
Abstract
T-cell engager (TCE)-based immunotherapy is clinically validated in hematological cancers. However, application in solid tumors faces hurdles including T cell penetration, the immunosuppressive tumor microenvironment, and toxicity. We develop an mRNA-encoded TCE (MTS105) targeting Glypican-3, the hepatocellular carcinoma antigen, delivered via lipid nanoparticles directly to liver tissue. In mice, rats, and cynomolgus monkeys, MTS105 exhibits higher liver exposure versus plasma. Liver-orthotopic tumor-bearing mice achieve complete, dose-dependent regression, with fast intratumoral T cell activation owing to sustained higher liver and tumor functional TCE exposure versus conventional antibody-based TCE. In vivo, MTS105 induces intratumoral CD8 cell precursor and terminally differentiated memory subsets with high activation scores. In cynomolgus monkeys, MTS105 displays favorable, linear plasma pharmacokinetics including mRNA, ionizable lipid, and translated TCE following single and repeated-four-weekly dosing (up to 45 μg/kg). No severe adverse effects or gross pathology were observed. Our results thus support the advancement of MTS105 into clinical trials, with a first-in-human study currently underway.
Similar content being viewed by others

Introduction
Bispecific T-cell engagers (TCEs) have demonstrated clinical efficacy, particularly in hematological malignancies, as evidenced by US FDA-approved therapies such as blinatumomab, mosunetuzumab, glofitamab, and teclistamab1,2,3,4. These TCEs function by bridging T cells and tumor cells, forming an immunological synapse through simultaneous binding to the CD3 receptor on T cells and tumor-associated antigens (TAAs) on tumor cells. The application of these therapies in solid tumors presents distinct challenges due to physical barriers, complex suppressive microenvironments, and the need for significantly higher peripheral drug exposure to achieve an effective intratumoral concentration5. Higher TCE exposure levels result in significant off-target toxicities; TCEs thus require careful engineering and dosing6. Tarlatamab, used for the treatment of small cell lung cancer, is currently the only TCE that has achieved clinical proof of concept in solid tumors7, which may be explained by a relatively small molecular size facilitating intratumoral penetration8. Other approaches to optimize the potency of TCEs under early clinical development include the local activation of T cells with a pro-drug, strategies to enhance TCE potency with fine-tuning of the affinity balance of CD3 and TAA binders, armoring TCEs with additional signals to facilitate intratumoral T cell trafficking, and optimizing the half-life (T1/2) of TCEs6,9.
Hepatocellular carcinoma (HCC) is a leading cause of cancer-related death worldwide10. Although early-stage disease may be curable by resection, transplantation, or ablation, most patients present with unresectable disease and have a poor prognosis11. Currently, therapeutic strategies for advanced HCC are limited, with immunotherapy such as atezolizumab plus bevacizumab for the first-line treatment of HCC associated with a relatively short overall survival and median progression-free-survival12, and an objective response rate below 30%13. Glypican-3 (GPC3) is a member of the heparan sulfate (HS) proteoglycan family on the cell surface, which is comprised of six subtypes (GPC1–6)14,15. During embryonic development, GPC3 is widely expressed across various tissues; however, its expression is strictly controlled and suppressed in most adult tissues16,17. In contrast, the upregulation of GPC3 has been reported in HCC with a prevalence of over 70% in various studies18,19,20,21, making it an excellent potential target for immunotherapeutic approaches. To date, various immunotherapies targeting GPC3 have been under investigation, including GPC3-targeting antibodies, GPC3-derived peptide/DNA vaccines, immunotoxins, genetic therapies, and chimeric-antigen-receptor modified cells22,23,24,25,26,27,28.
Based on the hypothesis that tissue-specific delivery of a TCE can enhance antitumor efficacy, we developed a novel liver-targeted mRNA-LNP encoding a GPC3-targeting TCE. This approach enables in situ expression of the TCE within the tumor microenvironment, which promotes localized T-cell activity and demonstrates potent antitumor effects in preclinical models of hepatocellular carcinoma. This strategy not only provides a promising therapeutic candidate for a cancer with limited treatment options but also establishes a versatile platform for solid tumor immunotherapy. The mRNA-LNP delivery platform could be broadly adapted to target other solid tumors by encoding TCEs against different tumor-associated antigens, potentially overcoming a major barrier in the field.
Results
Validation of MLX0473-based LNPs for mRNA delivery to the liver
Based on our ionizable lipid library, we identified a novel lipid (MLX0473)-based LNP for liver-tropic delivery in mice. To evaluate the efficiency of its hepatic delivery in vivo, MLX0473-based LNP encapsulating luciferase (Luc) mRNA was injected intravenously into C57BL/6J mice to evaluate bioluminescence distribution versus Lipid5, a widely used ionizable amino lipid for liver-targeted mRNA delivery29. In vivo imaging system (IVIS) imaging revealed that the MLX0473-based LNP group exhibited 7-fold higher luciferase activity in the liver compared to the Lipid5-based LNP group (Fig. 1A, B). We also compared the delivery efficiency of MLX0473-based LNP and GalNAc-conjugated LNP, which enable targeted delivery through ASGPR receptor-mediated endocytosis. Our MLX0473-based LNP showed significantly enhanced hepatic delivery, clearly outperforming both Lipid5 and its GalNAc-conjugated LNP counterpart (Supplementary Fig. 1). We next evaluated the tissue-specific persistent protein expression potential encoded by mRNA in rats. Human Erythropoietin (hEPO) mRNA encapsulated in MLX0473-LNP showed a much higher, sustained expression than Lipid5-based LNPs after a total of five repeated doses (Fig. 1C).

A Representative images of bioluminescence distribution in C57BL/6J mice treated with Lipid5-based LNP-Luc and MLX0473-based LNP-Luc for 6 h. Color bars denote the radiance (photons/s/cm2/sr) scale. B Quantification of luciferase expression in the liver. A two-sided Student’s t-test was used for comparing the Lipid5 and MLX0473 groups, **P < 0.01 (mean ± SD; n = 3 mice per group). C Left panel, treatment schedule of the study. Sprague-Dawley rats were intravenously administered 0.5 mg/kg hEPO mRNA delivered by Lipid5 or MLX0473-based LNPs (five-weekly). Serum was collected at 6, 24, and 72 h after administration. Right panel, hEPO concentration in serum was measured by ELISA (mean ± SEM; n = 4 mice per group).
In vitro functional validation of MTS105-encoded TCE
After establishing the efficiency of the liver-tropic MLX0473-based LNP in vivo, we evaluated the delivery of an mRNA-encoded TCE to the liver in vitro, for potential future application in the treatment of liver-associated diseases. We designed a TCE by fusing single-chain variable fragments (ScFvs) targeting HCC antigen GPC3 and CD3, separated by a flexible peptide linker. Due to the slow-release nature of mRNA–protein translation, we omitted the Fc element in the TCE with the aim of achieving an optimal stimulation window for T cells. The mRNA-encoding TCE was optimized through codon usage modifications to balance the Codon Adaptation Index (CAI) and Minimal Free Energy (MFE), taking into account the entire mRNA sequence, including untranslated regions (UTRs) and the coding sequence (CDS). The mRNA was encapsulated in MLX0473-based LNPs for efficient liver-tropic delivery, forming the mRNA-LNP complex (MTS105; Fig. 2A). To assess target specificity, flow cytometric binding assays were performed. MTS105-encoded TCE bound human CD3 on Jurkat cells and hGPC3 on stably expressing HEK293 cells in a concentration-dependent manner (Supplementary Fig. 2A). To evaluate whether the optimized mRNA was able to encode the functional TCE, 293T cells were transfected with MTS105, and the cell supernatant containing mRNA-encoded TCE was collected. ELISA analysis showed a strong expression of TCE in the cell supernatant in a dose-dependent manner (Supplementary Fig. 2B). The functionality of MTS105-encoded TCE was further assessed. Two protein controls were used to compare the functionality of mRNA-translated TCEs: the Fc-domain containing antibody-based TCE (analog of ERY974, Chugai Pharmaceutical Co., Ltd., Supplementary Fig. 2C) and the MTS105-sequence-identical recombinant TCE (rTCE). MTS105 mRNA-translated TCE showed potent lytic functionality when co-cultured with human peripheral blood mononuclear cells (PBMCs) and GPC3-positive (GPC3+) Hepa1-6-hGPC3-luc cell lines, equivalent to ERY974 and rTCE (Fig. 2B), and T cells were activated by the TCE to secrete cytokines IFN-γ and TNF (Fig. 2C). No appreciable lysis of GPC3-negative MC38-luc cells was detected at any concentration, indicating target-dependent activation of T cells by the TCEs.

