
Abstract
Apaf-1-interacting protein (APIP) has been implicated in inflammation-related processes, including myocardial infarction and cancer progression. However, its role in systemic inflammation remains elusive. Here, we investigate the APIP-mediated regulation of inflammasome activity in mice and human macrophages. Loss of APIP in the myeloid lineage (Apip cKO mice) compromises the activation of canonical NLRP3 and non-canonical caspase-11 inflammasomes, reducing pyroptosis in bone marrow-derived macrophages (BMDM). Conversely, these inflammatory responses are enhanced in BMDMs from APIP-transgenic mice. Consistently, APIP knockdown in human macrophages inhibits the activation of NLRP3 and caspase-4 inflammasomes. Mechanistically, APIP binds to TRAF6, activating downstream NF-κB and JNK signaling and facilitating the priming of both inflammasomes. Importantly, systemic inflammation induced by LPS or bacterial infection is attenuated in Apip cKO mice but exacerbated in APIP-transgenic mice. Thus, our findings suggest that APIP is crucial in regulating both canonical and non-canonical inflammasomes, presenting a potential therapeutic target for inflammatory diseases.
Similar content being viewed by others


Introduction
Macrophages, key white blood cells, are essential for defending against bacterial and viral infections through phagocytosis and inflammatory responses1. Central to their function are inflammasomes, which detect pathogens or danger signals, and facilitate the release of cytokines through pyroptosis, a specialized form of cell death that triggers intense inflammation2. Among these, the NLRP3 inflammasome has been extensively studied, and comprises NLRP3, ASC, and caspase-13. Once activated, caspase-1 cleaves IL-1β, IL-18, and gasdermin D, forming pores in the cell membrane that enable cytokine release and initiate pyroptosis3.
The NLRP3 inflammasome activation occurs in two stages: priming and activation4. Priming, typically induced by pathogen-associated molecular patterns (PAMP) like lipopolysaccharide (LPS) of gram-negative bacteria, increases the expression of pro-IL-1β, NLRP3, etc. The subsequent activation stage, such as K+ efflux, leads to inflammasome assembly and cytokine release. Moreover, non-canonical inflammasomes involving caspases-4 and -5 in humans and caspase-11 in mice are also essential for immune responses against LPS5. These caspases directly bind to LPS in the cytoplasm, get activated, and subsequently cleave gasdermin D to trigger pyroptosis. This process amplifies inflammatory responses and further activates the NLRP3 inflammasome6.
While inflammasomes are protective against infections, their excessive activation can cause severe inflammation, leading to autoimmune diseases like cryopyrin-associated periodic syndrome (CAPS), multiple sclerosis, and rheumatoid arthritis7. They also play a role in cancer, metabolic, and neurological disorders, making them promising therapeutic targets8. Among the various diseases involving inflammasomes, sepsis is of particular interest9,10. Sepsis, a fatal disease caused by infection, leads to systemic inflammation and multiple organ failure, remaining a serious global health problem despite advances in medical hygiene11.
Apaf-1-interacting protein (APIP) has emerged as a potential regulator in sepsis-related inflammasome pathways12,13. Beyond its role in inflammation, APIP is a ubiquitous protein involved in multiple cellular processes, including apoptosis, metabolism, and cancer progression. Functionally, APIP inhibits apoptosis by binding to APAF1, thereby blocking cytochrome c- and caspase-9-mediated cell death14. APIP also promotes cell survival under hypoxic conditions through sustained activation of the AKT and ERK pathways, a mechanism linked to myocardial infarction protection15,16. In addition to its role in apoptosis, APIP is crucial for methionine metabolism, catalyzing an intermediate step in the methionine salvage pathway, which is essential for maintaining cellular methylation reactions and polyamine biosynthesis17. Furthermore, APIP has also been implicated in cancer progression by modulating ERBB3–ERBB2 interactions in gastric cancer18 and promoting prostate cancer proliferation via ERK activation19.
Recent genetic studies have suggested a potential link between APIP and sepsis susceptibility. Genome-wide association studies (GWAS) from the ‘Seattle SIRS’ and the ‘Kenyan Bacteremia’ have identified two single-nucleotide polymorphisms (SNPs) near the APIP gene12,13. These SNPs are associated with reduced APIP expression and correlate with lower sepsis incidence and mortality in SIRS patients, as well as decreased incidence of non-typhoidal Salmonella (NTS) bacteremia. Current evidence suggests that APIP deficiency leads to the accumulation of 5’-methylthioadenosine (MTA) in the methionine salvage pathway, which promotes pyroptosis and enhances bacterial clearance12,13. However, the role of APIP in regulating sepsis-related inflammasomes and systemic inflammation has not yet been elucidated.
Here, we show that APIP regulates both NLRP3 and caspase-11/4 inflammasomes under various inflammatory conditions in mice and human macrophages. APIP stimulates the priming step of inflammasomes by binding to TRAF6. These effects of APIP exacerbate systemic inflammation in mice, underscoring the importance of its role in inflammatory diseases like sepsis.
Results
APIP promotes the activation of the NLRP3 inflammasome
To investigate the role of APIP in inflammation, we generated Apip knockout (KO) mice using a gene trap assay, but found that Apip homozygous KO mice were embryonic lethal (Supplementary Table 1). Therefore, we generated macrophage-specific Apip conditional KO mice (Apip cKO, Lyz2-Cre; Apipfl/fl) using Apip-floxed mice targeting exons 4 and 5 (Supplementary Fig. 1a, b) and myeloid lineage-specific Cre recombinase mice (Lyz2-Cre)20, and assessed the effects of Apip KO on inflammation in primary macrophages. With western blotting and qRT-PCR assays, we found undetectable Apip expression in bone marrow-derived macrophages (BMDM) isolated from Apip cKO mice compared to control (Apipfl/fl) mice, regardless of LPS treatment (Fig. 1a, b). Apip expression levels in other tissues, such as the heart and liver, were comparable between the groups (Supplementary Fig. 1c).

a, b APIP expression in BMDMs from Apipfl/fl (control) and Lyz2-cre; Apipfl/fl (Apip cKO) mice, untreated (NT) or LPS-stimulated (200 ng/ml, 3 h), analyzed by western blotting (a) and qRT-PCR (b). c–e IL-1β secretion and caspase-1 activation in control and Apip cKO BMDMs after LPS (200 ng/ml, 3 h) and ATP (3 mM, 30 min) treatment. L + A; LPS + ATP. Supernatants (Sup) and lysates (Cell) were analyzed by western blotting (c), and IL-1β (d) and caspase-1 (e) signals quantified. Data represent mean ± SD (n = 4 independent cultures; panels b, d, e). f, g APIP expression in WT and APIPTG/+ BMDMs analyzed by western blotting (f) and qRT-PCR (g). Data represent mean ± SD (n = 3 independent cultures). h–j IL-1β secretion and caspase-1 activation in WT and APIPTG/+ BMDMs after LPS (500 ng/ml, 3 h) and ATP (3 mM, 15 min or 30 min) treatment (WT, n = 5; APIPTG/+, n = 6 independent cultures) (h, i). Cells treated with LPS (1 μg/ml, 3 h), and ATP (3 mM, 30 min), and FAM-YVAD-FMK were stained with Hoechst and observed under a fluorescence microscope (j). FAM-YVAD-FMK–positive cells were quantified from 4 (NT) or 10 (LPS + ATP) random microscopic fields (technical replicates). Data represent mean ± SD. nd, not detected. k, l Pyroptosis in Apip cKO or APIPTG/+ BMDMs stimulated with LPS and ATP. Control and Apip cKO BMDMs were treated with LPS (200 ng/ml, 3 h) and ATP (3 mM, 1 h) (n = 4–6 independent cultures; exact n values shown on the graphs/Source Data) (k). WT and APIPTG/+ BMDMs were treated with LPS (500 ng/ml, 3 h) and ATP (3 mM, 30 min) (n = 3 independent cultures) (l). Cells were stained with calcein-AM and PI, and PI–positive cells were quantified by fluorescence microscopy. Data represent mean ± SEM. Source data are provided as a Source Data file. Statistical tests: Unpaired two-tailed Student’s t-test (d, e, g, i), two-way ANOVA with Holm–Sidak’s (b), with Tukey’s (j), or with Sidak’s multiple comparison test (k, l). ns, non-significant; arb. units, arbitrary units.
When BMDMs were treated with LPS and ATP, the levels of IL-1β in culture medium and active form of caspase-1 in cell lysates decreased by more than 50% in Apip cKO BMDMs compared to control BMDMs (Fig. 1c–e), indicating an inhibition of the NLRP3 inflammasome activation21. In parallel, when the APIP expression was reduced by siRNA in J774A.1 murine macrophage cell line, levels of secreted IL-1β and IL-18, as well as caspase-1 activation as determined by western blotting and FAM-YVAD-FMK staining, were reduced upon LPS and nigericin treatment (Supplementary Fig. 2a–c, e–g). Similarly, APIP knockdown reduced caspase-1 activation in response to LPS and nigericin treatment in both differentiated THP-1 macrophages and primary human macrophages (Supplementary Fig. 2k, o).
In addition, we generated APIP transgenic mice (TG) expressing human APIP under the CMV early enhancer/chicken beta actin (CAG) promoter16,18, which ubiquitously express higher levels of human APIP compared to mouse APIP (Fig. 1f, g). Mouse and human APIP share 94% sequence identity and 97% similarity, indicating a high degree of conservation. A similar set of experiments was conducted in BMDMs isolated from wild-type (WT) and APIPTG/+ mice. The results demonstrated that compared to WT BMDMs, levels of secreted IL-1β and IL-18, as well as caspase-1 activation, significantly increased in APIPTG/+ BMDMs (Fig. 1h–j and Supplementary Fig. 15b, d–f). Collectively, these results indicate that APIP plays a crucial role in regulating the NLRP3 inflammasome in macrophages.
