
Abstract
Dry reforming of methane is an effective approach to convert two major greenhouse gases, methane and carbon dioxide, into high-value syngas, used as a feedstock for bulk and fine chemical synthesis. However, catalyst deactivation and carbon deposition under harsh conditions hinder its industrialization process. Herein, we present a Ce-modified and Ni-exsolved perovskite catalyst, 0.2Ce-La0.97Ni0.4Cr0.6O3, for achieving highly efficient and robust CH4-CO2 reforming with CH4 and CO2 conversions of 87.4% and 92.9% at 800 °C, respectively. Moreover, this unique catalyst exhibits remarkable stability, maintaining its superior activity over 800 h. Characterization and density functional theory reveal that two Ce species are present: surface oxygen vacancy-moderate CeO2-x (Cesurf) and bulk lattice Ce (Cebulk). These play a specific role in methane dry reforming, where the Cesurf promotes CO2 adsorption and hinders carbon deposition, while Cebulk induces lattice strain and Ni exsolution, key factors contributing to the high activity and stability.
Similar content being viewed by others

Introduction
Dry reforming of methane (DRM) is a critical process for simultaneously transforming two greenhouse gases, methane (CH4) and carbon dioxide (CO2), into syngas, a mixture of H2 and CO with a near-equimolar H2/CO ratio1,2. The generated syngas is a crucial feedstock for producing chemicals such as methanol and C2+ via Fischer-Tropsch synthesis, and it can be further processed into high-value chemicals such as methanol and dimethyl ether3,4. Consequently, DRM enables both the efficient use of C1 resources and the reduction of carbon emissions. However, the DRM process is constrained by the stability and energy consumption required to break the strong C-H (439.3 kJ mol−1) and C=O (750 kJ mol−1) bonds, posing significant operational challenges5.
Catalysts play a pivotal role in lowering reforming temperature and energy consumption in DRM. Until now, significant progress has been made with non-noble metal-supported catalysts, such as Ni/MgO6, Ni/MgFeAlO47, Ni/LaZrO28, Co-La/Mg-Al2O39, Ni@Co/CeO210, Co-Mg/TiO2-Al2O311, Ni3Fe1/Al2O312, 0.3%Fe-Ni/Al2O313. Nevertheless, to achieve high activity while simultaneously suppressing catalyst deactivation over the above-mentioned Fe-, Co-, and Ni-based catalysts remains a considerable challenge. Moreover, the occurrence of side reactions, including carbon deposition and reverse water-gas shift (rWGS), also lowers the H atom utilization efficiency and the overall efficiency of the DRM process.
Perovskite oxides, with their thermally stable crystal structures, have been extensively studied for catalytic applications at elevated temperatures. Particularly, exsolved perovskite materials, with B-site doping, exhibit excellent oxygen transport properties, with highly dispersed and reactive metal nanoparticles anchored on their surfaces that resist sintering14. Doping of B-sites in ABO3-type perovskites with transition metals that exhibit low segregation energies promotes these metals to exsolve from the bulk lattice and anchor on the perovskite surface under reduction conditions15,16. This exsolution process generates reactive metal nanoparticles with strong metal-support interactions, which is to the benefit of sustaining catalytic activity17,18,19. Moreover, doping-induced lattice distortion would enhance the oxygen mobility of perovskite materials. This promotes the generation of more oxygen vacancies, thereby hindering carbon deposition during DRM due to the promotion of O2−lattice transformation and the generation of surface O* species from CO2 dissociation20.
The A-site cations in perovskites, such as calcium21, strontium22, barium23, lanthanum24,25, cerium26, praseodymium27,28, neodymium29, and others, typically do not directly participate in catalysis, but influence oxygen vacancy content and modulate the electronic properties of the B-site cations through charge balance control. Research has primarily focused on perovskites doped with B-site elements, including nickel30, cobalt31, and iron32, as well as noble metals like ruthenium33, rhodium34, platinum35, and iridium36. Nevertheless, further investigation is required to elucidate the regulation of particle size and density distribution of B-site transition metal exsolved nanoparticles, as well as the impact of A-site doping on the catalytic activity.
In this study, we developed a series of Ce-modified and Ni-doped La0.97Ni0.4Cr0.6O3 perovskite catalysts, with the Ni exsolved (Niex) La0.97Ce0.03Ni0.4Cr0.6O3 catalysts obtained after hydrogen reduction. Cerium oxides (CeO2-x), due to their Ce3+/Ce4+ redox pair, are a potential oxygen storage-release material for DRM27,37,38. Additionally, as far as we are aware, few studies have explored the impact of Ce modification on the exsolution and the catalytic properties of perovskites. Our study revealed that the Ce species exist in two distinctive forms within the perovskite catalyst: A-site lattice Ce, which induces lattice distortion and promotes Ni exsolution, and surface oxygen vacancy (Ov)-abundant Ce species (CeO2-x), which provides oxygen storage and a release cycle microenvironment, which inhibits carbon accumulation and promotes CO2 activation. Here, we demonstrate the synergistic effect of these two forms of Ce in enhancing DRM activity and stability.
Results and discussion
Catalyst performance
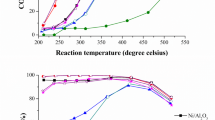
The DRM performance of Niex-La0.97Ce0.03Ni0.4Cr0.6O3 catalyst with varying Ce doping ratios is shown in Fig. 1a. With increasing Ce ratio from 0 to 0.2, CH4 and CO2 conversion rates improved to 91.1% and 93.3%, respectively. However, excessive Ce (R-0.4Ce) resulted in a sharp decline in the respective conversions to 54.0% and 67.2%. This trend reflects a balance between optimal Ce doping and excessive coverage of Ni-active sites by CeO2-x. H2 and CO selectivity, as well as the H2/CO ratio also exhibit a similar volcanic trend with increasing Ce doping (Fig. 1b). Reactions at different temperatures (650–850 °C) over R-0.2Ce demonstrated a significant increase of H2 (83.8% → 96.7%), CO (95.6% → 98.8%) selectivity, and the H2/CO ratio (0.85 → 0.95) from 650 to 750 °C, while the increase gradually slows down at a temperature > 750 °C (Figs. 1c and S1–S3). This phenomenon can be explained by the occurrence of rWGS (H2 + CO2 → CO + H2O, ΔH = + 41 kJ/mol) equilibrium, and Ce doping modulates its dynamic equilibrium as well as acts to increase the H utilization efficiency (Figs. S4 and S5). The CH4 conversion of 95.2% and CO2 conversion of 98.8% with a H2/CO ratio of 1.0 were achieved at 850 °C, a relatively greater performance in comparison to previously reported examples.

a CH4, CO2 conversion rates over various samples with different Ce doping ratios in the DRM reaction. b H2, CO selectivity and H2/CO ratio. c The conversion rates of CH4 and CO2, selectivity of H2 and CO, and H2/CO ratio at different temperatures for sample 0.2Ce. d The conversion rates of CH4 and CO2, selectivity of H2 and CO, and H2/CO value at different WHSV for sample 0.2Ce at 750 °C. e Stability test of R-0.2Ce and R-0Ce for 800 h and 120 h. Evaluated conditions: 800/750 °C, CH4: CO2: N2 = 1: 1: 1, WHSV = 30,000 mL g−1 h−1. Arrhenius plot in terms of the rate of product of f H2 and g CO catalyzed by the R-0Ce and R-0.2Ce catalysts. 25 mg of each catalyst was loaded into the quartz tube. h Comparison of the DRM performance with literature (Table S1).
