycelium, a concentration of sporangia at the base, and alterations in melanin production75.
One of the most interesting insights provided by the DAP-seq data is the opportunity to study gene regulation and evolutionary conservation of the targets. Having identified six direct WC-2C targets in this study, we analyzed whether white-collar proteins also regulate the orthologs of these genes in other fungi. To do this, we identified orthologous genes of each target in the 303 fungal genomes available in FungiDB76 and searched for the GATC motif in their promoters. In addition to Mucoromycota, we detected a high frequency of the WC-2C-recognized motif in the Ascomycota and Basidiomycota, especially for genes ID: 1927246, 1878327, and 1888704, suggesting a possible regulation of these genes by WC-2 in other fungal species (Fig. 5f). Moreover, we found a high frequency of the GATC site in the promoters of crgA (ID 1913569) orthologs, including those from Chytridiomycota (Fig. 5f), indicating strong conservation of the crgA regulation by WC-2 across fungal clades.
Exploring evolutionary conservation of promoter architecture across fungal lineages
The conservation of targets observed for WC-2C and crgA prompted us to examine the conservation of the promoter structure across distinct fungal clades more closely. We examined WC-2C motif occurrence and found that similar to the peak detected in R. microsporus, other Mucoromycota representatives also contained the GATC motif located 300 bp upstream the TSS (Fig. 6a). Interestingly, the specific distribution of this motif was also shared by some Basidiomycota and Ascomycota species, such as Kwoniella dejecticola and Aspergillus carbonarius, suggesting a certain degree of conservation in promoter architecture. In contrast, distinct promoter architecture signatures were observed for each clade. Ascomycota displayed two GATC sites located ~150 bp upstream of the TSS, separated by 20–30 bp (Fig. 6a), consistent with previous characterizations of WC-2 binding to Neurospora promoters32. In basidiomycete orthologs, GATC sites were located around 100–150 bp upstream of the TSS, with closer spacing compared to those detected in Ascomycota (Fig. 6a). In the chytrid Batrachochytrium dendrobatidis, only one GATC site was detected, coinciding with site distribution in Basidiomycota.

a Analysis of the crgA promoter across the fungal kingdom. A schematic diagram is shown on the left for R. microsporus on the top and representative species for each phylum. Distribution of WC-2 motifs for each promoter is indicated on the right panel. The orange line indicates the distribution of the GATC site conserved across all Mucoromycota and bound by WC-2C in R. microsporus according to DAP-seq data. Sites are color-coded according to the FIMO score. Complete species names (top to bottom): Rhizopus microsporus, Kwoniella dejecticola, Melampsora laricis, Aspergillus carbonarius, Hortaea werneckii, Batrachochytrium dendrobatidis. b HLH Myc9 (top) and FK4 (bottom) motif distribution on its own promoters. The orange line indicates the distribution of the HLH Myc9 and FK4 sites conserved in Mucoromycota and bound by these TFs in R. microsporus according to DAP-seq data. Sites are color-coded according to the FIMO score. Information on the genomes analyzed can be found in Supplementary Data 1. Complete species name (top to bottom): Cryptococcus gatii, Cryptococcus neoformans, Kwoniella dejecticola. Source data are provided as a Source Data file.
Transcriptional autoregulation is one of the simplest yet most efficient mechanisms of gene expression regulation. Autoregulation loops are highly conserved throughout vertebrate evolution77 and are present from the lambda phage to higher complex eukaryotes78. We scanned the promoters of each TF studied in this work for signs of autoregulation (Supplementary Fig. 9a, b). To investigate the conservation of these autoregulation mechanisms and characterize promoter architecture conservation, we focused on two TFs, HLH Myc9 and FK4, which showed evidence of autoregulation. The promoter of the HLH Myc9 TF displayed a robust peak with two binding sites (Supplementary Fig. 9a and Fig. 6b). The promoter HLH Myc9 ortholog in Mucoromycota also contained a binding site located near the TSS, although the presence of a second motif was more variable (Fig. 6b). Notably, this second site was also missing in the basidiomycete Kwoniella dejecticola, though the site located ~100 bp upstream of the TSS was conserved (Fig. 6b). In contrast, the promoters of HLH Myc9 orthologs in Cryptococcus species showed a completely different architecture, with the C. gattii clade displaying a site located 200 bp upstream of the TSS that was absent in the C. neoformans clade, indicating inter-species variability in promoter architecture, which could also suggest functional divergencies in the TF orthologs. Orthologs of FK4 were only found in Mucoromycota fungi, and all their promoters contained the motif bound by FK4 (Fig. 6b), suggesting that the autoregulation loop of this TF is well-conserved within this early diverging fungal phylum.
Discussion
The comprehensive characterization of cistromes has long been hindered by technical challenges and the limited scalability of procedures for identifying TFBS8. However, the recent development of the DAP-seq technique has significantly increased the scalability and reliability of fungal cistrome characterizations, democratizing its application to groups of organisms that have historically been understudied13,15,31,79,80. A notable example is the EDF, a fungal subkingdom that has traditionally received less attention than Dikarya, a disparity largely due to difficulties in genetic manipulation81 and the complexity of laboratory handling34. Despite these challenges, the limited knowledge of their biology makes EDF particularly valuable for understanding the evolution of eukaryotic organisms. Coupled with their immense diversity35, EDF provide ideal models for exploring new regulatory mechanisms. In this context, the characterization of 58 TFs from R. microsporus represents a major step forward in understanding regulatory mechanisms in the early branches of fungal evolution. This work provides information on the TFBS of a diverse set of fungal TFs, along with insights into their associated regulatory networks. Although the TFs used in the DAP-seq experiments were expressed in vitro and therefore lack PTMs, which can be important for DNA binding in some cases82,83, the data presented here constitute a highly valuable resource for enabling more detailed functional studies of the TFs analyzed in this study.
