
Abstract
T-box riboswitches regulate gene expression by sensing tRNA aminoacylation status, but their dynamic mechanisms remain elusive. Here, we present single-molecule FRET studies of a full-length translational ileS T-box, showing that its decoding domain folds independently, and the initial tRNA anticodon binding promotes proper folding of the discriminator domain. Subsequent uncharged tRNA binding stabilizes the Antisequestrator (AntiS) conformation and GAG linker, resulting in translation initiation. Conversely, charged tRNA binds to a distinct T-box conformation, of which the linker is highly flexible and stem III is away from AntiS, rendering irreversible transition to the Sequestrator conformation if downstream sequences are transcribed, leading to translation inhibition. Collectively, both steps of tRNA binding are in a conformational selection manner, and the GAG linker, especially G96, acts as the main linchpin in tRNA 3΄-termini sensing. An elaborate model is proposed to understand how cotranscriptional folding, stepwise tRNA binding and dynamic conformational transitions coordinate T-box regulation.
Similar content being viewed by others

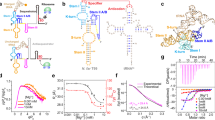

Introduction
T-box riboswitches are important riboregulators that regulate the expression of genes involved in amino acid synthesis, transport and tRNA aminoacylation through binding to specific tRNAs and sensing their aminoacylation status1,2,3,4. Typical T-box riboswitches are modular RNA devices that consist of a 5΄ decoding or aptamer domain (stems I, II and IIA/B) and a 3΄ discriminator domain (stems III and Antiterminator/AntiSequestrator (AntiT/AntiS)), which are connected by a single-stranded linker (Fig. 1a). The decoding domain containing a specifier loop in stem I is mainly responsible for specific binding to the anticodon of tRNA, while the discriminator domain could sense the charging state of tRNA 3΄NCCA end and then undergo conformational switching between the AntiT/AntiS and Terminator/Sequestrator stems, resulting in transcription attenuation or translation initiation of downstream genes5,6,7,8. Though structural organization of the decoding domain such as the length of stem I and the existence of stem II and IIA/B varies, the discriminator domain is highly similar across different T-box riboswitches9,10,11. T-box riboswitches have been found to be widely spread in Gram-positive bacteria, including some pathogenic bacteria but absent in humans2,3,12,13, therefore, they have been developed as novel antimicrobial targets with reduced drug resistance4,14.

a Sequence and secondary structure of the full-length ileS T-box riboswitch and tRNAIle construct used in this study. Red lines represent the interaction between tRNAIle and T-box riboswitch (Anticodon and Specifier loop, NCCA 3’-end and AntiS bulge). The secondary structure of the mutually exclusive sequestrator domain is predicted by Mfold46. b Schematic diagram for the T-box constructs of different lengths used in this study. c Crystal structure of the complex formed between the core region of ileS T-box and tRNA (PDB: 6UFG) as colored in (a). d Schematic representation of the total internal reflection fluorescence microscopy (TIRFM)-based smFRET analysis of the core ileS T-box riboswitch and its interaction with tRNA. Cy3 and Cy5 were shown as green and blue stars, respectively.
Recently, multiple structural techniques including X-ray crystallography and cryo-EM have been applied to resolve the high-resolution structures of the isolated decoding domains, the core regions (including the decoding, linker and discriminator domains but lack of the Terminator/Sequestrator stems and their downstream sequences) of both transcriptional and translational T-box riboswitches in complex with their cognate tRNA ligands9,10,11,15,16. These structures provide important insights into the mechanisms of specific and high-affinitiy tRNA recognition, binding and amino acid sensing by T-box riboswitches. For example, the cocrystal structure of the complex of core region of a translational ileS T-box from Mycobacterium tuberculosis and its cognate tRNA ligand reveals two binding sites9 (Fig. 1c). The first binding site between specifier loop in stem I and tRNA anticodon is laterally reinforced by the interactions between stems I and II and also the coaxially stacked stem IIA/B. Besides, the second binding site between the tRNA 3΄NCCA end and the highly conserved 5΄-UGGN-3΄ sequence of discriminator domain via base pairing interaction is further stabilized by the coaxially stacked tRNA acceptor stem and the bottom helix of AntiS. The stem III and nearby linker region are highly involved in the formation of functional aminoacylation sensing module by contributing key tertiary interactions. Interestingly, two distinct models regarding the discrimination of tRNA aminoacylation state by T-box riboswitches were proposed. The Zhang lab supported the idea that the discriminator domain is rigid to sterically reject the aminoacylated tRNA10, whereas the Ke lab considered that the discriminator domain is conformationally flexible to accommodate even charged tRNA, which then disrupts the tertiary interactions that drive the formation of AntiS and favors the formation of Sequestrator conformation9. Although both models are supported by current biochemical and structural evidences, accordingly, the authentic mechanism for discrimination of tRNA aminoacylation state remains unclear and requires further studies.
The tRNA recognition, binding and aminoacylation state discrimination by T-box riboswitches are a series of conformationally dynamic processes. As the available high-resolution structures only provide static snapshots of the truncated T-box riboswitches in holo form9,10,11,15,16, the structural and dynamic information for a full-length T-box riboswitch in apo form is missing, whether and how the conformation dynamics of T-box induced by binding to tRNA with different aminoacylation states exerts gene regulation is less understood. Recently, studies on the decoding domain of a translational ileS T-box riboswitch by single-molecule fluorescence resonance energy transfer (smFRET) have revealed that tRNA anticodon binding induces significant intra- and inter-stem dynamics within the decoding domain17,18. smFRET analyses on the core regions of a transcriptional GlyQS T-box and a translational ileS T-box mainly have demonstrated a two-step binding kinetics for charged and uncharged tRNA to T-box riboswitch19,20,21. However, the structural dynamics of a full-length T-box riboswitch induced by tRNA 3΄-end binding, especially in the discriminator domain and nearby regions, remains largely unknown. This is partly due to the limitations of the schemes used for fluorescent labeling in these studies which only apply to 5΄- or 3΄-ends of RNAs19,20,21.
RNAs are known to fold while they are being transcribed, causing an additional layer of complexity in elucidating structural dynamics of T-box riboswitches upon binding to charged or uncharged tRNA. In this study, RNA transcripts of various lengths are used to mimick the co-transcriptional folding intermediates of a translational ileS T-box riboswitch from M. tuberculosis (Fig. 1b). Empowered with the recently developed approaches for site-specific fluorescent labeling based on the NaM-TPT3 unnatural base pair (UBP) system17,22, we introduced multiple pairs of donor and acceptor dyes at internal positions across the RNA transcripts. We then utilize smFRET technique to probe the intra- and interdomain conformational dynamics of the T-box riboswitches induced by uncharged or a mimic of charged tRNA (Fig. 1d). We find that the first step binding of tRNA anticodon to the decoding domain of ileS T-box enhances the docking of stems I and II and promotes the proper folding of discriminator domain. The second step, binding of uncharged tRNA 3΄NCCA end to the AntiS stabilizes the flexible linker region to form a stable T-box/tRNA complex, rendering the exposure of downstream SD and AUG sequences to allow translation initiation. Conversely, the discriminator domain can still bind to a mimic of charged tRNA through a distinct conformation, in which the linker region becomes more flexible and stem III is away from AntiS domain, rendering dynamic transition of the AntiS to the Sequestrator conformations which inhibits translation initiation. The essential role of linker region in discriminating the tRNA charging state is further supported by analyzing the single-point or deletion mutants. Once the Sequestrator conformation is formed, it barely transits to the AntiS conformation, even in the presence of uncharged tRNA. Taken together, we propose a plausible model delineating how a translational ileS T-box riboswitch recognizes tRNA stepwisely, discriminates the aminoacylation state and dynamically switches its conformations to regulate translation initiation cotranscriptionally.
Results
The core T-box forms distinct complexes with tRNA upon stepwise tRNA binding
Previous smFRET studies showed that the core regions of both a transcriptional GlyQS and a translational ileS T-box riboswitches bind to their cognate tRNAs in two steps, in which the T-box stem I recruits tRNA through the specifier-anticodon interaction first followed by sensing and binding the 3΄NCCA end with its discriminator domain19,20,21, but how the global and local conformations of core T-boxes change in response to tRNA binding remains largely unknown. To better understand the mechanisms of stepwise tRNA binding, we first tested the binding of core ileS T-box riboswitch from M. tuberculosis (T166, 166 denotes the length of the T-box construct used) to three tRNA constructs with different 3΄ termini (tRNA∆, tRNAIle and tRNA78) by electrophoretic mobility shift assay (EMSA). T166 consists of stem I, II, IIA/B, linker region, stem III and AntiS domain but doesn’t contain the sequence that encodes the thermodynamically more favorable sequestrator structure and the following SD sequence (Fig. 1b), which thus is a minimal construct to study the two-step tRNA binding. While wild type tRNAIle has both the anticodon and the free 3΄-end binding sites (Fig. 1a), the tRNA∆ construct only reserves the first but is deprived of the second binding site of tRNAIle by deletion of the GCCA 3΄-end (Supplementary Fig. 1a). Given that aminoacylated tRNAs are reported to be unstable and prone to be hydrolyzed under experimental conditions and the aminoacylation efficiency is usually not high enough in vitro23,24,25, tRNA78 containing an extra nucleotide C at the 3΄-end of tRNAIle was used to mimick the aminoacylation state (Supplementary Fig. 1b). T166 alone showed a smeared band in the native gel, indicating that the core ileS T-box is conformationally flexible and doesn’t fold properly in apo form (Fig. 2a). While addition of tRNA∆ produced a less but somewhat smeared band for the complex, addition of tRNAIle produced a dense and narrow band with increased mobility, consistent with the formation of a stable and compact T-box-tRNAIle complex through two contact sites (Fig. 2a). These results suggest that loss of the second tRNA binding is detrimental to the folding of core ileS T-box, even though the first contact site is formed. Interestingly, addition of tRNA78 could also produce a dense and narrow band, though the band migrated slower than that for tRNAIle. It is likely that the charged tRNA (mimicked by tRNA78) can establish two contact sites with the core ileS T-box to form a complex though with distinct conformation (Fig. 2a).

