
Abstract
HIV-1 envelope glycoproteins (Env) from primary HIV-1 isolates typically adopt a pretriggered “closed” conformation that resists to CD4-induced (CD4i) non-neutralizing antibodies (nnAbs) mediating antibody-dependent cellular cytotoxicity (ADCC). CD4-mimetic compounds (CD4mcs) “open-up” Env allowing binding of CD4i nnAbs, thereby sensitizing HIV-1-infected cells to ADCC. Two families of CD4i nnAbs, the anti-cluster A and anti-coreceptor binding site (CoRBS) Abs, are required to mediate ADCC in combination with the indane CD4mc BNM-III-170. Recently, new indoline CD4mcs with improved potency and breadth have been described. Here, we show that the lead indoline CD4mc, CJF-III-288, sensitizes HIV-1-infected cells to ADCC mediated by anti-CoRBS Abs alone, contributing to improved ADCC activity. When administrated along with the anti-CoRBS 17b, CJF-III-288 delayed viral rebound after ART interruption in HIV-1-infected humanized mice, demonstrating potential for eliciting ADCC in vivo. Structural and conformational analyses reveal that CJF-III-288, in combination with this anti-CoRBS Abs, potently stabilizes an asymmetric “open” State-3 Env conformation. This Env conformation orients the anti-CoRBS Ab to improve ADCC activity and therapeutic potential.
Similar content being viewed by others


Introduction
Combination antiretroviral therapy (cART) can inhibit multiple stages of the HIV-1 life cycle, suppressing viral replication and preventing the progression to AIDS in people living with HIV-1 (PLWH)1,2. However, cART alone does not eradicate the virus. Due to the persistence of a latent reservoir, lifelong cART is required to prevent viral rebound3,4,5,6. New strategies aimed at targeting and eliminating HIV-1-infected cells are needed to achieve a cure. Antibody-dependent cellular cytotoxicity (ADCC) represents one of the primary effector mechanisms used by the immune system to clear infected cells. This response relies on the ability of antibodies (Abs) to bind viral antigens on the surface of infected cells, thereby facilitating their recognition and destruction by immune effector cells such as natural killer (NK) cells. The HIV-1 envelope glycoprotein (Env) trimer represents the only viral protein exposed on the surface of HIV-1-infected cells and therefore serves as the primary target for ADCC-mediating antibodies7,8.
Env is derived from a gp160 precursor, which is synthesized, trimerized, and glycosylated in the host cell’s endoplasmic reticulum7,9. Trimeric Env is subsequently cleaved by host furin-like proteases as it traffics through the Golgi apparatus on its way to the cell surface10,11,12. The cleaved mature Env trimer is composed of three exterior gp120 subunits non-covalently associated with three transmembrane gp41 subunits13,14,15. This trimeric Env is dynamic, transitioning from a pretriggered “closed” (State 1) to a “fully open” (State 3) conformation upon interaction with the viral receptor CD416,17,18. During viral entry, CD4 engages the gp120 subunit by inserting phenylalanine 43 (Phe43) of domain 1 into a highly conserved pocket located at the interface of the inner and outer gp120 domains, known as the Phe43 cavity19. CD4 interaction stabilizes downstream Env conformations, leading to a progression from State 1, through an intermediate (State 2), to the fully open State-3 conformation16,17,20.
Unliganded Envs from most primary HIV-1 isolates naturally assume a “closed” State-1 conformation16, which is resistant to non-neutralizing antibodies (nnAbs) commonly elicited in the majority of PLWH21,22. Although these nnAbs have the potential to eliminate infected cells via ADCC, they target epitopes that only become accessible in “open” conformations, upon Env-CD4 interaction22,23. HIV-1 has evolved Nef and Vpu proteins that limit this possibility by downregulating CD4, thereby protecting HIV-1-infected cells from ADCC mediated by CD4-induced (CD4i) nnAbs22,23,24. By driving Env to downstream “open” conformation(s), CD4-mimetic compounds (CD4mcs) expose vulnerable CD4i Env epitopes and sensitize HIV-1-infected cells to ADCC mediated by Abs present in the plasma from PLWH25,26,27,28,29,30.
Two families of CD4i nnAbs, the anti-cluster A and anti-coreceptor binding site (CoRBS) Abs, have been shown to mediate potent ADCC when combined with BNM-III-170, a lead CD4mc compound of the indane class31,32. This sensitization of infected cells to ADCC requires a sequential opening of the Env trimer32. In contrast to membrane-bound CD4, BNM-III-170 is unable on its own to fully “open up” the trimer31. However, BNM-III-170 efficiently exposes the CoRBS and subsequent binding of anti-CoRBS Abs further opens the Env trimer, exposing the inner domain of gp120 and enabling the binding of anti-cluster A Abs32. Of note, it is well described in the literature that anti-CoRBS, on their own, poorly mediate ADCC32,33,34,35,36.
Recently, more potent CD4mcs have been developed26. These new CD4mcs, based on an indoline scaffold, exhibit increased potency and breadth in preventing viral entry over the previous lead indane CD4mc, BNM-III-170. This enhanced antiviral activity is likely due to more favorable π–π overlap from the indoline pose and better contacts with the vestibule of the CD4-binding pocket on gp12026. The lead indoline CD4mc, CJF-III-288, also potently sensitizes HIV-1-infected cells to ADCC mediated by plasma from PLWH 26, 30.
In the present study, we aimed to gain a deeper understanding of the mechanism underlying this improved activity. Unlike previous indane CD4mcs, we found that CJF-III-288 enables anti-CoRBS Abs to mediate ADCC in the absence of anti-cluster A Abs, contributing to its improved ADCC activity. Mechanistically, we found that CJF-III-288 binding is associated with the stabilization of an asymmetric “open” State-3 Env conformation and a modification of anti-CoRBS Ab binding orientation.
Results
CJF-III-288 enables anti-CoRBS Abs to mediate ADCC
To better characterize the ADCC-stimulating potential of the lead indoline CD4mc CJF-III-288, we compared its activity in combination with an anti-cluster A/anti-CoRBS Ab cocktail to that of the previous indane CD4mc BNM-III-17032. Activated human primary CD4 + T cells were infected with the transmitted/founder virus CH058 (CH058TF). Two days post-infection, the ability of anti-cluster A Abs (A32 or N5i5) and anti-CoRBS Ab (17b), to recognize and eliminate the HIV-1-infected cells by ADCC, alone or in combination, was evaluated by flow cytometry. As previously reported32,33, BNM-III-170 failed to expose the cluster A epitope on its own, but significantly improved 17b binding (Fig. 1a). Since 17b engagement exposes the cluster A epitope, more antibody binding was observed upon anti-cluster A addition. Despite efficiently recognizing HIV-1-infected cells in the presence of BNM-III-170, 17b failed to mediate ADCC alone (Fig. 1b). Elimination of infected cells by ADCC was only detected when 17b and an anti-cluster A Ab were combined together with BNM-III-170 (Fig. 1b). Intriguingly, despite similar levels of 17b binding upon CJF-III-288 addition, this antibody was able to mediate ADCC in the presence of this CD4mc alone (Fig. 1b). As observed with BNM-III-170, CJF-III-288 and 17b addition enabled anti-cluster A Ab interaction (Fig. 1a and Supplementary Fig. 1). Importantly, these phenotypes were consistent for three additional infectious molecular clones (IMC) (HIV-1JRFL, HIV-1AD8 and HIV-1JRCSF) (Fig. 1c, d) and for different anti-CoRBS Abs (N12i2, X5, C2, 412D, and 48D) (Fig. 1e, f). The capacity to sensitize HIV-1-infected cells to ADCC mediated by anti-CoRBS Abs was specific to the indoline CD4mc CJF-III-288 and was not observed with the indane (BNM-III-170) or the piperidine (TFH-I-116-D1) CD4mc37 (Fig. 1e, f). These results indicate that the indoline CD4mc CJF-III-288 allows anti-CoRBS Abs to mediate ADCC against HIV-1-infected cells.

a Recognition and b ADCC-mediated elimination of primary CD4 + T cells infected with HIV-1CH058TF by anti-cluster A Abs (either A32 or N5i5) (n = 9) and anti-CoRBS Abs (17b) (n = 10), alone or in combination (n = 9) (at 5 µg/ml total concentration), in the presence of 50 µM of indicated CD4mc or equivalent volume of DMSO. Data are presented as mean values +/- SEM. c Recognition and d ADCC-mediated elimination of primary CD4 + T cells infected with indicated IMCs by anti-cluster A (either A32 or N5i5) and anti-CoRBS (17b), alone or in combination, in the presence of 50 µM of indicated CD4mc or equivalent volume of DMSO. c, d Shown are the median fluorescence intensities (MFI) and percentage of ADCC obtained in at least 3 biological replicates for each IMC (HIV-1CH058TF n = 10, HIV-1JRFL n = 6, HIV-1AD8 n = 3, HIV-1JRCSF n = 5). e Recognition and f ADCC-mediated elimination of primary CD4 + T cells infected with HIV-1CH058TF by indicated anti-CoRBS Abs in the presence of 50 µM of indicated CD4mc or equivalent volume of DMSO. e, f Shown are the mean MFI and percentage of ADCC obtained from 4 biological replicates for each tested anti-CoRBS Abs (17b, N12i2, X5, C2, 412D, and 48D). Statistical significance was tested using a, b Two-way ANOVA test with a Tukey post-test, c–f one-way ANOVA with a Tukey post-test or Kruskal-Wallis test with a with a Holm-Sidak post-test based on the normality test. P values are shown in the graphs. Source data are provided as a Source Data file.
CJF-III-288 shows improved ADCC activity at low concentrations
We next compared the ability of CJF-III-288 and BNM-III-170 to induce recognition of infected cells and ADCC by anti-CoRBS Abs +/- anti-Cluster A Abs over a range of concentrations. Activated human primary CD4 + T cells were infected with the same four primary IMCs, and the ability of 17b and A32, alone or in combination, to recognize HIV-1-infected cells was assessed two days post-infection in the presence of increasing concentrations of each CD4mc. As presented in Fig. 2, both CD4mc similarly enhanced the ability of 17b alone or combined with A32, to recognize HIV-1-infected cells at the highest concentration tested (50 µM for JRFL, JRCSF and AD8). Of note, since CH058TF is more susceptible to CD4mc due to the presence of a threonine at position 37538, similar levels of binding were detected starting at 1 µM. At low concentrations (<10 µM for JRFL, JRCSF and AD8 and <1 µM for CH058TF), CJF-III-288 was more effective than BNM-III-170 (Fig. 2a, c) in enabling Abs to recognize infected cells. This improved potency was reflected in a significant increase in the area under the curve (AUC) of Ab binding for all tested IMCs (Fig. 2b, d). Supporting its improved potency, CJF-III-288 also exposed the CoRBS on Env from the clade A HIV-1BG505 virus, which was reported to be resistant to BNM-III-17026 (Supplementary Fig. 2). The improved potency of CJF-III-288 was also reflected in its capacity to sensitize infected cells to ADCC mediated by anti-CoRBS Abs alone or in combination with anti-Cluster A Abs. (Fig. 2e, g). This superior ADCC activity of CJF-III-288 against HIV-1CH058TF-infected cells was again reflected by a significant increase in the AUC (Fig. 2f, h). As expected, minimal recognition and little ADCC-mediated elimination of infected cells were observed with A32 in the absence of 17b (Supplementary Fig. 3).

a–d Recognition of primary CD4 + T cells infected with indicated IMC by anti-CoRBS Ab (17b) alone or in combination with anti-cluster A Ab (A32), in the presence of the indicated concentration of BNM-III-170 or CJF-III-288. a, c Graphs represent the median fluorescence intensities (MFI) obtained in at least 5 biological replicates with each IMCs (HIV-1CH058TF n = 8, HIV-1JRFL n = 5, HIV-1AD8 n = 5, HIV-1JRCSF n = 5). b, d Graphs represent the area under the curve (AUC) calculated from the MFI obtained in a and c. e–h ADCC-mediated elimination of CD4 + T cells infected HIV-1CH058TF by anti-CoRBS (17b) Abs alone (n = 4) or in combination with anti-cluster A Abs (A32) (n = 5), in the presence of indicated concentration of BNM-III-170 or CJF-III-288. e, g Shown are percentage of ADCC obtained in at least 4 experiments. f, h Shown are the area under the curve (AUC) calculated from the percentage of ADCC obtained in e and g. The dashed lines represent the mean binding or ADCC obtained in absence of CD4mc. Data are presented as mean values +/- SEM. Statistical significance was tested using a, c, e, g Two-way ANOVA test with a Holm-Sidak post-test, b, d, f, h two-sided paired t test or two-sided Wilcoxon test based on the normality test. P values are shown in the graphs. Source data are provided as a Source Data file.