APIP regulates the NLRP3 inflammasome-dependent pyroptosis
Following the assessment of the NLRP3 inflammasome activity, we further investigated the impact of APIP on the NLRP3 inflammasome-dependent pyroptosis. Cell death was evaluated by staining with calcein-AM and propidium iodide (PI), followed by counting PI-positive cells, and by measuring cytotoxicity using a lactate dehydrogenase (LDH) release assay. As reported22,23, cell death caused by LPS and ATP or nigericin treatment in macrophages was inhibited by MCC950, a potent NLRP3 inflammasome inhibitor, and partially by VX-765, a peptidomimetic inhibitor of caspase-1, but not effectively by inhibitors that are peptide-based or target apoptosis or necroptosis (Supplementary Fig. 3a–d), confirming that the type of cell death is NLRP3 inflammasome-dependent pyroptosis. In addition to PI staining and LDH assay, pyroptosis was further confirmed by detecting gasdermin D (GSDMD) cleavage. Notably, pyroptosis was significantly reduced in Apip cKO BMDMs (Fig. 1k), as well as in APIP-knockdown J774A.1 cells (Supplementary Fig. 2a, d, h–j), THP-1 cells (Supplementary Fig. 2l–n), and primary human macrophages (Supplementary Fig. 2p–r). On the other hand, the pyroptosis was enhanced in APIPTG/+ BMDMs compared to WT BMDMs (Fig. 1l and Supplementary Fig. 15a–c). These results indicate that APIP regulates the NLRP3 inflammasome-dependent pyroptosis.
We expanded our investigation on the role of APIP in inflammation and pyroptosis induced by various signals. The results revealed that APIP knockdown in J774A.1 cells reduced inflammatory responses, including IL-1β secretion and caspase-1 activation, in response to LPS and monosodium urate (MSU) crystals (Supplementary Fig. 4a–c), which activate the NLRP3 inflammasome and contribute to gout24. This highlights the role of APIP in regulating the inflammatory responses triggered by DAMPs and PAMPs, such as LPS, nigericin, ATP, and MSU.
APIP enhances the formation of ASC and NLRP3 oligomers
The oligomerization of ASC, an adapter protein of the NLRP3 inflammasome3, is considered a marker of the NLRP3 inflammasome formation25. We thus investigated whether APIP affects the formation of ASC oligomers. When BMDMs were stimulated with LPS and ATP and their lysates were incubated with disuccinimidyl suberate (DSS) cross-linker, the formation of ASC oligomers, including monomers, dimers, trimers, and oligomers, was readily observed in the ASC pellets of WT BMDMs (Fig. 2a). Intriguingly, the formation of ASC oligomers was reduced to 40% in Apip cKO BMDMs compared to control, while the total amounts of ASC in cell extracts were not changed (Fig. 2b). When we performed the same experiments in J774A.1 and THP-1 cells, we found that ASC oligomerization was similarly reduced in APIP knockdown cells compared to control (Supplementary Fig. 5a, b, d, e). Conversely, ASC oligomerization was increased in APIPTG/+ BMDMs compared to WT BMDMs (Fig. 2c). Furthermore, using a co-immunoprecipitation (Co-IP) assay, we found that the interaction between NLRP3 and ASC following LPS and ATP treatment was enhanced in APIPTG/+ BMDMs compared to WT BMDMs (Fig. 2d). However, as will be described in detail below, the increased ASC oligomerization and NLRP3–ASC interaction were suppressed by treatment with Takinib, a TRAF6 signaling inhibitor26.

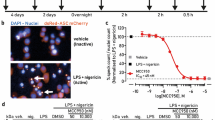
a–c Stimulatory effect of APIP on ASC oligomerization. Control and Apip cKO BMDMs were untreated (NT) or treated with LPS (200 ng/ml, 3 h) and ATP (3 mM, 30 min) (a, b). WT and APIPTG/+ BMDMs were treated with LPS (1 μg/ml, 3 h), Takinib (10 μM, 30 min), and ATP (3 mM, 30 min) (c). Cell lysates were incubated with DSS (2 mM, 1 h), and ASC pellets and lysates were analyzed by western blotting (a, c). Pellet signals were quantified and normalized to ASC levels in lysates (b). Data represent mean ± SD (n = 3 independent cultures). d Co-IP analysis of the ASC–NLRP3 interaction. WT and APIPTG/+ BMDMs treated as in (c) were subjected to Co-IP using an anti-ASC antibody. Input, 5%. e, f Stimulatory effect of APIP on ASC speck formation. Control and Apip cKO BMDMs were treated with LPS (1 μg/ml, 3 h) and ATP (3 mM, 30 min) (e). WT and APIPTG/+ BMDMs were treated with LPS (500 ng/ml, 3 h) and ATP (3 mM, 30 min) (f). Cells and nuclei were stained with anti-ASC antibody and Hoechst 33342, respectively. ASC speck–positive cells (magenta arrowheads; representative) were quantified from 2 (f, NT), 4 (e, NT), or 10 (LPS + ATP) random microscopic fields (technical replicates). Scale bar, 100 μm; nd, not detected. Data represent mean ± SD. Statistical tests: two-way ANOVA with Sidak’s multiple comparison test (b, e, f). *p < 0.05, **p < 0.01, ***p < 0.001. Exact p values are in the Source Data. Source data are provided as a Source Data file. arb. units, arbitrary units.
In addition, we analyzed whether APIP affects the formation of ASC specks, a visual hallmark of inflammasome activation25. Similar to the ASC oligomers, the formation of ASC specks following LPS and ATP treatment was significantly attenuated in Apip cKO BMDMs compared to control (Fig. 2e). Additionally, ASC specks were decreased in APIP knockdown J774A.1 cells upon exposure to LPS and nigericin (Supplementary Fig. 5c). Moreover, upon exposure to LPS and ATP, ASC specks significantly increased in APIPTG/+ BMDMs compared to WT BMDMs (Fig. 2f). These data suggest that APIP promotes the formation of ASC oligomers and specks in macrophages. Furthermore, Native-PAGE assay revealed that treatment with LPS and ATP or nigericin induced the formation of NLRP3 oligomers in BMDMs and J774A.1 cells (Supplementary Fig. 5f–h). On the other hand, this formation of NLRP3 oligomers was reduced in Apip cKO BMDMs and APIP knockdown J774A.1 cells, whereas it was enhanced in APIPTG/+ BMDMs (Supplementary Fig. 5f–h). These results underscore the importance of APIP in promoting the formation of NLRP3–ASC oligomers, which in turn facilitates the NLRP3 inflammasome activation.
APIP regulates the NLRP3 inflammasome and pyroptosis independently of MTA
In the methionine salvage pathway, it was reported that reduced expression of APIP leads to the accumulation of intermediate MTA within the cell, and this accumulation increases the NLRP3 inflammasome activation and pyroptosis12. Contrary to this report, we found that decreasing APIP expression in macrophages reduced the NLRP3 inflammasome activation and pyroptosis (Fig. 1c–e, k and Supplementary Fig. 2). Therefore, we decided to further investigate this discrepancy. Treatment with MTA in the presence of LPS and nigericin increased levels of secreted IL-1β in J774A.1 cells, but this increase was attenuated in APIP knockdown cells (Supplementary Fig. 6a, b). Additionally, under the same conditions, MTA treatment had no effect on the activation of caspase-1, whereas APIP knockdown consistently reduced it (Supplementary Fig. 6a, c). Thus, MTA together with LPS and nigericin increases the amounts of IL-1β, but not active caspase-1, independently of APIP.
We also evaluated whether MTA affects the NLRP3 inflammasome-dependent pyroptosis in J774A.1 cell. PI staining revealed that the LPS and nigericin-induced pyroptosis was reduced by APIP knockdown, as seen above, but was not affected by MTA treatment (Supplementary Fig. 6d). We extended our analysis to determine the effects of MTA on LPS-induced inflammatory responses. Assessment of cytokine mRNA levels with qRT-PCR revealed that additional treatment with MTA further increased IL-1β mRNA levels compared to cells treated solely with LPS (Supplementary Fig. 6e). On the other hand, mRNA levels of TNF and IL-18 decreased with MTA treatment under the same condition. Consistent with these findings, MTA treatment increased IL-1β secretion in response to LPS and nigericin, while reducing TNF secretion (Supplementary Fig. 6f, g). Together, MTA itself may enhance the NLRP3 inflammasome activity, as previously reported, by increasing pro-IL-1β levels in macrophages independently of APIP.
To assess whether the catalytic MtnB (methylthioribulose-1-phosphate dehydratase) activity of APIP influences IL-1β expression, we utilized the E139A mutant, which retains only ~1% of the catalytic activity of WT APIP17. Transient expression of either APIP WT or E139A in RAW264.7 cells followed by LPS treatment led to comparable increases in pro-IL-1β and phosphorylated NF-κB (Ser536) levels relative to the control (Supplementary Fig. 7a–c), suggesting that APIP enhances LPS signaling independently of its MtnB activity.
APIP does not affect the AIM2 inflammasome activity
Among the well-characterized canonical inflammasomes, the AIM2 inflammasome is notable for its activation upon recognition of double-stranded DNA from both microbial and host cells27. Similar to the NLRP3 inflammasome, the activation of the AIM2 inflammasome requires ASC and caspase-1. These findings prompted us to investigate whether APIP also affects the AIM2 inflammasome. We thus transfected BMDMs and J774A.1 cells with poly(dA:dT) to activate the AIM2 inflammasome28. Notably, we found that APIP did not affect the incidence of cell death and caspase-1 activation following AIM2 inflammasome activation in BMDMs, as observed in both Apip cKO and APIPTG/+ BMDMs (Supplementary Fig. 8a–h). Consistently, APIP knockdown in J774A.1 cells also did not alter IL-1β secretion, caspase-1 activation, and cell death upon AIM2 inflammasome activation (Supplementary Fig. 9a–g).