a Electrophoretic mobility shift assay (EMSA) showing the binding between different tRNA constructs and core region of T-box riboswitch (T166). The results were consistent across all three independent experimental replicates. b Dimensionless Kratky plots for T166 in complex with different tRNA constructs. c Schematic representation of the different fluorescent labeling constructs of T-box-tRNA complexes for smFRET analysis. The green and blue circles represent the Cy3 and Cy5 fluorophores, respectively. d Representative smFRET traces (left), FRET histograms (middle) and transition density plots (right) for the respective labeling constructs in c in the presence of 20 mM Mg2+ and three different tRNA constructs: tRNAΔ, tRNAIle and tRNA78. Green, blue and black lines represent the Cy3 intensity, Cy5 intensity and FRET efficiency, respectively. FRET histograms were well fitted with Guassion peaks, shown in red and blue for the low- and middle- or high-FRET, respectively. Transition density plots illustrate number of transition events per second. N denotes the total number of molecules used to generate the FRET histograms from three independent experiments (n = 3). Source data are provided as a Source Data file.
To directly visualize the conformational differences of complexes formed between T166 and different tRNA constructs, we probed their folding and global structures in the presence of 20 mM Mg2+ by small-angle X-ray scattering (SAXS) (Fig. 2b, Supplementary Fig. 2, Supplementary Table 1). Under such conditions, the specifier-anticodon interactions between T-box and different tRNA constructs are expected to be of high affinity11,17, which are tight enough to stabilize each of the complexes. The dimensionless Kratky plot, plotted as (qRg)2I(q)/I0 versus qRg, reflects the degree of folding and compactness of the molecules in solution. As shown in Fig. 2b, the dimensionless Kratky plot of T166/tRNA∆ complex displays a significant enrichment in the high scattering angles, characteristic of a molecule with large disordered regions. In contrast, both plots for complexes of T166/tRNAIle and T166/tRNA78 exhibit obvious curving down features at high scattering angles, consistent with folded complexes with two-site binding. As the curving down feature for T166/tRNAIle complex is more prominent, it is likely the global structure of T166/tRNAIle complex is more compact than that of T166/tRNA78 complex, which is also supported by the smallest structural parameters derived from the Pair distance distribution function (PDDFs), including the radius of gyration (Rg) and the maximum particle dimension (Dmax), for T166-tRNAIle complex (Supplementary Fig. 2, Supplementary Table 1).
To monitor the conformational rearrangements of T-box upon complex formation with high temporal and spatial resolution, we characterized the complexes with total internal reflection fluorescence microscopy (TIRFM)-based smFRET method. The 5΄-end of different tRNA constructs (5΄Cy3-tRNA∆, 5΄Cy3-tRNAIle and 5΄Cy3-tRNA78) were all labeled with the Cy3 dye. To label the internal site of T166 with the Cy5 dye, the most recently developed NaM-TPT3 UBP-based labeling strategy was utilized17,22. Briefly, the nucleotide U139 in the AntiS stem of T166 transcript was replaced with an alkyne-derived unnatural nucleotdide NaM (rNaMCO) by in vitro transcription, which was followed by click chemistry reaction to conjugate azide-modified Cy5 dye (T166-139Cy5) (Fig. 2c, Supplementary Fig. 1c). EMSA assay showed that UBP-based fluorescent labeling had minor effects on the binding affinity of T166-139Cy5 to tRNAIle (Supplementary Fig. 3). To enable TIRFM-based smFRET analysis, the 3΄-end of T166 was extended with a 21-nucleotides (nts) single-stranded sequence by in vitro transcription, which was used to anneal with a DNA oligonucleotide containing a biotin modification at the 5΄-end for surface immobilization (Fig. 2c, Supplementary Fig. 1c). The time-dependent Cy3 and Cy5 intensities were recorded under equilibrium conditions in the presence of 20 mM Mg2+, which is essential for high-affinity binding between T-box and the different tRNA constructs17,19. The FRET efficiency (EFRET) was calculated from the Cy3 and Cy5 intensities of thousands of individual molecules, which reflects the intramolecular distance between Cy3 and Cy5 labeling sites. The FRET trajectories were subjected to hidden Markov modeling analysis, which not only estimates the respective mean FRET values of the observed FRET states but also derives the relative frequencies of transitions between different FRET states through the transition density plots (TDPs).
In the presence of tRNA∆, T166 is assumed to form complex through the first step binding. While the T166/tRNA∆ complex samples a low- (E ~ 0.3) and a middle-FRET (E ~ 0.59) states, with most of the traces showing transition between low- and middle-FRET states, binding of tRNAIle to T166 results in a major high-FRET population with FRET value around ~0.89 which rarely transits to a low-FRET (E ~ 0.38) state (Fig. 2d). Based on the crystal structure of M. tub core T-box/tRNAIle complex, the distance between T-box 139U and tRNA 5΄-end is around ~38 Å, which should result in a high-FRET value. Therefore, the high-FRET (E ~ 0.89) is assigned to be the fully tRNAIle-bound state of T166 (with two contact sites formed) while the low- and middle-FRET correspond to the partially-bound state. The dynamic transitions for T166/tRNA∆ complex suggest that the relative orientation between tRNA∆ and the discriminator domain of T166 is conformationally flexible when only the first step binding is established. Furthermore, the high-FRET state for T166/tRNAIle complex indicates that the second site binding induces significant compaction in the complex. Intriguingly, the T166/tRNA78 complex also samples a high-FRET state but with a lower FRET value than that for T166/tRNAIle complex (0.75 vs. 0.89) and frequently transits to a low-FRET state, indicating that the 3΄NCCAC end of tRNA78 could also establish direct interactions with the discriminator domain of T166 though resulting in a distinct complex structure. To analyze the NCCA end binding kinetics, the dwell time distributions and transition rates between the middle- and high-FRET states for T166/tRNA78 and T166/tRNAIle complexes were extracted from the individual single-molecule trajectories. We found that dissociation of the 3΄-end of tRNA78 from the T166 AntiS (high- to middle-FRET transition) is much faster than that for tRNAIle (0.82 s-1 vs 0.27 s-1), indicating that the second site interaction of aminoacylated tRNA is more dynamic and less stable than that of 3΄-end free tRNA. In addition, TDP analysis also showed that T166/tRNA78 transits much more frequently between middle-FRET and high-FRET than T166/ tRNAIle (Fig. 2d).
Structural dynamics of the decoding domain of core T-box
Compared to the decoding domain of T-box from Nocardia farcinica (N. far T99) which structural dynamics have been analyzed by smFRET17,18, the stem II of the decoding domain of M. tub T-box is much shorter (37 vs 48 nt) (Supplementary Fig. 1d-e). To understand the structural dynamics of the decoding domain of M.tub T-box (T87), we first characterized the interstem motion between stems I and II of T87 alone (Fig. 3a, Supplementary Fig. 1e). Similar labeling sites as that in T99 of N. far ileS T-box for smFRET analysis were chosen17. Specifically, Cy3 and Cy5 dyes were covalently labeled to site C6 in stem I and site G46 in stem II using the UBP-based labeling strategy (T87-6Cy3/46Cy5) without disrupting the tRNA binding affinity (Fig. 3a, Supplementary Fig. 3). All smFRET data were collected in 20 mM Mg2+, and in the absence or presence of different tRNA constructs (tRNA∆, tRNAIle and tRNA78) (Fig. 3b). Different from N. far T99-6Cy3/54Cy5 which only samples a low-FRET state (E ~ 0.32) in apo form, M.tub T87-6Cy3/46Cy5 mainly samples a low-FRET state (E ~ 0.3) coexisting with a minor middle-FRET state (E ~ 0.6) under the same condition. Visual inspection of smFRET traces revealed that T87/6-46 displayed predominantly static heterogeneity, with most molecules occupying either the low-FRET state or middle-FRET state, showing rare transitions between them during observation (Fig. 3b, Supplementary Fig. 4a). From the crystal structure of tRNA-bound M.tub T-box, the distance between the labeling sites in stem II is ~40.0 Å, which is expected to give a middle-FRET signal. It is likely high Mg2+ alone is sufficient to promote the proper docking of stems I and II into a tRNA-bound conformation for high affinity tRNA binding and both the unbound and bound-like conformation coexist in M.tub T87 alone. The population of T87-6Cy3/46Cy5 middle-FRET increased prominently (from ~25% to ~75%) upon addition of saturating tRNA∆, tRNAIle or tRNA87, indicating that binding of tRNA anticodon to Specifier (first-step binding) selects the bound-like conformation and promotes the complex formation, which, however, is not affected by the tRNA 3΄-end. Thus, tRNA aminoacylation status has a minor effect on the first-step tRNA binding by the decoding domain, consistent with previous study on GlyQS T-box riboswitch19. Notably, these results are obviously different from that in N. far T99 which high Mg2+ facilitates the pre-docking between stems I and II to form a competent tRNA binding conformation, and subsequent tRNA binding induced the proper docking of stem I and stem II17,18.