The influence of different weight hourly space velocities (WHSVs) on DRM performance remained comparable across WHSVs from 12,000 to 48,000 mL g−1 h−1. High CH4 and CO2 conversions of 81.1% and 84.2%, respectively, were maintained below a WHSV value of 48,000 mL g−1 h−1 (Fig. 1d). The optimal R-0.2Ce catalyst was subjected to long-term stability measurements, which showed abrupt and obvious deactivation at the initial stage of the DRM reaction, and then recovered, possibly due to surface reconstruction in a dynamic equilibrium between CH4 and CO2 (Figs. S6–S11). Furthermore, the R-0.2Ce catalyst was subjected to 800 h long-term stability test under more severe operating conditions (Figs. 1e and S12). It demonstrates excellent stability, maintaining a CH4 conversion rate of 87.4% and a CO2 conversion rate of 92.9% after 800 h, with negligible deactivation compared to its initial performance. In contrast, the R-0Ce sample exhibits a gradually declining performance after ~65 h, with pronounced deactivation observed after switching to 750 °C at 80 h. This resulted in reduced CH4 and CO2 conversion rates of 61.7% and 73.6%, respectively.
As shown in Fig. 1f, g, the apparent activation energies for H2 and CO yields exhibited differences in the rate control steps for the same samples, suggesting the distinct rate-determining steps for H2 and CO production. Furthermore, R-0.2Ce demonstrated lower apparent activation energies compared to R-0Ce, indicating the enhancement in Ce modification. This phenomenon may be attributed to an increase in the interfacial redox capacity via CeOx ↔ CeOx-δ looping according to the following characterizations. The R-0.2Ce catalyst exhibits higher apparent methane activity and lower carbon formation rate at the same DRM temperatures, which demonstrates obvious cutting-edge compared with previously reported catalysts (Fig. 1h and Table S1).
Structural characterizations
Powder XRD patterns of the calcined perovskite oxides, LaCrO3, LaNi0.4Cr0.6O3, and CeO2-x-La0.97Ce0.03Ni0.4Cr0.6O3, all display a similar perovskite phase containing Cr6+ (La2CrO6) after B-site Ni doping (Fig. S13, and see Table S2 for their ICP-OES results). We consider that this arose from maintaining the charge balance of the bulk phase. (TEM images of C-0Ce and C-0.2Ce as shown in Figs. S14 and S15). In the R-0.2Ce sample, a peak belonging to metallic Ni(111) is observed at 44.5°, while the main perovskite peak at 32.6° shifts to a lower angle compared with the calcined one (Fig. S16). This implies that part of the B-site Ni has exsolved from the bulk phase to form Ni0 nanoparticles on the surface.
Transmission electron microscopy (TEM) images of the R-0.2Ce sample reduced at 900 °C confirmed Ni nanoparticle exsolution (Fig. 2a). Additionally, energy-dispersive X-ray spectroscopy (EDS) mapping confirms the homogeneous distribution of La, Ce, Cr, and O elements, evidencing the formation of the La-Ce-Ni-Cr-O perovskite. The Ni nanoparticle, exposed (111) crystal plane, was proportionally embedded within the parent perovskite, according to the HR-TEM (Fig. 2b). AFM further confirmed the exsolved Ni nanoparticles distributed on the perovskite surface (Fig. S17). In addition, a partially reduced CeO1.75(220) species was also observed on the perovskite surface layer.

a1 HAADF-STEM image of R-0.2Ce. a2 EDS mapping of La, Ce, Ni, Cr, and O elements. b HR-SEM image of the exsolved Ni nanoparticle of R-0.2Ce. c SEM images of R-0Ce. d SEM images of R-0.4Ce. e Particle size distribution of exsolved nanoparticles of three samples (R-0Ce, R-0.2Ce, R-0.4Ce). f AC-HAADF-STEM images of R-0.2Ce and EDS mapping of Ce and Cr elements. g AC-HAADF-STEM image of R-0.2Ce and h corresponding showcasing diagram of perovskite A-site content in three layers.
The effect of Ce doping on the particle size and distribution of exsolved structure was investigated (Fig. 2c–e). Among the investigated samples, R-0.2Ce possessed the smallest average particle size and the highest dispersion of exsolved Ni nanoparticles. It was demonstrated that Ce doping is conducive to the exsolution of Ni nanoparticles when compared with R-0Ce, possibly because Ce doping into the A-site lattice induces lattice distortion and reduces the energy barrier for Ni exsolution. Nevertheless, excessive Ce doping leads to significant increase in the average particle diameters (~95.8 nm) of the exsolved Ni (Fig. S18). Excessive Ce modification in the R-0.4Ce sample leads to CeO2-x aggregation on the perovskite surface, which hinders Ni segregation and aggregates the nanoparticles on the surface, resulting in a significant decrease in the density of active sites and an increase in the particle size.
Prior research has demonstrated that the reduction temperature exerts a pronounced influence on the dimensions and distribution of the exsolved nanoparticles39,40. The LaMer model proposed by Sugimoto et al.41 and the findings from O’Leary et al.42 suggest that high-temperature reduction increases Ni segregation and transformation rates. Nevertheless, the growth barrier is meanwhile reduced, which results in a decrease in the density and an increase in the size of the exsolved Ni particles, leading to less dispersed nanoparticles. Therefore, we examined the evolution of surface morphology with the R-0.2Ce sample as a function of reduction temperature from 700 to 1000 °C (Figs. S19–S22). The results are in accordance with the previous work, i.e., high reduction temperatures promote Ni exsolution but also cause nanoparticle aggregation, while low temperatures inhibit the exsolution process. The DRM test further confirmed that the sample reduced at 900 °C exhibited the highest catalytic performance, attributed to its smaller particle sizes and higher dispersion (Figs. S23–S25). Specifically, this sample achieved the highest particle density of ~9.3 NPs/μm2, with an average particle diameter of 37.9 nm. The DRM test also demonstrated the best performance over the sample reduced at 900 °C (Fig. S26).
Using aberration-corrected high-angle annular dark-field scanning transmission electron microscopy (AC-HAADF-STEM) combined with EDS mapping (Fig. 2f), we confirmed Ce doping at the A-site and Ni doping at the B-site of the LaCrO3 perovskite. Additionally, a proportion of Ce was enriched as a secondary phase on the perovskite surface. EDS mapping of La/Ni shows non-overlapping distributions, testifying to their occupations of A- and B-sites, respectively. Some Ni undergoes segregation and aggregates in the central region (Fig. S27). Comparison of La/Ni and La/Cr mappings reveals that Ni occupies the B-site perovskite together with Cr. Ce/Cr mappings further reveal the co-existence of two Ce species: Ce doped into the A-site and thin CeO2-x nanolayers present on the surface and bulk. The measured lattice parameters (a = 0.558 nm, c = 0.572 nm) are consistent with those derived from XRD refinement (Pnma space group). The atomic intensity signals within the orange boxes were analyzed, confirming the doping of Ce at the A-site of the perovskite structure (Fig. 2g, h).
