
Abstract
DNA G-quadruplexes (G4s), formed by guanine-rich sequences in mammalian genomes, are non-canonical structures implicated in gene regulation. However, their strand-specific genomic distribution and mechanistic roles in transcription remain poorly understood. Here, we report a strand-specific G4 sequencing (ssG4-seq) method for global profiling of G4 structures across multiple mammalian genomes. This method faithfully recapitulates known G4 structures and identifies thousands of previously unannotated G4s in human K562 cells. Remarkably, over 95% of G4s are located at enhancers and promoters across species, with promoters containing dual-strand G4s exhibiting significantly stronger transcriptional activation compared to those with single-strand G4s. Mechanistically, we identify SP1 as a potent G4 reader that facilitates transcription by modulating enhancer-promoter chromatin looping. Furthermore, we demonstrate that cancer-associated mutations can destabilize G4 structures, impair SP1-mediated chromatin interactions, and contribute to tumorigenesis. Our study demonstrates the power of ssG4-seq in elucidating G4 functions in gene regulation and disease.
Similar content being viewed by others


Introduction
DNA G-quadruplexes (G4s) are non-canonical secondary structures formed in guanine-rich regions of DNA, characterized by the stacking of two or more G-tetrads stabilized by Hoogsteen hydrogen bonding and monovalent cations such as potassium or sodium1,2. These structures are highly dynamic and mainly form in single-stranded DNAs, particularly in regions with high GC content, such as telomeres and gene promoters1,2,3,4. G4s have been linked to fundamental biological processes, including transcription4,5,6,7, replication8,9,10,11,12, and telomere maintenance13,14,15, making them key players in cellular homeostasis and disease processes16, including cancer17,18,19 and neurodegenerative disorders20.
In promoter regions, G4 structures are implicated in transcriptional regulation7,21, likely mediated by G4-binding proteins and helicases. Potential G4-binding proteins, including transcription factors (TFs) and chromatin-associated proteins22, such as YY123, recognize and bind to G4s, modulating their stability and influencing transcription. Additionally, G4 helicases such as DHX3624,25, BLM26,27, and WRN28,29 unwind G4 structures, resolving topological constraints and potentially influencing transcription machinery activity. By regulating the formation and resolution of G4s, these proteins play conditional roles in fine-tuning gene expression, particularly at promoters. However, the identities of G4 readers at promoters and other regulatory regions, as well as their precise mechanisms of action, remain poorly understood.
To study G4s in cells, several antibodies and an engineered protein—such as sty330, 1H631, D132, BG433, and G4P34—have been developed. These antibodies and the engineered protein specifically recognize and bind to G4s with high affinity and selectivity, enabling their visualization in fixed and live cells. By leveraging these antibodies, a state-of-the-art method, BG4 ChIP-seq (chromatin immunoprecipitation and high-throughput sequencing), was developed for mapping endogenous G4 structures across various cell types and tissues35. More recently, advanced methods such as G4-CUT&Tag36, snG4-CUT&Tag37, Chem-map38, and G4access39 were further developed to map native G4 structures at high resolution in bulk and single cells. While these methods have significantly advanced our ability to detect G4s genome-wide within the nucleus, they lack strand-specific resolution, limiting insights into the orientation of these structures.
To address this limitation, we developed a strand-specific G4 sequencing (ssG4-seq) method for high-resolution, genome-wide profiling of G4s in human, mouse, African green monkey, and Drosophila cells. Using this method, we found that G4 structures are predominantly located at enhancers and promoters, where the presence of G4s on both strands activates target gene transcription more potently than G4s on a single strand. Additionally, we demonstrated that the transcription factor SP1 functions as a potent G4 reader, orchestrating transcription by modulating enhancer-promoter looping. Furthermore, we found that cancer-associated mutations in G4 structures directly contribute to tumorigenesis by disrupting SP1-mediated enhancer-promoter interactions. Our study not only provides a powerful tool for investigating G4 structures and their regulatory roles but also sheds light on their functional significance in gene regulation and disease.
Results
Overview of ssG4-seq method
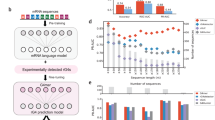
G4 structures predominantly form in single-stranded DNA (ssDNA)1,2. Guided by this fact and aiming to comprehensively map G4s across ssDNAs, we developed the ssG4-seq method for strand-specific profiling of G4s by digesting double-stranded DNAs (dsDNAs) after chromatin immunoprecipitation (ChIP) with a G4-specific antibody BG4, a G4-selective scFv antibody33. As outlined in Fig. 1a, K562 cells were first crosslinked with formaldehyde to stabilize G4 structures and chromatin within the nucleus. Chromatin was then sonicated into 100–300 bp fragments, and a high-purity Flag-tagged BG4 antibody was applied to the soluble chromatin fraction to bind G4s in ssDNA regions, as well as G4s flanked by dsDNAs on one or both ends (Fig. 1a, and Supplementary Fig. 1a). G4-containing fragments were subsequently pulled down using anti-Flag M2 magnetic beads, and a concentration-optimized duplex-specific nuclease (DSN) was employed to digest dsDNAs on the beads. The remaining G4-associated ssDNAs were eluted from beads and converted into strand-specific libraries for deep sequencing (see Supplementary Fig. 1b, Methods and Supplementary Data 1). We observed that DSN effectively digests non-G4 dsDNAs, which contain G4 levels comparable to random shuffle sequences but markedly lower than those in retained G4 regions (Supplementary Fig. 1c–e).

