
Abstract
Understanding how cells differentiate to their final specialized fates is a fundamental problem in biomedical science. Single-cell multi-omic profiling provides an opportunity to identify dynamic molecular changes, but new computational approaches are needed to realize this potential. In particular, previous methods for RNA velocity inference lack support for multi-lineage, multi-sample, and multi-omic single-cell data and cannot be used to identify differential dynamics. To overcome these challenges, we introduce MultiVeloVAE, a probabilistic framework for multi-sample RNA velocity inference that integrates single-cell RNA and multi-omic data. MultiVeloVAE models gene expression and chromatin accessibility on a shared time scale, performs multi-sample inference from datasets with partially overlapping modalities, accounts for lineage bifurcations, and enables statistical testing of velocity parameters among cell types and over time. Using newly generated 10X Multiome datasets from human embryoid bodies and differentiating macrophage cells, we demonstrate that MultiVeloVAE provides novel insights into chromatin accessibility and gene expression dynamics during development.
Similar content being viewed by others


Introduction
A multicellular organism contains diverse cell types that perform unique functions and express unique combinations of genes. A central goal in cellular and developmental biology is to understand the gene regulatory mechanisms that establish and maintain the identities of cell types. Single-cell RNA sequencing–and more recently, single-cell multi-omic profiling–have become pivotal tools for investigating these questions. However, because single-cell sequencing technologies destroy the cell, measuring individual cells longitudinally is not feasible. Instead, computational methods are required to assemble snapshots of cells into a coherent trajectory of dynamic changes across differentiation stages.
Early efforts to reconstruct trajectories from scRNA data focused on pseudotime inference1,2,3, but recently approaches based on RNA velocity have gained popularity4,5,6,7. RNA velocity uses mechanistic assumptions about the transcription and splicing process to infer the directions and rates of gene expression changes. This, in turn, avoids the need for specifying a starting cell state, as is required in pseudotime inference. La Manno et al.4 first formulated the notion of RNA velocity and developed a way to solve the proposed ordinary differential equations through steady-state assumptions. Later work5 relaxed the steady-state assumption, allowing all cells (not just those at steady state) to be used in parameter estimation. Bergen et al. also introduced the notion of latent time, which can be inferred while fitting the parameters of the RNA velocity model and used to rank cells by differentiation stage in a manner similar to pseudotime inference methods. Conveniently, RNA velocity methods rely on only conventional scRNA data, rather than metabolic labeling; they simply quantify nascent and mature RNA molecules separately using reads that indicate whether introns have been spliced out of a transcript. The concept of RNA velocity has subsequently been extended to incorporate additional molecular markers of the gene expression life cycle, including protein expression8 and chromatin accessibility9. We previously showed that incorporating chromatin accessibility and gene expression into a joint model improved the accuracy of cell fate estimation and identified cell states in which epigenome and transcriptome are temporarily out of sync9.
These first RNA velocity inference methods have inherent limitations: each gene is modeled on a separate time scale; gene expression is modeled as a discrete on or off state; and a single cell type is assumed, neglecting lineage bifurcations. Several new methods have been developed to address these issues: UniTVelo10 introduces a shared time scale with a more flexible parametric model, while DeepVelo11 and cellDancer12 infer cell-specific transcription, splicing, and degradation rates, offering better performance on complex datasets. Bayesian approaches like VeloVI13 and PyroVelocity14 enhance RNA velocity estimation by learning cell time distributions and modeling discrete RNA counts, respectively. However, each approach has drawbacks. For instance, UniTVelo does not model multiple cell types and provides limited biochemical interpretability, whereas DeepVelo and cellDancer rely on post hoc cell time inference, leading to potential inconsistencies between inferred time and velocity.
Additionally, these previous approaches lack key capabilities that are required to make RNA velocity useful for biological discovery: multi-sample inference; the ability to model multi-omic data; incorporating different types of data; and differential testing. Many important situations require inferring RNA velocity from multiple samples, including case-control studies, multi-subject genetic association studies, and developmental atlases. Single-cell integration methods such as Seurat15, LIGER16, and scVI17 can integrate single-cell datasets, correct batch effects, identify biologically similar cells, and find differentially expressed genes across datasets. However, there is no analogous way to perform RNA velocity inference on multiple samples. A related challenge is that many such studies feature a combination of data types–for example, gene expression and chromatin accessibility are co-profiled in some cells, while only gene expression is measured in other cells. Additionally, existing methods do not allow users to identify statistically significant changes in RNA velocity or other parameters between cell types, between biological conditions, or over time.
To address these limitations, we introduce MultiVeloVAE, a robust probabilistic model that enables multi-sample velocity inference from single-cell RNA and/or single-cell multi-omic data. Crucially, MultiVeloVAE also enables statistical testing to identify differential velocity, models all genes on a common time scale, and models developmental lineage bifurcation by allowing rate parameters to vary continuously with cell state. MultiVeloVAE also extends the notion of epigenomic priming and coupling (introduced in our previous work9) to a continuous and multi-lineage setting.
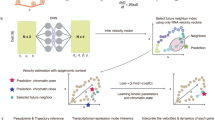
We have summarized the unique features of MultiVeloVAE compared to previous methods in Fig. 1b. We generated new experimental datasets from human in vitro differentiated macrophages and embryoid bodies and used MultiVeloVAE to discover genes with cell-type-specific velocity and decoupling. We also used MultiVeloVAE to characterize how these cell-type-specific dynamics in macrophages are distributed within transcription factor regulatory networks. Finally, we performed in silico perturbations of key hematopoietic differentiation factors.

a Diagrams of multi-omic dynamics assumption9 (Top) and MultiVeloVAE neural network architecture (Bottom). The network takes chromatin accessibility (c), unspliced RNA (u), and spliced RNA (s) values as input, along with optional sample covariates such as batch (b). The encoder network infers cell state and cell time for each cell from (c, u, s). c is optional, allowing inference from scRNA, multiome, or both kinds of data. Note that the latent time t is shared across all genes. The decoder network infers cell-specific and gene-specific chromatin state kc and transcription rate ρ from cell state and latent time (and optionally sample covariates b). The decoder then reconstructs (c, u, s) using the analytical solution to an ODE. b Table summarizing the advantages of MultiVeloVAE over existing velocity inference methods. c When integrating multiple datasets, MultiVeloVAE can integrate samples that differ in terms of technical effects such as library size or sequencing time, identify corresponding cell states across different biological contexts, and infer joint dynamics for all cells.
Results
MultiVeloVAE infers cell times, cell states, and rate parameters from multi-sample multi-omic data
MultiVeloVAE extends our previous methods, MultiVelo and VeloVAE, to support multi-omic and multi-batch data, employing a Bayesian framework for velocity inference. The inputs to MultiVeloVAE are single-cell measurements of chromatin accessibility (c) and both mature spliced (s) and nascent unspliced (u) transcripts, along with a categorical variable indicating the identity of the sample from which the cell was derived (Fig. 1a). The c measurements are optional–MultiVeloVAE can process either scRNA or multi-omic profiles, or a combination of both. The model then estimates latent variables that describe the dynamics of gene expression and chromatin accessibility, including cell time (t), cell state (z), and biochemical rate parameters (θ), through posterior inference. The key model underlying this inference process is a system of ordinary differential equations (ODEs) that describe the relationship between chromatin accessibility and gene expression, as in our previous work9. These equations capture the biochemical assumptions that chromatin near a gene must open before the gene is transcribed, and the gene must first be transcribed as unspliced pre-mRNA before it is spliced into mature mRNA. We interpret the latent time t in our model as an estimate of the real time at which the cell was sequenced. If real time labels are available, we can use these as a statistical prior for the latent times. In this case, we can assign real temporal units (e.g., hours) to latent time based on the real time labels used for the prior.
Our model is designed to address several shortcomings of existing RNA velocity approaches that limit their utility for biological discovery (Fig. 1b). First, instead of assuming a single binary transcription state for each gene across all cells, we allow a continuous state-specific transcription rate ρ for each cell, ranging between 0 (fully repressed) and 1 (fully induced) for each gene. Similarly, we model the chromatin state kc as a continuous and cell-specific parameter. These modifications enable MultiVeloVAE to flexibly model gene-wise lineage bifurcations and transcriptional boosts6. Second, we model all genes on a common time scale t, ensuring that t remains consistent across all genes during training. Third, we can perform multi-sample inference, including on samples with partially overlapping modalities such as single-cell RNA and single-cell multiome data. And fourth, we perform this parameter inference within a setting that allows hypothesis testing, such as differential velocity analysis.
To fit our model using real data, we use auto-encoding variational Bayes18, in which an autoencoder neural network estimates the posterior distributions of latent variables. Through this model, we jointly train both the inference and generative components, enabling the inference of latent variables from observed data and the prediction of outcomes for new data (Fig. 1a). The inference model (encoder network) takes values of (c, u, s) as inputs and estimates posterior distributions for cell time and cell state parameters. Concurrently, the generative model (decoder network) predicts gene-specific chromatin state (kc) and transcription rate (ρ) values for each cell based on its inferred low-dimensional latent state. The values of (c, u, s) for each cell can then be reconstructed using the analytical solution of the velocity ODEs. Intuitively, MultiVeloVAE simultaneously trains the neural network while optimizing the differential equation parameters, ensuring consistency in underlying dynamics across all genes. We train the model by maximizing the evidence lower bound (ELBO) of the marginal likelihood.
This inference framework also provides a powerful way to perform multi-sample inference, including when samples have partially overlapping modalities. We can incorporate batch effect correction capability directly into our model by conditioning latent distributions and rate parameters on each cell’s known batch labels, using a conditional autoencoder approach. Conditioning on sample-level covariates has been shown to be effective in a variety of single-cell data integration tasks17,19. By conditioning the encoder and decoder on the known sample label b of each cell (represented as a categorical, one-hot-encoded variable), the cell state latent variable z and cell time t become independent of the sample. Note that this strategy removes the sample effect from the latent space, but not the values of individual genes. To align samples in the high-dimensional gene expression space, we can choose a “reference batch” br and decode all latent cell states z using br. That is, after training, the model can be used to generate counterfactual “corrected” (c, u, s) counts in the high-dimensional gene space by taking cell states from one sample and decoding them with the parameters from another sample (Fig. 1c). Finally, we can further extend this multi-sample inference strategy by modifying the encoder and decoder networks to infer shared latent state and latent time values from partially overlapping modalities and impute unmeasured values.
MultiVeloVAE outperforms previous methods when using RNA only
We implemented an RNA-only mode for MultiVeloVAE, allowing head-to-head comparison with previous RNA velocity methods. To do this, we can set chromatin accessibility (c) to 1, thus eliminating the effects of chromatin openness on transcription dynamics. In RNA-only mode, MultiVeloVAE can accept (u, s) only inputs, such as those from scRNA-seq or snRNA-seq. We then assessed MultiVeloVAE’s performance on 10 real scRNA-seq datasets of varying sizes and complexities20,21,22,23,24,25,26,27,28,29 (Fig. 2, Supplementary Fig. 1). For example, bone marrow mononuclear cells (BMMC) (Fig. 2a) and subsampled mouse brain (Fig. 2b) are complex, multi-lineage datasets that have traditionally posed challenges for RNA velocity methods. In the BMMC dataset, the streams derived from MultiVeloVAE velocities originate from the HSC cluster in the right island of UMAP and naive T cell populations in the left. In contrast, other methods generate varying levels of backflows and inaccurate temporal predictions (Supplementary Fig. 2).

a Inferred velocity stream on UMAP coordinates, colored by cell types (Top) and latent time (Bottom) in a BMMC dataset. b Inferred velocity stream on UMAP coordinates, colored by cell types and latent time (Left two figures) in a subset mouse brain dataset. The uncertainty of cell states and true captured times are plotted on the same UMAP (Right two figures). c Density and scatter plots show the distribution and relationship between cell-state uncertainty and inferred latent time. The bar plot inset shows the regression coefficient between cell-state uncertainty and latent time for each cell type. The top-left, top-right, and bottom-right figures are colored by cell-type annotations, whereas the scatter plot at the bottom-left is colored by cell cycle score. d Inferred lineages on UMAPs of mouse gastrulation (Left) and human bone-marrow hematopoietic cells (Right). e Illustration of transcription boost and example MURK gene phase portraits (Smim1 and Hba-x). f Benchmarking results of the Spearman correlation between latent time and true time, GCBDir, and Mann–Whitney U statistic of direction correctness on 10 scRNA-seq datasets. Dashed horizontal lines show means, and solid horizontal lines show medians. The boxes represent the interquartile ranges (IQRs), and whiskers extend to 1.5 times the IQRs. Source data are provided as a Source Data file.
In another example, we applied MultiVeloVAE to a developing mouse brain dataset24 (Fig. 2b), with subsampled cell types from the neural tube and neural crest to ensure connected lineages. The inferred velocities and latent times identify neural tube and neural crest cells as root cells, with smooth transitions toward more differentiated cell types, such as neurons, glioblasts, and fibroblasts. The uncertainty of the cell state is highest for the least differentiated cells, suggesting that our model learns the biological uncertainty in uncommitted cells (Fig. 2c). Distinct cell types also display unique uncertainty patterns over time; for instance, neurons retain high uncertainty as they mature, while fibroblast and oligodendrocytes quickly gain fate determination upon differentiation, as can be seen from the regression coefficients of the state uncertainty values of each cell type along latent time (Fig. 2c). In general, we found that the uncertainty of the cell state inversely correlates with the cell cycle scores. This dataset also has many real-time points–cells were profiled daily from embryonic days 7 to 18 and pooled together, providing us with a ground-truth temporal reference. Plotting the actual capture time alongside inferred latent time demonstrates the model’s temporal accuracy (Fig. 2b).