The TF landscape in fungi shows remarkable diversity both between EDF and Dikarya and within the phyla included in EDF. This diversification highlights the evolutionary dynamics of TFs across fungal lineages, offering new insights into their regulatory complexity. The generation of genome-wide binding profiles for 58 TF belonging to 11 different families using DAP-seq has led to the identification of the DNA motifs recognized by these TFs, significantly expanding the catalog of fungal TF motifs, which has been dominated by motifs from Dikarya48. Analysis of the binding profiles and peak distribution across the genome revealed interesting differences between TF families, suggesting the presence of specific regulatory mechanisms, a phenomenon observed in higher organisms84. Moreover, the cistrome characterization of R. microsporus has further elucidated the complex interactions between TFs and their genomic targets (Fig. 3a). Functional classification of these targets revealed a robust clustering of TFs belonging to the same family (Fig. 3b), highlighting the existence of functional specialization among TF families. It should be noted that some TFs showed a high degree of overlap in their target genes, such as the pairs GATA Znf4 and GATA Znf8, or bZip3 and bZip6 (Supplementary Data 3). Notably, both members of each pair showed homology with SreA and AtfB from Ascomycetes, respectively, suggesting they may represent duplicated copies in Mucorales. Nevertheless, further characterization of the TFs analyzed in this study will be essential to more precisely determine the potential coregulatory activity of some of these TFs in future research.
6mA is an essential epigenetic modification in R. microsporus and is associated with transcriptionally active genes42. In recent years, the biological implications of this epigenetic modification have garnered significant interest, with studies spanning from humans and mice to plants and fungi43,44,45,85,86,87,88,89. Although ~30% of the genes in R. microsporus are directly regulated by 6mA42, the indirect effects of this modification, particularly through the methylation of genes encoding TFs and the regulatory networks that they control, have remained largely unexplored. In this study, we show that 58 TFs collectively target 4272 genes, representing 40% of the R. microsporus genome. This finding suggests that 6mA exerts indirect control over a substantial portion of the genome through its regulation of TFs. The essential nature of 6mA complicates efforts to analyze how this epigenetic modification affects the expression of TFs it regulates. However, a conditional mutant for the catalytic subunit of the 6mA methyl transferase complex shows a ~30–40% reduction in 6mA under restrictive conditions42. When we examined the methylation of genes encoding the 58 TFs studied, a subset, likely the most sensitive to the loss of methyltransferase activity, showed reduced methylation and decreased expression. Interestingly, the expression of a significant number of target genes regulated by these TFs was altered, with downregulation observed more frequently. These results provide strong evidence that 6mA indirectly controls target genes through its regulation of TFs, highlighting its pivotal role in the transcriptional regulatory networks of R. microsporus.
The regulatory influence of 6mA extends even further when considering both direct targets and indirect targets, encompassing nearly three-quarters of the genes in the R. microsporus genome. This extensive control likely explains why the loss of symmetric 6mA in this fungus is lethal. Additionally, a proportion of methylated genes are targeted by TFs regulated by 6mA. Interestingly, the methylation status of target genes varies significantly among different TFs and families, with HLH-type TFs showing the strongest preference for binding to methylated genes. This differential affinity for methylated versus unmethylated genes has already been demonstrated in a large-scale study using DAP-seq to investigate genes with and without 5mC in A. thaliana13.
One of the interesting aspects of the DAP-seq data generated in this study is its ability to evaluate any genomic region for binding by a wide range of TFs. By using this information, we expand our understanding of biologically relevant processes that have been poorly or scarcely characterized in EDF. Although several light-responsive genes have been characterized in Mucorales65,66,90 and changes in gene expression associated with loss-of-function mutations in white-collar proteins have been described67,71,72,91, it has been the integration of gene expression data from light and dark growth conditions42 with DAP-seq data for WC-2C, one of the TFs analyzed in this study, that has enabled the identification and validation of several target genes. This approach provides a better understanding of the role of this photoreceptor subunit in the transcriptional response to light, including its regulation of the carotenogenic repressor crgA. Similarly, while zinc-regulated systems have been exploited to develop regulatable expression systems in R. microsporus42, the TFs involved in this regulation remained largely unknown. The characterization of the Znf1, a Zap1-like TF involved in zinc-related transcriptional regulation in yeasts60, reveals its role in regulating several genes involved in zinc metabolism in R. microsporus. However, the orthologs of some Zap1 targets are not regulated by Znf1 in R. microsporus, highlighting differences in regulatory mechanisms and suggesting the involvement of other, yet unidentified, transcription factors in the regulation of these genes. On another note, despite the significant peak found for bZip5 in the promoter of the sexP gene, further studies are required to confirm the involvement of this TF in regulating the mating locus, particularly using R. microsporus strains with known mating conditions and compatible partners are available92.
This study is limited, as target validation using alternative approaches such as ChIP-seq was not feasible due to technical constraints associated with working on non-model organisms, including the lack of commercial antibodies and difficulties in tagging TFs. Nevertheless, replicated DAP-seq experiments provide valuable information on TFBS, not only for uncovering additional targets within the same organism but also for predicting regulatory mechanisms in other species and exploring the conservation of promoter structures across the fungal kingdom. By focusing on the high conservation of autoregulation loops77,78, we have identified both common regulatory elements and specific signatures across different fungal phyla. Overall, these results highlight the broad relevance of the data generated, not only for advancing our understanding of R. microsporus biology but also for enriching our knowledge of gene regulation mechanisms across the fungal kingdom.
