
Abstract
SYNGAP1 regulates synaptic plasticity through interactions with scaffold proteins and modulation of Ras and Rap GTPase signaling. Human SYNGAP1 mutations are linked to intellectual disability, epilepsy, and autism. In mice, Syngap1 haploinsufficiency causes impaired LTP, premature maturation of dendritic spines, learning disabilities, and seizures, reflecting the human phenotypes of SYNGAP1 syndrome. Recently, SYNGAP1 was shown to influence cortical neurogenesis and progenitor proliferation in human organoids. Here, we show that Syngap1 absence or haploinsufficiency does not alter neocortical progenitors and their cellular output in mice. Despite careful analysis of cortical progenitor properties, we fail to replicate the main findings from human organoids. This discrepancy suggests species-specific or methodological differences and raises questions about the broader relevance of SYNGAP1’s role in neurogenesis. The absence of cortical progenitor deficits in haploinsufficient mice, which exhibit cognitive deficits and seizures, indicates these arise from SYNGAP1’s regulation of synapse function rather than its role on neurogenesis.
Similar content being viewed by others


Introduction
Synaptic Ras GTPase-activating protein 1 (SYNGAP1) is a critical postsynaptic protein predominantly expressed in the brain, where it plays a crucial role in synaptic plasticity, learning, and memory1,2. Since its discovery in the late 1990s3,4, SYNGAP1 has been identified as a major regulator of synaptic strength through its interaction with synaptic scaffold proteins and modulation of Ras and Rap GTPase signaling pathways. It is one of the most abundant proteins in the postsynaptic density (PSD)5,6,7, where it interacts with PSD-95 and other critical scaffolding proteins, positioning it as a pivotal component in regulating excitatory synapses. SYNGAP1’s GAP activity modulates Ras-ERK and PI3K-Akt signaling pathways, which can regulate synaptic plasticity mechanisms, including long-term potentiation (LTP)8.
Syngap1 heterozygous loss-of-function (LoF) mice present strongly impaired LTP, premature maturation of dendritic spines, and learning disabilities9,10,11. Later studies identified spontaneous seizure activity and seizure susceptibility in these mice12,13. Homozygous knockout mice died within days of birth9,10,11 coincident with the time frame when the expression of Syngap1 dramatically increases in the brain14,15,16. These structural and functional abnormalities underline the importance of SYNGAP1 in postnatal neurodevelopment and synapse regulation, supporting its role as a critical molecular hub in synaptic function. Following the identification of SYNGAP1’s role in synaptic plasticity, heterozygous LoF mutations in human SYNGAP1 were linked to a highly penetrant and severe neurodevelopmental disorder (SYNGAP1 syndrome; MRD5; SYNGAP1-related intellectual disability; SYNGAP1 encephalopathy)17,18,19. Patients with SYNGAP1 mutations often present with intellectual disability, epilepsy, and autism spectrum disorder. With more than 1400 genetically diagnosed patients worldwide and an estimated prevalence at around 6 out of 100,000 individuals20, SYNGAP1 has become a key target for understanding the molecular mechanisms that contribute to neurodevelopmental diseases and cognitive impairment.
The majority of these studies have focused on developmental stages after neuronal migration is complete and when excitatory synapses have started to emerge. More recently, studies have begun exploring the potential involvement of SYNGAP1 in earlier stages of neurodevelopment, including cortical development21,22. SYNGAP1 depletion leads to an increased ratio of neural progenitor cells to neurons and a reduction of ventricle and telencephalon size in Xenopus21. In human cortical organoids, SYNGAP1 haploinsufficiency accelerated neuronal maturation, which decreased the ratio of progenitor cells to neurons and resulted in a reduced ventricular area22. These findings suggest that SYNGAP1 regulates developmental processes during brain development at much earlier stages than previously thought.
In this study, we sought to determine the extent to which these results extend to cortical neurogenesis in mice. Using histological analysis of genetically modified mice carrying heterozygous and homozygous knock-out alleles for Syngap1 (Syngap1KO), we did not observe significant alterations in the properties of cortical progenitors and the generation of neocortical cell layers. A comprehensive analysis of progenitor numbers, proliferative capacity, and division mode, along with quantifications of overall neocortical thickness and neuronal and glial cell counts at different developmental ages, did not replicate the key findings observed in human organoids. We thus conclude that Syngap1 is not essential for neocortical development in mice that otherwise recapitulate most aspects of SYNGAP1-related intellectual disability9,10,11,12,13. Our findings highlight potential species-specific or methodological differences and raise important questions about the broader applicability of SYNGAP1’s role in neurogenesis.
Results
Syngap1 levels do not affect the number of neocortical progenitor cells in mice
A recent study has proposed that SYNGAP1 plays a role in cortical neurogenesis by affecting the proliferation and differentiation of late progenitor cells in human organoids22. Cortical progenitors have been broadly divided into two classes named apical progenitors (APs = PAX6-positive cells) that undergo mitosis in the ventricular zone (VZ), and intermediate progenitors (IPs = TBR2-positive cells) that undergo mitosis in the subventricular zone (SVZ)23,24,25,26,27,28. Although SYNGAP1 is mainly expressed in neurons at postnatal ages14,15,29, we show by RNAscope in situ hybridization that some Syngap1 mRNA is detectable in Pax6-positive murine neocortical progenitor cells already at embryonic days 14.5 and 18.5 (E14.5 and E18.5; Supplementary Fig. 1a; Fig. 1a). However, we failed to detect the high expression in ventricle wall cells previously reported22. Analyses of previously published single-cell RNA sequencing datasets have confirmed that Syngap1 is expressed at very low levels during embryonic development in both mouse30 and human31 neocortex (Supplementary Fig. 1b–f’).