a Schematic representation for the respective fluorescent labeling constructs. The green and blue circles represent the Cy3 and Cy5 fluorophores, respectively. Representative smFRET traces (left), FRET histograms (middle) and transition density plots (right) for each smFRET construct in (a) in four different conditions: in the absence of tRNA, in the presence of tRNAΔ, tRNAIle and tRNA78. Notably, the tRNAs are at saturating concentrations in (b, c), but at much lower concentrations in (d, e). Green, blue and black lines represent the Cy3 intensity, Cy5 intensity and FRET efficiency, respectively. FRET histograms were well fitted with Guassion peaks, shown in red and blue for the low- and middle- or high-FRET, respectively. N denotes the total number of molecules used to generate the FRET histograms from three independent experiments (n = 3). Transition density plots illustrate number of transition events per second. Scale bar for the transition density plots in (d, e) are the same and are shown on the top side. Source data are provided as a Source Data file.
To understand if the first-step tRNA binding to the decoding domain is affected by the discriminator domain, we also probed the dynamics of decoding domain in the context of core T-box (T166) (Fig. 3c, Supplementary Fig. 4b). Similar phenomena was observed for T166-6Cy3/46Cy5 under the same conditions. While T166-6Cy3/46Cy5 alone samples a major low-FRET (E ~ 0.3) and a minor middle-FRET (E ~ 0.6) states in apo form, in the presence of saturating tRNA∆, tRNAIle or tRNA78, the populations for middle-FRET corresponding to the tRNA-bound state were substantially increased from 25% to 75%, which are independent on the tRNA 3΄-end. These results suggest that the first-step binding may not be thermo-dynamically affected by the second-step binding.
It is reported that interaction of the tRNA 3΄-end with the Antiterminator of the transcriptional GlyQS T-box (second-step binding) further stabilizes the tRNA/T-box complex by decreasing the tRNA dissociation kinetics19. To substantiate this point on ileS core T-box, we also conducted smFRET experiments for T166-6Cy3/46Cy5 and T87-6Cy3/46Cy5 in the presence of different concentrations of tRNAs (Fig. 3d, e, Supplementary Fig. 5). The occupancy of middle-FRET fraction increases until the tRNAIle concentration reaches 100 nM (Supplementary Fig. 5). However, the middle-FRET fraction for T166-6Cy3/46Cy5 in the presence of only 10 nM tRNAIle is close to that in the presence of saturating tRNAIle. These results support the conclusion that the second-step binding contributes to the stability of T-box/tRNA complex.
Structural dynamics of the discriminator domain of core T-box
To map the conformational changes in the discriminator domain and nearby region of core T-box upon stepwise tRNA binding, we further characterized three labeling constructs of T166 in complex with tRNAs of different aminoacylation states. Among these constructs, the Cy3 was labeled at site of 89 in the linker (T166-89Cy3), site 107 in stem III (T166-107Cy3) or site 148 in AntiS (T166-148Cy3), respectively, and Cy5 was coupled to the 3΄-end of the DNA oligonucleotide complementary to T-box RNA (D-3΄Cy5) (Fig. 4a–c). All these labeled constructs form stable complexes with the tRNAIle (Supplementary Fig. 3), and smFRET data were collected under the same conditions as above.

a–c Schematic representation for the respective fluorescent labeling constructs for smFRET analysis. The green and blue circles represent the Cy3 and Cy5 fluorophores, respectively. d–f Representative smFRET traces (left), FRET histograms (middle) and transition density plots (right) for each smFRET construct in (a) in four different conditions: in the absence of tRNA, in the presence of tRNAΔ, tRNAIle and tRNA78. Green, blue and black lines represent the Cy3 intensity, Cy5 intensity and FRET efficiency, respectively. FRET histograms were well fitted with Gaussian peaks, shown in red and blue for the low- and middle- or high-FRET, respectively. Transition density plots illustrate number of transition events per second. N denotes the total number of molecules used to generate the FRET histograms from three independent experiments (n = 3). Source data are provided as a Source Data file.
The T166-89Cy3/D-3΄Cy5 construct was used to characterize the relative motions between the linker and the bottom helix of AntiS in response to stepwise tRNA binding (Fig. 4d). In 20 mM Mg2+ and the absence of tRNA, T166-89Cy3/D-3΄Cy5 mainly samples a middle-FRET (E ~ 0.5) state which occasionally transits to a high-FRET (E ~ 0.85), revealing a dynamic and flexible linker in solution. While addition of tRNA∆ has a minor effect on the RNA which still fluctuates frequently between the middle- (E ~ 0.5) and high-FRET (E ~ 0.85), the RNA mostly stays in the high-FRET state which occupancy increases substantially and rarely transits to middle-FRET upon addition of tRNAIle. From the crystal structure of M. tub T-box/tRNA complex, the distance between the labeling sites of T166-89Cy3/D-3΄Cy5 is ~26.1 Å, which is expected to give a high-FRET signal. Thus, the middle- and high-FRET states presumably correspond to the tRNA NCCA-unbound and -bound conformation, respectively. It is likely the linker region of T166 alone at high Mg2+ samples both the unbound and fully bound conformation, while the first-step binding by tRNA∆ cause minor effects to the linker region, the two-site binding by tRNAIle strongly stabilize the fully bound conformation. Interestingly, in the presence of tRNA78, T166-89Cy3/D-3΄Cy5 exhibits a highly populated (60%) middle-FRET coexisting with the high-FRET state (40%) (Fig. 4d). Both of the transition occupancy density plots (TODP) and TDPs showed that the majority of molecules exhibit frequent transitions between the middle- (E ~ 0.56) and high-FRET (E ~ 0.85) states (Fig. 4d, Supplementary Fig. 6), indicating that the linker region becomes much more flexible and is no longer stabilized in the fully bound conformation upon sensing the charged tRNA 3΄-end. Notably, tRNA78-bound T166-89Cy3/D-3΄Cy5 samples a distinct middle-FRET state from that bound by tRNA∆ (E ~ 0.56 vs E ~ 0.49), supporting that the linker region is responsive to the NCCA 3΄-end of charged tRNA.
The T166-107Cy3/D-3΄Cy5 construct was used to characterize the dynamics of stem III in response to stepwise tRNA binding (Fig. 4e). In 20 mM Mg2+ and the absence of tRNA, T166-107Cy3/D-3΄Cy5 displays a broad FRET distribution and transits frequently between a low-FRET (E ~ 0.39) and middle-FRET (E ~ 0.65), indicating that the orientation of stem III relative to AntiS is flexible in apo form. The distance between the labeling sites of T166-107 and the 3΄-end estimated from the crystal structure is about 48.5 Å, which should give a middle-FRET value. In the presence of tRNA∆, T166-107Cy3/D-3΄Cy5 samples both the low- (E ~ 0.39) and middle-FRET (E ~ 0.65) states, of which the middle-FRET state became predominant and its occupancy increased from 44% to 56%, suggesting that the first-step binding of tRNA anticodon may promote the structural reorganization of stem III and AntiS into the NCCA 3΄-end bound conformation. In the presence of tRNAIle, T166-107Cy3/D-3΄Cy5 mainly occupies the middle-FRET (E ~ 0.65) state, whose population increased from 60% to 70%. These results indicate that the two-site binding further stabilizes the complex. By contrast, in the presence of tRNA78, T166-107Cy3/D-3΄Cy5 mainly samples the low-FRET state (increase from 34% to 48%) which transits frequently to the middle-FRET state, indicating that stem III moves away from the bottom helix of AntiS upon binding to the aminoacylated tRNA.