Previous studies have highlighted that certain cell-type marker genes, especially in erythrocytes (Fig. 2d), exhibit transcriptional boosts during maturation6,30, which challenges RNA velocity analyses due to an additional inflection in the induction phase (Fig. 2e, left). This complexity arises because genes with convex-shaped phase portraits are often initialized as repression genes, causing discrepancies in gene alignment. To address this, we train the generative process of each gene using a categorical random variable that represents combinations of inductive and repressive phases, or bases. The induction base starts from zero expression, while the repression base descends from a predicted steady-state upper-bound. After training, each gene is assigned to the basis with the highest probability (see Methods for details). We tested MultiVeloVAE with this multi-basis approach on datasets referenced in a review article6 and observed accurate lineage predictions in blood cells (Fig. 2d). The precise time point of transcriptional boost of a gene can be located by finding where the acceleration of unspliced counts reaches the closest point near zero (Supplementary Fig. 3a–d). For the top multiple-rate kinetics (MURK) genes identified by the authors30, MultiVeloVAE assigns an induction basis and reconstructs transcriptional boosts in unspliced and spliced phase portraits (Fig. 2e, right). We further show genes with the highest fit likelihoods that were assigned as induction-only or repression-only to demonstrate the accuracy of assignment (Supplementary Fig. 3e). We note that transcriptional boosts are often less problematic as dataset lineage diversity increases, due to the ability of the model to see all genes simultaneously.
We benchmarked MultiVeloVAE’s RNA-only mode against existing RNA velocity methods, including scVelo5, UniTVelo10, DeepVelo11, VeloVI13, PyroVelocity14, and cellDancer12. We evaluated performance based on correlations with known time point labels, the accuracy of predicted cell-type transitions, and model fit quality (Fig. 2f, Supplementary Fig. 4). We propose an extended version of cross-boundary direction correctness10 (CBDir). Originally designed to quantify how accurately estimated cell transitions align with known cell-type transitions, we generalize CBDir to incorporate k-step neighbors and time ordering in the evaluation. We also subtract a background CBDir derived from a random walk to normalize the metric relative to random guessing. Besides GCBDir, we computed Mann–Whitney U statistics to test whether velocity vectors transition to cells from the correct cluster (based on prior knowledge) more often than to a randomly selected set of neighbors. For MultiVeloVAE, DeepVelo, VeloVI, and PyroVelocity, we calculated data fit metrics on a held-out test set. As scVelo and UniTVelo lack out-of-sample prediction capabilities, we excluded them from this test evaluation. Additionally, as cellDancer does not explicitly reconstruct data but optimizes cosine similarity instead, we excluded it from the MSE and MAE comparisons. MultiVeloVAE achieves better performance across these metrics, fitting the data accurately, aligning well with true time point labels, and generalizing well to test held-out test sets.
In general, the latent time from velocity methods lacks a meaningful unit because it is linearly associated with the simultaneously inferred rate parameters. Our model can be further guided by ground-truth time labels to improve the alignment of velocity time and actual time scale, as well as provide the time with real-world time measures such as hours or days. This is achieved by using true cell capture time as a statistical prior for latent time. We show examples of velocity and time inference with and without time prior information on a mouse embryonic fibroblast (MEF) dataset with six time points31,32 (Supplementary Fig. 5). In both scenarios, MultiVeloVAE accurately identifies the successful and unsuccessful reprogramming outcomes marked by induced endoderm progenitor marker Apoa1 and MEF marker Col1a232. Importantly, when trained with time-prior information, the latent time distribution of cells of each time point better aligns with the true time range from 0 to 28 days (Supplementary Fig. 5f).
We further provide detailed gene-specific predictions in the Pancreas dataset, known for its popularity in RNA velocity modeling (Supplementary Fig. 6). Genes in this dataset exhibit differing induction-repression switch points5. Methods that fit each gene independently, such as scVelo, UniTVelo, and VeloVI, produce conflicting gene dynamics, whereas DeepVelo, cellDancer, and MultiVeloVAE capture fate transitions for each marker gene on a shared temporal axis.
MultiVeloVAE improves velocity inference from multi-lineage single-cell multi-omic data
In our previous work, MultiVelo9, we developed a model of how chromatin accessibility influences transcription rate. We observed two ways in which chromatin accessibility and gene expression can be “out of sync”: priming, when chromatin opens before transcription begins; and decoupling, when chromatin closing and transcriptional repression begin at different times. We also observed two possible orderings of chromatin and transcriptional events when a gene is repressed (Model 1 and Model 2). Using single-cell data, MultiVelo jointly estimates ODE parameters and assigns each cell to one of four states (primed, coupled on, decoupled, or coupled off) based on the relationship between chromatin accessibility and gene expression. Its ability to quantify the temporal dynamics and coordination between chromatin remodeling and transcriptional regulation at the single-cell level provided an important advancement to the RNA velocity field. However, MultiVelo assumed a single set of parameters for all cells in a population and assigned cells to discrete states, neglecting continuous state changes and the emergence of multiple cell types during differentiation.
A key motivation for developing MultiVeloVAE is to generalize MultiVelo to multi-lineage differentiation settings. This will allow identification of emerging differences among the transcription and chromatin opening rates as multipotent cells differentiate into multiple lineages, as well as cell-type-specific priming or decoupling differences. For example, three germ layers (endoderm, ectoderm, and mesoderm) emerge early in embryonic development through a series of molecular changes. Because MultiVeloVAE estimates cell-specific and gene-specific chromatin opening and transcription rates, it should allow improved velocity inference from single-cell multi-omic data in such multi-lineage contexts.
We generated a new 10X Multiome dataset from 7-day-old human embryoid bodies (EBs). Human induced pluripotent stem cells placed in microwell culture plates self-assemble to form EBs, which are 3D embryo-like in vitro systems. EBs can give rise to more than 70 cell types, including early members of nearly all human cell lineages, within 21 days. Thus, our new EB data provides an opportunity to both study the transcriptomic and epigenomic changes of early human differentiation and evaluate MultiVeloVAE on a multi-lineage system.
We performed multi-omic velocity inference on our new EB dataset using both MultiVeloVAE and our previous method MultiVelo. MultiVeloVAE accurately positions NANOG+ pluripotent cells as root cells and predicts differentiation into mesendoderm and ectoderm cell types33,34 (Fig. 3a). In contrast, MultiVelo’s result displays unexpected backflows. A key advantage of MultiVeloVAE’s design lies in inferring a single time per cell across all genes, thus avoiding issues with conflicting cell times that limit the accuracy of previous methods, including MultiVelo (Fig. 3b). Furthermore, while MultiVelo assumes that there is a single induction and repression phase for each gene, MultiVeloVAE can model lineage bifurcations using cell-wise chromatin and transcription rate parameters. This allows much more accurate fitting for genes whose expression and accessibility progressively diverge during cell fate determination. For example, differentiation markers such as PAX6, ENC1, and SAT1 exhibit heightened dynamics toward the early ectoderm, neuroectoderm, and mesendoderm lineages, respectively, while running parallel to other lineages along the inferred timeline. This enhanced dynamic modeling allows for clearer distinctions between cell-type transitions, which are intermingled in MultiVelo.

a Inferred velocity streams on a UMAP colored by cell types (Top) or latent time (Bottom), from MultiVeloVAE (Left) or MultiVelo (Right). b Generated spliced mRNA values of PAX6, ENC1, and SAT1 as functions of latent time, from either MultiVeloVAE (Left) or MultiVelo (Right). c MultiVeloVAE inferred velocity streams on the HSPC UMAP colored by cell types (Left) or latent time (Right). d MultiVelo latent time inference result for HSPCs. e Uncertainty of cell states captured by MultiVeloVAE via variational inference. f Example modality priming pattern and distribution captured by MultiVeloVAE for Wnt3 gene in a mouse skin dataset. The arrows on UMAPs indicate the expected differentiation order. From left to right, the UMAPs are colored by the original chromatin accessibility, unspliced counts, spliced counts, and cell-state difference computed as kc − ρ. g Generalized CBDir scores across four datasets over 5 steps of neighbors (higher is better). The means are shown as solid lines, and credible intervals are shown as ribbons. The metric is computed on either the entire gene space (Left) or embedded space (Right) h Runtime comparison (n = 5) with MultiVelo. The boxes represent the interquartile ranges (IQRs), the middle line represents the median, and whiskers extend to 1.5 times the IQRs. Source data are provided as a Source Data file.
MultiVeloVAE also shows significant advantages over MultiVelo on several additional 10X Multiome datasets from our previous paper. MultiVeloVAE successfully predicts cell-type transitions and latent time in the mouse brain dataset, capturing the developmental trajectories from radial glia to neurons or astrocytes, consistent with MultiVelo (Supplementary Fig. 7a, c). Notably, when generating a UMAP embedding based on the cell states inferred by MultiVeloVAE rather than the principal components of gene expression, we observe an improved separation of cell types and lineages (Supplementary Fig. 7a, bottom). For example, this UMAP based on the cell state distinctly separates oligodendrocyte progenitor cells (OPC) from astrocyte and neuronal groups, reflecting the established notion that oligodendrocytes are not attached to neuronal and astrocyte lineages5. As in the EB dataset, we observe better separation of lineage branches for neuronal marker genes, including Satb1, Gria2, and Grin2b, compared to MultiVelo (Supplementary Fig. 7b). The predicted cell transitions based on the noisier chromatin accessibility profiles (c) and inferred chromatin velocities (\(\frac{dc}{dt}\)) also better align with expectation, demonstrating the model’s denoising ability, unlike MultiVelo (Supplementary Fig. 7d).
In MultiVeloVAE, cell transitions and cell times are jointly encoded, which not only improves lineage prediction accuracy in the hematopoietic stem and progenitor cell (HSPC) sample9 but also corrects the latent time produced previously by MultiVelo (Fig. 3c, d, Supplementary Fig. 7e). Additionally, variational inference models the stochasticity around latent variables, allowing an assessment of the uncertainty in cell states. By overlaying this uncertainty, represented as multivariate normalized variation, on the UMAP embedding, we find that stem-like and multipotent progenitor cells exhibit higher uncertainty in lineage commitment, indicating the biological relevance of the inferred latent states. Moreover, MultiVeloVAE can accommodate complex cell-type compositions, as shown in a multi-omic human embryonic brain dataset35 (Supplementary Fig. 7f, g). In this instance, the model identifies the global stem cell type as a cycling population and correctly directs cells toward different developmental paths, while MultiVelo mistakenly designates mGPC/OPC as the root cell, resulting in erroneous backflows across major lineages.
MultiVeloVAE accurately orders cells in a mouse skin dataset36 (Supplementary Fig. 7h) and reconstructs marker expressions for inductive lineages, including the inner root sheath (IRS), medulla, and cuticle/cortex (Supplementary Fig. 7i). Previously, we identified a key regulatory gene in hair development with a distinct chromatin priming pattern, and we quantitatively measured delay in each time window by binning the cells along the latent time to assess chromatin accessibilities and spliced counts9. Since MultiVeloVAE models chromatin opening and transcription rates as continuous values rather than discrete phases, we can quantify the priming property on a per-cell basis by calculating the difference between these rate parameters. We mapped these differences onto the UMAP, alongside Wnt3 accessibility and expression levels (Fig. 3f). Indeed, this difference accurately captures the chromatin activation region preceding gene expression onset. In contrast, MultiVelo cannot model lineage branching, nor can it capture continuous priming patterns, causing it to incorrectly infer Wnt3 priming for all the cells differentiating toward the IRS lineage due to relatively low RNA count. In contrast, MultiVeloVAE correctly infers separate priming states for the IRS lineage and the lineage that truly has Wnt3 priming. (Supplementary Fig. 7j). Interestingly, MultiVeloVAE’s inferred latent time exhibits the highest correlation with diffusion-based pseudo-time (used by the authors to illustrate trajectories) compared to both scVelo and MultiVelo (Supplementary Fig. 7k).
We inspected the cell-specific chromatin opening and transcription rate parameters inferred by MultiVeloVAE. We observed that the parameters accurately reflect the induction and repression stages of a gene and recapitulate the priming and decoupling patterns we previously reported for genes like Satb2 and Gria2 (Supplementary Fig. 8a). The inferred velocities, when plotted along latent time, can be qualitatively examined to confirm that they are consistent with the derivatives of their associated modalities. (Supplementary Fig. 8b) In addition, how well the velocities of individual genes conform to global directionality can be assessed by computing a coherence score between the gene’s velocity and the expected spliced count displacement towards the next-step neighbors, as implemented in VeloVI13. As expected, genes with low coherence scores towards both neuronal and astrocyte lineages show phase portraits with more ambiguous patterns (Supplementary Fig. 8c).
To quantitatively compare our method with MultiVelo, we computed k-step CBDir and Mann–Whitney U statistics in both the original gene expression space and the embedded UMAP space across five multi-omic datasets (Fig. 3g, Supplementary Fig. 8d). MultiVeloVAE achieves higher mean cell-type transition accuracy than MultiVelo across up to five neighboring steps. Additionally, MultiVeloVAE runs significantly faster than MultiVelo by leveraging GPU-accelerated gradient descent (Fig. 3h).
Multi-sample multi-omic velocity inference removes technical variation and preserves biological variation
Many important biological research settings, such as case-control studies, multi-subject genetic association studies, and developmental atlases, require comparing multiple single-cell datasets. These multi-sample comparisons are challenging due to the presence of both important biological variation and nuisance technical variation. Various single-cell integration methods have been developed and are widely used as standard procedures in the field15,16,17,37. However, these methods are not designed for trajectory or velocity inference, requiring users to perform ad hoc procedures. For example, in our previous MultiVelo paper, we performed a two-step procedure where we “corrected” the (c, u, s) values from two samples with an integration method, then fit MultiVelo on the corrected values. However, chaining single-cell integration and velocity inference methods in this way does not properly incorporate sample covariates into the velocity inference process, does not allow statistical hypothesis testing for sample comparison, and can accumulate errors. Thus, a velocity model that incorporates sample covariates directly into the inference process offers significant advantages.