a RNAscope for Syngap1 (red) and Pax6 (gray) in the somatosensory cortex of wild-type mice at E18.5. Low and top lines represent the limits of the ventricular zone (VZ) and cortical plate (CP), respectively. Boxed area at higher magnification on the right. Circles highlight the cells that show co-expression of Syngap1 and Pax6 in the VZ. These results were confirmed in three independent mice. b Overview of midbrain coronal sections of control and homozygous Syngap1 knockout (Syngap1KO) mice analyzed by PAX6 immunolabeling at E18.5. Lines represent the limits of the ventricle and CP. c Control (CT), heterozygous (+/−) and homozygous (−/−) Syngap1KO brains analyzed by PAX6 and TBR2 immunolabeling in the somatosensory cortex at E18.5 (n = 3 mice/genotype; one-way ANOVA Dunnett’s multiple comparisons test: PAX6 – CT vs −/− p = 0.8805, CT vs +/− p = 0.7600, F2,6 = 0.2097; TBR2 – CT vs −/− p = 0.2206, CT vs +/− p = 0.6551, F2,6 = 1.507). d Cortical thickness in the somatosensory cortex of Control (CT), heterozygous (+/−), and homozygous (−/−) Syngap1KO brains at E18.5 (n = 3 mice/genotype; one-way ANOVA Dunnett’s multiple comparisons test: CT vs −/− p = 0.2708; CT vs +/− p = 0.6740, F2,6 = 1.242). DL deep layer; UL upper layer, cc corpus callosum; Values are mean ± SEM; ns not significant; Scale bars: 50 µm (lower magnification), 10 µm (higher magnification), 500 µm (b, overview). Source data are provided as a Source data file.
Quantitative analysis of histological sections revealed that at E18.5 the number of PAX6+ APs and TBR2+ IPs was not altered in the somatosensory cortex of heterozygous and homozygous Syngap1KO mice10 (Fig. 1b, c; rostral → caudal axis – Supplementary Fig. 2a). The number of neocortical progenitor cells was also normal in mice carrying the GAP-AL mutation which eliminates the GAP activity of Syngap1 (Syngap1GAP*; Supplementary Fig. 2c)8. In addition, the number of PAX6+ APs was not altered in the motor cortex of heterozygous and homozygous Syngap1KO mice (Supplementary Fig. 2b), and the cortical thickness remained unaltered in Syngap1KO and Syngap1GAP* mice at E18.5 (Fig. 1d; Supplementary Fig. 2d).
Syngap1 levels do not affect the properties of progenitor cells in mice
To further investigate the potential role of SYNGAP1 in progenitor function, we injected EdU into pregnant mice at E18.5 and analyzed EdU incorporation into DNA 1 h later in the somatosensory cortex of heterozygous and homozygous Syngap1KO mice (Fig. 2a, a’). Consistent with an unaltered number of progenitor cells, the number of EdU+ cells was also unaffected in the somatosensory cortex of heterozygous and homozygous Syngap1KO mice at E18.5 (Fig. 2a). Next, we investigated cell cycle exit by injecting EdU into pregnant mice at E17.5 and analyzing the number of EdU+/Ki67− cells 24 h later (Fig. 2b, b’). The number of EdU+/Ki67− cells was not altered in the somatosensory cortex of heterozygous and homozygous Syngap1KO mice at E18.5 (Fig. 2b, b’). We conclude that the cell cycle and proliferative capacity of neocortical progenitors are normal in heterozygous and homozygous Syngap1KO mice.

a, a’ EdU (gray) incorporation in the somatosensory cortex of Control (CT), heterozygous (+/−) and homozygous (−/−) Syngap1KO mice after 1 h EdU injection at E18.5 (n = 3 CT, n = 4 +/− and −/− mice; one-way ANOVA Dunnett’s multiple comparisons test: CT vs −/− p = 0.9674; CT vs +/− p = 0.9666, F2,8 = 0.1020). b, b’ EdU (gray) and Ki67 (red) immunolabeling in the somatosensory cortex of Control (CT), heterozygous (+/−) and homozygous (−/−) Syngap1KO mice after 24 h EdU injection at E17.5 (n = 3 CT and −/−, n = 4 +/− mice; one-way ANOVA Dunnett’s multiple comparisons test: CT vs −/− p = 0.6438; CT vs +/− p = 0.9984, F2,7 = 0.4912). c Cleavage plane analysis by PH3 (green) and γ-TUBULIN (red) immunolabeling in Control (CT), heterozygous (+/−) and homozygous (−/−) Syngap1KO ventricular zone (VZ) at E18.5 (n = 3 CT, n = 4 −/−, n = 5 +/− mice; one-way ANOVA Dunnett’s multiple comparisons test: CT vs −/− p = 0.8913; CT vs +/− p = 0.7559, F2,9 = 0.1909). c’ Cleavage plane analysis by PH3 (green) and γ-TUBULIN (red) immunolabeling in Control (CT) and Brn1/2-cKO VZ at E14.5 (n = 3 mice/genotype; two-sided unpaired t-test: CT vs cKO p = 0.0308). d, d’ PAX6 (red), TBR2 (gray), and Ki67 (red) immunolabeling in the somatosensory cortex of Control (CT) and Syngap1−/− mice at E14.5 (n = 3 mice/genotype; two-sided unpaired t-test: PAX6 p = 0.9060; TBR2 p = 0.5374; Ki67 p = 0.7142). e, e’ TBR2 (gray) and EdU (red) immunolabeling in the somatosensory cortex of control (CT) and Syngap1−/− mice at E14.5 (n = 3 CT, n = 4 −/− mice; two-sided unpaired t-test: p = 0.2898). f Cleavage plane analysis in Control (CT) and homozygous (−/−) Syngap1KO ventricular zone (VZ) at E14.5 (n = 3 CT, n = 4 −/− mice; two-sided unpaired t-test: CT vs −/− p = 0.3010). Low and top lines represent the limits of the VZ and cortical plate (CP), respectively. DL deep layer, UL upper layer, SVZ subventricular zone. Values are mean ± SEM; ns not significant; *p < 0.05; Scale bars: 50 µm (lower magnification), 5 µm (c). Source data are provided as a Source data file.
