
Abstract
Ferroptosis, a regulated form of cell death driven by lipid peroxidation, holds promise for targeting treatment-resistant cancer cells. Using a panel of melanoma cell lines, we uncover variability in the timing of ferroptosis onset upon exposure to iron and polyunsaturated fatty acids (PUFAs). This heterogeneity is linked to differences in PUFA sequestration into lipid droplets (LDs) and their subcellular distribution, particularly near lipid-metabolizing organelles such as mitochondria. In late-onset models, ferroptosis is delayed by peripheral LD retention and triggered by nutrient deprivation and AMP-activated protein kinase (AMPK) activation, which promotes LD trafficking toward mitochondria. Early responders bypass this mechanism. Our findings identify nutrient status and LD dynamics as key modulators of PUFA- and iron-induced ferroptosis, offering insights for therapeutic exploitation in cancer.
Similar content being viewed by others
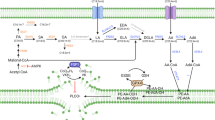


Introduction
The ultimate goal of therapeutic cancer intervention is to eliminate cancer cells with minimal effects on normal healthy tissue. Most current strategies are directed toward the selective induction of regulated cell death (RCD) in cancer cells. For a long time, apoptosis has been considered the primary mechanism of RCD and, therefore, selected as a critical target of many anticancer drugs. Cancer cells, however, often exploit mechanisms to evade apoptosis, leading to poor responses and contributing to therapy resistance1. The recent identification of other RCD mechanisms opens new avenues towards cancer treatment. One of these mechanisms is ferroptosis. Ferroptosis is an iron-dependent cell death propagated by polyunsaturated fatty acids (PUFA) peroxidation. A specific class of ferroptosis inducers, such as Erastin and RSL3, can induce this process. These compounds target the System Xc− cystine/glutamate antiporter SLC7A11 and glutathione peroxidase GPX4, which are involved in redox control and alleviating lipid peroxidation2,3,4,5. Since the discovery that ferroptosis contributes to tumor cell killing by several treatment modalities, including chemotherapy, radiotherapy, and immunotherapy, and that many treatment-resistant cancers remain responsive to ferroptosis inducers6,7,8,9, ferroptosis has come into the spotlight as a potential alternative strategy in cancer therapy and as a way to overcome drug resistance.
Emerging evidence reveals the relationship between melanoma differentiation status, sensitivity to ferroptosis, and lipid metabolism in the context of BRAF inhibition. Differentiated melanoma cells often possess enhanced antioxidant defenses, making them more resilient to ferroptosis despite exposure to BRAF inhibitors. In contrast, undifferentiated melanoma cells exhibit heightened sensitivity due to metabolic vulnerabilities, including altered iron metabolism and ineffective counteraction of oxidative stress10,11,12,13. Lipid metabolism further influences this sensitivity; undifferentiated cells may have impaired regulation of lipid peroxidation, leading to toxic lipid accumulation that triggers ferroptosis. Conversely, differentiated melanoma cells may leverage lipid saturation and storage to reduce oxidative stress effects14,15. Hence, modulation of this balance by increasing the cancer PUFA content may induce a critical vulnerability in undifferentiated cells. This has great therapeutic potential, as these cells have been shown to drive resistance to standard-of-care treatments, including targeted therapy and immune checkpoint blockade16.
In this study, to gain more insight into the mechanisms that contribute to cancer cells’ sensitivity to ferroptosis and to identify critical vulnerabilities that can be exploited in new combination cancer treatments, we examine the effect of iron and PUFA supplementation in a panel of drug-resistant and drug-sensitive melanoma cell lines. Interestingly, the sensitivity of melanoma cells to PUFA/iron-induced ferroptosis varies significantly depending on how they handle supplemented PUFAs. The sequestration of PUFAs within lipid droplets (LDs) and the subcellular distribution of these LDs relative to organelles, such as mitochondria, are key factors influencing the ferroptotic response to iron and PUFA supplementation. Notably, this response is affected by starvation-induced AMPK (AMP-activated protein kinase) activation, LD redistribution, and LD-mitochondria association. These findings highlight the roles of nutrient status and AMPK-mediated LD dynamics as modulators of PUFA- and iron-induced ferroptosis, offering insights into strategies to sensitize therapy-resistant cancer cells to cell death induction.
Results
Differential effect of PUFA and iron supplementation on ferroptosis induction in a panel of biologically diverse melanoma cell lines
To explore to what extent melanoma cells harboring distinct differentiation states and responsiveness to BRAF inhibitors (Table S1) are sensitive to natural inducers of ferroptosis, we exposed a panel of 8 well-characterized melanoma cell lines13,17 to a mixture of arachidonic acid (AA; 20-4 ω−6) and docosahexaenoic acid (DHA; 22-6 ω−3) to induce lipid polyunsaturation. We treated them with ferric ammonium citrate (Fe3+), a physiological form of non-transferrin-bound iron, which causes intracellular iron overload and induces ferroptosis in various biological systems18,19. Measurement of cell death at different time points using the SYTOX Green dead cell stain revealed remarkable differences in the time courses of cytotoxic response induction among the various cell lines, with M257, M229, M238 and M238R being early responders (>50% cell death after 48 h) and M249, M249R, 451Lu and 451LuR responding later or less severely (Fig. 1A). Notably, at the compound concentrations used, only the combination of PUFA and Fe3+ induced significant lethality (Fig. S1A), with no considerable difference in the uptake of PUFA and iron between groups (Fig. S1B). Notably, there was an inverse correlation between the differentiation status of the indicated melanoma cell lines and the time-dependent response to PUFA/Fe3+-induced cell death (Fig. S1C). Dedifferentiated cells with phenotypic plasticity, which confer them an adaptive drug tolerance/resistance, were susceptible to cytotoxicity induced by PUFA/Fe3+ 20,21. No association with resistance to BRAF-targeted treatment was observed.

A Cell death measurement (SYTOX) in melanoma cells treated with PUFA (10 µM DHA and ARA) and Fe3+ (10 µM) at 24, 48, and 72 h. Data represent the mean ± SD from n = 3 independent experiments comprising 3 wells each, analyzed using a two-way ANOVA. B Lipid peroxidation (BODIPY 581/591 C11) in melanoma cell lines +/− PUFA/Fe3+ at 24 h (early responders) and 48 h (late responders). Data represent the mean ± SD from n = 3 independent experiments comprising 3 wells each, analyzed using a two-tailed two-way ANOVA with Dunnett’s test (****P < 0.0001). C Malondialdehyde (MDA) accumulation (TBARS) in M257 cells (24 h) and 451LuR cells (48 h) +/− PUFA/Fe3+. Data represent the mean ± SD from n = 3 independent experiments comprising 3 wells each, analyzed using a two-tailed two-way ANOVA with Šídák correction (****P < 0.0001). D Cell death measurement (SYTOX) in melanoma cell lines +/− PUFA/Fe3+, +/− ferroptosis inhibitors Ferrostatin (5 µM) or iron chelator Deferoxamine (20 µM), apoptosis inhibitor Z-VAD (10 µM), or necroptosis inhibitor Nec-1s (10 µM) at 48 h or 72 h. Data represent the mean ± SD from n = 3 independent experiments comprising 3 wells each, analyzed using a two-tailed two-way ANOVA with Dunnett’s test (****P < 0.0001). E Growth and cell death analysis (SYTOX) of 3D 451LuR tumor spheroids +/− PUFA/Fe3+, +/− Ferrostatin (5 µM) at 72 h. Representative images from n = 3 independent experiments. F Time course (Incucyte ZOOM) of SYTOX staining of 3D 451LuR tumor spheroids +/− PUFA/Fe3+, +/− Ferrostatin (5 µM). Data represent the mean ± SD from n = 3 independent experiments comprising 3 wells each, analyzed using a two-tailed one-way ANOVA (****P < 0.0001). Source data are provided as a Source Data file.
Lipid peroxidation-mediated ferroptosis was supported by the accompanying increase in lipid peroxides, as measured by BODIPY 581/591 C11 staining, and the accumulation of malondialdehyde (MDA), a secondary product of lipid peroxidation (Fig. 1B, C). The induction of cell death was inhibited by either ferrostatin-1 or deferoxamine but not by the apoptosis inhibitor Z-VAD and the necroptosis inhibitor Nec-1s (Fig. 1D). This 2D cell line data was corroborated by monitoring the cytotoxicity of PUFA and iron supplementation in a 3D spheroid model generated from 451LuR cells. Treatment with PUFA/Fe3+ resulted in a gradual increase in SYTOX intensity over time, which was entirely prevented by ferrostatin (Fig. 1E, F).
Sequestration of PUFA into lipid droplets and their differential distribution determines the timing of ferroptosis induction
In exploring the molecular underpinnings of ferroptosis induced by PUFA/Fe3+, RNA sequencing across a comprehensive panel of models revealed that glycerolipid metabolism is among the top three enriched pathways. This finding was accompanied by a notable upregulation of triglyceride (TAG) synthesis genes in treated cells compared to controls (Figs. 2A, S2A). Given the established protective role of TAG-rich lipid droplets22,23,24, we investigated whether the differential sequestration of PUFA into LD-associated neutral lipids, primarily TAGs, contributes to the varying responses observed in melanoma models. Mass spectrometry-based lipidomics revealed that supplementation of cell cultures with dietary PUFA resulted in a limited change in the polyunsaturation degree of membrane phospholipids. In contrast, PUFA-containing neutral lipids, particularly DHA-TAGs and DHA-Cholesterol ester (CEs), increased dramatically across all melanoma cells (Figs. 2B, S2B). Similarly, the unbound fraction of PUFA significantly rose in both groups at an equal rate (Fig. 2C). The preferential partitioning of exogenous PUFA into LDs was further corroborated by Stimulated Raman Scattering (SRS) microscopy using a deuterated isotope of docosahexaenoic acid (DHA-d5), which demonstrated that exogenously administered PUFA was sequestered mainly in LDs rather than in plasma membranes, lysosomes, ER, or mitochondria (Fig. S2C, D). To further investigate the dynamics between saturated and unsaturated fatty acids within lipid droplets upon PUFA supplementation, we assessed changes in two additional Raman wavenumbers. The band around 1660 cm⁻¹, which indicates C=C stretching vibrations, serves as a marker for unsaturated fatty acids, while the 1440 cm⁻¹ band corresponds to the scissoring of the CH2–CH3 banding in saturated carbon chains25. As shown in Fig. S2E, F, signals for both saturated and unsaturated fatty acids were co-localized with lipid droplets. Notably, the ratio of unsaturated to saturated fatty acids increased following PUFA treatment compared to the control condition in both early and late-responder models (M257 and 451LuR). However, the difference in unsaturation levels between M257 and 451LuR was not statistically significant.