a Schematic diagram of ssG4-seq method. Key steps include G4 immunoprecipitation using the BG4 antibody, double-stranded DNA digestion by DSN nuclease, construction of single-strand-specific libraries, and deep sequencing. Pink dots indicate nucleotide modifications in linkers. b Pearson correlation analysis showing high reproducibility between three ssG4-seq biological replicates (Rep) in K562 cells. c IGV snapshot of G4 signal in K562 cells. Shaded regions represent high-confidence peaks detected by ssG4-seq. d Comparison of FRiP scores across G4 mapping methods. Each data point represents a biological replicate, except for the G4 CUT&Tag-Li data, which is from a single replicate. Error bars represent mean ± s.d. P-values were determined by a two-tailed unpaired Student’s t-test. e ssG4-seq (right) showing a higher signal-to-noise ratio compared to BG4 ChIP-seq (left). f Venn diagram showing the overlap between BG4 ChIP-seq and ssG4-seq peaks. BG4 ChIP-seq specific peaks, n = 420; ssG4-seq specific peaks, n = 6122. g ssG4-seq (pink) showing higher signal around overlapped peaks, ssG4-seq-specific peaks, BG4-ChIP-specific peaks compared to BG4 ChIP-seq (blue). Randomly selected genomic regions serve as a negative control.
To preserve the strand-specific information of G4 structures in the sequencing libraries, an adaptase-based ssDNA library preparation kit was used40. Briefly, the eluted ssDNAs were first treated with T4 PNK to phosphorylate the 5′ end and hydroxylate the 3′ end. A short poly(C) tail, with a median length of 4–8 bases, was then added to the 3′ end of each ssDNA by terminal transferase (TdT). The poly(C) tail subsequently base-paired with a G-overhang on truncated adapter 1, generating blunt-ended dsDNAs with the assistance of DNA polymerase. Following this, truncated adapter 2 was ligated to the 5′ end of the dsDNA using T4 DNA ligase. Finally, ssG4-seq libraries were prepared by PCR amplification with index primers for paired-end sequencing (~330 bp in length, Supplementary Fig. 1b).
Comparisons to G4 mapping methods
We generated a total of 76.89 million mappable reads from three highly correlated biological replicates in K562 cells (R = 0.98), demonstrating reproducible G4 mapping, as illustrated in Fig. 1b. After peak calling with MACS241, we identified 13,820 high-confidence G4 peaks present in all three replicates. To assess the reliability of our method, we compared ssG4-seq peaks with those identified by established G4 mapping methods, including BG4 ChIP-seq42, G4 Chem-map38, G4access39, G4 CUT&Tag-Li36, and G4 CUT&Tag-Hui37. As illustrated in the IGV (Integrative Genomics Viewer)43 snapshot (Fig. 1c), ssG4-seq, G4access, and G4 CUT&Tag-Hui all successfully recapitulated well-known G4 peaks at the Myc promoter. Moreover, ssG4-seq and G4access not only captured all BG4 ChIP-seq peaks but also uniquely identified G4s in the promoters of TSPAN31, CCDC28B, IQCC, TMEM234, and EIF3I (Fig. 1c), which were undetected by other methods. Additionally, ssG4-seq revealed a novel G4 peak in the MARCHF9 promoter.
To evaluate the background levels of our method, we calculated the fraction of reads in peaks (FRiP score), a critical metric for assessing the quality and specificity of ChIP-seq and similar assays44. FRiP quantifies the proportion of sequencing reads that fall within identified peaks relative to the total number of mapped reads in the experiment. A higher FRiP score indicates a better signal-to-noise ratio and greater specificity of the assay. By computing FRiP scores, we found that ssG4-seq has a score of ~0.09, which is sixfold higher than that of BG4 ChIP-seq (Fig. 1d). Similarly, ssG4-seq was comparable to G4 CUT&Tag and G4access, though it showed slightly lower performance than Chem-map (Fig. 1d). Notably, both G4access and Chem-map exhibited high variability. Furthermore, ssG4-seq signals showed stronger enrichment at G4 peaks (Fig. 1e and Supplementary Fig. 1f), indicating that ssG4-seq generally provides lower background noise than the other G4 mapping methods.
For a direct comparison, we identified 13,820 high-confidence ssG4-seq peaks present in all three biological replicates (Supplementary Data 2). Among these, ~8000 peaks overlapped with BG4 ChIP-seq peaks (Fig. 1f), while 6122 peaks were specific to ssG4-seq and 420 peaks were specific to BG4 ChIP-seq. This analysis revealed that ssG4-seq successfully recapitulated 95% of the BG4 ChIP-seq peaks (7930/8350). We further calculated BG4 ChIP-seq signals around the centers of overlapping peaks, ssG4-seq-specific peaks, and BG4 ChIP-seq-specific peaks. Compared to randomly selected genomic regions, ssG4-seq exhibited stronger signal intensity than BG4 ChIP-seq across all three peak categories (Fig. 1g). In addition, we observed that ssG4-seq peaks overlapped with 53.7% of G4 CUT&Tag-Li peaks, 53.9% of G4 CUT&Tag-Hui peaks, and 51% of Chem-map peaks (Supplementary Fig. 1g). For direct comparison with G4access, which generates the largest number of relatively narrow peaks, 82.5% of ssG4-seq peaks overlapped with G4access peaks (Supplementary Fig. 1g).
Potential G4 structures are typically characterized by guanine-rich motifs, often following the pattern G₃₋₅N₁₋₇G₃₋₅N₁₋₇G₃₋₅N₁₋₇G₃₋₅, where “G” represents guanine and “N” represents any nucleotide (A, T, C, or G)45. These potential quadruplex sequences (PQS) are frequently cross-referenced with experimental data to validate G4 formation. By analyzing the proportion of PQS within identified peaks, we found that BG4 ChIP-seq, G4 CUT&Tag-Hui, and ssG4-seq peaks were more enriched for PQS. This enrichment declined more gradually with decreasing peak read numbers than in G4 CUT&Tag-Li and Chem-map (Supplementary Fig. 1h). Representative snapshots (Supplementary Fig. 1i) further show that, compared with other G4 detection methods, ssG4-seq provides a higher signal-to-noise ratio than G4 CUT&Tag-Li and BG4 ChIP-seq, and is comparable to Chem-map, G4 CUT&Tag-Hui, and G4access. These results collectively demonstrate that ssG4-seq is a robust and reliable method for genome-wide G4 identification.
