
Abstract
Tetrahydropyridine skeletons serve as ubiquitous structural motifs which are predominant in natural products, bioactive and drug molecules. Thus, the development of efficient and general synthetic methods for the synthesis of tetrahydropyridine frameworks with different substitutions has attracted great attention from the community. Moreover, the control of regioselectivity and site-divergency remains an unmet challenge. Herein, we report a regioselective and site-divergent reductive hydroarylation of pyridinium salts with aryl nucleophiles, enabling direct access to diverse tetrahydropyridine derivatives with aryl substitution at different positions under mild conditions. Mechanistic studies support that the reaction underwent a CoH-mediated double-reduction process of pyridinium salts.
Similar content being viewed by others

Introduction
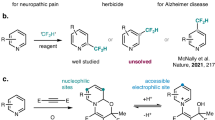
Tetrahydropyridine derivatives represent an important class of six-membered nitrogen-containing molecules, which were ubiquitous in natural products, pharmaceuticals, agrochemicals, and bioactive molecules with potential anticancer, anti-inflammatory, and analgesia activity (Fig. 1a)1,2,3,4,5,6. To this end, numerous efforts have been paid to developing efficient and straightforward synthesis of tetrahydropyridine skeletons from simple and readily-available starting materials under mild conditions7,8,9,10. Classic methods heavily rely on the construction of six-membered tetrahydropyridine ring by intramolecular or intermolecular annulation process, such as classic aza-Prins cyclization11,12,13, aza-Diels-Alder reaction14,15,16,17,18, (aza-)Michael addition19,20,21,22, carbonyl-olefin-metathesis23,24. Nevertheless, multiple synthetic steps, harsh reaction conditions, and precious metals are required. Thus, the development of efficient synthetic routes to avoid above mentioned limitations remains highly desirable. On the other hand, pyridines serve as readily-available and cost-effective aromatic precursors for six-membered nitrogen-containing rings. Over past years, dearomatization of pyridines have made significant progress in synthesizing diverse functionalized piperidine derivatives25,26,27,28. The challenge associated with this dearomatization protocol for the synthesis of tetrahydropyridines is that tetrahydropyridines are more reactive than corresponding parent pyridines. Accordingly, over-reduction29,30,31,32,33,34 and products with poor- selectivity control35 were commonly generated (Fig. 1b), and limited examples are achieved for the access to the target tetrahydropyridines via direct reductive strategy of simple pyridines36,37,38,39,40. Antonchick group disclosed a chiral phosphoric acid catalyzed asymmetric reduction of pyridines by using Hantzsch ester as reducing reagents, affording various tetrahydropyridine derivatives with good enantioselectivities37. Unfortunately, very limited scope of pyridines could be applied. On the other hand, pyridinium cations, which are easily derived from pyridines, serve as a privileged alternative of pyridines with significantly increased reactivity yet impose more challenges on selectivity control. Recently, dearomative functionalization of N-activated pyridinium species by site-selective nucleophilic addition of nucleophiles has been developed as an efficient strategy for the rapid construction of 2,3-dihydro-4-pyridones or dihydropyridine derivatives41. In particular, Feringa42 and Doyle43 have developed Cu- and Ni-catalyzed asymmetric C2-addition to pyridinium salts with organozinc reagents. However, only 4-methoxypyridinium salts could be applied with preformed organozinc species. Yoo44 reported Cu-catalyzed C2-selective dearomative [5 + 1] cycloaddition of N-enamine pyridinium salts with alkynes to furnish various dihydropyridine derivatives with good yields and stereoselectivities. In addition, Karimov group disclosed Cu- and Rh-catalyzed asymmetric C2-addition to pyridinium salts with alkyl or aryl boron precursors45,46. Later, Karimov group also developed a light-promoted, Ni-catalyzed C2-addition of pyridinium salts to 2-arylated dihydropyridines with aryl iodides with sacrificial zinc47. Recently, visible-light mediated C4-alkylation of pyridinium salts were also established to deliver 4-alkylated dihydropyridines48,49. Lam50 and Harutyunyan51 reported Au- and Cu-catalyzed nucleophilic C4-alkylation of pyridinium salts to give dihydropyridines with good yields. Furthermore, organocatalytic C2- and C4-selective functionalization of pyridinium salts have been developed to deliver C2- and C4-functionalized dihydropyridine derivatives (Fig. 1b)52,53,54,55,56,57,58,59,60,61. To date, existing strategies provide access to dihydropyridines with limited regioselectivities and no example enabling access to C6-selectivity has been developed. Twofold reductive functionalization of pyridinium salts to tetrahydropyridines remains an unmet challenge. Moreover, access to regiodivergent isomer of tetrahydropyridine derivatives in a controlled manner from identical precursors imposed additional challenges on reductive functionalization of pyridinium salts. Herein, we report a controlled regioselective and site-divergent reductive hydroarylation of pyridinium salts to access diverse tetrahydropyridine scaffolds with different substitution patterns (Fig. 1c). Co-catalysis allows for the reductive C4-arylated tetrahydropyridines and stoichiometric metal-free conditions lead to C6-arylated tetrahydropyridines, providing a straightforward protocol for efficient synthesis of different tetrahydropyridines from identical starting materials.