Methods
DAP-seq
DAP-seq experiments were performed following a previously established procedure13,14 with minor modifications15. Briefly, genomic DNA from R. microsporus was fragmented to an average size of 150 bp (Covaris LE220-Plus) and sequencing libraries were prepared using the KAPA HyperPrep kit (Roche). In parallel, genes encoding transcription factors were codon refactored to overcome synthesis constraints (Supplementary Data 4) and assembled from synthetic DNA fragments (Twist Biosciences, CA), cloned into pIX_HALO plasmid backbone yielding an in-frame N-terminus HALO tag, confirmed via sequencing, and used for PCR amplification to yield linear PCR fragments containing the Halo-TF fusion protein driven by a T7 promoter. The PCR product was purified with SPRI beads, and the correct size of each was verified using a Fragment Analyzer (Agilent Technologies). For in vitro protein expression the TnT T7 Quick for PCR DNA System (Promega L5540) was used, with each reaction containing 10 µL of each TF PCR product at a minimum concentration of ~100 ng/µL, in a total volume of 100 µL, and with an incubation time of 2 h at 30 °C. Magne HaloTag Beads (Promega G7282) were washed and resuspended in binding/wash buffer (1× phosphate buffered saline + 0.005% NP40 detergent). To bind TFs to magnetic beads, 20 µL of washed beads were resuspended in a total volume of 50 µL binding/wash buffer and incubated with 100 µL of each expressed at room temperature for 1 h on a rotator. The HaloTag-TF-bound beads were gently washed in 150 µL of the same binding/wash buffer 3×, using a magnetic plate adapter to pellet the beads and discard the wash buffer between each wash. The beads were then combined with the 20 ng of the prepared gDNA fragment library and 2000 ng of salmon sperm DNA to mitigate non-specific binding, brought to a total volume of 150 µL using binding/wash buffer, and incubated on a rotator at room temperature for 1 h. Mock in vitro protein expression product, generated without any TF expression template, was used as a negative control. After the final wash, the beads were resuspended and boiled in 22 µL of diluted index primers15 at 95 °C for 15 min, and 20 µL of eluate from each well was transferred to a new plate and mixed with an equal volume of KAPA HiFi HotStart ReadyMix for 10 cycles of amplification. Equal volumes of each reaction were pooled across rows of the plate and then purified by gel purification. The resultant DAP-seq libraries were pooled for sequencing on a NovaSeq using the S4 flowcell (Illumina).
DAP-seq data processing
DAP-seq reads were mapped to the R. microsporus reference genome (https://mycocosm.jgi.doe.gov/Rhimi59_2/) using Bowtie293. MACS394 (v3.0.0a6) was used to call the peaks and motifs were generated using MEME95 with the 100 most significant peaks, as scored by signal value in column 7 of the narrowPeak files. TFs were filtered, discarding those with FRIP (Fraction of Reads in Peaks) < 5%. The top motif for each TF was clustered and visualized using the R package MotifStack (v1.48.0)96. Peak distribution was assessed using Bedtools (v2.30.0)97 and visualized with deepTools (v3.1)98. Whole-genome genome comparisons and binding profiles over regions of interest were performed using deepTools (v3.1)98 using a 10 kb bin size for whole-genome coverage comparisons and 10 bp for binding plots. TF targets were predicted using the RnaChipIntegrator tool99, filtering only those with a peak located at −500 + 100 bp from TSS. For each TF pair, we calculated the number of shared target genes and the jaccard similarity index, defined as the size of the intersection divided by the size of the union of their respective target sets. The gene network of TF and TF targets was constructed using Cytoscape (v3.10.2)100. GO annotations were retrieved from the R. microsporus v2 annotation available at Mycocosm (https://mycocosm.jgi.doe.gov/Rhimi59_2). A P-value of 0.05 was used as a cutoff for the identification of enriched terms (Fisher’s exact test). The top 2 most enriched GO Biological Process terms were considered for each TF-target set. Motifs were compared against fungal motifs in the JASPAR database using TomTom (MEME suite v 5.5.5)95. The best hit for each TF was retained and the P-value of the significance was used for plotting. Motif frequency was analyzed in MACs and as well as in an equal number (6083) of random regions of the same average length generated using the complete R. microsporus genome. Contingency tables of motif presence/absence in MACs and random regions were evaluated with Fisher’s exact test. Promoters were scanned using FIMO (MEME suite v 5.5.5)95, and only motifs with scores >0 were retained. Scores were used to code the motifs found in color.
Western blot
5 µL (10% of the synthesis reaction) protein was loaded into a NuPAGE 4–12% Bis-Tris and transferred to a nitrocellulose membrane using the Semi-Dry Electroblotting Unit System (Sigma-Aldrich). HaloTag fused TF were detected using the specific anti HaloTag monoclonal antibody (Promega, catalog number G9211, lot number 0000661251, dilution 1:1000). HRP-labeled secondary anti mouse antibody was added (Cytiva, catalog number RPN2108, lot number, 24K3127, dilution 1:5000). The Amersham ECL detection kit (RPN2108) was used for peroxidase reaction and results visualization.