In human cortical organoids, SYNGAP1 haploinsufficiency affects the division mode of progenitor cells22. In the VZ, depending on their cleavage plane, APs undergo either symmetric proliferative divisions (vertical = 60–90° plane) or asymmetric differentiative divisions (oblique = 30–60° plane and horizontal = 0–30° plane, respectively)32,33,34,35. In addition, APs engage in two modes of neurogenesis termed direct and indirect neurogenesis23,24,25,26,27,28. During direct neurogenesis, APs divide asymmetrically to self-renew and to generate one neuron, while during indirect neurogenesis, APs divide asymmetrically to self-renew and generate an IP that then gives rise to neurons23,24,25,26,27. Although indirect neurogenesis generates neurons for all cortical layers, it is the predominant neurogenic mode that produces upper-layer projection neurons (ULNs)24,25,36,37,38,39. Defects in the division mode of neocortical progenitors alter the identity and topography of cortical neurons and can cause several neurological and psychiatric disorders28,40,41. In SYNGAP1 haploinsufficient human organoids, there is an increased proportion of progenitors undergoing differentiative division22. To determine if a similar phenotype was observed in mice, we analyzed the division planes of mitotic ventricular APs (PH3-positive cells) in heterozygous and homozygous Syngap1KO mice at E18.5. Quantitative analysis of histological sections in the somatosensory cortex revealed a normal ratio of self-renewing and differentiative divisions in neocortical progenitors of heterozygous and homozygous Syngap1KO mice (Fig. 2c). As a positive control, we analyzed Brn1 and Brn2 conditional knockout (Brn1/2-cKO) mice. Brn1 and Brn2 have previously been shown to regulate the proliferative capacity of cortical progenitor cells and the balance between direct and indirect neurogenesis42. In agreement with earlier findings, the ratio of progenitors to neurons was significantly altered in Brn1/2-cKO mice (Fig. 2c’), demonstrating that our analysis tools are sufficiently sensitive to detect changes in the biology of neocortical progenitor cells.
Neocortical progenitors generate neurons for different cortical layers in sequential order. Deep-layer neurons (DLNs) are generated prior to ULNs, which only begin to emerge in mice around E14.5. Subsequently, these progenitors start to generate glial cells around E16.543. To determine the extent to which Syngap1 affects the properties of progenitor cells during earlier developmental stages at the beginning of ULN generation, we analyzed markers of progenitor cells (PAX6 and TBR2) and proliferation (Ki67) in the somatosensory cortex of homozygous Syngap1KO mice at E14.5. Immunohistochemical analysis revealed that the number of progenitor cells, as well as their proliferative capacity, was normal in homozygous Syngap1KO mice at E14.5 (Fig. 2d, d’). Additionally, as a readout of cells actively going through indirect neurogenesis, we analyzed the number of TBR2+ cells that have incorporated EdU 1 h after injection. Levels of indirect neurogenesis were unaltered in the somatosensory cortex of homozygous Syngap1KO mice at E14.5 (Fig. 2e, e’). Quantitative analysis also revealed a normal ratio of self-renewing and differentiative divisions in neocortical progenitors from the somatosensory cortex of heterozygous and homozygous Syngap1KO mice at E14.5 (Fig. 2f; Supplementary Fig. 1g). Altogether, these results show that the neocortical properties of progenitor cells are normal in the absence of Syngap1 in mice.
Syngap1 levels do not affect the number of neurons and glial cells generated in mice
To further determine the extent to which Syngap1 may have essential roles during the formation of neocortical cell layers, we analyzed numbers of ULNs, DLNs, and astrocytes in heterozygous and homozygous Syngap1KO mice at P0 and P13. Immunohistochemical analysis of CTIP2 (highly expressed in DLNs) and BRN2 (highly expressed in ULNs at P0) did not reveal any differences in the numbers of CTIP2+ and BRN2+ neurons in the somatosensory cortex of heterozygous and homozygous Syngap1KO mice compared to wild-type at P0 (Fig. 3a). Numbers of CTIP2+ and BRN2+ neurons were also normal in Syngap1GAP* mice at P0 (Supplementary Fig. 2e, e’). In addition, we observed normal numbers of astrocytes (SOX9-positive cells) in the somatosensory cortex of heterozygous and homozygous Syngap1KO mice at P0 (Fig. 3b, b’). Immunohistochemistry analysis of CTIP2 and CUX1 (highly expressed in ULNs at P13) revealed normal numbers of DLNs and ULNs in the somatosensory cortex of heterozygous Syngap1KO mice at later postnatal ages (P13; Fig. 3c). Numbers of astrocytes were also unaltered at P13 in these mice (Fig. 3d; Supplementary Fig. 2f).

a, a’ CTIP2 (red) and BRN2 (green) immunolabeling in the somatosensory cortex of Control (CT), heterozygous (+/−) and homozygous (−/−) Syngap1KO mice at P0 (n = 5 CT, n = 4 −/− and +/− mice; one-way ANOVA Dunnett’s multiple comparisons test: CTIP2 – CT vs −/− p = 0.3673, CT vs +/− p = 0.2418, F2,10 = 1.486); BRN2 – CT vs −/− p = 0.9595, CT vs +/− p = 0.8484, F2,10 = 0.2509). Arrowheads highlight migration defects for BRN2-positive cells. b, b’ SOX9 (red) immunolabeling in the somatosensory cortex of Control (CT), heterozygous (+/−) and homozygous (−/−) Syngap1KO mice at P0 (n = 3 CT, n = 4 −/− and +/− mice; one-way ANOVA Dunnett’s multiple comparisons test: CT vs −/− p = 0.9856; CT vs +/− p = 0.6821, F2,8 = 0.3347). c, c’ CTIP2 (red) and CUX1 (gray) immunolabeling in the somatosensory cortex of Control (CT) and Syngap1+/− mice at P13 (n = 4 mice/genotype; two-sided unpaired t-test: CTIP2 – CT vs +/− p = 0.9317; CUX1 – CT vs +/− p = 0.8127). d, d’ EdU (gray) and CUX1 or SOX9 (red) immunolabeling at P13 in the somatosensory cortex of Control (CT) and Syngap1+/− mice after EdU injection at E18.5 (n = 4 mice/genotype; two-sided unpaired t-test: EdU+CUX1+ CT vs +/− p = 0.7904; EdU+SOX9+ CT vs +/− p = 0.0690). DL deep layer, UL upper layer, VZ ventricular zone, WM white matter. Values are mean ± SEM; ns not significant. Scale bars: 50 µm (P0), 100 µm (P13). Source data are provided as a Source data file.