APIP enhances the non-canonical inflammasome activity in macrophages
Because LPS also activates the non-canonical inflammasome when internalized into the cytoplasm29, we examined whether APIP affects the non-canonical inflammasome. BMDMs were primed with Pam3CSK4, a synthetic triacylated lipopeptide that activates the TLR2/TLR1 heterodimer, instead of using LPS to prevent unnecessary TLR4 induction. Cells were then treated with MCC950 and transfected with LPS. As reported30, the cytotoxicity markedly increased by LPS transfection in Pam3CSK4-treated BMDMs, regardless of MCC950 treatment (Fig. 3a). Intriguingly, this cytotoxicity and GSDMD cleavage were significantly decreased in Apip cKO BMDMs and increased in APIPTG/+ BMDMs, suggesting that APIP promotes the non-canonical inflammasome activation (Fig. 3a–f).

a–f Effect of APIP on non-canonical inflammasome activation. Control and Apip cKO BMDMs (a–c), or WT and APIPTG/+ BMDMs (d–f) were treated with Pam3CSK4 (1 μg/ml, 4 h) and MCC950 (10 μM, 1 h), and transfected with LPS (2.5 μg/ml, 16 h). Supernatants were analyzed by LDH assay (a, d). Cell lysates were analyzed by western blotting (b, e), and blot signals quantified by densitometry (c, f). LPS (tf), LPS transfection; NT-GSDMD, N-terminal GSDMD. Data represent mean ± SD. (n = 4 independent cultures; c n = 5). g–l Effect of APIP on caspase-11 expression. Control and Apip cKO BMDMs (g–i), or WT and APIPTG/+ BMDMs (j–l) were treated with LPS (1 μg/ml, 3 h). Cell lysates were analyzed by western blotting (g, j), and signals of pro-CASP11 (p38, arrow) and APIP were quantified by densitometry (h, i, k, l). Data represent mean ± SD. (n = 4 independent cultures; h n = 3). Statistical tests: two-way ANOVA with Tukey’s multiple comparison test (a, h, k) or unpaired two-tailed Student’s t-test (c, d, f, i, l). ns, non-significant. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. Exact p values are in the Source Data. Source data are provided as a Source Data file. arb. units, arbitrary units.
Together with the results from BMDMs, APIP knockdown in J774A.1 cells, THP-1 cells, and primary human macrophages resulted in reduced cell death following the non-canonical inflammasome activation (Supplementary Figs. 10a and 11a–c, e, f). Immunoblot analysis revealed that Pam3CSK4 treatment and LPS transfection increased pro-caspase-11, a key effector in the non-canonical inflammasome, in J774A.1 cells (Supplementary Fig. 10b, c). APIP knockdown significantly attenuated the increase of pro-caspase-11 and IL-1β in supernatants, as well as pro-caspase-11 and caspase-1 in cell lysates. Under the same condition, ectopic expression of APIP increased cell death and the levels of pro-caspase-11 and pro-IL-1β in RAW264.7 cells that do not express ASC31 (Supplementary Fig. 10d–f). Collectively, APIP increases not only the activity of the NLRP3 inflammasome but also that of the non-canonical inflammasome.
To delineate the role of APIP in the non-canonical inflammasome pathway, we further evaluated the effect of APIP on caspase-11 expression. Western blot analysis revealed that LPS treatment increased pro-caspase-11 levels, which were decreased in Apip cKO BMDMs and increased in APIPTG/+ BMDMs compared to control cells (Fig. 3g, h, j, k). LPS treatment also elevated the levels of both endogenous mouse APIP and exogenous human APIP (Fig. 3i, l). Consistently, APIP knockdown attenuated the LPS-induced increase in pro-caspase-11 levels in J774A.1 cells (Supplementary Fig. 10g, h), whereas ectopic expression of APIP enhanced it in RAW264.7 cells (Supplementary Fig. 10i, j). Moreover, APIP knockdown reduced pro-caspase-4 levels in both THP-1 cells and primary human macrophages under basal conditions (Supplementary Fig. 11b, d, e, g). Notably, treatment with phorbol 12-myristate 13-acetate (PMA)32 or macrophage-colony stimulating factor (M-CSF)33 during differentiation was sufficient to induce pro-caspase-4 expression, likely via Protein Kinase C (PKC)-mediated NF-κB activation34, with TRAF6 involvement. Thus, APIP acts as a key regulator of the non-canonical inflammasome in macrophages, affecting both pyroptosis and the expression of essential inflammatory proteins such as caspase-11 and caspase-4.
APIP promotes the priming of NLRP3 inflammasome via the TRAF6-TAK1 axis
In addition to stimulating both the NLRP3 and caspase-11 inflammasomes, APIP increases pro-IL-1β and pro-caspase-11 levels (Supplementary Fig. 10e–j). We thus explored the priming step of inflammasome activation. Treatment of J774A.1 cells with LPS markedly increased mRNA levels of IL-1β, TNF, and IL-18, and this increase was significantly attenuated by APIP knockdown (Fig. 4a–d). Similarly, compared to control BMDMs, mRNA levels of TNF and IL-1β were reduced in Apip cKO BMDMs upon LPS treatment (Fig. 4e, f). Furthermore, we examined the mitogen-activated protein kinase (MAPK) pathway, as well as NF-κB activation in J774A.1 cell. LPS treatment increased levels of phospho-JNK1 (p46), phospho-JNK2 (p54), pro-IL-1β, and phospho-IκBα (S32/S36) (Fig. 4g, h). Whereas this induction was all reduced in APIP knockdown cells. These results suggest that APIP enhances the activation of both NF-κB and JNK, which are downstream of the TRAF6-TAK1 signaling35.

a–f qRT-PCR analysis of LPS-induced inflammation. J774A.1 cells were transfected with negative control siRNA (NC) or siApip (100 nM, 48 h) and left untreated (NT) or treated with LPS (500 ng/ml, 3 h) (a–d). Control and Apip cKO BMDMs were treated with LPS (200 ng/ml, 3 h) (e, f). Total RNAs were analyzed by qRT-PCR. Data represent mean ± SD (n = 3 independent cultures). g–k Western blot analysis of LPS-induced inflammation. J774A.1 cells were transfected with NC or siApip (100 nM, 48 h) and treated with LPS (500 ng/ml, 3 h) (g, h). RAW264.7 cells were transfected with Mock or APIP for 24 h and treated with LPS (1 μg/ml) alone or with Takinib (10 μM) for 3 h (i–k). Cell lysates were analyzed by western blotting (g, i), and blot signals quantified by densitometry (h, j, k). Data represent mean ± SD (n = 4 independent cultures; h n = 5 for pro-IL-1β and n = 3 for p-IκBα). Statistical tests: two-way ANOVA with Tukey’s multiple comparison test (a–d, j), unpaired two-tailed Student’s t-test (e, f), multiple unpaired t-test with Holm–Sidak’s method (h), or one-way ANOVA with Tukey’s multiple comparison test (k). ns, non-significant. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. Exact p values are in the Source Data. Source data are provided as a Source Data file. arb. units, arbitrary units.
Since the NLRP3 phosphorylation at Ser198/194, a key priming event facilitated through JNK1 downstream of TLRs, promotes self-association of NLRP336, we examined whether APIP regulates the NLRP3 phosphorylation. With IP assays, we confirmed that LPS treatment induced NLRP3 phosphorylation at serine in J774A.1 cells (Supplementary Fig. 12). Although we could not identify the exact phosphorylation site due to the lack of available antibodies, this serine phosphorylation was reduced by APIP knockdown in J774A.1 cells (Supplementary Fig. 12). Additionally, NLRP3 phosphorylation at tyrosine, especially at Tyr861/859, which is known to inhibit NLRP3 inflammasome activation37, was not affected by APIP knockdown. These results suggest that APIP may regulate the NLRP3 inflammasome via the JNK1-mediated phosphorylation of NLRP3.
Conversely, we assessed the effect of APIP overexpression on the NLRP3 inflammasome priming. Although RAW264.7 cells do not express ASC, the priming step of the NLRP3 inflammasome, particularly the NF-κB pathway, can be studied38. When RAW264.7 cells were treated with LPS, we found that levels of pro-IL-1β were further enhanced by ectopic expression of APIP (Fig. 4i, j). However, this increase was negated by treatment with Takinib, a potent TAK1 inhibitor. These results indicate that the TRAF6-TAK1 signaling is necessary for the stimulatory effect of APIP on pro-IL-1β expression, the priming step of NLRP3 inflammasome activation. Interestingly, levels of both endogenous and ectopic APIP repeatedly increased with LPS treatment (Figs. 1b, 3i, l, and 4k). This implies that APIP is regulated through a yet unidentified mechanism beyond transcriptional control under inflammatory conditions, given the different promoters of endogenous and exogenous (CMV) APIP. However, ectopic APIP expression decreased with Takinib co-treatment (Fig. 4k). These findings suggest a possible positive feedback relationship between APIP expression and NF-κB signaling.