A KEGG enrichment analysis of significantly differentially expressed genes (RNA-seq) in melanoma cell lines (M257, M238, 451Lu, 451LuR) +/− PUFA/Fe3+ (18 h, 36 h). padj <0.001, n = 3 different wells, analyzed using a two-tailed DESeq2 with Benjamini–Hochberg correction. B Lipidomics profiling of phospholipids (PC, PE, PG, PI, PS) and neutral lipids (CE, TAG, DAG) in melanoma cell lines +/− PUFA/Fe3+. Data represent the mean ± SD from n = 3 different wells. C Measurement of free DHA and ARA in early- and late-responding models (ER and LR, respectively) +/− PUFA/Fe3+ (24 h). Box plot showing median of n = 3 independent experiments, analyzed using a two-tailed unpaired t-test (FC = Fold change). D Lipid droplet (LD) density (BODIPY 493/503 staining) in melanoma cells +/− PUFA/Fe3+ (24 h and 48 h). n = 10 images, analyzed using a two-way ANOVA with Dunnett’s test. E Lipid droplet (LD) (BODIPY 493/503, green) and nuclei (Nucstain, blue) staining of melanoma cells +/− PUFA/Fe3+ (24 h). Representative images from n = 10. F Quantitative analysis of LD distribution (BODIPY 493/503) in melanoma cell lines. Mean ± SD of n = 3 independent experiments (n > 100 cells/condition). Two-way ANOVA with Šídák correction. G, H Effect of DGAT inhibitors T863 and PF-06424439 on cytotoxicity (SYTOX) and lipid peroxidation (BODIPY 581/591 C11) in melanoma cell lines +/− PUFA/Fe3+. Mean ± SD from n = 3 independent experiments comprising 3 wells each, analyzed using a two-way ANOVA with Dunnett’s (****P < 0.0001). I Effect of DGAT KD compared to control (ND KO) M257 and 451LuR cells on cell death induction (SYTOX) by PUFA/Fe3+ (72 h). Mean ± SD from n = 3 independent experiments comprising 3 wells each, analyzed using a two-way ANOVA with Šídák correction (****P < 0.0001). J Effect of KIF5B (48 h), Spastin (SPAST) (72 h) KD versus control (NC KO) on LD distribution (BODIPY 493/503) in 451LuR after PUFA/Fe3+ treatment. n = 10. K, L Impact of KIF5B (48 h), Spastin (SPAST) (72 h) KD versus control (NC KO) on lipid peroxidation (C11BODIPY) and cell death (SYTOX) at 72 h. Mean ± SD from n = 3 independent experiments comprising 3 wells each, analyzed using a two-way ANOVA with Dunnett’s test (****P < 0.0001). Source data are provided as a Source Data file.
Fluorescent staining of LDs with BODIPY 493/503 (green) revealed a substantial increase in LD accumulation across all cell lines, irrespective of group classification (Fig. 2D). Consistent with RNA sequencing data indicating differential, time-dependent regulation of TAG synthesis genes, the analysis of 100 cells per cell line demonstrated distinct temporal dynamics in LD synthesis. Specifically, early responders (ER) showed a significant decrease in LD synthesis after 24 h, while late responders (LR) maintained a stable synthesis rate even at the 48-h mark (Fig. S2A, D). Further analysis of the BODIPY 493/503 fluorescence images revealed unique LD distribution patterns among the different models. Early responders presented numerous small, punctate LDs evenly distributed throughout the cell, with a notable concentration in the perinuclear region. In contrast, late responders showed a tendency for bigger LDs to cluster at the cell periphery (Fig. 2E, F). Interestingly, over time, these clustered LDs gradually dispersed into smaller droplets and migrated toward the perinuclear area (Fig. S2G, H). This observation suggests a dynamic regulation of LD distribution that may have important implications for cellular metabolism.
To investigate the role of lipid droplets in initiating ferroptosis, we modulated the accumulation of neutral lipids in these droplets by inhibiting LD biogenesis. Specifically, our study involved blocking diacylglycerol acyltransferases (DGAT1i T863 and DGAT2i PF-06424439) and utilizing RNAi-mediated knockdown of DGAT1 and DGAT2, which effectively eliminated LD formation (Figs. S2I, J, S6A). This intervention, particularly using the DGAT2 knockdown (KD) model, partially rescued cells from PUFA/Fe3+-induced lipid peroxidation and cell death, particularly in late responders (Figs. 2G–I, S2K, S2L). This observation aligns with our lipidomics data (Fig. S2M) and highlights the role of LDs in modulating cellular responses to PUFAs in melanoma models, particularly among late responders.
To clarify the causal relationship between lipid droplet (LD) distribution and sensitivity to ferroptosis, especially in late responders, we investigated LD movement by knocking down two genes: KIF5B, a motor protein that facilitates LD transport to the periphery in nutrient-rich conditions26,27, and spastin (SPAST gene), which facilitates inward dispersion of LDs during glucose starvation28. Image analysis revealed that KIF5B knockdown decreased LD clustering, while spastin knockdown increased it in 451LuR cells exposed to PUFA/Fe3+ (Figs. 2J, S6G). These changes in LD distribution were linked to significant alterations in lipid peroxidation and ferroptosis-related toxicity, as shown in Fig. 2K, L. Overall, these findings highlight that modulating LD dynamics and distribution significantly affects ferroptosis sensitivity in late-responder cells.
Energy stress-mediated AMPK activation promotes ferroptotic cell death by affecting lipid droplet dynamics
Given the well-established role of LDs as energy reservoirs, we investigated whether their differential and time-dependent translocation in late responders was linked to the cells’ energy demands. Quantification of oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) revealed significantly higher activity in both oxidative phosphorylation (OXPHOS) and glycolysis pathways in late responders compared to early responders. Early responders, however, exhibited a lower overall bioenergetic capacity, which was slightly reduced by PUFA/Fe3+ exposure in both groups (Fig. 3A, B). This insight, combined with the gradual hydrolysis of PUFA-TAG over time (Fig. S3A), indicates that the varying adaptability of LDs in late responders is intricately connected to spatial energy metabolism and the onset of starvation processes. To further explore the impact of starvation and glucose depletion on LD translocation and ferroptosis induction, we treated M257 and 451LuR cells with PUFA/Fe3+ and replenished the culture medium with or without glucose after 20 h. Glucose deprivation led to decreased density of LD and increased cytotoxicity, whereas glucose supplementation notably alleviated cytotoxic effects in 451LuR late-responder cells but not in M257 early model (Fig. S3B, C).

A Heatmap of metabolic parameters (Seahorse) at baseline vs PUFA/Fe3+ in melanoma cells (n = 3 independent samples). B OXPHOS vs glycolytic parameters in melanoma cells at baseline. Mean ± SD from n = 3 independent experiments comprising 3 wells each. (OCR: oxygen consumption rate). C Cell death (SYTOX) in M257 (48 h) and 451LuR (72 h) after 20 h PUFA/Fe3+ treatment and medium refreshment +/− glucose (Glu). Mean ± SD from n = 3 independent experiments comprising 3 wells each, two-way ANOVA with Dunnett’s test (****P < 0.0001). D Western blot of P-AMPK (MW: 62 kDa, red: 70 kDa), total AMPK (MW: 62 kDa, red: 70 kDa), GAPDH (MW: 37 kDa, blue: 35 kDa) in 451LuR +/− PUFA/Fe3+, +/− AMPK activators (A769662, EX-229), ComC at 24/48 h (L = Ladder). n = 3 independent experiments. E, F Effect of AMPK knockout (AMPK KO) versus control (AAVS KO) on cell death induction (SYTOX) and lipid peroxidation (BODIPY) by PUFA/Fe3+ in 451LuR cells +/− ZVAD, NEC-1S or Ferrostatin. Mean ± SD from n = 3 independent experiments comprising 3 wells each, two-way ANOVA, with Šídák (****P < 0.0001). G, H Effect of AMPK modulators Compound C (ComC), A769662, and glucose deprivation -Glu) on lipid peroxidation (BODIPY) and cell death induction (SYTOX) by PUFA/Fe3+ in 451LuR. Mean ± SD from n = 3 independent experiments comprising 3 wells each, analyzed using a one-way ANOVA with Dunnett’s test (****P < 0.0001). I, J Effect of MUFA (oleate) supplementation on lipid peroxidation (BODIPY) and cell death induction (SYTOX) by PUFA/Fe3+ in 451LuR. Mean ± SD from n = 3 independent experiments comprising 3 wells each, one-way ANOVA with Dunnett’s test (****P < 0.0001). K Effect of AMPK modulators (EX-229, Compound C, and AMPK KO) on lipid droplet (LD) accumulation (BODIPY 493/503) in 451LuR cells. Representative images from n = 10. L Lipid droplet (LD) size quantification (ImageJ 2.0) in 451LuR cells +/− PUFA/Fe3+ +/− EX-229. Scatter dot plot indicating the median from n = 15 images in 3 independent experiments, analyzed using one-way ANOVA with Dunnett’s test (****P < 0.0001). M Effect of AMPK activators A769662 or glucose deprivation on TAG levels in 451LuR cells +/− PUFA/Fe3+ (n = 3 independent wells). N, O Effect of AMPK modulators A769662 and Compound C on PUFA/Fe3+-induced cytotoxicity (SYTOX) in 3D 451LuR spheroids (SYTOX). Mean ± SD from n = 3 independent experiments comprising 3 wells each, analyzed using a two-way ANOVA (****P < 0.0001). Source data: Source Data file.
In response to starvation, our RNA sequencing data revealed activation of AMPK signaling, particularly highlighted by increased expression of PRKAA1 (AMP-activated protein kinase) mRNA in late responders. Notably, this expression was further upregulated following PUFA treatment compared to early responders (Fig. S3C). Although we did not detect significant changes in the levels of phosphorylated AMPK (P-AMPK) or total AMPK protein among the groups (Fig. S3D, E), the observed upregulation of the mTOR gene and downregulation of both downstream targets of the AMPK pathway suggest that these cells have an enhanced ability to adapt metabolically under stress. This adaptability is further supported by the observed time-dependent upregulation of AMPK, which coincides with LD hydrolysis (Figs. 3D, S3F).
To demonstrate AMPK’s role in inducing ferroptosis, CRISPR-Cas9-mediated AMPK knockout significantly diminished PUFA/Fe3+-induced lipid peroxidation and cell death and increased AMPK KO proliferation (Figs. 3E, F, S3G). In contrast, activating AMPK with the activator A769662 increased lipid peroxidation and intensified PUFA/Fe3+-induced cytotoxicity, mimicking the effects of glucose deprivation. The AMPK inhibitor Compound C replicated the effects of the knockout (Fig. 3G, H). Notably, AMPK activation did not promote ferroptosis in 451LuR cells supplemented with monounsaturated fatty acid (MUFA), specifically oleate, rather than PUFA, highlighting the specific role of PUFA in this process (Fig. 3I, J). Similar outcomes were observed in other late-responder cell lines (Fig. S3H, I).