To further investigate if the late progenitors are generating normal levels of neurons and glial cells, we analyzed cell fate by injecting EdU at E18.5 and quantifying the number of EdU-positive cells expressing the indicated cellular markers in the somatosensory cortex of control and heterozygous Syngap1KO mice at P13. Syngap1 deficiency did not affect the ratio of EdU-positive ULNs (EdU+CUX1+ cells) or EdU-positive glial cells (EdU+SOX9+ cells) in mice (Fig. 3d, d’). In addition, analysis of a previously published whole-brain RNA sequencing dataset16 shows that the expression of neurodevelopmental marker genes and cortical cell layer/type-specific marker genes is unaltered in Syngap1+/− and Syngap1+/c.3583-9G>A haploinsufficient adult mice (Supplementary Fig. 3).
Overall, these results show that heterozygous and homozygous Syngap1KO mice have normal numbers of progenitor cells that proliferate and differentiate to generate appropriate numbers of neurons and glia. Interestingly, the migration of ULNs was slightly affected in both homozygous Syngap1KO and homozygous Syngap1GAP* mice (arrowheads on Fig. 3a and Supplementary Fig. 2e; Supplementary Fig. 2g, h), suggesting that Syngap1 may play a role in the migration and circuit integration of ULNs and that this effect is GAP activity dependent.
Discussion
Recent studies have begun to explore the function of SYNGAP1 during the early stages of neurodevelopment. A study using CRISPR-Cas9 unilateral injections in Xenopus21 showed that Syngap1 deficiency results in a decrease in brain size and in the area occupied by neural progenitor cells. They observed an increased ratio of neural progenitor cells to neurons, suggesting that Syngap1-deficient progenitors differentiate less in this species21. More recently, a study in human cortical organoids demonstrated that SYNGAP1 influences neocortical neurogenesis by accelerating the differentiation of human progenitor cells22. In this context, SYNGAP1 haploinsufficiency also leads to a reduced ventricular area, but the ratio of progenitor cells to neurons is decreased as Syngap1-deficient progenitors are more differentiative in human organoids. Our findings present a significant contrast to these studies. While we observed minor ULN migration defects in mice with Syngap1 haploinsufficiency, we did not observe any alterations in the numbers and proliferative capacity of their neocortical progenitor cells. The results from the SYNGAP1 study in human organoids suggested that SYNGAP1-related brain disorders may arise through non-synaptic mechanisms22, but our results showing the absence of cortical progenitor phenotypes in Syngap1 haploinsufficient mice provide strong evidence that cognitive deficits and seizures observed in these mice9,10,11,12,13,16 do not require differences in neurogenesis and are more likely dependent on the SYNGAP1’s regulation of synaptic function.
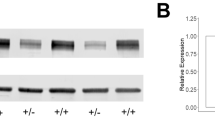
Although the ratio of radial glial progenitors to neurons was reported to be reduced in Syngap1KO mice mimicking the increased differentiating capacity of human radial glial progenitors in SYNGAP1 haploinsufficient human organoids22, we have found that the number of progenitors and the cortical thickness occupied by these progenitors is normal across the rostral-caudal axis in both Syngap1KO mice and in Syngap1GAP* mice when compared to the corresponding rostral-caudal axis in their control littermates. We showed that the number of self-renewing and differentiative murine progenitors remains normal in Syngap1KO at two different developmental ages. Importantly, the mice we have used in this study are under the same genetic background as the ones used in the previous study22. As previously characterized16, there is likely little protein arising from the deleted allele in these mice. Syngap1+/− mice were previously generated by deleting the core exons 6 and 7, which shifts the reading frame and leads to reduction of Syngap110. Western blotting of protein extracted from the whole brain of Syngap1+/− mice revealed approximately 50% less Syngap1 protein expression, and Northern blots with a Syngap1 probe targeting exons 8–11 show a similar reduction of Syngap1 expression with no significant other isoform bands16. This data suggests little compensation for Syngap1 function in these mice. Importantly, cortical thickness and the numbers of neurons and glial are also not altered in either Syngap1KO or Syngap1GAP* mice compared to their control littermates. In line with our results in Syngap1 haploinsufficient mice and despite severe intellectual disabilities, only a small subset of human SYNGAP1 haploinsufficiency patients (~2%) are diagnosed with microcephaly or structural brain abnormalities18,19. This supports the idea that the patient’s cognitive impairments and seizures are more likely related to SYNGAP1’s role in regulating neuronal physiology rather than neurogenesis and consequently, brain size.
Nonetheless, the reported discrepancies emphasize the complexity of SYNGAP1’s role in neurodevelopment and suggest a nuanced interaction with species-specific developmental pathways. SYNGAP1’s involvement in the biology of progenitors could be cell-specific. The previous studies and our work mainly focus on the biology of apical progenitors residing in the VZ22, but SYNGAP1 might play important roles in outer radial glial cells (oRGCs). oRGCs are neocortical progenitors that reside in the outer SVZ and, although their number and contribution to neurogenesis is very reduced in lissencephalic mice, they are very abundant and account for most neurons in more evolved species with a more complex neocortex27,34,44,45,46,47,48,49. The biology of these cells can be recapitulated in human organoids50, and therefore, this might explain some of the differences we observe in mice, where oRGCs are present in very limited numbers. Although single-cell RNA sequencing analysis shows that the expression of Syngap1 is very low during embryonic development in both mouse and human neocortex—including in oRGCs (Supplementary Fig. 1b–f’)—further investigations are necessary to understand the mechanistic pathways influenced by SYNGAP1 across different species, cell types, and developmental contexts.
In conclusion, our study contributes to the growing body of literature exploring the multifaceted roles of SYNGAP1 in neurodevelopment. While our results differ from the previous studies21,22, they emphasize the need for continued research into the diverse mechanisms of SYNGAP1 across species. Understanding these differences will be critical for developing targeted therapeutic strategies for neurodevelopmental disorders associated with SYNGAP1 mutations. Collaboration and dialog within the scientific community will enhance our understanding of these complex processes, and we look forward to further investigations that will bridge the findings from human and animal studies.
Methods
All the experiments reported in this study adhered to the ethical regulations and guidelines of the National Institute of Health Guide for Care and Use of Laboratory Animals. The experiments involving mice were approved by the Institutional Animal Care and Use Committee at Johns Hopkins University School of Medicine under the study protocol numbers: MO22M217, M023M70, and MO23M52.