a Representative biologically active molecules containing tetrahydropyridine scaffolds. b State-of-the-art: Site-selective dearomative functionalization of pyridine or pyridinium salts for the synthesis of tetrahydropyridines. c This work: Regiodivergent reductive dearomative hydroarylation of pyridinium salts.
Results and discussion
We initiated the investigation by selecting pyridinium salt (1a) and 1-methylindole (2a) as model substrates to test the feasibility of the reaction. After extensive initial trials, dearomative hydroarylation of pyridinium salt (1a) was detected in the presence of Co(salen) complex ([Co-1]) and tert-butyl peroxybenzoate (TBPB), and dimethylphenylsilane (PhMe2SiH) in acetonitrile (CH3CN) at room temperature, affording reductive C-4 arylated product 3a and C-6 arylated product 4a in 8% and 10% yields, respectively (Table 1, entry 1). The encouraging initial results prompted us to further investigate different dimensions of parameters. Addition of sub-stoichiometric potassium phosphate increased the formation of 3a in 50% yield with 18% of 4a (Table 1, entry 2). The use of other base could also promote the desired reductive-oxidative relay enabled reductive hydroarylation process, albeit in lower efficiency (Table 1, entries 3 and 4). Next, a series of oxidants were examined, including di-tert-butyl peroxide (DTPB), dibenzoyl peroxide (BPO), m-chloroperoxybenzoic acid (m-CPBA), affording the desired products (3a and 4a) in inferior yields and selectivities (Table 1, entries 5–7). Notably, the use of Mn(OAc)2 as additive further improved the yields of 3a (70% yield) and 4a (30% yield), although with poor selectivity (Table 1, entry 8). Evaluation of catalyst revealed that Co(salen) complex [Co-2] decreased the yield of the reaction yet increased the selectivity of 3a versus 4a, affording 3a in 60% yield (Table 1, entry 9). Switching the additive to Mn(OAc)2 tetrahydrate, which may facilitate dearomative addition of cobalt-hydride species to pyridinium salts, improved the yield of 3a to 70% (Table 1, entry 10), which may serve as base to promote the reaction (Supplementary Tables 1, 6). Further optimization furnished 3a in 95% yield with excellent regioselectivity (Table 1, entry 11). To our delight, the use of silver trifluoroacetate in the absence of cobalt catalyst completely shut down the formation of C-4 arylated product 3a, indication the formation of 3a is a Co-catalyzed process. Instead, C-6 arylated product 4a was exclusively formed in 48% yield (Table 1, entry 12). Testing other inorganic salts revealed silver free inorganic salts failed to deliver the desired product (Table 1, entries 13–16), indicating silver salt may serve as oxidant. Then, sulfonyl fluoride derivatives were evaluated as organic oxidant as well as hydride transfer promoter (Table 1, entries 17–20). C-6 arylated product 4a was exclusively formed in 99% yield as single regioisomer.
Scope for the reaction
With the optimized reaction conditions in hand, the scope for this regioselective and site-divergent dearomative reductive hydroarylation of pyridinium salts by reductive-oxidative relay with aryl nucleophiles was examined. First, the scope for Co-catalyzed C-4 selective hydroarylation was examined and the results are summarized in Fig. 2. Various arenes could efficiently react with pyridinium salts to give desired reductive C-4 arylated products 3 in good yields and excellent regioselectivities. Free indole was used as nucleophile to deliver 3b in 79% yield with 8.4:1 rr. This promising result prompted us to investigate substituent effects on the indole ring for the reaction. Substituents at 4-, 5-, 6-, or 7-positions of indole ring, including various electron-withdrawing (F, Cl, Br, I, and CO2Me) and electron-donating (Me, OMe and OBn) groups, were all compatible under the Co-catalysis conditions, affording corresponding C-4 arylated products of pyridinium salt (1a) in 49%–98% yields with 8.6:1 to >20:1 rr (3c–3l). Notably, more steric hindered free indoles with substituents at 2-position were well-tolerated in the reaction to deliver desired products in 39% and 76% yields, respectively (3m and 3n). Additionally, various N-alkylated indole derivatives bearing different functional groups, such as cyano, acetal, chloride, and ester, were all good substrates for Co-catalyzed reductive C-4 arylation of pyridinium salts, furnishing desired products (3o–3t) in high yields (77%–92%) with good to excellent levels of regioselectivity. It is noteworthy that the use of 1,3,5-trimethoxybenzene as nucleophile also resulted in C-4 arylated product 3u in 61% yield with >20:1 rr. More structurally complex cholesterol-derived unprotected indole was successfully transformed to corresponding C-4 arylated product 3v in 64% yield with excellent regioselectivity. In addition, methyl acetoacetate could also react with pyridinium salts (1a) to deliver the C-4 selective hydroalkylation product 3w in 50% yield with >20:1 rr and 1.2:1 dr. Other heterocycles, such as 2-methoxythiophene, furnished the C-4 selective reductive hydroarylation product (3x) in 44% yield with >20:1 rr.