To explore the structural reorganization of AntiS domain comprised of two helix and a heptanucleotide bulge, we also analyzed the T166-148Cy3/D-3΄Cy5 construct (Fig. 4f). The distance between the labeling sites estimated from the crystal structure of the M. tub core T-box/tRNA complex is about 43.5 Å, which will give a middle-FRET signal. In 20 mM Mg2+ and the absence of tRNA, T166-148Cy3/D-3΄Cy5 samples a low-FRET (E ~ 0.2) and a middle-FRET state (E ~ 0.45), corresponding to the partially folded and folded state, respectively. Interestingly, even in the presence of tRNA∆, the occupancy of middle-FRET state (E ~ 0.45) increased substantially, indicating that the first-step tRNA binding may promote the proper folding of AntiS. Subsequent recognition and binding by the NCCA 3΄-end of tRNAIle further stabilized the middle-FRET state and T166-148Cy3/D-3΄Cy5 prevalently samples the middle-FRET state. Similarly, in the presence of tRNA78, T166-148Cy3/D-3΄Cy5 predominantly stays in the middle-FRET state, suggesting that the folding of AntiS domain is less sensitive to the tRNA 3΄-end aminoacylation status.
Taken together, these smFRET data depict a delicate picture showing how the individual domain of core T-box responds to stepwise tRNA binding with different aminoacylation states, providing further evidence that charged tRNA binding stabilizes a structural intermediate distinct from that in the T-box/tRNAIle complex, in which the single-stranded linker region dynamically transits to a conformational state away from the discriminator domain and subsequently leads to structural rearrangement of stem III.
The role of linker region in the 3΄-end discrimination of tRNA
Both in vitro structural and in vivo studies have shown that the linker region, especially the conserved GAG (nucleotides 94-96) sequence, is important to sense and discriminate the aminoacylation status of tRNA NCCA 3΄-end9. Besides the conserved GAG sequence, the linker also contains a seven-nucleotides (7-nts) single-stranded region which is not involved in tertiary interactions. To further understand the role of linker region in tRNA 3΄-end discrimination mechanism, two single-point mutants (G94U and G96C) were constructed. Additionally, we also constructed a linker deletion mutant (∆88-93) which the 7-nt single-stranded region was deleted.
We first probed the mutational effects of these mutants on the binding of core T-box with different tRNA constructs by EMSA (Fig. 5a). Similar to T166 wild type, all the mutants in the presence of 20 mM Mg2+ only displayed a smeared band in the native gel. The G96C mutant shifted much more slowly, likely due to misfolding of the T-box into a less compact structure in its apo form. In the presence of 20 mM Mg2+ and different tRNAs, all the mutants form stable T-box/tRNA complexes, but the complexes formed between G96C and tRNA constructs display a band shifting slower than the complexes formed between T166 and tRNA constructs (Fig. 5a), suggesting that the G96C/tRNAIle and G96C/tRNA78 complexes presumably adopted a distinct and less compact conformation.

a Electrophoretic mobility shift assay (EMSA) showing the binding between different tRNA constructs and T166 linker mutants. The results were consistent across all three independent experimental replicates. b Schematic representation of the different fluorescent labeling constructs of T-box-tRNA complexes for smFRET analysis. The green and blue circles represent the Cy3 and Cy5 fluorophores, respectively. c–e Representative smFRET traces (left), FRET histograms (middle) and transition density plots (right) for the respective labeling constructs in (c) in the presence of 20 mM Mg2+ and three different tRNA constructs: tRNAΔ, tRNAIle and tRNA78. Green, blue and black lines represent the Cy3 intensity, Cy5 intensity and FRET efficiency, respectively. FRET histograms were well fitted with Gaussian peaks, shown in red and blue for the low- and middle- or high-FRET, respectively. Transition density plots illustrate number of transition events per second. N denotes the total number of molecules used to generate the FRET histograms from three independent experiments (n = 3). Source data are provided as a Source Data file.
We further characterized the conformational differences of the complexes formed between these mutants and different tRNA constructs by using the T166-139Cy5/tRNA-5΄Cy3 labeling construct (Fig. 5b). In the presence of 20 mM Mg2+ and tRNA∆, all the mutants exhibit similar FRET distribution patterns as T166 wild type, indicating that the mutations at the linker region cause little effects on the first site binding. In the presence of 20 mM Mg2+ and tRNAIle or tRNA78, the G94U and ∆88-93 mutants exhibit similar FRET distribution patterns as T166 wild type (Fig. 5d, e), though the occupancy of low-FRET corresponding to the tRNA NCCA 3΄-end-unbound state slightly decreases for ∆88-93 mutant, indicating that these two mutants have a minor effect on the second site binding. By contrast, the complex of G96C/tRNAIle displayed a minor population (40%) of low-FRET (E ~ 0.3) and a major population of high-FRET (E ~ 0.72), which is similar to the FRET population for G96C/tRNA78 (Fig. 5c). These smFRET data suggest that the conformations of G96C mutants in complex with either tRNAIle or tRNA78 are similar, in other words, G96C mutant abolished the discrimination ability of tRNA 3΄-end aminoacylation. However, the G94U and ∆88-93 mutants have minor effects on the 3΄-end discrimination ability.
Given that tRNA78 binding mostly induced structural rearrangement of the linker region and stem III, we next utilized the labeling constructs of T166-107Cy3/D-3΄Cy5 to monitor the effects of these mutants on the relative motions between stem III and the bottom helix of AntiS (Fig. 6, Supplementary Fig. 7). smFRET data were collected under the same conditions as that for T166-107Cy3/D-3΄Cy5. T166-G94U-107Cy3/D-3΄Cy5 mutant exhibited a two-FRET state population centered at ~0.45 and ~0.61 in the absence of tRNA, which is slightly different from that in T166-107Cy3/D-3΄Cy5 probably due to the misfolding caused by mutations. However, T166-G94U-107Cy3/D-3΄Cy5 resembles T166 binding to different tRNA constructs in all other three conditions (Fig. 6b), suggesting that G94U has a minor effect on the recognition and discrimination of tRNA, which is consistent with previous T-box induction assay that G94U only decreases the T-box induction by 2–3 folds9. T166-G96C-107Cy3/D-3΄Cy5 displayed a dominant (70%) low-FRET state (E ~ 0.4) and a minor middle-FRET state (E ~ 0.7) in the absence of tRNA or in the presence of tRNA∆ (Fig. 6c). In the presence of tRNAIle, the low-FRET state shifted to a lower FRET value (E ~ 0.33) but remained highly populated (70%). However, addition of tRNA78 only slightly increased the occupancy of low-FRET state, suggesting that G96C mutation leads to the disruption of tertiary interactions. Intriguingly, T166-∆88-93-107Cy3/D-3΄Cy5 also resembles T166-107Cy3/D-3΄Cy5 in all conditions, but the molecules occupying low-FRET state is larger than that for T166-107Cy3/D-3΄Cy5 (60% vs 50%) (Fig. 6d).

a Schematic representation for the secondary structure of each smFRET construct. The green and blue circles represent the Cy3 and Cy5 fluorophore, respectively. b–e Representative smFRET traces (left), FRET histograms (middle) and transition density plots for each smFRET construct in (a) in four different conditions: in the absence of tRNA, in the presence of tRNAΔ, tRNAIle and tRNA78. Green, blue and black lines represent the Cy3 intensity, Cy5 intensity and FRET efficiency, respectively. FRET histograms were well fitted with Gaussian peaks, shown in red and blue for the low- and middle- or high-FRET, respectively. Transition density plots illustrate number of transition events per second. N denotes the total number of molecules used to generate the FRET histograms from three independent experiments (n = 3). Source data are provided as a Source Data file.
Additionally, we also investigated the G96C mutational effect on the structural dynamics of linker region using T166-G96C-89Cy3/D-3΄Cy5 construct (Fig. 6e). In the absence of tRNA or in the presence of tRNA∆, T166-G96C-89Cy3/D-3΄Cy5 transits frequently between a middle-FRET state and a high-FRET state (Fig. 6e, Supplementary Fig. 7d), but the population of high-FRET state (E ~ 0.77) is lower than that of the T166-89Cy3/D-3΄Cy5. Strikingly, the population of middle-FRET state remains highly populated even in the presence of tRNAIle, indicating that G96C mutation abolished the key tertiary contacts of linker region with AntiS and tRNA NCCA 3΄-end, as a result, the G96C lost the tRNA 3΄end discrimination ability. Since G96 forms a long-range base-pairing interaction with C131 in the T-box/tRNA complex structure, we then constructed a G96C-C131G mutant to investigate whether the discrimination ability could be rescued. As expected, the T166-G96C-C131G-89Cy3/D-3΄Cy5 construct mainly samples a high-FRET state in the presence of tRNAIle but transits to a major middle-FRET upon addition of tRNA78, suggesting that this rescue mutation restores the 3΄-end discrimination ability (Supplementary Fig. 8).
Collectively, these data suggest that the linker region serves a pivotal role in recognition and discrimination of tRNA NCCA 3΄end by contributing key tertiary contacts with the AntiS domain.