Mice
Mice were maintained on a 14 h light/10 h dark cycle. Both male and female mice were used, and no obvious differences between the sexes were noted. All mice were group-housed in pathogen-free facilities with regulated temperature and humidity and given ad libitum access to food and water. All mice used were seemingly free of infection, health abnormalities, or immune system deficiencies, and were employed independently of their gender. None of the mice used had been used for previous experiments. The date of the vaginal plug detection was designated E0.5, and the date of birth P0. Syngap1+/− heterozygous knockout mice10, Syngap1 GAP mutants (+/GAP*, GAP*/GAP*)8, and WT (Control) littermates were maintained on a mixed of C57/B6J and 129S background. To maintain mutants on the mixed genetic background, we chose breeders randomly from within a colony and backcrossed the offspring to F1 hybrids between the two strains that comprise the mixed background (in this case, C57BL/6 and 129 F1 hybrids) about every 10 generations. Emx1-Cre [B6.129S2-Emx1tm1(cre)Krj]51 and Brn1/2 conditional knockout mice have been described42. CD1 time-pregnant wild-type mice were obtained from Charles River Laboratories.
Animal euthanasia
We used the most humane methods for euthanasia following the recommendations of the panel report on Euthanasia of the American Veterinary Medical Association and the guidelines of the National Institute of Health Guide for Care and Use of Laboratory Animals. Different methods were used for mice of different ages, according to the experiments that needed to be performed: neonatal and adult mice—isoflurane inhalation followed by cervical dislocation; newborn mice—chilling and decapitation; embryos 14 and 18 days post coitum—chilling and decapitation; newborn to adult mice—cardiac perfusion under full anesthesia.
RNAscope multiplex in situ hybridization
Brain sections were processed for multiplex fluorescent in situ hybridization RNAscope52 following the manufacturer’s instructions (#323110, Advanced Cell Diagnostics). Gene-specific probes: Syngap1 (417381), Pax6 (412821-C2).
Single-cell RNA sequencing analysis
To analyze the expression of Syngap1 in mice, we reanalyzed the available dataset from mouse cerebral cortex30. The same Uniform Manifold Approximation and Projection (UMAP) coordinates from the publication were used for the feature plot. For the cell type classification, apical progenitors from the original paper were classified as ventricular radial glial cells (vRG), intermediate progenitors were classified as IP, and the neurons were classified as excitatory neurons (EN.) The total Unique Molecular Identifiers (UMI) in each cell was scaled by a factor of 10000, and the count was log-transformed after adding a pseudo count of 1 for the feature plot. The color bar of the feature plot was scaled by the minimum and maximum expression of a certain gene. To analyze the expression of SYNGAP1 in humans, we reanalyzed the available dataset from human neocortex31. The cell identity was assigned by the original metadata, with all the excitatory neuronal cell types grouped as EN and all other non-neuronal cell types grouped as other. A new UMAP was learned to visualize the feature plot of cell identities (vRG, outer radial glial cells (oRG), IP, EN, and others). The total UMI in each cell was scaled by the median total UMI among all cells, and the count was log-transformed after adding a pseudo count of 1. The normalized counts were scaled to zero mean and unit variance to learn 50 PCAs. Among them, the top 16 were used to learn the UMAP with 30 nearest neighbors and a min_dist of 0.3. The log-transformed data were used to represent each gene in the feature plot, and the color bar of the feature plot was scaled by the minimum and maximum expression of a certain gene. For the dotplots, the size of each circle was scaled to the proportion of cells with positive gene expression. The expression level of each gene was normalized within each cell group and shown by different colors of the circle. To analyze the expression of cellular marker genes in two different Syngap1 haploinsufficiency mouse models, we reanalyzed the available dataset from Araki et al.16. Differential expression at the gene level was determined using DESeq2 with a false discovery rate of 0.1 (Benjamini–Hochberg correction) and absolute log2 fold change value threshold of 0.1, correcting for rRNA ratio and sex.
Immunohistochemistry
Embryonic brains were fixed in 4% paraformaldehyde (PFA) in phosphate-buffered saline (PBS) for 2–4 h at 4 °C. Postnatal mice were transcardially perfused with 20 ml ice-cold 4% PFA using a peristaltic pump at a rate of 2 ml/min. Brains were removed from the skull and post-fixed in 4% PFA overnight at 4 °C. Embryonic brains were cryopreserved with 30% sucrose, embedded in Tissue-Tek O.C.T. compound (#4583, SAKURA), and frozen at −80 °C. They were then sectioned coronally at 16 µm with a cryostat (CM3050 S, Leica). Postnatal brains were embedded in 3% low-melting-point agarose (#R0801, Thermo Fisher Scientific) in PBS and sectioned coronally at 60 µm with a vibrating microtome (VT1200S, Leica). For immunohistochemistry, brain sections were permeabilized in PBS containing 0.2% Triton X-100 and blocked in 10% goat serum for 1 h at room temperature. Brain slices were then incubated with primary antibodies in blocking solution overnight (or 48 h, depending on the thickness of the section and antibody used) at 4 °C and subsequently incubated with the appropriate secondary antibodies diluted in blocking solution for 2 h at room temperature. To label all cell nuclei, the fluorescent nuclear dye DAPI (1 μg/ml, #D9542, Sigma-Aldrich) was included with the secondary antibody solution. The sections were mounted with ProLongTM Gold (#P36930, Thermo Fisher Scientific). Primary antibodies: anti-CTIP2 (1:1000, rat monoclonal, ab18465 Abcam, clone 25B6, current lot: GR322373-4); anti-CUX1 (1:1000, rabbit polyclonal, 11733-1-AP Proteintech, current lot: 00050220); anti-BRN2 (1:1000, rabbit monoclonal, 12137 Cell Signaling, clone D2C1L); anti-Ki67 (1:500, rabbit polyclonal, ab15580 Abcam, current lot: GR3285096-1 or 1:500, rat monoclonal, 14-5698-82 Thermo Fisher Scientific, clone SolA15, current lot: 2496198); anti-PAX6 (1:250, mouse monoclonal, MA1-109 Thermo Fisher Scientific, clone 13B10-1A10, current lot: XD341788 or 1:500, rabbit polyclonal, 901301 Biolegend, clone Poly19013, current lot: B343290); anti-TBR2 (1:500, rat monoclonal, 14-4875-82 Thermo Fisher Scientific, clone Dan11mag, current lot: 2488481 or 1:500, rabbit polyclonal, ab183991 Abcam, clone EPR19012, current lot: GR3237130-1); anti-SOX9 (1:1000, rabbit monoclonal, ab185966 Abcam, clone EPR14335-78 current lot: GR3194558-4); anti-PH3 (1:1000, rat monoclonal, ab10543 Abcam, clone HTA28, current lot: 1052214-1); anti-γ-TUBULIN (1:500, goat polyclonal, A. Holland gift53); anti-Centrin (1:500, rabbit polyclonal, A. Holland gift54). Secondary antibodies: Alexa 555, 647, 488 anti-rat (A21434; A21247, A11006), anti-rabbit (A21430; A21246), and anti-mouse (A21425; A21237) and Alexa 488 anti-rabbit (A11070); all diluted 1:500 and all from Thermo Fisher Scientific. Antigen retrieval55 was performed for all immunohistochemistry at embryonic ages. The method used here was a heat-induced citrate method55. The sections were then mounted with ProLongTM Gold (P36930, Thermo Fisher Scientific).