MultiVeloVAE addresses these challenges by introducing multi-sample inference for both cell states and ODE parameters. In this approach, the latent cell embedding variable is conditioned on known sample covariates, enabling the neural network to identify cell state variation independent from sample covariates. Additionally, gene-specific rate parameters are modeled separately for each sample, allowing inference of sample-specific effects. This training process results in an integrated cell state space that is jointly learned from multiple samples. Note that, as with related approaches such as scVI, the integration occurs only in the latent space–high-dimensional (c, u, s) values will still reflect differences among samples. After training, we can generate counterfactual “corrected” (c, u, s) counts in the high-dimensional gene space by taking cell states from one sample and decoding them with the parameters from another sample.
We demonstrate MultiVeloVAE’s multi-sample inference capability on two 10X Multiome datasets generated from human hematopoietic stem and progenitor cells (HSPCs). Both samples were isolated from human bone marrow and cultured for seven days, but the samples came from different donors, and their library preparation and sequencing were performed months apart. This led to substantial batch effects despite comparable cell-type composition (Fig. 4a, top). With the presence of batch effects, single-sample-only velocity methods such as MultiVelo can be easily biased by the library size and unspliced-spliced ratio differences between samples (Supplementary Fig. 9a). Performing batch correction using separate approaches prior to velocity inference risks disrupting the intricate relationship between modalities. For instance, we attempted to use Scanorama38 to extract batch corrected modality counts jointly from the two samples; however, the corrected modalities no longer support successful multi-omic velocity prediction afterwards (Supplementary Fig. 9b, c).

a The UMAP coordinates obtained from original concatenated gene expression of the two samples (Top) and latent cell embedding from MultiVeloVAE after batch correction (Bottom). The same sets of UMAPs are colored by cell types, batch labels, inferred latent time, and CD133 expression. Velocity streams denote batch-corrected lineage predictions. b Phase portraits and dynamic plots of several lineage marker genes colored by batch label and cell types, before (Top) and after (Bottom) batch correction. c Distributions of rate parameters for the two batches. d Performance benchmarking of batch effect removal metrics from scIB. e Performance benchmarking of biological variance conservation metrics from scIB. Source data are provided as a Source Data file.
After multi-sample velocity inference with MultiVeloVAE, previously disjoint cell types merged cohesively as seen on a UMAP based on latent cell states. The multi-sample velocity inference also correctly identified hematopoietic stem cells as precursors of dendritic cells, granulocytes, erythrocytes, and megakaryocytes39,40 (Fig. 4a, bottom). The earliest cells align with expression of CD133, a hematopoietic stem cell marker. Detailed examination of marker gene dynamics shows that integration merges gene expression profiles without compromising the ability to model complex multi-lineage differentiation (Fig. 4b). Additional batch-corrected phase portraits of chromatin accessibility and transcription dynamics for high-likelihood genes further validate the quality of results (Supplementary Fig. 10). Reassuringly, the distributions of rate parameters across the two batches largely overlap, which is expected due to the similar cell type composition of the two samples (Fig. 4c).
Variation between samples involves both important biological signals and unwanted technical artifacts. Thus, an ideal integration method should balance between removing technical effects and preserving biological variation. To assess this aspect of our approach, we benchmark MultiVeloVAE against the two best-performing single-cell integration methods from a recent benchmarking paper, Scanorama38 and scVI17. We benchmarked performance using the same evaluation code and metrics used in the benchmarking paper19 (Fig. 4d, e). MultiVeloVAE demonstrates comparable performance with Scanorama in batch effect removal and outperforms it on graph integration metrics, including the local inverse Simpson’s Index (iLISI) and the k-nearest-neighbor batch effect test (kBET) when evaluated in the embedded space (Fig. 4d). Although it scores lower in batch effect removal metrics compared to scVI, MultiVeloVAE ranks consistently higher in biological conservation metrics than scVI (Fig. 4e). These results indicate that MultiVeloVAE retains biological variation while integrating samples with technical variation, achieving our goal of multi-sample inference of gene dynamics via biochemically principled differential equations.
MultiVeloVAE infers state-specific decoupling patterns
In our previous MultiVelo paper, we characterized two different regulatory relationships between chromatin accessibility and gene expression: priming, in which a gene becomes accessible before it is transcribed, and decoupling, in which chromatin accessibility and transcription change at different times during gene repression. We also identified two possible orders of events–Model 1 and Model 2, which differ by their decoupling phases. In brief, both types of genes experience discrete phases where chromatin and transcription are simultaneously on (coupled induction) or off (coupled repression). In a Model 1 gene, chromatin starts to close before transcription terminates, while in a Model 2 gene, transcription is terminated before chromatin begins closing. These out-of-sync regulatory patterns can have different biological implications—a recent publication has experimentally studied the interplay between the two modality states and found that distinct cell states can affect splicing outcomes41.
MultiVeloVAE now models chromatin accessibility and transcription with continuous, cell-specific rate parameters kc and ρ. Comparing the values of kc and ρ also allows us to identify the priming and decoupling phenomena we reported in the MultiVelo paper, as well as the Model 1 vs. Model 2 distinctions. Now, however, the notions of coupled and decoupled states are no longer discrete but continuous and cell-specific. That is, rather than inferring a single value for the amount of coupling or decoupling for a gene across all cells, the chromatin accessibility and transcription of a gene can now be coupled or decoupled to varying degrees across a population of cells. This allows us to identify cell-type-specific variation in gene regulation.
We define a cell’s decoupling factor δ for a gene as the difference between the cell’s chromatin opening rate (kc) and its transcription rate (ρ). We also define the coupling factor κ as their centered sum (see Methods for details). The decoupling factor ranges from −1 to 1. A value of δ = 1 indicates that the chromatin opening rate is greater than the transcription rate, analogous to the “priming” state of MultiVelo as well as the “decoupling” state in Model 2 of MultiVelo. A value of −1 is analogous to the decoupling phase of Model 1 genes. The coupling factor κ also ranges from −1 to 1. A value of κ = 1 indicates coupled induction (analogous to the coupled-on state of MultiVelo) and a value of −1 indicates coupled repression (analogous to the coupled-off state of MultiVelo). Thresholding δ and κ gives discrete states analogous to MultiVelo’s discrete states. Note that δ and κ are now also different for every cell and every gene, so that we can identify cell-type-specific priming and decoupling.
We examined the coupling and decoupling factors inferred by MultiVeloVAE for some lineage markers in the HSPC datasets. Following the multi-sample velocity inference from the previous section, we obtained batch-corrected modality counts and velocity profiles of the two HSPCs. We observed that genes often had κ ≈ 1 (coupled induction) primarily in cells differentiating toward a specific cell fate and κ ≈ −1 (coupled repression) in cells differentiating toward the other fates (Fig. 5a). Interestingly, when comparing the multi-lineage coupling and decoupling patterns together, the decoupling factors show more intermediate stories of how the lineage bifurcations occur (Fig. 5b). For example, δ ≈ 1 during the down-regulation of HDC, similar to the decoupling phase of a Model 1 gene. Genes like AZU1 and LYZ show phases with δ ≈ −1, similar to the decoupling phase of a Model 2 gene. Many genes exhibit both δ > 0 and δ < 0 in different cell types.

a The dynamics of chromatin, unspliced RNA, and spliced RNA for several lineage markers, colored by continuous coupling factors, range from −1 (coupled repression) to 1 (coupled induction). The diverging colormap centers around 0. The inset shows the RNA expression of the marker genes on the UMAP embedded cell states from Fig. 4a. b Similar to a but colored by continuous decoupling factors, range from −1 (kc = 0, ρ = 1) to 1 (kc = 1, ρ = 0). c Scenic + gene regulatory network (GRN) containing all transcription factors (TFs) and genes participated in velocity inference. The TF nodes are labeled with the TF names. Region nodes regulated by the TFs are colored by their differential accessibility on a log2 scale in the specified cell type. The edges linking regions to target genes are colored by accessibility-expression correlations and transparent by region-to-gene importance from Scenic+. The target gene nodes are colored by cell-type-specific mean coupling factors and transparent by the magnitudes. For each cell type (Top-Platelet and Bottom-DC), the activated TF-regulated triplets are circled. d The Spearman correlation coefficients of coupling (Left) and decoupling (Right) factors of target genes with the RNA expressions of the TFs. e The mean predictions and credible intervals of posterior-sampled cells along with a zero line colored by cell types. f Example normalized dynamics plots of a TF's RNA, the associated region accessibility, and the target gene’s chromatin, unspliced, and spliced counts plotted along inferred latent time for specified lineages. Modality counts were reconstructed by MultiVeloVAE. Source data are provided as a Source Data file.
We next wanted to investigate how coupled and decoupled states relate to gene regulatory interactions. To do this, we ran Scenic + 42 to infer enhancer-gene regulatory networks from one of the HSPC samples. Scenic + predicted transcription factors (TFs) that regulate target genes through chromatin accessibility peaks. We focused on 16 TFs that were in both MultiVeloVAE results and Scenic+ results and were expressed at multiple HSPC differentiation stages (Fig. 5c and Supplementary Fig. 11a). Most regulatory interactions predicted by Scenic + are positive interactions, but there are some negative linkages as well. We then labeled region nodes with accessibility logFC in a specific cell type and marked gene nodes with average coupling factors in the cell type. In Platelet and DC, the genes linked to the highlighted genomic regions show overall patterns of coupled-induction compared to the other nodes in the regulatory network, suggesting that the TFs promote differentiation toward a particular fate. In contrast, the same genes show mostly positive decoupling values in cells from the HSC cluster, indicating that these cells are in an initial priming state (Supplementary Fig. 11b).
During erythroid differentiation, the dominant effect of TFs gradually switches from GATA2 to GATA143,44. When plotting CMP, MEP, and terminal Erythrocyte cell types sequentially for genes that are positively differentially expressed in Erythrocyte lineage, the coupling factors of genes linked to GATA1 show notable increases, while genes linked to GATA2 show minor change (Supplementary Fig. 11c). However, when we examine the decoupling factors of several genes linked to GATA2, such as Granulocyte marker HDC, they obtain negative decoupling patterns as cells mature into erythrocytes, indicating that they have been epigenomically repressed. While the coupling factors tell overall patterns of a gene’s association with a TF or a lineage, the decoupling factors display the details of multi-omic gene regulation. In general, we found that for TF-gene pairs identified by the regulatory network grouped by positive or negative regulations, the coupling factors of cells subset to non-background lineages of a gene (see Methods for details) are positively associated with the regulatory effects of TFs (Fig. 5d). In contrast, the decoupling factor is, in general, inversely associated with the TF’s RNA level. This makes sense because a TF with a positive regulatory effect can quickly prime a gene at the very beginning towards the induced trajectory, causing a high decoupling status initially, while it quickly returns to a more stable coupled-induced status later on with both chromatin open and transcription activated. A TF with a negative regulatory effect will cause a gene to be epigenomically repressed at later stages, like genes down-regulated by GATA2. Both scenarios would result in a positive association with the TF’s expression level. We also inspected housekeeping genes, which need to be constitutively expressed. One would not expect these genes to be epigenomically repressed through the differentiation timeframe, and indeed, their decoupling status remains positive (see Methods for details) (Fig. 5e). We show several detailed normalized dynamics of several TF-region-gene dynamics within different categories of regulations to demonstrate the TFs’ effects on downstream target genes both epigenomically and transcriptomically, including initial priming and chromatin-wise repression (Fig. 5f, Supplementary Fig. 12).
Our approach models c, which is the sum of accessibility across all peaks linked to a gene. However, individual cis-regulatory elements can play important and diverse roles in regulating transcription kinetics. Although MultiVeloVAE does not directly model the effects of individual cis-regulatory elements, we can perform downstream analyses to investigate the influence of individual peaks. To do this, we can correlate the accessibility of individual peaks with the rate parameters inferred by MultiVeloVAE. As an example, we look at a Granulocyte marker gene SRGN and its associated chromatin accessibility peaks (Supplementary Fig. 13a). The peaks that are highly correlated with either promoter accessibility or gene transcription levels are boxed. We can identify peaks whose accessibility is associated with a gene’s transcription by calculating mutual information (MI). MI accurately identifies the strongly connected peaks and gives the 10th peak a high score. Looking more closely, this peak is specifically correlated with the GMP lineage of the gene, which can be visually examined on cell-type-specific coverage plots in Signac. This peak may contribute to the gene’s moderate expression levels in GMP.
To investigate the histone marks associated with coupled vs. decoupled states across the multiple cell types in our datasets, we examined ENCODE ChIP-seq data. Additionally, we computed the correlation between the accessibility of each gene-linked peak and the decoupling factor of each gene (Supplementary Fig. 13b). A higher correlation value for a peak indicates that accessibility of that peak is associated with a decoupled state for the gene. Similarly, we calculated the correlation between the accessibility of each peak and the coupling factor of its linked gene. Next, we intersected our peak locations with chromatin state annotations from ChromHMM45. This allowed us to ask the following question: Which ChromHMM states are associated with a high or low coupling or decoupling factor?
A handful of chromHMM states are positively associated with decoupling: BivProm1, EnhWk4, HET4, PromF4, PromF5, Quies5, TSS1, and TSS2. The associations with Prom and TSS states make sense because our model assumes that promoter and TSS accessibility are positively associated with chromatin opening, thus boosting the decoupling or priming factor in our model when a gene is first induced. The association with heterochromatin (HET4) and quiescent (Quies4) states may indicate that decoupling occurs when peaks in heterochromatin regions are first opened. Bivalent promoter (BivProm1) and weak enhancer (EnhWk4) associations may indicate that decoupling happens when cis-regulatory regions are being established by histone modification changes. Similarly, several ChromHMM annotations show the highest association with coupled states: Acet1; BivProm1,2, and 3; PromF3, 4, and 5; Polycomb repressed (ReprPC1); and TSS 1 and 2. Although more work is needed to investigate these connections, our results suggest that there are interesting histone mark differences among peaks that are related to decoupled vs. coupled states.