A MTS105 is a novel LNP for efficient liver-tropic delivery with a size of 65–95 nm. mRNA encapsulated in MTS105 was optimized for improved intracellular stability and translational efficiency, including UTRs and CDS. The mRNA-encoded TCE was composed of ScFvs targeting GPC3 and CD3, separated by a flexible peptide linker. B T cell-dependent cellular toxicity assay (TDCC). Human PBMCs and Hepa1-6-hGPC3-luc or MC38-luc tumor cell lines were co-cultured for 48 h with supernatants from the MTS105-transfected 293T cell line at a 5:1 E:T ratio. rTCE and ERY974 were used as a positive control. Specific lysis was assessed via bioluminescence. C Cytokines released after MTS105-encoded TCE-induced specific lysis. IFN-γ and TNF were measured with ELISA using the supernatants from the TDCC assay (B). rTCE and ERY974 were used as a positive control. D TDCC assay. Human PBMCs and Hepa1-6-hGPC3-luc or MC38-luc tumor cell lines were co-cultured for 48 h with MTS105-encoded TCE containing mouse serum at a 5:1 E:T ratio. rTCE and ERY974 were used as a positive control. Specific lysis was assessed via bioluminescence. E MTS105-mediated CD8 T cell activation after 24 h of incubation of human PBMCs as effector cells with Hepa1-6-hGPC3-luc or MC38-luc target cells at a 5:1 E:T ratio. The concentration of 1111 pM for MTS105-transfected supernatant, rTCE and ERY974 was shown in the assay. T cell activation was assessed by measuring CD25, CD69 and CD107a levels on CD4 and CD8 T cells by flow cytometry. F MTS105-containing serum mediated cytotoxicity after coincubation of human PBMCs with GPC3+ HepG2 cells and GPC3- HL-60 cells as target cells. Cytotoxicity assays were performed at a 10:1 E:T ratio. Serum of cynomolgus monkey was collected after MTS105 administration, serially diluted, and added to the cocultures, followed by incubation for 24 h. The OD value detected with cell cytotoxicity by LDH was measured as readout. All data are expressed as mean ± SD (n = 3 biological replicates). Differences between the two groups were assessed by a two-sided Student’s t-test, while comparisons across multiple groups were performed using one-way ANOVA. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
Subsequently, we evaluated whether administering MTS105 intravenously in animals led to the in vivo generation of functional TCE molecules. Analysis of serum from C57BL/6J wild-type mice treated with MTS105 demonstrated potent lysis of Hepa1-6-hGPC3-luc tumor cells in a dose-dependent manner, similar to the activity of rTCE or ERY974, while GPC3-negative MC38-luc cells were not lysed (Fig. 2D). We also observed that the expression of CD25, CD69 and CD107a, general markers of T cell activation, were significantly elevated in CD8 and CD4 T cells incubated with Hepa1-6-hGPC3-luc cells, but not with MC38-luc cells (Fig. 2E; Supplementary Fig. 2D). Similarly, serum from cynomolgus monkeys treated with MTS105-mediated potent killing of GPC3+ HepG2 cells, but not to GPC3- HL-60 cell lines (Fig. 2F), These data suggest that intravenous injection of MTS105 can correctly translate the functional TCE in vivo, the activity of which is dependent on the binding of human CD3 and GPC3.
MTS105 demonstrated strong antitumor activity in an orthotopic HCC model
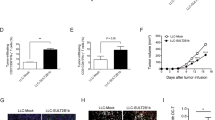
To evaluate the antitumor activity of MTS105 in vivo, we established a liver orthotopic tumor-bearing immunocompetent mouse model. Since MTS105-encoded TCE is not cross-reactive to murine Cd3e, we used a human CD3 transgenic mouse in which all three components of the Cd3 complex, Cd3e, Cd3d, and Cd3g, were replaced by their human counterparts, CD3E, CD3D, and CD3G. CD3 humanized mice were surgically inoculated with mouse Hepa1-6-hGPC3-luc cells at the liver site. After confirmation of engraftment by bioluminescence imaging (BLI) 8 days post-inoculation, the mice received a three-weekly intravenous injection of 0.05, 0.15, or 0.5 µg/mouse MTS105 or matched control: Tris buffer as vehicle control, MLX0473-based empty LNP, or ERY974 (Fig. 3A). As expected, MTS105 was more enriched in the liver tissue than the periphery, in contrast with the antibody-based control ERY974 (Fig. 3D); even at the highest dose of 1 µg/mouse, MTS105 did not match the maximum plasma concentration (Cmax) of ERY974. Interestingly, MTS105-translated TCEs were detected in the orthotopic liver tumor, while ERY974 did not penetrate the tumor (Fig. 3D). This tumor-specific enrichment of TCE by MTS105 indicates a more potent antitumor effect than with the conventional antibody-based approach with ERY974, despite its shorter T1/2 via the removal of the Fc element (Fig. 3B, C). Notably, MTS105 induced complete tumor regression, with no recurrence observed during an 84-day follow-up period. Our data suggest that liver-specific delivery of mRNA-translated TCE may create ample opportunity for efficient T cell engagement in the tumor microenvironment and resultant targeted tumor cell death. Indeed, MTS105 induced rapid intratumoral T cell infiltration and strong Granzyme B release in tumor tissue at 24 h post-injection, with results more pronounced at 72 h (Fig. 3E; Supplementary Fig. 3A). We did not observe any body weight loss over 21 days post-treatment (Supplementary Fig. 3B), nor did we see major cytokine releases in the tumor-bearing mice at 6 and 24 h post-injection (Supplementary Fig. 3C). Notably, MTS105-treated mice exhibited significantly lower ALT levels compared to the LNP empty control group, while no significant differences were observed among the other treatment groups (Supplementary Fig. 3D). To further validate T cell function, we performed an IFN-γ ELISpot assay to assess antigen-specific T cell responses in splenocytes isolated from MTS105-treated mice co-cultured with hGPC3+ Hepa1-6 cells. As shown in Fig. 3F, MTS105 treatment led to a statistically significant increase in IFN-γ-secreting T cells compared to untreated controls.

A Treatment schedule of the in vivo efficacy study. Effect of MTS105 in humanized CD3EDG mice bearing mouse Hepa1-6-hGPC3-luc tumors. After grouping, mice were treated with MTS105 (0.5, 0.15, 0.05 μg/mouse), ERY974 (1 mg/kg), LNP empty (0.5 μg/mouse), or Tris vehicle. B Tumor growth was monitored by means of bioluminescence imaging. n = 5 mice per group. C Tumor growth for individual mice. Color bars denote the radiance (photons/s/cm2/sr) scale at each time point. D MTS105-encoded TCE concentration was quantified by ELISA in serum, liver and tumor from orthotopic HCC mice at 6 h post-dosing. n = 4 biologically independent samples. E Representative H&E and IHC images of human CD3, GPC3, and mouse Granzyme B staining (×4 magnification) in Hepa1-6-hGPC3-luc orthotopic tumors from MTS105-treated mice euthanized 24 and 72 h after treatment. Reddish brown staining indicates a positive IHC signal. Scale bar: 200 μm. n = 3 mice per group. F Splenocytes from MTS105-treated and untreated mice were co-cultured with hGPC3+ Hepa1-6 cells or hGPC3- 293T cells overnight. IFN-γ SFU were detected and enumerated. PMA was used as positive control, and culture medium (blank) was used as negative control. n = 3 biologically independent samples. G Hepa1-6-hGPC3-luc cells were re-challenged in mice that achieved tumor regression following MTS105 treatment, with a 56-day interval before re-inoculation (n = 8 mice). Naïve CD3EDG mice were inoculated using the same tumor cells as a control (n = 5 mice). Tumor growth was monitored using bioluminescence imaging. H Cell subtype frequency analysis for (G) at Day 28. Data are expressed as mean ± SD. Differences between the two groups were assessed by two-sided Student’s t-test, while comparisons across multiple groups were performed using one-way ANOVA. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
We next evaluated whether the mRNA-translated TCE could induce memory cell formation in immunocompetent mice. Tumor-free mice from the MTS105-treated group (at day 56 post-tumor regression) did not exhibit tumor growth following rechallenge with Hepa1-6-hGPC3-luc cells, while tumor growth was uninhibited among the controlled treatment naïve mice (Fig. 3G; Supplementary Fig. 3E). We performed immunophenotype analysis on T cells in the peripheral blood and spleen at the endpoint of the re-challenge study (Fig. 3H). MTS105 treatment significantly enhanced CD8 T cells in peripheral blood, accompanied by an increase of effector memory CD8 T cells and a decrease of naive subsets. Notably, a similar expansion of effector memory CD8 T cells was detected in the spleen. These findings suggest that the observed shift toward an effector memory phenotype in CD8 T cells may contribute to the durability of antitumor activity of MTS105.
Based on the well-characterized synergistic mechanism between CD3 bispecific antibodies and PD-1 blockade, we conducted additional efficacy studies evaluating the combination of MTS105 with a murine anti-PD-1 antibody in an immunocompetent mouse model. To detect a potential treatment benefit of this combination, MTS105 was administered at a suboptimal dose (0.15 µg/mouse), below its single-agent therapeutic threshold. The combination therapy demonstrated significantly enhanced antitumor efficacy compared to either agent as monotherapy, indicating a synergistic effect and supporting the potential of MTS105 in combination with an immune checkpoint inhibitor (Supplementary Fig. 3F).
To better evaluate the therapeutic advantages of mRNA-based MTS105, we conducted a systematic comparison with an equivalent protein-based TCE (rTCE). Pharmacokinetic (PK) analysis (Supplementary Fig. 4A) showed that the peripheral Cmax of rTCE at a 3 μg dose was comparable to that of MTS105 at 0.5 µg. In the orthotopic HCC model, MTS105 achieved significantly greater antitumor efficacy despite a less frequent dosing regimen (once-weekly vs. three times weekly for rTCE), with neither treatment inducing body weight loss (Supplementary Fig. 4B, C). Immunophenotyping performed on Day 10 post-treatment highlighted several key advantages of MTS105 over rTCE: a higher CD8/CD4 T cell ratio in both peripheral blood and tumor tissue and rTCE treatment induced higher PD-1 expression on intratumoral T cells, while both groups showed similar levels of activation (CD69) and proliferation (Ki-67) markers (Supplementary Fig. 4D). Importantly, MTS105 increased levels of effector memory CD8 T cells in both peripheral blood and spleen (Supplementary Fig. 4E). Collectively, these findings underscore the superior pharmacodynamic profile of MTS105, likely contributing to its antitumor activity.