To further elucidate the mechanism by which AMPK mediates late-onset ferroptosis, we assessed the effect of AMPK modulation on the distribution of LDs. Confocal microscopy imaging revealed that depletion of AMPK, either pharmacologically or genetically, reinforced the clustering of bigger LDs in the peripheral extremities of 451LuR cells and prevented their dispersion in the cell body (Figs. 3K, S3J, K, S6B, C), while AMPK activation by EX-229 evoked the redistribution of LDs toward the cell center. Moreover, AMPK activation led to a significant increase in the size of lipid droplets and enhanced their subsequent hydrolysis over time (Figs. 3L, M, S3L, M). These effects were replicated in 3D 451LuR tumor spheroids treated with A769662 and Compound C (Fig. 3N, O). Collectively, these findings link lipid droplet translocation with energy demand and starvation, highlighting the role of AMPK activation in enhancing ferroptosis post-PUFA/Fe3+ supplementation.
Activated AMPK boosts LD-MT tethering through detyrosinated tubulin
Since starvation-mediated AMPK activation enhances both the dispersion and hydrolysis of LDs, we explored potential differential interactions between LDs and fatty acid-metabolizing organelles, such as mitochondria, peroxisomes, and lysosomes, across different groups. Confocal imaging of M257 and 451LuR cells with antibodies against mitochondria (MT), peroxisomes (PER), and lysosomes (LYS) revealed a time-dependent distribution pattern and organellar association of LD-associated PUFAs (Fig. 4A). In early responder cells, LDs localized closely with mitochondria and, to a lesser extent, with peroxisomes at early time points (12 h) after PUFA loading. Conversely, in late responders, LDs initially localized far from mitochondria and peroxisomes, only redistributing towards mitochondria-rich areas at later time points (48 h). Image-based quantification confirmed this time-dependent shift in LD-MT colocalization in late-responder cells (Fig. 4B). Live imaging using SRS-confocal microscopy of 451LuR cells treated with DHA-d5 and Fe3+ during starvation, co-stained with Mitotracker, CellLight Peroxisome-GFP, and Lysotracker, was consistent with fixed imaging results. DHA-d5 initially localized to lipid droplets at the cell periphery, subsequently migrating towards the perinuclear region and eventually overlapping with mitochondrial areas, particularly upon AMPK activation (Fig. S4A). Treatment with Etomoxir, a CPT1 inhibitor, reduced DHA-d5 accumulation in mitochondria in 451LuR cells at the starvation time point (48 h), with a slight increase in localization towards peroxisomes and lysosomes (Fig. S4B, C). Transmission electron microscopy (TEM) further revealed numerous close LD-MT appositions under stimulated conditions (Fig. 4C). TEM images revealed increased electron density in PUFA-loaded LDs, likely due to enhanced osmium staining associated with the unsaturated nature of DHA. Notably, upon AMPK activation, the lipid droplets appeared darker. This is likely attributable to the higher PUFA content, including DHA, which resulted in greater osmium binding and, consequently, increased electron density. Most dispersed LDs were in contact with mitochondria, and quantification confirmed that AMPK activation increased these LD-MT contact sites upon PUFA loading (Fig. 4D). This proximity promoted mitochondrial morphological changes, including decreased cristae and swollen mitochondria, particularly under ferroptosis-exacerbating conditions triggered by AMPK activation (Fig. 4E).

A Time-dependent redistribution of LDs and colocalization with lipid-metabolizing organelles (MT; mitochondria, PER; peroxisomes, LYS; lysosomes) in M257 and 451LuR cells + PUFA/Fe3+ (BODIPY 493/503 (green) for LDs; anti-TOMM20 (Mitochondria), anti-PMP70 (Peroxisome), anti-Lamp2A (Lysososme); Nucstain (blue)). Representative images from n = 10 from 3 independent experiments. B Colocalization analysis of lipid droplets (LD) with mitochondria (MT), peroxisomes (PER), and lysosomes (LYS) using NIS-Elements AR 5.21 (FC = Fold change). n = 10 images. C TEM images of LD-MT contacts (<70 nm, arrows) in 451LuR cells +/− PUFA/Fe3+, +/− EX-229 (48 h). Representative images from n = 10 from 2 independent experiments. D Quantification of LD-MT contacts (<70 nm) using ImageJ 2.0. A scatter dot plot shows the median. Two-tailed one-way ANOVA. n = 10 images from 2 independent experiments. E TEM images of mitochondrial morphology of 451LuR cells +/− PUFA/Fe3+ (48 h). F Western blot analysis and quantification of detyrosinated tubulin (DT-Tubulin) (MW: 55 kDa, blue: 55 kDa) and GAPDH (MW: 37 kDa, blue: 35 kDa) in 451LuR cells loaded with PUFA/Fe3+, +/− AMPK activator A769662 at 24 h and 48 h (L = Ladder). Box plots show the median from 2 independent experiments, analyzed using a two-tailed one-way ANOVA with Šídák correction. Uncropped blots in Source Data. G Schematic of Parthenolide action (TCP tubulin carboxypeptidase, TTL tubulin–tyrosine ligase). Created in BioRender. https://BioRender.com/zssov4o. H Confocal images of LD (BODIPY 493/503, PLIN2), MT (anti-TOMM20), anti-detyrosinated tubulin (DT-Tubulin), and nuclei (NucBlue) in 451LuR cells (48 h) treated with PUFA/Fe3+, +/− A769662, +/− tubulin carboxypeptidase inhibitor Parthenolide. Representative images from 2 independent experiments. I LD-MT colocalization quantified by NIS-Elements AR 5.21. The scatter dot plot shows the median from n = 10 images from 2 independent experiments, analyzed using a one-way ANOVA with Dunnett’s. J, K Effect of AMPK activators (A769662 and EX-229) and tubulin carboxypeptidase inhibitor Parthenolide on PUFA/Fe3+-induced lipid peroxidation (BODIPY 581/591 C11) and cell death (SYTOX) in 451LuR. Data represent the mean ± SD from n = 3 independent experiments comprising 3 wells each, analyzed using a one-way ANOVA with Dunnett’s test (****P < 0.0001). Source data are provided as a Source Data file.
Building on earlier observations that induced AMPK to relocate LDs and mitochondria on detyrosinated microtubules, enhancing mitochondrial oxidation of LD-PUFA, we examined the impact of dietary PUFA and AMPK activator on tubulin termination. RNA sequencing analysis revealed significant differences in the expression of tubulin-related genes between the two groups, with VASH1—encoding a tyrosine carboxypeptidase (TCP) responsible for tubulin detyrosination—showing higher levels in late responders compared to early responders, particularly when treated with PUFA/Fe3+ (Fig. S4D, E).
PUFA/Fe3+ treatment showed a temporal downregulation of detyrosinated tubulin, followed by upregulation upon starvation or treatment with the AMPK activator A769662 (Fig. 4F). Interestingly, Parthenolide (PTN), an inhibitor of tubulin carboxypeptidase (TCP) (Fig. 4G), blocked LD dispersion and its interaction with mitochondria, partially reducing lipid peroxidation and rescuing cells from cytotoxicity in both groups (Figs. 4H–K, S4F). In summary, these findings underscore the crucial interplay between LDs and mitochondria in PUFA-induced ferroptosis.
Channeling of PUFAs between lipid droplets and mitochondria facilitates the induction of ferroptosis
Given the proximity of LDs and mitochondria, which facilitates the transfer of PUFAs into mitochondria, along with the observed accumulation of supplemented iron in mitochondria across both cell groups (Fig. S5A), we investigated whether mitochondria play a role in ferroptosis-induced cytotoxicity. SRS imaging-colocalization results and the upregulation of mRNA expression for genes involved in LD-mitochondria interaction (PLIN2, PLIN5), mitochondrial tethering (MFN1, MFN2), lipid import into mitochondria (CPT1), and beta-oxidation (ACADL) upon PUFA/Fe3+ loading (Figs. 5A, S5B) indicate a potential role of mitochondria. In contrast, genes involved in lysosomal or peroxisomal lipid metabolism (LAMP2, ACOX2, PEX5) were not affected by PUFA/Fe3+ treatment.

A mRNA expression of proteins involved in lipid droplet/mitochondria/peroxisome/lysosome interactions in 451LuR +/− PUFA/Fe3+ (48 h) (n = 3 different wells in one experiment). B Effect of Etomoxir, Antimycin A, and MitoTempo on PUFA/Fe3+-induced cell death induction (SYTOX) in 451LuR cells (72 h). Mean ± SD from n = 3 independent experiments comprising 3 wells each, analyzed using a one-way ANOVA with Dunnett’s test (****P < 0.0001). C Effect of CPT1 KO versus control (AAVS KO) on PUFA/Fe3+-induced expression of enzymes in mitochondrial fatty acid oxidation (flow cytometry). Mean ± SD from n = 3 independent experiments comprising 3 wells each, analyzed using a two-way ANOVA with Dunnett’s test. D, E Effect of CPT1 KO versus control (AAVS KO) on PUFA/Fe3+-induced mitochondrial lipid peroxidation (MitoPerOx) and cell death (SYTOX) in 451LuR cells. Mean ± SD from n = 3 independent experiments comprising 3 wells each, analyzed using a two-way ANOVA with Šídák correction (****P < 0.0001). F Imaging of TMRE (red) and SYTOX (green) of 451LuR cells +/− PUFA/Fe3+, +/− mitochondrial depolarizer carbonyl cyanide chlorophenylhydrazone (CCCP). n = 4 independent wells, Representative experiment of the 3 performed. different wells. G, H Quantification of SYTOX (green) and TMRE (red) (Incucyte ZOOM) in 451LuR cells +/− PUFA/Fe3+, +/− CCCP. Mean ± SD from n = 4 independent wells, analyzed using a two-tailed two-way ANOVA with Dunnett’s test (****P < 0.0001). I, J Mitochondrial lipid peroxidation (MitoPerOx) and cell death (SYTOX green) in 451LuR cells with AMPK activator (A76966 or EX-229) and MitoTempo (48 h). Mean ± SD from n = 3 independent experiments comprising 3 wells each, analyzed using a one-way ANOVA with Dunnett’s test. K Mitochondrial oxygen consumption rate (OCR) in 451LuR cells +/− PUFA/Fe3+, +/− AMPK activator A769662 (48 h). n = 5 different wells, Representative curve from n = 2 independent experiments. L Effect of AMKP modulators A769662 and Compound C (ComC) on Mitochondrial membrane potential (TMRM) in 451LuR cells treated with PUFA/Fe3+ (36 h). Data represent the mean ± SD from n = 3 independent experiments, each comprising 3 wells, analyzed using a one-way ANOVA with Dunnett’s test. M, N Effect of DGAT inhibition, DGATi, MitoTempo, AMPK activator (A76966), and Parthenolide on PUFA/Fe3+-induced cytotoxicity in 3D 451LuR spheroids (SYTOX). Data represent the mean ± SD from n = 3 independent experiments comprising 3 wells each. Two-tailed one-way ANOVA. Source data are provided as a Source Data file.