Reaction conditions: the reaction was conducted using 1a (0.50 mmol) and 2 (0.20 mmol). a Yield based on recovery of starting materials.
Subsequently, we proceeded to explore the scope of aryl nucleophiles for reductive C-6 selective arylation of pyridinium salts under metal-free conditions. As shown in Fig. 3, the N-methylindole substrate exhibited excellent reactivity and regioselectivity, affording 4a in 94% yield with >20:1 rr. The structure of C6-arylation of pyridinium salts was unambiguously confirmed by the X-ray crystallographic analysis of 4a. Notably, the use of unprotected indoles as nucleophile smoothly resulted in reductive C-6 arylated of tetrahydropyridine 4b with 3-position of free indole in 86% yield with >20:1 rr. Next, various free unprotected indoles bearing different functional groups on C4-, C5-, C6-, and C7-positions were investigated for the dearomative reductive C6-arylation of pyridinium salt (1a). For example, C4-substituted unprotected indoles with electron-withdrawing or electron-donating groups gave C6-arylated products 4c–4e in 43%-75% yields. Moreover, C5-substituted free indoles bearing different substituents were good substrates for metal-free reductive C6-arylation, delivering corresponding products 4f–4h in 71%–82% yields. In addition, C6- and C7-substituted unprotected indole substrates were all successfully transformed to corresponding C6-arylated tetrahydropyridines (4i–4l) at 3-position of free indoles in 56%–94% yields. It was found that more steric hindered free indoles with substitution at C2-position worked well for the reaction, affording corresponding C6-arylated tetrahydropyridines (4m and 4n) in 96% and 84% yields, respectively. Next, a series of N-alkylated indoles were tested. A wide range of functional groups, such as nitriles, acetals, chlorides, esters, alkynes, and alkenes, were well-tolerated and remained untouched, affording corresponding C6-arylated tetrahydropyridines (4o–4w) at 3-position of free indoles in 60–98% yields. To our delight, other electron-rich arenes, such as 1,3,5-trimethoxybenzene, was also a feasible substrate, leading to formation of desired product 4x in 40% yield. Notably, unprotected indole based on complex molecules was smoothly transformed to give desired product 4y in 72% yield.

The reaction was conducted using 1a (0.40 mmol) and 2 (0.20 mmol).
Next, the scope of pyridinium salts was further investigated for both C4- and C6-selective reductive hydroarylation of pyridinium salts using 1-methylindole 2a (Fig. 4). N-Benzyl pyridinium salts bearing methyl or fluoride at the ortho or meta positions of the benzene ring worked smoothly to generate C-4 selective reductive hydroarylation products 5a and 5b in 90% and 91% yield with good regioselectivity. Meanwhile, corresponding C-6 selective reductive hydroarylation products 6a and 6b were also obtained in 32% and 89% yield, respectively. Moreover, N-2-naphthylmethyl pyridinium salt was efficiently transformed to C-4 selective reductive hydroarylation product 5c in 84% yield with > 20:1 rr and C-6 selective reductive hydroarylation product 6c in 91% yield. In addition, N-methyl pyridinium salt also reacted efficiently to generate C4- and C6-selective reductive hydroarylation products 5d and 6d in 44% and 72% yields. Notably, replacing the nitriles to other electron-withdrawing groups, such as esters and sulfones on pyridine ring were also tolerated, giving corresponding products 5e, 6e, and 6f in 49–98% yields. Substituents on different positions of pyridine were tested. The C5-methyl-substituted pyridinium salt showed good reactivity, affording corresponding products 5g and 6g in synthetic useful yields with moderate to good levels of regioselectivity. In comparable, the C6-methyl-substituted pyridinium salt gave C-6 selective reductive hydroarylation product 6h with a quaternary carbon center in 68% yield, yet no C-4 selective reductive hydroarylation product 5h was obtained. Unfortunately, pyridinium salts without electron-withdrawing group failed to deliver C-4 or C-6 selective reductive hydroarylation products.