MTS105 effectively eradicated GPC3+ xenograft tumors in human PBMC-engrafted mice
To evaluate the antitumor activity of MTS105 among human immune cells, we used NOG mice engrafted with human PBMC as an experimental model. Tumor growth was monitored by BLI after in situ inoculation of Hep3B-luc cells in the liver. Eleven days after tumor inoculation, mice were injected with a three-weekly intravenous dose of 0.1 or 0.5 µg/mouse MTS105, empty LNP control, 0.3 mg/kg ERY974, or Tris buffer. Both MTS105 and ERY974 effectively eradicated the tumors (Fig. 4A–C), and ERY974 had a higher serum concentration than MTS105 after intravenous administration (Fig. 4D). No drug-related weight loss was observed.

A Treatment schedule of the in vivo efficacy study. Effect of MTS105 in NOG mice engrafted with human PBMCs and bearing mouse Hep3B-luc tumors. After grouping, mice were treated with MTS105 (0.5 and 0.1 μg/mouse), ERY974 (0.3 mg/kg), LNP empty (0.5 μg/mouse), or Tris vehicle. B Tumor growth was monitored by means of bioluminescence imaging. n = 6 mice per group. C Tumor growth for individual mice. D MTS105-encoded TCE concentration was quantified by ELISA in serum from orthotopic HCC mice at 6 h after the first treatment (n = 6 biologically independent samples). Data are expressed as mean ± SD. A one-way ANOVA was used to compare groups. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
MTS105 induced intratumoral CD8 T cell precursors and terminally differentiated memory subsets
To more comprehensively identify the mechanism by which MTS105 and ERY97 TCEs regulate the tumor microenvironment, we performed single-cell RNA sequencing (scRNA-seq) on immune cells isolated from tumors. We clustered the tumor-infiltrating immune cells and non-immune cells into 16 populations, and identified notable increases in T cell populations among MTS105-treated tumors compared to those treated with Tris and ERY974 (Fig. 5A, B; Supplementary Fig. 5A). We next analyzed the population of five distinct T cell subsets (Fig. 5C; Supplementary Fig. 5B). Compared to treatment with Tris and ERY974, MTS105 significantly upregulated CD8/CD4 T cell populations and decreased the proportions of Treg, Pro_T as well as Xcl1+T cell populations (Fig. 5D). MTS105 treatment was associated with a higher activation gene signature, indicating T cell activation (Fig. 5E; Supplementary Fig. 5C).

A UMAP showing the tumor-infiltrating immune cells and non-immune cells from mice (n = 3 mice per group) treated with Tris vehicle control, MTS105 (0.5 μg/mouse) or ERY974 (1 mg/kg). B Cell ratio and cell numbers of clusters identified in (A). C UMAP showing T cell clusters in tumors treated with Tris, MTS105, and ERY974. D Comparison of the proportions of five T subsets, and E comparison of T cell activation score in Tris, MTS105, and ERY974 groups. F UMAP showing CD8 T cell clusters in tumors treated with Tris, MTS105 and ERY974. G Comparison of the proportions of 12 CD8 T clusters in Tris, MTS105 and ERY974 groups. H Comparison of the cell ratio of C2, C4, C5 subsets in Tris and MTS105-treated tumors. I Differentiation trajectory of CD8 T cells. Color scale indicates the ordering of cells in pseudotime. J Heat map of pseudotime-correlated genes. Color scale indicates gene expression level. K B2m and Ccl5 expression in pseudotime trajectory analysis. In the box plot, the lower and upper edges represent the first (Q1, 25%) and third (Q3, 75%) quartiles, respectively, with the central line indicating the median (Q2, 50%). Statistical analysis was performed using two-sided Wilcoxon rank-sum tests.
To further explore the profile of gene expression among the CD8 T cell subset, 12 T cell clusters were validated by marker genes and distinct cell state-enriched gene expression (Supplementary Fig. 5D). We found that MTS105 induced significant expansion of the C2 population, which highly expressed Hspa1b, Hspa1a and Nr4a3 (Fig. 5F–H). A pseudotime trajectory analysis revealed a transcriptional transition from C4 and C5 toward C2 state along the axis (Fig. 5I). A heat map drawn based on the extracted pseudotime-correlated genes (Fig. 5J) revealed that the genes were clustered into four patterns, with gene expression in pattern 4 increasing over pseudotime. C2 was identified as the main cell type in pattern 4, which expressed B2m and Ccl5, indicating that these genes altered their transcriptional expression after MTS105 treatment (Fig. 5K).
MTS105 exhibited a favorable pharmacokinetic profile and tolerability in nonhuman primates
The protein sequence of TCE demonstrated similar binding affinities between cynomolgus and human GPC3 and CD3 (Supplementary Table 1). To extrapolate the PK and safety profile of in vivo MTS105-translated TCE in a species of higher relevance to humans, we conducted a 4-week, repeated-dose GLP toxicology study in cynomolgus monkeys. Following a 30-min infusion of MTS105 at 45 µg/kg (n = 10, mixed gender), the concentration-time profile of MTS105-translated TCE is presented in Fig. 6A, B. The time to peak plasma concentration (Tmax) was 6–9 h after dose, indicating rapid protein expression after MTS105 administration. The terminal elimination T1/2 was 13.7 ± 5.05 h in females and 13.6 ± 0.92 h in males after the first dose, notably longer than the general BiTE format of TCE (e.g., the T1/2 of blinatumomab was 2.05 h in chimpanzees and 2.10 h in humans30,31). The AUC0-t and Cmax female-to-male ratio of MTS105-translated TCE were in the range of 0.5 to 2.0 both on D1 and D22, with no significant gender differences. After 4-week repeated dosing in monkeys, the AUC0-t and Cmax D22 to D1 ratio of MTS105-translated TCE were almost in the range of 0.5 to 2.0 both in females and males, with no significant accumulation (Supplementary Table 2).

A Schedule of the repeated-dose PK study. The cynomolgus monkeys were given 4-weekly intravenous administration of 45 μg MTS105, indicated by blue arrows. Blood was collected at 0.25, 6, 12, 24, 48, 72 and 168 h post D1 and D22 treatment and at 12 and 168 h post D8 and D15 treatment, indicated by yellow arrows. B MTS105-translated TCE concentration in cynomolgus monkey serum samples measured by MSD (n = 10, mixed gender). C MTS105-translated TCE concentration in serum or tissue samples from all the tested animals. Mice, rats, and cynomolgus monkeys were intravenously administered 50 μg/kg MTS105 at 24 h post-dose (mice, n = 4, female; rats, n = 10, mixed gender; cynomolgus monkeys, n = 2, mixed gender). D A time-course analysis in rats (n = 10, mixed gender). Rats were intravenously administered MTS105 at a dose of 50 μg/kg, with plasma and organ samples collected at 6, 24, 48, 72 and 168 h post-dose for protein concentration analysis using MSD. E Serum IL-6 concentration over time in a repeated-dose study described in (A). Serum samples were collected at pre-dose, D2, D7 and D30) for analysis (n = 10, mixed gender). F Hepatic and renal function over time in a repeated-dose study described in (A). Levels of ALT, AST, ALP, TBIL, DBIL, UREA and CREA were measured at D1, D2, D7, D28 and D57 (n = 10, mixed gender). Data are expressed as mean ± SD.
To explore the translatability of tissue distribution of mRNA-encoded TCE protein across species, a 50 µg/kg dose of MTS105 was delivered via systemic injection in mice, rats and cynomolgus monkeys. The highest concentrations of TCE protein were consistently detected in the liver at 24 h post-treatment across all three species (Fig. 6C). The other organs where TCEs could be detected were the spleen and kidney. A time-course analysis of tissue biodistribution was performed in rats, in which MTS105-translated TCE exhibited the highest accumulation in the liver, followed by plasma, spleen and lymph nodes (Fig. 6D). The higher ratio of liver to plasma concentration of TCE was confirmed in cynomolgus monkeys, suggesting a favorable therapeutic window when considering the peripheral cytokine release-related adverse effects associated with many TCEs32,33,34,35. Moreover, to evaluate the long-term potential adverse effects of MTS105, our GLP toxicology study in cynomolgus monkeys included a 4-week recovery period following the 4-week repeated-dose regime. No clinically significant adverse effects were observed in gross pathology. IL-6 levels increased in all MTS105-treated groups at 24 h post-first dose (D1) but returned to baseline by D7 (Fig. 6E). Liver and renal function were closely monitored, with only transient elevations in hepatic (ALT, AST, ALP), biliary (TBIL, DBIL), and renal (UREA, CREA) biomarkers detected after the first dose. All values returned to baseline levels by the 2nd weekly dose (Fig. 6F).
Histopathological analysis showed mild inflammatory cell infiltration in the liver at the end of the dosing period, with the animals exhibiting complete recovery by the end of the recovery period. No inflammatory response was observed in the spleen or kidney. (Supplementary Fig. 6). Together, these PK and safety profiles warrant a first-in-human study of MTS105.