Co-transcriptional regulation of tRNA 3΄-end recognition by full-length T-box
As the core ileS T-box (T166) lacks the sequences to form Sequestrator and mainly adopts the AntiS conformation in the presence of free 3΄-end tRNA (tRNAIle), how the binding to tRNAs with different aminoacylation status drives conformational switching to regulate translation initiation in the full-length ileS T-box is largely unknown. According to the predicted secondary structure, transition between the AntiS and Sequetrator conformations in full-length ileS T-box riboswitch (T229) requires large-scale secondary structure rearrangement, which involves the breaking of multiple base pairs and is thus energetically unfavorable to occur (Fig. 1a). It is speculated that translational T-box dynamically interconvert in equilibrium between AntiS and Sequestrator conformations depending on the tRNA ligand concentrations4, which however hasn’t been experimentally observed.
To better understand the mechanism of translational regulation by full-length ileS T-box, we prepared the T229-139Cy5/166Cy3 and T166-139Cy5/166Cy3 labeling constructs to monitor AntiS/Sequestrator conformational switching (Fig. 7a, b). The tRNAIle-bound core T-box (T166) was used as a positive control of AntiS conformation which two-site contact is formed. From the crystal structure of core T-box/tRNA complex (AntiS conformation), the predicted distance between labeling sites is ~35.5 Å and should give a high-FRET signal. From the predicted secondary structure of full-length T-box in the Sequestrator conformation, a long stem-loop helix will separate the labeling sites further (Fig. 1a), thus resulting in smaller FRET values in T229-139Cy5/166Cy3. Consistently, T166-139Cy5/166Cy3 in 20 mM Mg2+ exhibited a broad FRET population with a major peak at E ~ 0.8 in apo form, and the high-FRET state became more prominent upon addition of tRNAIle. Intriguingly, T229-139Cy5/166Cy3 displayed a broad FRET population with a major peak at E ~ 0.55 under both conditions, suggesting that T229 adopted a distinct conformation from T166 which is marginally affected by tRNAIle binding. To substantiate this phenomenon, we also prepared the labeling constructs of T229-148Cy5/166Cy3 and T166-148Cy5/166Cy3, in which the predicted distance between labeling sites in the AntiS conformation is ~43.5 Å. Similarly, the major FRET peak value of T229-148Cy5/166Cy3 (E ~ 0.3) is much lower than that for T166-148Cy5/166Cy3 (E ~ 0.58) under both conditions, indicative of a longer distance between the labeling sites and a more extended conformation of T229. We speculated that the in vitro synthesized T229 mainly adopts the Sequestrator conformation in apo form, which presumably impairs the recognition of tRNA NCCA 3΄-end due to the high energy barrier to transit from the Sequestrator to the AntiS conformation.

a Schematic representation for the secondary structure of smFRET construct used to probe the structural dynamics of AntiS/sequestrator domain. b FRET histograms for each smFRET construct in a with different lengths in the absence or presence of tRNAIle. N denotes the total number of molecules used to generate the FRET histograms from three independent experiments (n = 3). c Schematic representation for the secondary structure of smFRET construct used in co-transcriptional and post-transcriptional assay. The green and blue circles represent the Cy3 and Cy5 fluorophore, respectively. d FRET histograms for smFRET construct in (c) with different lengths prepared by post-transcriptional manner in the presence of tRNAΔ or tRNAIle. e Schematics for the sample prepared by post-transcriptional vs co-transcriptional manner used for smFRET assay. f FRET histograms for full-length T-box/tRNA complex in (c) prepared by post-transcriptional or co-transcriptional manner in the presence of tRNAΔ, tRNAIle, or tRNA78. Source data are provided as a Source Data file.
As RNA folding generally occurs faster than transcription, nascent RNA can start to fold immediately after emerging from RNA polymerase26. During co-transcriptional folding, interactions with ligands can direct RNA to form alternative structures which can critically influence cellular RNA functions27,28,29,30,31. To assess the potential effects of cotranscriptional folding on AntiS/Sequestrator transtion, we analyze tRNA-induced post-transcriptional conformational changes of various T-box constructs at different lengths (T166, T209, T215, T229) (Fig. 7c, d). Cy3 was attached to the 5΄end of tRNAs with different 3΄-termini, and Cy5 was linked to different T-box constructs by replacing nucleotide 139U of T-box with rTPT3A followed by conjugation with NHS-modifed Cy5 dye. smFRET data were collected in the presence of tRNA∆ or tRNAIle. T166-139Cy5 samples a major low-FRET (E ~ 0.3) state in the presence of tRNA∆, whereas a middle-FRET (E ~ 0.6) state emerged and highly populated upon addition of tRNAIle, which presumably corresponds to the tRNA NCCA 3΄-end-bound state. As the length of T-box riboswitch increases, the FRET population in the presence of tRNA∆ mostly resembles that in T166, however, in the presence of tRNAIle, the FRET population became more broader and the middle-FRET state became less prominent, especially for T229 (Fig. 7d), suggesting that as the transcript lengths increase, the post-transcriptionally synthesized T-box is less sensitive in response to the recognition of tRNA NCCA 3΄-end, in other words, the populations for AntiS conformation become less.
To understand whether the middle-FRET population could be affected by tRNA binding during the transcription of T-box, we prepared the co-transcriptionally synthesized T229/tRNA complexes by adding Cy5-modified rTPT3TP, Cy3-labeled tRNA∆, tRNAIle or tRNA78 into the transcription buffer (Fig. 7e), mimicking the cotranscriptional folding and tRNA binding with the T-box riboswitch. As expected, the FRET populations in the presence of tRNA∆ resemble that of T229 prepared posttranscriptionally, whereas the FRET histogram became much narrower and the middle-FRET (E ~ 0.6) state became more prominent for T229-139Cy5/5΄Cy3-tRNAIle prepared co-transcriptionally than posttranscriptionally (Fig. 7f), suggesting that the folding of T229 is presumably regulated by cotranscriptional recognition and binding of tRNA NCCA 3΄-end. Interestingly, a major high-FRET (E ~ 0.8) population emerged for T229 in the presence of tRNA78 under both conditions, which is quite different from that in the presence of tRNA∆ and tRNAIle. Together with the smFRET data on T166 (Fig. 4), we speculate that the extra terminus moiety of tRNA78 destabilize the tertiary interactions among AntiS, linker and NCCA end and nucleates a series of conformational changes in stem III and linker region, which finally leads to the formation of alternative sequestrator conformation and brings the 5΄Cy3 of tRNA close to the 139Cy5 in T-box. Collectively, our data elucidated the tRNA aminoacylation discrimination mechanism by ileS T-box riboswitch at the single-molecule level and provided important insights into the co-transcriptional regulation mechanism.
Discussion
T-box riboswitches are unique riboregulators which respond to two chemical inputs including tRNAs and their attached amino acids. Exploring the structure and conformational dynamics of T-box riboswitches provides valuable insights into not only the RNA-centered regulatory mechanisms but also RNA-targeted therapeutics. Despite the recently solved multiple high-resolution structures of T-box complexes and relevant smFRET-based dynamic studies9,10,11, the dynamic mechanisms underlying tRNA recognition and amino acid sensing by a full-length T-box riboswitch are not yet clearly understood, partly due to the lack of efficient internal fluorophore labeling methods for large RNAs. Empowered with our recently developed UBP-based site-specific fluorescent labeling methods for RNA molecules17,22, we here have mapped in-detail the local and global structural dynamics of a full-length translational ileS T-box riboswitch in response to tRNA with different 3΄-termini by smFRET and SAXS, and have depicted a picture on how tRNA-dependent conformational dynamics and cotranscriptional folding coordinately regulate its translation initiation (Fig. 8).

The decoding domain comprised of stem I, II and IIA/B is mainly responsible for recruiting specific tRNAIle, including charged and uncharged tRNAs. When the aminoacylation sensing module was transcribed from RNA polymerase, the AntiS domain folds into preorganized conformation for efficient tRNA NCCA end recognition but the linker region is conformationally dynamic and flexible. If the tRNA is uncharged, the NCCA end will establish stable interactions with AntiS and the linker region stably docked at its preferred conformation. If the tRNA is charged, NCCA end still base pairs with AntiS but the linker region transit frequently from the docked to undocked conformation and in turn leads to the away of stem III from AntiS. Without the stabilization by coaxially stacked stem III and the tertiary contacts contributed by linker region, tRNA NCCA end is easier to dissociate from AntiS. As the SD sequence is transcribed, T-box will probably rearrange the secondary structure to form sequestrator conformation due to the destabilization of AntiS and finally inhibit the translation initiation.