aReaction was conducted using 1 (0.5 mmol, 2.5 equiv), 2a (0.2 mmol, 1.0 equiv), [Co-2] (0.006 mmol, 3 mol%), TBPB (0.6 mmol, 3.0 equiv), Mn(OAc)2.4H2O (0.12 mmol, 0.6 equiv) and PhMe2SiH (4.0 equiv) in CH3CN (2.0 mL) at rt for 12 h. bReaction was conducted using 1 (0.4 mmol, 2.0 equiv), 2a (0.2 mmol, 1.0 equiv), A (0.4 mmol, 2.0 equiv) and PhMe2SiH (4.0 equiv) in CH3CN (2.0 mL) at rt for 12 h.
Mechanistic studies and proposed mechanism
Next, preliminary mechanistic investigations have been conducted to probe possible mechanisms for regiodivergent reductive hydroarylations of pyridinium salts under both Co-catalyzed and stoichiometric metal-free conditions. As depicted in Fig. 5, the reaction of pyridinium salt 1a and 2-methyl indole 2n in the presence of Ph2SiD2 produced the corresponding reductive C-4 selective hydroarylation product 7 with four deuterium atoms incorporated on the C5 and C6-positions of piperidine skeleton with equal distribution, indicating that iterative migratory insertion of Co-H to pyridinium ring may be involved without face selectivity. In addition, the reductive C-6 selective hydroarylation product 8 under stoichiometric metal-free conditions was successfully formed in 94% yield from the reaction of pyridinium salt 1a and 1-methyl indole 2a in the presence of Ph2SiD2 with only one deuterium atom incorporated on the C5-position of piperidine ring.

Control experiments to probe the reaction mechanism.
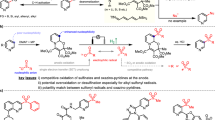
Based on literature precedence and experimental results62,63,64,65,66, possible cycles for Co-catalyzed reductive C4-selective hydroarylation and metal-free reductive C6-selective hydroarylation of pyridinium salts are proposed and depicted in Fig. 6. In terms of Co-catalyzed reductive C4-selective hydroarylation, the reaction begins with the formation of cobalt (III) intermediate D from cobalt (II) via SET process using TBPB as the oxidant, which was accompanied by production of a tert-butoxy radical. Transmetalation of D with silane delivers a Co(III)-H species E65. Subsequently, site- and regioselective addition of Co(III)-H E to pyridinium salt 1 gives a dearomative intermediate F. Then, intermediate F further undergoes a regioselective migratory insertion with Co(III)-H species, providing an allylic cobalt(III) intermediate G62,63,64. Next, cobalt(IV) complex H could be formed via a SET oxidation process by oxygen radical63,64,65,66,67. Further nucleophilic displacement reaction of H in the presence of aryl nucleophiles 2 furnishes the final product 3 and regenerates Co(II) catalyst to close the catalytic cycle. Another alternative reaction pathway involved an extended iminium ion intermediate I from intermediate G, which was further attacked by indole 2 in the presence of a base to give the final product 3 is also possible. In terms of metal-free reductive C4-selective hydroarylation of pyridinium salts, the reaction begins with the facile decomposition of methyl 2,2-difluoro-2-(fluorosulfonyl)acetate to give difluoro(fluorosulfonyl)methanide resulting in formation of difluorocarbene, which can subsequently convert to trifluoromethyl anion and bromodifluoromethyl anion in the presence of halides, and then undergoes 1,4-addition of pyridinium salts 1, followed by SN2’ substitution with aryl nucleophiles 2 to provide intermediate J. Re-aromatization of J gives intermediate K. Next, the intermediate K undergoes hydrosilylation in the presence of silane to give intermediate L. Following desilylation of intermediate L in the presence of fluoride affords the reductive C-6 selective hydroarylation product 468,69.

A possible mechanism for C4- and C6-selective reduction hydroarylation of pyridinium salts under Co-catalysis and metal-free conditions.
In summary, a regioselective and site-divergent dearomative hydroarylation of pyridinium salts with arenes to tetrahydropyridines with different substitution patterns has been developed. C-4 selective reductive hydroarylation of pyridinium salts has been realized by Co-catalysis, and metal-free conditions enabled C-6 selective reductive arylation of pyridinium salts. The reaction features the switchable site-selectivity by using different reaction conditions, providing efficient access to arylated tetrahydropyridine derivatives with substitution at different positions in good yields with excellent levels of regioselectivity from identical starting materials.
Methods
General procedure 1
In a nitrogen-filled glovebox, [Co-2] (5.5 mg, 0.006 mmol, 3 mol%), pyridinium salt 1 (0.5 mmol, 2.5 equiv), Mn(OAc)2•4H2O (29.4 mg, 0.12 mmol, 0.6 equiv) were dissolved in CH3CN (2.0 mL) in a screw-cap Schlenk tube equipped with a magnetic stirrer. To the mixture was added nucleophile 2 (0.2 mmol, 1.0 equiv), and the mixture was stirred at room temperature for 5 min, followed by addition of TBPB (105.9 μL, 0.6 mmol, 3.0 equiv) and PhMe2SiH (122.0 μL, 0.8 mmol, 4.0 equiv). The resulting mixture was stirred at room temperature for 12 h. The reaction was quenched with water (10.0 mL) and extracted with ethyl acetate (3 × 10.0 mL). The combined organic phases were dried over Na2SO4, filtered, concentrated under reduced pressure, and purified by flash chromatography with silica gel to give the pure product.