Strand-specific G4s in transcriptional regulation
Having validated the ssG4-seq methodology, we next aimed to evaluate the strand-specific mapping capability of our approach. We separated the reads into Watson and Crick strands based on their directionality and performed peak calling independently (see Methods). Compared to unstranded (combined reads from both strands) G4 peaks, we observed clear differences in the peaks identified by ssG4-seq on the two strands (Fig. 2a), demonstrating that G4 structures exhibit strand specificity. Next, we used MEME-ChIP46 to identify DNA motifs enriched in unstranded G4 peaks, as well as in Watson and Crick strand G4 peaks. This analysis revealed nearly identical G-rich DNA motifs across all three categories (Fig. 2b), highlighting the central role of guanines in peak formation. Notably, 88.3% of ssG4-seq unstranded peaks contained the corresponding motif, compared with 75.1% for Watson strand peaks and 71.1% for Crick strand peaks. To further assess the strand-specific mapping capability of our approach, we incorporated the concept of PQS for predicting G4 structures. As expected, we found that Watson G4s are highly enriched in Watson strand PQS but markedly depleted in Crick strand PQS (Fig. 2c). Similarly, Crick G4s showed the opposite pattern. These findings collectively validate the strand-specific mapping accuracy of our ssG4-seq method.

a IGV snapshot of strand-specific G4 structures identified by ssG4-seq. Watson and Crick strands of G4 structures are shown in red and green, respectively. BG4 ChIP-seq signals are shown in cyan color. Unstrand track refers to sequencing reads obtained without distinguishing between the Watson and Crick strands in the ssG4-seq library. b Enriched motifs identified in the unstranded, Watson, or Crick strand ssG4-seq peaks. c Heatmap of Watson- or Crick-strand-specific ssG4-seq signals centered on PQS for each strand. d Representative ssG4-seq peaks showing strong (left, Class I, n = 3405) or weak (right, Class II, n = 10415) strand-specificity in promoter regions. e Pol II occupancy distribution around ssG4-seq peaks (Class I, blue; Class II, pink). f Gene expression levels for promoters lacking G4s versus those containing Watson- and/or Crick-strand G4s. The numbers of genes are 5549, 1959, 2447 and 21,492 for Neither, Crick, Watson, and Both, respectively. The boxplot displays the median as the center line, the first and third quartiles (Q1 and Q3) as the box borders, and the whiskers extend to the most extreme data points within 1.5 times the interquartile range (IQR). P-values were determined by a two-tailed unpaired Student’s t-test. g Genomic distribution of Watson- and Crick-strand G4s.
To quantitatively characterize the strand bias of peaks identified by ssG4-seq, we introduced a K value, calculated as the ratio of Watson strand signal to Crick strand signal (K = Watson strand signal/Crick strand signal), which reflects the G4-forming propensity. A higher K value indicates a greater likelihood that G4 formation occurs exclusively on the Watson strand, while a lower K value suggests a preference for the Crick strand. When K approximates 1, it implies that both strands have comparable G4-forming ability. Based on the K value, G4 peaks were classified into two categories (Class I versus Class II, Fig. 2d). Approximately 3405 peaks were assigned to Class I (K > 2 or K < 0.5), which exhibits a strong strand bias, indicating that G4s predominantly form on one strand of the dsDNA. In contrast, about 10,415 peaks were categorized as Class II (0.5 ≤ K ≤ 2), which shows a milder bias, suggesting that both strands are capable of forming G4 structures. Notably, two slightly shifted G4s with a typical spacing of 37 bp were observed in Class II peaks (Supplementary Fig. 2a), indicating the presence of multiple G4s folded on opposite strands; however, this spacing appears unrelated to gene transcription (Supplementary Fig. 2b). Moreover, the GC skew of Class II was significantly lower than that of Class I (Supplementary Fig. 2c), indicating a more pronounced GC content difference between Watson and Crick strands in Class I. This result further supports the strand-specific capability of ssG4-seq, as G4s in Class I are predominantly localized to one strand.
Next, we investigated the relationship between G4s and gene expression levels. We confirmed a positive correlation, consistent with a recent report21, in which higher gene expression levels were associated with increased G4 content (Supplementary Fig. 2d). To determine whether Class I and Class II G4s differ in their regulatory roles, we analyzed the distribution of RNA polymerase II (Pol II)47 on these peaks. Pol II was significantly more enriched on Class II peaks than Class I (Fig. 2e). Similarly, transcription factors showed a stronger binding preference for Class II peaks (Supplementary Fig. 2e). Notably, genes with both Watson and Crick G4s at promoter regions displayed the highest RNA expression levels (n = 21,492), while those with only Watson or Crick G4s showed intermediate expression levels (n = 4406, Fig. 2f and Supplementary Fig. 2f). Genes without G4s were associated with the lowest expression levels (Neither, n = 5549, Fig. 2f and Supplementary Fig. 2f). Furthermore, genome-wide distribution analysis of G4s revealed their predominant association with promoters (83%), enhancers (14%), and intergenic regions (0.6%) (Fig. 2g). These findings underscore the functional relevance of G4s in gene regulation and highlight the strand-specific resolution provided by ssG4-seq.
Predominant location of G4s at promoters and enhancers across species
As ssG4-seq revealed predominant G4 locations at promoters and enhancers in K562 cells (Fig. 2g), we next explored whether this distribution pattern is conserved across different cell types and species. To this end, we conducted ssG4-seq in human cell lines (K562, HeLa, HepG2, U2OS, and Ramos), monkey Vero cells, and murine CH12F3 cells (Supplementary Data 1 and 2). A Circos plot illustrates the considerable conservation of G4s across these species and cell lines, with notable syntenic regions such as human chromosome 14, which aligns with monkey chromosomes 24 and 29, as well as mouse chromosomes 12 and 14 (Fig. 3a, b). Statistical analysis revealed that G4s are not randomly distributed but are instead enriched in specific genomic regions, particularly promoters and enhancers (Fig. 3c, d), suggesting a regulatory role in transcription. Furthermore, G4 signals were more significantly enriched in super-enhancers (SEs) than in typical enhancers (TEs) (Supplementary Fig. 3a). A similar pattern was observed in Drosophila, with representative tracks were shown in Supplementary Fig. 3b.