To further clarify the effect of Ce doping on the Ni exsolution, XRD Rietveld refinement of C-0Ce and C-0.2Ce samples was carried out. Results confirm the Pnma space group (Figs. S28 and S29) and a reduced cell volume when compared to LaCrO3, confirming partial replacement of B-site Cr by smaller-radius Ni (Tables S3 and S4). Ce doping also reduces the cell volume further (233.92 Å3 → 233.58 Å3), as Ce3+ (0.134 nm) and Ce4+ (0.114 nm) have smaller ionic radii than La3+ (0.136 nm)26. The LaNi0.4Cr0.6O3 perovskite yielded a tolerance factor of 0.97, indicating the presence of lattice distortion after B-site Ni doping, see detailed introduction in supplementary information.
After reduction, the average cell volumes of both R-0Ce and R-0.2Ce remain smaller than that of LaCrO3, representing successful substitution of Cr by Ni due to its smaller radius. The reduced samples were fitted by Pnma and R-3c space groups (Tables S3 and S4). The exsolved Ni nanoparticles of R-0Ce and R-0.2Ce account for 3.51 wt% and 6.79 wt%, respectively, resulting in a 93.4% promoted Ni exsolution after Ce doping. Furthermore, the average cell volume of R-0.2Ce (282.44 Å3) is obviously smaller than that of R-0Ce (309.41 Å3), further demonstrating the enhancement of Ni exsolution under Ce doping43. Furthermore, the increased percentage of asymmetry orthorhombic (Pnma) structures44 after Ce doping (59.2% for R-0.2Ce and 35.9% for R-0Ce) indicates greater lattice distortion, which facilitates Ni exsolution (Figs. 3a, b and S30).

a XRD refinement data of R-0.2Ce. b XRD refinement data of R-0Ce. c Normalized Ni K-edge XANES data of the R-0.2Ce and R-0Ce. d Ni K-edge EXAFS data of R-0.2Ce and R-0Ce in R-space. e Fourier transform of the k3-weighted EXAFS curves (empty dots) and fit model (red line) for the reduced R-0.2Ce samples. The light gray shadow indicates the fitted region. f R-0.2Ce k3-weighted wavelet transform plots of Ni K-edge EXAFS spectra. g R-0Ce k3-weighted wavelet transform plots of Ni K-edge EXAFS spectra.
X-ray absorption spectroscopy (XAS) characterization was further conducted to determine the chemical and electronic states of Ni and Ce. We observed the presence of a shoulder at the high-energy flank of the white line (8.353 keV) and the plateau period between 8.357 keV and 8.363 keV for the normalized Ni K-edge X-ray absorption near edge structure (XANES) of the R-0Ce and R-0.2Ce samples, which can be regarded as distinctive signatures of Ni incorporation into the lattice of perovskite, confirming the substitution of Ni (Fig. 3c)45,46. The absorption threshold E0 of both samples is situated between Ni and NiO, indicating that their average valence states are between 0 and +2. The slightly lower E0 energy of R-0.2Ce than that of R-0Ce further suggests the formation of more embedded Ni0 exsolved nanoparticles in R-0.2Ce.
Fourier-transformed R-space from Extended X-ray absorption fine structure (EXAFS) spectra of R-0Ce and R-0.2Ce further confirms the lattice distortion caused by B-site Ni doping. According to the curve fitting of EXAFS data, the R-0Ce and R-0.2Ce possess two forms of Ni-O bonds, Ni-O1 and Ni-O2 paths, which correspond to bond lengths of 1.89 and 2.02 Å, and 1.92 and 2.06 Å, respectively (Figs. 3d, e, and S31, Table S5). The prolonged Ni-O bond lengths after Ce doping point to its role in enhancing the lattice oxygen mobility, along with the two longer Ni-O paths of R-0.2Ce, which show lower coordination numbers, also support the existence of more oxygen vacancies. Further, the R-0.2Ce sample shows a stronger Ni-Ni signal (at ~2.49 Å), which implies the promotion of B-site Ni exsolution after doping with Ce. Results from XANES linear combination fitting corroborate this speculation (Fig. S32 and Table S6). The scattering peak of the second shell layer is assigned to Ni-La or Ni-Ce paths. The bond lengths of R-0.2Ce (3.28 Å) are slightly shorter than those of R-0Ce (3.33 Å), contributing to the A-site substitution of La by Ce.
To differentiate the backscattering atoms, wavelet transform analysis was conducted (Figs. 3f, g, S33 and S34). The intensity centers in k-space (strongest oscillation) of both R-0Ce and R-0.2Ce are between Ni foil (8.3 Å−1) and Ni-O coordination (6.1 Å−1), revealing the possible existence of Ni-Cr coordination47. Combined with XRD and XAS results, two Ce species, CeO2-x with abundant oxygen vacancies and lattice Ce within the A-site perovskite structure, were therefore confirmed. (Other relevant XAS data can be found in Figs. S35 and S36).
XPS analysis demonstrated a decrease in the Cr3+/(Cr3++Cr4+) ratio upon Ce doping, suggesting that the Ce4+ may promote the conversion of Cr3+ to Cr4+ through redox balance for its high oxidation capacity. (Figs. 4a–c and S37, Table S7). This alters the charge balance and could lead to electron accumulation for B-site cations48. The peak binding energy shifts of B-site elements in perovskite structures reflect variations in B−O bond strength49. Results from Cr 2p and Ni 3p indicate the gradual increase and decrease in the binding energy with increasing amounts of Ce introduced, suggesting a strengthening of the Cr-O bond and a weakening of the Ni-O bond. This also indicates that the metallic Ni is in an electron-rich state, and the electron cloud density can be modulated by the regulation of the Ce doping amount. The shift of the O 1s binding energy decreases first and then increases, which reflects an average B-O bond strength with Ce doping, as confirmed by the Raman spectroscopy (Fig. S38). The surface Ce3+ content in the R-0.4Ce sample decreased to 8.5%, likely due to the aggregation of surface CeO2-x species at higher Ce concentrations, which favors the formation of more stable Ce4+ species. Additionally, the XPS signal intensity of metallic Ni0 was significantly reduced in the R-0.4Ce sample, while the Ni2+ peak was notably enhanced. These results suggest that the excess CeO2-x phase promotes the chemical oxidation of surface Ni while physically hinders the exposure of metallic Ni0 through surface coverage. As confirmed by TEM results, it is revealed that excessively high Ce doping is not conducive to exsolving Ni nanoparticles. XPS under different atmospheres was applied to elucidate the alterations in the electronic structure and oxidation state at the catalyst interface in methane and carbon dioxide atmospheres, as shown in Figs. S39–S41, Table S8.

XPS spectra of a Ce 3d. b Ni 3p. c O 1s. d The EPR spectra of R-0Ce, R-0.2Ce, and R-0.4Ce. e H2-TPR profiles of calcined catalysts. MS signal of CH4/CO2 alternately pulse experiments at 750 °C on f R-0Ce and g R-0.2Ce. CH4-TPR of h R-0Ce, and i R-0.2Ce.
EPR results also supported the enhancement in oxygen vacancy formation with elevated Ce doping, and the amount of the oxygen vacancies formed follows the order of R-0.4Ce > R-0.2Ce > R-0Ce (Fig. 4d). Combined with XPS results, we conclude that a moderate surface and bulk oxygen vacancy distribution promises optimal carbon resistance and CO2 activation performance. H2-TPR tests were carried out to examine the reducibility of the perovskite catalysts (Fig. 4e). H2-TPR profiles of four Ce-modified La0.97Ni0.4Cr0.6O3 samples correspond to the transformation of Ni3+→Ni2+, Cr6+→Cr4+/3+, Ni2+→Ni0, and Ce4+→Ce3+26. Among them, R-0.2Ce exhibits the lowest preliminary reduction temperature of ~240 °C, confirming its exceptional lattice oxygen mobility.