General procedure 2
In a nitrogen-filled glovebox, pyridinium salt 1 (0.4 mmol, 2.0 equiv) and nucleophile 2 (0.2 mmol, 1.0 equiv) were dissolved in CH3CN (2.0 mL) in a screw-cap Schlenk tube equipped with a magnetic stirrer. To the mixture were added methyl 2,2-difluoro-2-(fluorosulfonyl)acetate (47.5 μL, 0.4 mmol, 2.0 equiv) and PhMe2SiH (122.0 μL, 0.8 mmol, 4.0 equiv). The resulting mixture was stirred at room temperature for 12 h. The reaction was quenched with water (10.0 mL), extracted with ethyl acetate (3 × 10.0 mL). The combined organic phases were dried over Na2SO4, filtered, concentrated under reduced pressure, and purified by flash chromatography with silica gel to give the pure product.
Data availability
The authors declare that all other data supporting the findings of this study are available within the article and Supplementary information files, and also are available from the corresponding author upon request. The X-ray crystallographic coordinates for structures reported in this study have been deposited at the Cambridge Crystallographic Data Centre (CCDC), under deposition numbers CCDC 2296932 (4a). These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.
References
Nakao, A. et al. Tetrahydropyridine derivatives with inhibitory activity on the production of proinflammatory cytokines: part 1. Bioorg. Med. Chem. Lett. 19, 4607–4610 (2009).
Wichitnithad, W., O’Callaghan, J. P., Miller, D. B., Train, B. C. & Callery, P. S. Time-dependent slowly-reversible inhibition of monoamine oxidase A by N-substituted 1,2,3,6-tetrahydropyridines. Bioorg. Med. Chem. 19, 7482–7492 (2011).
Chang, J. W. et al. Selective inhibitor of platelet-activating factor acetylhydrolases 1b2 and 1b3 that impairs cancer cell survival. ACS Chem. Biol. 10, 925–932 (2015).
Zhu, Y.-J. et al. Approach to thiazole-containing tetrahydropyridines via aza–Rauhut–Currier reaction and their potent fungicidal and insecticidal activity. RSC Adv. 6, 112704–112711 (2016).
Wu, W. et al. The discovery of tetrahydropyridine analogs as hNav1.7 selective inhibitors for analgesia. Bioorg. Med. Chem. Lett. 27, 2210–2215 (2017).
Halicki, P. C. B. et al. Intramacrophage potential of a tetrahydropyridine: a promising compound in combating Mycobacterium tuberculosis. Tuberculosis 136, 102252 (2022).
Felpin, F.-X. & Lebreton, J. J. C. Synthesis of 2, 6-dialkyl-1, 2, 5, 6-tetrahydropyridines and their applications in total synthesis. Curr. Org. Synth. 1, 83–109 (2004).
Khan, M. M., Khan, S., Saigal, S. & Iqbal, S. Recent developments in multicomponent synthesis of structurally diversified tetrahydropyridines. RSC Adv. 6, 42045–42061 (2016).
Dudognon, Y., Rodriguez, J., Constantieux, T. & Bugaut, X. Organocatalytic enantioselective synthesis of tetrahydropyridines. Eur. J. Org. Chem. 2018, 2432–2442 (2018).
Verma, A., Bharti, R. & Sharma, R. Effect of methods and catalysts on the one-pot synthesis of tetrahydropyridine derivatives: a mini-review. Orbital: Electron. J. Chem. 13, 335–349 (2021).
Cheng, J., Tang, X., Yu, Y. & Ma, S. FeCl3-catalyzed cyclization of α-sulfonamido-allenes with aldehydes-the substituent effect. Chem. Commun. 48, 12074–12076 (2012).
Subba Reddy, B. V., Nair, P. N., Antony, A., Lalli, C. & Grée, R. The aza-Prins reaction in the synthesis of natural products and analogues. Eur. J. Org. Chem. 14, 1805–1819 (2017).
Talavera-Alemán, A., Marrot, J., Dagousset, G. & Thomassigny, C. Synthesis of nitrogen-and oxygen-containing heterocycles by Prins cyclization in continuous flow. Synthesis 53, 1478–1488 (2021).
Larsen, S. D. & Grieco, P. A. Aza Diels-Alder reactions in aqueous solution: cyclocondensation of dienes with simple iminium salts generated under Mannich conditions. J. Am. Chem. Soc. 107, 1768–1769 (1985).