To further corroborate the involvement of mitochondria, we explored whether mitochondrial inhibition would impact sensitivity to ferroptosis in this group. Treatment with Etomoxir, which reduced the colocalization of PUFAs in mitochondria, also diminished cell death induction (Figs. 5B, S5C). Similarly, Antimycin A, an inhibitor of complex III of the oxidative phosphorylation pathway, and MitoTempo, a mitochondrial-targeted antioxidant, reduced cell death induction in 451LuR cells but not in M257 cells. Inhibitors of peroxisomal oxidation (3AT), lysosomal activity (CQ), or COX activity (LOR) had little effect individually but showed impact when combined with MitoTempo (Fig. S5D). In contrast to 451LuR cells, combined inhibition of all organelles, including lysosomes and mitochondria, provided only partial protection against PUFA/Fe3+-induced cytotoxicity in M257 cells (Fig. S5D).
Supporting the involvement of mitochondrial lipid utilization, CRISPR/Cas9-mediated knockout of CPT1 decreased the expression of enzymes involved in beta-oxidation (Figs. 5C, S6D, E), reduced the mitochondrial PUFA peroxide levels in 451LuR cells (Fig. 5D), and decreased their susceptibility to PUFA/iron-induced cell death (Fig. 5E). To further validate the role of mitochondria in PUFA-induced ferroptosis, we depleted mitochondrial function in 451LuR cells with the mitochondrial depolarizer carbonyl cyanide chlorophenylhydrazone (CCCP). We monitored mitochondrial function and cell death using SYTOX and tetramethylrhodamine ethyl ester (TMRE), which are sequestered by active mitochondria. Cell time-lapse imaging revealed that CCCP-induced mitochondrial dysfunction significantly reduced PUFA-mediated ferroptotic sensitivity (Fig. 5F–H).To further evaluate mitochondrial function, we measured mitochondrial lipid peroxidation, membrane potential, and oxidative phosphorylation (OCR). The AMPK activator significantly enhanced mitochondrial lipid peroxidation, as evidenced by the MitoPerOx probe and colocalization of oxidized BODIPY 581/591 C11 with MitoTracker (Figs. 5I, S5E, F). This was accompanied by a decreased oxygen consumption rate (OCR) and reduced mitochondrial membrane potential upon co-incubation of 451LuR cells with PUFA/Fe3+ (Fig. 5K, L). These data, alongside increased levels of mitochondrial TCA metabolites such as citric acid (Fig. S5G), indicate that mitochondrial perturbations are linked to ferroptosis induction. The observed decrease in mitochondrial functionality corresponded with increased cell death, which could be mitigated by MitoTempo (Fig. 5J). Inhibition of lysosomal autophagy (and the endosomal pathway) and peroxisomal activity with chloroquine and 3-Amino-1,2,4-triazole (3-AT) did not significantly impact lipid peroxidation or cell death following AMPK activation (Fig. S5H, S5I). Similar results were observed when 451LuR cells in 3D culture were co-treated with modulators of AMPK, LD, and mitochondrial function (Fig. 4M, N). Together, these observations suggest that the mitochondrial interaction with LDs is pivotal in facilitating lipid peroxidation and ferroptosis, making mitochondria a key target for modulating ferroptosis-related cell death.
ER-MT-LD membrane contact site is a potential hub for lipid peroxidation
Given the relocation of LDs towards mitochondria triggered by AMPK activation, we explored the temporal mobilization of PUFAs from TAG to other lipid classes more profoundly using tracer lipidomics. Remarkably, our observations revealed that 13C-labeled DHA initially localized in TAGs but eventually accrued in phosphatidylcholine (PC) under conditions of nutrient deprivation (Fig. 6A). Notably, inhibiting DGAT resulted in diminished accumulation of DHA in both TAG and PC. Given the pivotal role of PC-PUFAs in mitochondrial homeostasis29,30,31, our subsequent analysis aimed to explore whether this lipid remodeling led to a heightened pro-ferroptotic lipid composition within the mitochondrial membranes. Lipidomics analysis of mitochondrial membrane fractions 48 h post-PUFA/Fe3+ supplementation revealed an increased normalized incorporation of PUFAs into mitochondrial PC, particularly upon AMPK activation (Fig. 6B). This was accompanied by an increase in lipid peroxidation at the level of the mitochondrial membrane, as revealed by BODIPY 581/591 C11 staining of mitochondria in cells transfected with the mCherry-TOMM20 plasmid, which stains the mitochondrial membrane, as observed by super-resolution microscopy (Fig. 6C, D).

A 13C-DHA incorporation into PC and TAG in 451LuR +/− PUFA/Fe3+ +/− DAGT inhibition (DGATi) at 24/48 h (n = 3 independent wells). (PC = phosphatidylcholine, TAG = triglyceride). B Lipidomics of mitochondrial membrane TAG, DAG, PC, PE in 45LuR cells +/− PUFA/Fe3+ +/− AMPK activator EX-229 (48 h). Data represent the mean ± SD from n = 3 independent wells, analyzed using a two-way ANOVA with Dunnett’s test. (DAG = Diacylglycerol, PE = Phosphatidylethanolamine). C SIM images of oxidized BODIPY 581/591 C11 and mCherry-TOMM20 (outer mitochondrial membrane) in 451LuR cells treated with PUFA/Fe3+ and EX-229. Representative image from n = 10. White line, location of line profile in (D). D Line profile of oxidized BODIPY 581/591 C11 and mCherry-TOMM20 (48 h PUFA/Fe3++ EX-229). E Schematic of the CDP-DAG pathway of PUFA incorporation into mitochondrial membranes. Created in BioRender. https://BioRender.com/katmuk5. F TEM of LD-MT contacts in 451LuR cells + PUFA/Fe3+. Arrows indicate ER tubules. Representative image from n = 10. G Effect of CHPT1, CEPT1, MFN1, and MFN2 knockdown (KO) versus control (NC) on DAG levels in isolated mitochondrial membranes from 451LuR 48 h after PUFA/Fe3+ treatment. Data represent mean ± SD from n = 3 independent wells, analyzed using two-tailed two-way ANOVA with Dunnett’s test. H, I Effect of CHPT1, CEPT1, MFN1, and MFN2 knockdown (KD) versus control (NC) on mitochondrial lipid peroxidation (MitoPerOx) and cell death (SYTOX) in 451LuR +/− PUFA/Fe3+. Data represent mean ± SD from n = 3 independent experiments comprising 3 wells each, analyzed using a two-tailed one-way ANOVA (****P < 0.0001). J Live images of effects of CHPT1 and MFN2 knockdown (KD) versus control (NC) on the distribution of lipid droplets (BODIPY 493/503), mitochondria (MitoTracker FM deep red), and mitochondrial membrane potential (TMRE). Arrows indicate peripheral LD locations. Representative image from n = 10. K, L Effects of CHPT1 and MFN2 knockdown (KD) versus control (NC) on TMRE intensity (Median from n = 15 images) and MT-LD colocalization in 451LuR based on immunofluorescence stainings as in (J). Scatter dot plot shows the median from n = 6 images, analyzed using a two-tailed one-way ANOVA (****P < 0.0001). Source data are provided as a Source Data file.
It is known that the transfer of PUFAs from TAGs to PC in mitochondrial membranes is mediated by a network of proteins at the interface of LDs, ER, and mitochondria32,33,34,35,36 (Fig. 6E). This involves inter-organellar tether proteins, including PLIN1, MFN1, and MFN2, which are upregulated upon PUFA/Fe3+ treatment (Fig. 5A), as well as enzymes involved in PC synthesis, i.e., choline phosphotransferase 1 (CHPT1) and choline/ethanolamine phosphotransferase 1 (CEPT1) (Fig. 6E). Transmission electron microscopy confirmed a close interaction between these organelles upon PUFA/Fe3+ treatment (Fig. 6F). To explore the role of these interactions in PUFA-mediated ferroptosis, we suppressed the expression of CHPT1, CEPT1, MFN1, and MFN2. Notably, knockdown of CHPT1 and MFN2 led to the accumulation of PUFA-DAG, an increase in lipid droplets at the cell periphery away from mitochondria, restoration of membrane potential (as measured by TMRE fluorescence), reduced mitochondrial lipid peroxidation (MitoPerOx), and protection of PUFA/Fe3+-induced cell death (SYTOX) (Figs. 6G–L, S6F). Collectively, these findings indicate that the mitochondrial membrane, particularly at contact sites with the ER and lipid droplets, serves as a correlative hotspot for ferroptosis induction after PUFA/Fe3+ treatment.
Discussion
Given the well-established hallmark of many cancers to evade apoptosis, the induction of ferroptosis has emerged as an exciting and promising alternative strategy to treat tumors that progress despite standard-of-care therapies37,38. Here, to further explore the efficacy of combining PUFA and iron supplementation in inducing ferroptosis, we observed significant differences in the timeline of ferroptosis induction in a panel of diverse pre-clinical melanoma cell lines. Interestingly, we established a link between these differences and the differential sequestration of PUFA in LDs and their subcellular distribution, depending on the cell’s energy and metabolic status, as shown in Fig. S7. We demonstrated that low-energy early responder cells—often resembling undifferentiated melanoma phenotypes—rely more on lipid oxidation for energy. These cells possess a higher fraction of free PUFA, with PUFA-loaded LDs positioned near fatty acid-utilizing organelles such as mitochondria and peroxisomes. This proximity allows for efficient consumption of fatty acids (FAs) for oxidation, resulting in early sensitivity to ferroptosis. In contrast, late responders, which more often represent differentiated melanoma cells, exhibit elevated glycolysis rates and reduced dependence on lipid oxidation. They sequester exogenous PUFA in remotely clustered LD complexes, limiting accessibility to lipid-metabolizing organelles. Only when nutrient depletion occurs do these LDs translocate toward mitochondria, leading to late-onset ferroptosis. Only when the media becomes nutrient-depleted and the cells starve later do LDs translocate toward the MT, resulting in late-onset ferroptosis. The central involvement of LDs in this process is illustrated by the finding that PUFA/Fe3+-mediated ferroptosis is partially reversed by inhibitors of LD formation in both early and late ferroptotic cells. These findings support a dual role for LDs by functioning both as a buffer to prevent cytosolic lipotoxicity of exogenous PUFA39,40,41 and as a reservoir of PUFAs for phospholipid synthesis, consequently increasing membrane oxidative damage, as demonstrated in our study and as corroborated by previous research29. Our lipidomics data confirmed that TAG-associated PUFA in LD is converted into mitochondrial PC upon time-dependent starvation and PUFA/Fe3+-exposure, leading to mitochondrial stress, membrane lipid peroxidation, and depolarization. In further support of this concept, inhibition of CHPT1, a significant player in the Kennedy pathway involved in PC synthesis, increased DAG accumulation and attenuated the induction of cell death. These data imply a role for PUFA-PC as a proximal lipid mediator triggering ferroptosis, which is in line with a recent report by Qui et al.29 and prior studies show that exogenous PUFAs impact mitochondrial structure, oxidative phosphorylation, and the concentration of unsaturated lipids42,43. In further support of the transfer of PUFA from LD-TAG to MT-PC, the knockdown of critical players in LD-ER-MT contacts reduces the ferroptosis-inducing potential of PUFA/Fe3+ administration. Notably, although proteins like PLIN2 and PLIN5 were upregulated at the lipid droplet-mitochondria interface, specifically knocking down MFN2 significantly reversed the ferroptotic phenotype. While initially known for its function in mitochondrial fusion, MFN2 plays diverse roles, including its involvement in mitochondrial autophagy (mitophagy), mitochondrial movement, and lipid exchange processes. It acts as a connector between organelles such as MT, ER, and LD44. Recent research has associated MFN2 with increased susceptibility to ferroptosis induction in various diseases, in part through ER-MT contacts44,45,46,47. The pivotal role of mitochondria and their membranes as the prime site of the late-onset ferroptotic process is underscored by our findings that ferroptosis is inhibited by the CCCP depolarizer, blocking PUFA transfer, disrupting the ETC chain, or adding mitochondrial antioxidants.