To confirm these findings with primary clinical samples, CD4 + T cells were expanded from PLWH under ART, as previously described29,31,39. Viral replication was monitored over time by intracellular p24 staining. Upon expansion, CD4 + T cells were stained with nnAbs in the presence of the indicated CD4mc. Expanded endogenously-infected cells were also used as target cells and autologous peripheral blood mononuclear cells (PBMCs) as effectors using a FACS-based ADCC assay. Consistent with the results obtained with IMC infection, superior antibody binding and ADCC responses were observed with CJF-III-288 relative to BNM-III-170 at different concentrations (Fig. 3).

a–d Recognition of ex vivo-expanded CD4 + T cells from four PLWH under ART by anti-CoRBS Abs (17b) alone or in combination with anti-cluster A Abs (A32), in the presence of indicated concentration of BNM-III-170 or CJF-III-288. a, c Graphs represent MFI obtained with 4 donors (4 biological replicates). b, d Graphs represent the AUC calculated from the MFI obtained in a and c. e–h ADCC-mediated elimination of ex vivo-expanded CD4+T cells from three PLWH under ART by anti-CoRBS Abs (17b) alone or in combination with anti-cluster A Abs (A32), in the presence of the indicated concentration of BNM-III-170 or CJF-III-288. e, g Shown are percentage of ADCC obtained with 3 donors (3 biological replicates). f, h, Shown are the area under the curve (AUC) calculated from the percentage of ADCC obtained in e and g. The dashed lines represent the mean binding or ADCC obtained in the absence of CD4mc. Statistical significance was tested using a, c, e, g.Two-way ANOVA test with a Holm-Sidak post-test, b, d, f, h two-sided paired t test or two-sided Wilcoxon test based on the normality test. P values are shown in the graphs. Source data are provided as a Source Data file.
Anti-CoRBS Abs drive PLWH plasma-mediated ADCC in the presence of CJF-III-288
We next extended our analysis to plasma from PLWH. As observed with nnAbs, CJF-III-288 was more effective than BNM-III-170 in enhancing plasma binding and ADCC activity at low concentrations (Fig. 4a–d). To evaluate the contribution of anti-CoRBS Abs to the improved ADCC activity mediated by PLWH plasma in the presence of CJF-III-288, we performed a Fab fragment competition assay. Briefly, HIV-1-infected cells were preincubated with 17b Fab fragments in the presence of CJF-III-288 prior to incubation with autologous PBMCs and PLWH plasma in the FACS-based ADCC assay. In this experimental setting, the 17b Fab masks the CoRBS exposed by CJF-III-288 and prevents the binding and ADCC activity of anti-CoRBS Abs present in PLWH plasma. Preincubation with 17b Fab substantially reduced the ADCC responses mediated by PLWH plasma in the presence of CJF-III-288, thus confirming the key contribution of anti-CoRBS Abs in this response (Fig. 4e).

a, b Recognition of primary CD4 + T cells infected with indicated IMC by PLWH plasma, in the presence of indicated concentration of BNM-III-170 or CJF-III-288. a Shown are median fluorescence intensities obtained with plasma from at least 6 PLWH (6 biological replicates) (HIV-1CH058TF n = 8, HIV-1JRFL n = 6, HIV-1AD8 n = 6, HIV-1JRCSF n = 6). b Shown are the area under the curve (AUC) calculated from the MFI obtained in a. c, d ADCC-mediated elimination of HIV-1CH058TF-infected primary CD4 T cells by plasma from 8 PLWH in the presence of the indicated concentration of BNM-III-170 or CJF-III-288. c Shown are the percentages of ADCC obtained with plasma from 8 PLWH (8 biological replicates). d Shown are the AUC calculated from the ADCC values presented in c. The dashed lines represent the mean MFI or ADCC obtained in the absence of CD4mc. e ADCC-mediated elimination of HIV-1CH058TF-infected primary CD4 T cells preincubated or not with 17b Fab fragments, by plasma from 8 PLWH in the presence of 1 µM of BNM-III-170 or CJF-III-288 (8 biological replicates). Data are presented as mean values +/- SEM. Statistical significance was tested using a, c Two-way ANOVA test with a Holm-Sidak post-test, b, d two-sided paired t test or two-sided Wilcoxon test based on the normality test, e One-way ANOVA with a Holm-Sidak post-test. P values are shown in the graphs. Source data are provided as a Source Data file.
17b Ab, when combined with CJF-III-288, delays viral rebound after antiretroviral therapy interruption in HIV-1-infected humanized mice
To extend our in vitro findings to an in vivo setting, we evaluated viral rebound dynamics following analytical antiretroviral treatment interruption (ATI) in HIV-1JRCSF-infected humanized mice treated with CD4mc in combination with the anti-CoRBS Ab 17b. One day before ATI, humanized mice (fully suppressed for viremia for >3 weeks) were administered either BNM-III-170 or CJF-III-288 with 17b (BNM-III-170+17b and CJF-III-288+17b, respectively). Plasma viral loads were monitored longitudinally to assess the impact of these treatments on viral rebound in relation to a control cohort of mice that was mock-treated (Fig. 5a, schematic).

a Experimental outline. Cartoons were created in BioRender. Kumar, P. (2025) https://BioRender.com/y37a8as. Humanized NSG-Tg(IL15) mice were intraperitoneally infected with HIV-1JRCSF and assessed for plasma viral loads (PVLs) by quantitative real-time PCR. At PVLs of 104-105 copies/ml, mice were subcutaneously administered combination antiretroviral therapy (ART, consisting of tenofovir disoproxil fumarate, emtricitabine, and dolutegravir). PVLs were stably suppressed for >3 weeks on ART. A day before ART interruption, mice were divided into three groups: Mock-treated (black) (n = 2), BNM-III-170 + 17b (purple) (n = 4), and CJF-III-288 + 17b (cyan) (n = 3). The CD4mc were administered at 3, 3, 1, 1, and 1 mg on consecutive days from day 0 (D0) to day 4 (D4), and 17b antibody was administered at 1.5 mg on D0 and 0.75 mg on D2 and D4. b PVLs obtained post-ATI in individual mice from the mock-treated (black), BNM-III-170 + 17b (purple), and CJF-III-288 + 17b groups (cyan). PVL measurements for individual mice are shown as dashed lines, and mean values for each regimen are shown as solid lines. Source data for b are provided as a Source Data file.
Mock-treated mice exhibited rapid viral rebound, with viremia exceeding 10⁴ copies/ml plasma within the first five days of ATI (Fig. 5b). Mice in the BNM-III-170 + 17b-treated group showed relatively comparable viral rebound dynamics, with viremia detected within 5 days of ATI in two mice, mirroring the mock-treated group. The remaining two mice experienced viral rebound on days 9 and 13 post-ATI, respectively (Fig. 5b).
In contrast, CJF-III-288+17b-treated mice demonstrated a clear delay in viral rebound (Fig. 5b). Two of the mice in this group showed viral rebound at 13 days post-ATI, and a third mouse exhibiting an even more prolonged delay, with viral rebound occurring at 17 days post-ATI. These findings support the in vitro observations that CJF-III-288 sensitizes HIV-1-infected cells to anti-CoRBS Ab-mediated ADCC, contributing to delayed viral rebound following ATI in vivo.
CJF-III-288, in combination with anti-CoRBS Abs, stabilizes the State-3 Env conformation
Our functional data indicated that CJF-III-288 could stabilize Env in a conformation distinct from that induced by other CD4mc, including BNM-III-170. To determine the impact of CJF-III-288 on Env conformation and how it differs from the one induced by BNM-III-170, we used a modified version of a well-established single-molecule Förster Resonance Energy Transfer (smFRET) imaging assay16. Briefly, one fluorophore was attached to the V1 loop of HIV-1JRFL gp120 using amber stop codon suppression to introduce a non-natural amino acid (nnAA). The nnAA was then labeled by copper-free click chemistry with a tetrazine-conjugated fluorophore40. A second fluorophore was attached to the A1 peptide inserted into the V4 loop of gp12028. Pseudovirion infectivity assays demonstrated that these modifications to Env, including attachment of the fluorophores, did not affect Env function (Supplementary Fig. 4). Native virions incorporating a single labeled Env protomer were immobilized on quartz microscope slides and imaged using internal reflection fluorescence (TIRF) microscopy. Hidden Markov modeling (HMM) of the smFRET trajectories indicated the existence of four non-zero FRET states, and a 0-FRET state that corresponds to photobleaching (Supplementary Fig. 5). As previously reported16,17,28,31,41,42, smFRET analysis of unbound Env showed a predominant low-FRET state, consistent with the adoption of a “closed” State-1 Env conformation (Fig. 6a, c). Incubation of the virions with the CD4mcs BNM-III-170 or CJF-III-288 led to destabilization of State 1 and a shift towards more “open” downstream conformations, including the high-FRET States 2 and 2 A, and the intermediate-FRET State 3. This reduction of State 1 occupancy was significantly greater upon incubation with CJF-III-288 than with BNM-III-170 (Fig. 6c). Incubation with the anti-CoRBS Ab 17b alone had no effect on Env conformation. The combination of 17b and BNM-III-170 only modestly reduced State 1 occupancy, consistent with our previous results31. However, when combined with CJF-III-288, 17b significantly modified Env’s conformation, further reducing State 1 occupancy and favoring State 3 (Fig. 6a, c). Similarly, incubation of virions with CJF-III-288 in combination with PLWH plasma resulted in improved State 3 stabilization relative to BNM-III-170 (Fig. 6b, d). Stabilization of State 3, as well as State 1 destabilization by PLWH plasma correlated with ADCC (Fig. 6e). Of note, in presence of CJF-III-288, we noted a wide range of ADCC activities for a similar level of downstream conformational distribution (Fig.6e); this could be associated to varying levels of other classes of CD4i nnAbs present in plasma from PLWH, as recently reported30. Altogether, these results indicate that CJF-III-288, in combination with anti-CoRBS Abs or plasma from PLWH, shifts the conformational landscape from State 1 towards the fully open State 3 conformation. Stabilization of the State-3 conformation could sensitize HIV-1-infected cells to ADCC mediated by anti-CoRBS Abs alone.

a, b Histograms of FRET values compiled from the population of individual HIV-1JRFLEnv trimers on the virion surface in the absence or presence of CD4mc (BNM-III-170 or CJF-III-288), the anti-CoRBS Ab 17b, or PLWH plasma. Histograms are presented as the mean determined from three technical replicates with error bars reflecting the standard error. Overlaid on the histograms are Gaussian distributions determined from HMM analysis of the individual FRET trajectories. Conformational state labels (States 1, 2, 2 A, and 3), which have been previously identified16,31, are indicated on the corresponding Gaussian. c, d The occupancies in States 1 and State 3 were calculated from the HMM analysis for each trace and represented with violin plots (from three technical replicates). c The mean and median occupancy are shown as horizontal lines and circles, respectively. Vertical lines reflect the 25–75% quantiles. e Correlation between State 1 and State 3 occupancy and ADCC mediated by plasma from PLWH d, e Each shape represents a different plasma from PLWH. Statistical significance was tested using (c, d) one-way ANOVA with a Holm-Sidak post-test and (e) two-sided Spearman rank correlation test. P values are shown in the graphs. Source data for c, d, and e are provided as a Source Data file.
CJF-III-288 induces an asymmetric opening of Env that modifies the binding orientation of anti-CoRBS Abs
Previous studies have revealed that the ability of Abs to bind Fcγ receptors (FcγR) and activate ADCC is influenced by their binding orientation36,43. Based on this observation, we hypothesized that CJF-III-288 could stabilize Env in a conformation that modifies the orientation of anti-CoRBS Abs binding, thereby enhancing their ADCC activity. To gain molecular-level insights into potential differences in Env trimer architecture, specifically the geometry of trimer assembly and the orientation of anti-CoRBS antibodies bound to Env triggered by CJF-III-288, we employed molecular imaging methods, including single-particle Cryo-EM and CryoET. Using Cryo-EM, we determined a 3.5-Å structure of the BG505 sgp140 SOSIP.664 trimer in complex with CJF-III-288 and 17b Fab (Supplementary Table 1, Supplementary Figs. 6–10). We selected 17b as a representative anti-CoRBS Ab in the Cryo-EM studies to allow direct comparison to the complex of BNM-III-170-BG505 SOSIP.664-17b Fab determined previously using the same method by the Bjorkman laboratory44. Figure 7 shows the overall structure of the CJF-III-288-BG505 SOSIP.664-17b Fab complex that we determined using C1 symmetry. The overall density of gp120-gp41 protomers was well-defined with the some poorly resolved regions corresponding to the 17b Fab (Supplementary Fig. 7). There was high-resolution density within the Phe43 cavity in all gp120 protomers for CJF-III-288 (Supplementary Fig. 7d). The CJF-III-288-BG505 SOSIP.664-17b Fab trimer is an asymmetric assembly of the three protomers that differ mainly in the conformation of gp41 and the 17b Fab (only the variable portions of the Fabs were built into the model due to poor densities for the constant regions). Overall, the three CJF-III-288-BG505 SOSIP.664-17b Fab protomers can be superimposed with an average root-mean-square deviation (RMSD) for Cα atoms of 2.52 Å (the RMSD between protomer A and B, protomer A and C, and protomer B and C are 2.69 Å, 1.73 Å, and 3.14 Å respectively, Supplementary Fig. 10a). On the whole, the gp120 protomers are the best-defined portions of the complex with the conformation of residues forming the CJF-III-288 binding pocket within each protomer being well preserved (the RMSD between residues of the CJF-III-288 pockets of protomers A and B, protomers A and C, and protomer B and C are 0.565 Å, 0.563 Å, and 0.524 Å, respectively; Supplementary Fig. 10b). As previously described for CJF-III-288 in its complex with the gp120 coree26, the CJF-III-288 binding pocket includes the highly conserved S375 and D368, a continuous segment from G472 to R476, a continuous β20-β21 loop from I424 to I430, and the H105 of α1, which makes contacts with the propyl group of the compound (Fig. 7a, left blow-up view). Of note, there is a high degree of similarity between the BG505 SOSIP.664 and gp120 coree CJF-III-288 binding pockets (Supplementary Fig. 11).