Discussion
The development of TCEs for solid tumors has faced great challenges, partly due to the difficulty of achieving sufficient intratumoral penetration to effectively engage T cells and tumor cells. Our data show that a tissue-specific LNP delivery system for mRNA-encoded TCEs enabled high in situ enrichment of the in vivo-assembled TCE protein directly in the liver and surrounding liver-related tumors. This localized expression enhances therapeutic efficacy by overcoming the delivery and penetration limitations commonly associated with TCEs in solid tumors.
In two liver-orthotopic tumor animal models, namely an immunocompetent mouse model and a humanized xenograft model, MTS105 demonstrated consistently high potency, achieving complete tumor remission at relatively low mRNA doses. This was accompanied by rapid intra-tumor T cell infiltration and activation, attributed largely to the high TCE concentration in the liver and tumor, enabled by liver-targeted LNP delivery and efficient mRNA–protein translation. Compared to the conventional antibody-based approach, the high liver-to-plasma ratio of TCE achieved by mRNA-LNP delivery may reduce peripheral toxicity caused by non-specific T cell activation, thereby significantly expanding the therapeutic window for this class of therapeutic.
The major hurdle in clinical development for TCE has been the on-target/off-tumor toxicity, which limits the therapeutic window32,33,34,35. Importantly, neither mice nor cynomolgus monkeys exhibited signs of adverse effects following MTS105 administration at or higher than efficacious doses. In the 4-week repeated-dose GLP toxicology study in cynomolgus monkeys, all animals survived until the scheduled necropsies at the end of the dosing period and the recovery period. No MTS105-related gross pathological abnormalities or statistically significant organ weight changes were observed at either the end of the dosing period or the recovery period. Transient elevations in hepatic (ALT, AST, ALP), biliary (TBIL, DBIL), and renal (UREA, CREA) function biomarkers, along with increased IL-6 levels, were observed within 24 h after the first dose. All parameters returned to baseline levels by the end of the recovery period. Histopathological analysis showed mild inflammatory cell infiltration in the liver at the end of the dosing period, with the animals exhibiting complete recovery by the end of the recovery period. Although MTS105-translated TCE was detected in the spleen and kidney, histopathological evaluation following repeated dosing revealed no drug-related tissue damage in these organs. These results further support the favorable safety profile of MTS105 and suggest that its biodistribution does not pose a significant risk for clinical application.
Balancing drug exposure and dosing frequency for TCEs remains a key focus in clinical development. During the design of our TCE, we intentionally excluded any T1/2 extending elements, such as Fc regions, to prevent prolonged T cell over-activation. In our mRNA-LNP therapeutic MTS105, the PK profile of the TCE is strictly governed by mRNA translation and subsequent clearance. Once transfected, cells steadily translate the mRNA into proteins until the mRNAs are completely degraded, resulting in extended protein exposure. For MTS105, this translates to a sustained 48–72 h of TCE exposure in both tissue and plasma. We hypothesized that the mRNA-translated TCE could provide an optimal therapeutic window sufficient to effectively activate T cells without driving them into exhaustion or activation-induced cell death (AICD). This hypothesis was supported by scRNA-seq data, which revealed that mRNA-translated TCE rendered more CD8 T cells with a much higher activation gene signature and tissue resident memory T cells expressing hspa1a and hspa1b, and Nr4a, a signature that has been identified previously36. A pseudotime trajectory analysis indicated that these T cell subsets were differentiated from other subsets by acquiring B2M and CCL5.
Cross-species translatability has been a significant challenge in LNP discovery. While nonhuman primates are generally considered the most predictive models for human responses, ethical constraints have led to the majority of LNP optimization being performed in rodents. However, translating LNP potency data from rodents to nonhuman primates has proven difficult, particularly for intravenous administration37,38. Our lipid was originally discovered in mice, demonstrating delivery efficiency approximately 10 times higher (based on luciferase signals) compared to a benchmark LNP validated in humans. We hypothesized that such high delivery efficiency could facilitate improved cross-species translatability. Supporting this, MTS105 exhibited consistent and persistent liver-specific expression of TCE and a stable liver-to-plasma ratio across mice, rats, and nonhuman primates. This suggests that high delivery efficiency can indeed bridge the translational gap between rodent and primate models. However, in human clinical trials, mRNA translational efficiency may be hindered by dose limitations, interindividual variability (e.g., genetic background and health status), and other confounding factors. Furthermore, the PEG component used in the LNP formulation has the potential to trigger immunogenicity in humans, which could affect the PK profiles of both the LNP and mRNA-translated protein. Our current preclinical assessments rely on transgenic animal models and CDX models, which may not be able to fully recapitulate the complexity of human hepatocellular carcinoma. These critical translational challenges will be carefully evaluated in the clinical setting.
In conclusion, our findings highlight the potential of MTS105 as an mRNA-based immunotherapy for treating GPC3+ cancers, offering a broad therapeutic window due to its unique tissue-specific delivery system. This preclinical study provides the foundation for initiating a first-in-human clinical trial to evaluate its safety and preliminary efficacy, which is currently underway (NCT06689540).
Methods
Animal experiments
Mouse and rat experiments
Animals were housed in a specific pathogen-free barrier (SPF) at the animal center of METiS Pharmaceuticals (Beijing, China) in an individual ventilated cage (IVC). The animals were maintained under controlled conditions: temperature at 20–26 °C, relative humidity at 40–70%, and a 12-h light/dark cycle. The animals had to be acclimatized for 7 days before the experiment. For animal studies, the animals were earmarked before grouping, and experimental/control animals were bred separately. At the end of animal experiments, the mice were euthanised by cervical dislocation. All studies in mice and rats were approved by the Institutional Animal Care and Use Committee of METiS Pharmaceuticals (ethical approval no. MN-IAC-20231200).
According to the requirements of our institutional ethical board, we have strictly adhered to the tumor progression limits in all experiments. Since the orthotopic liver tumor model used herein does not allow direct measurement of tumor volume, the early termination criteria were as follows: (1) the appearance of abnormal movement or paralysis; (2) body weight loss exceeding 20% from the initial treatment; (3) very poor body condition.
NHP study
The nonhuman primate study was conducted by InnoStar, Bio-tech Nantong Co., Ltd. It was reviewed by the Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC) in October 2022. The care and use of nonhuman primates in this study complied with the Guide for the Care and Use of Laboratory Animals (2011) issued by the National Research Council, USA, Laboratory Animal Administration (revised in 2017) issued by the State Science and Technology Committee, People’s Republic of China, and Laboratory Animal Administration Regulations (2008) issued by the People’s Government of Jiangsu Province. All studies were approved by the Institutional Animal Care and Use Committee of InnoStar (ethical approval no. IACUC-2024-M-465).
Cell lines and Human peripheral blood monocytes (PBMCs)
HEK293T cells (FH0244) were purchased from FuHeng Biology, and MC38-luc cells (YC-A002-Luc-P) from Genai Biology. HEK293T and MC38-luc cells were cultured in DMEM (Gibco, 11965092) supplemented with 10% fetal bovine serum (FBS, Thermo, A5669701) at 37 °C in a humidified atmosphere of 5% CO2. Hepa1-6-hGPC3-luc cells were constructed by Biocytogen Pharmaceuticals and maintained in vitro as a monolayer culture in DMEM supplemented with 10% FBS, 1% NEAA (Gibco, 11140-050), 1% L-Glutamine (Gibco, 25030-081), 1% pyruvate (Gibco, 11360070), 1% HEPES (Invitrogen, 15630080) and 1 µg/mL Puromycin (MCE, HY-K1057) at 37 °C in a humidified atmosphere of 5% CO2. HEK293-hGPC3 cells (GM-C15633) were purchased from Genomeditech and cultured in DMEM supplemented with 10% FBS at 37 °C in a humidified atmosphere of 5% CO2. The human liver carcinoma cell line Hep3B-luc (310678) was purchased from Biocytogen. Cells were maintained in MEM (Gibco, 11090081) supplemented with 10% FBS at 37 °C in a humidified atmosphere of 5% CO2. HepG2 (HB-8065) and HL-60 (CCL-240) cells were purchased from the American Type Culture Collection (ATCC). HepG2 cells were cultured in DMEM supplemented with 10% FBS at 37 °C in a humidified atmosphere of 5% CO2. HL-60 cells were cultured in IMDM (Gibco, 12440053) supplemented with 20% FBS at 37 °C in a humidified atmosphere of 5% CO2. Jurkat cells (CTCC-400-0075) were purchased from MeisenCTCC and cultured in RPMI1640 (Gibco, 11875093) supplemented with 10% FBS at 37 °C in a humidified atmosphere of 5% CO2.
PBMCs were purchased and isolated from Shanghai OriBiotech by Ficoll density gradient centrifugation from buffy coats of healthy human donors. All recruited volunteers provided written informed consent. The study was approved and authorized by the Ethics Committee of Shanghai Zhaxin Integrated Traditional Chinese and Western Medicine Hospital (Hospital Ethics Expedited Review No. 202428).