APIP binds to TRAF6 via its N-terminal region in macrophages
In a proteomic study using tandem affinity purification combined with mass spectrometry in HEK293 cells, APIP (also known as MMRP19) was detected at low frequency with TRAF1 and TRAF2, suggesting a possible association39. Moreover, using the ProteinPrompt program, we also predicted a high likelihood of interaction between APIP and TRAF1 (score = 0.9613/1). The recognition of LPS by TLR4 on the cell surface is known to increase the E3 ligase activity of TRAF6, leading to TAK1 activation and subsequent activation of both NF-κB and JNK40,41. We thus hypothesized that APIP may bind to TRAF6 to mediate both transcriptional and non-transcriptional priming of the NLRP3 inflammasome via TLR/IL-1R signals42,43. From IP assays, we found the interaction between HA-APIP and FLAG-TRAF6 in the transfected HEK293T cells (Supplementary Fig. 13a, b) and confirmed the interaction between endogenous APIP and TRAF6 in J774A.1 cells (Fig. 5a). Additionally, LPS treatment enhanced the interaction between APIP and TRAF6 in a time-dependent manner, with the increase observed up to 30 min (Fig. 5b). Under the same conditions, we could not clearly observe the interaction between APIP and TRAF1 or TRAF2. Therefore, these results suggest that APIP predominantly interacts with TRAF6, whereas its interactions with TRAF1 and TRAF2 are confounded by non-specific background signals. Notably, no interactions between APIP and NLRP3, ASC, or caspase-1 were detected in J774A.1 cells treated with LPS or in BMDMs treated with LPS and ATP (Supplementary Fig. 14a–c), suggesting that APIP does not directly interact with NLRP3 inflammasome components to promote their activation.

a, b Endogenous interaction between APIP and TRAF6 in macrophages. J774A.1 cells left untreated (NT) (a) or treated with LPS (1 μg/ml, 10 or 30 min) (b), were subjected to Co-IP using an anti-APIP antibody. Input, 5%; arrows, TRAF6; asterisks, non-specific signals. c Requirement of the N-terminal region of APIP for TRAF6 binding. HEK293T cells transfected with APIP WT and deletion (∆) mutants for 24 h (upper) were analyzed by Co-IP using an anti-TRAF6 antibody (lower). Input, 5%; asterisks, light chains. d–f Effect of APIP WT and ΔN60 mutant on pro-IL-1β and p-NF-κB. RAW264.7 cells transfected with Mock, APIP WT, or ΔN60 mutant for 24 h, treated with LPS (500 ng/ml, 3 h), were analyzed by western blotting (d). Signals of pro-IL-1β (e) and p-NF-κB (f) quantified by densitometry. Data represent mean ± SD (n = 4 independent cultures). g, h Effect of APIP on TRAF6 auto-ubiquitination. RAW264.7 cells transfected with FLAG-TRAF6 alone or with APIP for 24 h (g), and J774A.1 cells transfected with NC or siApip (100 nM, 48 h) (h), were treated with MG132 (5 μM, 4 h) and LPS (1 μg/ml, 30 min). Cells were analyzed by Co-IP with anti-FLAG M2 affinity gel (g) or anti-TRAF6 antibody (h), followed by immunoblotting with anti-ubiquitin (K63, K48, or total) antibodies. Input, 5%. i Inhibition of TRAF6 signaling attenuates APIP-enhanced pyroptosis. WT and APIPTG/+ BMDMs were treated with LPS (500 ng/ml, 3 h), MCC950 (5 μM) or Takinib (20 μM, 30 min), and ATP (3 mM, 1 h). Cells were stained with calcein-AM and PI, and PI–positive cells were quantified. Data represent mean ± SEM (n = 3–6 independent cultures; exact n values shown on the graph/Source Data) (i). Experiments in a–c, g, h were independently repeated three times with similar results. Statistical tests: one-way ANOVA with Tukey’s (e, f) or two-way ANOVA with Sidak’s multiple comparison test (i). ns, non-significant. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. Exact p values are in the Source Data. Source data are provided as a Source Data file. arb. units, arbitrary units.
To delineate the binding motifs in APIP and TRAF6, domain mapping experiments were conducted using various deletion mutants (∆). Co-IP assays using an anti-TRAF6 antibody demonstrated that deletion of amino acids 1–60 at the N-terminus (∆N60), but not other regions including the C-terminus and middle, of APIP significantly impaired its ability to bind TRAF6 (Fig. 5c). We further generated a series of additional deletion mutants at the N-terminus and found that APIP mutants lacking up to the N-terminal 50 amino acids (∆N20–∆N50) showed an unexpected increase in their binding to TRAF6 (Supplementary Fig. 13c). Unlike these mutants, APIP ∆N60 mutant almost lost the binding affinity. In addition, when we examined the TRAF6 domains, deletion of the RING domain or coiled-coil domain in TRAF6 abolished its interaction with APIP (Supplementary Fig. 13d).
APIP binds to TRAF6 to activate the TRAF6-TAK1 signaling
To investigate the impact of APIP–TRAF6 interaction on the activation of TRAF6, RAW264.7 cells were transfected with APIP WT or ΔN60 mutant and treated with LPS. Compared to the control LPS-treated group, ectopic expression of APIP WT increased both pro-IL-1β levels and NF-κB p65 (S536) phosphorylation, but APIP ΔN60 mutant failed to do so (Fig. 5d–f). Considering the role of TRAF6 in the NF-κB signaling upon LPS treatment44, these results suggest that the binding of APIP to TRAF6 enhances TRAF6 activity. We thus examined the E3 ligase activity of TRAF6, particularly its K63-linked auto-ubiquitination45. IP assay results revealed that APIP overexpression promoted the K63-linked ubiquitination, but not the K48-linked ubiquitination, of FLAG-TRAF6 in RAW264.7 cells upon exposure to LPS (Fig. 5g). Consistently, similar experiments in J774A.1 cells revealed that APIP knockdown led to a reduction in the K63-linked ubiquitination of TRAF6 by LPS (Fig.5h).
Because TRAF6 activation is followed by TAK1 activation to stimulate the NLRP3 inflammasome activity46, we further examined whether inhibition of the TRAF6-TAK1 signaling by Takinib alleviates the stimulatory effects of APIP on the NLRP3 inflammasome. PI staining, LDH assay, and GSDMD immunoblotting revealed that Takinib treatment abolished the increase of LPS and ATP-induced pyroptosis in APIPTG/+ BMDMs compared to WT BMDMs (Fig. 5i and Supplementary Fig. 15a–c). Furthermore, Takinib treatment also reduced the enhanced ASC oligomerization, NLRP3 oligomerization, ASC–NLRP3 interaction, caspase-1 activation, and secretion of IL-1β and IL-18 observed in LPS and ATP-treated APIPTG/+ BMDMs (Fig. 2c, d, Supplementary Figs. 5h and 15d–f). These results indicate that the stimulatory effects of APIP on the NLRP3 inflammasome are mediated through TAK1. In addition, the increased NLRP3 inflammasome activation observed in APIPTG/+ BMDMs was also attenuated by treatment with SP600125, a JNK inhibitor (Supplementary Fig. 15a–f). Given that JNK functions downstream of TRAF6–TAK1 signaling upon TLR activation47, these findings further indicate that APIP enhances the activation of NLRP3 inflammasome via TRAF6-TAK1 signaling.
The crucial role of APIP in animal models of systemic inflammation
Since APIP regulates the activation of inflammasomes dependent on caspase-1 and caspase-11 in macrophages, we investigated the role of APIP in mouse models of endotoxemia, a serious condition significantly influenced by both caspases48. We first investigated the effects of APIP on systemic inflammation under conditions similar to the GWAS data, where reduced APIP expression may correlate with decreased sepsis mortality12,13. We intraperitoneally injected a high dose of LPS into control and Apip cKO mice. Compared to control mice, Apip cKO mice showed reduced mortality and higher body temperature (Fig. 6a, b), aligning with the GWAS data. Furthermore, we observed that serum IL-1β and TNF levels were significantly lower in Apip cKO mice than in control mice (Fig. 6c, d). These results demonstrate that myeloid lineage-specific Apip KO in mice sufficiently reduced the systemic inflammation induced by LPS. In addition, the same experiments using LPS were performed in WT and APIPTG/+ mice. The results revealed that APIP overexpression in mice exacerbated mortality, markedly decreased body temperature (Fig. 6e, f), and increased serum IL-1β and TNF levels (Fig. 6g, h). Notably, the LPS-induced increase in serum IL-1β in APIPTG/+ mice was abolished by injection of C25-140, a TRAF6-Ubc13-specific inhibitor, as well as by injection of MCC950. These findings suggest that the effects of APIP on systemic inflammation is mediated through TRAF6 and NLRP3 inflammasome.

a–d Effects of APIP conditional-KO on systemic inflammation. Control and Apip cKO mice (10–11-weeks-old, n = 13 mice per group) were intraperitoneally injected with LPS (E. coli O111:B4, 15 mg/kg). Survival (a) and body temperature (b) were monitored. Control and Apip cKO mice (female, 10–12 weeks) were injected with LPS (15 mg/kg, 8 h), and serum IL-1β and TNF were quantified by ELISA (c, d). e–h Effects of APIP overexpression on systemic inflammation. WT and APIPTG/+ mice (male, 9 weeks, n = 6 mice per group) were injected with LPS (E. coli O55:B5, 20 mg/kg). Survival (e) and body temperature (f) were monitored. WT and APIPTG/+ mice (10–12 weeks) were injected with LPS (15 mg/kg, 8 h) alone or with C25-140 (14 mg/kg, three doses at 12 h intervals, last dose 4 h before LPS) (g, h). MCC950 (10 mg/kg) was administered 1 h before and 3.5 h after LPS, followed by 4.5 h incubation (g). Serum IL-1β (g) and TNF (h) were quantified by ELISA. i Serum IL-1β levels in mice co-injected with LPS and MSU. WT, control, and Apip cKO mice (10–12 weeks) were injected with LPS (1.5 mg/kg), followed 4 h later by MSU crystals (50 mg/kg). After 12 h, serum IL-1β was quantified by ELISA. j–m In vivo effects of APIP conditional-KO on bacterial sepsis. Control and Apip cKO mice (10–12 weeks, n = 8 mice) received fecal suspension injection (FSI, 1000 mg/kg). Survival (j) and body temperature (k) were monitored. Control and Apip cKO mice (10–12 weeks) received FSI (1000 mg/kg, 8 h), and serum IL-1β and TNF were quantified by ELISA (l, m). Data, except for survival graphs, represent mean + or ± SD (n = 3–13 mice per group; exact n values shown on the graphs/Source Data). Source data are provided as a Source Data file. Statistical tests: Log-rank test (a, e, j), two-way ANOVA with Sidak’s (b, f, k) or Tukey’s multiple comparison test (g, i), or unpaired two-tailed Student’s t-test (c, d, h, l, m).