a Left, the overall structure of CJF-III-288-BG505-17b Fab complex with a molecular surface displayed over gp120 and gp41, gp120 is colored dark green and gp41 is colored gray. The 17b Fab variable region is shown as a cartoon with darker and lighter shades of cyan for heavy and light chains, respectively. A side view of the complex is shown. Middle, a blow-up view shows the details of the CJF-III-288 binding pocket. Secondary structures are shown within the pocket. Residues forming the pocket are colored magenta and are labeled. Right, comparison of the two binding pockets of CJF-III-288 and BNM-III-170 bound to BG505 SOSIP.664 (as in PDB 7LO6)44. Pockets in protomer A in each complex were selected for the comparison. CJF-III-288 is shown as yellow sticks, and BNM-III-170 is shown as orange sticks. The gp120s of the CJF-III-288 and BNM-III-170 BG505 SOSIP.664 complexes are colored darker and lighter shades of green, respectively. b The residue-resolved buried-surface-area (BSA) of gp120 residues contributing to the CJF-III-288-protein and BNM-III-170-protein interfaces, as determined by PISA. BSA values represent the average of the three copies in the trimer. Residues present only in the CJF-III-288-BG505 SOSIP.664 complex are highlighted in magenta. Amino acids are named using the International Union of Pure and Applied Chemistry (IUPAC) code. Source data for b are provided as a Source Data file.
We next compared the binding modes of CJF-III-288 and BNM-III-170 within BG505 SOSIP.664 (Fig. 7a, right blow-up view) within a single gp120 protomer. The chemical structures of BNM-III-170 and CJF-III-288 are nearly identical except for the replacement of the indane in BNM-III-170 with an indoline in CJF-III-288 and the addition of a propyl carbamate in CJF-III-288; the replacement of the indane C3 with the indoline N3 affords the addition of this distinguishing propyl carbamate. The BG505 SOSIP.664-bound conformation of CJF-III-288 and BNM-III-170 is highly similar for the identical portions of both compounds with the exception of the positions of the dimethylamine attached to the C5 carbon and the guanidinium group attached to the C2 carbon that show different orientations. As a consequence of this conformational change, I430 and D474 of gp120 contribute more buried-surface area (BSA) to the CJF-III-288-BG505 SOSIP.664-17b Fab complex interface as compared to BNM-III-170 (Fig. 7b). As expected, CJF-III-288, which is modified with a propyl-carbamat attached to the N3 nitrogen of the indoline ring, contacts three more gp120 residues in its binding interface, specifically H105, Q428 and R476, that are not utilized by BNM-III-170. These contacts contribute an additional 30.71 Å2 of BSA to the CJF-III-288 binding interface. Overall, the complexes of CJF-III-288 and BNM-III-170 with the BG505 SOSIP.664-17b Fab are formed with a BSA of 980.0 Å2 and 818.4 Å2, respectively.
Next, we examined the details of the conformation of the trimer in the CJF-III-288-BG505 SOSIP.664-17b and BNM-III-170-BG505 SOSIP.664-17b complexes (Fig. 8). When the two complexes are aligned based upon gp120/gp41 protomers, protomer B and protomer C superimpose relatively well. However, protomer A did not align well, which confirms a noticeable difference in the trimer assembly and position of the 17b Fab. In the CJF-III-288-BG505 SOSIP.664-17b Fab complex, gp120 subunits displace away from the central gp41 helices as compared to trimer complex with BNM-III-170 (Fig. 8a, b). This displacement is evident for all three protomers to a variable extent, with the greatest rotation in protomer A. A hallmark of the trimer assembly changes includes the outward displacement of gp120 relative to the central gp41 helices, which can be easily measured by the change of position of the gp120 α0 helix that packs against the gp41 HR1 helix. To measure this displacement, we generated a series of centroids by calculating the average position of α-carbon atoms for residues 65-73 to represent the α0 helix in each protomer. In the CJF-III-288 structure, the α0 helix is displaced approximately 7.8 Å, 1.5 Å and 1.3 Å (for protomers A, B and C, respectively) away from corresponding α0 helix in the BNM-III-170 structure (Fig. 8b). We also created a centroid to represent gp41 center by calculating the average of the α-carbon positions of the central gp41 α7 helices, residues 570-595 from the three protomers. Using the gp41 center as a hinge point, we measured α0 helix angular displacement in the superposed CJF-III-288-BG505 SOSIP.664-17b and BNM-III-170-BG505 SOSIP.664-17b complexes. The angular displacements were 11°, 1.6°, and 0.1° in protomers A, B, and C, respectively. This confirms the dissimilarity of the overall trimer assembly and supports the notion that the CJF-III-288-BG505 SOSIP.66417b Fab trimer is more asymmetric and more ‘open’ as compared to its BNM-III-170-triggered counterpart. Most importantly, the differences in Env trimer assembly directly define the way in which the 17b IgG approaches the Env trimer. To quantify the differences in the angles of approach of the Fabs for each trimer protomer between the two complexes, we used the gp41 center as the hinge point and the centers of the Fabs to measure the angular displacements (Fig. 8a, b). The differences we measured vary for each gp120 protomer with angular 17b displacements for protomers A, B, and C of 10.4°, 1.5°, and 1°, respectively. It should be noted that the observed differences in the angle of 17b’s approach occur only in the context of the Env trimer as a whole and are the result of changes in the orientation of gp120 relative to the gp41 helices within the trimer. No changes were observed in the 17b epitope or its angle of approach to any of the three individual gp120 protomers when these were analyzed individually. The interactions of 17b Fab with gp120 are essentially identical to those observed in other Env-17b complex structures, including the BNM-III-170-BG505 SOSIP.664-17b complexes used here for comparison (Supplementary Fig. 12). Thus, this suggest that changes in the angle of approach of 17b to the Env trimer we describe here results solely from changes in the orientations of the gp120 protomers relative to the trimer gp41. Finally, we also compared the changes to the BG505 SOSIP trimer assembly induced by CJF-III-288 to those induced by other agents that target the CD4 binding site, including CD4 and other CD4mcs (Fig. 8c). The distance between the Phe43 pocket residue 375 and the angle of the gp120 protomer rotation in the CJF-III-288-BG505 SOSIP. 664-17b Fab complex differs from that in the BNM-III-170-BG505 SOSIP.664-17b complex. However, it closely resembles the asymmetric Env Conformation B of the CD4-BG505 SOSIP.664/E51 Fab complex, formed by E51, a tyrosine-sulfated CoRBS antibody that binds gp120, mimicking CCR5 interactions45. This analysis further demonstrates that CJF-III-288, in combination with 17b, stabilizes the Env trimer in a more asymmetric and “open” conformation compared to BNM-III-170 coupled with 17b. The observed changes in the overall trimer assembly are driven by alterations in the relative orientations of the gp120 protomers to gp41 within the BG505 SOSIP.664 trimer, with the most significant changes occurring in one protomer. Since the orientation of individual gp120 protomers within the trimer determines the angle of approach of 17b to the Env trimer, these angles can vary for each protomer.

a CJF-III-288-BG505 SOSIP.664-17b Fab and BNM-III-170-BG505 SOSIP.664-17b Fab (PDB:7LO6) complexes superimposed based upon gp120/gp41 protomers. Center, the gp120 and gp41 in the CJF-III-288 and BNM-III-170 complexes are shown as ribbons in darker and lighter shades of green/gray, respectively (the α0 helix is shown as red/blue). Right and left, blow-up views show the structural alignments of Protomers A, B, and C; the 17b Fab is colored light and dark cyan for the BNM-III-170 complex and red for the CJF-III-288 complex. The angle of approach for 17b in each complex is shown by a line drawn from the center of gp41 (calculated as the average of gp41 α-carbon positions for residues 570–595 in both structures) and the center of mass of the Fab variable domain (calculated as the average α-carbon position for heavy and light chain variable residues) with the distance between the Fab centers shown above by a black arrow. The relative Fab displacements for each protomer of the two aligned complexes were calculated as angles based upon three points, the centers of two adjacent Fabs and the center of gp41. The center of gp41 was used as the hinge point. b Changes to the opening of the timer induced by CJF-III-288 or BNM-III-170. The CJF-III-288-BG505 SOSIP.664 and BNM-III-170-BG505 SOSIP.664 trimers are shown superimposed based upon gp120/gp41 protomers (colored as in panel a). Relative bound positions of 17b Fab from each complex are shown with heavy- and light-chain (VH and VL) positions determined by the center of mass of their α-carbon atoms (displayed as balls). The distance between the centers of the α0-helices in the two structures (calculated as the average of α-carbon atoms for residues 65-73) in the superimposed trimers is shown above with a black arrow indicating the rotation of the helix. The relative α0-helix displacements for each protomer of the two aligned complexes were calculated as angles based upon three points, the centers of two adjacent α0-helices and the center of gp41. The center of gp41 was used as the hinge point. c Changes to overall Env trimer assembly, calculated based upon changes to the position of the α-carbon of S375 relative to the gp41 center (calculated as the average of the α-carbon positions of the central gp41 α7 helices, residues 570-595). Distances between the 375 Cα of each protomer (a–c), the 375Cα and the gp41 center (d–f) and the angle between the gp41 center and two neighboring 375 Cα’s (α, β, γ) are shown.
CryoET structure of HIV-1 Env in complex with CJF-III-288 and 17b Abs on virions confirms the asymmetric opening of Env in presence of CJF-III-288
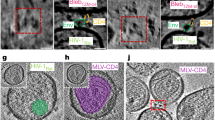
Next, we employed CryoET to assess 17b binding to Env on intact virions in the presence of CJF-III-288. AT-2 inactivated HIV-1BaL virus41,46,47 was incubated with 100 µM CJF-III-288, and subsequently incubated on ice with 100 µg/ml 17b Ab (full IgG) before being plunge frozen and analyzed by CryoET. 17b Ab binding to Env trimers was apparent in tomographic slices (Fig. 9a). Subtomogram averaging and classification was performed to solve the structure of Env bound to three 17b Abs (21.5 Å resolution, C1 symmetry) (Fig. 9b, c, Supplementary Fig. 13a–f, Supplementary Table 2). Due to high structural heterogeneity from using full IgG, thorough classification was needed to isolate Env trimers bound to three 17b Abs (Supplementary Fig. 13b, d). All three outwardly protruding V1/V2 loops characteristic of an open CD4-bound Env were clearly present in the average structure despite the absence of CD4 (Fig. 9b, c)20,41,48,49.