Our findings highlight the pivotal role of AMPK in regulating ferroptosis induction. Under conditions of energy stress, AMPK enhances cellular resilience against ferroptosis, enabling tumor cells to thrive in challenging environments. Contrarily, Song et al. have shown that AMPK, through BECN1 phosphorylation, actively promotes ferroptosis by forming a complex with SLC7A11, thereby inhibiting system Xc- activity. Our results are consistent with this notion, demonstrating that AMPK activation—induced by either glucose deprivation or AMPK activators—leads to tubulin detyrosination. This, in turn, increases LD intensity, enlarges LD size, and accelerates LD redistribution toward mitochondria.
AMPK is crucial in linking mitochondrial activity with ferroptosis. Activation of AMPK enhances mitochondrial function and mitophagy, facilitating the breakdown of PUFAs and increasing the availability of substrates for peroxidation. Thus, AMPK activation modulates ferroptosis sensitivity, particularly in contexts involving dietary PUFAs. Our data suggest that AMPK activation influences the dynamics of contact sites between lipid droplets, mitochondria, and the ER, which are essential for lipid exchange and metabolism.
Furthermore, the differentiation status of melanoma cells, as mediated by key regulatory factors such as MITF (Microphthalmia-associated transcription factor), significantly influences their metabolic profiles and ferroptosis sensitivity. MITF is integral to maintaining the differentiated phenotype of melanoma, regulating melanin production and melanosome biology. Differentiated melanoma cells, characterized by high MITF expression, tend to demonstrate enhanced resistance to ferroptosis through increased melanin synthesis and a higher ratio of MUFA/PUFA, which can mitigate oxidative stress effects and help regulate iron levels within cells48,49. Conversely, undifferentiated melanoma cells, often marked by lower MITF activity, are more susceptible to ferroptosis, potentially due to their reliance on free PUFAs and altered lipid metabolism, which render them vulnerable to lipid peroxidation.
The interplay between differentiation, BRAF inhibition, and ferroptosis sensitivity raises intriguing questions about the potential for targeted therapies that exploit the metabolic vulnerabilities associated with differentiation status. For instance, how might therapies that manipulate MITF and tyrosinase expression impact lipid metabolism and ferroptosis sensitivity? Additionally, understanding how differentiated and undifferentiated melanoma cells respond to BRAF inhibitors in terms of lipid dynamics and ferroptosis could provide valuable insights for developing combinatorial treatment strategies aimed at overcoming resistance in aggressive melanoma subtypes. Ultimately, elucidating these connections may enhance our ability to design effective therapeutic interventions that harness the unique characteristics of melanoma differentiation and lipid metabolism in inducing ferroptosis.
Methods
This research was conducted in accordance with all applicable ethical guidelines and regulations under SBB approval 219 2017/0767.
Cell culture
Cell lines M257, M229, M238, M238R, M249, M249R, 451Lu, and 451luR were kindly provided by Professor A. Ribas and R. Lo. All cell lines were propagated in DMEM High Glucose (Sigma-Aldrich), supplemented with 10% FBS (Gibco) and 4 mM glutamine (Thermo Fisher Scientific), and were periodically tested for mycoplasma contamination. Treatments were performed in DMEM High Glucose, supplemented with 2% FBS and the following chemicals: Docosahexaenoic acid (10 μM, Sigma-Aldrich), arachidonic acid (10 μM, Sigma-Aldrich), Docosahexaenoic Acid-d5 (10 μM, Sanbio), Docosahexaenoic Acid Alkyne (10 μM, Sanbio), Ferric ammonium citrate (10 μM, Sigma-Aldrich), Ferrostatin (5 μM, Sigma-Aldrich), Liproxtatin-1 (5 μM, Sigma-Aldrich), Z-VAD (10 μM, Sigma-Aldrich), Nec-1s (10 μM, Sigma-Aldrich), T863 (10 μM, S Sigma-Aldrich), PF-06424439 (10 μM, Sigma-Aldrich), Etomoxir (10 μM, Cayman), Antimycin A (1 μM, Sigma-Aldrich), Oligomycin (10 μM, Sigma-Aldrich), FCCP (5 μM, Sigma-Aldrich), A-769662(70 μM, Sigma-Aldrich), EX-229 (30 μM, Selleck Chemicals), Compound C (2 μM, Selleckchem), Rotenone (1 μM, Tocris), Mitotempo (10 μM, Sigma-Aldrich), Chloroquine (2 μM, Sigma-Aldrich), Lornoxicam (20 μM, Selleck Chemicals), 3-Amino-1,2,4-triazole; 3-AT (2 mM, Sigma-Aldrich), Parthnolide (2 μM, Sigma-Aldrich).
3D spheroid models
Spheroids were formed using 2 × 103 451LuR cells/well and were seeded in 96U bottom plate, Nunclon™ Sphera™ Microplates (Thermo Fisher Scientific) in DMEM High Glucose (Sigma-Aldrich) supplemented with 2% FBS and 4 mM glutamine. The next day, spheroids were treated with the desired compounds. SYTOX green, at a final concentration of 1 μM, was also added to the wells. Spheroid growth and green fluorescence were monitored using the IncuCyte ZOOM system (Essen BioScience) for at least 3 days. Images were taken at 2-h intervals. The intensity of SYTOX green was measured as the total green object integrated intensity (GCU × µM²/image) using Incucyte software.
Generation of knockout cell lines
For the knockout of a specific gene, sgRNAs were cloned into the pLentiCRISPRv2 plasmid (Addgene 52961) following the standard cloning protocol. A pool of four sgRNAs for CPT1 KO and AMPK KO was selected from the Brunello genome-wide knockout library. HEK293T cells were seeded in T150 (TPP) flasks with 40 mL of DMEM containing 10% fetal bovine serum at 45% confluency to generate lentivirus particles and incubated overnight. After 1 day, cells were transfected using X-TremeGENE 9 (Roche) with the pLentiCRISPRv2 KO plasmids, together with the packaging plasmids psPAX2 and envelope plasmid pMD2.G, and incubated overnight to form lentiviral particles coated with the VSV-G protein. ~16 h post-transfection, the medium was replaced by serum-free growth medium (DMEM + 1.1 g/100 mL BSA and 20 µg/mL gentamicin). The lentivirus supernatant was harvested ~48 h after transfection and stored at −80 °C. Knockout cells were generated by transducing them with the lentiviral pool of 4 sgRNAs and selecting them with puromycin (1 µg/ml) for 3 days. KO confirmation was conducted using Western blot and qPCR.
Generation of knockdown cell lines
451LuR cells were forward transfected with 10 nM of Silencer-Select Negative Control siRNA (Thermo Fisher Scientific), two sequences targeting DGAT1 (s16568, s16569), DGAT2 (s85667, s85668), CHPT1 (s32552, s32553), CEPT1 (s20316, s20317), MFN1 (s31218, s31219), MFN2 (s19260, s19261), KIF5B (s731, s732), and SPASTIN (s13348, s13349) (Silencer-Select siRNA, Thermo Fisher Scientific) with lipofection according to the manufacturer’s recommendations (Lipofectamine RNAiMAX, Thermo Fisher Scientific). KD confirmation was conducted using qPCR.
RNA extraction and RT-qPCR
The cell lysate was extracted using the PureLink RNA Mini Kit (Invitrogen) according to the manufacturer’s protocol. 1 μg RNA from each sample was reverse-transcribed to cDNA using a high-capacity cDNA Reverse Transcriptase Kit (Invitrogen, 18080044). Quantitative PCR reactions were performed using Fast SYBR Green Master Mix (Thermo Fisher Scientific, 4385612) with duplicate measurements on the LightCycler 96 Real-Time PCR instrument (Roche Life Science). 18S was used as an internal reference. Differences in mRNA levels compared with 18 s were computed between vehicle and experimental groups using the ∆∆Ct method. Primers used were:
hACADL: Fw 5‘-CTTGCATGGCGAAATATTGGG-3‘, Rv 5‘-ATAGATTGGCTGAACTCTGGC-3‘,
hACOX2: Fw 5‘-ACATGGCAAGAACAGCCTAC-3‘, Rv 5‘-CATAACAGCCAAGTGCTGAATTT-3‘,
hPRKAB1: Fw 5‘-ATTGATGATGAAATTACAGAAGCCA-3‘, Rv 5‘-GCATCTGAATCACTCCTTTGGC-3,
hATG5: Fw 5‘-TTTGCATCACCTCTGCTTTC-3‘, Rv 5‘-TAGGCCAAAGGTTTCAGCTT-3‘,
hATGL: Fw 5‘-GGCCAGACCCAGCTTCTTC-3‘, Rv 5‘-TAGGCCAAAGGTTTCAGCTT-3‘,
hCEPT1: Fw 5‘-ACC TTT TTG GCA ATC TAT GAT TCC A −3‘, Rv 5‘-TTG CCA ACA CCA CCT GTG AA-3‘,
hCHPT1: Fw 5‘-GCATGTTGAGATTTGGAAAAGTGG−3‘, Rv 5‘-TCCAAGAACTGGAAGGATCTTCA-3‘,
hCD36: Fw 5‘-CAAAACGGCTGCAGGTCAAC-3‘, Rv 5‘-CCTCTGTGAGCCGCCTG-3‘,
hCPT1: Fw 5‘-GGAGACAGACACCATCCAGC-3‘, Rv 5‘-GCCAGACCTTGAAGTAGCGT-3‘,
hDGAT1: Fw 5‘-CACTGGGAGCTGAGGTGC-3‘, Rv 5‘-AGCATCACCACACACCAGTT-3‘,
hDGAT2: Fw 5‘-CATGGTCGAGGCCTCTTCTC-3‘, Rv 5‘-GGCTCTCCCACAACAGTG-3‘,
hLAMP2A: Fw 5‘-GGCTCCGTTTTCAGCATTGC-3‘, Rv 5‘-ACAGTCTGCTCTTTGTTGCAC-3‘,
hMFN1: Fw 5‘-AGTACATGGAGGATGTGCGC-3‘, Rv 5‘-CGAGCTTCCGACGGACTTAC-3‘,
hMFN2: Fw 5‘-GGGAAGGTGAAGCGCAATGT-3‘, Rv 5‘-AGTGGGGATGCATTCACCTC-3‘,
hPEX5: Fw 5‘-GGCGCTCCGCGAGAG−3‘, Rv 5‘-CGAGCTTCATGAGCGGGTT-3‘,
hPEX13: Fw 5‘-CAGCATCAACTGGGCAAGTG−3‘, Rv 5‘-GCACTTTGGGTTGTTGTTCTTTG−3‘,
hPLIN2: Fw 5‘-ATCAGCCATCAACTCAGATTGT-3‘, Rv 5‘-CCCCAGTCACAGTAGTCGTC-3‘,
hPLIN3: FW 5‘-CAGGTGACAGTGGAAGAACC-3‘, RV 5‘-CAGTCTTGATGTGCGGGTAG-3‘,
hPLIN5: FW 5‘-CTGGCCTACGAGCACTCTG-3‘, RV 5‘-TGCATGTGGTCTATCAGCTCC-3‘,
hPTGS2: Fw 5‘-GTTCCACCCGCAGTACAGAA-3‘, Rv 5‘-GCAGACATTTCCTTTTCTCCTGT-3‘,
hRAB7A: Fw 5‘-CTCGCTTCTGTCCTCCGTTT-3‘, Rv 5‘-AGGTCATCCTTCAAACGCGG-3‘.