a Circos plot displaying G4 signals (1 Mb bins) across chromosomes in human (K562, HeLa, HepG2, U2OS, Ramos), monkey (Vero), and mouse (CH12F3) cells. Color-coded ribbons link syntenic regions between murine (chr12, chr14) and monkey (chr24, chr29) chromosomes to human chr14. b Magnified view of the circos plot with annotated genes. Metaplot showing that G4 signal are largely accumulated in promoter (c) and enhancer (d) across various species and cell lines. e Heatmap showing the Pearson correlation of ssG4-seq signals in promoter and enhancer regions across different species and cell lines.
To quantify the conservation of G4s, we calculated Pearson correlations for G4s in promoter and enhancer regions across species and cell lines (Fig. 3e). The results show that the degree of conservation is closely tied to evolutionary relationships, with closely related species generally showing higher G4 correlation and distantly related species exhibiting lower correlation (Fig. 3e). For example, in human cell lines (Ramos, K562, U2OS, HeLa, and HepG2), promoter-associated G4s show strong conservation, with correlations ranging from ~0.67 to 0.76. Similarly, promoter G4 correlations between human and monkey cell lines are moderately high (~0.64–0.72). In contrast, correlations between more distantly related species, such as Vero (monkey) and CH12F3 (mouse), are notably lower (~0.54) (Fig. 3e). Furthermore, cell lines derived from the same tissue origin across species exhibited higher G4 similarity in promoter regions—for instance, Ramos and CH12F3 (Fig. 3e). These findings highlight the evolutionary conservation of G4s and underscore their potential functional significance in transcription across species.
SP1 binds DNA G4s in enhancer and promoter regions
To identify TFs with a strong binding preference for G4 structures—potentially playing a significant role in transcriptional regulation through G4s—we established three criteria: (1) a higher G content in the TF’s DNA binding motif, (2) a larger number of overlapping peaks between TF binding sites and G4 peaks, and (3) a higher proportion of overlapping peaks relative to the total binding sites. A higher G content in the motif suggests an increased likelihood of G4 formation, while a larger number and proportion of overlaps indicate a more prominent regulatory role for G4s in the TF’s function. We selected K562 cells as our model system due to the availability of ChIP-seq datasets for hundreds of TFs in the ENCODE database48.
We analyzed ChIP-seq data for 344 TFs from ENCODE49, ranking their binding motifs by GC enrichment. Notably, SP1 emerged as the top candidate (Fig. 4a), with a G content exceeding 80% in its motif. In contrast, the positive control YY1, a known G4 binding protein23, showed about 31% G content, while the negative control ZNF507 contained only 20.6% (Supplementary Fig. 4a). Furthermore, SP1 displayed ~10,000 overlapping peaks with G4s, accounting for 61% of its total binding sites and representing about 72% of the G4 peaks identified by ssG4-seq (Fig. 4b, c). While some proteins, such as FUS, showed a high proportion of overlapping peaks (nearly 80%), the absolute number of overlaps was only around 1000, suggesting a more limited role in G4-mediated gene regulation.

a Transcription factors ranked by GC content of their binding motifs (inset: SP1 motif from JASPAR). b Venn diagram showing the overlap between SP1 binding sites and ssG4-seq peaks. c Scatter plot showing the numbers versus the percentage of TF binding peaks that overlapped with G4s. A known G4-binding protein YY1 and negative control ZNF507 are marked in red. d Heatmap showing the occupancy of G4s, SP1, and YY1 around enhancer (E) and promoter (P) regions, spanning 2.5 kb upstream to 2.5 kb downstream from the center of each region. e Representative tracks of ssG4-seq, SP1, YY1, and ZNF507 ChIP-seq signals around enhancers and promoters.
Compared with YY1, SP1 exhibited stronger ChIP-seq enrichment at ssG4-seq peaks, whereas ZNF507 showed minimal overlap (Fig. 4b and Supplementary Fig. 4b). Given that G4s are predominantly located at promoters and enhancers, we observed that SP1 binding signals were significantly enriched in these regions compared to YY1 and ZNF507 (Fig. 4d). This finding further supports the idea that SP1 may interact with G4 structures within key regulatory regions, as previously suggested by in vitro50 and in vivo21 studies. A specific genomic example illustrates this conclusion (Fig. 4e), where SP1 exhibits more peaks and stronger signals at G4 loci compared to YY1. Taken together, these findings strongly suggest that SP1 is a genome-wide G4-binding protein, highlighting its potential role in G4-mediated gene regulation.
SP1 exhibits stronger binding affinity to G4s than dsDNA
To assess the overall distribution of SP1 and G4 in vivo, we conducted high-resolution immunofluorescence (IF) experiments using structured illumination microscopy (SIM) and calculated the average distance between adjacent fluorescence spots. The IF results revealed that SP1 loci are predominantly colocalized with G4 sites (Fig. 5a), with a mean distance between SP1 and BG4 of ~130 ± 40 nm. This distance is significantly shorter than those observed for YY1 (153 ± 43 nm) and ZNF507 (611 ± 293 nm), suggesting that SP1 has a stronger tendency to associate with G4s in vivo.

a SIM analysis showing SP1 preferably co-localize with G4s compared with YY1 and ZNF507. The spatial distance between fluorescence dots were calculated and illustrated on the right. Green, G4 signal; Red, SP1, YY1 or ZNF507; blue, DAPI. Dash line denoted the mean disatance between G4 and the analyzed transcription factors. b Schematic of G-rich ssDNA annealing into G4 structures (KCl buffer) or dsDNA (LiCl buffer). c Circular dichroism spectra characterizing G4 DNA (blue), ssDNA (pink), and dsDNA (green) conformations. d ELISA quantification of SP1 binding affinity (G4 > ssDNA > dsDNA). Data represent mean ± SD from three biological replicates. Data in d represent results from three independent experiments. Source data are provided as a Source Data file.