Alternate CH4-CO2 pulse experiments were conducted to reveal the capability of R-xCe in C-H bond activation and lattice oxygen mobility at 750 °C (Figs. 4f, g and S42). Among the three tested samples, R-0.4Ce shows inferior performance due to the suppression of Ni exsolution by the highly oxidized state of the surface and the coverage of active sites by CeO2-x species, resulting in a low methane conversion; while over R-0Ce a much higher H2/CO ratio was achieved, demonstrating the influence of increased carbon deposition. We anticipate that the superiority of R-0.2Ce is due to: (i) high lattice oxygen mobility supplied by surface CeO2-x species and bulk-phase perovskite. This hinders carbon deposition with abundant oxygen vacancy generated; (ii) high CO2 activation and splitting capability. This is actuated by the formed oxygen vacancies, thus inducing sustainable O2−lattice release-supply looping.
To further confirm the impact of lattice oxygen mobility of exsolved perovskite on methane activation and conversion, CH4-TPR tests were carried out (Figs. 4h, i and S43). The R-0.2Ce and R-0.4Ce samples detected an increased CO2 signal at temperatures as low as 230 °C and 247 °C, respectively, demonstrating their capability in low-temperature CH4 oxidation and enhancement in lattice oxygen mobility. H2 signals emerged subsequently at 405 °C, accompanied by CO evolution. In contrast, over R-0Ce, H2 signals were recorded at 464 °C, followed by CO2 and CO signals at approximately 482 °C, corresponding to much weaker peak intensities. Notably, the H2 peaks observed over the Ce-doped samples reached their highest value at approximately 750 °C, followed by a decline and subsequent rise. This phenomenon could be attributed to the reduction of a substantial quantity of bulk-phase oxygen in CeO2-x at elevated temperatures (H2-TPR of CeO2), which then combines with H2 to generate H2O. This process also results in the attenuation of the methane partial oxidation reaction due to the decreased H2 and CO signals. The CO signal intensity of the R-0Ce samples is markedly lower than that of the Ce-containing samples, which can be assigned to the lower surface oxygen vacancies formed. In summary, two forms of Ce species, surface Ov-abundant CeO2-x and A-site lattice Ce, concurrently contribute to highly efficient CH4 oxidation, CO2 dissociation, and exsolution of Ni nanoparticles, thereby enhancing CH4-CO2 reforming with a highly efficient and robust catalyst.
The structural, morphological, and surface properties of the as-prepared R-0Ce and R-0.2Ce samples and after long-term DRM testing were characterized by XRD, XPS, Raman, etc. Powder XRD results indicate that the perovskite phase and exsolved Ni remain basically unchanged after long-term DRM test, demonstrating its stability (Figs. 5a and S44). XPS spectra of reacted R-0.2Ce for Ce 3d, O 1s, Ni 3p, and Cr 2p are displayed in Fig. 5b–e, which demonstrated a notable increase in the Ce3+ species. Ce3+ exhibits strong redox capabilities that promote the dynamic capture and release of lattice oxygen, thereby facilitating the removal of surface carbon. The observed increase in Ni0 content is attributed to the reducing environment of DRM, which facilitates the continuous segregation of Ni ions from the perovskite bulk phase, forming a larger population of Ni0 species. This dynamic migration contributes to the sustained enhancement in catalytic activity during the initial reaction stage, reflecting a dynamic equilibrium within the catalytic system under DRM conditions.

a XRD pattern of the reacted samples. XPS spectra of b Ce 3d. c O 1s. d Ni 3p. e Cr 2p. f TG of 120h-reacted R-0Ce and 800 h-reacted R-0.2Ce.
Thermogravimetric analysis (Fig. 5f) revealed that the carbon deposition on R-0.2Ce after 800 h of reaction led to only a 1.6% weight loss, while R-0Ce showed a 6.4% loss after just 120 h, underscoring the superior carbon resistance of R-0.2Ce. Raman spectroscopy further confirmed the reduced carbon deposition after Ce doping (Fig. S45).
SEM and TEM characterizations (Figs. S46 and S47) showed that the post-reaction morphology of R-0.2Ce closely resembles that of the fresh sample, with only minimal formation of carbon nanotubes. In contrast, R-0Ce displayed significant sintering of the perovskite surface and agglomeration of Ni nanoparticles, along with more pronounced carbon nanotube formation. Quantitatively, the average Ni particle size in R-0.2Ce increased moderately from 37.9 nm to 61.8 nm after 800 h, whereas in R-0Ce it grew substantially from 69.0 nm to 116.9 nm. These results confirm that Ce doping not only mitigates carbon deposition but also improves the anti-sintering stability of the catalyst.
To gain insights into the intermediate species and the reaction pathways during the DRM process, in-situ DRIFTS experiments were conducted (Figs. 6a, b and S48). Introducing 10 vol% CH4 into the reactor at 500 °C resulted in a pronounced CH4 peak at approximately 3016 cm−1, while peaks representing CO ( ~ 2200 cm−1) and CO2 ( ~ 2360 cm−1) emerged due to the partial and complete oxidation of CH450. The weak CO peak observed on the R-0Ce sample aligns with the preceding CH4-TPR results, indicating its limited capacity to activate CH4 at 500 °C. Additionally, two bands corresponding to the deformation vibrations of CHx* (x ≤ 2) and CH3* were observed at ~1335 cm−1 and ~1350 cm−151,52, resulting from the dissociation of C-H bonds at the three-phase interface of Niex, CeO2-x, and La0.97Ce0.03Ni0.4Cr0.6O3 with the assistance of lattice oxygen activation. Upon introducing 10 vol% CO2, the vibration of the hydroxyl group (OH*) was detected at ~3600-3800 cm−153, while the prominent peak at ~2360 cm−1 was attributed to the gaseous CO2. Oxygen vacancies adsorb and dissociate CO2 to produce O* species, leading to the generation of bidentate carbonates, HCOO*, and CHxO* at ~1580 cm−1, 1550 cm−1, and 1390 cm−1, respectively54,55,56,57. Notably, the R-0Ce sample exhibited a weaker peak for CHxO, elucidating a relatively low capability for CO2 activation. As the atmosphere shifted to 10 vol% CH4-10 vol% CO2, the CHx* vibration signal (~1330 cm−1) was significantly weakened and slightly shifted to a higher wavelength, corresponding to the CH3* vibration (~1350 cm−1). This suggests that the catalysts we designed effectively hinder methane decomposition during the DRM process, leading to the combination of CHx with O* or H* with COx* to produce key oxygen-containing intermediate species28, such as CHxO*, HCOO*, OH*, and bidentate carbonates.

a R-0.2Ce and b R-0Ce at 500 °C, expose to 10% CH4 for 20 min, 10% CO2 for 20 min, and 10% CH4-10% CO2 for 20 min.
Figure 7a shows that the segregation energy required for Ni exsolved from the B-site of C-0.2Ce catalyst decreases significantly with increasing Ce doping. When Ce is doped into the first layer, the segregation energy decreases by 7.3%, from −1.23 eV to −1.32 eV. When Ce is doped into the third layer, the segregation energy decreases from −1.23 eV to −2.36 eV. This indicates that A-site Ce doping significantly promotes the in-situ exsolution of Ni, which well explains the phenomenon of greater density distribution of surface Ni nanoparticles and enhanced DRM activity in our experiments.