In vivo expression of luc mRNA
C57BL/6J mice (Cat. NO. 219, 6–8 weeks old, female, obtained from Beijing Vital River Laboratory Animal Technology, n = 3) were intravenously injected with MLX0473 or Lipid5-based LNPs containing 10 μg luciferase mRNA. Six hours after injection, 100 μL D-Luciferin (PerkinElmer, 122799) 20 mg/mL was intraperitoneally injected into the mice. After 10 min, the mice were imaged using IVIS (PerkinElmer).
mRNA sequence design
The mRNA sequence for MTS105 was designed using the proprietary algorithm MrNatty, developed by METiS Pharmaceuticals. This algorithm optimizes the entire mRNA sequence, including the 5’ UTR, CDS, and 3’ UTR regions. MrNatty integrates advanced RNA structure prediction methodologies and natural language processing techniques to achieve a balance between mRNA stability and translational efficiency. This design approach ensures optimal codon usage and sequence stability, enhancing the overall efficiency of mRNA translation into the target protein.
Generation of MTS105
Template synthesis
The mRNA sequences for MTS105 were incorporated into plasmids featuring a T7 promoter at the 5’ terminus and a 120 A polyA tail at the 3’ terminus, followed by a restriction site (e.g., BsaI). These plasmids served as templates for in vitro transcription. The plasmid constructs were prepared by GenScript BioTech Corp.
mRNA synthesis
Synthesis of mRNA was performed via in vitro transcription by T7 RNA polymerase described in detail as follows:
-
1)
Template linearization: 100 μg plasmid DNA templates were linearized with BsaI restriction enzyme (New England Biolabs, R3733V), and purified by adding 20 μL of 3 M sodium acetate (Thermo, AM9740) and 200 μL 100% isopropanol (MACKLIN, 67-63-0). Upon incubation at 37 °C for 3 h, samples were centrifuged at 14,000 × g at 4 °C for 15 min. The resulting DNA pellets were washed twice in 80% alcohol, then air-dried and resuspended in Nuclease-Free Water (Invitrogen, AM9937) for downstream experiments.
-
2)
T7-mediated in vitro transcription: mRNA synthesis was performed using in vitro transcription mediated by T7 RNA polymerase (Novoprotein, E121). The transcription process included the co-transcriptional incorporation of CleanCap AG (TriLink, N-7113-10) as a trinucleotide Cap 1 analog at a final concentration of 4 mM, and the substitution of uridine with N1-pseudouridine (Hongene, R5-064) to enhance mRNA stability and translational efficiency. All nucleotides were used at a final concentration of 7.5 mM in the transcription reaction. After incubation at 37 °C for 3 h, Dnase I (New England Biolabs, M0303S) was used to digest the DNA template. Then, IVT-mRNAs were purified using RNA Clean Beads (Vazyme, N412-02) according to the manufacturer’s instructions.
-
3)
IVT-mRNA characterization: the integrity and concentration of the synthesized mRNA were analyzed using UV/Vis spectroscopy (NanoDrop 2000) and capillary electrophoresis (5200 Fragment Analyzer System, Agilent), ensuring the quality of MTS105 for subsequent therapeutic applications.
LNP formulation
The ionizable lipid MLX0473 was designed and synthesized in-house at Metis, while Lipid5 (MCE, HY-138171), DSPC (AVT, S01005), cholesterol (AVT, O01001), and DMG-PEG 2000 (Sinopeg, 6020112402) were obtained commercially. Lipid nanoparticles (LNPs) were prepared at an N:P ratio of 6 using a standardized lipid composition (50 mol% ionizable lipid, 38.5 mol% cholesterol, 10 mol% DSPC, and 1.5 mol% DMG-PEG 2000) dissolved in ethanol. Through microfluidic mixing at a 3:1 aqueous-to-organic phase ratio, the ethanolic lipid solution was combined with mRNA dissolved in 10 mM citrate buffer (pH 4.0). The resulting LNP dispersion underwent buffer exchange into DPBS (pH 7.4) for neutralization, followed by sterile filtration (0.2 μm) and aliquoting for storage at −20 °C until further use. The detailed physicochemical characterization of the LNPs for clinical trials is shown in Supplementary Table 3.
Recombinant reference proteins
ERY974 analog was prepared by Universal Biol as previously described39,40. MTS105-sequence-identical recombinant TCE (rTCE) was prepared by Novoprotein (Shanghai, China). Both proteins were expressed using Expi293 cells and purified according to the respective manual. Purified proteins were tested for endotoxin contamination and stored in PBS at −80 °C.
MTS105 transfection
2.5 × 105 293T cells were seeded in each well on a 24-well plate and incubated overnight at 37 °C. After incubation, 293T cells were treated with MTS105 (LNP-mRNA) for 24 h. At the end of the incubation period, the supernatant was collected and centrifuged to remove the cell pellet before using ELISA for quantification of protein expression.
Cell binding assays
To assess GPC3-specific binding, 5 × 105 HEK293-hGPC3 cells were added to individual flow cytometry tubes. The cells were stained for Live/Dead (Zombie Aqua Fixable Viability Kit, BioLegend, 423101) in PBS for 15 min at room temperature. MTS105-encoded TCE at a concentration of 86.8–2777.5 pM was then added to each tube, followed by 1 h of incubation at 4 °C. The cells were then washed and centrifuged. Cell staining buffer containing G4S Linker Rabbit mAb (CST, 71645S) was added to each tube, incubated at 4 °C for 30 min, and protected from light; followed by the addition of anti-rabbit IgG (H + L), F(ab’)2 fragment (Alexa Fluor 488 Conjugate) (CST, 4412) and incubation under the same conditions for an additional 30 min. The cells were then washed twice with cell staining buffer.
To assess CD3-specific binding, 5 × 105 Jurkat cells were added to individual flow cytometry tubes. The cells were stained for Live/Dead (Zombie Aqua Fixable Viability Kit, BioLegend, 423101) in PBS for 15 min at room temperature. MTS105-encoded TCE at a concentration of 30–7690 nM was then added to each tube, followed by 1 h of incubation at 4 °C. The cells were then washed and centrifuged. Cell staining buffer containing FITC-labeled Human GPC3 Protein (ACRO, GP3-HF258) was added to each tube, incubated at 4 °C for 30 min, and protected from light. The cells were then washed twice with cell staining buffer.
Flow cytometry was performed using a Cytoflex S (BECKMAN) instrument, and the data were analyzed with FlowJo_v10.8.1.
T cell-dependent cellular cytotoxicity assays
After quantification of mRNA-encoded TCE in the supernatant of 293T cells or mouse serum, the supernatant, ERY974, or rTCE were sequentially diluted to nine doses. In the cell killing experiment, the total experimental system was 100 μL, including 20 μL drug, 40 μL target cells and 40 μL effector cells. Target cells Hepa1-6-hGPC3-luc and MC38-luc were digested with pancreatic enzymes from petri dishes, then resuspended with AM medium (RPMI1640, 1% NEAA, 10 mM HEPES, 50 µM β-mercaptoethanol, 1 mM sodium pyruvate, 100 U/mL P/S, 5% hiFBS) and counted by cell counter (Bobo Ju, JSY-FL-049). A suspension of 2.5 × 105 cells/mL was obtained by AM medium. Frozen PBMCs were removed from liquid nitrogen and melted in a water bath (Jinghong, DK-8B) at 37 °C, then added to a centrifuge tube containing an appropriate amount of preheated AM medium and centrifuged at 250 × g for 15 min. The cells were directly resuspended with 10 mL AM medium and then resuspended into 1.25 × 106 cells/mL suspension. The drug, target cells and effector cells were added to the 96-well plate with a white background, and cultured at 37 °C for 48 h. Cell death was detected using the Steady-Glo Luciferase Assay System (Promega, E2520), by adding 100 μL Luciferase reagent, waiting in the dark for 5 min, and measuring luminescence with a microplate reader (BioTek, SYNERGY H1). The percentage of cell killing was calculated as follows: Cytotoxicity% = (1-Lum sample/ Lum control) x 100%.
To assess the cytotoxicity of MTS105 produced in nonhuman primates, serum from cynomolgus monkeys injected with MTS105 (50 µg/kg) was sequentially diluted to eight doses, with blank cynomolgus monkey serum as the negative control. HepG2 was used as GPC3-positive target cells, and HL-60 was used as GPC3-negative target cells. Human PBMCs (2 × 105 cells/well) and target cells (2 × 104 cells/well) were co-cultured for 24 h with diluted serum in a 96-well round-bottom plate, at a 10:1 E:T ratio. The OD value was detected using the Cytotoxicity LDH Assay Kit-WST (Dojindo Molecular Technologies, CK12) as per the manufacturer’s instructions.
In vitro T cell activation assay
Human PBMCs (5 × 104 cells/well) and target cells (Hepa1-6-hGPC3-luc and MC38-luc, 1 × 104 cells/well) were co-cultured at a 5:1 E:T ratio on 96-well tissue culture plates. MTS105-transfected 293T supernatant (first concentration of 1111 pM), rTCE (first concentration of 5555 pM) and ERY974 (first concentration of 5555 pM) were sequentially diluted to five doses and added into the test and control wells, and a blank control group with only effector cells and target cells was set. Assay plates were then incubated at 37 °C with 5% CO2 for 24 h. Thereafter, the contents of each plate were transferred to a 96-well round-bottom plate and the two parallel holes were merged, washed with FACS staining buffer (PBS containing 2% heat-inactivated FBS), and centrifuged (500 × g, 5 min) to collect cell precipitates. Next, the cells were stained for Live/Dead in FACS staining buffer for 15 min at room temperature using FVS510 (BD, 2257424), then stained for surface markers for 30 min at RT in FACS staining buffer after washing using anti-human CD45 Alexa Fluor 700 (BD, 560566), CD4 BV605 (BD, 562658), CD8 APC-Cy7 (BD, 557834), CD69 PE (BD, 555531), CD25 BB515 (BD, 564467). Flow cytometry was performed using a Cytoflex S (BECKMAN) instrument, and the data were analyzed with FlowJo_v10.8.1. The total T cell activation was determined as follows: Activated T cell = % Parent CD25++% Parent CD69+-% Parent CD25+ CD69+. The gating strategy is shown in Supplementary Fig. 7.