Given the greater contribution of caspase-11 than caspase-1 to endotoxic shock induced by LPS at doses as high as ~54 mg/kg29, we employed a LPS concentration as low as 1.5 mg/kg to minimize caspase-11 activation, and subsequently injected mice with MSU crystals to specifically activate the NLRP3 inflammasome in vivo24. We found that serum IL-1β levels in mice injected with both LPS and MSU were significantly higher than in those injected with LPS alone (Fig. 6i). Interestingly, serum IL-1β levels were significantly lower in Apip cKO mice compared to control mice following the injection of LPS and MSU (Fig. 6i). These results indicate that APIP also functions in systemic inflammation that is predominantly mediated by NLRP3 inflammasome.
To evaluate the role of APIP in bacterial sepsis with translational relevance, we employed the fecal suspension intraperitoneal injection (FSI) model developed by Tsuchida 49, which induces sepsis by injecting a suspension of mouse feces into the peritoneal cavity. FSI (administered at 1000 or 700 mg/kg) induced sepsis in both control and Apip cKO mice; however, Apip cKO mice showed significantly lower mortality and maintained higher body temperatures compared to control mice (Fig. 6j, k and Supplementary Fig. 16a, b). In addition, serum IL-1β and TNF levels measured 8 h after FSI were lower in Apip cKO mice (Fig. 6l, m). In contrast, when FSI was administered at 500 mg/kg, APIPTG/+ mice exhibited higher mortality and a more pronounced decrease in body temperature compared to WT mice (Supplementary Fig. 16c, d). Collectively, these results indicate that APIP plays a crucial role in LPS-induced endotoxemia and in bacterial sepsis, which has greater pathophysiological relevance, as well as in the specific activation of NLRP3 inflammasome in vivo.
Discussion
As APIP has been implicated as a modulator of inflammation in GWAS, this study elucidates the crucial role of APIP in inflammatory responses in macrophages, particularly its regulatory influence over both canonical and non-canonical inflammasomes (Fig. 7). APIP levels in macrophages and mice affect the NLRP3 inflammasome activity, including IL-1β and IL-18 secretion, caspase-1 activation, and pyroptosis. Furthermore, APIP’s role in regulating the non-canonical inflammasome, especially through the caspase-11/4 expression, expands its range of functions in stimulating inflammatory responses. On the other hand, APIP does not affect the activity of the double-stranded DNA-induced AIM2 inflammasome, pointing to a selective mechanism of action in inflammasome regulation.

APIP binds to TRAF6 and enhances its activity upon LPS exposure. This interaction promotes the NF-κB pathway, driving transcriptional priming of the NLRP3 inflammasome and caspase-11/4 non-canonical inflammasomes, and the JNK pathway, contributing to non-transcriptional priming of the NLRP3 inflammasome. Together, these processes amplify pyroptosis in response to danger signals and exacerbate systemic inflammation in mice. This figure was created in BioRender. Kwangmin (2025) https://BioRender.com/i6tv231.
An important question, then, is how APIP regulates these inflammasomes. The ability of APIP to regulate the inflammasomes would be attributed to its interaction with TRAF6, a key player in the NF-κB and JNK pathways that are activated by LPS and other inflammatory stimuli. These pathways regulate NLRP3 inflammasome through transcriptional priming and non-transcriptional priming, such as NLRP3 phosphorylation36,43,46. TRAF6 also affects the non-canonical inflammasome by increasing caspase-11 expression through NF-κB50,51. Among many types of inflammasomes, those that are highly responsive to TRAF6 signaling upon LPS treatment or transfection are likely susceptible to regulation by APIP. Additionally, not only the NLRP3 inflammasome but also the caspase-11 non-canonical inflammasome are regulated by LPS-TLR4 signaling52,53. Even more, the former promotes the latter through IL-18-induced IFN-γ54 and vice versa through K+ efflux55. Given that these two inflammasomes are closely related, the influence of APIP on them is likely to be noticeable.
There is increasing evidence that some microRNAs regulate inflammation, probably by targeting APIP. MicroRNA-146a (miR-146a) is upregulated in response to inflammatory stimuli and targets IRAK1, IRAK2, and TRAF6 to suppress NF-κB signaling56,57. MiR-146a-5p targets these proteins to contribute to the negative feedback regulation of inflammation, protecting tissues from damage58,59. Interestingly, miR-146a-3p, another strand of the miR-146a precursor, targets APIP19. Our findings suggest that miR-146a-3p regulates inflammasome activation and pyroptosis by inhibiting APIP. This dual targeting by miR-146a indicates a close functional relationship between TRAF6 and APIP, potentially within the same pathway or protein complex60.
Our current findings contradict the previous studies showing that APIP inhibits pyroptosis12,17. We believe that they have methodological limitations; those studies used HeLa and HEK293T cells, which lack functional inflammasomes and are therefore unsuitable to study inflammasome-dependent processes61,62. Moreover, distinguishing cell death types in those studies often remains unclear. For example, capase-1 overexpression induces apoptosis depending on the circumstances63,64. Cell death following Salmonella infection can induce not only pyroptosis but also apoptosis or necroptosis65. Thus, confirming the type of cell death requires appropriate inhibitors. However, the studies did not use inhibitors after caspase-1 overexpression17 and used only the YVAD peptide-based caspase-1 inhibitor, which is ineffective in blocking pyroptosis23, after Salmonella infection12. Moreover, the studies used indirect methods, such as administering metabolites like MTA in the methionine salvage pathway12,13, instead of directly manipulating APIP levels to examine its effects on inflammasomes.
Our results from in vivo mouse experiments indicate that APIP plays a crucial role in systemic inflammation. The positive correlation of APIP expression levels with sepsis severity observed in mouse models aligns well with the GWAS results12,13, suggesting that reducing APIP expression mitigates susceptibility to sepsis and improves outcomes. By regulating both NLRP3 and non-canonical inflammasomes, targeting APIP offers promise for drug development strategies aimed at balancing immune responses, particularly in pathological conditions characterized by excessive inflammation, such as SIRS and sepsis. Further investigations to elucidate the precise interactions between APIP and TRAF6 and to manipulate TRAF6 expression are necessary. Moreover, elucidating the role of APIP in regulating other inflammasomes, such as the flagellin-induced NAIP/NLRC4 inflammasome2, is also critical. These investigations will broaden our understanding of APIP’s influence across different inflammasomes, potentially paving the way for novel therapeutic strategies for inflammation-related diseases.
Methods
This research complies with all relevant ethical regulations. All experiments involving mice were conducted with the approval of IACUC at Seoul National University (SNU-210125-4, SNU-240122-2). Primary human CD14+ monocytes (70035.2, Stemcell Technologies) were commercially obtained from de-identified, consented donors under IRB-approved protocols. Thus, IRB review and additional consent were not required.
Chemicals and reagents
The following chemicals and reagents were used in this study: LPS (L4524 and L2630), ATP (A2383), Nigericin (N7143), MTA (D5011), PI (P4170), DSS (S1885), Hoechst 33342 (H3570), Takinib (SML2216), Ac-YVAD-cmk (SML0429), IDN-6556 (SML2227), and Necrostatin-1 (Nec-1, N9037) from Sigma-Aldrich; Pam3CSK4 (tlrl-pms), MCC-950 (inh-mcc), VX-765 (inh-vx765i-1), MSU Crystals (tlrl-msu-25), Poly(dA:dT) (tlrl-patn), and PMA (tlrl-pma) from Invivogen; Calcein-AM (C1430, Invitrogen); MG132 (BML-PI102, Enzo Biochem Inc.); Z-DEVD-fmk (sc-311558, Santa Cruz Biotechnology); FAM-YVAD-FMK (97, Immunochemistry Technology); C25-140 (HY-120934, MedChemExpress); M-CSF (RP01221, ABclonal).
Mice
The APIPTG/+ mice were generated as previously described in ref. 18. Briefly, human APIP cDNA was subcloned into the pCAGGS vector, expressing APIP under the CMV early enhancer/chicken beta actin (CAG) promoter. The transgenic cassette was isolated and injected into C57BL/6 mouse embryos, resulting in the generation of APIPTG/+ mice. The Apipflox/neo+ mice, targeting exons 4 and 5 of Apip, were generated in the C57BL/6 N background following the procedures described in Supplementary Fig. 1a, b and the subsequent section of Methods. The Apipflox mice were obtained by crossing the FLPe transgenic mice (stock# 005703, The Jackson Laboratory) with the Apipflox/neo+ mice. The Apipflox/flox mice were then crossed with Lyz2-Cre mice (stock# 004781, The Jackson Laboratory; generously gifted by Dr. C. Kang, Seoul National University, Korea) to create Lyz2-Cre; Apipfl/fl (Apip cKO) mice. Mice were housed in a specific pathogen-free (SPF) barrier facility at Seoul National University under standard institutional conditions, including controlled temperature and humidity, and a 12 h light/dark cycle. Animals were provided food and water ad libitum, and experimental and control groups were co-housed. Animals were euthanized by CO2 inhalation. In most experiments, both sexes were used without distinction. In Fig. 6c, only female mice were included, as they showed a more pronounced response to LPS at that time point. In Fig. 6e, only male mice were used because of limited animal availability at the time the experiment was performed.