Western blotting
Western blotting was carried out on melanoma cell lines. Following ice-cold PBS washes, cells were collected in sample buffer (Thermo Fisher) supplemented with DTT (Sigma), sonicated, and boiled for 5 min. Equal amounts of protein were loaded onto precast gels (NuPAGE, Thermo Fisher), transferred to nitrocellulose membranes, and incubated with antibodies against phospho-AMPK (1/1000 dilution) (Cell Signaling, #2535), AMPK (1/1000 dilution) (Cell Signaling, #2532), and Anti-β-Tubulin Antibody (1/500 dilution) (Sigma-Aldrich, #AB3201). Representative uncropped blots are presented in the source data file.
TBARS assay (MDA)
3 × 106 cells were harvested after the desired incubation with PUFA/Fe3+. Malondialdehyde levels in sonicated cell lysates, tissue lysates, and serum were determined using a TBARS Assay Kit (Cayman, 700870) following the manufacturer’s instructions. The result was normalized to total cellular protein (BCA assay).
Iron assay
1 × 106 cells were harvested following 24-h treatment with PUFA/Fe3+. The relative amount of iron was measured using a colorimetric iron assay kit (Abcam, ab83366) according to the manufacturer’s protocol. A microplate reader was utilized to measure the absorbance at 593 nm for each well.
Fatty acid uptake assay
12 × 103 cells were plated in a 96-well black-sided plate. Following the desired incubation with PUFA/Fe3+, fatty acid uptake was measured using a fluorometric fatty acid uptake kit (Sigma-Aldrich, MAK156). Fluorescence intensity was quantified using excitation/emission wavelengths of 488/530 nm with a microplate reader.
Fatty acid oxidation assay
8 × 105 cells were seeded in 10 cm petri dishes. Upon stimulation with treatment, the cells were trypsinized and stained with three primary antibodies.; ACADVL, ACADM, and HADHA following the manufacturer’s protocol (Abcam, ab118183). The expression was measured by flow cytometry (BD FACSVerse, BD Biosciences)(excitation/emission = 488/594), and at least 1 × 104 cells were analyzed per sample using FlowJo.
Flow cytometric analysis
Briefly, 1.5 × 105 cells were seeded in a 6-well plate. The next day, cells were treated with stimuli and harvested in 300 μL HBSS containing 5 μM BODIPY 581/591 C11 or 500 nM MitoPerOx, and 500 nM Tetramethylrhodamine ethyl ester perchlorate (TMRE). They were then incubated for 30 min at 37 °C. Cells were then resuspended in 300 μL of fresh HBSS and analyzed using flow cytometry (BD FACSVerse, BD Biosciences) with excitation/emission settings of 488/520 nm and 488/575-590 nm, respectively. At least 1 × 104 cells were analyzed per sample using FlowJo. Cells were initially gated (P1) based on forward scatter (FSC-A) and side scatter (SSC-A) to eliminate debris and doublets. A second gate (P2) using FSC height (FSC-H) versus FSC width (FSC-W) was applied to eliminate cell aggregates and isolate single cells. Within the P2 gate, target populations were identified based on fluorescence intensity in the relevant channels. To ensure accurate gating, Fluorescence Minus One (FMO) controls were used to assess fluorescence spillover, while unstained controls accounted for background autofluorescence.
Cell viability assessment using SYTOX™ Green
1 × 104 cells were seeded in a 96-well black cell carrier plate. The next day, cells were co-treated with stimuli and SYTOX green at a final concentration of 1 μM for at least 30 min at 37 °C. The fluorescent signal was measured using a PerkinElmer microplate reader every 24 h with Excitation/Emission: 504/523 nm. At any desired measurement time points, 4 μl of 10× lysis solution (Promega) was added to each plate well to achieve maximal lysis and was incubated for 20 min at 37 °C. The final measurement was performed using a microplate reader, and the cell death percentage was calculated as follows: % Cell Death = ((Fluorescence measured before lysis - Blank) / (Fluorescence measured after lysis - Blank)) × 100.
Proliferation assays
Proliferation curves were generated using an IncuCyte ZOOM system (Essen BioScience) on cells seeded in microplates (TPP or Nunc Edge) based on phase-contrast images taken at 2 h intervals for the duration of the experiments.
Immunoblotting analysis
Following ice-cold PBS washes, cells were collected in sample buffer (Thermo Fisher) supplemented with DTT (Sigma-Aldrich), sonicated, and boiled for 5 min. Equal amounts of protein were loaded onto precast gels (NuPAGE, Thermo Fisher), transferred to nitrocellulose membranes, and incubated with antibodies against CPT1A (ab128568, Abcam, 1:500), Phospho-AMPKα (2535S, Cell Signaling Technology, 1:1000), AMPKα (2532, Cell Signaling Technology, 1:1000), ATGL (sc-365278, Santa Cruz Biotechnology, 1:1000), GAPDH (5174, Cell Signaling, 1:5000), detyrosinated tubulin (AB-3201, Sigma-Aldrich, 1:1000), Protein Ladder (26619, Thermo Fisher).
Immunofluorescence imaging
1.5 × 105 Cells were seeded on 17-mm coverslips in 6-well plates. After treatment at various time points, washed cells in HBSS were incubated with optimized dyes, BODIPY 493/503 (2 μM), for 30 min at 37 °C. Then, coverslips were fixed by PFA 3.7% in PBS and were blocked with PBS containing 0.1% saponin and 5% goat serum for 1 h, following primary incubation with antibodies against TOMM20 (11802-1-AP, Proteintech, 1:200), PMP70 (ab3421, Abcam, 1:200), and LAMP2A (ab18528, Abcam, 1:200) overnight. After incubation with a secondary antibody, they were finally mounted in ProLong™ Glass Antifade Mountant with NucBlue™ Stain. Images were analyzed using high-resolution confocal Zeiss LSM900 Airyscan2 with a 63× oil objective. 10 images were taken per coverslip, and at least three independent biological replicates were performed for each condition. We used the software NIS-Elements AR 5.21 (Nikon) for intensity and overlap analysis of the Airyscan images. For lipid droplet segmentation, the same threshold was applied per cell line. For mitochondria segmentation, the threshold values were manually adjusted per image.
Plasmid transfection
The plasmid, mCherry-TOMM20-N-10, was a gift from Michael Davidson (Addgene plasmid # 55146). For transfection into 451LuR cells, DNA plasmids were propagated in the Escherichia coli DH5α (TAKARA) strain and purified using the Plasmid Plus Midi Kit (QIAGEN). 1.5 × 105 cells were seeded in 35-mm dishes. The cells were transfected with mCherry-TOMM20-N-10 on the same day of treatment using Lipofectamine LTX Reagent with PLUS Reagent, as described in the manufacturer’s manual. Briefly, 1 µg of the plasmid DNA was mixed with 9 μl Lipofectamine LTX and 10 µl PLUS Reagent in 200 μl OptiMEM, vortexed, incubated at room temperature (RT) for 10 min, and added dropwise to the cells and incubated for 24–36 h before imaging of the cells.
Fluorescent staining of live cells
1.5 × 105 Cells were seeded in 35 mm imaging dishes. After the desired time points of treatment, they were once washed with HBSS and were incubated with trackers LysoTracker deep red (100 nM, Invitrogen), MitoTracker CMXRos (100 nM, Invitrogen), MitoTracker deep red (100 nm, Invitrogen) and ER-Tracker™ Green (BODIPY™ FL Glibenclamide) (1 μM, Invitrogen), or CellLight™ Peroxisome-GFP, BacMam 2.0 (30 particles per cell(PPC)), BODIPY 581/591 C11 (5 μM, Invitrogen), Tetramethylrhodamine ethyl ester perchlorate (TMRE)(500 nM, Sigma-Aldrich) all diluted in HBSS and were incubated for 30 min at 37 °C with coverslips.
SRS imaging analysis
1.5 × 105 melanoma 451LuR cells from ATCC were seeded into 35 mm imaging dishes with a polymer coverslip bottom (No. 1.5H, ibidi) and treated with DHA-d5 (10 µM, Sanbio) and other desired inhibitors for a specified time. Cells were washed with HBSS (+CaCl2, +MgCl2, Gibco) and stained with LysoTracker deep red (100 nM, Invitrogen), MitoTracker CMXRos (100 nM, Invitrogen), and CellLight™ Peroxisome-GFP, BacMam 2.0 (30 particles per cell(PPC)) (Invitrogen) according to the manufacturer’s protocol. Cells were imaged in DMEM with no phenol red (4.5 g/l, Gibco) for fluorescent channels and SRS imaging.
SRS imaging was performed on an inverted Leica Stellaris 8 Coherent Raman Scattering laser-scanning microscope with the SRS option (Leica Microsystems, Mannheim, Germany). The PicoEmerald S Optical Parametric Oscillator (APE, Berlin, Germany) provided the fixed Stokes beam wavelength at 1031.306 nm and the tuneable pump beam wavenumber of 2129 cm−1 for DHA-d5 and 2852 cm−1 for lipid droplets. At the moment of imaging, the Stokes laser power was 0.1510 W, and the pump laser power was 0.2950 W; both were attenuated to 50% intensity. The pump and Stokes beams were spatially and temporally overlapped using two dichroic mirrors and a delay stage inside the laser system. They were then coupled into the laser-scanning microscope with optimized near-IR throughput. Unidirectional SRS images were acquired using a 40× objective, HC PL IRAPO 40×, N.A. 1.10 water immersion lens (Leica Microsystems, Mannheim, Germany) with 3.1625 μs pixel dwell time over a 512 × 512 frame and a speed of 400 Hz. The resulting pixel size was 0.19 µm. The signal intensity was collected using an oil condenser (1.4 NA, Leica Microsystems, Mannheim, Germany) in the forward direction and demodulated using a Lock-In Amplifier (Zürich Instruments, Zürich, Switzerland). The F-SRS detector was used with a gain of 10. The imaging system was controlled by the software LasX version 4.6.1.27508.