Phylogenetic and protein conservation analyses
InterProScan101 domain data was retrieved from sequenced and annotated genomes available at JGI. The complete list of TF families and species included in the analysis is indicated in Supplementary Data 1. The species tree was generated after analyzing the 62 proteomes with OrthoFinder (Inflation factor, -I 1.5)102. The white-collar phylogenetic tree was constructed aligning known Mucor lusitanicus and Phycomyces blakesleeanus white-collar proteins64 with the predicted R. microsporus WC-2 proteins using MAFFT103. The phylogenetic tree was obtained using RAxML104 (PROTGAMMAWAGF substitution model) with 100 bootstrap replicates.
RT-qPCR
The expression of the potential TF target genes was analyzed by RT-qPCR at different zinc concentrations (ZnSO4) (0 mg/L and 20 mg/L) and in dark growth and with light exposure (24 h). Amplicons were generated with specific primers (Supplementary Data 5). Total RNA was extracted using the RNeasy Mini kit (Qiagen) and submitted to a DNase treatment (Sigma, On-Column DNaseI treatment set). cDNA was synthesized from 1 μg of total RNA using the iScriptTM cDNA synthesis kit (Biorad). cDNA amplification was carried out in triplicate using a reaction mixture containing 2× Power SYBR® Green Master Mix (Applied biosystem, Waltham, MS, USA), 150 nM of gene-specific primers, and 100 ng of cDNA. The real-time PCR was carried out using the QuantStudioTM 1 real-time PCR system (Applied Biosystems) according to the established experiment template in the equipment. The melting curve and non-template controls were also measured to discern non-specific amplifications. The relative expression of target genes was normalized with the amplification levels of the constitutively expressed mitC (Mitochondrial carrier) gene (ID:1927801) (Supplementary Data 5) and calculated using the 2−∆∆CT method.
Mutant strain generation and validation
R. microsporus mutant strains were generated via CRISPR-Cas9–mediated disruption of target genes using microhomology-directed repair DNA templates75,105. For wc-2c (ID: 1921537) and znf1 (ID: 1799646) disruption, a template DNA containing the pyrF (ID: 1869696) gene flanked by 38 nt homology regions was used to disrupt the targeted gene. Recombination was enabled through the directed double-strand break (DSB) provoked by Cas9 in an uracil-auxotrophic strain (UM1)75. Designed crRNAs (Alt-RTM CRISPR-Cas9 crRNA) used to target the CRISPR/Cas9 system were purchased from IDT (Supplementary Data 5). crRNAs were coupled with tracRNA (Alt-RTM CRISPR-Cas9 tracRNA, IDT) to form the gRNA, which was assembled in vitro with Cas9 (Alt-RTM S.p. Cas9 nuclease, IDT) to form the ribonucleoprotein complex. Gene disruptions and homo/heterokaryosis were validated by PCR using primers flanking the disruption site that amplifies both WT and mutant nuclei if present. All primers used for construct amplification and PCR validation are listed in Supplementary Data 5. DNA was extracted following a phenol/chloroform extraction106, and all PCR amplifications were performed with the Herculase II Fusion DNA polymerase (Agilent), adapting the annealing temperature to each primer pair following the manufacturer’s recommendations.
Fungal growth conditions
All the fungal strains used and generated in this work derive from Rhizopus microsporus ATCC 11559. Transformants of the auxotrophic strain UM1 strain75 using the pyrF marker were grown in Minimal Media with Casamino acids (MMC)107. During transformation, electroporated protoplasts were resuspended in ice-cold Yeast Peptone Glucose (YPG) media +0.5 M Sorbitol for 90 min and then centrifugated at 80 × g and resuspended in YNB media +0.5 M Sorbitol. All strains were grown at 30 °C under light or dark conditions as specified. DAP-seq and gene expression samples were collected 24 h after inoculating 1·106 spores per plate. To analyze the expression of the zinc-regulated genes, the medium was supplemented with 20 mg/L of ZnSO4.
Phenotypic characterization
Dark and light exposure comparisons were performed by growing 1·104 spores distributed in five points of the plate in YPG medium pH 4.5. Plates were incubated at 30 °C for 72 h, exposed to continuous white light, or covered in aluminium foil. Plates were imaged using a Stemi 305 Zeiss microscope (integrated camera). For sporulation quantification, 1·106 spores were inoculated in the center of a plate of YPG medium pH 4.5. Plates were incubated at 30 °C for 72 h. A chunk of agar of 1 cm2 (one per plate, three for each strain) was transferred to a 50 ml tube containing 10 mL of PBS 1X. The tube was vigorously vortexed to release spores. Spores were counted in a Neubauer chamber to determine the concentration, and total spore production was calculated.
For znf1Δ phenotypic characterization, 1·106 spores were inoculated in a flask with media with and without ZnSO4 supplementation (20 mg/L). Biomass was collected after 72 h of growth in a rotary shaker at 30 °C and lyophilized. Dry weight was obtained from three independent growth experiments. Spore production was performed as described above but with media supplemented with ZnSO4 when indicated.
Statistical analysis
Statistical details are detailed in the Results, Figures, and Figure legends, including the number of biological and technical replicates as well as the dispersion and precision measures (mean and SD). Statistical analyses were performed using GraphPad Prism 9 (https://www.graphpad.com). Data normality was analyzed using the Shapiro–Wilk normality test with a significance level (alpha) of 0.05. Direct comparisons between the expression levels of methylated and unmethylated genes were analyzed using Welch’s test. Pearson correlation factors, P-values cut-off for Fisher exact tests, and Welch’s tests for direct comparisons are indicated in the respective figure legends. To assess the significance of downregulated genes of unmethylated TF target genes, contingency tables with upregulated and downregulated genes were generated for target and non-target genes and Fisher exact test was conducted with a significance level (alpha) of 0.05 (two-tailed). Differences in TF proportions between species were evaluated by creating contingency tables and performing Chi-square comparisons. Two-tailed P values were calculated, setting 0.05 as the threshold for significant differences.