Our study highlights the advantages of NaM-TPT3 UBP-based site-specific labeling strategies in smFRET analysis of long RNAs. Site-specific incorporation of extrinsic fluorophores into long RNAs is prerequisite for smFRET measurements, which, however, remains challenging for traditional labeling methods, including chemical synthesis and splint-directed enzymatic ligation32,33. Recently, there has been a surge of interest in the dynamic mechanisms of both transcriptional and translational T-box riboswitches6,17,18,19,21. For examples, intermolecular smFRET measurements have been performed for the core regions of B. subtilis glyQS T-box and M. tub ileS T-box facilitated with 5′-end Cy5 labeling of tRNAs and complementary DNA oligonucleotide carrying Cy3 at its 3′-end which is annealed to the T-box RNAs6,19,21. Though such studies provide kinetic frameworks to understand how specific RNA elements dictate the high binding affinity and specificity and support a two-step binding model, the specific local and global structural rearrangements triggered by tRNA binding is lacking due to the sparsity of labeling sites used. In this study, we have achieved site-specific internal fluorescent labeling across a full-length translational T-box riboswitch and its subconstructs at multiple sites, allowing for a comprehensive mapping of the intra- and interdomain conformational dynamics across RNA variants of different lengths. Since all the labeling sites are located in the single-stranded regions and not involved in any tertiary interactions, they should have a minor effect on global folding and the tRNA binding affinity of T-box (Supplementary Fig. 3). Facilitated with the UBP-based strategy, one noticeable finding in our work is that the detailed structural dynamics of discriminator domain induced by charged tRNA mimic was captured. In addition to post-transcriptional labeling, Cy5-labeled TPT3s have also been used to prepare the co-transcriptionally tRNA-bound T-box in this work (Fig. 7), providing valuable toolbox to shed lights on the cotranscriptional regulation mechanism. It is expected that the UBP-based labeling strategy will have broader applications in the preparation of labeled long RNAs within native elongation complexes for co-transcriptional studies.
Intriguingly, we found that the molecular mechanism underpinning cognate tRNA decoding by the M. tub ileS T-box is different from that by the N. far ileS T-box, whose secondary structures are highly homologous except that N. far stem II is 11 nts longer. Riboswitches might recognize their cognate ligands through lock and key, induced-fit or conformational selection mechanisms, which can be regulated by Mg2+ due to its role in promoting RNA folding34. Our recent smFRET analysis on the decoding domain of N. far ileS T-box supports an induced-fit recognition mechanism that Mg2+-induced pre-docking of stems I and II is a prerequisite for efficient initial tRNA contact to the specifier, but not sufficient to induce the proper docking of stems I and II into the tRNA-bound conformation, which, however, can be only achieved by subsequent cognate tRNA binding17. By contrast, the decoding domain of M. tub ileS T-box exhibits large conformational heterogeneity in the absence of tRNA, which samples both the open (undocked) and closed (docked) conformations, and the first-step specifier-anticodon binding which is independent on the tRNA 3′-end aminoacylation status enhances the population of the closed conformation. Thus, the decoding domain of M. tub ileS T-box recognizes its cognate tRNA in a conformational selection manner. Similarly, the inter-stem motions between the linker region and AntiS, between stem III and AntiS which reflect the structural dynamics of the discriminator domain all sample multiple conformations in the absence of tRNA, while the first-step binding by tRNA∆ only marginally affects their conformational ensembles, the two-site binding by cognate tRNAIle increases the population of the docked conformation for all the labeling constructs (Fig. 4). These results support that the discriminator domain establishes the second-step binding also through the conformational selection mechanism, which may enable a faster and more efficient tRNA recognition and translation regulation since the bound-like conformations already exist in apo form and no additional energy penalty to drive conformational change as that in the induced-fit mechanism. As the M. tub stem II is only 11 nts shorter than the N. far stem II, we speculate that shorter stem II may have a weaker stacking interaction with stem IIA/B and the orientation between stem I and stem IIA/B is relatively flexible, thus resulting in a dynamic conformation equilibrium in apo form. tRNA binding shifts the equilibrium toward docked conformation. However, this hypothesis requires further experiment investigation.
Our results showed that tRNA binding and M. tub T-box folding are directly coupled, the first-step tRNA anticodon-specifier binding enhances not only the folding of decoding domain, but promotes the proper arrangement of stem III and AntiS in the discriminator domain, which are essential for the second-step tRNA 3΄-terminus recognition (Fig. 4). As the 5΄ decoding domain is synthesized before the 3΄ discriminator domain, it is expected to fold independently, which samples both the open and closed conformations at high Mg2+ alone, thus its folding is less dependent on the tRNA. These observations are consistent with the sequential two-step binding model, which emphasizes the significance of the first anticodon-specifier binding in sensing the 3΄-end aminoacylation status. It is thus not unexpected that the in vitro synthesized full-length T-box presumably adopts a misfolded conformation showing smeared band in the native gel in the absence of tRNA (Fig. 2a). Our work also highlights G96 in the linker region as a key nucleotide to sense the aminoacylation status of tRNA. Previous structural studies demonstrated that the conserved GAG sequence in the linker establishes specific tertiary interactions with the uncharged tRNA NCCA end9. The results in this work show that the G94U and ∆88-93 mutants cause marginal effects to the 3΄-end discrimination, but the G96C mutant completely abolishes its 3΄-end discrimination capacity which exhibits enhanced conformational flexibility.
Transcription and translation are intimately coupled in bacteria, in which the ribosome is capable to initiate translation once the SD sequence and start codon have been transcribed by RNA polymerase35,36. Therefore, translational riboswitches would start to fulfill their functions while they are being synthesized as those transcriptional riboswitches37,38. Our smFRET data on ileS T-box riboswitch constructs with different lengths suggested that the folding and tRNA 3΄-terminus recognition of a translational T-box riboswitch is cotranscriptionally regulated (Fig. 7). Our results showed that the tRNA NCCA 3΄-end efficiently binds T-box only after the AntiS domain is transcribed but before the transcription of SD sequence (T166-T215). The fully synthesized ileS T-box riboswitch (T229) predominantly adopted a sequestrator conformation in apo form, which blocks the recognition of tRNA 3΄-terminus. Another translational thiamine pyrophosphate riboswitch has also been revealed to be kinetically controlled and the transition between Sequestrator and AntiS conformation becomes more difficult when the full-length riboswitch is transcribed37, indicating that kinetic control mechanisms are widely spread in both transcriptional and translational T-box riboswitches.
Taken together, we depict a picture on how tRNA-dependent conformational dynamics and folding coordinately regulate the translation initiation by the M. tub full-length translational T-box riboswitch (Fig. 8). When the decoding domain emerges from the RNA polymerase, it begins to fold and samples a heterogenous conformational ensemble. The closed conformation can be strongly stablized upon binding to charged or uncharged cognate tRNA. As the aminoacylation sensing module is transcribed from RNA polymerase, the AntiS domain folds into preorganized conformation for efficient tRNA NCCA end recognition, but the linker region is conformationally dynamic and flexible. If the tRNA is uncharged, the NCCA end will establish stable tertiary interactions with AntiS and the linker region and promote its preferred docked conformation, which is not disturbed even when the SD sequence is transcribed and thus allows the binding of ribosome to initiate translation. If the tRNA is charged, the NCCA end still base pairs with AntiS, but the linker region transits frequently from the docked to undocked conformation, which in turn leads to separation of stem III from AntiS. Without the stabilization by coaxially stacked stem III and the tertiary contacts contributed by linker region, tRNA NCCA end is easier to dissociate from AntiS (Fig. 2d). As the SD sequence is transcribed, the T-box in the undocked conformation rearrange to form sequestrator conformation which is more thermodynamically stable than the AntiS, finally resulting in inhibition of the translation initiation.
Methods
RNA sample preparation
The wild-type T-box and its mutants, tRNA constructs with different 3΄-end termini, were generated as follows. Plasmids encoding an upstream T7 promoter and full-length T-box or tRNA were gene-synthesized and sequenced by Wuxi Qinglan Biotechnology Inc, Wuxi, China. All the other mutants were generated using the Transgen’s Fast Mutagenesis System. The double-stranded DNA fragment templates for in vitro RNA production were generated by PCR using an upstream forward primer targeting the plasmids and a downstream reverse primer specific to respective cDNAs. The RNAs were transcribed in vitro using T7 RNA polymerase and purified by preparative, non-denaturing or denaturing polyacrylamide gel electrophoresis, the target RNA bands were cut and passively eluted from gel slices into buffer containing 0.3 M sodium acetate, 1 mM EDTA, pH 5.2 overnight at 4 °C. The RNAs were further passed through the size exclusion chromatography column to the final buffer conditions for further use. The sequences for all the constructs and the mutants are listed in Supplementary Table 2. The primer sequences used in this study are summarized in Supplementary Table 3.
Electrophoretic mobility shift assay (EMSA)
RNA samples used for EMSA were prepared as described above and stored in a buffer containing 50 mM HEPES (pH 7.5), 100 mM KCl. Dye-labeled or unlabeled T-box (2.4 μM) was incubated with varying concentrations of tRNA (0.24−7.2 μM) supplemented with 20 mM Mg2+ in 37 °C for 20 min. Then the mixtures were loaded on 8% native polyacrylamide gel containing 10 mM Mg2+ and the gels were run at room temperature for ~60 min. The gels were then stained by Gelsafe dye and imaged using gel imager. EMSA for different labeling constructs were conducted on independent biological triplicates.