He, L. et al. Highly enantioselective aza-Diels-Alder reaction of 1-azadienes with enecarbamates catalyzed by chiral phosphoric acids. Angew. Chem. Int. Ed. 125, 11294–11297 (2013).
Wu, Y.-K., Rawal, V. H. & Chemistry, B. Rapid construction of tetrahydropyridine scaffolds via formal imino Diels-Alder reactions of Schiff bases and Nazarov reagents. Org. Biomol. Chem. 17, 8827–8831 (2019).
Pandya, V. G. & Mhaske, S. B. Construction of tetrahydrobenzo [f] quinoline scaffolds via polar [4 + 2]-cycloaddition reaction with arynes as dienophiles. Tetrahedron Lett. 101, 153901 (2022).
Yang, D. et al. Catalyst-free inverse-electron-demand aza-Diels-Alder reaction of 4, 4-dicyano-2-methylenebut-3-enoates and 1, 3, 5-triazinanes: access to polysubstituted tetrahydropyridines. Org. Biomol. Chem. 21, 5413–5418 (2023).
Huo, L., Ma, A., Zhang, Y. & Ma, D. Assembly of 4-substituted 3-nitro-1,2,3,4-tetrahydropyridines via organocatalytic Michael addition. Adv. Synth. Catal. 354, 991–994 (2012).
Roy, T. K., Parhi, B. & Ghorai, P. Cinchonamine squaramide catalyzed asymmetric aza-Michael reaction: dihydroisoquinolines and tetrahydropyridines. Angew. Chem. Int. Ed. 57, 9397–9401 (2018).
Song, Y. X. & Du, D. M. Recent advances in catalytic asymmetric aza-Michael addition triggered cascade reactions. Adv. Synth. Catal. 363, 4667–4694 (2021).
Shi, S. et al. C(sp3)-H fluorosulfonylvinylation/aza-Michael addition approach to FSO 2-functionalized tetrahydropyridines. Org. Chem. Front. 10, 3805–3810 (2023).
Ma, L. et al. FeCl3-catalyzed ring-closing carbonyl-olefin metathesis. Angew. Chem. Int. Ed. 55, 10410–10413 (2016).
Rykaczewski, K. A. et al. Tetrahydropyridines via FeCl3-catalyzed carbonyl-olefin metathesis. Org. Lett. 22, 2844–2848 (2020).
Ding, Q., Zhou, X. & Fan, R. Recent advances in dearomatization of heteroaromatic compounds. Org. Biomol. Chem. 12, 4807–4815 (2014).
Aleksiev, M. & García Mancheño, O. Enantioselective dearomatization reactions of heteroarenes by anion-binding organocatalysis. Chem. Commun. 59, 3360–3372 (2023).
Ji, P. et al. Photochemical dearomative skeletal modifications of heteroaromatics. Chem. Soc. Rev. 53, 6600–6624 (2024).
Escolano, M. et al. Recent strategies in the nucleophilic dearomatization of pyridines, quinolines, and isoquinolines. Chem. Rev. 124, 1122–1246 (2024).
Glorius, F., Spielkamp, N., Holle, S., Goddard, R. & Lehmann, C. W. Efficient asymmetric hydrogenation of pyridines. Angew. Chem. Int. Ed. 43, 2850–2852 (2004).
Piras, L., Genesio, E., Ghiron, C. & Taddei, M. J. S. Microwave-assisted hydrogenation of pyridines. Synlett 8, 1125–1128 (2008).
Liu, Y. & Du, H. Metal-free borane-catalyzed highly stereoselective hydrogenation of pyridines. J. Am. Chem. Soc. 135, 12968–12971 (2013).
Tian, J. J. et al. Borane-catalyzed chemoselective and enantioselective reduction of 2-vinyl-substituted pyridines. Angew. Chem. Int. Ed. 59, 18452–18456 (2020).
Clarke, J. J., Maekawa, Y., Nambo, M. & Crudden, C. M. Borenium-catalyzed reduction of pyridines through the combined action of hydrogen and hydrosilane. Org. Lett. 23, 6617–6621 (2021).
Yang, Z.-Y., Luo, H., Zhang, M. & Wang, X.-C. Borane-catalyzed reduction of pyridines via a hydroboration/hydrogenation cascade. ACS Catal. 11, 10824–10829 (2021).
Arrowsmith, M., Hill, M. S., Hadlington, T., Kociok-Köhn, G. & Weetman, C. Magnesium-catalyzed hydroboration of pyridines. Organometallics 30, 5556–5559 (2011).
Lei, A., Chen, M., He, M. & Zhang, X. Asymmetric hydrogenation of pyridines: enantioselective synthesis of nipecotic acid derivatives. Eur. J. Org. Chem. 19, 4343–4347 (2006).
Rueping, M. & Antonchick, A. P. Organocatalytic enantioselective reduction of pyridines. Angew. Chem. Int. Ed. 46, 4562–4565 (2007).