a AT-2 inactivated HIV-1BaL was incubated with 100 µM CJF-III-288 before addition of 100 µg/ml 17b Abs and subsequent plunge freezing for cryoET analysis. Tomographic slices of 17b Abs binding to Env on virions in the presence of CJF-III-288 are shown (blue: 17b, green: Env). Representative tomographic slices from three different tomograms collected from one sample preparation are shown. b Subtomogram averaging was performed on Env in complex with CJF-III-288 and 17b Abs (EMDB: 46646). A central slice along the density is shown. Panels to the right show three slices along the length of the structure. 17b Fabs and V1/V2 loops are indicated by arrows. c Isosurface rendering of the structure. d The atomic model of BG505 SOSIP.664 in complex with CJF-III-288 and 17b Fab (PDB:9CF5, this paper) was rigidly fit into the cryoET density. e The atomic model of BG505 SOSIP.664 in complex with BNM-III-170 and 17b Fab (PDB: 7LO6)44 was superimposed onto the gp41 helices of the CJF-III-288 atomic model. The three 17b Fab domains from the BNM-III-170 structure (blue) and the CJF-III-288 structure (red) are compared for their agreement with the cryoET density. The gp120 (green) and gp41 (gray) subunits from the CJF-III-288 structure and BNM-III-170 structure are shown in darker and lighter shades, respectively. f Focused classification revealed heterogeneity in the 17b fab position. Two representative subclasses are shown (pink and yellow) and compared to the combined average (gray). g The angle between the 17b Fabs from subclass 1 and subclass 2 relative to the approximate position of N335 (green sphere) is shown for each protomer. h Focused classification on the membrane region revealed subclasses at different tilting angles. Two representative subclasses (pink and yellow) are compared with the combined average density (gray). i Per-particle tilting analysis. The polar plot coloring indicates the normalized frequency of angles (n = 3512). j Histogram and box-and-whisker plot of the data in (i) showing the distribution of Env tilting (n = 3512). The median, range, and interquartile ranges (IQR) are shown. The mode is indicated by a red circle and a dashed line. Source data for i, j are provided as a Source Data file.
Structures of BG505 SOSIP.664 trimers bound to 17b Fab and either CJF-III-288 (PDB :9CF5, Fig. 7 this manuscript) or BNM-III-170 CD4mc44 (PDB:7LO6) were compared with the cryoET density map (Fig. 9d, e). The CJF-III-288 structure was first rigidly fit into the cryoET density map before superimposing the BNM-III-170 structure by the gp41 residues. Upon examining the three protomers bound to 17b Fabs from both structures, the BG505 SOSIP.664 structure bound with CJF-III-288 best agreed with the cryoET density (Fig. 9e). Consistent with the analysis of differing asymmetry between the two SOSIP structures, one 17b Fab in the BNM-III-170 adopted a different orientation than the respective cryoET density as a result of the difference in gp120 displacement (Fig. 9e).
We assessed the heterogeneity of the cryoET structure bound to three 17b molecules through focused classification. Classification on the 17b Fab with weaker density revealed subclasses with different 17b positions (Fig. 9f, Supplementary Fig. 13g). This suggests that the asymmetric gp120 protomers adopt a range of conformations when bound to 17b in complex with CJF-III-288 (Supplementary Movie 1), which is consistent with the relatively lower resolution observed on one 17b Fab bound to the CJF-III-288 SOSIP.664 structure (Supplementary Fig. 7) as well as the smFRET data indicating dynamics (Fig. 6). The orientation of this Fab was found to vary by approximately 13° relative to N355 which is near the V4 loop, while the orientation of the other two Fabs remained within ~5° of each other (Fig. 9g). Classification on the membrane region resulted in subclasses showing Env at different tilting angles (Fig. 9h and Supplementary Fig. 13h), similar to previous observations of unliganded Env tilting50. We next assessed Env tilting on a per-particle basis using focused membrane-refinement to determine perpendicular membrane vectors at each particle position. Focused angular refinement on the unrefined membrane region resulted in strong lipid bilayer density and unresolved trimer density (Supplementary Fig. 14a, b). Env tilt angle was determined by comparing the angles of the aligned trimer vector and the angle of the perpendicular membrane vector (Supplementary Fig. 14a). Calculated tilt angles were confirmed visually in tomograms (Supplementary Fig. 14c) and by mapping membrane refinement vectors along with aligned trimers back to tomograms (Supplementary Fig. 14d). Per-particle tilting analysis revealed a right-skewed distribution with a median Env tilt at approximately 23.9°, a mode of 19.2°, and a range of approximately 0-76° (Fig. 9i, j). This analysis reveals the flexibility of the Env MPER region and Fab binding orientation when Env is in complex with CJF-III-288 and 17b Ab.
Discussion
HIV-1 Env conformation significantly impacts the susceptibility of HIV-1-infected cells to ADCC. The unliganded Env trimer presents at the surface of HIV-1-infected cells predominantly assumes a “closed” State-1 conformation that protects infected cells from ADCC-mediated by nnAbs typically elicited in PLWH16,22,23. CD4 downmodulation by accessory proteins Nef and Vpu, along with multiple intermolecular interactions within the Env trimer, helps to maintain this “closed” conformation, thus preventing the exposure of CD4i Env epitopes18,22,23,51,52,53. The use of CD4mcs to “open up” the Env trimer and expose these vulnerable epitopes has been proposed as a new strategy to eliminate HIV-1-infected cells25,26,29,31,32,37,54. Rational, structure-based design has led to the generation of indoline CD4mcs showing broader and more potent viral inhibition and ADCC activity compared with previous indane CD4mcs26. Here, we report that the lead indoline CD4mc, CJF-III-288, has gained the capacity to sensitize HIV-1-infected cells to ADCC mediated by anti-CoRBS Abs, contributing to its improved ADCC activity in vitro and in vivo.
This property is notable because the well-characterized anti-CoRBS Abs are unable to mediate potent ADCC against infected cells exposing either “closed” or “open” Env conformations. When combined with indane CD4mcs, anti-CoRBS Abs were reported to contribute to the ADCC response by enabling anti-cluster A Abs to engage the Env trimer, at which point the Fc-portion of both families of Abs contribute to the ADCC response31,32,33,54. The exceptional ability of CJF-III-288 to enable anti-CoRBS to mediate ADCC against cells infected with multiple primary viruses, even at low concentrations of CD4mc (Figs. 1–3), underscores the therapeutic potential of the indoline CD4mcs. In contrast to the indane CD4mc BNM-III-170, CJF-III-288, when combined with anti-CoRBS, was able to delay viral rebound in HIV-1-infected humanized mice (Fig.5).
The improved ADCC activity of CJF-III-288 compared with that of the indane CD4mcs likely relates to its superior ability to shift the Env conformational landscape from State 1 to “open” downstream conformations, which are more susceptible to ADCC mediated by nnAbs and PLWH plasma31,53. Compared to BNM-III-170, treatment with CJF-III-288 led to a more pronounced reduction of State 1 occupancy, even in the absence of 17b (Fig. 6). The combination of CJF-III-288 and 17b further destabilized State 1 and stabilized State 3, again to a greater extent than BNM-III-170 and 17b. The maintained conformational dynamics of Env bound to CJF-III-288 and 17b likely contribute to the variability in 17b orientation seen in our structural analyses. Although CJF-III-288 slightly increased the occupancy of State 2 A, under no condition did State 2 A become predominant, as previously seen in the presence of anti-cluster A Abs31. This indicates that State 3 is also a viable substrate for ADCC. The asymmetry in Env conformation revealed through our structural analyses is likely insufficient in amplitude to be detected by smFRET with the current fluorophore attachment sites. Overall, our findings suggest that CJF-III-288’s ability to stabilize State 3 contributes to its activity in sensitizing HIV-1-infected cells to ADCC mediated by anti-CoRBS and PLWH plasma.
Structural analyses helped delineate the mechanism behind the improved ADCC activity of anti-CoRBS Abs in the presence of CJF-III-288. The structure of the CJF-III-288-BG505 SOSIP.664-17b complex obtained by cryo-EM revealed significant differences in the trimer assembly relative to the previously published BNM-III-17-BG505 SOSIP.664-17b complex44 (Fig. 8). This is evidenced by a substantial outward rotation of the gp120 subunits, particularly in protomer A, where the α0 helix shifts significantly further away from the central gp41 helices compared to the BNM-III-170 complex. The CJF-III-288-BG505 SOSIP.664-17b closely resembles the asymmetric Env conformation B of the CD4-BG505 SOSIP.664/E51 Fab complex45. E51 is a tyrosine-sulfated CoRBS antibody that was shown to induce an asymmetric opening of the Env trimer, leading to rearrangements of the relative positions of gp120/gp41 protomers within the Env trimer. It has been suggested that E51-bound Env conformations represent structural intermediates of Env, wherein the CoRBS is being formed before transitioning into a fully open state. Thus, our data suggest that CJF-III-288 is able, on its own, to promote the conformational changes required to form the coreceptor binding site. Indeed, both indane and indoline CD4mcs must be able to induce coreceptor binding, based on activation of HIV-1 infection of CD4-negative, coreceptor-expressing cells26,55. However, BNM-III-170 and CJF-III-288 induce distinct asymmetric conformations in the Env trimer, orienting the CoRBS differently. These structural distinctions would be expected to influence how the anti-CoRBS Abs approach one of the gp120 subunits of the trimer (Fig. 8). Although changes in the Ab approach were noticed for each gp120 protomer in the trimer, the largest deviation was observed for one (protomer A) where a shift of 13.4 Å for the center of mass of the Fab variable domain of the anti-CoRBS Ab was detected. Cryo-ET analysis of the membrane-bound Env trimer confirmed the asymmetric opening of the trimer, as the CJF-III-288-BG505 SOSIP.664-17b structure fits relatively well into the cryo-ET density map (Fig. 9). Assessment of the heterogeneity of the cryo-ET structure revealed the flexibility of the Env MPER and anti-CoRBS Ab binding when Env is in complex with CJF-III-288. CJF-III-288 may allow anti-CoRBS Fabs to adopt a range of conformations, notably with a variation of ~13° within the protomer A, consistent with cryo-EM findings. Furthermore, the observed variation of the Env trimer’s tilting angle also has the potential to facilitate Fc receptor engagement on effector cells by accommodating diverse antibody/Env orientations, thereby improving alignment with the Fc receptor’s angle of approach.
Overall, both functional and structural data strongly support the notion that CJF-III-288 induces “open”, asymmetric Env conformations that enable anti-CoRBS Abs to bind in orientations that allow better Fc receptor engagement, boosting their ADCC activity in vitro and in vivo (Fig. 10). This highlights CJF-III-288’s potential as a therapeutic agent, capable of harnessing the full potential of non-neutralizing CD4i antibody responses to target HIV-1-infected cells through Fc-effector activities.

The indane CD4mc, BNM-III-170, in combination with the anti-CoRBS Abs 17b, stabilizes more “open” Env conformations. However, these conformations prevent proper orientation of the Fc region of 17b, impairing efficient interaction with Fc receptors on effector cells and resulting in a low ADCC response both in vitro and in vivo. In contrast, the more potent indoline CD4mc, CJF-III-288, when combined with 17b, stabilizes a more open, asymmetric State-3 Env conformation, enabling 17b to adopt orientations that promote Fc receptor engagement. Env flexibility and the binding of 17b in the context of CJF-III-288 may facilitate Fc receptor engagement on effector cells by accommodating diverse antibody/Env orientations. This mechanism contributes to enhanced ADCC activity in vitro and delays viral rebound following antiretroviral therapy interruption (ATI) in vivo in humanized mice. Cartoons were created in BioRender. Marchitto, L. (2025) https://BioRender.com/rdvwtec.
Methods
Cell lines and primary cells
293 T human embryonic kidney cells (obtained from ATCC) were maintained at 37 °C under 5% CO2 in Dulbecco’s Modified Eagle Medium (DMEM) (Wisent, St. Bruno, QC, Canada), supplemented with 5% fetal bovine serum (FBS) (VWR, Radnor, PA, USA) and 100 U/mL penicillin/streptomycin (Wisent). Human peripheral blood mononuclear cells (PBMCs) from 7 HIV-negative individuals (6 males, 1 female) (Supplementary Table 3) and 4 PLWH (4 males) (Supplementary Table 4) obtained by leukapheresis and Ficoll-Paque density gradient isolation were cryopreserved in liquid nitrogen until further use. CD4 + T lymphocytes were purified from resting PBMCs by negative selection using immunomagnetic beads per the manufacturer’s instructions (StemCell Technologies, Vancouver, BC) and were activated with phytohemagglutinin-L (10 µg/mL) for 48 h and then maintained in RPMI 1640 (Thermo Fisher Scientific, Waltham, MA, USA) complete medium supplemented with rIL-2 (100 U/mL).
Antibody production
FreeStyle 293 F cells (Thermo Fisher Scientific) were grown in FreeStyle 293 F medium (Thermo Fisher Scientific) to a density of 1 × 106 cells/mL at 37 °C with 8% CO2 with regular agitation (150 rpm). Cells were transfected with plasmids expressing the light and heavy chains of each mAb using ExpiFectamine 293 transfection reagent, as directed by the manufacturer (Thermo Fisher Scientific). One week later, the cells were pelleted and discarded. The supernatants were filtered (0.22-μm-pore-size filter), and antibodies were purified by protein A affinity columns, as directed by the manufacturer (Cytiva, Marlborough, MA, USA). Antibodies were dialyzed against phosphate-buffered saline (PBS) and stored in aliquots at −80 °C. To assess purity, antibodies were loaded on SDS-PAGE polyacrylamide gels in the presence or absence of β-mercaptoethanol and stained with Coomassie blue. The anti-CoRBS 17b Ab Fab fragments were prepared from purified IgG (10 mg/mL) by proteolytic digestion with immobilized papain (Pierce, Rockford, IL) and purified using protein A, followed by gel filtration chromatography on a Superdex 200 16/60 column (Cytiva).