Construction of Apip flox targeting vectors
A bacterial artificial chromosome (BAC) clone containing the Apip gene (RP23-16F14) was used to construct a targeting vector. The 5’ homology arm of 5.65 kb and the 3’ homology arm of 4.35 kb were retrieved from the BAC clone and inserted into the 5’ entry clone (pD5UE-2) and the 3’ entry clone (pD3DE-2), respectively, using the Quick and Easy BAC modification kit (Gene Bridges, Dresden, Germany). A 1.45 kb DNA fragment carrying Apip exons 4 and 5 was amplified by PCR and subcloned into KpnI/SacI sites of a modified middle entry clone [pDME-1; a DNA fragment of loxP sequence and pgk-Neo cassette flanked by two FLP recognition target (FRT) sites was sequentially located at the site 235 bp upstream of the exon 4, while the other loxP sequence was placed at the site 236 bp downstream of the exon5]. For targeting vector assembly, the three entry clones were recombined to a destination vector plasmid (pDEST-DT; containing a CAG promoter-driven diphtheria toxin gene) by using MultiSite Gateway Three-fragment Vector Construction Kit (Invitrogen). The targeting vector linearized with SalI was electroporated into the mouse embryonic stem (ES) cell line, RENKA, which was derived from the C57BL/6N strain. Homologous recombinant ES clones were identified by Southern blot analysis. Culture of the ES cell and generation of chimeric mice (Apipflox/neo+ mice) were performed as described previously in ref. 66.
Cells
J774A.1 (40067, Korean Cell Line Bank), RAW264.7 (40071, Korean Cell Line Bank), and HEK293T (CRL-3216, ATCC) cells were cultured in DMEM (LM 001-05, Welgene) supplemented with 10% (v/v) fetal bovine serum (12483020, Gibco), 10 μg/ml gentamycin (15710060, Gibco), and 100 U/ml penicillin-streptomycin (15140122, Gibco). THP-1 cells (40202, Korean Cell Line Bank) and primary human CD14+ monocytes (70035.2, Stemcell Technologies) were cultured in RPMI-1640 (SH30027.01, Cytiva) supplemented with 10% (v/v) fetal bovine serum and 100 U/ml penicillin-streptomycin. For differentiation into macrophages, THP-1 cells were treated with PMA (100 nM) for 72 h, and primary human monocytes were cultured with M-CSF (25 ng/ml) for 5–7 days. All cells were maintained at 37 °C in a humidified incubator with 5% CO2. For subculturing, J774A.1 and RAW264.7 cells were gently detached using a cell scraper, HEK293T cells were dissociated using trypsin-EDTA, and THP-1 cells were passaged by centrifugation and resuspension. Cells were seeded at a 1:5 to 1:10 ratio, depending on confluency.
Transfection with siRNA or plasmid DNA
For siRNA transfection, J774A.1 and differentiated THP-1 cells were incubated with 100 nM of either negative control siRNA (NC, SN-1002, Bioneer, Korea) or siRNA targeting APIP (J774A.1: siApip, 56369-2; THP-1: siAPIP, 51074-1; primary human macrophages: siAPIP#1, 51074-1, siAPIP#2, 51074-2; Bioneer, Korea) using Lipofectamine RNAiMAX (13778100, Invitrogen) according to the manufacturer’s protocol. The siRNA sequences are listed in Supplementary Table 2. Cells were incubated for 24–48 h before further treatments. For DNA transfection, RAW264.7 and HEK293T cells were transfected with the indicated DNA constructs using Lipofector-pMAX (AB-LF-M100, AptaBio, Korea) following the manufacturer’s protocol and incubated for 24 h before further experiments. For transfection, DMEM without antibiotics was used. Recombinant DNA constructs were generated in previous studies16,18 (pcDNA3 HA-APIP, p3xFLAG CMV10-APIP, p3xFLAG CMV10-APIP ΔN20, p3xFLAG CMV10-APIP ΔN30, p3xFLAG CMV10-APIP ΔN40, p3xFLAG CMV10-APIP ΔN50, p3xFLAG CMV10-APIP ΔN60, p3xFLAG CMV10-APIP ΔC62, p3xFLAG CMV10-APIP Δ123-180, and pcDNA3 HA-APIP E139A) or in this paper (pFLAG CMV-TRAF6, pFLAG CMV-TRAF6 ΔRING, pFLAG CMV-TRAF6 ΔZincFinger, pFLAG CMV-TRAF6 ΔCoiledCoil, and pFLAG CMV-TRAF6 ΔTRAF-C). The pFLAG CMV-TRAF6 plasmid was kindly provided by Dr. K. Ahn (Seoul National University, Korea). TRAF6 deletion mutants were generated by PCR-based site-directed mutagenesis using primers targeting specific regions, and the resulting constructs were verified by DNA sequencing.
Generation of bone marrow-derived macrophages
BMDMs were from the femurs and tibias of Apipfl/fl (control), Lyz2-Cre; Apipfl/fl (Apip cKO), WT, and APIPTG/+ mice. A 10 ml syringe was used to flush the bone marrow with PBS from the trimmed ends of the bones into 50 ml conical tubes. The extracted cells were thoroughly resuspended and filtered using a cell strainer (70 µm). The cells were centrifuged (244 g, 5 min), after which the supernatant was discarded. RBC lysis buffer (1 ml) was added to the pellet and pipetted thoroughly, followed by the addition of RBC lysis buffer (9 ml). The mixture was gently inverted and left for more than 1 min before centrifugation (244 g, 5 min). The supernatant was then discarded, and the pellet was resuspended in DMEM. The cells were plated at a concentration of 6 million cells per 100 mm culture dish for subsequent experiments. The cells were cultured in DMEM with 10% FBS and 20% L929-conditioned medium for 7 days to induce macrophage differentiation.
Treatments for activation of inflammasomes
To stimulate the NLRP3 inflammasome, BMDMs, J774A.1 cells, THP-1 cells, and primary human macrophages were treated with LPS (100–1000 ng/ml) for 3–4 h and nigericin (5 μM) or ATP (3 mM) from 15 min to 2 h. For experiments involving MSU crystals, J774A.1 cells were treated with LPS (1 μg/ml) for 3 h and MSU (100 μg/ml) for 6 h. To stimulate the non-canonical inflammasome, BMDMs, J774A.1 cells, RAW264.7 cells, THP-1 cells, and primary human macrophages were treated with Pam3CSK4 (1 μg/ml) for 4 h followed by MCC950 (10 μM) for 1 h or not, and transfected with LPS (2.5 μg/ml) for 14–24 h. To stimulate the AIM2 inflammasome, BMDMs and J774A.1 cells were treated with LPS (1 μg/ml) for 3 h, and transfected with poly(dA:dT) (1 or 2 μg/ml) for 4 or 5 h. The transfection of LPS and poly(dA:dT) was performed using Lipofectamine 2000 (11668019, Invitrogen).
Western blot
Cell lysates and supernatants were collected, and both were lysed in 2× sample buffer (100 mM Tris-Cl, pH 6.8, 4% SDS, 20% glycerol, 10% β-mercaptoethanol, 0.01% bromophenol blue). For mouse tissues, following grinding with a tissue grinder and centrifugation (10,000×g, 10 min, 4 °C), the supernatants were mixed with a 4-fold volume of 2× sample buffer. Proteins were separated by SDS-PAGE and transferred to PVDF membranes (AE-6667-P, Atto) using the Semi-Dry transfer system (WSE-4025, ATTO, Japan). Membranes were blocked with 3% BSA (A0100-010, GenDEPOT) in TBS-T (10 mM Tris-Cl, pH 8.0, 150 mM NaCl, and 0.5% Tween-20) at room temperature for 1 h and incubated overnight at 4 °C with the indicated primary antibodies (1:1000–5000), as detailed in Supplementary Table 3. The membranes were washed with TBS-T and incubated with the corresponding HRP-conjugated secondary antibodies (1:20,000) for 2 h at room temperature, and the blots were visualized using ECL substrate. Densitometric analysis was performed using ImageJ software. Protein signals on the blots were quantified and normalized to TUBA, ACTB, or the indicated reference protein. For the analysis of native samples, cells were lysed in non-denaturing lysis buffer (50 mM Tris-Cl, pH 6.8, 10% glycerol, 0.01% bromophenol blue). Native-PAGE was performed using non-denaturing gels and Tris-glycine buffer (6 g Tris, 28.8 g glycine, and 2 l distilled water). After electrophoresis, western blotting was performed as above using a primary antibody against NLRP3.
Fluorescence microscopy for pyroptosis assessment
After the activation of inflammasomes, cells were stained with calcein-AM (2 μM) and PI (5 μg/ml) for 2 min. Calcein-positive cells, representing live cells, and PI-positive cells, indicating cell death, were counted using a fluorescence microscope (Olympus IX71, Japan). The percentage of cell death was determined by dividing the number of PI-positive cells by the total number of cells.
LDH assay for cytotoxicity assessment
LDH release in the culture supernatants was measured using the EZ-LDH assay kit (DG-LDH1000, DoGenBio, Korea) according to the manufacturer’s instructions. Briefly, 100 μl of reaction mixture was added to 10 μl of supernatant in a 96-well plate, incubated at room temperature for 30–60 min in the dark, and absorbance was measured at 450 nm using a microplate reader.
Co-immunoprecipitation
Cells were lysed in RIPA buffer (50 mM Tris-Cl, pH 7.5, 150 mM NaCl, 1% Triton X-100, 1% sodium deoxycholate, 0.1% SDS) or PBST buffer (0.5% Triton X-100 in PBS), passed through a 26 G needle 15 times, and centrifuged (15,000×g, 15 min, 4 °C). The supernatants were incubated overnight at 4 °C with antibodies against APIP, TRAF6, ASC, NLRP3, HA, rabbit IgG, or mouse IgG (1:500), or with FLAG M2 affinity gel (20 μl). The mixtures with antibodies were incubated with Protein G Sepharose (20 μl) for 6 h at 4 °C. Immunocomplexes were collected by centrifugation (2000×g, 3 min, 4 °C), washed with PBS, and analyzed by western blotting.