a (111) oriented slab model and the segregation energy defined as the energy change upon swapping Ni and Cr between the first and third layers. Free energy profiles for the reactions related to CH2* intermediate on the b R-0Ce and c R-0.2Ce catalysts. Density of states and d-band center for d R-0Ce and e R-0.2Ce catalyst surfaces. f The differential charge density of R-0.2Ce catalyst. Cyan and yellow regions present the electron accumulation and depletion, respectively. La, Ce, Cr, Ni, and O atoms are shown in red, yellow, purple, green, and blue, respectively.
Our experimental characterization confirms that CH4 is initially activated through C-H bond cleavage to form CH3*, which is then dehydrogenated to a CH2* intermediate54. From this point, two potential reaction pathways were investigated: oxidation of CH2* to CH2O*, and further dehydrogenation of CH2* to CH* (Figs. 7b and S49, S50). On the R-0Ce catalyst, CH2O* formation has a high activation free energy of 2.00 eV and a reaction free energy of 0.58 eV, whereas CH2* dehydrogenation to CH* is kinetically more favorable, with an activation free energy of 1.02 eV and a reaction free energy of 0.49 eV. Therefore, CH* formation dominates on R-0Ce, leading to a higher risk of carbon accumulation. In contrast, on the R-0.2Ce catalyst (Figs. 7c and S49, S50), CH2* more readily reacts with active oxygen to form CH2O*, with a lower activation free energy of 1.57 eV, while the competing dehydrogenation pathway to CH* requires 1.74 eV. This suggests that Ce doping suppresses excessive CH2* dehydrogenation, thus inhibiting carbon deposition and promoting CH2O* formation. Additionally, the oxidation of CH2* to CH2O* on R-0.2Ce is more kinetically favorable compared to R-0Ce (1.57 eV vs. 2.00 eV), further confirming the role of Ce in mitigating carbon buildup.
As shown in Fig. 7d, e, the εd value of R-0.2Ce (−1.84 eV) is closer to the Fermi level than that of R-0Ce (-2.03 eV), indicating stronger CO2 adsorption and activation on R-0.2Ce. This facilitates C-O bond cleavage and the generation of adsorbed CO and reactive oxygen species. These species can effectively react with carbon derived from CH2* dehydrogenation, thereby suppressing carbon accumulation. Moreover, differential charge density and Bader charge analysis (Fig. 7f, Tables S9 and S10) reveal electron transfer from the CeO2-x cluster to the Ni cluster, significantly enhancing the metal-support interaction. This results in stronger anchoring of exsolved Ni nanoparticles to the perovskite surface, improving their thermal stability and reducing sintering during DRM, as confirmed by SEM and TEM analyses. The electron donation also increases the d-orbital electron density of Ni, potentially boosting the adsorption and activation of both CH4 and CO2.
In summary, we report a Ce-modified and Ni-exsolved Niex-La0.97Ni0.4Cr0.6O3 perovskite catalysts that demonstrates superior DRM activity and satisfactory stability. The 0.2Ce-La0.97Ni0.4Cr0.6O3 catalyst exhibits exceptional CH4 and CO2 conversions of 87.4% and 92.9%, respectively, along with a desirable H2/CO ratio of 0.93, over 800 h at 800 °C. Characterizations including XRD refinement, AC-HADDF-STEM, XPS, XAS, etc. confirmed that the two Ce species exploited a synergistic effect on DRM activity promotion. Firstly, the A-site bulk lattice Ce induces lattice distortion, reducing the segregation energy required for the exsolution of Ni at the B-site of perovskite. It promotes the ability of the catalyst to activate C-H bonds. Secondly, surface oxygen vacancy-moderate Ce species (CeO2-x) exists beyond the perovskite surface and acts as a looping carrier, enhancing CO2 activation and suppressing carbon deposition. The modulation of two distinct forms of Ce offers novel insights for designing high-performance exsolved perovskite catalysts with enhanced DRM efficiency.
Methods
Catalyst synthesis
All the chemicals used in this study were received from Sinopharm Chemical Reagent Co. Ltd. The catalysts were synthesized by the Pechini method58. Five samples, LaNi0.4Cr0.6O3, xCe-La0.97Ni0.4Cr0.6O3 (x = 0.1, 0.2, 0.3, or 0.4), were synthesized according to different Ce: La: Ni: Cr ratios of x: 0.97: 0.4: 0.6. In a typical synthesis, stoichiometric amounts of lanthanum nitrate hexahydrate (La(NO3)3 6H2O), cerium nitrate hexahydrate (Ce(NO3)3 6H2O), nickel nitrate hexahydrate (Ni(NO3)2 6H2O), chromic nitrate (Cr(NO3)3 9H2O) were dissolved in deionized water to obtain a solution of 0.25 M. After that, citric acid and ethylene glycol were added into the solution with a mole ratio of metal ions (Ce4+, La3+, Ni2+, and Cr3+): citric acid: ethylene glycol equal to 1.0: 1.5: 3.0. Then, the solution was stirred in a water bath at 80 °C until a gel was formed. The formed gel was then dried in a drying oven at 150 °C for 12 h, and the obtained solid was subsequently ground to fine powder within a particle size range of 0.15 ~ 0.20 mm. The collected powder was calcined in a muffle furnace in air for 6 h at 850 °C with a heating rate of 2 °C/min. Finally, the calcined sample was reduced by 10 vol% H2 for 3 h under various temperatures (700, 800, 900, or 1000 °C). The samples xCe-La0.97Ni0.4Cr0.6O3 samples before and after reduction are abbreviated as C-xCe and R-xCe, respectively (see Table S11 for detailed List of Abbreviations).
Catalyst characterization
The contents of La, Ce, Ni, and Cr in each sample were measured by inductively coupled plasma optical emission spectroscopy (ICP-OES, PerkinElmer Avio-500).
The crystal structures of the samples were performed by an X-ray diffractometer (PANalytical Empyren) using Cu Kα (λ = 1.5406 Å) radiation 45 kV and 40 mA, operating on a continuous scan mode. The X-ray patterns was recorded in a range of 5-90° at a scanning rate of 5°/min. XRD refinement data were processed by a Fullprof software59.
Scanning electron microscopy (SEM, TESCAN MIRA4 LMH) with an energy-dispersive X-ray spectroscopy detector (EDS mapping) to characterize the morphology of exsolved perovskite samples and analyze the distribution of Ce, La, Ni, Cr, and O elements.
FEI Tecnai G2 F20 TEM was used to characterize the surface morphology of the catalyst and the element distribution. The samples used for TEM measurements were put into ethanol to obtain a suspension (1 mg mL−1) and then treated by ultrasonic for 5 min.
High-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) was performed using a JEOL JEM-ARM200F instrument.
Bruker Dimension edge Atomic Force Microscope (AFM) was used to explore the surface morphology and the roughness of the R-0.2Ce catalysts.
X-ray photoelectron spectroscopy (XPS) measurements were performed on a Thermo Scientific TM K-Alpha TM+ spectrometer equipped with a monochromatic Al Kα X-ray source (1486.6 eV) to analyze the chemical states of the catalysts. The binding energies were calibrated by taking C1s peak (284.8 eV) as a reference.