Plasmids and proviral constructs
Transmitted/founder infectious molecular clones (IMCs) of patient CH058 was previously described56,57. IMCs encoding HIV-1 reference strains JRFL, JR-CSF, AD8 (Vpu + ), and BG505 (T332N) were described elsewhere58,59,60,61. The vesicular stomatitis virus G (VSV-G)-encoding plasmid was previously described62.
Viral production, infections and ex vivo amplification
For in vitro infection, vesicular stomatitis virus G (VSV-G)-pseudotyped HIV-1 viruses were produced by co-transfection of 293 T cells with an HIV-1 proviral construct and a VSV-G-encoding vector using the calcium phosphate method. Two days post-transfection, cell supernatants were harvested, clarified by low-speed centrifugation (300 × g for 5 min), and concentrated by ultracentrifugation at 4 °C (100,605 × g for 1 h) over a 20% sucrose cushion. Pellets were resuspended in fresh RPMI, and aliquots were stored at −80 °C until use. Viral preparations were titrated directly on primary CD4 + T cells to achieve similar levels of infection among the different IMCs tested. Viruses were then used to infect activated primary CD4 + T cells from HIV-1 negative donors (Supplementary Table 3) by spin infection at 800 × g for 1 h in 96-well plates at 25 °C (Figs. 1, 2, 4, 6 and Supplementary Figs. 1–3). All experiments using VSV-G-pseudotyped HIV-1 isolates were done in a biosafety level 3 laboratory following manipulation protocols accepted by the CRCHUM Biosafety Committee, which respects the requirements of the Public Health Agency of Canada.
To expand endogenously infected CD4 + T cells, primary CD4 + T cells were isolated from PBMCs obtained from 4 PLWH (Supplementary Table 4) by negative selection as described above. Purified CD4 + T cells were activated with PHA-L at 10 μg/mL for 48 h and then cultured for at least 6 days in RPMI 1640 complete medium supplemented with rIL-2 (100 U/ml) to reach greater than 5% infection for the ADCC assay.
Antibodies and human plasma
The following anti-Env Abs were used as primary mAbs to stain HIV-1-infected primary CD4 + T cells: anti-cluster A A32, N5i5; anti-co-receptor binding site 17b, C2, N12i2, 48D, 412D, X5; anti-gp120 outer domain 2G12. Goat anti-human IgG (H + L) (Thermo Fisher Scientific) pre-coupled to Alexa Fluor 647 were used as secondary antibodies in flow cytometry experiments. The A32 mAbs was conjugated with Alexa-Fluor 647 (Thermo Fisher Scientific) as per the manufacturer’s protocol and was also used for cell-surface staining of HIV-1-infected cells. Mouse anti-human CD4 (Clone OKT4, FITC-conjugated; Biolegend, San Diego, CA, USA) and anti-p24 mAb (clone KC57; PE-Conjugated; Beckman Coulter) or Mouse anti-human CD4 (Clone OKT4, PE-conjugated; Biolegend, San Diego, CA, USA) and anti-p24 mAb (clone KC57; FITC-Conjugated; Beckman Coulter) were used to identify the productively-infected cells as previously described24. Plasma from PLWH, obtained from the FRQS-AIDS and Infectious Diseases Network (the Montreal Primary HIV Infection Cohort63,64), were collected, heat-inactivated, and conserved as previously described29.
Small CD4-mimetics
The small-molecule CD4-mimetic compounds (CD4mc) BNM-III-170, CJF-III-288 and TFH-I-116-D1 were synthesized as described previously26,37. The compounds were dissolved in dimethyl sulfoxide (DMSO) at a stock concentration of 10 mM and diluted in phosphate-buffered saline (PBS) for cell-surface staining or in RPMI-1640 complete medium for ADCC assays.
Flow cytometry analysis of cell-surface staining
Cell surface staining was performed at 48 h post-infection. Mock-infected or HIV-1-infected primary CD4 + T cells were incubated for 45 min at 37 °C with anti-Env mAbs (5 µg/mL) or plasma from PLWH (1:1000) in the presence or absence of different concentrations of CD4mc (BNM-III-170, TFH-I-116-D1, CJF-III-288) or equivalent volume of DMSO. Cells were then washed twice with PBS and stained with the appropriate Alexa Fluor 647-conjugated secondary antibody (2 µg/mL) and the viability dye staining (Aqua Vivid; Thermo Fisher Scientific) (1:1000 dilution) for 20 min at room temperature. Cells were then stained with FITC- or PE-conjugated mouse anti-CD4 Abs (1:100 dilution). After two PBS washes, cells were fixed in a 2% PBS-formaldehyde solution. Infected cells were then permeabilized using the Cytofix/Cytoperm Fixation/ Permeabilization Kit (BD Biosciences, Mississauga, ON, Canada) and stained intracellularly using PE or FITC-conjugated mouse anti-p24 mAb (1:100 dilution). The percentage of productively-infected cells was determined by gating on the living CD4lowp24high cell population as previously described24 and presented in Supplementary Fig. 15. Samples were acquired on an FORTESSA flow cytometer (BD Biosciences), and data analysis was performed using FlowJo v10.5.3 (Tree Star, Ashland, OR, USA). Area under the curve (AUC) values were calculated from the median fluorescence intensities obtained at different CD4mc concentrations (log10-transformed) using GraphPad Prism software.
Antibody-dependent cellular cytotoxicity (ADCC) assay
Measurement of ADCC using a fluorescence-activated cell sorting (FACS)-based infected cell elimination (ICE) assay was performed at 48 h post-infection. Briefly, HIV-1-infected primary CD4 + T cells were stained with the viability dye (Aqua Vivid; Thermo Fisher Scientific)(1:1000 dilution) and the cell proliferation dye eFluor670 (Thermo Fisher Scientific) (0.5 µM final concentration) and used as target cells. Cryopreserved autologous PBMC effectors cells, stained with cell proliferation dye eFluor450 (Thermo Fisher Scientific) (0.5 µM final concentration), were added at an effector: target ratio of 10:1 in 96-well V-bottom plates (Corning, Corning, NY). Target cells were treated with CD4mc (CJF-III-288, BNM-III-170 or TFH-I-116-D1) at indicated concentrations or equivalent volume of DMSO. Anti-Env mAbs (5 µg/mL) or PLWH plasma (1:1000 dilution) were added to appropriate wells and cells were incubated for 5 min at room temperature. For blocking experiments, infected cells were pre-incubated with the anti-CoRBS Ab 17b Fab fragment at 10 µg/mL in the presence of CD4mc before adding PLWH plasma. The plates were subsequently centrifuged for 1 min at 300 × g, and incubated at 37 °C, 5 % CO2 for 5 h. before being stained with FITC- or PE-conjugated Mouse anti-CD4 Abs. After one PBS wash, cells were fixed in a 2% PBS-formaldehyde solution. Infected cells were then permeabilized using the Cytofix/Cytoperm Fixation/ Permeabilization Kit (BD Biosciences, Mississauga, ON, Canada) and stained intracellularly using PE or FITC-conjugated mouse anti-p24 mAb (1:100 dilution). Productively-infected cells were identified based on p24 and CD4 detection as described above. Samples were acquired on an FORTESSA flow cytometer (BD Biosciences), and data analysis was performed using FlowJo v10.5.3 (Tree Star, Ashland, OR, USA). The percentage of ADCC was calculated with the following formula: [(% of CD4lowp24+ cells in Targets plus Effectors) − (% of CD4lowp24+ cells in Targets plus Effectors plus plasma or mAbs) / (% of CD4lowp24+ cells in Targets) × 100] by gating on infected living target cells. All negative value were set to 0. The gating strategy is presented in Supplementary Fig. 16. Area under the curve (AUC) values were calculated from the percentage of ADCC obtained at different CD4mc concentrations (log10-transformed) using GraphPad Prism software.
Studies in humanized mice
The mice were bred and maintained under specific pathogen-free conditions with a 12-hour light/dark cycle, at an ambient temperature of 20–24 °C and relative humidity of 45–65%. All animal studies were performed with authorization from the Institutional Animal Care and Use Committees (IACUC) of Yale University. Humanized NSG-Tg(IL15) mice (Jackson Laboratory, Strain #030890) were generated as previously described54. Briefly, human CD34+ hematopoietic stem cells were isolated from umbilical cord blood by magnetic separation, and 10⁵ CD34+ HSCs transplanted intrahepatically into 1- to 3-day-old NSG-Tg(IL15) mice (both male and female) pre-irradiated at 150 cGy in an X-ray irradiator. After 10 weeks of engraftment, human immune cell reconstitution was assessed by flow cytometry. A total of 100 μl of peripheral blood was collected via retroorbital bleeding, and PBMC were isolated using Ficoll density gradient centrifugation; stained with fluor-conjugated anti-human CD45 (BD Biosciences, Cat#: 555485), CD3 (Biolegend, Cat#: 300424), CD4 (Biolegend, Cat#: 317432), CD8 (BD Biosciences, Cat#: 561617), and CD56 (Biolegend, Cat#: 362508) antibodies (all at a 1:1000 dilution), and analyzed by flow cytometry to confirm engraftment.
Humanized mice (approximately 4 month old) were intraperitoneally challenged with 30,000 PFU of HIV-1JRCSF. Infection profile was analyzed through weekly retro-orbital bleeds. RNA extracted from plasma using the Qiagen RNeasy Mini kit was used for quantifying viral RNA using iTaq Universal One-Step RT-qPCR kit with qPCR performed on a Bio-Rad CFX96 real-time PCR machine with a sensitivity of reliably detecting 300 copies per ml of plasma.
When PVLs reached 104-105 copies/ml, mice were treated with daily subcutaneous injections of a three-drug ART regimen: tenofovir disoproxil fumarate (TDF, 5.1 mg/kg), emtricitabine (FTC, 40 mg/kg), and dolutegravir free base (DTG, 2.5 mg/kg). ART was administered for over four weeks, with stable viral suppression to viral loads below the limit of detection achieved within a week’s time of ART treatment in all cohorts.
A day before ART interruption, mice were grouped (2–4 mice per group, each group including both male and female mice) and subcutaneously injected with BNM-III-170 plus 17b or CJF-III-288 plus 17b as outlined in the main figure of the text. BNM-III-170 and CJF-III-288 were dissolved in DMSO at a concentration of 200 mg/ml, diluted in PBS, and administered subcutaneously at 3 mg per mouse for two consecutive days, followed by 1 mg per mouse for the next three consecutive days. The 17b antibody was injected subcutaneously three times, with an initial dose of 1.5 mg per mouse alongside CD4mc treatment, followed by two subsequent doses of 0.75 mg per mouse on alternate days. Mice treated with vehicle (DMSO) served as mock-treated group (see also Fig. 5a for a schematic timeline).
smFRET imaging
For smFRET imaging, pseudovirions were formed with the HIV-1NL4-3 core and HIV-1JRFL Env. To facilitate fluorophore attachment, an amber stop codon was introduced at position N135 in the V1 loop by site-directed mutagenesis, and the A4 peptide was inserted into the V4 loop of Env by overlap-extension PCR, as described28. Briefly, HEK-293T FIRB cells65 were co-transfected with plasmids encoding an aminoacyl tRNA synthetase and corresponding suppressor tRNA (NESPylRSAF/hU6tRNAPyl)66, eRF1-E55D, pNL4-3 ΔRT ΔEnv16, and a 20:1 mass ratio of wild-type HIV-1JRFL Env to tagged Env. Pseudovirus was collected 48 hours post-transfection and pelleted through a 10% sucrose cushion in PBS by ultracentrifugation for 2 h at 35,000 RPM. Pellets was then resuspended in labeling buffer (50 mM HEPES pH 7.0, 10 mM CaCl2, 10 mM MgCl2) and incubated overnight at room temperature with 5 μM LD650-coenzyme A (Lumidyne Technologies, New York,NY, USA), and 5 μM acyl carrier protein synthase (AcpS), which attaches the fluorophore to the A4 peptide in V4 of gp120, as described16,31,42. The virus was then incubated with 0.5 µM Cy3-tetrazine (Jena Biosciences, Jena, Germany) for 30 min at room temperature, followed by addition of 60 μM DSPE-PEG2000-biotin (Avanti Polar Lipids, Alabaster, AL, USA) and incubation for an additional 30 min at room temperature. Finally, virus was purified by ultracentrifugation on a 6–30% OptiPrep (Sigma-Aldrich, MilliporeSigma, Burlington, MA, USA) density gradient for 1 hour at 35,000 RPM at 4 °C using a SW40Ti rotor (Beckman Coulter Life Sciences, Brea, CA, USA). Gradient fractions were collected, analyzed by western blot, aliquoted, and stored at -80 °C until their use in imaging experiments.