Ethics statement
Experiments and procedures complied with all relevant ethical regulations and were approved by the Biosafety and Ethics Committee of the Universidad de Murcia.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Raw data used in this study are available at Sequence Read Archive (SRA) under the accession numbers SRP536317, SRP496795, SRP496796, SRP496797, SRP496798, SRP496799, SRP496800, SRP496816, SRP496817, SRP496818, SRP496819, SRP496820, SRP496821, SRP496822, SRP496823, SRP496824, SRP496825, SRP496830, SRP496831, SRP496832, SRP496833, SRP497170, SRP497171, and SRP497172, and in the Gene Expression Omnibus (GEO) under the accession numbers GSE285378. The Genome of R. Microsporus ATCC 11559 used in this study is available at DDBJ/ENA/GenBank under the accession JBFXLZ010000000 [https://www.ncbi.nlm.nih.gov/datasets/genome/GCA_046865755.1/]. Source data are provided with this paper.
Code availability
Custom scripts generated for data processing are available at the Github repository (https://doi.org/10.5281/zenodo.17143943)108.
References
Lupien, M. & Brown, M. Cistromics of hormone-dependent cancer. Endocr. Relat. Cancer 16, 381–389 (2009).
Rimoldi, M. et al. DNA methylation patterns of transcription factor binding regions characterize their functional and evolutionary contexts. Genome Biol. 25, 146 (2024).
Kaluscha, S. et al. Evidence that direct inhibition of transcription factor binding is the prevailing mode of gene and repeat repression by DNA methylation. Nat. Genet. 54, 1895–1906 (2022).
Mattick, J. S. et al. Long non-coding RNAs: definitions, functions, challenges and recommendations. Nat. Rev. Mol. Cell Biol. 24, 430–447 (2023).
Zheng, R. et al. Cistrome Data Browser: expanded datasets and new tools for gene regulatory analysis. Nucleic Acids Res. 47, D729–D735 (2019).
Oki, S. et al. ChIP-Atlas: A data-mining suite powered by full integration of public ChIP-seq data. EMBO Rep. 19, e46255 (2018).
Kheradpour, P. & Kellis, M. Systematic discovery and characterization of regulatory motifs in ENCODE TF binding experiments. Nucleic Acids Res. 42, 2976–2987 (2013).
Kidder, B. L., Hu, G. & Zhao, K. ChIP-Seq: technical considerations for obtaining high-quality data. Nat. Immunol. 12, 918–922 (2011).
Berger, M. F. & Bulyk, M. L. Universal protein-binding microarrays for the comprehensive characterization of the DNA-binding specificities of transcription factors. Nat. Protoc. 4, 393–411 (2009).
Jolma, A. et al. Multiplexed massively parallel SELEX for characterization of human transcription factor binding specificities. Genome Res. 20, 861–873 (2010).
Xie, Z., Hu, S., Blackshaw, S., Zhu, H. & Qian, J. hPDI: a database of experimental human protein-DNA interactions. Bioinformatics 26, 287–289 (2009).
Jolma, A. et al. DNA-binding specificities of human transcription factors. Cell 152, 327–339 (2013).
O’Malley, R. C. et al. Cistrome and epicistrome features shape the regulatory DNA landscape. Cell 166, 1598 (2016).
Bartlett, A. et al. Mapping genome-wide transcription-factor binding sites using DAP-seq. Nat. Protoc. 12, 1659–1672 (2017).
Baumgart, L. A. et al. Persistence and plasticity in bacterial gene regulation. Nat. Methods 18, 1499–1505 (2021).
Zhang, H. et al. Scanning iron response regulator binding sites using DAP-seq in the Brucella genome. PLOS Neglected Trop. Dis. 17, e0011481 (2023).
Rhijn, N. et al. Beyond bacteria: The growing threat of antifungal resistance. Lancet 404, 1017–1018 (2024).
Niego, A. G. T. et al. The contribution of fungi to the global economy. Fungal Divers. 121, 95–137 (2023).
Iliev, I. D. et al. Focus on fungi. Cell 187, 5121–5127 (2024).
Shelest, E. Transcription factors in fungi. FEMS Microbiol. Lett. 286, 145–151 (2008).
Smith, K. M. et al. Transcription factors in light and circadian clock signaling networks revealed by genomewide mapping of direct targets for Neurospora white collar complex. Eukaryot. cell 9, 1549–1556 (2010).
Chung, D. et al. ChIP-seq and in vivo transcriptome analyses of the Aspergillus fumigatus SREBP SrbA reveals a new regulator of the fungal hypoxia response and virulence. PLoS Pathog. 10, e1004487 (2014).
Reuwsaat, J. C. V. et al. The transcription factor Pdr802 regulates titan cell formation and pathogenicity of Cryptococcus neoformans. mBio 12, e03457–20 (2021).
Badis, G. et al. A library of yeast transcription factor motifs reveals a widespread function for Rsc3 in targeting nucleosome exclusion at promoters. Mol. Cell 32, 878–887 (2008).
Zhu, J. & Zhang, M. Q. SCPD: a promoter database of the yeast Saccharomyces cerevisiae. Bioinform 15, 607–611 (1999).