Raman measurements were conducted using a Renishaw inVia Raman spectrometer with a 532 nm laser wavelength. Powder samples were applied to a glass slide and flattened for consistency. Spectra were acquired from various positions on each sample, ensuring that each sample underwent a minimum of three tests.
The detection of oxygen vacancies was performed using electron paramagnetic resonance (EPR) spectroscopy, tested on BRUKER EMXPLUS EPR in continuous wave electron paramagnetic resonance.
In-situ diffuse reflectance infrared Fourier transform spectroscopy (In-situ DRIFTS) was conducted by using a Nicolet iS50 spectrometer equipped with an In-situ diffuse reflectance cell and a mercury-cadmium-telluride detector. In a typical measurement, the perovskite catalyst was initially heated from room temperature to 500 °C with a heating rate of 10 °C/min and maintained for 30 min under N2 (30 mL/min), and the background spectrum was collected after stabilization. Afterwards, 10 vol% CH4 in N2, 10 vol% CO2 in N2, and a mixture of 10 vol% CH4 and 10 vol% CO2 in N2 were successively introduced into the cell for 20 min. The spectra were collected every 5 minutes, which lasted for 60 minutes in total.
The temperature-programmed reaction of methane (CH4-TPR) measurements were performed by using a chemisorption analyzer (Micromeritics AutoChem II 2920) connected with a mass spectrometer (MS, Hiden-20 R&D). In a typical experiment, the sample was placed into the U-type quartz reactor and heated at 300 °C for 30 min under He (20 mL/min) with a heating rate of 10 °C/min. The sample was then cooled to room temperature, followed by the introduction of 10 vol% CH4 in He (20 mL/min). Meanwhile, MS is conducted to record the CH4 signal. Once the CH4 curve is stabilized, the reactor was heated from room temperature to 900 °C with a heating rate of 10 °C/min. The outlet products, H2, CH4, H2O, CO, and CO2, were detected by MS according to the m/z results of 2, 16, 18, 28, and 44.
The CH4-CO2 transient alternating pulse measurements were conducted by using a chemisorption analyzer (Micromeritics AutoChem II 2920). In a typical measurement, 100 mg sample was put into the reactor and pretreated by He (20 mL/min) for 30 min at 300 °C with a heating rate of 20 °C/min. Then, the CH4-CO2 transient alternating pulse experiments were performed at 750 °C by switching the reaction atmosphere between 10 vol% CH4 in He and 10 vol% CO2 in He for every 7 min. The outlet products were also detected by MS.
H2-TPR measurements were conducted by using the same chemisorption analyzer. In a typical test, 100 mg calcined sample was pretreated in Ar (40 mL/min) at 300 °C for 30 min, and the temperature was then reduced to 50 °C. Once the baseline stabilized, the temperature was increased from 50 to 900 °C with a heating rate of 10 °C/min, meanwhile switching the atmosphere to 10 vol% H2 in Ar (40 mL/min). The hydrogen consumption signals were detected using a thermal conductivity detector.
The XAS (Ni K-edge) were collected at BL14W beamline in Shanghai Synchrotron Radiation Facility (SSRF). The storage rings of SSRF were operated at 3.5 GeV with a stable current of 200 mA. Using Si(111) double-crystal monochromator, the data collection was carried out in fluorescence mode using a Lytle detector. All spectra were collected in ambient conditions. XAS data were analyzed using the Demeter software package60. A linear function was subtracted from the pre-edge region, then the edge jump was normalized using the Athena software. The χ(k) data were isolated by subtracting a smooth, third-order polynomial approximating the absorption background of an isolated atom. The k3-weighted χ(k) data were Fourier transformed after applying a Hanning window function (Δk = 1.0). The global amplitude EXAFS (CN, R, σ2 and ΔE0) were obtained by nonlinear fitting, with least-squares refinement, of the EXAFS equation to the Fourier-transformed data in R-space, using Artemis software.
Catalytic performance
Experimental tests were performed in a fixed-bed reactor (600 mm in length and 8 mm in inner diameter). In a typical experiment, 300 ± 5 mg (60 mg for long-term test) of reduced catalyst was placed into the isothermal temperature interval of a quartz tube, as supported by an appropriate amount of quartz wool. Before each test, the sample-loaded quartz tube was purged with 30 mL/min N2 for 10 minutes. Then, the furnace is heated from room temperature to the target temperature (650, 700, 750, 800, or 850 °C). Once the temperature reaches the target one and stabilizes, a total flow rate of 25 vol% CH4 and 25 vol% CO2 in N2 was introduced (CH4: CO2: N2 = 1: 1: 1 for long-term test). The long-term experimental test was conducted for 800 h. The outlet products, H2, CO, CO2, and CH4, were determined by a GC (INFICON Micro GC Fusion).
The DRM performances were evaluated by CH4, CO2 conversion rates, H2, CO selectivity, and H2/CO mole ratio. Total flow rate of the outlet gas (Ftotal, mol s−1) is defined as:
where Cout(N2) refers to the outlet concentration of N2, Fin(y) represents the feeding rate of y, y = N2, CH4, or CO2. The outlet flow rate of gas i (Fout(i), mol s−1) is calculated as:
where i refers to possible components of CH4, CO2, CO, or H2. The conversion rates of CH4 and CO2, X(CH4, %) and X(CO2, %), were calculated as:
The H2/CO ratio were calculate by the follow equations:
The error bars in Figs. 1, S1–S3, and S26 represent the standard deviation (SD) obtained from three independent performance tests.
DFT calculation
Density functional theory (DFT) calculations are implemented in the Vienna ab initio Simulation Package (VASP 5.4.4)61. Perdew-Burke-Ernzerhof with generalized gradient approximation (GGA) was used to describe the exchange-correlation functional62. A vacuum layer of 15 Å was introduced to avoid interactions between periodic images. A cutoff energy of 400 eV for the plane-wave basis sets and a 3 × 3 × 1 grid generated by the Monkhorst-Pack method were adopted for geometric optimization calculations63. Meanwhile, the k-point grid in the Brillouin zone was set to be 8×8×1 by the Monkhorst-Pack scheme when calculating the electronic structure. Structure optimizations were performed until the total energies converged to 5 × 10–6 eV and the forces acting on the relaxed ions were less than 0.05 eV/Å. To counteract erroneous electron delocalization, GGA + U methods were applied to La, Cr and Ni atoms. Values of ULa = 5 eV, UCr = 2 eV, UNi = 3.5 eV, UCe = 6 eV were utilized64,65,66,67. The climbing-image nudged elastic band (CI-NEB) method68 was employed to search for the transition states (TS), then, the dimer method was used to optimize the located TS69. Moreover, the TS structure was confirmed with only one imaginary frequency.
Both R-0Ce and R-0.2Ce catalyst models were constructed based on the experimental characterization results that the (111) crystal plane was the dominantly exposed crystallographic surface. As shown in Fig. S50, The R-0Ce catalyst was modeled using a three-layer p(2 × 1) LaCrO3(111) supercell, in which the bottom two layers were fixed. In this model, a Ni4 cluster was located on the surface, with an adjacent oxygen vacancy surrounding the Ni4 cluster. The R-0.2Ce model was obtained by loading a CeO2 cluster on the R-0Ce surface and creating an oxygen vacancy within the cluster by removing one oxygen atom, resulting in a CeO2-x cluster structure. To calculate Ni segregation energy on the (111) plane, a model for Ni-doped LaCrO3 catalyst was constructed using a five-layer p(1 × 1) supercell with the bottom two layers fixed.