MultiVeloVAE identifies genes with differential dynamics during macrophage differentiation
A key goal of analyzing single-cell data from differentiating cells is to identify dynamic changes in gene expression among different cell populations during cell fate transition. However, previous RNA velocity approaches cannot perform such differential dynamic analyses because they fit shared parameters for all cells (no cell-specific parameters) and/or do not use a statistically principled approach that allows hypothesis testing. MultiVeloVAE overcomes these limitations to enable statistically principled differential dynamic analysis of single-cell multi-omic data. This capability enables exciting new types of analyses, such as finding genes whose transcription rate or chromatin opening rate diverge as two differentiated cell types emerge from a single progenitor population.
Building on the variational Bayes inference framework of MultiVeloVAE, we developed a Bayesian differential testing approach to identify genes with differential dynamics between two groups of cells. We followed a common Bayesian approach to hypothesis testing by calculating Bayes factors to determine the relative probability of two hypotheses based on both the prior distribution and the observed data. Intuitively, our testing approach consists of sampling repeatedly from the posterior distributions of cell state for cells from two groups, then comparing the corresponding variables from the sampled cell states. Our testing framework can assess any of the variables estimated by MultiVeloVAE, including chromatin and transcription rate parameters, chromatin accessibility, spliced and unspliced mRNA expression levels, and chromatin or RNA velocity. For variables that are negative or are constrained between 0 and 1, we utilize a log-difference (LD) scale for group comparisons instead of the more conventional log fold change (LFC) scale. To account for multiple hypothesis testing, we control the false discovery proportion expected under the posterior. Further details of the differential test framework are provided in Methods.
Having developed this new differential dynamics framework, we used it to study gene expression and chromatin accessibility changes during macrophage differentiation. We followed a previously published protocol to generate a new 10X Multiome dataset from human HSPCs. We further cultured an additional group of HSPC cells for seven more days under cytokine treatment to induce progenitor cells toward macrophage differentiation. We then integrated these more differentiated cells with one of the HSPC samples. Comparing the UMAP embeddings before and after integration (Fig. 6a), we observed that cell types form biologically meaningful connections across the two samples. As anticipated, the cytokine-treated sample contains a much higher proportion of cells on the macrophage-associated lineage due to directed induction. The main cell types in this lineage include lymphoid-myeloid-primed progenitors (LMPPs), granulocyte-macrophage progenitors (GMPs), monocyte-dendritic-cell progenitors (MDPs), monocytes, and polarized M1/M2 macrophages46 in the expected differentiation sequence47 (Supplementary Fig. 14a). In contrast, only a few cytokine-treated cells align with the DC lineage, represented by progenitor DCs (Prog DC) and DCs, which share common progenitors with macrophages. The inferred latent time aligns with expected maturation stages; specifically, lineages toward both macrophages (D14 monocyte, M1 macrophage, and M2 macrophage) and DCs (Prog DC and DC) exhibit progressively higher median cell-time predictions (Supplementary Fig. 14b). For cell types shared between the two samples, such as erythrocytes, granulocytes, mast cells, and platelets, cells from the cytokine-treated sample are positioned later in latent time (Supplementary Fig. 14c). We used CellRank32 to explicitly delineate the terminal states given MultiVeloVAE’s batch-corrected velocities, and the connectivities between cell types align well with biological expectations (Supplementary Fig. 14d). The distribution of rate parameters between the two samples is largely overlapping (Supplementary Fig. 14e), suggesting that the differences between the samples lie in numerous small shifts in chromatin and transcriptional rates for individual genes, while the parameters lie in similar ranges in both samples.

a UMAP plots of newly generated 10X Multiome data from human hematopoietic stem and progenitor cells before cytokine treatment (blue) and 7 days after treatment with pro-macrophage cytokines (orange). The top panel shows unintegrated cells, and the bottom panel shows cells after multi-sample velocity inference. Both panels are colored by the same cell type annotations. b Top: Volcano plot of genes with significant differential velocity between 5000 posterior-sampled cells generated from each of macrophage (n=850) and dendritic cell (n = 221) clusters. Gene groups with p < 0.05 and log differences smaller than −3 or larger than 3 are colored green or blue, respectively. The displayed P-values were not adjusted for multiple testing, but all genes shown have been verified to have a False Discovery Rate < 0.05. Bottom: UMAP plots of cells differentiating toward either macrophage or DC fates, colored by velocity for each of the significant genes from the top panel. c, d Genes with differential dynamics between macrophages and dendritic cells. Each column shows the differential dynamics results for a single gene. Each row corresponds to a different parameter: chromatin opening rate kc, transcription rate ρ, velocity \(\frac{ds}{dt}\), and spliced count s. The x-axis of each plot represents latent time, and the y-axis is the log difference or fold change between two populations being compared (macrophages vs. DCs in this case). The color of each point is the Bayes factor for a change between populations within a time bin. The gray shaded area is the 95% credible interval for a Gaussian process fit to the log difference or fold change across latent time. The p-value is from a likelihood ratio test with one degree of freedom for whether the Gaussian process fits better than a null model not including time as a predictor. The inset scatter plots show integrated c, u, and s values (y-axis) as a function of latent time (x-axis), colored by cell type, for the corresponding gene. c Comparing macrophages against DCs. d Similar plots to c but comparing DCs against macrophages.
Following integration of the HSPC and macrophage samples, we identified genes with differential induction along the macrophage lineage vs. DC lineage. We selected genes with significantly different RNA velocity (p < 0.05 and FDR < 0.05). Reassuringly, many of the top-ranked genes with differential kinetics in macrophages vs. DCs are marker genes specific to either macrophage or DC populations (Fig. 6b). Similarly, we identified genes with differential velocities in erythrocyte and megakaryocyte lineages (Supplementary Fig. 15a) as well as for each of the three EB lineages (Supplementary Fig. 16).
The parameters fit by our model provide an unprecedented opportunity to study the differential dynamics of genes over time. Single-cell technology enables us to examine each gene with high temporal resolution rather than limiting comparisons to two static conditions, as in bulk sequencing. For example, we can compare differentiating cells to find genes whose transcription rates diverge as two distinct cell fates emerge. To achieve this, we first sampled pseudo-cells from the two conditions through posterior sampling with batch correction, creating two continuous pools of cells in latent time. We then calculated a test statistic (either log difference, LD, or log fold change, LFC) assessing the difference between two cell populations at each latent time point. We also used Gaussian process regression to test whether the LD or LFC values showed a time-varying trend by performing a likelihood ratio test between a time-varying model and a time-constant null model. Intuitively, genes selected by this procedure show a time-dependent divergence between two groups of cells, such as would be expected for a lineage-specifying gene that is activated in one cell type while it is repressed in another. These analyses revealed many genes with iterative priming patterns along latent time, in which chromatin opening rate (kc), transcription rate (ρ), spliced velocity (\(v=\frac{ds}{dt}\)), and spliced counts (s) show coordinated changes over time (Fig. 6c). The genes we identified show not only temporal shifts in their rate parameters but also temporally varying LD and LFC between cell types. For example, the PROS1 gene shows the highest difference in chromatin opening and RNA transcription rates between macrophages and DCs early in latent time, the highest velocity difference in the middle of latent time, and the highest fold change in spliced count late in latent time. This indicates that the PROS1 gene is primed by early chromatin accessibility and transcription, then establishes stable high expression in macrophages late in the latent time. Similar trends are also seen for LGMN and LGALS3. We performed a similar analysis to identify genes differentially primed toward the DC lineage compared to macrophages (Fig. 6d) and performed two-way comparisons between erythrocyte and megakaryocyte lineages (Supplementary Fig. 15b, c). We further demonstrate another application of our approach by TFs with differential velocity between lineages, which represent candidate driver TFs. In Supplementary Fig. 15d, we used the human TF list provided by the cisTarget database42 to select TFs with differential velocity among the aforementioned lineages. All of these TFs display clear branching events, consistent with possible roles as driver TFs. Interestingly, the chromatin dynamics of ZNF385D and ARID5B show decoupling patterns towards Platelet and Mast Cell lineages, respectively, compared to their own unspliced and spliced dynamics.
We can also use our differential testing framework to identify cell-type-specific decoupling between chromatin accessibility and transcription. To do this, we can simply compare δ and κ between two groups of cells (Supplementary Fig. 17a). We focus on three genes used in our previous study9 from the 10X Genomics multi-omic mouse brain sample: Robo2, Satb2, and Gria2, which show different epigenomic and transcriptional dynamics inside the neuronal lineage. The same patterns of priming and decoupling that we previously identified are recognizable in the dynamics of chromatin and unspliced counts. Specifically, the later neuronal cell types show coupled repression of Robo2 and Satb2, whereas Gria2 shows a decoupled pattern with open-chromatin and repressed transcription. We test each category of the two factors within each cell type against the surrounding background cell types within the lineage, for example, testing Upper Layer cells against combined V-SVZ and Deeper Layer cells, to find genes differentially enriched for a certain factor within the cell type. Robo2 shows coupled induction in the V-SVZ layer, and Satb2 shows coupled induction in the Upper Layer. Robo2 switches to coupled repression in both Upper Layer and Deeper Layer, whereas Satb2 shows coupled repression only beginning at the Deeper Layer. Gria2 is differentially decoupled when testing posterior-sampled cells from both Upper Layer and Deeper Layer together against V-SVZ, and this is consistent with the story in the dynamics plots. The relationship between the states can also be directly examined in the Cartesian coordinates of kc and ρ.
MultiVeloVAE generates unobserved multi-omic profiles from partially overlapping modalities and in silico perturbations
A key advantage of the MultiVeloVAE framework is its ability to reconstruct multi-sample, multi-omic data from a latent representation. This allows two related capabilities, which we demonstrate in this section: (1) multi-sample velocity inference from partially overlapping modalities such as 10X Multiome and scRNA, and (2) prediction of velocity after in silico perturbation. Although single-cell multi-omic measurements offer direct linkage between chromatin and transcription profiles within the same cell, making them ideal for studying epigenomic regulation, single-cell RNA-seq remains the more widely used sequencing method, with a vast number of publicly available datasets across diverse cell and tissue types. Here, we demonstrate for the first time that scRNA-seq data and single-cell multi-omic samples can be used for joint velocity inference and prediction of missing chromatin information for RNA-only samples.
This functionality is enabled by two key features of our model: (1) the conditional VAE architecture, which incorporates multi-sample data to separate shared biological variation from sample-specific effects, and (2) the multi-omic velocity ODE framework, which provides the mechanistic link between chromatin and RNA dynamics. By integrating both multi-omic and RNA-only samples, we can jointly learn gene dynamics from all samples and predict chromatin accessibility profiles for RNA-only samples.
As a demonstration, we processed a public scRNA-seq bone marrow sample from a healthy donor48 and integrated it with two of our multi-omic HSPC datasets. Following integration, most cell types annotated separately in each dataset align well in a unified cell-state UMAP plot (Fig. 7a), with the exception of the progenitor B-cell cluster (Prog B), which comes exclusively from the bone marrow sample. The predicted lineage tree originates from an HSC cluster, validated by CD133 expression. Principal components (PCs) of the latent cell-state variable confirm effective integration and preservation of cell-type distinctions (Supplementary Fig. 18). To assess integration quality, we examined 3-dimensional phase portraits of marker genes for erythrocytes (HBB), granulocytes (HDC), and dendritic cells (LYZ) (Fig. 7b). Remarkably, these genes align across all three dimensions, including the chromatin axis, indicating a successful generation of chromatin accessibility values for the scRNA-seq sample.

a Integration results similar to Figs. 4a and 6a. b 3D phase portraits of original and batch-corrected c-u-s values of three lineage marker genes. c Dynamic plots of generated chromatin, unspliced, and spliced values along latent time for only scRNA-seq cells. The chromatin values are unobserved in the input. d Generated chromatin values by time with arrows indicating inferred velocities of randomly selected cells. The Top plot shows only scRNA-seq cells colored by cell types. The Bottom plot shows cells from all three datasets colored by batch labels. e In silico knockout (KO) of SPI1 (PU.1). The mRNA expression of SPI1 is shown on the batch-corrected UMAP. The UMAP on the top right shows the predicted perturbation force as velocity arrows. The bottom-right UMAPs show the first principal component of the absolute differences between perturbed and unperturbed cell states, as well as the absolute differences between the inferred latent time before and after perturbation. f Similar to e but for GATA1 knockout. g Change of fate probabilities from CellRank before and after perturbation. Sample size (cell number) n for each group: Platelet=651, Erythrocyte=3043, Mast Cell = 476, Granulocyte = 1211, DC = 294, and Prog B = 668. The boxes represent the interquartile ranges (IQRs), the middle line represents the median, and whiskers extend to 1.5 times the IQRs. h CellOracle inferred identity shift after perturbation. Source data are provided as a Source Data file.
Furthermore, the generated chromatin profiles for the scRNA-seq sample exhibit dynamic properties. For example, predicted profiles of lineage markers HBB, HDC, LYZ, and PF4 (platelet marker) for the scRNA-seq data all display modality priming patterns, with chromatin accessibility preceding transcriptional activation (Fig. 7c). The chromatin velocity vectors for these genes show expected differentiation directions, accurately capturing multi-lineage trajectories (Fig. 7d, top). Interestingly, comparing chromatin dynamics across the three samples reveals that the bone marrow scRNA sample has a shorter inferred temporal duration than the two multi-omic HSPC datasets (Fig. 7d, bottom).
We benchmarked the ATAC-seq prediction quality against scButterfly49, scCross50, and MultiVI51, by treating one of the multi-omic HSPC samples as RNA-only and integrating it with the other one. The predicted and true accessibilities of top-likelihood genes after velocity inference from MultiVeloVAE show overall high Pearson correlation coefficients (Supplementary Fig. 19a). The performance of our model is on par with the scButterfly and better than both scCross and MultiVI in terms of correlation and mean squared errors (Supplementary Fig. 19b, c), though scButterfly does not provide any functionality to do trajectory inference. We ran MultiVeloVAE with each method’s inferred ATAC profile. The dynamic chromatin plots of individual genes, such as the HSC and LMPP marker SPINK2, allow us to qualitatively assess each method’s abilities to retain velocity-guided biological variation (Supplementary Fig. 19d). MultiVeloVAE-generated ATAC counts have the clearest lineage separation. In addition, MultiVeloVAE achieves the highest GCBDir score compared to the other methods and the baseline that uses ground-truth ATAC-seq values (Supplementary Fig. 19e).