Li, M. et al. Double DAP-seq uncovered synergistic DNA binding of interacting bZIP transcription factors. Nat. Commun. 14, 2600 (2023).
Zhu, J. et al. ZmEREB57 regulates OPDA synthesis and enhances salt stress tolerance through two distinct signalling pathways in Zea mays. Plant, Cell Environ. 46, 2867–2883 (2023).
Franco-Zorrilla, J. M. & Prat, S. DAP-Seq identification of transcription factor-binding sites in potato. Methods Mol. Biol. 2354, 123–142 (2021).
Hebdon, S. D., Gerritsen, A. T., Chen, Y.-P., Marcano, J. G. & Chou, K. J. Genome-wide transcription factor DNA binding sites and gene regulatory networks in Clostridium thermocellum. Front. Microbiol. 12, 695517 (2021).
Huberman, L. B. et al. DNA affinity purification sequencing and transcriptional profiling reveal new aspects of nitrogen regulation in a filamentous fungus. Proc. Natl Acad. Sci. 118, e2009501118 (2021).
Wu, V. W. et al. The regulatory and transcriptional landscape associated with carbon utilization in a filamentous fungus. Proc. Natl Acad. Sci. 117, 6003–6013 (2020).
Sancar, C. et al. Combinatorial control of light induced chromatin remodeling and gene activation in Neurospora. PLoS Genet. 11, e1005105 (2015).
Todd, R. B., Wong, K. H. & Goldman, G. H. Editorial: Transcription factors and regulation of transcriptional programs in fungi. Front. Fungal Biol. 3, 1117910 (2022).
Berbee, M. L., James, T. Y. & Strullu-Derrien, C. Early diverging fungi: diversity and impact at the dawn of terrestrial life. Annu. Rev. Microbiol. 71, 1–19 (2017).
Voigt, K. et al. H. B. Early-diverging fungal phyla: taxonomy, species concept, ecology, distribution, anthropogenic impact, and novel phylogenetic proposals. Fungal Divers. 109, 59–98 (2021).
Partida-Martinez, L. P. & Hertweck, C. Pathogenic fungus harbours endosymbiotic bacteria for toxin production. Nature 437, 884–888 (2005).
Pawlowska, T. E. Symbioses between fungi and bacteria: from mechanisms to impacts on biodiversity. Curr. Opin. Microbiol. 80, 102496 (2024).
Hachmeister, K. A. & Fung, D. Y. C. Tempeh: a mold-modified indigenous fermented food made from soybeans and/or cereal grains. Crit. Rev. Microbiol. 19, 137–188 (1993).
Roden, M. M. et al. Epidemiology and outcome of zygomycosis: a review of 929 reported cases. Clin. Infect. Dis. 41, 634–653 (2005).
Lax, C., Nicolás, F. E., Navarro, E. & Garre, V. Molecular mechanisms that govern infection and antifungal resistance in Mucorales. Microbiol. Mol. Biol. Rev. 88, e0018822 (2024).
Verma, R. et al. Rare case of primary cutaneous mucormycosis of the hand caused by Rhizopus microsporus in an immunocompetent patient. Int. J. Dermatol 53, 66–69 (2014).
Lax, C. et al. Symmetric adenine methylation is an essential DNA modification in the early-diverging fungus Rhizopus microsporus. Nat. Commun. 16, 3843 (2025).
Lax, C. et al. Symmetric and asymmetric DNA N6-adenine methylation regulates different biological responses in Mucorales. Nat. Commun. 15, 6066 (2024).
Mondo, S. J. et al. Widespread adenine N6-methylation of active genes in fungi. Nat. Genet. 49, 964–968 (2017).
Chaturvedi, A. et al. The methylome of the model arbuscular mycorrhizal fungus, Rhizophagus irregularis, shares characteristics with early diverging fungi and Dikarya. Commun. Biol. 4, 901 (2021).
Shelest, E. Transcription factors in fungi: TFome dynamics, three major families, and dual-specificity TFs. Front. Genet. 8, 53 (2017).
Merényi, Z. et al. Genomes of fungi and relatives reveal delayed loss of ancestral gene families and evolution of key fungal traits. Nat. Ecol. Evol. 7, 1221–1231 (2023).
Rauluseviciute, I. et al. JASPAR 2024: 20th anniversary of the open-access database of transcription factor binding profiles. Nucleic Acids Res. 52, D174–D182 (2023).
Rep, M. et al. The Saccharomyces cerevisiae Sko1p transcription factor mediates HOG pathway-dependent osmotic regulation of a set of genes encoding enzymes implicated in protection from oxidative damage. Mol. Microbiol. 40, 1067–1083 (2001).
Nehlin, J. O., Carlberg, M. & Ronne, H. Yeast SKO1 gene encodes a bZIP protein that binds to the CRE motif and acts as a repressor of transcription. Nucleic Acids Res. 20, 5271–5278 (1992).
Garcia-Gimeno, M. A. & Struhl, K. Aca1 and Aca2, ATF/CREB Activators in Saccharomyces cerevisiae, are important for carbon source utilization but not the response to stress. Mol. Cell. Biol. 20, 4340–4349 (2000).
Cai, M. & Davis, R. W. Yeast centromere binding protein CBF1, of the helix-loop-helix protein family, is required for chromosome stability and methionine prototrophy. Cell 61, 437–446 (1990).
Löhning, C. & Ciriacy, M. The TYE7 gene of Saccharomyces cerevisiae encodes a putative bHLH-LZ transcription factor required for Ty1-mediated gene expression. Yeast 10, 1329–1339 (1994).