Small angle X-ray scattering
All the parameters for SAXS data collection are summarized in Supplementary Table 5. Briefly, SAXS measurements were carried out at room temperature at the beamline BL19U2 of the National Center for Protein Science Shanghai (NCPSS) and Shanghai Synchrotron Radiation Facility (SSRF). The scattered X-ray photons were recorded with a PILATUS 100k detector (Dectris) at BL19U2. The setups were adjusted to achieve scattering q values of 0.009 < q < 0.415 Å−1, where q = (4π/λ)sin(θ), and 2θ is the scattering angle. Thirty-two-dimensional images were recorded and reduced for each buffer or sample and no radiation damage was observed. Scattering profiles of the RNAs were calculated by subtracting the background buffer contribution from the sample-buffer profile using the program PRIMUS3.2 following standard procedures39. Guinier analysis was performed to calculate the forward scattering intensity I(0) and the radius of gyration (Rg), which were also estimated from the scattering profile with a broader q range of 0.006–0.30 Å−1 using the indirect Fourier transform method implemented in the program GNOM4.640, along with the PDDF, p(r), and the maximum dimension of the RNA, Dmax.
5΄-end fluorophore labeling of RNAs
5΄-end fluorophore labeling of RNAs was performed by following the protocols developed in previous studies with a minor improvement20,41. Briefly, purified RNA was precipitated by ethanol and resuspended in 0.1 M MES (pH 6.0). This was followed by the addition of N-(3-Dimethylaminopropyl)-N0-ethylcarbodiimide hydrochloride (EDC) along with ethylene diamine and 0.1 M imidazole solution (pH 6.0). The mix was incubated at 37 °C for 3 h and followed by ethanol precipitation for at least three times to remove residual EDC. Then the EDC-treated tRNA was resuspended in 0.1 M NaHCO3 (pH 8.0) and mixed with 30-fold NHS-Cy3 dye, incubated at room temperature for 4–6 h. Then Cy3-labeled tRNA was precipitated by ethanol and resuspended in buffer containing 50 mM HEPES (pH 7.5) and 100 mM KCl, stored at −80 °C for further smFRET experiments.
Site-specific internal labeling of T-box RNAs using UBP system
The deoxyribonucleotide phosphoramidites (dTPT3-CEP and dNaM-CEP, for DNA primer synthesis), the triphosphorylated deoxyribonucleotides (dTPT3TP and dNaMTP, for PCR) and ribonucleotides (rTPT3ATP, rTPT3COTP, rNaMCOTP for transcription), were custom-synthesized as described17,42. Reversed primers containing unnatural nucleotides were synthesized and used for overlapping PCR to introduce the dNaMTP and dTPT3TP into the DNA template. Then the PCR products containing dNaM and dTPT3 at specific sites were used as the DNA templates for in vitro transcription and purified as described above. The purified T-box products modified with rNaMCO and rTPT3A at specific sites were precipitated with cold ethanol (2.5 volumes) in the presence of sodium acetate (0.3 M) at –80 °C for at least 0.5 h. After centrifuging at 4 °C, the ethanol was removed and the pellet was washed with ethanol (75%) three times, then the pellet was dried for 10 min. Finally, the product was resuspended in DEPC-treated water and subjected to Cy5 fluorescent labeling. RNAs (0.3 mM, 10.5 μL) were mixed with 33.5 μL 1.5 × Click chemistry buffer, 5 μL Sulfo-cyanine5 azide (20 mM in DMSO), 1 μL of sodium ascorbate (50 mM). The resulting mixture was incubated at room temperature overnight. Then the labeled RNAs were precipitated by ethanol as described above and resuspended in 0.1 M NaHCO3 (pH 8.0), then the Cy5 labeled T-boxs were mixed with 5 μL NHS-Cy3 (20 mM in DMSO) and incubated at room temperature for 4–6 h. Then Cy5-Cy3 double-labeled T-box RNAs were precipitated by ethanol as described above and resuspended in buffer containing 50 mM HEPES (pH 7.4) and 100 mM KCl, stored at −80 °C for further smFRET experiments. The labeling efficiency was calculated by measuring the absorption of RNAs at 260 nm, 546 nm and 650 nm, respectively. The labeling efficiencies for Cy3 and Cy5 are ~70% and ~90%, respectively.
Preparation of T229/tRNA complex in co-transcription manner
First, we prepared the Cy5-labeled rTPT3TP for in vitro co-transcription. rTPT3ATP was mixed with 30-fold NHS-Cy5 dye and incubated at room temperature for 4–6 h. Then the mixture was subjected to HPLC to remove free Cy5 dye, and the purified TPT3A-Cy5 was lyophilized and resuspended in buffer containing 50 mM HEPES (pH 7.4) and 100 mM KCl. For the preparation of cotranscriptional T229/tRNA complex, DNA templates prepared by PCR, TPT3A-Cy5, NTPs, 5’-biotin modified DNA linker, and T7 RNA polymerase was mixed and incubated at 37 °C for 0.5 h. Then the mixture was directly used for smFRET experiments without further purification.
Single-molecule FRET experiments
For single-molecule experiments, 450 nM labeled T-boxes were mixed with 300 nM biotin-modified DNA and different tRNA constructs in 50 mM HEPES (pH 7.5), 100 mM KCl by incubating the mixture at 95 °C for 2 min, then fast cooling on the ice and add Mg2+ to the final concentration of 20 mM, and finally equilibrated at 37 °C for 20 min. Samples were diluted 1200 times in the buffer containing 50 mM HEPES, 100 mM KCl and 20 mM Mg2+ and immobilized on the slides by biotin-streptavidin interactions. Then the imaging buffer containing 3 mg/mL glucose, 100 μg/mL glucose oxidase, 40 μg/mL catalase, 1 mM cyclooctatetraene (COT), 1 mM 4-nitrobenzylalcohol (NBA) and 1.5 mM 6-hydroxy-2,5,7,8-tetramethyl-chromane-2-carboxylic acid (Trolox) was flowed into the channel. smFRET experiments were conducted at 25 °C by using a home-built objective-type TIRF microscope. The time resolution of each movie was 100 ms/frame and we collected 800 frames for each movie. In each field of view, about 50–60% molecules were selected for further analysis. 3–4 movies were collected for each condition. All of the experiments were repeated at least three times, from which experimental errors were estimated.
Single-molecule FRET data analysis
smFRET data were analyzed by the custom-made software program. Single-molecule movies were collected by Cell Vision software (Beijing Coolight Technology) and then analyzed by a custom-made software program developed as an ImageJ 1.43 u plugin (http://rsb.info.nih.gov/ij). Fluorescence spots were fitted by a 2D Gaussian function within a 9-pixel by 9-pixel area, matching the donor and acceptor spots using a variant of the Hough transform43. The background-subtracted total volume of the 2-D Gaussian peak was used as raw fluorescence intensity I. FRET trajectories containing donor and acceptor and displayed anticorrelation behaviors were picked and analyzed. Histograms of FRET efficiency for T-boxes in different conditions were built by using the frames of all traces before photobleaching (more than 400 molecules, on average >250 frames per molecule). The histograms were divided by the total number of FRET frames and the frequencies were labeled as ‘probability’ in the y-axis. smFRET traces were further analyzed by the two-state Hidden Markov Model-based software to extract the kinetics information44. The dwell times of different FRET states were calculated from idealized traces predicted by the two-state Hidden Markov Model and the dwell time distribution was fitted to a single or two-exponential function to calculate the dwell times and transition rates (Supplementary Table 4). TDPs depict the fraction of counts transition from a specific initial FRET state to a final FRET state among total FRET counts and show the frequency of transitions between specific FRET states. TODPs show the fraction of molecules that exhibit a specific transition at lease once45. Both of TDPs and TODPs were constructed from the idealized dynamic smFRET traces determined by the Hidden Markov Model.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The data supporting the findings of this study are available from the corresponding authors upon request. The study used PDB ID: 6UFG. Source data for the figures and Supplementary Figs. are provided as a Source Data file. Source data are provided with this paper.
Code availability
Custom scripts used to analyze the single-molecule FRET data are available upon reasonable request.
References
Zhang, J. & Ferre-D’Amare, A. R. Structure and mechanism of the T-box riboswitches. Wiley Interdiscip. Rev. RNA 6, 419–433 (2015).
Vitreschak, A. G., Mironov, A. A., Lyubetsky, V. A. & Gelfand, M. S. Comparative genomic analysis of T-box regulatory systems in bacteria. RNA 14, 717–735 (2008).
Gutierrez-Preciado, A., Henkin, T. M., Grundy, F. J., Yanofsky, C. & Merino, E. Biochemical features and functional implications of the RNA-based T-box regulatory mechanism. Microbiol. Mol. Biol. Rev. 73, 36–61 (2009).
Suddala, K. C. & Zhang, J. An evolving tale of two interacting RNAs-themes and variations of the T-box riboswitch mechanism. IUBMB Life 71, 1167–1180 (2019).
Sherwood, A. V., Grundy, F. J. & Henkin, T. M. T box riboswitches in Actinobacteria: translational regulation via novel tRNA interactions. Proc. Natl Acad. Sci. USA 112, 1113–1118 (2015).