Having established that MultiVeloVAE can generate reasonable out-of-sample predictions, we explored whether this capability allows predictions of genetic perturbation effects. To do this, we performed in silico perturbation of two key hematopoietic transcription factors: SPI1 (PU.1) and GATA152,53. These factors are regulated by mutual inhibition and orchestrate the early priming of multipotent cells into distinct fates. To simulate a knockout (KO) in silico, we set the c, u, s values to 0 while leaving other values unchanged, following a previous study54,55,56. Passing the modified input through the pre-trained model yielded new latent cell states and time variables and allowed us to predict outcomes post-perturbation. We computed “perturbation force” as the shifts in RNA velocity induced by the simulated KO (Fig. 7e, f). The KO of SPI1 reversed differentiation flows toward GMP-associated lineages, such as DCs (Fig. 7e), while GATA1 KO disrupted downstream lineages of MEP, including megakaryocytes and erythrocytes (Fig. 7f). These results are similar to those reported in the Dynamo paper using single-cell RNA-seq data52. Principal component analysis (PC1) of the differences in perturbed and original cell states (z) as well as the differences in latent times both highlight these fate disruptions, particularly in the promoted cell types. We applied CellRank32 to explicitly compute the probabilities of transitioning towards each terminal cell state before and after KO of two genes (Fig. 7g). The change in fate probabilities shows an increase in MEP-associated lineages, such as Platelet and Erythrocyte, and a drop in DC lineage, and vice versa for GATA1. We also compared with CellOracle’s53 perturbation predictions. Because CellOracle does not have an innate ability to integrate multiple samples, we supplied batch-corrected counts to it, and its inferred identity shifts align with MultiVeloVAE’s perturbation force vectors well (Fig. 7h). These results show that the in silico knockdown effects are qualitatively reasonable and consistent with the known roles of SPI1 and GATA1.
Discussion
The MultiVeloVAE framework uses variational Bayesian inference to model molecular changes during cell differentiation in a statistically principled fashion. Our approach not only improves gene fitting in many cases but also resolves some of the key limitations of previous RNA velocity methods. The accurately reconstructed measurements of individual genes on a unified time scale enable direct comparisons of epigenomic and transcriptional dynamics within and across genes. MultiVeloVAE adapts to multi-modal kinetics across complex biological systems while retaining interpretability via a differential equation model. The latent chromatin and transcription states capture lineage-specific properties while also accounting for uncertainty in single-cell measurements. MultiVeloVAE also generalizes the notions of decoupled and coupled states, allowing us to model cell-type-specific relationships between chromatin accessibility and transcription in multi-lineage differentiation processes. MultiVeloVAE can also perform multi-sample inference when conditioned on known batch labels, opening new possibilities for studying dynamic changes between conditions. Furthermore, the integration is robust and flexible as it handles datasets with only partially overlapping modalities, such as 10X Multiome and scRNA-seq.
There are, however, notable challenges and areas for future development. Like other velocity-based methods, MultiVeloVAE’s inference outcome depends on the quality of unspliced and spliced RNA measurements, which can lead to difficulty in mature cell types with reduced differentiation potential, such as PBMCs. For effective integration across datasets, reliable velocity inference is needed for every dataset to ensure consistent results. Additionally, current velocity inference methods, including deep-learning-based approaches, rely on de novo training. Recent efforts to integrate new data with atlas-level datasets have shown promise for cell-type annotation. Developing pre-trained and validated parameter sets for known cell types could benefit applications that require generic velocity inference on similar, low-quality samples. In the future, it may be promising to extend the approach to additional types of single-cell data, such as CITE-seq. Overall, MultiVeloVAE represents a significant advancement in RNA velocity analysis, with enhanced inference accuracy, multi-sample support, and interpretable kinetic parameters. We anticipate that MultiVeloVAE will be especially valuable for exploring gene dynamics in settings requiring multi-sample comparisons, such as case-control studies, developmental atlases, and studies of genetic variation.
Methods
Problem setup
We formulate the computational problem of modeling cellular dynamics using mathematical equations of biochemical processes. In general, single-cell experiments produce cell-by-feature count profiles. RNA reads are further classified as either unspliced or spliced, corresponding to two matrices. Each observation (cell), indexed by i, can be represented by a vector xi(ti) parameterized by unobserved time ti. We seek to infer the cell time ti and predict the velocity of changes dxi/dti.
We denote cg, ug, and sg as the accessibility and expression values of the g-th gene. Extending upon our previous work57, we define (1) the feature vector of a cell as \({{{\bf{x}}}}={[{c}_{1},{c}_{2},\ldots,{c}_{G},{u}_{1},{u}_{2},\ldots,{u}_{G},{s}_{1},{s}_{2}\ldots,{s}_{G}]}^{T}\), (2) the kinetic equation of the g-th gene as a system of ordinary differential equations relating changes in cg, ug, and sg over time. The RNA velocity of the gene is defined as ds/dt4.
Modeling chromatin accessibility and gene expression kinetics
As first proposed in 20184, the kinetic equation of RNA velocity is modeled by a system of two linear ODEs:
with model parameters ρ × α, β, and γ correspond to gene-specific transcription rate, splicing rate, and degradation rate, respectively. Note that α is a scale factor controlling the maximum possible transcription level for each gene and is constant across cells. In contrast, ρ is cell-specific. The original RNA velocity formulation by La Manno et al.4 does not make any assumption about the transcription rate except for cells in steady states and cannot explicitly estimate α, cell-specific or otherwise. The RNA velocity formulation in the scVelo paper5 can be seen as a special case of this equation, in which ρ is restricted to be an indicator function: \(\rho :={I}_{\{t < {t}_{off}\}}\). The scVelo model assumes that two discrete phases can occur in the gene expression process: (1) induction, when new unspliced RNA molecules are being transcribed, and (2) repression, when the transcription process stops and no new unspliced molecules are made. The induction phase is assumed to start at ton = 0, and the transition from induction to repression occurs at a later time point toff. In our previous VeloVAE paper57, we explicitly estimated ρ as a cell-specific, real-valued relative transcription rate in [0, 1] (Supplementary Fig. 20a). Given an initial condition u(0) = u0, s(0) = s0, the analytical solution to the ODE is
The steady states of the system are given by the limiting values of the equations as time grows without bound:
In our previous work, MultiVelo9, we added a single variable c ∈ [0, 1] to represent the chromatin accessibility associated with a gene (Supplementary Fig. 20a). Such chromatin accessibility is obtained by combining the openness of a gene’s promoter and correlated, nearby enhancer regions as an indication of how easily transcription machinery can bind. We link this epigenomic property to the rate of unspliced mRNA production by multiplying it by the gene’s transcription rate.
Solving these equations analytically, we obtain:
Note that α and αc are gene-specific scale factors shared across cells. Analogous to the cell-specific transcription rate ρ, the parameter kc models the cell-specific chromatin opening rate. In the MultiVelo paper, we modeled kc as a binary value to indicate whether chromatin is opening (1) or closing (0). This is conceptually similar to scVelo’s approach. We also previously proposed a second-order ODE to describe chromatin accessibility as a continuous-time stochastic Markov process in which accessibility rapidly transitions between two discrete theoretical boundary states {fully-closed, fully-open} (Supplementary Fig. 20a). Instead of attempting to solve this complex stochastic ODE to describe the chromatin accessibility analytically, we now ask the neural network to directly approximate the most likely conformation within a short interval given four transitions: {opening, closing, remain open, remain closed}. The specific transition a cell experiences depends on the relationship between kc and its initial value c0. Note that kc can be alternatively interpreted as the expectation of a genomic region’s steady state as time goes to infinity based on a cell’s instantaneous regulation patterns (Supplementary Fig. 20b).
We allow kc to take on real values in [0, 1] and infer it as a function of the cell states, similar to ρ. Note that both the cell-specific transcription rate ρ and chromatin opening rate kc share the same range of values and convey similar linear associations with the steady state values of their corresponding modalities: a large value indicates a high future potential.
Neural network architecture
The inference process is performed by an encoder parameterized by a neural network (Supplementary Fig. 21a). For cell time t and cell state z, we assume a Gaussian posterior as in ref. 57. Besides the unspliced and spliced mRNA counts, chromatin accessibilities of all genes are also fed into the neural network. The generative model first maps latent cell states to cell-specific instantaneous chromatin opening state kc and relative transcription rate ρ. We assume the prior p(z, t) = p(z)p(t) is a multi-variate Gaussian distribution where p(z) and p(t) are isotropic. The model can optionally take an informative time prior \({p}^{{\prime} }(t) \sim {{{\mathcal{N}}}}({t}_{true},{\sigma }_{0}^{2})\) if a cell-wise capture time ttrue is available. In this case, σ0 is proportional to the time interval between two adjacent capture time points. Next, the model deploys ODE solutions as parametric functions to generate means of c, u, and s, assuming they are all Gaussian random variables. The assumptions used to compute the likelihood of our probabilistic model given the ODE parameters of each gene are carried over from MultiVelo. In short, we assume the residuals of each modality form an independent multivariate Gaussian. Latent time is inferred using a separate neural network module and incorporated into the ODE solutions for final data reconstruction. Both the encoder and decoder networks of MultiVeloVAE are represented by multi-layer perceptrons (MLPs). We train MultiVeloVAE using the standard mini-batch stochastic gradient descent.
Multi-sample inference
Batch effects often arise when data generation steps are performed under different conditions. If strong batch effects are present, the samples separate predominantly by batch artifacts, hindering our ability to infer velocity and lineage relationships from multiple samples accurately. Even in the absence of batch effects, it is often of interest to identify shared and distinct biological variation among samples. Hence, we designed a conditional VAE (cVAE) model to solve this common problem while jointly inferring cellular dynamics.
CVAE58, being an extension of traditional VAE, performs variational inference conditioned on control variables. It has been shown to successfully remove batch effects in single-cell settings17. During training, the one-hot encoded sample labels are concatenated to the multi-omic count matrices as input to the encoder of MultiVeloVAE. This conditional information is also imposed on the input of the decoder, which has been reparameterized from the latent cell-state variables, thus making the latent variable z conditioned on batch covariates. The latent time variable t does not participate in the conditional inference process; instead, we modify the decoder ODE network by allowing genes to use different rate parameter sets across different conditions (batches). Under this setting, the conditional marginal log-likelihood is maximized in the objective function. The multi-omic collapsed evidence lower bound (ELBO) is formulated as
Besides conditioning on batch labels, continuous covariates can also be regressed out through the cVAE by concatenating them along the inputs of the encoder and decoder17. For instance, we found that supplying the total unspliced counts to the model can help mitigate over-fitting, which benefits batch correction.
Because genes from one sample may be lowly expressed in the others, due to biological reasons like stem cell markers in maturing populations or technical reasons such as dropouts, training ODE parameters on the other samples can be very unstable. Therefore, we allow the model to train different gene sets gb across different samples by masking out the genes that are not expressed or not highly variable in a given sample. A small regularization term consisting of differences between ODE parameters across samples is also added to the loss for two reasons: (1) to help update untrained ODE parameter sets due to gene masking, and (2) to encourage similar gene-specific ODE parameters across samples and place the larger burden of resolving batch differences onto the powerful neural network cell-state variable z.
where θ: = [αc α β γ] and r is the reference batch.
After training, the batch-corrected chromatin accessibilities and RNA expressions can be generated by designating the same batch label to all cells. By default, the model uses a user-specified reference batch to unify the other batches. The network is also able to learn the optimal scaling and offsetting parameters for non-reference batches that can best homogenize the reconstructed counts.
Velocity vectors are computed from batch-corrected expression values after training, and the cosine similarity of cell-to-cell transition5 in this case is given by
We used metrics provided in scIB19 to benchmark with other methods. For feature-level comparisons, we ran principal component analysis (PCA) on the concatenated (un)corrected c-u-s matrix. For comparisons in embedded space, we used UMAP for the uncorrected dataset, the latent cell-state variable (z) for MultiVeloVAE, and Xemb layer in the Anndata objects for other methods. Outputs used for comparison are in the normalized and log-transformed space.
RNA-only and heterogeneous mode
As shown in our previous work9, the multi-omic ODE model can be easily reduced to the canonical RNA velocity ODE equations by enforcing c0 to be 1 and αc to be 0. This sets the chromatin accessibility of all genes to be fully open and remain in a steady state indefinitely. When training on a single RNA-only sample, the input and output of MultiVeloVAE resemble those of VeloVAE. In this case, we explicitly set kc to be 1 and disable kc network parameter update.
In more advanced usage, such as when integrating a combination of scRNA-seq and multi-omic samples, the missing chromatin information of RNA-only samples can be inferred through batch correction when a multi-omic reference sample is supplied. Pseudocounts of 1’s are used for placeholder input chromatin values of the neural network as well as predicted output. The RNA-only samples are trained via previously mentioned RNA-only settings, except that kc is no longer artificially set but rather simply gradient frozen with regard to RNA-only cells. This reflects the intuition that we not only want the network parameters of kc to be optimized on real chromatin patterns but also acknowledge its downstream effects when generating unspliced and spliced values.
The reconstruction log-likelihood for mixed RNA-only approach with partial overlapping features, ignoring pseudo-chromatin counts, therefore becomes
where Nmul + Nrna = N, and x is formed as [c,u,s] for multi-omic cells or [1,u,s] for RNA-only cells.