Wang, X.-B., Zeng, W. & Zhou, Y.-G. Iridium-catalyzed asymmetric hydrogenation of pyridine derivatives, 7,8-dihydro-quinolin-5 (6H)-ones. Tetrahedron Lett. 49, 4922–4924 (2008).
Tang, W. J. et al. Highly enantioselective hydrogenation of quinoline and pyridine derivatives with iridium-(P-Phos) catalyst. Adv. Synth. Catal. 352, 1055–1062 (2010).
Tang, W. et al. Highly efficient and enantioselective hydrogenation of quinolines and pyridines with Ir-difluorphos catalyst. Org. Biomol. Chem. 8, 3464–3471 (2010).
Bull, J. A., Mousseau, J. J., Pelletier, G. & Charette, A. B. Synthesis of pyridine and dihydropyridine derivatives by regio- and stereoselective addition to N-activated pyridines. Chem. Rev. 112, 2642–2713 (2012).
Fernández-Ibáñez, M. Á, Maciá, B., Pizzuti, M. G., Minnaard, A. J. & Feringa, B. L. Catalytic enantioselective addition of dialkylzinc reagents to N-acylpyridinium salts. Angew. Chem. Int. Ed. 41, 9339–9341 (2010).
Chau, S. T., Lutz, J. P., Wu, K. & Doyle, A. G. Nickel-catalyzed enantioselective arylation of pyridinium ions: harnessing an iminium ion activation mode. Angew. Chem. Int. Ed. 52, 9153–9156 (2013).
De, N. et al. Cu (I)-catalyzed enantioselective [5 + 1] cycloaddition of N-aromatic compounds and alkynes via chelating-assisted 1, 2-dearomative addition. ACS Catal. 10, 10905–10913 (2020).
Nallagonda, R. & Karimov, R. R. Copper-catalyzed regio-and diastereoselective additions of boron-stabilized carbanions to heteroarenium salts: synthesis of azaheterocycles containing contiguous stereocenters. ACS Catal. 11, 248–254 (2020).
Robinson, D. J., Spurlin, S. P., Gorden, J. D. & Karimov, R. R. Enantioselective synthesis of dihydropyridines containing quaternary stereocenters through dearomatization of pyridinium salts. ACS Catal. 10, 51–55 (2019).
Nallagonda, R., Musaev, D. G. & Karimov, R. R. Light-promoted dearomative cross-coupling of heteroarenium salts and aryl iodides via nickel catalysis. ACS Catal. 12, 1818–1829 (2022).
Yang, Y., Xu, C.-H., Teng, F. & Li, J.-H. Catalysis dearomatization-enabled visible-light-induced 1,2-alkylsulfonylation of alkenes using sodium sulfinates and pyridinium salts. Adv. Synth. Catal. 362, 3369–3373 (2020).
Xu, C.-H., Li, J.-H., Xiang, J.-N. & Deng, W. Merging photoredox/nickel catalysis for cross-electrophile coupling of aziridines with pyridin-1-ium salts via dearomatization. Org. Lett. 23, 3696–3700 (2021).
O’Brien, L., Argent, S. P., Ermanis, K. & Lam, H. W. Gold(I)-catalyzed nucleophilic allylation of azinium ions with allylboronates. Angew. Chem. Int. Ed. 61, e202202305 (2022).
Somprasong, S., Castiñeira Reis, M. & Harutyunyan, S. R. Grignard reagent addition to pyridinium salts: a catalytic approach to chiral 1,4-dihydropyridines. ACS Catal. 14, 13030–13039 (2024).
Comins, D. L. & Abdullah, A. H. Synthesis of 1-acyl-1,4-dihydropyridines via copper hydride reduction of 1-acylpyridinium salts. J. Org. Chem. 49, 3392–3394 (1984).
Comins, D. L. & Mantlo, N. B. Stereocontrolled preparation of cis- and trans-2,6-dialkylpiperidines via 1-acyldihydropyridine intermediates. Synthesis of (±)-solenopsin A and (±)-dihydropinidine. J. Org. Chem. 56, 2506–2512 (1991).
Comins, D. L., Zhang, Y.-M. & Joseph, S. P. Enantiopure N-acyldihydropyridones as synthetic intermediates: asymmetric synthesis of benzomorphans. Org. Lett. 1, 657–660 (1999).
Chia, W.-L., Shen, S.-W. & Lin, H.-C. Novel synthesis of liquid crystalline compounds of 5-substituted 2-(4-alkylphenyl) pyridines. Tetrahedron Lett. 42, 2177–2179 (2001).
Sun, Z., Yu, S., Ding, Z. & Ma, D. Enantioselective addition of activated terminal alkynes to 1-acylpyridinium salts catalyzed by Cu-bis (oxazoline) complexes. J. Am. Chem. Soc. 129, 9300–9301 (2007).