ASC oligomerization and speck formation
For ASC oligomerization, cells were lysed and cross-linked with DSS (2 mM) for 1 h, and analyzed by western blotting. For ASC speck formation, cells were fixed using 4% paraformaldehyde for 10 min and permeabilized using 0.2% Triton X-100 for 15 min. They were then blocked with 5% BSA for 1 h, stained with ASC antibody (1:400) overnight at 4 °C, incubated with Alexa Fluor 488 anti-rabbit antibody (1:1000) for 1 h, and stained with Hoechst 33342 for 2 min. They were observed using a fluorescence microscope (Olympus IX71, Japan). The percentage of cells containing ASC specks was quantified across multiple microscopic fields.
FAM-FLICA staining for Caspase-1 activity detection
Cells were incubated with FAM-YVAD-FMK (97, Immunochemistry Technology) for 30 min and washed twice with 1× Apoptosis Wash Buffer. After counterstaining nuclei with Hoechst 33342 (0.5% v/v) for 10 min, cells were observed using a fluorescence microscope (Olympus IX71, Japan). The percentage of cells containing caspase-1 specks was quantified across multiple microscopic fields.
qRT-PCR
Total RNA from cells was extracted using TRI Reagent (TR 118, MRC) according to the manufacturer’s protocol. Typically, one µg of RNA was reverse-transcribed using an oligo(dT) primer (O1024-050, GenDEPOT) and M-MLV reverse transcriptase (RT001S, Enzynomics). qRT-PCR was conducted using SYBR Green (4367659, Applied Biosystems) with primers specific for Nlrp3, Tnf, Il1b, Il18, Apip, Ppia, APIP, and GAPDH (Supplementary Table 4). The analysis was performed in QuantStudio3 (Applied Biosystems).
Mouse models of systemic inflammation
WT and APIPTG/+ C57BL/6 mice (9–11-weeks-old) and control and Apip cKO C57BL/6 mice (10–12-weeks-old) were intraperitoneally injected with LPS (for WT and APIPTG/+, L4524, Sigma-Aldrich, 20 mg/kg; for control and Apip cKO, L2630, Sigma-Aldrich, 15 mg/kg) to induce systemic inflammation. Body temperature and survival rates were then monitored. To measure cytokine levels in the serum, control and Apip cKO mice (10–12-weeks-old) were euthanized 8 h after injection of LPS (L2630, 15 mg/kg). WT and APIPTG/+ mice (10–12-weeks-old) were subjected to the same LPS treatment and euthanized 8 h later. For the C25-140 + LPS group, C25-140 (14 mg/kg) was administered intraperitoneally three times at 12-h intervals, with the final dose given 4 h prior to LPS injection. For the MCC950 + LPS group, MCC950 (10 mg/kg) was administered intraperitoneally 1 h before and 3.5 h after LPS injection. Mice were euthanized 4.5 h after the second MCC950 administration. Blood samples were collected via cardiac puncture, allowed to clot for 40–60 min, and centrifuged (2000×g, 15 min, 4 °C). IL-1β, TNF, or IL-18 levels were quantified using ELISA (88-7013A-86, 88-7324-22, 88-50618-22, Invitrogen).
To activate the NLRP3 inflammasome in vivo, WT, Control, and Apip cKO mice (10–12-weeks-old) were first intraperitoneally injected with LPS (L2630, 1.5 mg/kg) for priming. After 4 h, they either received an injection of MSU crystals (50 mg/kg) or PBS, and euthanized 12 h later. Serum was obtained as above, and IL-1β levels were quantified using ELISA.
To induce bacterial sepsis, Control, Apip cKO, WT, and APIPTG/+ mice (10–12-weeks-old) were intraperitoneally injected with fecal suspension (FSI; 500–1000 mg/kg body weight) as previously described49. The fecal suspension was prepared by homogenizing freshly collected mice feces in sterile saline at a concentration of 50–100 mg/ml, followed by two rounds of filtration through a 70 µm cell strainer. For survival and body temperature analysis, mice were monitored at the indicated timepoints. For cytokine measurement, Control and Apip cKO mice were euthanized 8 h after FSI, and serum IL-1β and TNF levels were quantified by ELISA.
Statistical analyses
ASC immunostaining and FAM-YVAD-FMK staining data were obtained from a single experiment using randomly selected microscopic fields, whereas all other experiments were independently performed with data derived from distinct biological replicates. Specifically, experiments using primary human macrophages were independently performed with cells derived from a single donor vial, differentiated on different days to provide independent biological replicates. Data are presented as mean ± SD or SEM, and statistical significance was determined using unpaired two-tailed Student’s t-test, multiple unpaired t-test with Holm–Sidak’s method, two-way ANOVA with Tukey’s, Sidak’s, or Holm–Sidak’s multiple comparison tests, one-way ANOVA with Dunnett’s or Tukey’s multiple comparison tests, or Log-rank test using GraphPad Prism 10. Statistical significance is expressed as follows: *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. Exact p values are indicated in the Source Data file or graphs.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The data that support the findings of this study are available within the article and its Supplementary Information files. Source Data for all graphs and uncropped blots are provided with this paper. Mice lines used in this study have been deposited as sperm to the Laboratory Animal Resource Center (LARC) of Korea Research Institute of Bioscience and Biotechnology (KRIBB), resource numbers; PX0005222 (Apip cKO), PX00005223 (APIPTG/+). Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Yong-Keun Jung (ykjung@snu.ac.kr). Source data are provided with this paper.
References
Weiss, G. & Schaible, U. E. Macrophage defense mechanisms against intracellular bacteria. Immunol. Rev. 264, 182–203 (2015).
Franchi, L., Eigenbrod, T., Munoz-Planillo, R. & Nunez, G. The inflammasome: a caspase-1-activation platform that regulates immune responses and disease pathogenesis. Nat. Immunol. 10, 241–247 (2009).
Paik, S., Kim, J. K., Silwal, P., Sasakawa, C. & Jo, E. K. An update on the regulatory mechanisms of NLRP3 inflammasome activation. Cell Mol. Immunol. 18, 1141–1160 (2021).
Swanson, K. V., Deng, M. & Ting, J. P. The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 19, 477–489 (2019).
Yi, Y. S. Functional crosstalk between non-canonical caspase-11 and canonical NLRP3 inflammasomes during infection-mediated inflammation. Immunology 159, 142–155 (2020).
Pfalzgraff, A. & Weindl, G. Intracellular lipopolysaccharide sensing as a potential therapeutic target for sepsis. Trends Pharm. Sci. 40, 187–197 (2019).
Guo, H., Callaway, J. B. & Ting, J. P. Inflammasomes: mechanism of action, role in disease, and therapeutics. Nat. Med. 21, 677–687 (2015).
Voet, S., Srinivasan, S., Lamkanfi, M. & van Loo, G. Inflammasomes in neuroinflammatory and neurodegenerative diseases. EMBO Mol. Med. https://doi.org/10.15252/emmm.201810248 (2019).
Wu, R., Wang, N., Comish, P. B., Tang, D. & Kang, R. Inflammasome-dependent coagulation activation in sepsis. Front. Immunol. 12, 641750 (2021).
He, W., Dong, H., Wu, C., Zhong, Y. & Li, J. The role of NLRP3 inflammasome in sepsis: a potential therapeutic target. Int. Immunopharmacol. 115, 109697 (2023).
Rudd, K. E. et al. Global, regional, and national sepsis incidence and mortality, 1990–2017: analysis for the Global Burden of Disease Study. Lancet 395, 200–211 (2020).
Ko, D. C. et al. Functional genetic screen of human diversity reveals that a methionine salvage enzyme regulates inflammatory cell death. Proc. Natl. Acad. Sci. USA 109, E2343–E2352 (2012).
Wang, L. et al. Human genetic and metabolite variation reveals that methylthioadenosine is a prognostic biomarker and an inflammatory regulator in sepsis. Sci. Adv. 3, e1602096 (2017).
Cho, D. H. et al. Induced inhibition of ischemic/hypoxic injury by APIP, a novel Apaf-1-interacting protein. J. Biol. Chem. 279, 39942–39950 (2004).
Cho, D. H. et al. Suppression of hypoxic cell death by APIP-induced sustained activation of AKT and ERK1/2. Oncogene 26, 2809–2814 (2007).
Lim, B. et al. Cardioprotective role of APIP in myocardial infarction through ADORA2B. Cell Death Dis. 10, 511 (2019).
Kang, W. et al. Structural and biochemical basis for the inhibition of cell death by APIP, a methionine salvage enzyme. Proc. Natl. Acad. Sci. USA 111, E54–E61 (2014).
Hong, S. H. et al. APIP, an ERBB3-binding partner, stimulates erbB2-3 heterodimer formation to promote tumorigenesis. Oncotarget 7, 21601–21617 (2016).
Wang, N. et al. Apaf-1 interacting protein, a new target of microRNA-146a-3p, promotes prostate cancer cell development via the ERK1/2 pathway. Cell Biol. Int. 46, 1156–1168 (2022).
Clausen, B. E., Burkhardt, C., Reith, W., Renkawitz, R. & Forster, I. Conditional gene targeting in macrophages and granulocytes using LysMcre mice. Transgenic Res. 8, 265–277 (1999).
Lopez-Castejon, G. & Brough, D. Understanding the mechanism of IL-1β secretion. Cytokine Growth Factor Rev. 22, 189–195 (2011).