Neural network and ODE parameter initialization
Network weights were initialized with Xavier uniform59. ODE parameters were initialized according to MultiVelo9 for multi-omic samples or scVelo5 for RNA-only samples. An inherent risk of fitting any RNA velocity model is that cells with lower counts tend to be grouped at early time points because the model has difficulty distinguishing true lowly expressed cells from low-quality ones in certain genes. To mitigate low-quality genes’ disruption to training, we clip the lower bound of each gene’s standard deviation used in likelihood calculation to a certain cutoff. To find a suitable cutoff, we first find genes that contain satisfactory dynamic information by fitting an ellipse to the cell mass of every gene on the 2D unspliced-spliced plane under linear time. Note that this generalizes from the steady-state approach used in previous methods4,5 where only a single line is fitted to presumably oval-shaped phase portraits. In this case, the longer axis of the ellipse approximates the linear steady-state RNA velocity function.
Let the spliced counts be x and the unspliced counts be y, the conic equation is defined as
The following criteria need to hold to qualify as a satisfactory gene under the RNA velocity framework.
-
1.
B2 − 4AC < 0 (Cells form a standard ellipse, not a hyperbola or a parabola.)
-
2.
\(\begin{array}{c}0 < \theta=\left\{\begin{array}{ll}\arctan (B/A-C)/2\quad \hfill& A > C\\ \arctan (B/A-C)/2+\pi /2\quad &\,{{\mbox{otherwise}}}\end{array}\right. < \pi /2\\ ({{{\rm{The}}}}\;{{{\rm{major}}}}\;{{{\rm{axis}}}}\;{{{\rm{is}}}}\;{{{\rm{positively}}}}\;{{{\rm{correlated}}}}\;{{{\rm{with}}}}y=x.)\end{array}\)
The minimum standard deviations of these satisfactory genes are then used as lower bounds for all genes.
With each ellipse fit, a gene’s unspliced-spliced phase portrait is partitioned into four quantiles by the long and short axes of the ellipse. Each cell of a gene is then labeled with the quantile information using a two-bit binary vector (i, j) ∈ [(0, 0), (0, 1), (1, 0), (1, 1)]. Intuitively, this labeling system denotes a cell’s position when the corresponding gene’s u-s coordinates have been transformed into the axes of the ellipse. These cell labels represent a coarse description of a gene’s splicing dynamics. Importantly, using two-bit labeling instead of one alleviates the common issue of mislabeling cells as binary phases in the low-quantile region of the phase portraits.
Using BasisVAE to model genes as a mixture of ODEs
BasisVAE60 was originally designed to account for feature-level clustering. It aims at clustering different features (dimensions) of a high-dimensional observation \(x={[{x}_{1},{x}_{2},\ldots,{x}_{d}]}^{T}\in {{\mathbb{R}}}^{d}\) via a variational auto-encoder. A potential application is to cluster genes with similar patterns.
The model assumes each feature is generated by one of K basis functions, i.e.,
Here, z is a low-dimensional latent variable generating the observation xi. The clustering information is stored in \({{{{\boldsymbol{w}}}}}_{i}={[{w}_{1}^{(i)},\ldots,{w}_{K}^{(i)}]}^{T} \sim\) Categorial(π1, …, πK) and π: = [π1, …, πK] ~ Dirichlet(ψ1, …, ψK). According to Märtens and Yau60, we can marginalize π using collapsed inference. The ELBO is given by
Here, the second term has an analytical form of
where \({n}_{k}:={\sum }_{i=1}^{d}{\psi }_{k}^{(i)}\) and Γ( ⋅ ) is the gamma function.
In the context of RNA velocity and multi-omic velocity, we let the solutions to the following set of ODEs be the basis functions of our VAE model. These basis functions represent multiple potential data-generating processes of each gene. Specifically, two ODEs denoting induction and repression are used for RNA velocity, whereas a total of four ODEs are used to represent the coupled and decoupled scenario between RNA and chromatin changes previously illustrated in MultiVelo: coupled induction, two possible orders of decoupled repression, and coupled repression. By introducing a latent variable w, we distinguish genes by their dynamic processes and promote gene-level clustering.
The generative process for the variational mixture of ODE models for the RNA-only model includes
Similarly, the generative process for the multi-omic model consists of
Here, F is the kinetic function of all genes and Σr is a diagonal covariance matrix.
To initialize the latent variables, we make use of a simple but critical fact: genes generated by the same underlying generative process should have similar dynamical behavior. We cluster the genes based on the aforementioned ellipse quantile-based cell vectors. By default, we group genes into 7 clusters comprising all possible combinations of the three types of gene trajectories: induction-only, repression-only, and complete. Next, for each cluster y, we perform two-sided Kolmogorov–Smirnov tests between \({{{{\mathcal{W}}}}}_{y}=\{{w}_{g}:{y}_{g}=y\}\) and Dirichlet(5.0, 5.0) and label all genes in the cluster as either induction or repression. The weights of decoupled chromatin-RNA bases are then initialized with half of the weights of the corresponding RNA basis functions.
Estimating cell-state uncertainty
We quantify the variation of each cell by normalizing the latent cell state standard deviation with the Euclidean norm of each dimension of z.
Estimating ODE parameter uncertainty
Optionally, the VAE model can be extended to account for uncertainty in the kinetic rate parameters of the ODE system. Let θ be the set of all ODE parameters. We assume that some prior distribution pλ(θ) generates the parameters. Similar to the (intractable) problem of inferring the latent cell time and state variables, we can use a variational approximation of the posterior qϕ(θ). The marginal likelihood of the input features can be bounded by
For the prior pλ(θ), we choose a factorized log-normal distribution, i.e., each rate parameter αc, α, β or γ is a random variable drawn from a log-normal distribution.
Training details
Initial conditions are important for accurately predicting the future state of each cell. To improve the accuracy of future state prediction, we first train MultiVeloVAE to convergence using default initial conditions of two ODE settings so that latent times and cell states are accurate, then determine the initial conditions for a cell at time t.
Specifically, training is performed in a two-stage process. The main difference between the two stages is how we set the initial conditions of each modality. During the first stage, a global tuple of values (c0, u0, and s0) for each gene is fitted, whereas, in the second stage, we aggregate cells at earlier time points as ancestors of later cells based on the proximity of their inferred latent cell-state z and time t variables. We perform an expectation-maximization (EM) algorithm in Stage 2:
-
(i)
E: Update the initial values based on ancestor cells and current ODE parameters.
-
(ii)
M: Optimize ODE parameters given initial values.
We find the initial conditions of cells in Stage 2 by averaging the (u, s) values observed in an immediately preceding time interval [t − δ1, t − δ2]. We then fine-tune the ODE parameters using these updated initial conditions, keeping latent time and cell state fixed. The second stage helps clean up and aggregate cells onto lineage trees by capturing continuous cellular transitions, from the noisy point-cloud estimations generated in the first stage.
All ODE parameters except the offset parameters are constrained to the positive range using a Softplus function (a smooth approximation of ReLU). To use it, a Softplus inverse function is first performed on the initialized values. During training, learning rates are dynamically adjusted by a cosine annealing scheduler to promote convergence and generalization.
Depending on the mode of training, certain parameters are ignored or masked to specific values to comply with the ODE assumption. For batch correction, scales for genes of reference spliced counts are fixed to 1’s and offsets of the three modalities of the reference batch to 0’s. For RNA-only mode, parameters related to chromatin are all masked. For mixed RNA-only mode, a combination of the previous two configurations is used.
The model would stop training once the change of initial values (c0, u0, and s0) stagnates (relative to the variance of each modality). The performance of the model on the holdout validation dataset is tracked during training, and the best model is retrieved at the end of each EM iteration to prevent over-fitting.
Generalizing cross-boundary direction correctness (GCBDir)
In a previous work10, the authors quantitatively measured the accuracy of velocity flow on any 2D visualization. Denote \({{{\mathcal{C}}}}\) as the set of all cell types. For any two cell types \(A,B\in {{{\mathcal{C}}}}\), we define (A, B) to be a transition pair if and only if B is a descendant cell type of A in a cell developmental process. The CBDir metric takes a set of known transition pairs \({{{\mathcal{T}}}}=\{(A,B):A,B\in {{{\mathcal{C}}}}\}\) as input and computes an average directional accuracy via cosine similarity. For any cell in c ∈ A with transition pair (A, B), its CBDir is defined as:
Here, vc is the projection of RNA velocity to a low-dimensional space, xc is the coordinates of c in the same space, and N(c) is the set of all neighbors of c in a KNN graph built from gene expression similarity. The overall CBDir of all known transition pairs is defined as
The CBDir metric is the first one to directly measure the correctness of velocity flow. It connects the visual perception of velocity flow to a quantity. We think it is beneficial to extend it for its following limitations:
-
1.
The velocity in CBDir is only a projection to a low-dimensional space. Hence, it’s not a direct measure of velocity at a gene expression level.
-
2.
Only the direct neighbors in the KNN graph are considered targets, i.e., future transcriptomic states. However, this assumption might not hold due to stochastic noise in the dataset. In addition, often, directly connected neighbors on a KNN graph are too similar at the transcriptomic level to be considered a future direction.
-
3.
CBDir does not take the ordering of cells into account. Time order is important because the dynamical system must proceed with increasing time.
-
4.
It remains unclear whether the velocity flow from one cell type to another is statistically significant.
We devised k-step CBDir (kCBDir) metric. Given a cell c ∈ CA with transition pair (A, B), kCBDir is defined as
where \(b:={I}_{\{{t}_{{c}^{{\prime} }}\ > \ {t}_{c}\wedge {{{{\boldsymbol{v}}}}}_{{{{\boldsymbol{c}}}}}^{T}({{{{\boldsymbol{x}}}}}_{{{{{\boldsymbol{c}}}}}^{{\prime} }}-{{{{\boldsymbol{x}}}}}_{{{{\boldsymbol{c}}}}})\ > \ 0\}}\). Here, we replace direct neighbors with k-step neighbors, Nk(c), on a KNN graph. Besides, xc and vc can be from either a low-dimensional embedding or the spliced counts. We further defined the generalized CBDir (GCBDir) by averaging over multiple different step sizes and subtracting an offset computed by taking a random walk on the cell KNN graph:
Here, kCBDir(c; Nk(c)) is the kCBDir between cell c and its k-step neighbors from its descendant cell type, and Rk(c) is the set of cells reached by randomly traversing a KNN graph starting from c.
Coupling and decoupling factors
We generalize the coupling and decoupling notions from MultiVelo to continuous states and define a decoupling factor as the difference between the two variables kc and ρ, and a coupling factor as the centered sum between the two variables kc and ρ. This direct comparison is meaningful because kc and ρ lie in the same range and both have a linear relationship with the steady state value of the ODE solution for their corresponding modalities.
The decoupling factor δ ranges from −1 to 1. A value of 1 indicates that the relative chromatin opening rate is greater than the relative transcription rate, analogous to the priming state of MultiVelo as well as the decoupling state in Model 2 of MultiVelo. A value of −1 is analogous to the decoupling phase of Model 1 genes. The coupling factor κ also ranges from −1 to 1. A value of 1 indicates coupled induction (analogous to the coupled-on state of MultiVelo) and a value of −1 indicates coupled repression (analogous to the coupled-off state of MultiVelo). Thresholding δ and κ gives discrete states analogous to MultiVelo’s discrete states. Note that δ and κ are now also different for every cell and every gene, so that we can identify cell-type-specific priming and decoupling. Importantly, in MultiVeloVAE, a cell simultaneously possesses both properties, unlike in MultiVelo.
Differential dynamics testing
Bayesian differential testing has been shown to provide valuable insights into finding genes of different transcriptomic profiles across conditions17,61. Our variational inference approach to RNA velocity also provides a unique opportunity to study the differential dynamics of genes in the form of \(\frac{ds}{dt}\). Because velocity can be a negative quantity, we can compare its absolute magnitude, in which case it is similar to mRNA counts and log(2) fold-change (LFC) is a viable metric. For example, when we want to find genes whose expression is fast changing regardless of direction as a measurement of population stability, the former approach is appropriate. We can also subtract two velocities using their original values and find what we term the log(2) difference (LD).
where s2 is the spliced value for the subtrahend cell.
We also conduct differential testing on kc, ρ, c, u, and s values. We use log differences for the bounded latent variables kc and ρ with default norm equals 0.1. We use LFC to measure the chromatin, unspliced, and spliced changes.
We follow a similar Bayes factor (BF)62 approach to compare the posterior distributions between populations A and B. Let the measured difference quantity be \({{{\mathcal{D}}}}\) (log fold-change (LFC) or log difference (LD)), null and alternative hypotheses are formed17,61.
The Bayes factor is computed as the odds ratio
When computing the posterior, pairs of samples are drawn from each population and fed into the VAE model via Monte Carlo sampling. Batch effects are corrected following the approach mentioned previously. In the special case where contrasting conditions span the same set of batches (samples), each pair of samples is enforced to be drawn from the same batch to bypass any batch effect. Generated expressions and velocities are collected by passing reparameterized latent variables through the decoder and ODE modules.
P-values of differential testing results from the “change” mode are controlled for multiple testing by means of false discovery proportions (FDP)61,63. The number of top genes ranked by probability of having differential dynamics k is selected so that the posterior expected false discovery proportion is below a certain threshold (default to 0.05).
Moreover, we extend the bulk-level comparisons to binned windows along the inferred time. One benefit of using a variational inference approach is that we can generate unseen pseudo-cells to boost statistical power or to reduce computational burden, depending on our choice of total cells. We first draw sample cells from the posterior state and time variables for both contrasting conditions σz, μz, σt, μt = F(X):
where G denotes the generative network and ODE equations.
For each condition, we then split these cells into fifty quantiles of small time windows containing equal numbers of the generated cells, and the means of the bins are recorded. The cells in the same time bin are subject to Bayes factor differential testing. The fifty data points are then smoothed by Gaussian Process regression with RBF kernel. In addition, we perform likelihood ratio tests64 (LRT) to compute the significance scores of the overall differential dynamics for each variable or modality against null hypotheses of no changes. We use a constant zero-line (for LD) or one-line (for LFC) as the null model. The intuition behind this choice of null hypothesis is that we want to identify genes that are different between two groups of cells over time. There are two ways this could happen: (1) the LD or LFC could show a time-varying trend or (2) the LD or LFC could show a constant nonzero trend over time (less common, but possible–see ρ LD for CSF2RB in Fig. 6d). In both hypotheses, random noise is modeled by a white noise kernel. P-values are obtained based on χ2 distribution with one degree of freedom.