EdU injections and labeling
EdU (5-ethynyl-2′-deoxyuridine; #A10044, Thermo Fisher Scientific) was diluted at 10 mg/ml in 0.9% NaCl (#114-055-101, Quality Biological) and administered in pregnant females at the specified embryonic pregnancy stage at 50 mg/Kg body weight. For proliferation analysis, a single intraperitoneal EdU injection was administered at E18.5, embryos were fixed after 1 h, and the number of EdU+ cells was quantified. For cell cycle exit calculation, a single intraperitoneal EdU injection was administered at E17.5; embryos were fixed after 24 h, and the percentage of EdU+ cells labeled with Ki67 was calculated. For cell-fate analysis, a single intraperitoneal EdU injection was administered at E18.5, and the number of EdU+ cells expressing the indicated cellular markers were analyzed at P13. EdU was labeled using Click-iT® EdU imaging kits with Alexa 555 (#C10638, Thermo Fisher Scientific). Briefly, brain sections were post-fixed with 3.7% formaldehyde and permeabilized with 0.5% Triton X-100 in PBS. The Click-iT® reaction cocktail was then added, and the sections incubated in the dark for 30 min at room temperature. After the washes, the brain sections were stained for the different proteins of interest using the immunohistochemistry protocol previously described.
Cleavage plane analysis
PH3 immunostaining was used to identify mitotic cells. γ-TUBULIN immunostaining was used to determine the position of both centrosomes (γ-TUBULIN positive) in mitotic cells and to mark the cleavage plane in relation to the ventricular surface. The angle of the cleavage plane was measured using ImageJ.
Imaging
All immunohistochemistry and ISH RNAscope data were acquired in an equivalent latero-medial level of the cortex from at least 3 independent animals per condition or genotype. Images were captured using a Zeiss LSM 800 confocal microscope. Maximum intensity projections were generated in ImageJ or Imaris software. In all fluorescence microscopy figures, different channels of image series were represented in pseudocolor, and contrast and brightness were adjusted manually using ImageJ or Adobe Photoshop software. All markers were counterstained with DAPI to allow visualization of overall cellular density.
Histological analysis
A series of z-stack (depth of 10 μm) confocal tiled images were used for cell quantifications; 150 μm-wide images (embryonic brains) or 300 μm-wide images (postnatal brains) comprising the entire extension of the cortex were analyzed. All quantifications were made in an equivalent latero-medial level of the cortex from at least 3 independent animals per condition or genotype. The number of cells expressing the markers of interest was quantified in radial sections of the same latero-medial level and adjusted per area. Cortical thickness was analyzed by measuring the length from the lower limit of the VZ to the upper limit of the CP. PAX6+ or TBR2+ cortical thickness was analyzed by measuring the length occupied by PAX6+ or TBR2+ cells from the lower limit of the VZ into the CP. To analyze the distribution of BRN2+ cells, we divided the cortex into longitudinal bins, and a rectangle was drawn from the border of the white matter with layer VI to the pial surface. This rectangle was then divided into ten equal bins, and the number of cells within each bin was quantified.
Statistical analysis
Statistical analyses of the immunohistochemistry experiments were done using GraphPad Prism software. Statistically significant differences were assessed by Student’s unpaired t-test or one-way analysis of variance (one-way ANOVA Dunnett’s multiple comparisons test; FDFn, DFd (DFn: degrees of freedom in the numerator; DFd: degrees of freedom in the denominator)), comparing two or more groups, respectively. To compare the distribution of BRN2+ cells in the different bins of the cortex for the different genotypes, we used two-way analysis of variance (two-way ANOVA-Šídák’s multiple comparisons test; FDFn, DFd). P < 0.05 was considered a significant difference. All values represent individual animals’ mean ± standard error of the mean (SEM). The statistical test used and the statistical significance are indicated in figure legends.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All the data generated in this study are provided in the main text or the Supplementary Information/Source data file. Source data are provided with this paper. All the raw data included in this study are stored in The Johns Hopkins Research Data Repository https://doi.org/10.7281/T1K3HJNO56. Source data are provided with this paper.
Materials availability
Correspondence should be addressed to R.L.H., S.B. or I.H. and requests for materials should be addressed to R.L.H.
Code availability
The code for the single-cell RNA sequencing analysis presented in this manuscript is available on: 10.5281/zenodo.16914738. The raw data for the single-cell RNA sequencing analysis in mice were previously published30 and are available in the GEO repository database under the accession code GSE153164. The raw data for the single-cell RNA sequencing analysis in humans were previously published31 and are available in the dbGaP repository database under the accession code phs001836 (https://www.omicsdi.org/dataset/dbgap/phs001836).
References
Gamache, T. R., Araki, Y. & Huganir, R. L. Twenty years of SynGAP research: from synapses to cognition. J. Neurosci. 40, 1596–1605 (2020).
Jeyabalan, N. & Clement, J. P. SYNGAP1: mind the gap. Front. Cell. Neurosci. 10, 32 (2016).