Horak, C. E. et al. Complex transcriptional circuitry at the G1/S transition in Saccharomyces cerevisiae. Genes Dev. 16, 3017–3033 (2002).
Solís, E. J. et al. Defining the essential function of yeast Hsf1 reveals a compact transcriptional program for maintaining eukaryotic proteostasis. Mol. Cell 69, 534 (2018).
Imazu, H. & Sakurai, H. Saccharomyces cerevisiae heat shock transcription factor regulates cell wall remodeling in response to heat shock. Eukaryot. Cell 4, 1050–1056 (2005).
Lorenz, M. C. & Heitman, J. Regulators of pseudohyphal differentiation in Saccharomyces cerevisiae identified through multicopy suppressor analysis in ammonium permease mutant strains. Genetics 150, 1443–1457 (1998).
Staats, C. C., Kmetzsch, L., Schrank, A. & Vainstein, M. H. Fungal zinc metabolism and its connections to virulence. Front. Cell. Infect. Microbiol. 3, 65 (2013).
Zhao, H. & Eide, D. The yeast ZRT1 gene encodes the zinc transporter protein of a high-affinity uptake system induced by zinc limitation. Proc. Natl Acad. Sci. 93, 2454–2458 (1996).
Wu, C.-Y. et al. Differential control of Zap1-regulated genes in response to zinc deficiency in Saccharomyces cerevisiae. BMC Genom. 9, 370 (2008).
Lyons, T. J. et al. Genome-wide characterization of the Zap1p zinc-responsive regulon in yeast. Proc. Natl Acad. Sci. 97, 7957–7962 (2000).
Lee, S. C. & Idnurm, A. Fungal sex: the mucoromycota. Microbiol. Spectr. 5, https://doi.org/10.1128/microbiolspec.funk-0041-2017 (2017).
Idnurm, A., Walton, F. J., Floyd, A. & Heitman, J. Identification of the sex genes in an early diverged fungus. Nature 451, 193–196 (2008).
Corrochano, L. M. & Garre, V. Photobiology in the Zygomycota: Multiple photoreceptor genes for complex responses to light. Fungal Genet. Biol. 47, 893–899 (2010).
Navarro, E. et al. A negative regulator of light-inducible carotenogenesis in Mucor circinelloides. Mol. Genet. Genom. 266, 463–470 (2001).
Nicolás, F. E. et al. A RING-finger photocarotenogenic repressor involved in asexual sporulation in Mucor circinelloides. FEMS Microbiol. Lett. 280, 81–88 (2008).
Silva, F. et al. A RING-finger protein regulates carotenogenesis via proteolysis-independent ubiquitylation of a White Collar-1-like activator. Mol. Microbiol. 70, 1026–1036 (2008).
Velayos, A., Blasco, J. L., Alvarez, M. I., Iturriaga, E. A. & Eslava, A. P. Blue-light regulation of phytoene dehydrogenase (carB) gene expression in Mucor circinelloides. Planta 210, 938–946 (2000).
Corrochano, L. M. & Cerdá-Olmedo, E. Sex, light and carotenes: the development of Phycomyces. Trends Genet. 8, 268–274 (1992).
Cerdá-Olmedo, E. Phycomyces and the biology of light and color. FEMS Microbiol. Rev. 25, 503–512 (2001).
Pérez-Arques, C. et al. A Mucoralean White Collar-1 photoreceptor controls virulence by regulating an intricate gene network during host interactions. Microorg 9, 459 (2021).
Navarro, E., Peñaranda, A., Hansberg, W., Torres-Martínez, S. & Garre, V. A White Collar 1-like protein mediates opposite regulatory functions in Mucor circinelloides. Fungal Genet. Biol. 52, 42–52 (2013).
Froehlich, A. C., Liu, Y., Loros, J. J. & Dunlap, J. C. White Collar-1, a circadian blue light photoreceptor, binding to the frequency promoter. Science 297, 815–819 (2002).
Linden, H. & Macino, G. White collar 2, a partner in blue-light signal transduction, controlling expression of light–regulated genes in Neurospora crassa. EMBO J. 16, 98–109 (1997).
Lax, C. et al. Stable and reproducible homologous recombination enables CRISPR-based engineering in the fungus Rhizopus microsporus. Cell Rep. Methods 1, 100124 (2021).
Basenko, E. Y. et al. What is new in FungiDB: a web-based bioinformatics platform for omics-scale data analysis for fungal and oomycete species. Genetics 227, iyae035 (2024).
Kiełbasa, S. M. & Vingron, M. Transcriptional autoregulatory loops are highly conserved in vertebrate evolution. PLoS One 3, e3210 (2008).
Crews, S. T. & Pearson, J. C. Transcriptional autoregulation in development. Curr. Biol. 19, R241–R246 (2009).
Zhou, L. et al. Gene duplication drove the loss of awn in sorghum. Mol. Plant 14, 1831–1845 (2021).
Hale, B. et al. Gene regulatory network inference in soybean upon infection by Phytophthora sojae. PLOS One 18, e0287590 (2023).
Lax, C. et al. Genetic manipulation in Mucorales and new developments to study mucormycosis. Int. J. Mol. Sci. 23, 3454 (2022).
Roy, D. & Sadanandom, A. SUMO mediated regulation of transcription factors as a mechanism for transducing environmental cues into cellular signaling in plants. Cell. Mol. Life Sci. 78, 2641–2664 (2021).
Filtz, T. M., Vogel, W. K. & Leid, M. Regulation of transcription factor activity by interconnected post-translational modifications. Trends Pharmacol. Sci. 35, 76–85 (2014).