New EB library preparation
Human iPSC culture
Human induced pluripotent stem cells (iPSCs) were cultured in mTeSR1 medium (STEMCELL, Cat. No. 85850) on Growth Factor Reduced, Matrigel Basement Membrane Matrix (Corning; Cat. No. 354230) coated dishes. mTeSR1 was replaced every day. When cells reached 70–80% confluency, they were passaged by aspirating media, washing with DPBS (Gibco; Cat. No. 14190144), and incubating with StemPro Accutase (Gibco; Cat. No. A1110501) at 37C for 5 min. Cells were mixed with DPBS and centrifuged at 300 g for 3 min. The supernatant was aspirated, resuspended in mTeSR1 medium supplemented with 10 nM y-27632 dihydrochloride ROCK inhibitor (STEMCELL; Cat. No. NC1678100), and counted with a Countess 3 Automated Cell Counter (ThermoFisher Scientific).
EB formation and maintenance
EBs were formed using the STEMCELL Aggrewell400 protocol (DX21732). Briefly, the AggreWell400 24-well plate was coated with AggreWell Rinsing Solution (STEMCELL; Cat. No. 07010). The dissociated iPSCs were seeded in AggreeWell EB Formation Medium (STEMCELL; Cat. No. 05893) into the AggreWell400 plate at 1000 cells per microwell, totaling 1.2 x 106 per well. After 24 h, half the media change was performed. After 48 h, EBs were harvested using a 40-um Strainer (ThermoFisher Scientific; Cat. No. 22-363-547) and transferred to an ultra-low attachment six-well plate in Essential E6 media (Gibco; Cat. No. A1516401). Media was replaced every other day and flash-frozen on day 7 in E6 media.
Nuclei isolation
Flash-frozen D7 EBs were thawed on ice. Nuclei isolation was performed according to the 10X Genomics demonstrated protocol ‘Nuclei Isolation from Complex Tissues for Single Cell Multiome ATAC + Gene Expression Sequencing’ (CG000375 Rev B). Briefly, EBs were homogenized using a 1000 μL wide-bore tip in lysis buffer and incubated on ice for 5 minutes. Cells were then filtered through a 70 um strainer and centrifuged at 500 g for 5 min at 4 °C. The supernatant was removed and 1 ml of PBS + 1% BSA + 1U/μL RNase inhibitor (Roche; Cat. No. 3335402001) was added and incubated on ice for 5 minutes. The pellet was resuspended using a wide bore tip and centrifuged at 500g for 5 minutes at 4 °C. The pellet was then resuspended in 100 μL 0.1X lysis buffer and incubated on ice for 2 min. 1 ml of buffer was added to mix the pellet and centrifuged at 500 g for 5 min at 4 °C. The supernatant was removed and resuspended in chilled diluted nuclei buffer (10X Genomics). The nuclei were diluted to 3000–8000 nuclei/ul by using a Countess 3 Automated Cell Counter and proceeded to Chromium Next GEM Single Cell Multiome ATAC + Gene Expression.
10X Multiome
Single nuclei libraries were generated using the 10X Genomics Chromium Controller following the manufacturer’s protocol for Multiome (ATAC+GEX) analysis. In brief, transposase was added to the nuclei suspension and incubated, before adding barcode and RT master mix and loading into the Chromium Controller chip with appropriate gel beads. Following the generation of single-cell gel bead-in-emulsions (GEMs), reverse transcription of the RNA and barcoding of the fragmented DNA were performed. The resulting product was cleaned, and both the cDNA and barcoded, fragmented DNA were amplified. One fraction of this product made the ATAC library via indexing PCR. The other fraction was amplified again to isolate the cDNA further, which was then quantified by Qubit (Invitrogen) and assessed for size on the TapeStation 4200 (Agilent). It underwent enzymatic fragmentation, size selection, adapter ligation, and indexing PCR. Final library quality was assessed using the LabChip GX Touch HT(Revvity) and was quantified by Qubit. Pooled libraries were then subjected to paired-end sequencing according to the manufacturer’s protocol (Illumina NovaSeq6000). BclConvert software (Illumina) was used to generate de-multiplexed Fastq files, and the CellRanger ARC Pipeline (10X Genomics) was used to align reads and generate count matrices.
New HSPC and macrophage library preparation
Ethics statement
Anonymized mobilized PBMCs from Fred Hutch Hematology were purchased after obtaining informed consent. Studies using these cells were determined to be exempt from human studies requirements by the University of Michigan Institutional Review Board because the project involves only biological specimens that cannot be linked to a specific individual by the investigator(s) directly or indirectly through a coding system.
Cell culture
Cell cultures were maintained at 37 ∘C in a 5% CO2 humidified atmosphere. CD34+ hematopoietic stem cells were acquired from the Fred Hutch Hematology core. Frozen CD34+ cells were thawed in serum-free RPMI plus 10mM HEPES, then cultured at 1 × 106 cells/mL of STIF medium [Stemspan II medium supplemented with 100 ng/mL stem cell factor, 100 ng/mL thrombopoietin, 100 ng/mL Flt3 ligand (all from Stemcell Technologies), and 100 ng/mL insulin-like growth factor binding protein 2 (R&D Systems)]. Cells were expanded at two and four days post-thaw. Seven days post-thaw, cells were processed for single-cell submission or transferred from STIF into macrophage-stimulating medium (MSM) [SFEM supplemented with 1x Myeloid Expansion Supplement II (Cat# 02694, StemSpanTechnologies) and 20 μg/mL IL-6 (R&D Technologies)] for in vitro-differentiated macrophages. MSM was adapted from previously published protocols65,66,67. Differentiating cells were cultured for an additional 7 days in MSM. HSCs were expanded in MSM on day 7 by seeding cells at 250k/well in 24-well plates in 500ul MSM. On day 10, cells were divided four times and maintained in 500 μL MSM. On day 14, cells were collected for single-cell analysis.
Single-cell sequencing
All cells were prepared according to the manufacturer’s ‘10x Genomics Nuclei Isolation Single Cell multi-ome ATAC + Gene Expression Sequencing’ demonstrated protocol. Briefly, cells were washed in 1 x PBS-/- supplemented with 0.04% BSA and sorted using the BigFoot cell sorter (Thermo Fisher) on days 7 and 14. For in vitro-differentiated adherent cells, cells were lifted using enzyme-free cell dissociation buffer (Cat# 13151014, Thermo Fisher) before washing and sorting. Nuclei were isolated following the ‘Low Cell Input Nuclei Isolation’ sub-protocol and immediately processed using the Chromium Next GEM Single Cell Multiome + Gene Expression kit following the manufacturer’s instructions.
Automated data preprocessing
After reading scRNA-seq datasets (some of which are provided by UniTVelo10) into AnnData68 format, we used Scanpy69 to select highly variable genes, normalize, and scale the gene expression counts. Next, we followed the scVelo preprocessing pipeline by performing PCA on the expression data and then smoothing the unspliced and spliced expression levels among k-nearest neighbors.
For the multi-omic mouse brain, human brain35, and single time-point HSPC datasets, the same AnnData objects used in MultiVelo9 were loaded into Python for running MultiVeloVAE. For the SHARE-seq36 mouse skin dataset, the order of the RNA normalization and the shared gene selection between ATAC and RNA steps during preprocessing was flipped to remove the confounding effect of total counts biasing variational inference.
A comprehensive and automated preprocessing procedure has been developed for our newly acquired EB, HSPC, and macrophage datasets. We chose to quantify unspliced and spliced count matrices using STARsolo70 –a variant of the STAR aligner for single-cell data, due to its ability to resolve multi-mapped reads and quantify mitochondrial reads, in addition to its rapid mapping speed. High-quality cell filtering and gene selection are jointly based on 10X CellRanger ARC output as well as STARsolo output, with the numbers of genes expressed, total mRNA counts, percentages of counts in the top 20% genes, mitochondrial RNA percentages, ribosomal protein RNA percentages, total unspliced counts, and total spliced counts as quality control criteria. Outliers are automatically determined and removed using median absolute deviations (MAD) with the same thresholds used across all datasets, adapted from sc-best-practices.org (Supplementary Fig. 22a).
Following joint filtering of RNA and ATAC matrices, we next set to regress out cell cycle effects from total RNA expressions, as we have observed that cell cycle effects prevented unsupervised clustering that reflects cell type characteristics in the blood cell datasets. We would also want to remove these effects in unspliced and spliced counts to avoid confounding lineage inference6. We do so by excluding genes that are predominantly driven by or correlated with cell cycles via selecting highly variable genes after adjusting for cell cycle effects. To make highly variable gene function viable, we added regression intercepts back to the count matrices after regression. We then applied Leiden71 clustering to find cell types. A list of canonical cell marker genes was used to annotate the clusters (Supplementary Fig. 22b–e).
To prepare for multi-sample inference, we first found genes that have been labeled as highly variable in each dataset from single-sample preprocessing steps and extracted the union of all such genes from the quality-controlled and normalized count matrices. Then we labeled genes that have more than 10% of cells expressed from each dataset matrix as highly variable as well to boost the number of overlapped genes across conditions. Expressions and accessibilities of all samples were merged into two AnnData68 objects. A similar set of preprocessing steps used for individual samples was performed on the joint object.
For the EB dataset, 4240 cells passed all quality control criteria. After highly variable gene selection, 3138 genes were left to be used for training. For the HSPC-HSPC integration, 17667 cells and 892 genes formed the input Anndata object after joint preprocessing of both samples illustrated above. For the HSPC-macrophage integration, the input contains 9908 cells and 929 genes. For the two HSPC and one BMMC partial integration example, we have 27,841 cells with 1044 genes.
Running Scenic+ and peak-level analyses
First, pycisTopic was run with default steps for the newly sequenced HSPC sample. Then the output, together with the preprocessed RNA anndata object with raw counts, was supplied to the Scenic + pipeline with default parameters. Only genes that participated in integrative velocity analyses were included to extract transcription factors and gene lists, and to plot the gene regulatory network. To avoid results biased by zero counts from repressed branches, we selected non-background cells for each gene for correlation analyses; only cells with either kc > 0.5 or ρ > 0.5 cutoffs were kept. For GATA2-GATA1 network analyses, only genes positively associated with the Erythrocyte cell type were kept–filtered with log2 fold-change above 0.
The latent time of the two HSPC samples was divided into fifty quantiles of time bins. From each bin, 200 posterior-sampled pseudo-cells were generated, and their mean decoupling factors and timepoints were computed. These were used to build a Gaussian Process regressor with an RBF kernel.
To normalize the TF RNA, region accessibility, and target gene modalities to comparable ranges, cells belonging to the entire lineage from HSC to the specified terminal cell type were extracted and min-max normalized. For instance, the Platelet lineage consists of HSC, CMP, MEP, Prog MK, Megakaryocyte, and Platelet cell types.
Peaks were visually examined in IGV and overlaid with ChromHMM full-stack annotation45. All ATAC peaks were then labeled with ChromHMM full-stack-annotated regions using bedtools, and Spearman correlation coefficients were computed between the factors and accessibilities for each category of annotations. Certain annotation groups are described to be enriched for blood cells, while the others are not. The detailed descriptions of each category can be found on the authors’ GitHub at ernstlab/full_stack_ChromHMM_annotations.
Multi-sample downstream analyses
At the end of training, the model outputs integrated latent variables, modality profiles, and unified parameters. An integrated UMAP coordinate is obtained from the batch-corrected latent cell-state variable z. We recomputed neighborhood relationships across samples using latent cell-state and time variables before building a Markov chain transition graph that was used for the velocity stream predictions.
For perturbation analyses, we computed the differences between perturbed and unperturbed values of three modalities as
These were treated as velocity vectors to obtain a transition map of cells similar to RNA velocity analysis5. The aggregated velocity streams on UMAP are referred to as the perturbation forces.
Cross-modality generation benchmarks
Normalized RNA and ATAC counts before neighborhood smoothing from MultiVeloVAE preprocessing steps were supplied to scButterfly, and inferred ATAC counts were neighborhood-smoothed after out-of-sample prediction to prepare for velocity inference. Raw RNA and ATAC counts and their low-dimensional embeddings–X_PCA and X_LSI were supplied to scCross, and the predicted raw ATAC counts were TFIDF normalized and neighborhood-smoothed to prepare for velocity inference. The HSPC sample for testing is treated as an expression-only sample and merged with the other multi-omic HSPC sample in MultiVI. Unnormalized counts from MultiVeloVAE preprocessing steps were supplied as input, and the get_accessibility_estimates() function was used to generate imputed accessibilities, which were then neighborhood-smoothed to prepare for velocity inference and benchmark metrics.
Development and testing environment
The package was developed on an Arch Linux system using an i3-12100F, RTX 3060 12GB, and 64GB RAM configuration.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The newly sequenced 10X Genomics Multiome HSPC and macrophage data in this study have been deposited in GEO GSE284047. The raw data for these samples are available under restricted access in dbGaP phs002915.v2.p1 [https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs002915.v2.p1] for patient privacy concerns. The new 10X Genomics Multiome EB data have been deposited in GEO GSE284047. The post-processed AnnData objects are shared through figshare at DOI: 10.6084/m9.figshare.30280333 [https://doi.org/10.6084/m9.figshare.30280333]. All other multi-omic and scRNA-seq datasets analyzed in the paper have been previously published and freely available. Source data are provided with this paper.
Code availability
MultiVeloVAE is implemented in Python and uses the NVIDIA CUDA framework. The code used to develop the model, perform the analyses, and generate results in this study is publicly available and has been deposited on GitHub at github.com/welch-lab/MultiVeloVAE under BSD-3-Clause license. and can be installed through PyPI. The specific version of the code associated with this publication is archived in Zenodo and is accessible via https://doi.org/10.5281/zenodo.1726825472.