SP1 is a zinc finger transcription factor that binds to GC-rich motifs in tens of thousands of promoters51,52 and in thousands of enhancers53. To quantitatively determine the binding affinity of SP1 protein toward G4 structures, ssDNAs, and dsDNAs, the enzyme-linked immunosorbent assay (ELISA) was adopted with well-known G4 structures identified in the genome. We first chose G-rich ssDNA from the Sμ region54 for ELISA assay. The Sμ ssDNA was folded into intermolecular G4 structures and isolated by gel filtration (Fig. 5b). After verifying the purity by circular dichroism spectroscopy and PAGE gel electrophoresis (Fig. 5c and Supplementary Fig. 5a, b), ELISA was performed with purified full-length SP1 protein from baculovirus insect cells (Supplementary Fig. 5c), the results showed that the dissociation constant (Kd) of SP1 for G4 structures is 7.5 ± 0.71 nM (Fig. 5d), which is about half that for ssDNA (13.48 nM) and more than five times lower than for dsDNA (46.46 nM). Similarly, we performed ELISA using another three intramolecular G4 DNAs, including hTEL G4, cKIT G4, and cMYC G4 (Supplementary Fig. 5d, e). Compared to ssDNA and dsDNA, SP1 exhibited lower Kd values for G4 structures (13.3 ± 1.58 nM for hTEL, 4.26 ± 0.4 nM for cKIT, 6.62 ± 1.2 nM for cMYC), indicating a higher binding affinity. These findings suggest that SP1 preferentially binds to G4 structures in vivo and in vitro.
SP1 modulates enhancer-promoter looping through G4s
As G4 structures are predominantly enriched in promoters and enhancers and exhibit significant overlap with SP1 binding sites, we hypothesize that SP1 protein may function directly in enhancer-promoter looping by binding to G4 structures. To test this hypothesis, we first analyzed H3K27ac HiChIP data in K562 cells55. Promoters and enhancers were categorized into four groups: those containing both G4 and SP1 (G4+ SP1+), those with only G4 (G4+ SP1−), those with only SP1 (G4− SP1+), and those lacking both (G4− SP1−). The enhancer-promoter interaction frequencies between these categories were then calculated. The heatmap revealed that the strongest interactions occur between G4+ SP1+ promoters and G4+ SP1+ enhancers (Fig. 6a), a pattern strikingly distinct from the negative control ZNF507 (Supplementary Fig. 6a). Since protein-mediated chromatin looping often relies on dimerization, as seen with CTCF and YY156,57, we next investigated whether SP1 also forms dimers, as previously reported58,59,60. FLAG-tagged and HA-tagged SP1 plasmids were co-transfected into 293T cells, and dimerization was assessed. Notably, SP1 dimerization persisted even after DNase I and RNase A treatment (Fig. 6b), indicating that SP1 dimer formation is independent of DNA or RNA.

a Heatmap of pairwise enhancer-promoter interactions showing co-occurrence of G4 structures and SP1 binding sites. G4+ SP1+, promoters or enhancers which contain G4 and SP1 binding sites simultaneously; G4+ or SP1+, promoters or enhancers which only contain G4 or SP1 binding sites, respectively. b Co-IP and western blot analysis demonstrating SP1 dimerization in K562 cells, independent of nucleic acid binding. Source data are provided as a Source Data file. c Classification of high-confidence interactions identified by SP1 HiChIP in K562 cells. d Three distinct types (I–III) of SP1-mediated enhancer-promoter interactions. E/P: enhancer or promoter. e IGV tracks showing SP1-mediated looping between enhancers and promoters at indicated loci. Genes with both SP1 binding and G4 structures exhibit higher expression levels (f) and stronger looping interactions (g) compared to control groups. The numbers of SP1-associated DNA-DNA loops for type I, type II, and type III are 4627, 4701, and 2099, respectively. The boxplot displays the median as the center line, the first and third quartiles (Q1 and Q3) as the box borders, and the whiskers extend to the most extreme data points within 1.5 times the interquartile range (IQR). The red dashed line represents the median of Type III. P-values were determined by a two-tailed unpaired Student’s t-test. h Working model showing that SP1 binds G4 structures to mediate enhancer-promoter looping and transcriptional activation.
Next, we conducted SP1 HiChIP to map genome-wide enhancer-promoter loops mediated by SP1 in K562 cells. A total of 20,895 high-confidence SP1-associated DNA-DNA loops were identified (Supplementary Data 3). Among these, 19,520 loops were located at enhancer and promoter regions, with SP1-associated enhancer-promoter loops accounting for 41% of the total loops (Fig. 6c). Notably, G4 structures were significantly enriched at the anchor points of these enhancer-promoter loops, with ~80% of interaction anchor points containing G4s in both sides (Type I, Fig. 6d) or one side (Type II, see left an example in Supplementary Fig. 6b), while only 18.4% lacked G4s (Type III, see right an example in Supplementary Fig. 6b). For example, a super-enhancer, SE641, interacts with the promoters of ARL4A and VWDE through G4 peaks identified by ssG4-seq (left, Fig. 6e). In another instance, a typical enhancer, TE1140, located within the intron of the CDC14A gene, interacts with two distal promoters via enriched G4 structures, potentially regulating their transcription (right, Fig. 6e). In addition, we found that the type I and type II SP1-associated DNA-DNA loops exhibit strong interaction frequency and higher RNA expression levels than type III loops without G4 structures (Fig. 6f, g).
Based on these findings, we propose a model in which SP1 homodimers mediate enhancer-promoter interactions (Fig. 6h). When both the promoter and enhancer contain G4s, SP1 preferentially binds to G4s with high affinity, leading to stronger interactions and higher target gene expression levels. When G4s are present in either the promoter or enhancer, SP1 exhibits a slightly lower affinity for chromatin DNA, resulting in moderate interaction strength and intermediate gene expression levels. In the absence of G4s at both sites, SP1 has the lowest affinity for chromatin DNA, mediating weak interactions and correspondingly low target gene expression levels. This model highlights the critical role of G4 structures in modulating SP1-mediated transcription regulation.