Donohoe, T. J., Connolly, M. J. & Walton, L. Regioselective nucleophilic addition to pyridinium salts: a new route to substituted dihydropyridones. Org. Lett. 11, 5562–5565 (2009).
Bertuzzi, G. et al. Nucleophilic dearomatization of pyridines under enamine catalysis: regio-, diastereo-, and enantioselective addition of aldehydes to activated N-alkylpyridinium salts. Org. Lett. 19, 834–837 (2017).
Flanigan, D. M. & Rovis, T. Enantioselective N-heterocyclic carbene-catalyzed nucleophilic dearomatization of alkyl pyridiniums. Chem. Sci. 8, 6566–6569 (2017).
McLaughlin, C., Bitai, J., Barber, L. J., Slawin, A. M. & Smith, A. D. Catalytic enantioselective synthesis of 1, 4-dihydropyridines via the addition of C (1)-ammonium enolates to pyridinium salts. Chem. Sci. 12, 12001–12011 (2021).
Patra, K., Reddy, M. K., Mallik, S. & Baidya, M. Divergent reaction of activated pyridines with α,α-difluorinated gem-diols: regioselective synthesis of gem-difluorinated dihydropyridines and dihydropyridones. Org. Lett. 24, 4014–4018 (2022).
Discolo, C. A., Touney, E. E. & Pronin, S. V. Catalytic asymmetric radical-polar crossover hydroalkoxylation. J. Am. Chem. Soc. 141, 17527–17532 (2019).
Ebisawa, K. et al. Catalyst- and silane-controlled enantioselective hydrofunctionalization of alkenes by cobalt-catalyzed hydrogen atom transfer and radical-polar crossover. J. Am. Chem. Soc. 142, 13481–13490 (2020).
Qin, T. et al. Cobalt-catalyzed radical hydroamination of alkenes with N-fluorobenzenesulfonimides. Angew. Chem. Int. Ed. 60, 25949–25957 (2021).
Miao, H., Guan, M., Xiong, T., Zhang, G. & Zhang, Q. Cobalt-catalyzed enantioselective hydroamination of arylalkenes with secondary amines. Angew. Chem. Int. Ed. 62, e202213913 (2023).
Wilson, C. V. et al. Cobalt–carbon bonding in a salen-supported cobalt(IV) alkyl complex postulated in oxidative MHAT catalysis. J. Am. Chem. Soc. 144, 10361–10367 (2022).
Qin, T. et al. Cobalt-catalyzed asymmetric alkylation of (hetero) arenes with styrenes. Angew. Chem. Int. Ed. 61, e202201967 (2022).
Carreras, V. & Ollevier, T. Fluoride-triggered synthesis of 1-aryl-2,2-difluoroalkenes via desilylative defluorination of (1-aryl)-2,2,2-trifluoroethyl-silanes. J. Org. Chem. 86, 13160–13168 (2021).
Kubota, K., Kondo, K., Seo, T., Jin, M. & Ito, H. Solid-state mechanochemical cross-coupling of insoluble substrates into insoluble products by removable solubilizing silyl groups: uniform synthesis of nonsubstituted linear oligothiophenes. RSC Adv. 13, 28652–28657 (2023).
Acknowledgements
This work is dedicated to the memory of Prof. Qing-Yun Chen. Financial support from the National Natural Science Foundation of China (22171127, 22371115, 22373056), Natural Science Foundation of Sichuan Province (2025ZNSFSC0128), Shenzhen Science and Technology Innovation Committee (JCYJ20230807093522044, JCYJ20240813094226034), Opening Project of Innovation Center for Chenguang High Performance Fluorine Material (SCFY2404), Sichuan Science and Technology Program (2024ZYD0017), The Pearl River Talent Recruitment Program (2019QN01Y261), Guangdong Provincial Key Laboratory of Catalysis (2020B121201002) is sincerely acknowledged. We acknowledge the assistance of the SUSTech Core Research Facilities. We thank Hai-Wu Du for the X-ray analysis of 4a (CCDC 2296932) and Mr. Zhang-Yin Yuan (SUSTech) for reproducing the results of 3i and 5d.
Author information
Authors and Affiliations
Contributions
W.S. conceived and directed the project. S.W. discovered the reaction. X.F.W. and S.W. developed the reaction, performed the experiments, and collected the data. Q.Y. and Y.L.L. co-directed the investigations. W.S., X.F.W. and Q.Y. analyzed the data and wrote the manuscript with contributions from other authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wei, XF., Wang, S., Li, YL. et al. Regioselective and site-divergent synthesis of tetrahydropyridines by controlled reductive-oxidative dearomative hydroarylation of pyridinium salts. Nat Commun 16, 10712 (2025). https://doi.org/10.1038/s41467-025-65735-7
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-65735-7