Sherwood, A. V., Frandsen, J. K., Grundy, F. J. & Henkin, T. M. New tRNA contacts facilitate ligand binding in a Mycobacterium smegmatis T box riboswitch. Proc. Natl Acad. Sci. USA 115, 3894–3899 (2018).
Grundy, F. J. & Henkin, T. M. tRNA as a positive regulator of transcription antitermination in B. subtilis. Cell 74, 475–482 (1993).
Grundy, F. J., Winkler, W. C. & Henkin, T. M. tRNA-mediated transcription antitermination in vitro: codon-anticodon pairing independent of the ribosome. Proc. Natl Acad. Sci. USA 99, 11121–11126 (2002).
Battaglia, R. A., Grigg, J. C. & Ke, A. Structural basis for tRNA decoding and aminoacylation sensing by T-box riboregulators. Nat. Struct. Mol. Biol. 26, 1106–1113 (2019).
Li, S. et al. Structural basis of amino acid surveillance by higher-order tRNA-mRNA interactions. Nat. Struct. Mol. Biol. 26, 1094–1105 (2019).
Suddala, K. C. & Zhang, J. High-affinity recognition of specific tRNAs by an mRNA anticodon-binding groove. Nat. Struct. Mol. Biol. 26, 1114–1122 (2019).
Saad, N. Y. et al. Two-codon T-box riboswitch binding two tRNAs. Proc. Natl Acad. Sci. USA 110, 12756–12761 (2013).
Apostolidi, M. et al. A glyS T-box riboswitch with species-specific structural features responding to both proteinogenic and nonproteinogenic tRNAGly isoacceptors. RNA 21, 1790–1806 (2015).
Frohlich, K. M. et al. Discovery of small-molecule antibiotics against a unique tRNA-mediated regulation of transcription in gram-positive bacteria. ChemMedChem 14, 758–769 (2019).
Zhang, J. & Ferre-D’Amare, A. R. Co-crystal structure of a T-box riboswitch stem I domain in complex with its cognate tRNA. Nature 500, 363–366 (2013).
Grigg, J. C. & Ke, A. Structural determinants for geometry and information decoding of tRNA by T box leader RNA. Structure 21, 2025–2032 (2013).
Niu, X. et al. Structural and dynamic mechanisms for coupled folding and tRNA recognition of a translational T-box riboswitch. Nat. Commun. 14, 7394 (2023).
Suddala, K. C. et al. Direct observation of tRNA-chaperoned folding of a dynamic mRNA ensemble. Nat. Commun. 14, 5438 (2023).
Suddala, K. C. et al. Hierarchical mechanism of amino acid sensing by the T-box riboswitch. Nat. Commun. 9, 1896 (2018).
Zhang, J. et al. Specific structural elements of the T-box riboswitch drive the two-step binding of the tRNA ligand. eLife 7, e39518 (2018).
Campos-Chavez, E. et al. Translational T-box riboswitches bind tRNA by modulating conformational flexibility. Nat. Commun. 15, 6592 (2024).
Niu, X. et al. Pseudoknot length modulates the folding, conformational dynamics, and robustness of Xrn1 resistance of flaviviral xrRNAs. Nat. Commun. 12, 6417 (2021).
Putzer, H., Condon, C., Brechemier-Baey, D., Brito, R. & Grunberg-Manago, M. Transfer RNA-mediated antitermination in vitro. Nucleic Acids Res. 30, 3026–3033 (2002).
Ohtsuki, T., Yamamoto, H., Doi, Y. & Sisido, M. Use of EF-Tu mutants for determining and improving aminoacylation efficiency and for purifying aminoacyl tRNAs with non-natural amino acids. J. Biochem. 148, 239–246 (2010).
Zhang, J. & Ferre-D’Amare, A. R. Direct evaluation of tRNA aminoacylation status by the T-box riboswitch using tRNA-mRNA stacking and steric readout. Mol. Cell 55, 148–155 (2014).
Pan, T. & Sosnick, T. RNA folding during transcription. Annu. Rev. Biophys. Biomol. Struct. 35, 161–175 (2006).
Garst, A. D., Edwards, A. L. & Batey, R. T. Riboswitches: structures and mechanisms. Cold Spring Harb. Perspect. Biol. 3, a003533 (2011).
Landgraf, T., Volklein, A. E., Furtig, B. & Schwalbe, H. The cotranscriptional folding landscape for two cyclic di-nucleotide-sensing riboswitches with highly homologous aptamer domains acting either as ON- or OFF-switches. Nucleic Acids Res 50, 6639–6655 (2022).
Widom, J. R. et al. Ligand modulates cross-coupling between riboswitch folding and transcriptional pausing. Mol. Cell 72, 541–552 e6 (2018).
Helmling, C. et al. NMR structural profiling of transcriptional intermediates reveals riboswitch regulation by metastable RNA conformations. J. Am. Chem. Soc. 139, 2647–2656 (2017).
Chauvier, A. & Walter, N. G. Regulation of bacterial gene expression by non-coding RNA: it is all about time!. Cell Chem. Biol. 31, 71–85 (2024).
Caruthers, M. H. A brief review of DNA and RNA chemical synthesis. Biochem. Soc. Trans. 39, 575–580 (2011).
Liu, Y., Sousa, R. & Wang, Y. X. Specific labeling: an effective tool to explore the RNA world. Bioessays 38, 192–200 (2016).
Suddala, K. C., Wang, J., Hou, Q. & Walter, N. G. Mg(2+) shifts ligand-mediated folding of a riboswitch from induced-fit to conformational selection. J. Am. Chem. Soc. 137, 14075–14083 (2015).
Castro-Roa, D. & Zenkin, N. In vitro experimental system for analysis of transcription-translation coupling. Nucleic Acids Res. 40, e45 (2012).
Kohler, R., Mooney, R. A., Mills, D. J., Landick, R. & Cramer, P. Architecture of a transcribing-translating expressome. Science 356, 194–197 (2017).
Uhm, H., Kang, W., Ha, K. S., Kang, C. & Hohng, S. Single-molecule FRET studies on the cotranscriptional folding of a thiamine pyrophosphate riboswitch. Proc. Natl Acad. Sci. USA 115, 331–336 (2018).
Chatterjee, S., Chauvier, A., Dandpat, S. S., Artsimovitch, I. & Walter, N. G. A translational riboswitch coordinates nascent transcription-translation coupling. Proc. Natl Acad. Sci. USA 118, e2023426118 (2021).
Konarev, P. V., Volkov, V. V., Sokolova, A. V., Koch, M. H. J. & Svergun, D. I. PRIMUS - a Windows-PC based system for small-angle scattering data analysis. J. Appl Cryst. 36, 1277–1282 (2003).
Svergun, D. I. Determination of the regularization parameter in indirect-transform methods using perceptual criteria. J. Appl. Cryst. 25, 495–503 (1992).
Rinaldi, A. J., Suddala, K. C. & Walter, N. G. Native purification and labeling of RNA for single molecule fluorescence studies. Methods Mol. Biol. 1240, 63–95 (2015).
Wang, Y. et al. Posttranscriptional site-directed spin labeling of large RNAs with an unnatural base pair system under non-denaturing conditions. Chem. Sci. 11, 9655–9664 (2020).
Yang, M. et al. The conformational dynamics of Cas9 governing DNA cleavage are revealed by single-molecule FRET. Cell Rep. 22, 372–382 (2018).
McKinney, S. A., Joo, C. & Ha, T. Analysis of single-molecule FRET trajectories using hidden Markov modeling. Biophys. J. 91, 1941–1951 (2006).
Blanco, M. & Walter, N. G. Analysis of complex single-molecule FRET time trajectories. Methods Enzymol. 472, 153–178 (2010).
Zuker, M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 31, 3406–3415 (2003).
Acknowledgements
This work was supported by grants from the National Key R&D Program of China (2024YFA0916700 to C.C. and 2021YFA1301500 to X.F.), the Strategic Priority Research Program of the Chinese Academy of Science (No. XDB0570000 to X.F.), the National Natural Science Foundation of China (No. U1832215 to X.F., No. 22425701, 22277063 and 22061160466 to C.C.), the Beijing Frontier Research Center for Biological Structure to C.C., China Postdoctoral Science Foundation (No. 2022M711845) and Shuimu Tsinghua Scholar Program to X.N.
Author information
Authors and Affiliations
Contributions
X.F. conceived and designed the project. C.C. and X.F. supervised the single-molecule FRET experiments. X.N. and S.C. performed the single-molecule FRET experiments. X.N. analyzed the smFRET data. S.C. performed the SAXS experiments and analyzed the data. X.N., C.C., and X.F. wrote the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Nils Walter, who co-reviewed with Adrien Chauvier, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Niu, X., Cai, S., Wang, J. et al. tRNA-dependent conformational dynamics and folding coordinate translational regulation by a full-length T-box riboswitch. Nat Commun 16, 10438 (2025). https://doi.org/10.1038/s41467-025-65388-6
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-65388-6