Genetic variants disrupt G4 structures to affect SP1 binding
To investigate the pathological relevance of G4 structures identified by ssG4-seq, we compiled 47.7 million high-confidence risk variants from the International Cancer Genome Consortium (ICGC)61, of which 10.4% had an RS (Rejected Substitution) score62 >2, indicating evolutionary conservation (Supplementary Fig. 7a). These compiled variants tended to localize at or near ssG4-seq peaks, with SP1-bound G4s showing more mutations than non-bound G4s (Supplementary Fig. 7b). In total, 84,593 risk variants located within 9773 SP1-bound G4 peaks (Fig. 7a). Among these risk variants, PQS analysis revealed that 703 variants might promote G4 formation, whereas 3146 variants, particularly G-A and C-T mutations, showed the potential to disrupt G4 formation (Fig. 7b).

a Venn diagram illustrating the overlap between ICGC mutations and SP1-binding G4 peaks in human K562 cells. b Mutation classification showing G4-stabilizing variants (enhanced SP1 binding) versus G4-disrupting variants (reduced SP1 binding). c Risk variants affected genes and the corresponding mutations in promoters. d Diagram of luciferase reporter assay for characterizing the influences of G4 structures and the corresponding risk variants. G4-disrupting risk variants significantly reduce luciferase reporter activity (e) and the RNA expression levels (qPCR, f). g qPCR showing significantly reduced transcription of CNDP2 and TUSC2 genes in the homozygous mutants compared to wild-type (WT) cells. h Cell growth curve analysis showing significantly increased proliferation rate of the homozygous mutant cells. Reduced G4 formation (BG4 ChIP-qPCR, i) and SP1 binding (SP1 ChIP-qPCR, j) at endogenous promoters containing the risk variants. k, l Knockdown of SP1 or mutation of G4 structures impaired chromatin looping between the TUSC2-TE14707 and CNDP2-TE10132 pairs. n = 3 biological replicates. Data in e–l are the mean ± s.d., n = 3 biological replicates, two-tailed unpaired Student’s t-test. Source data are provided as a Source Data file.
To explore further how these risk variants influence SP1 binding, we utilized SP1 SELEX data63 to quantify the binding affinity of SP1 protein to reference and variant sequences. By integrating these datasets, we identified 66 variants that could enhance both G4 formation and SP1 binding affinity. Among them, single-nucleotide variants (SNVs) accounted for 47%, while insertions (Ins) constituted 53%. Conversely, we identified 453 variants predicted to suppress G4 formation and reduce SP1 binding affinity, comprising 81% SNVs, 0.4% insertions, 8.6% deletions (Del), and 10% multi-nucleotide variants (Fig. 7b). Notably, among the 334 genes affected by variants that disrupt G4 formation, 7.8% (n = 26) were tumor suppressor genes (TSGs, Supplementary Fig. 7c), while the remaining genes were functionally associated with processes such as localization, cellular function, viral response, and development (Supplementary Fig. 7d). These findings suggest that risk variants may directly contribute to tumorigenesis by influencing G4 structures.
To investigate the effects of G4 mutations on gene expression, we analyzed expression quantitative trait loci (eQTLs) from GTEx database (https://gtexportal.org/home/), which are genetic variants that influence the gene expression level in specific tissues or cell types64,65. We examined the distribution of eQTLs within G4 peaks and SP1-bound G4 peaks, revealing that SP1-bound G4s had a higher eQTL content (Supplementary Fig. 7e). This finding suggests a direct connection between G4 mutations and the regulation of gene expression. For instance, the AMFR gene locus contains both G4 structures and SP1 binding sites (Supplementary Fig. 7f). When a G-to-C mutation occurs, G4 formation is disrupted, leading to reduced SP1 binding and a significant decrease in gene transcription (Supplementary Fig. 7g).
To explore how risk variants influence transcription through G4 structures, we selected four TSGs whose promoter regions contain G4 peaks bound by SP1. Notably, these G4 peaks harbor risk variants that disrupt G4 formation (Fig. 7c). We cloned the promoter regions of these four genes and inserted them into a promoterless firefly luciferase reporter vector (Fig. 7d). After transfecting wild-type (WT fragment insertion) and mutant (MT fragment insertion) plasmids into 293T cells for 24 h, we measured luciferase activity. The results showed a significant reduction in firefly luciferase activity when the G4 structure in the promoter region was disrupted by the risk variants (Fig. 7e). This effect likely occurs at the transcriptional level, as qPCR analysis revealed a significant decrease in mRNA levels (Fig. 7f).
Next, we employed a CRISPR-Cas9-mediated homologous recombination strategy to successfully generate homozygous knock-in mutations in the promoters of two TSGs, TUSC2 (Tumor suppressor candidate 2, a G-to-T transversion) and CNDP2 (a G-to-A transition), in K562 cells (see genotyping in Supplementary Fig. 7h). qPCR analysis confirmed significantly reduced transcription of these genes in the homozygous mutants compared to wild-type (WT) cells (Fig. 7g). Additionally, we observed a significant increase in the proliferation rate of cells harboring these homozygous mutations (Fig. 7h). Moreover, G4 ChIP-qPCR revealed a significantly decreased BG4 occupancy at the mutant promoters (Fig. 7i), while SP1 binding was also markedly reduced by at least 49% at both sites (Fig. 7j).
Next, we investigated whether SP1 knockdown directly affects G4-mediated enhancer–promoter looping. 3C-qPCR (Chromosome Conformation Capture combined with quantitative PCR)66 revealed that shRNA-mediated SP1 knockdown significantly reduced looping frequency between the TUSC2-TE14707 and CNDP2-TE10132 pairs in WT cells (Fig. 7k, l). In cells carrying risk variants (MT) at promoter G4s, enhancer–promoter looping was already markedly reduced. SP1 knockdown further decreased this looping frequency to a level comparable to that in WT cells after knockdown (Fig. 7k, l). Collectively, these results demonstrate that human genetic risk variants can impair transcription by disrupting SP1-associated enhancer-promoter loops, likely through their destabilizing effects on G4 structures.
Discussion
In this study, we developed a strand-specific G4 mapping method for global profiling of G4 structures across multiple mammalian and invertebrate genomes. Such efforts revealed a general feature that G4s are predominantly enriched in promoters and enhancers, where G4s at both strands tend to correlate with higher transcriptional activation ability. Moreover, we found that transcription factor SP1 functions as a potent reader protein of G4, and it regulates transcription by modulating long-range enhancer-promoter looping through G4. Furthermore, we identified tens of thousands of cancer-associated risk variants in G4 structures, which probably can affect SP1-mediated enhancer-promoter looping, thereby contributing to tumorigenesis. Our study demonstrates the power and applicability of ssG4-seq in studying the distribution, stability, and functional roles of G4 structures.