Lambert, S. A. et al. The human transcription factors. Cell 172, 650–665 (2018).
Liu, J. et al. Abundant DNA 6mA methylation during early embryogenesis of zebrafish and pig. Nat. Commun. 7, 13052 (2016).
Liang, Z. et al. DNA N6-adenine methylation in Arabidopsis thaliana. Dev. Cell 45, 406–416.e3 (2017).
Xiao, C.-L. et al. N6-methyladenine DNA modification in the human genome. Mol. Cell 71, 306–318.e7 (2018).
Li, X. et al. The exploration of N6-deoxyadenosine methylation in mammalian genomes. Protein Cell 12, 756–768 (2021).
Beh, L. Y., Müller, M. M., Muir, T. W., Kaplan, N. & Landweber, L. F. DNA-guided establishment of nucleosome patterns within coding regions of a eukaryotic genome. Genome Res. 25, 1727–1738 (2015).
Velayos, A., Eslava, A. P. & Iturriaga, E. A. A bifunctional enzyme with lycopene cyclase and phytoene synthase activities is encoded by the carRP gene of Mucor circinelloides. Eur. J. Biochem. 267, 5509–5519 (2000).
Silva, F., Torres-Martínez, S. & Garre, V. Distinct white collar-1 genes control specific light responses in Mucor circinelloides. Mol. Microbiol. 61, 1023–1037 (2006).
Mondo, S. J. et al. Bacterial endosymbionts influence host sexuality and reveal reproductive genes of early divergent fungi. Nat. Commun. 8, 1843 (2017).
Langmead, B., Trapnell, C., Pop, M. & Salzberg, S. L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10, R25–R25 (2009).
Zhang, Y. et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137 (2008).
Bailey, T. L., Johnson, J., Grant, C. E. & Noble, W. S. The MEME Suite. Nucleic Acids Res. 43, W39–W49 (2015).
Ou, J., Wolfe, S. A., Brodsky, M. H. & Zhu, L. J. motifStack for the analysis of transcription factor binding site evolution. Nat. Methods 15, 8–9 (2018).
Quinlan, A. R. & Hall, I. M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842 (2010).
Ramírez, F. et al. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 44, W160–W165 (2016).
Briggs, Donaldson & Zeef. RnaChipIntegrator (version 3.0.0) (2024). https://github.com/fls-bioinformatics-core/RnaChipIntegrator.
Shannon, P. et al. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 13, 2498–2504 (2003).
Jones, P. et al. InterProScan 5: genome-scale protein function classification. Bioinformatics 30, 1236–1240 (2014).
Emms, D. M. & Kelly, S. OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome Biol. 20, 238 (2019).
Katoh, K. & Standley, D. M. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780 (2013).
Stamatakis, A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313 (2014).
Lax, C. et al. Transformation and CRISPR-Cas9-mediated homologous recombination in the fungus Rhizopus microsporus. Star. Protoc. 3, 101237 (2022).
Osorio-Concepción, M., Lax, C., Navarro, E., Nicolás, F. E. & Garre, V. DNA Methylation on N6-adenine regulates the hyphal development during dimorphism in the early-diverging fungus Mucor lusitanicus. J. Fungi 7, 738 (2021).
Nicolás, F. E., Haro, J. P. de, Torres-Martínez, S. & Ruiz-Vázquez, R. M. Mutants defective in a Mucor circinelloides dicer-like gene are not compromised in siRNA silencing but display developmental defects. Fungal Genet. Biol. 44, 504–516 (2007).
Tahiri, G. Unveiling a pervasive DNA adenine methylation regulatory network in the early-diverging fungus Rhizopus microsporus. Github, https://doi.org/10.5281/zenodo.17143943 (2025).
Acknowledgements
This research was supported by the grant PID2021-124674NB-I00 and PID2024-160088NB-I00 to F.E.N. and V.G, funded by MICIU/AEI/10.13039/501100011033 and by ERDF/EU. The work (https://doi.org/10.46936/10.25585/60001127) was conducted by the US Department of Energy Joint Genome Institute (https://ror.org/04xm1d337), a DOE Office of Science User Facility, supported by the Office of Science of the US Department of Energy under Contract No. DE-AC02-05CH11231.
Author information
Authors and Affiliations
Contributions
C.L. conducted most of the analyses, prepared the R. microsporus material for sequencing, performed bioinformatic analysis of the results, prepared the figures and tables, designed and coordinated the project, and wrote the manuscript with significant input from G.T., F.E.N., and V.G. L.A.B. participated in the processing of DAP-seq data. G.T. participated in RT-qPCR analyses and bioinformatic analyses. N.L.-M. participated in RT-qPCR analysis. Y.Z. performed DAP-seq. I.K.B. designed and synthesized transcription factors for the DAP-seq experiments. S.J.M and B.K built and developed the DAP-seq viewer, respectively. R.C.O. conceived and designed DAP-seq experiments. V.N. managed the project. E.N. managed the project and provided materials. I.V.G. supervised and coordinated the project. F.E.N. supervised the study. V.G. analyzed the results and designed, supervised, and coordinated the project.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Joseph Strauss and the other, anonymous, reviewer for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Lax, C., Baumgart, L.A., Tahiri, G. et al. Unveiling a pervasive DNA adenine methylation regulatory network in the early-diverging fungus Rhizopus microsporus. Nat Commun 16, 10277 (2025). https://doi.org/10.1038/s41467-025-65177-1
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-65177-1