In vitro measurement of cytokine concentrations
After quantification of mRNA-encoded TCE in the supernatant of 293T cells, ERY974, and rTCE, solutions were sequentially diluted to seven doses. Cells were cultured using 96-well plates, with a total experimental system of 100 μL, including 20 μL drug, 40 μL target cells, and 40 μL effector cells. Target cells Hepa1-6-hGPC3-luc and MC38-luc were digested with pancreatic enzymes from petri dishes, then resuspended with AM medium and counted by cell counter (Bobo Ju, JSY-FL-049). A suspension of 2.5 × 105 cells/mL was obtained by AM medium. Frozen PBMCs were removed from liquid nitrogen and melted in a water bath (Jinghong, DK-8B) at 37 °C, added to a centrifuge tube containing an appropriate amount of preheated AM medium and then centrifuged at 250 × g for 10 min. Cells were directly resuspended with 10 mL AM medium into a suspension of 1.25 × 106 cells/mL. The drug, target cells, and effector cells were added to the 96-well plate with a white background, and cultured at 37 °C for 48 h. After 48 h, the concentrations of IFN-γ and TNF were detected by ELISA after the cell supernatant was diluted five times (Beyotime, PI511; PT518).
ELISA for human EPO
Sprague-Dawley rats (Cat. NO. 101, 7–8 weeks old, 200–250 g, female, n = 4) were obtained from Beijing Vital River Laboratory Animal Technology and subjected to five-weekly intravenous injections with MLX0473 or Lipid5-based LNPs containing 10 μg hEPO mRNA (n = 4). Serum was collected at 6, 24, and 72 h after administration.
ELISA of hEPO in rat serum samples was conducted at Beijing Solarbio Science & Technology. hEPO concentration was measured using a Human Erythropoietin ELISA Kit (Solarbio, SEKH-0161) per the manufacturer’s instructions.
ELISA for MTS105-encoded TCE
Mouse serum and tissue samples were collected at different time points for sandwich ELISA. Firstly, human GPC3 (Novoprotein, CW66) was coated on ELISA plates in coating buffer (Solarbio, SEKF103) overnight at 4 °C at a concentration of 2 µg/mL. Plates were washed with washing buffer (Solarbio, SEKF102), and blocked with blocking buffer (Solarbio, SEKF116). rTCE was used as the reference standard for quantitation. Plates were placed in the dark for 90 min after adding biotinylated Human CD3 epsilon Protein (Acro, CDE-H8224). Streptavidin-HRP (Abcam, ab7403) was used to detect human antibodies after 50 min. After washing plates three times, 100 µL of TMB (Solarbio, PR1200) was added for 5–15 min. Plate reading was conducted using an ELISA reader (Thermo) within 15 min after adding stop solution (Solarbio, C1058).
Liver orthotopic humanized CD3EDG syngeneic model
For establishing the syngeneic mouse model, female humanized CD3EDG mice (C57BL/6Smoc-Cd3etm1(hCD3E)Cd3dtm1(hCD3D)Cd3gtm1(hCD3G)Smoc, Cat. NO. NM-HU-220120) aged 7–8 weeks were purchased from Shanghai Model Organisms Center, Inc. Humanized CD3EDG mice were injected in situ with Hepa1-6-hGPC3-luc tumor cells (1 × 106 cells/mouse) in 0.02 mL Matrigel (Corning, 354234) into the left lobe of the mouse’s liver for tumor development. On the eighth day after cell inoculation, 100 μL D-Luciferin 20 mg/mL was intraperitoneally injected into the mice. After 10 min, the mice were imaged using IVIS (PerkinElmer). Those mice with total flux ranging between 1 × 108 and 4 × 108 and body weights exceeding 20 g were randomly divided into 6 groups (n = 5). The mean total flux of each group was approximately 2 × 108. ERY974 (1 mg/kg) or LNP empty (0.5 µg/mouse), or MTS105 (0.5, 0.15 or 0.05 µg/mouse) was intravenously administered to mice once-weekly for 3 weeks. After grouping, mice were imaged twice a week using IVIS (PerkinElmer) to obtain imaging signal diagrams and signal intensity. Body weight was recorded at the start of the study and twice a week throughout the experiment.
For the combination efficacy study of MTS105/anti-mPD-1, mice were randomized to five groups (n = 5), and the mean total flux of each group was approximately 5 × 108. Each mouse was treated with MTS105 0.15 µg/mouse once-weekly by intravenous injection or anti-mPD-1 (BioXCell, BP0146) 10 mg/kg twice-weekly by intraperitoneal injection, or with the combination of both for a duration of 3 weeks. PBS and LNP empty served as the control groups.
For the comparison study between MTS105 and rTCE, mice were randomized to 3 groups (n = 5), the mean total flux of each group was approximately 5 × 108. Each mouse was treated with MTS105 0.5 µg/mouse once-weekly or rTCE 3 µg/mouse triweekly by intravenous injection for 3 weeks. PBS served as the control group. On day 10, peripheral blood, spleen, and tumor samples were collected from three mice per group for immunophenotyping analysis.
Tissue samples were processed into single-cell suspensions and stained, followed by flow cytometry data collection. The cells were stained for Live/Dead in FACS staining buffer for 15 min at room temperature using Zombie Aqua Fixable Viability Kit (Biolegend, 423101), then stained for surface markers for 30 min at room temperature in FACS staining buffer after washing using anti-mouse CD45-APC-Cy7 (Biolegend, 147718), CD4-BV605 (Biolegend, 100451), CD8a-PE (Biolegend, 100707), CD44-FITC (Biolegend, 156008), and CD62L-APC (Biolegend, 161218), TCR β chain- PE/Cyanine7 (Biolegend, 109222), CD279 (PD-1)-FITC (Biolegend, 135214), CD69-APC (Biolegend, 104514), Ki-67-Alexa Fluor 700 (Biolegend, 652420). Flow cytometry was performed using a Cytoflex S (BECKMAN) instrument, and the data were analyzed with FlowJo_v10.8.1. The gating strategy is shown in Supplementary Fig. 7.
Liver orthotopic human PBMC reconstituted CDX model
For establishing the humanized NOG model, female NOG mice (NOD.Cg-PrkdcscidIl2rgtm1Sug/JicCrl, Cat. NO. 408) aged 7–8 weeks were purchased from Beijing Vital River Laboratory Animal Technology. Human PBMCs were revived and diluted to 5 × 106 cells/0.1 mL with PBS and injected into NOG mice intravenously. Four days later, NOG mice were injected in situ with Hep3B-luc tumor cells (5 × 105 cells/mouse) in 0.02 mL PBS into the left lobe of the mouse’s liver for tumor development. On the 15th day after PBMC inoculation, mice were subjected to peripheral anticoagulation collection from the inner canthus. Analysis of erythrolysis was performed on peripheral blood, followed by staining with PerCP/Cyanine5.5 anti-mouse CD45 antibody (BioLegend, 157208) and FITC anti-human CD45 antibody (BioLegend, 304038) at 4 °C for 30 min and flow cytometric analysis, to examine human PBMC engraftment. Mice with >5% human CD45-positive cells were considered successfully engrafted. The total flux within the range of 2 × 108–2 × 109, body weight greater than 18 g, and successfully reconstructed mice were randomly divided into 5 groups with 6 mice each, with the mean total flux around 1 × 109 in each group. ERY974 (0.3 mg/kg) or LNP empty (0.5 µg/mouse), or MTS105 (0.5 or 0.1 µg/mouse) was intravenously administered to mice once-weekly for 3 weeks. After grouping, mice were imaged twice a week using a small animal in vivo imaging system to obtain imaging signal diagrams and signal intensity. Body weight was recorded at the start of the study and twice a week throughout the experiment.
Measurement of cytokines in liver orthotopic humanized CD3EDG syngeneic model
Cytokines in serum samples were measured by Luminex assay (Univ-bio, Shanghai, China). Serum concentrations of human IL-6, IFN-γ, TNF, IL-2, IL-4, IL-1b, IL-13, IL-8, IL-10, and IL-12p70 were quantified using the Luminex X-200 system (Luminex, Austin, TX, USA) following the manufacturer’s instructions (LXLBH10-1, LabEx Co., Shanghai, China). Serum concentrations of mouse IL-6, IFN-γ, TNF, IL-2, IL-4, IL-1b, IL-5, KC, IL-10, and IL-12p70 were quantified using the Luminex X-200 system (Luminex, Austin, TX, USA) per the manufacturer’s instructions (LXLBM10-1, LabEx Co., Shanghai, China).