Chen, H. J., Rojas-Soto, M., Oguni, A. & Kennedy, M. B. A synaptic Ras-GTPase activating protein (p135 SynGAP) inhibited by CaM kinase II. Neuron 20, 895–904 (1998).
Kim, J. H., Liao, D., Lau, L. F. & Huganir, R. L. SynGAP: a synaptic RasGAP that associates with the PSD-95/SAP90 protein family. Neuron 20, 683–691 (1998).
Sugiyama, Y., Kawabata, I., Sobue, K. & Okabe, S. Determination of absolute protein numbers in single synapses by a GFP-based calibration technique. Nat. Methods 2, 677–684 (2005).
Cheng, D. et al. Relative and absolute quantification of postsynaptic density proteome isolated from rat forebrain and cerebellum. Mol. Cell. Proteom. 5, 1158–1170 (2006).
Sheng, M. & Kim, E. The postsynaptic organization of synapses. Cold Spring Harb. Perspect. Biol. 3, a005678 (2011).
Araki, Y. et al. SynGAP regulates synaptic plasticity and cognition independently of its catalytic activity. Science 383, eadk1291 (2024).
Komiyama, N. H. et al. SynGAP regulates ERK/MAPK signaling, synaptic plasticity, and learning in the complex with postsynaptic density 95 and NMDA receptor. J. Neurosci. 22, 9721–9732 (2002).
Kim, J. H., Lee, H.-K., Takamiya, K. & Huganir, R. L. The role of synaptic GTPase-activating protein in neuronal development and synaptic plasticity. J. Neurosci. 23, 1119–1124 (2003).
Vazquez, L. E., Chen, H.-J., Sokolova, I., Knuesel, I. & Kennedy, M. B. SynGAP regulates spine formation. J. Neurosci. 24, 8862–8872 (2004).
Clement, J. P. et al. Pathogenic SYNGAP1 mutations impair cognitive development by disrupting maturation of dendritic spine synapses. Cell 151, 709–723 (2012).
Ozkan, E. D. et al. Reduced cognition in Syngap1 mutants is caused by isolated damage within developing forebrain excitatory neurons. Neuron 82, 1317–1333 (2014).
Araki, Y. et al. SynGAP isoforms differentially regulate synaptic plasticity and dendritic development. eLife 9, e56273 (2020).
Gou, G. et al. SynGAP splice variants display heterogeneous spatio-temporal expression and subcellular distribution in the developing mammalian brain. J. Neurochem. 154, 618–634 (2020).
Araki, Y. et al. Mouse models of SYNGAP1-related intellectual disability. Proc. Natl. Acad. Sci. USA 120, e2308891120 (2023).
Hamdan, F. F. et al. Mutations in SYNGAP1 in autosomal nonsyndromic mental retardation. N. Engl. J. Med. 360, 599–605 (2009).
Vlaskamp, D. R. M. et al. SYNGAP1 encephalopathy: a distinctive generalized developmental and epileptic encephalopathy. Neurology 92, e96–e107 (2019).
Lloyd Holder, J., Hamdan, F. F. & Michaud, J. L. SYNGAP1-related intellectual disability. GeneReviews® (2019).
López-Rivera, J. A. et al. A catalogue of new incidence estimates of monogenic neurodevelopmental disorders caused by de novo variants. Brain 143, 1099–1105 (2020).
Willsey, H. R. et al. Parallel in vivo analysis of large-effect autism genes implicates cortical neurogenesis and estrogen in risk and resilience. Neuron 109, 788–804.e8 (2021).
Birtele, M. et al. Non-synaptic function of the autism spectrum disorder-associated gene SYNGAP1 in cortical neurogenesis. Nat. Neurosci. 26, 2090–2103 (2023).
Noctor, S. C., Flint, A. C., Weissman, T. A., Dammerman, R. S. & Kriegstein, A. R. Neurons derived from radial glial cells establish radial units in neocortex. Nature 409, 714–720 (2001).
Haubensak, W., Attardo, A., Denk, W. & Huttner, W. B. Neurons arise in the basal neuroepithelium of the early mammalian telencephalon: a major site of neurogenesis. Proc. Natl. Acad. Sci. USA 101, 3196–3201 (2004).
Noctor, S. C., Martínez-Cerdeño, V., Ivic, L. & Kriegstein, A. R. Cortical neurons arise in symmetric and asymmetric division zones and migrate through specific phases. Nat. Neurosci. 7, 136–144 (2004).
Miyata, T. et al. Asymmetric production of surface-dividing and non-surface-dividing cortical progenitor cells. Development 131, 3133–3145 (2004).
Hansen, D. V., Lui, J. H., Parker, P. R. L. & Kriegstein, A. R. Neurogenic radial glia in the outer subventricular zone of human neocortex. Nature 464, 554–561 (2010).
Villalba, A., Götz, M. & Borrell, V. The regulation of cortical neurogenesis. Curr. Top. Dev. Biol. 142, 1–66 (2021).
Porter, K., Komiyama, N. H., Vitalis, T., Kind, P. C. & Grant, S. G. N. Differential expression of two NMDA receptor interacting proteins, PSD-95 and SynGAP during mouse development. Eur. J. Neurosci. 21, 351–362 (2005).
Di Bella, D. J. et al. Molecular logic of cellular diversification in the mouse cerebral cortex. Nature 595, 554–559 (2021).
Polioudakis, D. et al. A single-cell transcriptomic atlas of human neocortical development during mid-gestation. Neuron 103, 785–801.e8 (2019).
Chenn, A. & McConnell, S. K. Cleavage orientation and the asymmetric inheritance of Notch1 immunoreactivity in mammalian neurogenesis. Cell 82, 631–641 (1995).
Knoblich, J. A. Mechanisms of asymmetric stem cell division. Cell 132, 583–597 (2008).
Shitamukai, A., Konno, D. & Matsuzaki, F. Oblique radial glial divisions in the developing mouse neocortex induce self-renewing progenitors outside the germinal zone that resemble primate outer subventricular zone progenitors. J. Neurosci. 31, 3683–3695 (2011).
Matsuzaki, F. & Shitamukai, A. Cell division modes and cleavage planes of neural progenitors during mammalian cortical development. Cold Spring Harb. Perspect. Biol. 7, a015719 (2015).