Unlike previous methods37,38,39,42,67, ssG4-seq provides strand-specific information, allowing us to distinguish G4s on the Watson and Crick strands. This information is critical for understanding the asymmetric roles of G4s in transcription and other biological processes. Additionally, ssG4-seq combines the specificity of G4-binding proteins with the sensitivity of next-generation sequencing, enabling the detection of G4 structures at high resolution. Furthermore, ssG4-seq can be applied to various biological systems, including cell lines, tissues, and clinical samples. This versatility makes it a powerful tool for studying G4s in different physiological and pathological contexts.
G4 structures serve as binding hubs for TFs, with some TFs exhibiting comparable binding affinities to G4s as to dsDNA22. In this study, we identified SP1 as a top candidate with the strongest G4-binding tendency based on two key criteria: a higher overlapping rate and a larger number of overlapping sites between SP1 binding and G4 structures. Subsequent in vivo and in vitro biochemical experiments confirmed that SP1 preferentially binds to G4s. Using SP1 HiChIP assays, we demonstrated that promoters (SP1+ G4+) and enhancers (SP1+ G4+) with SP1-bound G4s mediate stronger enhancer-promoter interactions, driving high levels of expression in their target genes. Furthermore, we identified many risk variants and eQTLs residing within the identified G4 peaks, particularly those bound by SP1. This indicates that SP1 binding to these G4s has a substantial impact on gene expression and may contribute to disease mechanisms. Additionally, our study revealed that risk variants in TSGs can disrupt G4s to repress promoter activities, highlighting the functional importance of G4 structures in tumorigenesis.
SP1 is a ubiquitous TF traditionally associated with binding GC-rich dsDNA68, and showed substantial binding affinity to G4 in vitro50. However, our findings reveal its ability to preferentially bind G4 structures in vivo, consistent with a recent report showing that AP-1 and SP1 co-immunoprecipitate with G4-containing promoters21. This expands the functional repertoire of SP1 and highlights the versatility of TFs in recognizing non-canonical DNA structures. Our work also positions G4s as critical regulatory hubs in the genome, where TFs like SP1 can dynamically interact to modulate gene expression. This challenges the conventional view of G4s as mere structural elements and underscores their functional importance in transcriptional regulation. Moreover, we demonstrated that SP1-bound G4s at promoters and enhancers facilitate stronger enhancer-promoter interactions, likely driving high levels of gene expression. This provides a mechanistic link between G4 structures, TF binding, and 3D genome organization.
Our discovery of risk variants that influence G4 structure and function represents a significant conceptual advance in understanding the interplay between genetic variation, DNA secondary structures, and disease mechanisms. We demonstrated that specific risk variants, particularly SNVs, can disrupt or stabilize G4 structures. This establishes a direct link between genetic variation and the formation of non-canonical DNA structures, expanding our understanding of how sequence changes influence genome architecture. By showing that G4-disrupting variants can alter TSG expression, our work highlights the functional importance of G4 structures in maintaining proper transcriptional regulation. This provides a mechanistic explanation for how non-coding variants can impact gene function and contribute to disease.
While ssG4-seq represents a major advance in mapping G4 structure, there are some limitations to consider. For example, this method relies on BG4 nanobody, which can exhibit inherent biases or preferences for specific G4 structures and may occasionally bind non-canonical DNA structures, resulting in off-target signals. Additionally, experimental treatments or buffer conditions that stabilize G4 structures can alter BG4 binding, potentially exaggerating or biasing the observed G4 landscape. The resolution of ssG4-seq is limited by the efficiency of G4 capture and DSN digestion. Future improvements could include the development of more specific G4-binding proteins and optimized protocols for G4 detection. In summary, ssG4-seq is a powerful and versatile method for mapping single-stranded G4 structures genome-wide. By providing high-resolution, strand-specific insights into G4 biology, ssG4-seq has the potential to transform our understanding of these dynamic structures and their roles in gene regulation, genome stability, and disease, paving the way for new discoveries and therapeutic innovations.
Methods
Cell culture
K562 (ATCC, CCL-243) and Ramos (ATCC, CRL-1596) cells were cultured in RPMI 1640 medium (Gibco, C22400500BT) supplemented with 10% fetal bovine serum (FBS; Gibco, 16000044) and 1% penicillin-streptomycin (Gibco, 15140122). CH12F3 cells were maintained in RPMI-1640 medium containing 10% FBS, 10% GlutaMAX™ (Gibco, 35050061), 50 μM 2-mercaptoethanol (Sigma-Aldrich, M6250), 10 mM sodium pyruvate (Gibco, 11360070), and 1% non-essential amino acids (NEAA; Gibco, 11140050). HeLa (ATCC, CCL-2), U2OS (ATCC, HTB-96), and Vero (ATCC, CCL-81) cells were incubated in Dulbecco’s Modified Eagle medium (Gibco, 11995065) supplemented with 10% FBS and 1% penicillin-streptomycin. HepG2 (ATCC, HB-8065) cells were maintained in Minimum Essential medium (Gibco, 11095080) containing 10% FBS and 1% penicillin-streptomycin. All cells were cultured in a 37 °C humidified incubator with 5% CO2. Drosophila S2 cells were cultured at 25 °C in Schneider’s Drosophila Medium (Gibco, 21720024), supplemented with 10% heat-inactivated FBS, and 1% penicillin-streptomycin. All the cultured cells were routinely checked to avoid contamination of mycoplasma.