Where indicated, labeled pseudovirions were incubated with 50 µg/ml 17b mAb and 100 μM CD4mc for 1 hour at room temperature, followed by immobilization on streptavidin-coated quartz slides. Virions were then imaged on a custom-built wide-field prism-based TIRF microscope67,68. Imaging was performed in PBS pH 7.4, containing 1 mM trolox (Sigma-Aldrich, St. Louis, MO, USA), 1 mM cyclooctatetraene (COT; Sigma-Aldrich, St. Louis, MO, USA), 1 mM 4-nitrobenzyl alcohol (NBA; Sigma-Aldrich, St. Louis, MO, USA), 2 mM protocatechuic acid (PCA; Sigma-Aldrich, St. Louis, MO, USA), and 8 nM protocatechuate 3,4-deoxygenase (PCD; Sigma-Aldrich, St. Louis, MO, USA) to stabilize fluorescence and remove molecular oxygen. Where indicated, concentrations of mAb 17b and CD4mc were maintained during imaging. smFRET data were collected using Micromanager v2.069. at 25 frames/sec, processed, and analyzed using the SPARTAN software (https://www.scottcblanchardlab.com/software) in Matlab (Mathworks, Natick, MA, USA)70. smFRET traces were identified according to criteria previously described31, and traces meeting those criteria were verified manually. FRET histograms were generated by compiling traces from each of three technical replicates and the mean probability per histogram bin ± standard error was calculated. Traces were then idealized to the five-state HMM using the MPL algorithm71 implemented in SPARTAN as described31. The idealizations were used to determine the occupancies (fraction of time until photobleaching) in each FRET state, and construct Gaussian distributions, which were overlaid on the FRET histograms to visualize the results of the HMM analysis. The FRET state occupancies for each trajectory were used to construct violin plots in Matlab, as well as calculate mean and median occupancies, standard errors, and quantiles. smFRET trajectories were analyzed using HMM analysis, which resulted in the identification of the number of FRET states and the mean FRET values. Five different models were considered during HMM analysis of the smFRET trajectories. Model parameters were optimized by maximization of likelihood using the maximum point likelihood (MPL) algorithm71 implemented in SPARTAN. Model fitness was compared by correcting the maximized likelihood for different numbers of model parameters using the Akaike information criterium (AIC). Minimization of the AIC indicates the model that best represents the data. Based on this criterium, the 5-state model (4 non-zero FRET states, and a 0-FRET state) was selected (Supplementary Fig. 5a). Addition of a sixth state did not improve model fitness. We then confirmed the identification of the 5 FRET states with an alternative HMM approach involving Bayesian inference (ebFRET)72. Importantly, ebFRET does not require input of initial FRET states. ebFRET analysis led to the identification of 5 states with FRET values that are essentially indistinguishable from the states identified by MPL (Supplementary Fig. 5b).
Functional validation of fluorescently labeled Env
To evaluate the function of modified and fluorescently labeled Env, we employed a pseudovirion infectivity assay. This assay was performed exactly as previously described28, with the exception that here fluorescently labeled virions were also tested. To this end, fluorescently labeled pseudovirions were formed as described above. In this case, however, the modified Env was not diluted with wild-type Env. Viral supernatant was collected 48 hours post-transfection. Pseudovirions were then pelleted over a 10% sucrose cushion in PBS by ultracentrifugation for 2 h at 25,000 RPM using a SW32Ti rotor (Beckman Coulter Life Sciences, Brea, CA, USA). Viral pellets were resuspended in PBS and incubated with Cy3-tetrazine and LD655-CoA as described above, and used to infect TZM-bl cells. Infectivity was assayed through quantification of luciferase (Supplementary Fig. 4), as previously described73. Infectivity values are expressed as the percentage of wild-type Env and compared using a one-way ANOVA.
Protein expression and purification for Cryo-EM studies
BG505 SOSIP.664 was expressed in Expi293F GnTI- cells (Thermo Fisher Scientific, Catalog# A39240). One day prior to transfection, cells were diluted to a density of 1 × 106 cells/ml for a total volume of 90 ml. 50 μg BG505 SOSIP.664 plasmid and 10 μg furin plasmid were mixed and diluted into 5 ml Expi293™ Expression Medium (Thermo Fisher Scientific, Catalog# A1435101). 75 μl FectoPRO (Polyplus) transfection reagent was diluted into 5 ml Expi293™ Expression Medium. Plasmids and transfection reagent were then mixed and incubated at room temperature for 10 minutes prior to addition to the cells. Cells were then grown for another 5 days, and the supernatant harvested by centrifugation. The supernatant was filtered through a 0.22 μm membrane and BG505 SOSIP.664 was purified with a PGT145 affinity column (PGT145 IgG covalently linked to Protein A agarose). Briefly, supernatant was loaded onto the PGT145 affinity column, the column was then washed with PBS, and finally, the BG505 SOSIP.664 protein was eluted with 3M MgCl2. The eluted BG505 SOSIP.664 protein was immediately buffer exchanged into PBS. BG505 SOSIP.664 was further purified with a Superdex 200 10/300 GL gel filtration column (Cytiva).
17b IgG was expressed in Expi293F cells (Thermo Fisher Scientific, Catalog# A14528) with a transfection protocol similar to that of BG505 SOSIP.664. For each 100 ml volume transfection, 25 μg 17b heavy chain plasmid and 25 μg 17b light chain plasmid were mixed and diluted with 5 ml Expi293™ Expression Medium (Thermo Fisher Scientific, Catalog# A1435101). 75 μl transfection reagent FectoPRO (Polyplus) was diluted in 5 ml Expi293™ Expression Medium. Plasmids and transfection reagent were mixed and incubated at room temperature for 10 minutes before being added to 90 ml cells. Cells were grown for an additional 7 days, and then the supernatant was harvested by centrifugation. The supernatant was filtered through a 0.22 μm membrane, and 17b IgG was purified with a protein A affinity column. Briefly, the supernatant was loaded onto the protein A column, the column was washed with PBS, and 17b IgG was eluted with 0.1 M glycine-HCl, pH 2.7. Fabs were generated from an overnight papain digestion of IgG at 37 °C using immobilized papain agarose (Thermo Fisher Scientific). The resulting Fab was separated from Fc and undigested IgG by passage over protein A resin. Fab was further purified by gel filtration using a Superdex 200 10/300 GL column (GE Healthcare) before use in cryo-EM sample preparation.
Cryo-EM sample preparation
CJF-III-288 and BG505 SOSIP.664 trimer were mixed at a molar ratio of 10:1, then incubated at room temperature overnight. 17b Fab was added on the next day at a 10:1 molar ratio to BG505 SOSIP.664 and then incubated at room temperature for 4 h. The CJF-III-288-BG505 SOSIP.664-17b complex was purified on a Superdex 200 10/300 GL column (GE Healthcare) and fractions containing CJF-III-288-BG505-17b complex were concentrated to 1.5 mg/ml. n-dodecyl-β-D-maltopyranoside (DDM) was added to a final concentration of 0.091 mM prior to freezing. To prepare cryo-specimen grids, a 3 μL aliquot of each sample was applied onto a Quantifoil 400-mesh Cu R1.2/1.3 grid with a2nm carbon film over the holes (400-mesh Cu R1.2/1.3-2 nm, Quantifoil Micro Tools GmbH), which had been freshly glow-discharged for 15 s at 15 mA using a PELCO easiGLOW (TED PELLA). The sample was then plunged into liquid ethane using a Leica GP2 (Leica Biosystems) after blotting for 5 s at 4 °C under 95% relative humidity.
Cryo-EM data collection and processing
Data was acquired on a FEI Titan Krios electron microscope operating at 300 kV equipped with Gatan Bioquantum Image filter-K3 direct electron detector (Gatan Inc) with 20 eV energy slit. 50-frame dose fractionated movies in super resolution mode were collected at a nominal magnification of 105,000 corresponding to a calibrated physical pixel size of 0.832 Å/px (0.416 Å/px super resolution), with a total exposure dose of 54.2 e − / Å2. Automated data acquisition was done in SerialEM version 4.0.2774. CryoSPARC ver.4.4.175 was used to process the cryo-EM data. A total of 9458 movies were imported. After Patch Motion Correction and Patch CTF Estimation, 9431 micrographs were used for the following processing. An initial 150,000 particles were picked using the Blob Picker and then subjected to 2D Classification. Particles with side views were used as templates for Template Picking. An extraction box size of 400-pix was used to extract particles. After several rounds of 2D Classification, 215,095 particles with balanced side and top views were used to do Ab-initio Reconstruction. 7,078 junk particles were used to do Ab-initio Reconstruction and generate 5 junk models. The good Ab-initio model was low-pass filtered to 30 Å. Heterogeneous Refinement using the low-pass filtered good Ab-initio model and the 5 junk models as references was conducted several times to clean extracted particles. After Heterogeneous Refinement, 1,087,694 particles were used to do Non-uniform Refinement which generated a 3.5 Å density map. C1 was applied to all reconstruction and refinement steps.
Atomic model building and refinement
An initial fit to the CryoEM reconstruction was done using the BNM-III-170-BG505 SOSIP.664-17b complex as a starting model (PDB ID 7LO6)44. Briefly individual protomers were fit manually to the reconstruction in ChimeraX76. CJF-III-288 was then added to the model by superposition of the gp120 complex of CJF-III-288 (PDB ID 8FM3) onto each protomer of the trimer. The initial model was then manually fit into the reconstruction using COOT77 and then refined with real space refinement in PHENIX78. Several rounds of refinement and model building were done to finalize the model. Data collection and refinement statistics are located in Supplementary Table 1.
CryoET sample preparation
HIV-1BaL virus (lot P4338) was produced from a chronically infected SupT1 HIV-1 BAL/A66-R5 CL.1 CLN445 as previously described20,41,79. This cell line produces HIV-1 virions containing high levels of Env to facilitate structural analyses41. The HIV-1BaL/SUP-T1-CCR5 cells were grown at 37 °C, 5% CO2 using large roller bottles (up to 30 L culture volume) in RPMI-1640 medium supplemented with 10% fetal bovine serum, 2 mM L-glutamine, 100 U/ml penicillin, and 100 µg/ml streptomycin. The culture was filtered with 5 µM capsule filters to remove cells, followed by 0.45 µM filters to remove debris and microvesicles. The supernatant was treated with 2,2’-dithiodipyridine (aldrithiol-2; AT-2) to inactivate the virus but preserve Env function20,46,80. AT-2 treated viral supernatant was then further purified and concentrated using continuous-flow sucrose-density-gradient ultracentrifugation. AT-2 inactivated HIV-1BaL stocks were incubated at room temperature for 1 hour in the presence of 100 μM CJF-III-288. The virus was transferred to ice, and 100 μg/ml 17b antibody was subsequently added for an approximately 30-minute incubation. Copper Quantifoil holey grids (2/1, 200 mesh) were glow-discharged for 35 seconds at 25 mA and pre-blotted with 3 μl BSA-colloidal gold solution before adding 5 μl of the virus sample preparation. Grids were blotted from the front with filter paper and plunged into liquid ethane with a homemade gravity plunger.
CryoET data acquisition
CryoET grids containing plunge-frozen HIVBaL with 17b IgG and CJF-III-288 were imaged on a Titan Krios electron microscope (Thermo Fisher Scientific) equipped with a 300 keV field emission gun, K3 Summit direct electron detector (Gatan), Volta Phase Plate (to enhance contrast), and a Gatan Imaging Filter. Data was collected using SerialEM software74 at 64,000X magnification (1.346 Å/pixel) and -0.5 µm defocus using a single axis bidirectional tilt series scheme from -60° to 60° with a tilt step of 3° and a cumulative specimen dose of 120 e- /Å2. Dose fractionation mode was used to generate 9 frames per image for subsequent motion correction. 87 total tomograms were collected on two different days (2 grids) from one sample preparation.
Tomogram reconstruction
MotionCor2 (version 1.5.0) was used to process dose-fractionated images, which were then compiled into image stacks81. Motion-corrected image stacks were aligned with IMOD/Etomo (version 4.12.54) using gold fiducial markers74. For visualization and particle picking, aligned stacks were binned by 8x or 4x, and IMOD was used to generate tomograms with weighted back-projection followed by deconvolution filtering. CTFPlotter was used to estimate defocus and phase values82.