Confocal fluorescence images were simultaneously acquired using the same instrument and objective. Excitation settings included a 488 nm laser power of 2.0 and a gain of 5.0, 561 nm with a laser power of 2.0 and a gain of 5.0, and 638 nm with a laser power of 2.0. Signals were collected with two HyD S detectors in analog mode through a 77.2 µm pinhole with 1 AU. A line average of 4 was applied for both SRS and fluorescent channels.
Transmission electron microscopy (TEM)
Cells were cultured on 35 mm glass-bottom dishes or coverslips made of Aclar till they reached 70–80% confluency. They were prefixed for 10 min at 37 °C with prewarmed double-concentrated fixative (8% paraformaldehyde (PFA) + 1% glutaraldehyde (GA) in 0.2 M phosphate buffer (PB), pH 7.4), added directly into the culture dish in a 1:1 proportion to the amount of culture medium. Next, the cells were further fixed with 4% PFA + 2.5% GA in 0.1 M PB buffer (pH 7.4) for 2 h at room temperature (RT). After rinsing with 0,1 M PB (pH 7.4), 3 ×5 min, they were rinsed again with the same buffer 3 × 3 min, but this time on ice, and subsequently postfixed with 1% osmium tetroxide (OsO4) and 1.5% potassium ferrocyanide (K4Fe(CN)6) in 0.1 M PB (pH 7.4) for 60 min on ice. Then, they were washed five times for three minutes each with Milli-Q water on ice and incubated overnight in 1% aqueous uranyl acetate at 4 °C. The next day the specimens were washed with Milli-Q water 5 × 7 min, incubated en bloc with lead aspartate (Walton’s lead aspartate: 0.02 M lead nitrate in 0.03M L- aspartic acid, pH 5.5) for 30 min at +60 °C, wash with Milli-Q water 5 × 3 min and dehydrated with graded series of ethanol: 1 × 10 min each step (30, 50, 70, 90% ethanol) at RT followed by, 2 × 10 min dehydration with pure ethanol on ice. Next, samples were infiltrated with the hard composition of AGAR resin, first with a 2:1 mixture of pure ethanol and resin for 1 h, next with a 1:1 mixture for 1 h, and finally with a 1:2 mixture of pure ethanol and resin overnight, all at RT. The following day, the samples were infiltrated with pure resin for 6 h, embedded in fresh, pure resin with beem capsule blocks on top, and polymerized for 48 h at 60 °C.
Polymerized blocks were detached from the coverslips, and ultrathin sections (70 nm) were collected on formvar-coated copper grids. The sections were imaged at 80 kV using a Transmission Electron Microscope (JEOL JEM 1400; Tokyo, JN) equipped with an EMSIS Quemesa camera (11 Mpxl; Münster, DE).
Glutaraldehyde (GA) (25% solution, Electron Microscopy Services), Paraformaldehyde (PFA) (32% Aqueous solution, Electron Microscopy Services), Na2HPO4 (dibasic) and NaH2PO4 (monobasic) (Electron Microscopy Services), L-aspartic acid (Sigma-Aldrich), Lead nitrate (Electron Microscopy Services), Osmium tetroxide (OsO4) (4% Aqueus solution, Electron Microscopy Services), Potassium ferrocyanide (K4Fe(CN)6) (Sigma-Aldrich), Epon resin MNA (Agar scientific), Epon resin DDSA (Agar scientific), Epon resin Agar 100 (Agar scientific), Epon resin BDMA (Electron Microscopy Services), Uranyl acetate (Electron Microscopy Services).
Oxygen consumption rate measurements (OCR/ECAR)
2 × 105 cells were seeded on Seahorse Xp culture plates (103022-100, Agilent) in Seahorse XF Assay Medium (103335-100, Agilent), pH 7.4, containing 1 mM Sodium Pyruvate (S8636, Thermo Fisher Scientific), 4 mM Glutamine (G7513, Sigma-Aldrich), and 25 mM Glucose (8769, Sigma-Aldrich). Before analysis, cells were incubated for 1 h in a CO2-free incubator at 37 °C. OCR was measured after serial injections of 3 µM Oligomycin (O4876, Sigma-Aldrich), 2 µM phenylhydrazone (FCCP, sc-203578, Santa Cruz Biotechnology), and 1 µM Antimycin A (A8674, Sigma-Aldrich). ECAR was measured after serial injections of 80 mM glucose, 3 µM oligomycin, and 500 mM 2-deoxy-D-glucose (D-8375, Sigma-Aldrich). Analysis was performed using Seahorse Wave Desktop Software (Agilent).
Lipidomics
For lipid extraction an amount of cells or tissue containing 10 μg of DNA was homogenized in 700 μL of water with a handheld sonicator and was mixed with 800 μl HCl(1 M):CH3OH 1:8 (v/v), 900 μl CHCl3, 200 μg/ml of the antioxidant 2,6-di-tert-butyl-4-methylphenol (BHT; Sigma-Aldrich) and 3 μl of SPLASH® LIPIDOMIX® Mass Spec Standard (#330707, Avanti Polar Lipids). After vortexing and centrifugation, the lower organic fraction was collected and evaporated using a Savant Speedvac spd111v (Thermo Fisher Scientific) at room temperature, and the remaining lipid pellet was stored at −20 °C under argon.
Just before mass spectrometry analysis, lipid pellets were reconstituted in 100% ethanol. Lipid species were analyzed by liquid chromatography electrospray ionization tandem mass spectrometry (LC-ESI/MS/MS) on a Nexera X2 UHPLC system (Shimadzu) coupled with a hybrid triple quadrupole/linear ion trap mass spectrometer (6500 + QTRAP system; AB SCIEX). Chromatographic separation was performed on a XBridge amide column (150 mm × 4.6 mm, 3.5 μm; Waters) maintained at 35 °C using mobile phase A (1 mM ammonium acetate in water-acetonitrile 5:95 (v/v)) and mobile phase B (1 mM ammonium acetate in water-acetonitrile 50:50 (v/v)) in the following gradient: (0–6 min: 0% B → 6% B; 6–10 min: 6% B → 25% B; 10–11 min: 25% B → 98% B; 11–13 min: 98% B → 100% B; 13–19 min: 100% B; 19–24 min: 0% B) at a flow rate of 0.7 mL/min which was increased to 1.5 mL/min from 13 min onwards. SM, CE, CER, DCER, HCER, and LCER were measured in positive ion mode with a precursor scan of 184.1, 369.4, 264.4, 266.4, 264.4, and 264.4, respectively. TAG, DAG, and MAG were measured in positive ion mode with a neutral loss scan for one of the fatty acyl moieties. PC, LPC, PE, LPE, PG, PI, and PS were measured in negative ion mode by fatty acyl fragment ions. Lipid quantification was performed by scheduled multiple reactions monitoring (MRM), the transitions being based on the neutral losses or the typical product ions as described above. The instrument parameters were as follows: Curtain Gas = 35 psi; Collision Gas = 8 a.u. (medium); IonSpray Voltage = 5500 V and −4500 V; Temperature = 550 °C; Ion Source Gas 1 = 50 psi; Ion Source Gas 2 = 60 psi; Declustering Potential = 60 V and −80 V; Entrance Potential = 10 V and −10 V; Collision Cell Exit Potential = 15 V and −15 V.
The following fatty acyl moieties were taken into account for the lipidomic analysis: 14:0, 14:1, 16:0, 16:1, 16:2, 18:0, 18:1, 18:2, 18:3, 20:0, 20:1, 20:2, 20:3, 20:4, 20:5, 22:0, 22:1, 22:2, 22:4, 22:5 and 22:6 except for TGs which considered: 16:0, 16:1, 18:0, 18:1, 18:2, 18:3, 20:3, 20:4, 20:5, 22:2, 22:3, 22:4, 22:5, 22:6.
Peak integration was performed with the MultiQuantTM software version 3.0.3. Lipid species signals were corrected for isotopic contributions (calculated with Python Molmass 2019.1.1) and were quantified based on internal standard signals and adhere to the guidelines of the Lipidomics Standards Initiative (LSI) (level 2 type quantification as defined by the LSI). Unpaired T-test p-values and FDR corrected p-values (using the Benjamini/Hochberg procedure) were calculated in Python StatsModels version 0.10.1. Pathway enrichment analysis of the lipidomics data was conducted using the Lipid Ontology (LION) enrichment analysis web application.
Metabolomics
10 μl of each sample was loaded into a Dionex UltiMate 3000 LC System (Thermo Fisher Scientific, Bremen, Germany) equipped with a C-18 column (Acquity UPLC -HSS T3 1.8 μm; 2.1 × 150 mm, Waters) coupled to a Q Exactive Orbitrap mass spectrometer (Thermo Fisher Scientific) operating in negative ion mode. A step gradient was carried out using solvent A (10 mM TBA and 15 mM acetic acid) and solvent B (100% methanol). The gradient started with 5% of solvent B and 95% of solvent A and remained at 5% B until 2 min post-injection. A linear gradient to 37% B was carried out for 7 min and then increased to 41% until 14 min. Between 14 and 26 min, the gradient increased to 95% B and remained at 95% B for 4 min. At 30 min, the gradient returned to 5% B. The chromatography was stopped at 40 min. The flow rate was maintained at 0.25 mL/min, and the column was kept at 40 °C throughout the analysis. The MS operated in full scan mode (m/z range: [70.0000-1050.0000]) using a spray voltage of 4.80 kV, capillary temperature of 300 °C, sheath gas at 40.0, and auxiliary gas at 10.0. The AGC target was set at 3.0E + 006 using a resolution of 140,000, with a maximum IT fill time of 512 ms. Data collection was performed using the Xcalibur software (Thermo Fisher Scientific). The data analyses were performed by integrating the peak areas (El-Maven – Polly - Elucidata).
Gene expression
For gene expression, cells were seeded in 6-well plates at a density of 1.5 × 105 cells per well. On the second day, the cells were treated with PUFA/Fe3+ for 18 h and 36 h. RNA was isolated using the PureLink™ RNA Mini Kit (12183018A, Invitrogen). Libraries were constructed, and bulk RNA-seq was performed by Novogene (UK).
Statistical analysis
Data were analyzed using GraphPad Prism 9.5.0. Statistical tests included unpaired t-tests, one-way ANOVA, and two-way ANOVA. For multiple comparisons, the Holm–Sidak correction was applied. Non-Gaussian data were analyzed using the Kruskal–Wallis test. Results are presented as mean ± standard deviation (SD), unless otherwise specified. Statistical significance was defined as p < 0.05, with significance levels indicated as follows: *p < 0.05, **p < 0.005, ***p < 0.0005, ****p < 0.0001. No statistical method was used to predetermine sample size. No data were excluded from the analyses. The experiments were not randomized. The investigators were not blinded to allocation during experiments and outcome assessment.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All data are available in the main text or the supplementary materials. The mass spectrometry data generated in this study have been deposited in the Mendeley Data repository with the identifier https://doi.org/10.17632/f2sy6jc388.1. The bulk RNA-seq data generated in this study have been deposited at the NCBI Gene Expression Omnibus (GEO) under accession number GSE295451. Source data are provided with this paper.