Tarabykin, V., Stoykova, A., Usman, N. & Gruss, P. Cortical upper layer neurons derive from the subventricular zone as indicated by Svet1 gene expression. Development 128, 1983–1993 (2001).
Kowalczyk, T. et al. Intermediate neuronal progenitors (basal progenitors) produce pyramidal-projection neurons for all layers of cerebral cortex. Cereb. Cortex 19, 2439–2450 (2009).
Vasistha, N. A. et al. Cortical and clonal contribution of Tbr2 expressing progenitors in the developing mouse brain. Cereb. Cortex 25, 3290–3302 (2015).
Huilgol, D. et al. Direct and indirect neurogenesis generate a mosaic of distinct glutamatergic projection neuron types in cerebral cortex. Neuron 111, 2557–2569.e4 (2023).
Barkovich, A. J., Guerrini, R., Kuzniecky, R. I., Jackson, G. D. & Dobyns, W. B. A developmental and genetic classification for malformations of cortical development: update 2012. Brain 135, 1348–1369 (2012).
Fernández, V., Llinares-Benadero, C. & Borrell, V. Cerebral cortex expansion and folding: What have we learned? EMBO J. 35, 1021–1044 (2016).
Barão, S. et al. Conserved transcriptional regulation by BRN1 and BRN2 in neocortical progenitors drives mammalian neural specification and neocortical expansion. Nat. Commun. 15, 8043 (2024).
Greig, L. C., Woodworth, M. B., Galazo, M. J., Padmanabhan, H. & Macklis, J. D. Molecular logic of neocortical projection neuron specification, development and diversity. Nat. Rev. Neurosci. 14, 755–769 (2013).
Wang, X., Tsai, J. W., Lamonica, B. & Kriegstein, A. R. A new subtype of progenitor cell in the mouse embryonic neocortex. Nat. Neurosci. 14, 555–562 (2011).
Fietz, S. A. et al. OSVZ progenitors of human and ferret neocortex are epithelial-like and expand by integrin signaling. Nat. Neurosci. 13, 690–699 (2010).
Reillo, I. & Borrell, V. Germinal zones in the developing cerebral cortex of ferret: Ontogeny, cell cycle kinetics, and diversity of progenitors. Cereb. Cortex 22, 2039–2054 (2012).
Reillo, I., De Juan Romero, C., García-Cabezas, M. Á & Borrell, V. A Role for intermediate radial glia in the tangential expansion of the mammalian cerebral cortex. Cereb. Cortex 21, 1674–1694 (2011).
Lamonica, B. E., Lui, J. H., Hansen, D. V. & Kriegstein, A. R. Mitotic spindle orientation predicts outer radial glial cell generation in human neocortex. Nat. Commun. 4, 1665 (2013).
Betizeau, M. et al. Precursor diversity and complexity of lineage relationships in the outer subventricular zone of the primate. Neuron 80, 442–457 (2013).
Eichmüller, O. L. & Knoblich, J. A. Human cerebral organoids—a new tool for clinical neurology research. Nat. Rev. Neurol. 18, 661–680 (2022).
Gorski, J. A. et al. Cortical excitatory neurons and glia, but not GABAergic neurons, are produced in the Emx1-expressing lineage. J. Neurosci. 22, 6309–6314 (2002).
Wang, F. et al. RNAscope: a novel in situ RNA analysis platform for formalin-fixed, paraffin-embedded tissues. J. Mol. Diagn. 14, 22–29 (2012).
Levine, M. S. et al. Centrosome amplification is sufficient to promote spontaneous tumorigenesis in mammals. Dev. Cell 40, 313–322.e5 (2017).
Moyer, T. C. & Holland, A. J. PLK4 promotes centriole duplication by phosphorylating STIL to link the procentriole cartwheel to the microtubule wall. eLife 8, e46054 (2019).
Tang, X., Falls, D. L., Li, X., Lane, T. & Luskin, M. B. Antigen-retrieval procedure for bromodeoxyuridine immunolabeling with concurrent labeling of nuclear DNA and antigens damaged by HCl pretreatment. J. Neurosci. 27, 5837–5844 (2007).
Barão, S., Xu, Y., Hong, I., Müller, U. & Huganir, R. L. Data associated with the publication: Syngap1 and the development of murine neocortical progenitor cells. Johns Hopkins Research Data Repository, v1. https://doi.org/10.7281/T1K3HJNO (2025).
Acknowledgements
The authors thank members of the Huganir and Müller laboratories, including Yoichi Araki, for suggestions, reagents, and technical help; Andrew Holland for the g-TUBULIN and Centrin antibodies. The authors are grateful to Ashley Irving, Sarah Richardson, Michelle Monroe, Tajma Smith, and Trinity Walker for mouse maintenance and genotyping assistance. The authors thank Giorgia Quadrato for her suggestions and constructive discussions. This work was supported by National Institutes of Health grants R37NS036715 (R.L.H.), R01HG012357 and RF1MH121539 (U.M.); Brain Pool Plus (BP+) Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science and ICT (RS-2025-25403864, I.H.); and Foundation for Science and Technology grant 2023.11467.PEX doi.org/10.54499/2023.11467.PEX (S.B.).
Author information
Authors and Affiliations
Contributions
Conceptualization: S.B., I.H., U.M., and R.L.H.; methodology: S.B., I.H., Y.X., U.M., and R.L.H.; investigation: S.B., I.H., Y.X., U.M., and R.L.H.; validation: S.B., I.H., Y.X., and R.L.H.; visualization: S.B., I.H., and Y.X.; formal analysis: S.B., I.H., Y.X., U.M., and R.L.H.; data curation: S.B., Y.X., and I.H.; resources: R.L.H., U.M., and S.B.; supervision: U.M. and R.L.H.; project administration: R.L.H., U.M., and S.B.; funding acquisition: R.L.H., U.M., and S.B.; writing: S.B., I.H., Y.X., U.M., and R.L.H.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Barão, S., Xu, Y., Hong, I. et al. Syngap1 and the development of murine neocortical progenitor cells. Nat Commun 16, 11060 (2025). https://doi.org/10.1038/s41467-025-66069-0
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-66069-0