Subtomogram averaging
Subtomogram averaging was performed using PEET (version 1.16.0a)83. HIV-1BaL Env trimers bound with CJF-III-288 and 17b Abs were manually selected from tomograms binned by 8 using IMOD. A point was placed on each Env trimer and at the center of each virion (9,419 initial Env trimers). PEET program spikeInit was then used to generate a rough estimate of the particle orientations. Particles binned by 4 were first aligned using no translational or angular searching to generate an initial formless reference of a ball sitting on a membrane surface. The membrane and Ab domains not connected to trimers (Fc domains or free Fab domains) were subsequently masked out for further alignment. An initial model was generated with resolution limited to 45 Å by strong lowpass filtration and binning before randomly splitting the data into two halves for gold-standard refinement. PCA-based classification was used to remove junk particles and separate out two vs. three 17b-bound trimers by masking on the Fabs with noticeably weaker density84. Env trimers bound to three 17b Abs were further analyzed. Small 2x-binned 3D subvolumes containing individual Env particles were reconstructed with 3D CTF correction using IMOD program subtomosetup. One final refinement iteration was performed on these 2x binned particles (3,507 final Env trimers). Fourier shell correlation (FSC) curves were calculated using Relion 4.085 and plotted in Python (version 3.8.5, matplotlib and scipy libraries) to estimate resolution using FSC = 0.143.
CryoET heterogeneity analysis of 17b Fab position and Env tilting
Focused PCA-based classification was performed on the final Env structure bound to three 17b Abs to assess heterogeneity using PEET83,84. A spherical mask was drawn on the weakest density 17b Fab domain to investigate variations in 17b Fab positions. Env tilting was assessed using an ellipsoid mask drawn on the membrane and MPER region. Structures were visualized in ChimeraX (version 1.7)76 and different 17b Fab positions and orientations were displayed as a movie using the ChimeraX “vop morph” function.
Trimers bound to three 17b molecules were assessed for tilting on a per-particle basis. The angle between the Env orientation vector (determined through subtomogram averaging) and a vector perpendicular to the membrane at each respective Env position was determined as the Env tilting angle. The vector perpendicular to the membrane was calculated using focused angular refinement on the MPER-membrane region as previously described86. Membrane refinement resulted in a disordered Env ectodomain and a well-defined membrane density. Calculated Env trimer angles were confirmed visually through tomographic slices and by mapping the Env trimers along with the perpendicular membrane vectors back into tomograms using the ChimeraX ArtiaX plugin76,87. Tilt angles distributions were plotted as a polar plot in Python (version 3.13.2, matplotlib and numpy packages) and as a histogram using R (version 4.3.2, ggplot2 package). The distribution mode was determined using the “multimode” R package88.
Statistical analysis
Statistics were analyzed using GraphPad Prism version 9.1.0 (GraphPad, San Diego, CA, USA). Every data set was tested for statistical normality, and this information was used to apply the appropriate (parametric or nonparametric) statistical test. P values < 0.05 were considered significant; significance values are indicated in each graph.
Ethics statement
The research reported in this manuscript was conducted in accordance with the ethical guidelines of the CRCHUM and was reviewed and approved by its institutional review board (Ethics committee, approval number MP-02-2024-11734). Written informed consent was obtained from all participants. Sex was not included as a variable in the study design. Participants were selected based on HIV-1 status, and sex was determined by self-report. Animal studies were conducted following the standards of the Institutional Animal Care and Use Committees (IACUC) of Yale University (IACUC approval number 11264). Placental cord blood used for humanized mouse engraftment was obtained through the Yale University Reproductive Sciences (YURS) Biobank. The biobank coordinates written informed consent with Yale New Haven Hospital at the time of donation; however, consent documentation and donor-identifying information were not disclosed to tissue end-users. Donor sex or gender was not collected, and CD34⁺ hematopoietic stem cells were isolated and used without regard to sex. Research adhered to the standards indicated by the Declaration of Helsinki.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Source Data are provided with this paper. Cryo-EM and Cryo-ET data generated in this study have been deposited in the Electron Microscopy Data Bank (EMDB) under accession codes EMD-45530 and EMD-46646, respectively. Raw SPA Cryo-EM movies will be uploaded to the Electron Microscopy Public Image Archive (EMPIAR). The atomic coordinates for the CJF-III-288-BG505 SOSIP.664-17b Fab complex have been deposited in the Protein Data Bank (PDB) under accession code 9CF5. Source data are provided with this paper.
References
Antiretroviral Therapy Cohort, C Life expectancy of individuals on combination antiretroviral therapy in high-income countries: a collaborative analysis of 14 cohort studies. Lancet 372, 293–299 (2008).
Deeks, S. G., Lewin, S. R. & Havlir, D. V. The end of AIDS: HIV infection as a chronic disease. Lancet 382, 1525–1533 (2013).
Finzi, D. et al. Identification of a reservoir for HIV-1 in patients on highly active antiretroviral therapy. Science 278, 1295–1300 (1997).
Chomont, N. et al. HIV reservoir size and persistence are driven by T cell survival and homeostatic proliferation. Nat. Med 15, 893–900 (2009).
Chun, T. W. et al. Presence of an inducible HIV-1 latent reservoir during highly active antiretroviral therapy. Proc. Natl Acad. Sci. USA 94, 13193–13197 (1997).
Chun, T. W. et al. Quantification of latent tissue reservoirs and total body viral load in HIV-1 infection. Nature 387, 183–188 (1997).
Checkley, M. A., Luttge, B. G. & Freed, E. O. HIV-1 envelope glycoprotein biosynthesis, trafficking, and incorporation. J. Mol. Biol. 410, 582–608 (2011).
Richard, J., Prevost, J., Alsahafi, N., Ding, S. & Finzi, A. Impact of HIV-1 Envelope Conformation on ADCC Responses. Trends Microbiol 26, 253–265 (2018).
Willey, R. L., Bonifacino, J. S., Potts, B. J., Martin, M. A. & Klausner, R. D. Biosynthesis, cleavage, and degradation of the human immunodeficiency virus 1 envelope glycoprotein gp160. Proc. Natl Acad. Sci. USA 85, 9580–9584 (1988).
Earl, P. L., Doms, R. W. & Moss, B. Oligomeric structure of the human immunodeficiency virus type 1 envelope glycoprotein. Proc. Natl Acad. Sci. USA 87, 648–652 (1990).
Freed, E. O., Myers, D. J. & Risser, R. Mutational analysis of the cleavage sequence of the human immunodeficiency virus type 1 envelope glycoprotein precursor gp160. J. Virol. 63, 4670–4675 (1989).
McCune, J. M. et al. Endoproteolytic cleavage of gp160 is required for the activation of human immunodeficiency virus. Cell 53, 55–67 (1988).
Allan, J. S. et al. Major glycoprotein antigens that induce antibodies in AIDS patients are encoded by HTLV-III. Science 228, 1091–1094 (1985).
Robey, W. G. et al. Characterization of envelope and core structural gene products of HTLV-III with sera from AIDS patients. Science 228, 593–595 (1985).
Wyatt, R. & Sodroski, J. The HIV-1 envelope glycoproteins: fusogens, antigens, and immunogens. Science 280, 1884–1888 (1998).
Munro, J. B. et al. Conformational dynamics of single HIV-1 envelope trimers on the surface of native virions. Science 346, 759–763 (2014).
Ma, X. et al. HIV-1 Env trimer opens through an asymmetric intermediate in which individual protomers adopt distinct conformations. Elife 7, 10.7554/eLife.34271 (2018).
Herschhorn, A. et al. Release of gp120 Restraints Leads to an Entry-Competent Intermediate State of the HIV-1 Envelope Glycoproteins. MBio 7, https://doi.org/10.1128/mBio.01598-16 (2016).
Kwong, P. D. et al. Structure of an HIV gp120 envelope glycoprotein in complex with the CD4 receptor and a neutralizing human antibody. Nature 393, 648–659 (1998).
Li, W. et al. HIV-1 Env trimers asymmetrically engage CD4 receptors in membranes. Nature 623, 1026–1033 (2023).
Decker, J. M. et al. Antigenic conservation and immunogenicity of the HIV coreceptor binding site. J. Exp. Med 201, 1407–1419 (2005).
Veillette, M. et al. The HIV-1 gp120 CD4-bound conformation is preferentially targeted by antibody-dependent cellular cytotoxicity-mediating antibodies in sera from HIV-1-infected individuals. J. Virol. 89, 545–551 (2015).
Veillette, M. et al. Interaction with cellular CD4 exposes HIV-1 envelope epitopes targeted by antibody-dependent cell-mediated cytotoxicity. J. Virol. 88, 2633–2644 (2014).
Richard, J. et al. CD4 downregulation precedes Env expression and protects HIV-1-infected cells from ADCC mediated by non-neutralizing antibodies. mBio 16, e0176325 (2025).
Ding, S. et al. A New Family of Small-Molecule CD4-Mimetic Compounds Contacts Highly Conserved Aspartic Acid 368 of HIV-1 gp120 and Mediates Antibody-Dependent Cellular Cytotoxicity. J. Virol. 93 (2019).
Fritschi, C. J. et al. Indoline CD4-mimetic compounds mediate potent and broad HIV-1 inhibition and sensitization to antibody-dependent cellular cytotoxicity. Proc. Natl Acad. Sci. USA 120, e2222073120 (2023).
Laumaea, A. et al. Small CD4 mimetics sensitize HIV-1-infected macrophages to antibody-dependent cellular cytotoxicity. Cell Rep. 42, 111983 (2023).
Marchitto, L. et al. The combination of three CD4-induced antibodies targeting highly conserved Env regions with a small CD4-mimetic achieves potent ADCC activity. J. Virol., e0101624, 10.1128/JVI.01823-18 (2024).
Richard, J. et al. CD4 mimetics sensitize HIV-1-infected cells to ADCC. Proc. Natl Acad. Sci. USA 112, E2687–E2694 (2015).
Tauzin, A. et al. Three families of CD4-induced antibodies are associated with the capacity of plasma from people living with HIV to mediate ADCC in the presence of CD4-mimetics. J. Virol. e0096024, 10.1128/jvi.00960-24(2024).
Alsahafi, N. et al. An asymmetric opening of HIV-1 envelope mediates antibody-dependent cellular cytotoxicity. Cell Host Microbe 25, 578–587 e5 (2019).
Richard, J. et al. Co-receptor binding site antibodies enable cd4-mimetics to expose conserved anti-cluster A ADCC epitopes on HIV-1 envelope glycoproteins. EBioMedicine 12, 208–218 (2016).
Anand, S. P. et al. Two families of ENV antibodies efficiently engage Fc-gamma receptors and eliminate HIV-1-infected cells. J. Virol. 93, 10.1128/JVI.01823-18 (2019).
Richard, J. et al. Across functional boundaries: Making nonneutralizing antibodies to neutralize HIV-1 and mediate Fc-mediated effector killing of infected cells. mBio 12, e0140521 (2021).
Ding, S. et al. A Highly Conserved Residue of the HIV-1 gp120 inner domain is important for antibody-dependent cellular cytotoxicity responses mediated by anti-cluster A antibodies. J. Virol. 90, 2127–2134 (2015).
Tolbert, W. D. et al. Defining rules governing recognition and Fc-mediated effector functions to the HIV-1 co-receptor binding site. BMC Biol. 18, 91 (2020).
Ding, S. et al. Piperidine CD4-mimetic compounds expose vulnerable env epitopes sensitizing HIV-1-infected cells to ADCC. Viruses 15, 10.3390/v15051185 (2023).
Prevost, J. et al. The HIV-1 Env gp120 inner domain shapes the Phe43 Cavity and the CD4 Binding Site. mBio 11, 10.1128/mBio.00280-20 (2020).
Prevost, J. et al. HIV-1 Vpu restricts Fc-mediated effector functions in vivo. Cell Rep. 41, 111624 (2022).
Nikic, I. et al. Debugging eukaryotic genetic code expansion for site-specific click-PAINT super-resolution microscopy. Angew. Chem. Int Ed. Engl. 55, 16172–16176 (2016).
Li, Z. et al. Subnanometer structures of HIV-1 envelope trimers on aldrithiol-2-inactivated virus particles. Nat. Struct. Mol. Biol. 27, 726–734 (2020).
Lu, M. et al. Associating HIV-1 envelope glycoprotein structures with states on the virus observed by smFRET. Nature 568, 415–419 (2019).
Acharya, P. et al. Structural definition of an antibody-dependent cellular cytotoxicity response implicated in reduced risk for HIV-1 infection. J. Virol. 88, 12895–12906 (2014).
Jette, C. A. et al. Cryo-EM structures of HIV-1 trimer bound to CD4-mimetics BNM-III-170 and M48U1 adopt a CD4-bound open conformation. Nat. Commun. 12, 1950 (2021).
Yang, Z., Wang, H., Liu, A. Z., Gristick, H. B. & Bjorkman, P. J. Asymmetric opening of HIV-1 Env bound to CD4 and a coreceptor-mimicking antibody. Nat. Struct. Mol. Biol. 26, 1167–1175 (2019).