References
Trapnell, C. et al. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat. Biotechnol. 32, 381–386 (2014).
Haghverdi, L., Büttner, M., Wolf, F. A., Buettner, F. & Theis, F. J. Diffusion pseudotime robustly reconstructs lineage branching. Nat. Methods 13, 845–848 (2016).
Welch, J. D., Hartemink, A. J. & Prins, J. F. SLICER: inferring branched, nonlinear cellular trajectories from single cell RNA-seq data. Genome Biol. 17, 106 (2016).
La Manno, G. et al. RNA velocity of single cells. Nature 560, 494–498 (2018).
Bergen, V., Lange, M., Peidli, S., Wolf, F. A. & Theis, F. J. Generalizing RNA velocity to transient cell states through dynamical modeling. Nat. Biotechnol. 38, 1408–1414 (2020).
Bergen, V., Soldatov, R. A., Kharchenko, P. V. & Theis, F. J. RNA velocity-current challenges and future perspectives. Mol. Syst. Biol. 17, e10282 (2021).
Gorin, G., Fang, M., Chari, T. & Pachter, L. RNA velocity unraveled. PLOS Comput. Biol. 18, 1–55 (2022).
Gorin, G., Svensson, V. & Pachter, L. Protein velocity and acceleration from single-cell multiomics experiments. Genome Biol. 21, 39 (2020).
Li, C., Virgilio, M., Collins, K. L. & Welch, J. D. Multi-omic single-cell velocity models epigenome-transcriptome interactions and improves cell fate prediction. Nat. Biotechnol. 41, 387–398 (2023).
Gao, M., Qiao, C. & Huang, Y. UniTVelo: temporally unified RNA velocity reinforces single-cell trajectory inference. Nat. Commun. 13, 6586 (2022).
Cui, H. et al. DeepVelo: deep learning extends RNA velocity to multi-lineage systems with cell-specific kinetics. Genome Biol. 25, 27 (2024).
Li, S. et al. A relay velocity model infers cell-dependent RNA velocity. Nat. Biotechnol. 42, 99–108 (2023).
Gayoso, A. et al. Deep generative modeling of transcriptional dynamics for RNA velocity analysis in single cells. Nat. Methods 21, 50–59 (2023).
Qin, Q., Bingham, E., La Manno, G., Langenau, D. M. & Pinello, L. Pyro-Velocity: probabilistic RNA velocity inference from single-cell data. Preprint at https://doi.org/10.1101/2022.09.12.507691 (2022).
Hao, Y. et al. Dictionary learning for integrative, multimodal and scalable single-cell analysis. Nat. Biotechnol. 42, 293–304 (2024).
Welch, J. D. et al. Single-cell multi-omic integration compares and contrasts features of brain cell identity. Cell 177, 1873–1887.e17 (2019).
Lopez, R., Regier, J., Cole, M. B., Jordan, M. I. & Yosef, N. Deep generative modeling for single-cell transcriptomics. Nat. Methods 15, 1053–1058 (2018).
Kingma, D. P. & Welling, M. Auto-encoding variational Bayes. In 2nd International Conference on Learning Representations, Banff, AB, Canada, April 14-16, 2014, Conference Track Proceedings (eds Bengio, Y. & LeCun, Y.) (ICLR, 2014).
Luecken, M. D. et al. Benchmarking atlas-level data integration in single-cell genomics. Nat. Methods 19, 41–50 (2022).
Bastidas-Ponce, A. et al. Comprehensive single-cell mRNA profiling reveals a detailed roadmap for pancreatic endocrinogenesis. Development 146, dev173849 (2019).
Pijuan-Sala, B. et al. A single-cell molecular map of mouse gastrulation and early organogenesis. Nature 566, 490–495 (2019).
Schiebinger, G. et al. Optimal-transport analysis of single-cell gene expression identifies developmental trajectories in reprogramming. Cell 176, 928–943.e22 (2019).
Stuart, T. et al. Comprehensive integration of single-cell data. Cell 177, 1888–1902.e21 (2019).
La Manno, G. et al. Molecular architecture of the developing mouse brain. Nature 596, 92–96 (2021).
Hochgerner, H., Zeisel, A., Lönnerberg, P. & Linnarsson, S. Conserved properties of dentate gyrus neurogenesis across postnatal development revealed by single-cell RNA sequencing. Nat. Neurosci. 21, 290–299 (2018).
Setty, M. et al. Characterization of cell fate probabilities in single-cell data with Palantir. Nat. Biotechnol. 37, 451–460 (2019).
Kharchenko, P. V. The triumphs and limitations of computational methods for scRNA-seq. Nat. Methods 18, 723–732 (2021).
Battich, N. et al. Sequencing metabolically labeled transcripts in single cells reveals mRNA turnover strategies. Science 367, 1151–1156 (2020).
Qiu, Q. et al. Massively parallel and time-resolved RNA sequencing in single cells with scNT-seq. Nat. Methods 17, 991–1001 (2020).
Barile, M. et al. Coordinated changes in gene expression kinetics underlie both mouse and human erythroid maturation. Genome Biol. 22, 197 (2021).
Biddy, B. A. et al. Single-cell mapping of lineage and identity in direct reprogramming. Nature 564, 219–224 (2018).
Lange, M. et al. Cellrank for directed single-cell fate mapping. Nat. Methods 19, 159–170 (2022).
Rhodes, K. et al. Human embryoid bodies as a novel system for genomic studies of functionally diverse cell types. eLife 11, e71361 (2022).
Han, X. et al. Mapping human pluripotent stem cell differentiation pathways using high-throughput single-cell RNA-sequencing. Genome Biol. 19, 47 (2018).
Trevino, A. E. et al. Chromatin and gene-regulatory dynamics of the developing human cerebral cortex at single-cell resolution. Cell 184, 5053–5069.e23 (2021).
Ma, S. et al. Chromatin potential identified by shared single-cell profiling of RNA and chromatin. Cell 183, 1103–1116 (2020).
Korsunsky, I. et al. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Methods 16, 1289–1296 (2019).
Hie, B., Bryson, B. & Berger, B. Efficient integration of heterogeneous single-cell transcriptomes using Scanorama. Nat. Biotechnol. 37, 685–691 (2019).
Velten, L. et al. Human haematopoietic stem cell lineage commitment is a continuous process. Nat. Cell Biol. 19, 271–281 (2017).
Laurenti, E. & Göttgens, B. From haematopoietic stem cells to complex differentiation landscapes. Nature 553, 418–426 (2018).
Hu, W. et al. Combined single-cell profiling of chromatin–transcriptome and splicing across brain cell types, regions and disease state. Nat. Biotechnol. https://doi.org/10.1038/s41587-025-02734-5 (2025).
Bravo González-Blas, C. et al. SCENIC+: single-cell multiomic inference of enhancers and gene regulatory networks. Nat. Methods 20, 1355–1367 (2023).
Suzuki, M. et al. GATA factor switching from GATA2 to GATA1 contributes to erythroid differentiation. Genes Cells 18, 921–933 (2013).
Anderson, E. M. et al. GATA1 and GATA2 sense/antisense transcriptional switching in early hematopoietic development. Blood 142, 5578–5578 (2023).
Vu, H. & Ernst, J. Universal annotation of the human genome through integration of over a thousand epigenomic datasets. Genome Biol. 23, 9 (2022).
Yunna, C., Mengru, H., Lei, W. & Weidong, C. Macrophage M1/M2 polarization. Eur. J. Pharmacol. 877, 173090 (2020).
Weng, C. et al. Deciphering cell states and genealogies of human haematopoiesis. Nature 627, 389–398 (2024).
Ainciburu, M. et al. Uncovering perturbations in human hematopoiesis associated with healthy aging and myeloid malignancies at single-cell resolution. eLife 12, e79363 (2023).
Cao, Y. et al. scButterfly: a versatile single-cell cross-modality translation method via dual-aligned variational autoencoders. Nat. Commun. 15, 2973 (2024).
Yang, X., Mann, K. K., Wu, H. & Ding, J. scCross: a deep generative model for unifying single-cell multi-omics with seamless integration, cross-modal generation, and in silico exploration. Genome Biol. 25, 198 (2024).
Ashuach, T. et al. MultiVI: deep generative model for the integration of multimodal data. Nat. Methods 20, 1222–1231 (2023).
Qiu, X. et al. Mapping transcriptomic vector fields of single cells. Cell 185, 690–711.e45 (2022).
Kamimoto, K. et al. Dissecting cell identity via network inference and in silico gene perturbation. Nature 614, 742–751 (2023).
Adamson, B. et al. A multiplexed single-cell CRISPR screening platform enables systematic dissection of the unfolded protein response. Cell 167, 1867–1882.e21 (2016).
Dixit, A. et al. Perturb-seq: dissecting molecular circuits with scalable single-cell RNA profiling of pooled genetic screens. Cell 167, 1853–1866.e17 (2016).
Jaitin, D. A. et al. Dissecting immune circuits by linking CRISPR-pooled screens with single-cell RNA-seq. Cell 167, 1883–1896.e15 (2016).
Gu, Y., Blaauw, D. T., and Welch, J. Variational mixtures of ODEs for inferring cellular gene expression dynamics. In Proc. 39th International Conference on Machine Learning, volume 162 of Proceedings of Machine Learning Research (eds Kamalika, C. et al.) 7887–7901 (PMLR, 2022).
Sohn, K., Lee, H., & Yan, X. Learning structured output representation using deep conditional generative models. In C. Cortes, N. Lawrence, D. Lee, M. Sugiyama, and R. Garnett, editors, Advances in Neural Information Processing Systems, volume 28. Curran Associates, Inc. (2015).
Glorot, X. & Bengio, Y. Understanding the difficulty of training deep feedforward neural networks. In Proc. Thirteenth International Conference on Artificial Intelligence and Statistics, of Proceedings of Machine Learning Research volume 9 (eds Yee Whye Teh & Mike Titterington) 249–256 (PMLR, 2010).
Märtens, K. & Yau, C. BasisVAE: translation-invariant feature-level clustering with variational autoencoders. In Proc. Twenty Third International Conference on Artificial Intelligence and Statistics, volume 108 of Proceedings of Machine Learning Research (eds Silvia C. & Roberto C.) 2928–2937 (PMLR, 2020).
Boyeau, P. et al. An empirical Bayes method for differential expression analysis of single cells with deep generative models. Proc. Natl. Acad. Sci. USA 120, e2209124120 (2023).
Held, L. & Ott, M. On p-values and Bayes Factors. Annu. Rev. Stat. Appl. 5, 393–419 (2018).
Newton, M. A., Noueiry, A., Sarkar, D. & Ahlquist, P. Detecting differential gene expression with a semiparametric hierarchical mixture method. Biostatistics 5, 155–176 (2004).
Wilks, S. S. The large-sample distribution of the likelihood ratio for testing composite hypotheses. Ann. Math. Stat. 9, 60–62 (1938).
Stec, M. et al. Expansion and differentiation of CD14+CD16− and CD14++CD16+ human monocyte subsets from cord blood CD34+ hematopoietic progenitors. J. Leukoc. Biol. 82, 594–602 (2007).
Way, K. J. et al. The generation and properties of human macrophage populations from hemopoietic stem cells. J. Leukoc. Biol. 85, 766–778 (2009).
Clanchy, FelixI. L. & Hamilton, J. A. The development of macrophages from human CD34+ haematopoietic stem cells in serum-free cultures is optimized by IL-3 and SCF. Cytokine 61, 33–37 (2013).
Virshup, I., Rybakov, S., Theis, F. J., Angerer, P. & Wolf, F. A. anndata: Annotated data. Preprint at https://doi.org/10.1101/2021.12.16.473007 (2021).
Wolf, F. A., Angerer, P. & Theis, F. J. SCANPY: large-scale single-cell gene expression data analysis. Genome Biol. 19, 15 (2018).
Kaminow, B., Yunusov, D. & Dobin, A. STARsolo: accurate, fast and versatile mapping/quantification of single-cell and single-nucleus RNA-seq data. Preprint at https://doi.org/10.1101/2021.05.05.442755 (2021).
Traag, V. A., Waltman, L. & Van Eck, NeesJan From Louvain to Leiden: guaranteeing well-connected communities. Sci. Rep. 9, 1–12 (2019).
Li, C. et al. Inferring differential dynamics from multi-lineage, multi-omic, and multi-sample single-cell data with MultiVeloVAE. https://doi.org/10.5281/zenodo.17268254 (2025).
Acknowledgements
This work is funded by NIH grant R01AI149669 to K.L.C. and J.D.W., R01HG010883 and UM1MH130966 to J.D.W., F31AI177258 to C.L., and F31AI15504 to M.C.V. We thank Jian Shu, Koseki Kobayashi-Kirschvink, and Chengxiang Qiu for helping preprocess the iPSC reprogramming datasets. We would also like to thank the University of Michigan Flow Cytometry Core and Advanced Genomics Core for assistance with experiments, particularly Wenpu Trim and David Sheltraw.
Author information
Authors and Affiliations
Contributions
M.C.V. and K.L.C. generated the 10X Multiome HSPC and macrophage data. K.H.L generated the 10X Multiome EB data. J.D.W. and Y.G. developed VeloVAE, which is the predecessor of this work. C.L. and J.D.W. developed and implemented MultiVeloVAE, performed data preprocessing and post-training analyses, and wrote the manuscript. Y.G. and C.L. performed benchmarks with velocity inference methods. C.L. performed benchmarks with batch correction and cross-modality inference methods. All authors have read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Hongyu Dong, who co-reviewed with Yuhao Chen and Boqiang Hu, Jianhua Xing and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Li, C., Gu, Y., Virgilio, M.C. et al. Inferring differential dynamics from multi-lineage, multi-omic, and multi-sample single-cell data with MultiVeloVAE. Nat Commun 16, 11505 (2025). https://doi.org/10.1038/s41467-025-66287-6
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-66287-6