Coll, R. C. et al. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat. Med. 21, 248–255 (2015).
Schneider, K. S. et al. The inflammasome drives GSDMD-independent secondary pyroptosis and IL-1 release in the absence of Caspase-1 protease activity. Cell Rep. 21, 3846–3859 (2017).
Guo, H. & Ting, J. P. Inflammasome assays in vitro and in mouse models. Curr. Protoc. Immunol. 131, e107 (2020).
Fernandes-Alnemri, T. et al. The pyroptosome: a supramolecular assembly of ASC dimers mediating inflammatory cell death via caspase-1 activation. Cell Death Differ. 14, 1590–1604 (2007).
Totzke, J. et al. Takinib, a selective TAK1 inhibitor, broadens the therapeutic efficacy of TNF-alpha inhibition for cancer and autoimmune disease. Cell Chem. Biol. 24, 1029–1039 e1027 (2017).
Hornung, V. et al. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature 458, 514–518 (2009).
Zhu, Q., Zheng, M., Balakrishnan, A., Karki, R. & Kanneganti, T. D. Gasdermin D promotes AIM2 inflammasome activation and is required for host protection against Francisella novicida. J. Immunol. 201, 3662–3668 (2018).
Hagar, J. A., Powell, D. A., Aachoui, Y., Ernst, R. K. & Miao, E. A. Cytoplasmic LPS activates caspase-11: implications in TLR4-independent endotoxic shock. Science 341, 1250–1253 (2013).
Ruhl, S. & Broz, P. Caspase-11 activates a canonical NLRP3 inflammasome by promoting K(+) efflux. Eur. J. Immunol. 45, 2927–2936 (2015).
Ye, J. et al. Scutellarin inhibits caspase-11 activation and pyroptosis in macrophages via regulating PKA signaling. Acta Pharm. Sin. B 11, 112–126 (2021).
Pinto, S. M. et al. Comparative proteomic analysis reveals varying impact on immune responses in Phorbol 12-myristate-13-acetate-mediated THP-1 monocyte-to-macrophage differentiation. Front Immunol. 12, 679458 (2021).
Wang, Y. et al. M-CSF induces monocyte survival by activating NF-kappaB p65 phosphorylation at Ser276 via protein kinase C. PLoS One 6, e28081 (2011).
Yang, H. J. et al. NF-kappaB regulates caspase-4 expression and sensitizes neuroblastoma cells to Fas-induced apoptosis. PLoS One 10, e0117953 (2015).
Kishida, S., Sanjo, H., Akira, S., Matsumoto, K. & Ninomiya-Tsuji, J. TAK1-binding protein 2 facilitates ubiquitination of TRAF6 and assembly of TRAF6 with IKK in the IL-1 signaling pathway. Genes Cells 10, 447–454 (2005).
Song, N. et al. NLRP3 phosphorylation is an essential priming event for inflammasome activation. Mol. Cell 68, 185–197.e186 (2017).
Spalinger, M. R. et al. NLRP3 tyrosine phosphorylation is controlled by protein tyrosine phosphatase PTPN22. J. Clin. Invest. 126, 1783–1800 (2016).
Park, M. Y. et al. Scutellarein inhibits LPS-induced inflammation through NF-κB/MAPKs signaling pathway in RAW264.7 Cells. Molecules. https://doi.org/10.3390/molecules27123782 (2022).
Bouwmeester, T. et al. A physical and functional map of the human TNF-alpha/NF-kappa B signal transduction pathway. Nat. Cell Biol. 6, 97–105 (2004).
Sun, L., Deng, L., Ea, C. K., Xia, Z. P. & Chen, Z. J. The TRAF6 ubiquitin ligase and TAK1 kinase mediate IKK activation by BCL10 and MALT1 in T lymphocytes. Mol. Cell 14, 289–301 (2004).
Hamidi, A. et al. Polyubiquitination of transforming growth factor beta (TGFbeta)-associated kinase 1 mediates nuclear factor-kappaB activation in response to different inflammatory stimuli. J. Biol. Chem. 287, 123–133 (2012).
Bauernfeind, F. G. et al. Cutting edge: NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J. Immunol. 183, 787–791 (2009).
Xing, Y. et al. Cutting edge: TRAF6 mediates TLR/IL-1R signaling-induced nontranscriptional priming of the NLRP3 inflammasome. J. Immunol. 199, 1561–1566 (2017).
Guo, Q. et al. NF-kappaB in biology and targeted therapy: new insights and translational implications. Signal Transduct. Target Ther. 9, 53 (2024).
Wang, C. et al. TAK1 is a ubiquitin-dependent kinase of MKK and IKK. Nature 412, 346–351 (2001).
Gong, Y. N. et al. Chemical probing reveals insights into the signaling mechanism of inflammasome activation. Cell Res. 20, 1289–1305 (2010).
Totzke, J., Scarneo, S., Yang, K. & Haystead, T. TAK1: a potent tumour necrosis factor inhibitor for the treatment of inflammatory diseases. Open Biol. 10, 200099 (2020).
Kayagaki, N. et al. Non-canonical inflammasome activation targets caspase-11. Nature 479, 117–121 (2011).
Tsuchida, T. et al. Protocol for a sepsis model utilizing fecal suspension in mice: fecal suspension intraperitoneal injection model. Front. Med. 9, 765805 (2022).
Schauvliege, R., Vanrobaeys, J., Schotte, P. & Beyaert, R. Caspase-11 gene expression in response to lipopolysaccharide and interferon-gamma requires nuclear factor-kappa B and signal transducer and activator of transcription (STAT) 1. J. Biol. Chem. 277, 41624–41630 (2002).
Yoo, L., Hong, S., Shin, K. S. & Kang, S. J. PARP-1 regulates the expression of caspase-11. Biochem Biophys. Res. Commun. 408, 489–493 (2011).
Rathinam, V. A. et al. TRIF licenses caspase-11-dependent NLRP3 inflammasome activation by gram-negative bacteria. Cell 150, 606–619 (2012).
Shi, J. et al. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature 514, 187–192 (2014).
Aachoui, Y. et al. Canonical inflammasomes drive IFN-gamma to prime Caspase-11 in defense against a cytosol-invasive bacterium. Cell Host Microbe 18, 320–332 (2015).
He, Y., Hara, H. & Núñez, G. Mechanism and regulation of NLRP3 inflammasome activation. Trends Biochem. Sci. 41, 1012–1021 (2016).
Hou, J. et al. MicroRNA-146a feedback inhibits RIG-I-dependent Type I IFN production in macrophages by targeting TRAF6, IRAK1, and IRAK2. J. Immunol. 183, 2150–2158 (2009).
Magilnick, N. et al. miR-146a-Traf6 regulatory axis controls autoimmunity and myelopoiesis, but is dispensable for hematopoietic stem cell homeostasis and tumor suppression. Proc. Natl. Acad. Sci. USA 114, E7140–E7149 (2017).
Xie, Y. F. et al. MicroRNA-146 inhibits pro-inflammatory cytokine secretion through IL-1 receptor-associated kinase 1 in human gingival fibroblasts. J. Inflamm. 10, 20 (2013).
Wu, H. et al. Extracellular vesicles containing miR-146a attenuate experimental colitis by targeting TRAF6 and IRAK1. Int. Immunopharmacol. 68, 204–212 (2019).
Alshalalfa, M. miRNA regulation in the context of functional protein networks: principles and applications. Wiley Interdiscip. Rev. Syst. Biol. Med. 6, 189–199 (2014).
Wang, Y. et al. Inflammasome activation triggers Caspase-1-mediated cleavage of cGAS to regulate responses to DNA virus infection. Immunity 46, 393–404 (2017).
Shi, H., Murray, A. & Beutler, B. Reconstruction of the mouse inflammasome system in HEK293T cells. Bio Protoc. https://doi.org/10.21769/BioProtoc.1986 (2016).
Exline, M. C. et al. Microvesicular caspase-1 mediates lymphocyte apoptosis in sepsis. PLoS One 9, e90968 (2014).
Tsuchiya, K. et al. Caspase-1 initiates apoptosis in the absence of gasdermin D. Nat. Commun. 10, 2091 (2019).
Wemyss, M. A. & Pearson, J. S. Host cell death responses to non-typhoidal Salmonella infection. Front. Immunol. 10, 1758 (2019).
Mishina, M. & Sakimura, K. Conditional gene targeting on the pure C57BL/6 genetic background. Neurosci. Res. 58, 105–112 (2007).
Acknowledgements
We thank Dr. C. Kang (Seoul National University, Korea) for providing Lyz-Cre mice. This work was supported by the CRI grant (RS-2025-00519823 to Y.-K.J.), the Bio & Medical Technology Development Program (RS 2024-00439842 to Y.-K.J.) through the National Research Foundation (NRF) grant funded by the Korea Ministry of Science and ICT (MSIT), and the Korea Dementia Research Project (RS-2023-KH134817 to Y.-K.J.) through the Korea Dementia Research Center (KDRC), funded by the Ministry of Health & Welfare and MSIT, Korea.
Author information
Authors and Affiliations
Contributions
K.J. and Y.-K.J. designed the research. K.J. conducted the experiments, analyzed the data, and prepared the figures. M. Kawamura, M.A., K.S., and M. Komatsu were responsible for generating the targeting vector for Apip exons and the Apipflox/neo+ mice. B.L. maintained APIPTG/+ mice and constructed the APIP DNA constructs. Y.-K.J. supervised the project. K.J. and Y.-K.J. analyzed all experiments and wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Jung, K., Kawamura, M., Lim, B. et al. APIP regulates the priming of canonical NLRP3 and non-canonical Caspase-11/4 inflammasomes by binding to TRAF6. Nat Commun 16, 10866 (2025). https://doi.org/10.1038/s41467-025-65893-8
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-65893-8