Rossio, J. L. et al. Inactivation of human immunodeficiency virus type 1 infectivity with preservation of conformational and functional integrity of virion surface proteins. J. Virol. 72, 7992–8001 (1998).
Liu, J., Bartesaghi, A., Borgnia, M. J., Sapiro, G. & Subramaniam, S. Molecular architecture of native HIV-1 gp120 trimers. Nature 455, 109–113 (2008).
Wang, H. et al. Cryo-EM structure of a CD4-bound open HIV-1 envelope trimer reveals structural rearrangements of the gp120 V1V2 loop. Proc. Natl Acad. Sci. USA, 10.1073/pnas.1615939113 (2016).
Dam, K. A., Fan, C., Yang, Z. & Bjorkman, P. J. Intermediate conformations of CD4-bound HIV-1 Env heterotrimers. Nature 623, 1017–1025 (2023).
Mangala Prasad, V. et al. Cryo-ET of Env on intact HIV virions reveals structural variation and positioning on the Gag lattice. Cell 185, 641–653 e17 (2022).
Kwon, Y. D. et al. Unliganded HIV-1 gp120 core structures assume the CD4-bound conformation with regulation by quaternary interactions and variable loops. Proc. Natl Acad. Sci. USA 109, 5663–5668 (2012).
Herschhorn, A. et al. The beta20-beta21 of gp120 is a regulatory switch for HIV-1 Env conformational transitions. Nat. Commun. 8, 1049 (2017).
Prevost, J. et al. Envelope glycoproteins sampling states 2/3 are susceptible to ADCC by sera from HIV-1-infected individuals. Virology 515, 38–45 (2018).
Rajashekar, J. K. et al. Modulating HIV-1 envelope glycoprotein conformation to decrease the HIV-1 reservoir. Cell Host Microbe 29, 904–916 e6 (2021).
Madani, N. et al. Activation and Inactivation Of Primary Human Immunodeficiency Virus Envelope Glycoprotein Trimers By Cd4-mimetic Compounds. J. Virol. 91, 10.1128/JVI.01880-16 (2017).
Ochsenbauer, C. et al. Generation of transmitted/founder HIV-1 infectious molecular clones and characterization of their replication capacity in CD4 T lymphocytes and monocyte-derived macrophages. J. Virol. 86, 2715–2728 (2012).
Salazar-Gonzalez, J. F. et al. Genetic identity, biological phenotype, and evolutionary pathways of transmitted/founder viruses in acute and early HIV-1 infection. J. Exp. Med 206, 1273–1289 (2009).
O’Brien, W. A. et al. HIV-1 tropism for mononuclear phagocytes can be determined by regions of gp120 outside the CD4-binding domain. Nature 348, 69–73 (1990).
Koyanagi, Y. et al. Dual infection of the central nervous system by AIDS viruses with distinct cellular tropisms. Science 236, 819–822 (1987).
Krapp, C. et al. Guanylate Binding Protein (GBP) 5 Is an Interferon-Inducible Inhibitor of HIV-1 Infectivity. Cell Host Microbe 19, 504–514 (2016).
Sanders, R. W. et al. A next-generation cleaved, soluble HIV-1 Env Trimer, BG505 SOSIP.664 gp140, expresses multiple epitopes for broadly neutralizing but not non-neutralizing antibodies. PLoS Pathog. 9, e1003618 (2013).
Emi, N., Friedmann, T. & Yee, J. K. Pseudotype formation of murine leukemia virus with the G protein of vesicular stomatitis virus. J. Virol. 65, 1202–1207 (1991).
Fontaine, J., Chagnon-Choquet, J., Valcke, H. S., Poudrier, J. & Roger, M. High expression levels of B lymphocyte stimulator (BLyS) by dendritic cells correlate with HIV-related B-cell disease progression in humans. Blood 117, 145–155 (2011).
Fontaine, J. et al. HIV infection affects blood myeloid dendritic cells after successful therapy and despite nonprogressing clinical disease. J. Infect. Dis. 199, 1007–1018 (2009).
Mukherjee, S. et al. Mechanism and significance of cell type-dependent neutralization of flaviviruses. J. Virol. 88, 7210–7220 (2014).
Nikic, I., Kang, J. H., Girona, G. E., Aramburu, I. V. & Lemke, E. A. Labeling proteins on live mammalian cells using click chemistry. Nat. Protoc. 10, 780–791 (2015).
Blakemore, R. J. et al. Stability and conformation of the dimeric HIV-1 genomic RNA 5’UTR. Biophys. J. 120, 4874–4890 (2021).
Jain, A. et al. Regulation of Ebola GP conformation and membrane binding by the chemical environment of the late endosome. PLoS Pathog. 19, e1011848 (2023).
Edelstein, A. D. et al. Advanced methods of microscope control using muManager software. J. Biol. Methods 1, 10.14440/jbm.2014.36 (2014).
Juette, M. F. et al. Single-molecule imaging of non-equilibrium molecular ensembles on the millisecond timescale. Nat. Methods 13, 341–344 (2016).
Qin, F., Auerbach, A. & Sachs, F. A direct optimization approach to hidden Markov modeling for single channel kinetics. Biophys. J. 79, 1915–1927 (2000).
van de Meent, J. W., Bronson, J. E., Wiggins, C. H. & Gonzalez, R. L. Jr. Empirical Bayes methods enable advanced population-level analyses of single-molecule FRET experiments. Biophys. J. 106, 1327–1337 (2014).
Díaz-Salinas, M. A. et al. Conformational dynamics of the HIV-1 envelope glycoprotein from CRF01_AE is associated with susceptibility to antibody-dependent cellular cytotoxicity. bioRxiv https://doi.org/10.1101/2024.08.22.609179 (2024).
Mastronarde, D. N. Automated electron microscope tomography using robust prediction of specimen movements. J. Struct. Biol. 152, 36–51 (2005).
Punjani, A., Rubinstein, J. L., Fleet, D. J. & Brubaker, M. A. cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296 (2017).
Meng, E. C. et al. UCSF ChimeraX: Tools for structure building and analysis. Protein Sci. 32, e4792 (2023).
Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. Features and development of Coot. Acta Crystallogr D. Biol. Crystallogr 66, 486–501 (2010).
Adams, P. D. et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D. Biol. Crystallogr 66, 213–221 (2010).
Panico, M. et al. Mapping the complete glycoproteome of virion-derived HIV-1 gp120 provides insights into broadly neutralizing antibody binding. Sci. Rep. 6, 32956 (2016).
Arthur, L. O. et al. Chemical inactivation of retroviral infectivity by targeting nucleocapsid protein zinc fingers: a candidate SIV vaccine. AIDS Res Hum. Retroviruses 14, S311–S319 (1998).
Zheng, S. Q. et al. MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods 14, 331–332 (2017).
Mastronarde, D. N. Accurate, automatic determination of astigmatism and phase with Ctfplotter in IMOD. J. Struct. Biol. 216, 108057 (2024).
Nicastro, D. et al. The molecular architecture of axonemes revealed by cryoelectron tomography. Science 313, 944–948 (2006).
Heumann, J. M., Hoenger, A. & Mastronarde, D. N. Clustering and variance maps for cryo-electron tomography using wedge-masked differences. J. Struct. Biol. 175, 288–299 (2011).
Scheres, S. H. RELION: Implementation of a bayesian approach to cryo-EM structure determination. J. Struct. Biol. 180, 519–530 (2012).
Grunst, M. W. et al. Structure and inhibition of SARS-CoV-2 spike refolding in membranes. Science 385, 757–765 (2024).
Ermel, U. H., Arghittu, S. M. & Frangakis, A. S. ArtiaX: An electron tomography toolbox for the interactive handling of sub-tomograms in UCSF ChimeraX. Protein Sci. 31, e4472 (2022).
Ameijeiras-Alonso, J., Crujeiras, R. M. & Rodriguez-Casal, A. multimode: An R Package For Mode Assessment. J. Stat. Softw. 97, 1–32 (2021).
Acknowledgements
The authors thank the CRCHUM BSL3 and Flow Cytometry Platforms for technical assistance, Mario Legault from the FRQS AIDS and Infectious Diseases network for providing the human PBMCs and plasma. Placental cord blood for humanized mouse engraftment was sourced from the Yale University Reproductive Sciences (YURS) Biobank. We thank Dennis Burton (The Scripps Research Institute) Julie Overbaugh (Fred Hutchinson Cancer Research Center) Frank Kirchhoff (Ulm University Medical Center) and Beatrice H. Hahn (University of Pennsylvania) for kindly providing the infectious molecular clone (IMC) JR-FL, BG505 (T332N), AD8 (Vpu + ) and CH058TF respectively. We thank Shenping Wu (Yale CryoEM Resource Center) and John Heumann (University of Colorado Boulder) for technical and conceptual advice regarding cryoET data collection and analysis. We thank James Robinson for providing the plasmids to produce the A32 and 17b Abs. We thank NIH Intramural CryoEM Consortium (NICE) for microscopy resource and consultant. We thank John P. Moore (Cornell University) for kindly giving us the BG505 SOSIP.664 plasmid and Andrew Ward (Scripps Research) for providing the plasmids to produce the PGT145 Abs. We thank Julian Bess, Jr. and Jeff Lifson of the AIDS and Cancer Virus Program of the Frederick National Laboratory for Cancer Research for providing the SupT1 HIV-1 BAL/A66-R5 CL.1 CLN445 cell line produced virus (lot P4338) used in this study. Production of the virus was supported with federal funds from the National Cancer Institute, National Institutes of Health, under Contract No. 75N91019D00024/HHSN261201500003I. This study was supported by a CIHR Team grant #197728, a Canada Foundation for Innovation (CFI) grant #41027 to A.F and by the National Institutes of Health to A.F. (R01AI148379, R01AI176531), A.F. and J.B.M. (R01AI150322), A.F. and M.P. (R01AI174908,), A.F., P.K., M.P., J.S and A.B.S. (R01AI186809), M.P. (P01AI162242), P.K. (UM1AI164559, R01AI145164), W.M. (R37AI150560, R01AI176904, P01AI150471), and M.W.G. (F31 AI176650). Support for this work was also provided by UM1AI164562 (ERASE) to A.F., J.S., A.B.S., and P.K. A.F. was supported by a Canada Research Chair on Retroviral Entry RCHS0235 950–232424. This research was also supported by the Intramural Research Program of the Eunice Kennedy Shriver National Institute of Child Health and Human Development to D.M. (ZIA HD008998). G.B.B., M.B., E.B., and K.D. were supported by CIHR doctoral fellowships. E.B. was supported by a FRQS doctoral fellowship. M.W.G. was supported by NIH T32 AI055403 and is a recipient of the Gruber Science Fellowship. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. The views expressed in this manuscript are those of the authors and do not reflect the official policy or position of the Uniformed Services University, US Army, the Department of Defense, or the US Government.
Author information
Authors and Affiliations
Contributions
Conceptualization: J.R., M.W.G, L.N, M.A.D-S, L.Z., P.K. W.M., J.B.M., M.P. and A.F. Methodology: J.R., M.W.G, L.N, M.A.D-S, L.Z. W.D.T., W.L., H.K., S.T.B., Z.C.L., A.J.M., R.K.H., D.M., P.K. W.M., J.B.M., M.P. and A.F. Investigation: J.R., M.W.G, L.N, M.A.D-S, L.Z., P.K., J.B.M., W.D.T, L.M, F.Z., C.B., R.K.H., Resources: J.R., L.N, M.W.G, M.A.D-S, D.Y., L.M., T.J.C., H-C.C., M.B., G.B-B., K.D., E.B., D.C., H.M., W.A.H., J.S., A.B.S., D.M., P.K., W.M., J.M., M.P. and A.F. Supervision: J.R., L.Z. W.A.H., J.S., A.B.S., W.L., D.M., P.K. W.M., J.B.M., M.P. and A.F. Funding acquisition: W.A.H., J.S., A.B.S., J.B.M., W.M., M.P. and A.F. Writing original draft: J.R., M.W.G, L.N, M.A.D-S, W.M., J.B.M., M.P. and A.F. Writing & editing: review: J.R., L.N, M.W.G, M.A.D-S, P.K., W.D.T., L.M, F.Z., C.B., D.Y., T.J.C., H-C.C., M.B., G.B-B., S.G., W.L., K.D., E.B., D.C., H.M., W.A.H., J.S., R.K.H., D.M., A.B.S., P.K., W.M., J.B.M., M.P. and A.F.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Richard, J., Grunst, M.W., Niu, L. et al. The asymmetric opening of HIV-1 Env by a potent CD4 mimetic enables anti-coreceptor binding site antibodies to mediate ADCC. Nat Commun 16, 10419 (2025). https://doi.org/10.1038/s41467-025-65866-x
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-65866-x