IFN-γ ELISPOT assays
Spleens were harvested from MTS105-treated and untreated humanized CD3EDG tumor-bearing mice (n = 3), mechanically dissociated using a 70 μm sterile cell strainer (FALCON, 352350), and centrifuged at 300 × g for 5 min. Pellets were lysed with Red Blood Cell Lysis Buffer (Beyotime, C3702) to obtain erythrocyte-lysized single-cell suspensions. Mouse IFN-γ pre-coated ELISpot plates (DAKEWE, 2210006) were activated with 200 μL/well of Opti-MEM (Gibco, 31985070) for 10 min at room temperature. After removing the medium, splenocytes (2 × 10⁵ cells/100 μL/well) were seeded in duplicate onto the plates. The experimental group was co-cultured with Hepa1-6-hGPC3-luc cells (2 × 10⁵ cells/100 μL/well). Cell Activation Cocktail (BioLegend, 423301), composed of PMA and ionomycin, was used as positive control. Culture medium (blank) was used as a negative control. The culture plate was cultured overnight at 37 °C and 5% CO2. After 18 h, the chromogenic reaction was performed according to the manufacturer’s protocol and quantified using an automated ELISpot reader (AID ispot, ELR08IFL).
Processing of raw data from scRNA-seq
scRNA-seq analysis was conducted at Beijing Shengji Technology Limited. Cells with a gene number <500 or a mitochondrial gene ratio >5% were excluded as low-quality cells. DoubletFinder (2.0.3) software was used to detect and delete possible twin data in the technical artifact. After the expression matrix of each sample was normalized and scaled using the ‘SCTransfrom’ function, the features and anchors for downstream integration were selected using the “PrepSCTIntegratio” and “findinintegrationanchors” functions. All valid samples were integrated using the “IntegrateData” function according to the above features and scaled using the “ScaleData” function. After data integration and scaling, principal component analysis and clustering were performed using the “RunPCA” and “FindCluster” functions. Dimensionality reduction was then performed using the “RunUMAP” function. Cell clustering was performed by using the “FindNeighbors” and “FindClusters” functions. Marker genes of each cluster were calculated using the “FindAllMarkers” function (avg_log2 FC ≥ 0.25 and P_val_adj<0.05).
Differential expression analysis from scRNA-seq data
Differentially expressed genes (DEGs) between control and experiment groups were calculated using the Seurat function “FindMarkers” with the Wilcoxon signed-rank test. Genes with | avg_log2FC| ≥ 0.25 and P_val_adj <0.05 were identified as DEGs.
Inference of the pseudotime trajectory
Monocle3 (1.0.0) was used to study the developmental trajectories of CD8 T cells. The starting point was called again without modifying the root_state parameter to obtain the expected time trajectory of cell differentiation. Genes related to cell differentiation fate were compared and analyzed. The pseudotime score for each CD8 T cell, as well as the pseudotime-correlated genes, are listed in Supplementary Data 1 and 2.
Differential gene expression analysis was performed using the differentialGeneTest function from the Monocle package. For each gene, two generalized linear models (a ‘full’ model and a ‘reduced’ model) were fitted and compared using a likelihood ratio test (LRT) to obtain P-values. The resulting P-values were then adjusted for multiple testing using the Benjamini-Hochberg (BH) method to control the false discovery rate (FDR), yielding q-values.
Gene set score analysis
Gene sets were collected from MsigDB (https://www.gsea-msigdb.org/gsea/index.jsp). Genes in each gene set are listed in Supplementary Data 3. Gene set expression scores were calculated using the function “AddModuleScore” of Seurat and visualized through R package ggplot2.
Surface plasma resonance (SPR)
Binding of MTS105-sequence-identical recombinant TCE to human and cynomolgus monkey GPC3 or CD3 epitopes was conducted at GenScript ProBio (Nanjing, China) using a Biacore 8 K (Cytiva) per the manufacturer’s instructions. All data were processed using the Biacore 8 K Evaluation Software version 4.0. Flow cell 1 and blank injections of running buffer in each cycle were used as a double reference for Resonance Units subtraction.
Tissue distribution of mRNA-encoded TCE in mice and rats
C57BL/6J mice (6–8 weeks old, female, n = 4) and Sprague-Dawley rats (6–8 weeks old, 160–280 g, mixed gender, n = 10) were intravenously administered with 50 µg/kg of MTS105. Plasma and tissues were harvested from mice at 24 h post-dose for ELISA quantification, while the plasma and tissues were collected from rats at pre-dose, 6, 12, 24, 48, 72 and 168 h post-dose for subsequent MSD immunoassay.
NHP study
The nonhuman primate study was conducted by InnoStar, Bio-tech Nantong Co., Ltd. The cynomolgus monkeys (conventional grade) were group-housed by sex in stainless steel cages for the whole day. The room temperature was maintained at 16–26 °C, and the relative humidity was maintained at 30–70%. The room was supplied with no fewer than 8 air changes per hour with 100% fresh air on a 12-h light/dark cycle.
For the tissue distribution of mRNA-encoded TCE, 2–4 kg cynomolgus monkeys (3–4 years old, n = 2, mixed gender) were subjected to a single intravenous injection. Plasma and tissues were collected at 24 h post-dose for MSD immunoassay.
For the PK and toxicity study (GLP), the experiment was divided into 5 groups, with 10 animals in each group, half male and half female, 3 years old. They were given intravenous injections of 0.9% sodium chloride (vehicle control group), LNP empty, and MTS105 of 10, 30, and 45 μg/kg. Treatment was administered once a week for a total of 5 doses (D1, D8, D15, D22, and D29) with a volume of 5 mL/kg. Blood collection for each group of animals was conducted at pre-dosing, 0.25 h, 1 h, 6 h, 12 h, 24 h, 48 h, 72 h and 168 h after administration of D1, 12 h after administration of D8, pre-dosing and 12 h after administration of D15, pre-dosing, 0.25 h, 1 h, 6 h, 12 h, 24 h, 48 h, 72 h and 168 h after dosing of D22, and before dissection of D30. Whole blood was collected into tubes containing EDTA-K2 anticoagulant and plasma was prepared by centrifugation for toxicokinetic sample analysis. Detection of the concentration of MTS105-translated protein in cynomolgus monkey plasma was conducted by MSD immunoassay. Tmax, Cmax, and AUC(0-t) were calculated by WinNonlin (version 8.3.4) (non-compartment model). The plasma for cytokine analysis was collected at pre-dose, 24 h post-dosing on D1, D7, 24 h post-dosing on D29. IL-6 in plasma was assessed using a CBA method (Assay kit of plasma: LEGENDplex; Lot Number: B404362; BioLegend). The serum for clinical chemistry analysis was collected at D2, D7, and D28. Serum samples were analyzed by the BECKMAN AU5800 Automatic Clinical Analyzer. Animals were necropsied at the end of dosing (D30; 3 female, 3 male) and recovery (D59, 2 female, 2 male) periods, as well as gross examination performed. The main organs and abnormal tissues of all animals were trimmed, embedded in paraffin, sectioned, and stained with hematoxylin and eosin (H&E).
MSD immunoassay for MTS105-encoded TCE in rats and cynomolgus monkeys
The 96-well plate was coated with a reconstitution solution of Human GPC3 Protein (His Tag, premium grade), and incubated overnight at 2–8 °C for 16–20 h. The plate was then sealed with 5% Milk-PBST and the test sample was added, followed by the addition of SULFO-TAG™ CD3 was used as the detection solution, and MSD Read Buffer T (2x) was added to the plate. The values were read by an electrochemical luminescence instrument (MESO Sector S 600MM). The relevant parameters of the standard curve were fitted through the four-parameter regression model using the theoretical concentration as the abscissa and the difference between the measured mean value and the blank mean value as the ordinate. Data were collected using Methodical Mind, and sample concentration was calculated using SoftMax Pro. The lower limit of quantification (LLOQ) of this method in plasma, heart, liver, lung, kidney, and bone marrow is 500,000 pg/mL.
Immunohistochemical analysis
Liver orthotopic tumor xenografts from the syngeneic or PBMC-humanized mice sacrificed at the study endpoint were fixed in 4% paraformaldehyde fix solution (Beyotime, P0099) for immunohistochemistry (IHC). Tissues were dehydrated, paraffin-embedded, and stained with rabbit antibodies against human CD3 (Abcam, ab133628), Granzyme B (Abcam, ab255598) and human GPC3 (Abcam, ab207080). Whole tumor images were scanned with the slice digital fluorescence scanning system (Jiangfeng, KF-PRO-005) and edited with the K-viewer software.
Tissues of sacrificed cynomolgus monkeys treated with MTS105 were fixed in 4% paraformaldehyde fix solution (Beyotime, P0099) for IHC. Tissues were dehydrated, paraffin-embedded, and H&E staining was performed.
The detailed information of antibodies in this study is shown in Supplementary Table 4.
Statistical analysis
The statistical analyses were performed using GraphPad Prism version 10.3.0. Comparisons of continuous variables between two groups were assessed using two-sided Student’s t-tests. Comparisons across multiple groups were performed using one-way analysis of variance (ANOVA). A P-value of less than 0.05 was deemed statistically significant.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The scRNA-seq data generated in this study have been deposited in the GSA database under the CRA024227, https://ngdc.cncb.ac.cn/gsa/browse/CRA024227. All data are included in the Supplementary Information or available from the authors, as are unique reagents used in this Article. The raw numbers for charts and graphs are available in the Source Data file whenever possible. A related patent has been submitted to the China National Intellectual Property Administration (CNIPA) in 2025. Regardless of the Patent application status, the mRNA and/or their sequences will be made available for research purposes upon request following MTA approval. Source data are provided with this paper.