The adsorption free energy (Gads), activation free energy (Ga), and reaction free energy (∆G) were determined by the following equations70:
where Emol/surface, Emol, and Esurface were the total free energies of the adsorbed system, the clean surfaces, and the free molecules, respectively. Considering our optimal catalytic reaction conditions were 750 °C and 1.0 atm, all energies were corrected at 1023.15 K and 1.0 atm using VASPKIT software.
References
Kohn, M. P. et al. Biogas reforming for syngas production: the effect of methyl chloride. Appl. Catal. B. 144, 353–361 (2014).
Zhu, H. et al. Recent advances in promoting dry reforming of methane using nickel-based catalysts. Catal. Sci. Technol. 14, 1712–1729 (2024).
Santos, R. G. D. et al. Biomass-derived syngas production via gasification process and its catalytic conversion into fuels by Fischer Tropsch synthesis: a review. Int. J. Hydrog. Energy 45, 18114–18132 (2020).
Zhang, S. et al. Synthesis of alpha,beta-unsaturated carbonyl compounds by carbonylation reactions. Chem. Soc. Rev. 49, 3187–3210 (2020).
He, C. et al. Single-atom alloys materials for CO2 and CH4 catalytic conversion. Adv. Mater. 36, e2311628 (2024).
Liu, X. et al. Modulating proportion of Ni0 species stabilized by Ni2+ on Ni-MgO catalyst with superior stability for dry reforming of methane. Chem. Eng. J. 493, 152499 (2024).
Kumar DAS, S. et al. Evolution of low z-elements in a Ni/MgFeAlO4 catalyst during reaction: insight from in situ XRS. ACS Catal. 14, 1311–1323 (2024).
Jiao, H. et al. Dry reforming of methane on Ni/LaZrO2 catalyst under external electric fields: a combined first-principles and microkinetic modeling study. ACS Appl. Mater. Interfaces 16, 35166–35178 (2024).
Liu, Y. et al. Surface basic site effect on CoLa/m-Al2O3 catalysts for dry reforming of methane. Mol. Catal. 562, 114205 (2024).
Yang, E. et al. Coke resistant NiCo/CeO2 catalysts for dry reforming of methane derived from core@shell Ni@Co nanoparticles. Appl. Catal. B. 339, 123152 (2023).
Shah, M. et al. Study on the effect of promotors in CO2 utilization for syngas production via dry reforming of methane over Co-MOX/TiO2-Al2O3 (MOX = La, Ce, Mg, and K) catalysts. Fuel Process. Technol. 257, 108089 (2024).
Li, Y. et al. Structural evolution of robust Ni3Fe1 alloy on Al2O3 in dry reforming of methane: Effect of iron-surplus strategy from Ni1Fe1 to Ni3Fe1. Appl. Catal. B. 331, 122669 (2023).
Zhao, R. et al. Highly dispersed Fe-decorated Ni nanoparticles prepared by atomic layer deposition for dry reforming of methane. Int. J. Hydrog. Energy 48, 28780–28791 (2023).
Abdulrasheed, A. et al. A review on catalyst development for dry reforming of methane to syngas: recent advances. Renew. Sustain. Energy Rev. 108, 175–193 (2019).
Neagu, D. et al. In situ growth of nanoparticles through control of non-stoichiometry. Nat. Chem. 5, 916–923 (2013).
Najimu, M. et al. Co-exsolution of Ni-based alloy catalysts for the valorization of carbon dioxide and methan. Acc. Chem. Res. 56, 3132–3141 (2023).
Neagu, D. et al. Roadmap on exsolution for energy applications. J. Phys. Energy 5, 031501 (2023).
Kousi, K. et al. Emergence and future of exsolved materials. Small 17, e2006479 (2021).
Sun, X. et al. Progress of exsolved metal nanoparticles on oxides as high performance (electro) catalysts for the conversion of small molecules. Small 17, e2005383 (2021).
Tsiotsias, A. I. et al. Bimetallic exsolved heterostructures of controlled composition with tunable catalytic properties. ACS Nano. 16, 8904–8916 (2022).
Jo, S. et al. Self-regenerative Ni-doped CaTiO3/CaO for integrated CO2 capture and dry reforming of methane. Small 20, e2401156 (2024).
Jo, Y. R. et al. Growth kinetics of individual co particles ex-solved on SrTi0.75Co0.25O3-delta polycrystalline perovskite thin films. J. Am. Chem. Soc. 141, 6690–6697 (2019).
Dama, S. et al. Active and durable alkaline earth metal substituted perovskite catalysts for dry reforming of methane. Appl. Catal. B. 224, 146–158 (2018).
Das, S. et al. Effect of partial Fe substitution in La0.9Sr0.1NiO3 perovskite-derived catalysts on the reaction mechanism of methane dry reforming. ACS Catal. 10, 12466–12486 (2020).
Wei, T. et al. LaMnO3-based perovskite with in-situ exsolved Ni nanoparticles: a highly active, performance stable and coking resistant catalyst for CO2 dry reforming of CH4. Appl. Catal. A. 564, 199–207 (2018).
Wang, M. et al. Effects of Ce substitution at the A-site of LaNi0.5Fe0.5O3 perovskite on the enhanced catalytic activity for dry reforming of methane. Appl. Catal. B. 224, 214–221 (2018).
Peng, W. et al. Enhanced activity and stability of Ce-doped PrCrO3-supported nickel catalyst for dry reforming of methane. Sep. Purif. Technol. 303, 122245 (2022).
Yao, X. et al. Enlarging the three-phase boundary to raise CO2/CH4 conversions on exsolved Ni–Fe alloy perovskite catalysts by minimal Rh doping. ACS Catal. 14, 5639–5653 (2024).
Wang, Y. et al. Coke-resistant NdFe0.7Ni0.3O3 perovskite catalyst with superior stability for dry reforming of ethane. Appl. Catal. B. 337, 123010 (2023).
OH, J. et al. Precise modulation of triple-phase boundaries towards a highly functional exsolved catalyst for dry reforming of methane under a dilution-free system. Angew. Chem. Int. Ed. 61, e202204990 (2022).
Wang, H. et al. Dry reforming of methane over bimetallic Ni-Co catalyst prepared from La(CoxNi1-x)0.5Fe0.5O3 perovskite precursor: catalytic activity and coking resistance. Appl. Catal. B. 245, 302–313 (2019).
Chen, Z. et al. Enhanced performance of La1-xFeO3-δ oxygen carrier via A-site cation defect engineering for chemical looping dry reforming of methane. Fuel Process. Technol. 248, 107820 (2023).
Álvarez M.A. et al. CO2 reforming of methane over Ni-Ru supported catalysts: on the nature of active sites by operando DRIFTS study. J. Co2 Util. 24, 509–515 (2018).
Xu, D.-J. et al. Doping low amount of Zirconium in Rh-LTO to prepare durable catalysts for dry reforming of methane. Mol. Catal. 535, 112822 (2023).
De Caprariis, B. et al. Rh, Ru and Pt ternary perovskites type oxides BaZr1-xMexO3 for methane dry reforming. Appl. Catal. A. 517, 47–55 (2016).