References
Sharma, A., Boise, L. H. & Shanmugam, M. Cancer metabolism and the evasion of apoptotic cell death. Cancers 11, 1–20 (2019).
Yang, W. S. & Stockwell, B. R. Ferroptosis: death by lipid peroxidation. Trends Cell Biol. 26, 165–176 (2016).
Reed, J. C. & Pellecchia, M. Ironing out cell death mechanisms. Cell 149, 963–965 (2012).
Dixon, S. J. et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 149, 1060–1072 (2012).
Cao, J. Y. & Dixon, S. J. Mechanisms of ferroptosis. Cell. Mol. Life Sci. 73, 2195–2209 (2016).
Wu, Y. et al. Ferroptosis in cancer treatment: another way to Rome. Front. Oncol. https://doi.org/10.3389/fonc.2020.571127 (2020).
Guo, J. et al. Ferroptosis: a novel anti-tumor action for cisplatin. Cancer Res. Treat. 50, 445–460 (2018).
Soengas, M. S. & Lowe, S. W. Apoptosis and melanoma chemoresistance. Oncogene 22, 3138–3151 (2003).
Liang, C., Zhang, X., Yang, M., and Dong, X. Recent progress in ferroptosis inducers for cancer therapy. Adv. Mater. https://doi.org/10.1002/adma.201904197 (2019).
Talebi, A. et al. Pharmacological induction of membrane lipid poly-unsaturation sensitizes melanoma to ROS inducers and overcomes acquired resistance to targeted therapy. J. Exp. Clin. Cancer Res. 42, 1–16 (2023).
Talebi, A. et al. Sustained SREBP-1-dependent lipogenesis as a key mediator of resistance to BRAF-targeted therapy. Nat. Commun. 9, 1–11 (2018).
Rysman, E. et al. De novo lipogenesis protects cancer cells from free radicals and chemotherapeutics by promoting membrane lipid saturation. Cancer Res. 70, 8117–8126 (2010).
Tsoi, J. et al. Multi-stage differentiation defines melanoma subtypes with differential vulnerability to drug-induced iron-dependent oxidative stress. Cancer Cell 33, 890–904.e5 (2018).
Das, U. N. Saturated fatty acids, MUFAs and PUFAs regulate ferroptosis. Cell Chem. Biol. 26, 309–311 (2019).
Magtanong, L. et al. Exogenous monounsaturated fatty acids promote a ferroptosis-resistant cell state. Cell Chem. Biol. 26, 420–432.e9 (2019).
Pozniak, J. et al. A TCF4-dependent gene regulatory network confers resistance to immunotherapy in melanoma. Cell 187, 166–183.e25 (2024).
Akbani, R. et al. Genomic classification of cutaneous melanoma. Cell 161, 1681–1696 (2015).
Kaplan, J. & Ward, D. M. The essential nature of iron usage and regulation. Curr. Biol. 23, R642–R646 (2013).
Correnti, M., Gammella, E., Cairo, G., and Recalcati, S. Iron absorption: molecular and pathophysiological aspects. Metabolites. https://doi.org/10.3390/metabo14040228 (2024).
Pillai, M., Chen, Z., Jolly, M. K. & Li, C. Quantitative landscapes reveal trajectories of cell-state transitions associated with drug resistance in melanoma. iScience 25, 105499 (2022).
Diazzi, S., Tartare-Deckert, S. & Deckert, M. The mechanical phenotypic plasticity of melanoma cell: an emerging driver of therapy cross-resistance. Oncogenesis 12, 1–7 (2023).
Bailey, A. P. et al. Antioxidant role for lipid droplets in a stem cell niche of Drosophila. Cell 163, 340–353 (2015).
Petan, T., Jarc, E. & Jusović, M. Lipid droplets in cancer: guardians of fat in a stressful world. Molecules 23, 11–15 (2018).
Jarc, E. et al. Lipid droplets induced by secreted phospholipase A2 and unsaturated fatty acids protect breast cancer cells from nutrient and lipotoxic stress. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 1863, 247–265 (2018).
Czamara, K. et al. Raman spectroscopy of lipids: a review. J. Raman Spectrosc. 46, 4–20 (2015).
Singh, J., Sanghavi, P. & Mallik, R. Microtubule motor driven interactions of lipid droplets: specificities and opportunities. Front. Cell Dev. Biol. 10, 1–10 (2022).
Huang, J. et al. Equal distribution of lipid droplets in daughter cells is regulated by microtubules. Cell Cycle 22, 1421–1433 (2023).
Tadepalle, N. et al. Microtubule-dependent and independent roles of spastin in lipid droplet dispersion and biogenesis. Life Sci. Alliance 3, 1–19 (2020).
Qiu, B. et al. Phospholipids with two polyunsaturated fatty acyl tails promote ferroptosis. Cell 187, 1177–1190.e18 (2024).
Benador, I. Y., Veliova, M., Liesa, M. & Shirihai, O. S. Mitochondria Bound to lipid droplets: where mitochondrial dynamics regulate lipid storage and utilization. Cell Metab. 29, 827–835 (2019).
Ademowo, O. S., Dias, H. K. I., Burton, D. G. A. & Griffiths, H. R. Lipid (per) oxidation in mitochondria: an emerging target in the ageing process? Biogerontology 18, 859–879 (2017).
Moessinger, C. et al. Two different pathways of phosphatidylcholine synthesis, the Kennedy Pathway and the Lands Cycle, differentially regulate cellular triacylglycerol storage. BMC Cell Biol. https://doi.org/10.1186/s12860-014-0043-3 (2014).
Ma, X., Qian, H., Chen, A., Ni, H. M. & Ding, W. X. Perspectives on mitochondria–er and mitochondria–lipid droplet contact in hepatocytes and hepatic lipid metabolism. Cells 10, 2273 (2021).
Chen, L., Jin, Y., Wu, J. & Ren, Z. Lipid droplets: a cellular organelle vital for thermogenesis. Int. J. Biol. Sci. 18, 6176–6188 (2022).
Boutant, M. et al. Mfn2 is critical for brown adipose tissue thermogenic function. EMBO J. 36, 1543–1558 (2017).
Horibata, Y. & Sugimoto, H. Differential contributions of choline phosphotransferases cpt1 and cept1 to the biosynthesis of choline phospholipids. J. Lipid Res. https://doi.org/10.1016/J.JLR.2021.100100 (2021).
Zhang, C., Liu, X., Jin, S., Chen, Y. & Guo, R. Ferroptosis in cancer therapy: a novel approach to reversing drug resistance. Mol. Cancer 21, 1–12 (2022).
Deng, J. et al. Targeting cancer cell ferroptosis to reverse immune checkpoint inhibitor therapy resistance. Front. Cell Dev. Biol. 10, 1–16 (2022).
Herms, A. et al. AMPK activation promotes lipid droplet dispersion on detyrosinated microtubules to increase mitochondrial fatty acid oxidation. Nat. Commun. https://doi.org/10.1038/ncomms8176 (2015).
Lange, M. & Olzmann, J. A. Ending on a sour note: lipids orchestrate ferroptosis in cancer. Cell Metab. 33, 1507–1509 (2021).
Thiam, A. R. & Beller, M. The why, when and how of lipid droplet diversity. J. Cell Sci. 130, 315–324 (2017).
Herbst, E. A. F. et al. Omega-3 supplementation alters mitochondrial membrane composition and respiration kinetics in human skeletal muscle. J. Physiol. 592, 1341–1352 (2014).
Sullivan, E. M. et al. Mechanisms by which dietary fatty acids regulate mitochondrial structure-function in health and disease. Adv. Nutr. 9, 247–262 (2018).
Sassano, M. & Tyurina, Y. Endoplasmic reticulum-mitochondria contacts are apical hotspots of lipid peroxidation driving ferroptosis. Nat Cell Biol. 6, 902–917 (2024).
Wei, S. et al. Ferroptosis mediated by the interaction between Mfn2 and IREα promotes arsenic-induced nonalcoholic steatohepatitis. Environ. Res. 188, 109824 (2020).
Li, C., Liu, J., Hou, W., Kang, R. & Tang, D. STING1 promotes ferroptosis through MFN1/2-dependent mitochondrial fusion. Front. Cell Dev. Biol. 9, 1–11 (2021).
Zaman, M. & Shutt, T. E. The role of impaired mitochondrial dynamics in MFN2-mediated pathology. Front. Cell Dev. Biol. 10, 1–18 (2022).
Shi, X. et al. Increased melanin induces aberrant keratinocyte − melanocyte − basal − fibroblast cell communication and fibrogenesis by inducing iron overload and ferroptosis resistance in keloids. Cell Commun. Signal. 23, 141 (2025).
More, S. et al. Secreted Apoe rewires melanoma cell state vulnerability to ferroptosis. Sci. Adv. 10, eadp6164 (2024).
Acknowledgements
We thank Professors A. Ribas, R. Lo, and D. Peeper for kindly sharing the cell lines. We are also grateful to the VIB-KU Leuven BioImaging Core for their support in sample preparation, training, and assistance with image acquisition and analysis. We extend our gratitude to the Laboratory of Experimental Oncology (KU Leuven) for providing access to the flow cytometer instrument and to the Laboratory of Molecular Biology of Leukemia (VIB-KU Leuven) for sharing their materials and instruments. This work was supported by grants from Research Foundation Flanders (FWO) G0C2219N (awarded to J.V.S.), Stand Up To Cancer, the Flemish Cancer Society (Kom op tegen Kanker) (awarded to S.M.), KU Leuven C14/21/095 grant INTERACTION (awarded to J.V.S. and P.A.), the Foundation Against Cancer (Stichting tegen Kanker) infrastructure grant 2022-043 (ATE-2022/1871), KU Leuven Infrastructure and Incubation grants, the Flemish Resilience Project PRISMO, private donations and EU Interreg EMR23 EURLIPIDS.
Author information
Authors and Affiliations
Contributions
S.M. and J.V.S. conceptualized and designed the study. S.M., with assistance from F.V., N.R., and M.A.S., developed the methodology, conducted the experiments, and collected the data. S.M. analyzed and interpreted the data with support from SH.W., A.E.A., J.D., and J.I. S.M., with contributions from J.C.M., P.A., S.S., and J.V.S., wrote and revised the manuscript. J.V.S. supervised the study and provided grant support.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Motamedi, S., Ravoet, N., Dehairs, J. et al. AMP-activated protein kinase-driven lipid droplet dynamics govern melanoma sensitivity to polyunsaturated fatty acid and iron-induced ferroptosis. Nat Commun 16, 11252 (2025). https://doi.org/10.1038/s41467-025-66113-z
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-66113-z