References
Kantarjian, H. et al. Blinatumomab versus chemotherapy for advanced acute lymphoblastic leukemia. N. Engl. J. Med. 376, 836–847 (2017).
Gökbuget, N. et al. Blinatumomab for minimal residual disease in adults with B-cell precursor acute lymphoblastic leukemia. Blood 131, 1522–1531 (2018).
Budde, L. E. et al. Safety and efficacy of mosunetuzumab, a bispecific antibody, in patients with relapsed or refractory follicular lymphoma: a single-arm, multicentre, phase 2 study. Lancet Oncol. 23, 1055–1065 (2022).
Budde, L. E. et al. Single-agent mosunetuzumab shows durable complete responses in patients with relapsed or refractory B-cell lymphomas: phase I dose-escalation study. J. Clin. Oncol. 40, 481–491 (2022).
Dreher, M. R. et al. Tumor vascular permeability, accumulation, and penetration of macromolecular drug carriers. J. Natl. Cancer Inst. 98, 335–344 (2006).
Baeuerle, P. A. & Wesche, H. T-cell-engaging antibodies for the treatment of solid tumors: challenges and opportunities. Curr. Opin. Oncol. 34, 552–558 (2022).
Ahn, M. J. et al. Tarlatamab for patients with previously treated small-cell lung cancer. N. Engl. J. Med. 389, 2063–2075 (2023).
Giffin, M. J. et al. AMG 757, a half-life extended, DLL3-targeted bispecific T-cell engager, shows high potency and sensitivity in preclinical models of small-cell lung cancer. Clin. Cancer Res. 27, 1526–1537 (2021).
Arvedson, T. et al. Targeting solid tumors with bispecific T cell engager immune therapy. Annu. Rev. Cancer Biol. 6, 17–34 (2022).
Bray, F. et al. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 68, 394–424 (2018).
Lau, W. Y. et al. Preoperative systemic chemoimmunotherapy and sequential resection for unresectable hepatocellular carcinoma. Ann. Surg. 233, 236–241 (2001).
Finn, R. S. et al. Atezolizumab plus bevacizumab in unresectable hepatocellular carcinoma. N. Engl. J. Med. 382, 1894–1905 (2020).
Kulkarni, A. V. et al. Effectiveness and safety of atezolizumab-bevacizumab in patients with unresectable hepatocellular carcinoma: a systematic review and meta-analysis. EClinicalMedicine 63, 102179 (2023).
Filmus, J. & Selleck, S. B. Glypicans: proteoglycans with a surprise. J. Clin. Invest. 108, 497–501 (2001).
Filmus, J. Glypicans in growth control and cancer. Glycobiology 11, 19r–23r (2001).
Iglesias, B. V. et al. Expression pattern of glypican-3 (GPC3) during human embryonic and fetal development. Histol. Histopathol. 23, 1333–1340 (2008).
Haruyama, Y. & Kataoka, H. Glypican-3 is a prognostic factor and an immunotherapeutic target in hepatocellular carcinoma. World J. Gastroenterol. 22, 275–283 (2016).
Hsu, H. C., Cheng, W. & Lai, P. L. Cloning and expression of a developmentally regulated transcript MXR7 in hepatocellular carcinoma: biological significance and temporospatial distribution. Cancer Res. 57, 5179–5184 (1997).
Sung, Y. K. et al. Glypican-3 is overexpressed in human hepatocellular carcinoma. Cancer Sci. 94, 259–262 (2003).
Midorikawa, Y. et al. Glypican-3, overexpressed in hepatocellular carcinoma, modulates FGF2 and BMP-7 signaling. Int. J. Cancer 103, 455–465 (2003).
Yamauchi, N. et al. The glypican 3 oncofetal protein is a promising diagnostic marker for hepatocellular carcinoma. Mod. Pathol. 18, 1591–1598 (2005).
Ishiguro, T. et al. Anti-glypican 3 antibody as a potential antitumor agent for human liver cancer. Cancer Res. 68, 9832–9838 (2008).
Sawada, Y. et al. Phase II study of the GPC3-derived peptide vaccine as an adjuvant therapy for hepatocellular carcinoma patients. Oncoimmunology 5, e1129483 (2016).
Li, S. Q. et al. GPC3 DNA vaccine elicits potent cellular antitumor immunity against HCC in mice. Hepato-Gastroenterol. 61, 278–284 (2014).
Wang, C., Gao, W., Feng, M., Pastan, I. & Ho, M. Construction of an immunotoxin, HN3-mPE24, targeting glypican-3 for liver cancer therapy. Oncotarget 8, 32450–32460 (2017).
Wang, K. et al. Iron-oxide-based nanovector for tumor targeted siRNA delivery in an orthotopic hepatocellular carcinoma xenograft mouse model. Small 12, 477–487 (2016).
Gao, H. et al. Development of T cells redirected to glypican-3 for the treatment of hepatocellular carcinoma. Clin. Cancer Res. 20, 6418–6428 (2014).
Dargel, C. et al. T cells engineered to express a T-cell receptor specific for Glypican-3 to recognize and kill hepatoma cells in vitro and in mice. Gastroenterology 149, 1042–1052 (2015).
Sabnis, S. et al. A novel amino lipid series for mRNA delivery: improved endosomal escape and sustained pharmacology and safety in non-human primates. Mol. Ther. 26, 1509–1519 (2018).
Blincyto: pharmacology review(s) (2014). https://www.accessdata.fda.gov/drugsatfda_docs/nda/2014/125557Orig1s000PharmR.pdf.
Blincyto: clinical pharmacology biopharmaceutics review(s) (2014). https://www.accessdata.fda.gov/drugsatfda_docs/nda/2014/125557Orig1s000ClinPharmRedt.pdf.
Martin, T. G. et al. Detailed overview of incidence and management of cytokine release syndrome observed with teclistamab in the MajesTEC-1 study of patients with relapsed/refractory multiple myeloma. Cancer 129, 2035–2046 (2023).
Li, C. C. et al. A novel step-up dosage regimen for enhancing the benefit-to-risk ratio of mosunetuzumab in relapsed or refractory follicular lymphoma. Clin. Pharmacol. Ther. https://doi.org/10.1002/cpt.3445 (2024).
Hutchings, M. et al. Dose escalation of subcutaneous epcoritamab in patients with relapsed or refractory B-cell non-Hodgkin lymphoma: an open-label, phase 1/2 study. Lancet 398, 1157–1169 (2021).
Topp, M. S. et al. Safety and activity of blinatumomab for adult patients with relapsed or refractory B-precursor acute lymphoblastic leukaemia: a multicentre, single-arm, phase 2 study. Lancet Oncol. 16, 57–66 (2015).
Kurd, N. S. et al. Early precursors and molecular determinants of tissue-resident memory CD8+ T lymphocytes revealed by single-cell RNA sequencing. Sci. Immunol. https://doi.org/10.1126/sciimmunol.aaz6894 (2020).
Maier, M. A. et al. Biodegradable lipids enabling rapidly eliminated lipid nanoparticles for systemic delivery of RNAi therapeutics. Mol. Ther. 21, 1570–1578 (2013).
Hatit, M. Z. C. et al. Species-dependent in vivo mRNA delivery and cellular responses to nanoparticles. Nat. Nanotechnol. 17, 310–318 (2022).
Chichili, G. R. et al. A CD3xCD123 bispecific DART for redirecting host T cells to myelogenous leukemia: preclinical activity and safety in nonhuman primates. Sci. Transl. Med. 7, 289ra282 (2015).
Davies, B. & Morris, T. Physiological parameters in laboratory animals and humans. Pharm. Res. 10, 1093–1095 (1993).
Acknowledgements
The authors wish to thank Beijing Shengji Technology Limited for single-cell RNA sequencing analysis. This study was partially funded by the National Center of Technology Innovation for Biopharmaceuticals (NCTIB). Editorial support was provided by Phoebe Kennedy, MSc, and Jake Burrell, PhD, on behalf of Rude Health Consulting Ltd. This support was funded by METiS Pharmaceuticals.
Author information
Authors and Affiliations
Contributions
Yan Huang designed and executed the majority of experiments. Shaoli Liu and Jing Xie were responsible for the LNP work. Feng Shi took responsibility for liver-targeting lipid development. Xiaoju Zhang carried out the mRNA work. Bingxu Zhang supervised the nonhuman primate work. Hongya Han and Xiaoyun Ma contributed to the concept and experimental design. Jia Zhang and Shuaibo Shao performed the detection of in vivo samples. Jianqi Zhang, Hongxiaoying Yu, Yongchao Zhao, Jiazheng Jin, Dong Xu, Quanjun Du, and Yifeng Geng were responsible for establishing the animal models and performing the animal experiments. Liang Liu, Jianing Wang, Yu Tan, Lushuai Jin, and Kelu Xu conducted the in vitro functional assays. Andong Liu and Wei Xu co-supervised the study. Yan Huang and Wei Xu wrote the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Friedrich Foerster, Jean-Charles Nault and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s Note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Huang, Y., Liu, S., Zhang, X. et al. Organ-specific delivery of an mRNA-encoded bispecific T cell engager targeting glypican-3 in hepatocellular carcinoma. Nat Commun 16, 11111 (2025). https://doi.org/10.1038/s41467-025-66087-y
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-66087-y