Oh, J. H. et al. Importance of exsolution in transition-metal (Co, Rh, and Ir)-doped LaCrO3 perovskite catalysts for boosting dry reforming of CH4 using CO2 for hydrogen production. Ind. Eng. Chem. Res. 58, 6385–6393 (2019).
Laosiripojana, N. et al. Catalytic dry reforming of methane over high surface area ceria. Appl. Catal. B. 60, 107–116 (2005).
Sun Z. et al. Tailoring an Fe-OV-Ce-triggered phase-reversible oxygen carrier for intensified chemical looping CO2 splitting. Carbon Energy 7, e70011 (2025).
Kim, Y. H. et al. Nanoparticle exsolution on perovskite oxides: insights into mechanism, characteristics and novel strategies. Nano-Micro Lett. 16, 33 (2023).
Kim, J. K. et al. Exceptional tunability over size and density of spontaneously formed nanoparticles via nucleation dynamics. Acs. Appl. Mater. Interfaces 12, 24039–24047 (2020).
Tadao Sugimoto, F. S. et al. Spontaneous nucleation of monodisperse silver halide particles from homogeneous gelatin solution I: silver chloride. Colloids Surf. A. 164, 183–203 (2000).
O’leary, W. et al. Influence of Sr-site deficiency, Ca/Ba/La doping on the exsolution of Ni from SrTiO3. J. Am. Chem. Soc. 145, 13768–13779 (2023).
Li, B. et al. A Ce/Ru codoped SrFeO3−δ perovskite for a coke-resistant anode of a symmetrical solid oxide fuel cell. ACS Catal. 10, 14398–14409 (2020).
Graf, M. E. et al. Rhombohedral R-3c to orthorhombic Pnma phase transition induced by Y-doping in BiFeO3. J. Phys. Condens. Matter 30, 285701 (2018).
Steiger, P. et al. Nickel incorporation in perovskite-type metal oxides–implications on reducibility. Acta Mater. 164, 568–576 (2019).
Rudolph, B. et al. Nanoparticle exsolution from nanoporous perovskites for highly active and stable catalysts. Adv. Sci. 10, e2205890 (2023).
Zhang, T. et al. Regulating electron configuration of single Cu sites via unsaturated N,O-coordination for selective oxidation of benzene. Nat. Commun. 13, 6996 (2022).
He, J. et al. Subsurface A-site vacancy activates lattice oxygen in perovskite ferrites for methane anaerobic oxidation to syngas. Nat. Commun. 15, 5422 (2024).
Zhang, R. et al. Tailoring catalytic and oxygen release capability in LaFe1–xNixO3 to intensify chemical looping reactions at medium temperatures. ACS Catal. 14, 7771–7787 (2024).
Kim, S. et al. Yolk–shell Pt-NiCe@SiO2 single-atom-alloy catalysts for low-temperature dry reforming of methane. ACS Catal. 11, 8247–8260 (2021).
Jiang, S. et al. Insight into the reaction mechanism of CO2 activation for CH4 reforming over NiO-MgO: a combination of DRIFTS and DFT study. Appl. Surf. Sci. 416, 59–68 (2017).
Ni, J. et al. Carbon deposition on borated alumina supported nano-sized Ni catalysts for dry reforming of CH4. Nano Energy 1, 674–686 (2012).
Xiao, Y. et al. Active exsolved metal–oxide interfaces in porous single-crystalline ceria monoliths for efficient and durable CH4/CO2 Reforming. Angew. Chem., Int. Ed. 134, e202113079 (2022).
Wang, H. et al. Facilitating the dry reforming of methane with interfacial synergistic catalysis in an Ir@CeO2-x catalyst. Nat. Commun. 15, 3765 (2024).
Takano, H. et al. Highly active Ni/Y-doped ZrO2 catalysts for CO2 methanation. Appl. Surf. Sci. 388, 653–663 (2016).
Guo, Y. et al. Low-temperature CO2 methanation over CeO2-supported Ru single atoms, nanoclusters, and nanoparticles competitively tuned by strong metal–support interactions and H-spillover effect. ACS Catal. 8, 6203–6215 (2018).
Pozdnyakova, O. et al. Preferential CO oxidation in hydrogen (PROX) on ceria-supported catalysts, part I: Oxidation state and surface species on Pt/CeO2 under reaction conditions. J. Catal. 237, 1–16 (2006).
Huízar-Félix, A. M. et al. Sol–gel based Pechini method synthesis and characterization of Sm1−xCaxFeO3 perovskite 0.1≤x≤0.5. Powder Technol. 229, 290–293 (2012).
Rodríguez-Carvajal, J. Recent advances in magnetic structure determination by neutron powder diffraction. Phys. B. 192, 55–69 (1993).
Ravel, B. et al. Data analysis for X-ray absorption spectroscopy using IFEFFIT. J. Synchrotron Radiat. 12, 537–541 (2005).
Goedecker, S. et al. Separable dual-space Gaussian pseudopotentials. Phys. Rev., B 54, 1703 (1996).
Perdew, J. P. et al. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865 (1996).
Monkhorst, H. J. et al. Special points for Brillouin-zone integrations. Phys. Rev. B 13, 5188 (1976).
Webster, L. et al. Distinct spin-lattice and spin-phonon interactions in monolayer magnetic CrI3. Phys. Chem. Chem. Phys. 20, 23546–23555 (2018).
Yu, H. et al. Strain-triggered distinct oxygen evolution reaction pathway in two-dimensional metastable phase IrO2 via CeO2 loading. J. Am. Chem. Soc. 146, 20251–20262 (2024).
Yang, Z. et al. Effects of Zr doping on stoichiometric and reduced ceria: A first-principles study. J. Chem. Phys. 124, 224704 (2006).
Preciado, M. A. et al. Vibrational and electronic peculiarities of NiTiO3 nanostructures inferred from first principal calculations. RSC Adv. 5, 17396–17404 (2015).
Henkelman, G. et al. Improved tangent estimate in the nudged elastic band method for finding minimum energy paths and saddle points. J. Chem. Phys. 113, 9978–9985 (2000).
Henkelman, G. et al. A dimer method for finding saddle points on high dimensional potential surfaces using only first derivatives. J. Chem. Phys. 111, 7010–7022 (1999).
Clay, J. P. et al. DFT comparison of intrinsic WGS kinetics over Pd and Pt. J. Catal. 320, 106–117 (2014).
Acknowledgements
Z.S. acknowledges the support of the National Natural Science Foundation of China (52476144, 42441835). G.J.H. acknowledges the support of RCUK|Engineering and Physical Sciences Research Council (EPSRC)–EP/W014408/1.
Author information
Authors and Affiliations
Contributions
Z.S., Z.Q.S., and G.J.H. supervised this research and conceived the idea. C.C.H. and Z.S. executed the perovskite preparation and characterization measurements, analyzed the experimental data, and wrote the original manuscript. The DFT analysis was assisted by Z.Y.Q. and R.G.Z. The manuscript was revised by C.C.H., Z.Y.Q., L.R.S., N.F.D., H.F.Q., T.J.A.S., Z.P.Z., R.G.Z., Z.S., Z.Q.S., and G.J.H. All authors discussed the results and commented on the paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Mona Bahout, Guntae Kim, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hao, C., Qu, Z., Smith, L.R. et al. Ce-induced synergistic effect in exsolved perovskite catalyst for highly efficient and robust methane dry reforming. Nat Commun 16, 10630 (2025). https://doi.org/10.1038/s41467-025-65619-w
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-65619-w