ssG4-seq
Chromatin fragmentation
Approximately 10 million cells were cross-linked in 20 ml PBS containing 1% formaldehyde (Sigma-Aldrich, F8775) at RT for 10 min. The reaction was quenched by adding 1 ml of 2.5 M glycine (Amresco, 0167-5KG) to a final concentration of 0.125 M. After gentle rocking for 10 min, the fixed cells were pelleted by centrifugation at 1200 rpm (4 °C, 5 min) and washed three times with ice-cold PBS. The cell pellet was resuspended in 1 ml ice-cold cytoplasm lysis buffer (10 mM Tris-Cl, pH 8.0, 0.5% NP-40, 10 mM NaCl, 1× protease inhibitor cocktail (Sigma-Aldrich, P8340-5ML)) and incubated on ice for 10 min. Nuclei were pelleted at 3500 rpm (5 min, 4 °C), and the supernatant was discarded. The nuclear pellet was resuspended in 700 μl nuclear lysis buffer (50 mM Tris-Cl, pH 8.1, 1% SDS, 10 mM EDTA, 1× protease inhibitor cocktail) and incubated at 4 °C for 10 min. The nuclear lysate was divided into two aliquots in 1.5 ml Bioruptor microtubes (Pico, C30010016) and sheared to 100–300 bp fragments using a Picoruptor (Diagenode, 13–15 cycles, 30 s ON/OFF). The sonicated chromatin was clarified by centrifugation at 12,000 rpm (15 min, 4 °C), and the supernatant was pooled into a new 1.5 ml DNA LoBind tube (Eppendorf, 022431021) and kept on ice. Chromatin concentration was determined using the Qubit double-stranded DNA HS assay kit (Thermo Fisher Scientific, Q32851).
G4 immunoprecipitation
2 μg of fragmented chromatin was incubated in 500 μl blocking buffer (25 mM HEPES, pH 7.5, 10.5 mM NaCl, 110 mM KCl, 1 mM MgCl₂, 1% BSA) supplemented with 50 U RNase A (Thermo Fisher, EN0531) at 37 °C for 20 min with shaking (1200 rpm) to remove RNA G4 structures. Following RNA digestion, 0.5 μg BG4 antibody was added and the mixture was incubated at 16 °C for 1 h with shaking. Concurrently, 10 μl anti-FLAG M2 magnetic beads (Sigma, M8823) were pre-blocked in blocking buffer at 16 °C for 1 h. The blocked beads were then added to the chromatin-antibody mixture and incubated at 16 °C for 1 h to capture BG4-bound G4 structures. The immunoprecipitated complexes were washed three times with ice-cold wash buffer (10 mM Tris, pH 7.4, 100 mM KCl, 0.1% Tween 20), followed by treatment with Heat-Labile Double-Strand Specific DNase (DSN or HL dsDNase; Haigene, C5010) in rCutSmart buffer at 37 °C for 10 min to digest dsDNA. After a final wash with wash buffer at 37 °C for 10 min, DNA was eluted by incubating the beads in elution buffer (10 mM Tris-HCl, pH 8.0, 50 mM NaCl, 10 mM CaCl₂) containing 0.4 mg/mL proteinase K (Takara, 9034) at 37 °C for 1 h followed by 65 °C overnight. The eluted DNA was purified using Oligo Clean & Concentrator kit (Zymo, D4061) and finally eluted into 20 μl low-EDTA buffer (10 mM Tris-HCl, pH 8.0, 0.1 mM EDTA).
Single-stranded DNA library construction
The ssDNA library was prepared through five sequential steps: denaturation, poly(C)-assisted ligation, extension, blunt end ligation, and indexing PCR. First, 10 μM truncated adapters (P7-1/P7-2 and P5-1/P5-2) were prepared by annealing equimolar primers (20 μM) in low salt buffer (20 mM Tris-HCl, pH 8.0, 50 mM NaCl, 0.1 mM EDTA) using a thermal program of 95 °C for 5 min followed by gradual cooling (−0.1 °C/s) to RT. For denaturation, 15 μl DNA was heat-treated at 95 °C for 2 min and immediately chilled on ice for 2 min to generate ssDNA fragments. Poly(C)-assisted ligation was then performed by adding 25 μl of TdT mix [0.2 μl of Terminal transferase (NEB, M0315L), 0.2 μl of T4 Polynucleotide Kinase (NEB, M0201L), 0.2 μl of Thermostable FenI (NEB, M0645S), 1.2 μl of Escherichia coli DNA Ligase (NEB, M0205L), 0.2 μl of Klenow Fragment (3′−5′ exo-; NEB, M0212S), 4 μl of 10× reaction buffer (0.7 M Tris-HCl, pH 8.0,10 mM MgAc2, 0.26 mM NAD+, 10 mM DTT, 10 mM ATP, 0.5 M KAc,10 mM dCTP), 2.5 μl of 10 μM truncated adapter 1, and 16.5 μl of ddH2O] to the tube, enabling template-independent ligation of adapter 1 to the ssDNA fragments via poly C:poly G pairing. Extension followed with 46 μl elongation mix containing 3 μl of 25 μM extension primer E7 and 43 μl of 2× Phanta Max Master Mix, using PCR conditions of 98 °C/1 min, 60 °C/2 min, and 72 °C/5 min. The product was purified (1.2:1 Ampure XP beads) and eluted in 17 μl nuclease-free water. Subsequently, 23 μl ligation mix (1 μl of 10 μM truncated adapter 2, 2 μl of Quick Ligase, and 20 μl of 2×Quick Ligase Reaction Buffer (NEB, M2200)) was added and ligated the adapter 2 to the 5’ end of dsDNA at 25 °C for 10 min, followed by Ampure XP bead purification (1:1 ratio) and eluted into 20 μl of nuclease-free water. Finally, indexing PCR was performed with 30 μl reaction mix (25 μl of 2×KAPA HiFi HotStart ReadyMix (KK2602), 2.5 μl of 10 μM P5 primer, and 2.5 μl of 10 μM index P7 primer) using thermal cycling (98 °C/3 min; 9-11 cycles of 98 °C/15 s, 60 °C/20 s, 72 °C/1 min; 72 °C/5 min), with subsequent Ampure XP bead purification (1:1 ratio) and elution in 20 μl nuclease-free water.
Immunofluorescence
K562 cells were fixed with 4% paraformaldehyde (Sigma-Aldrich, F8775) and permeabilized with 0.3% Triton X-100 for 15 min. To eliminate potential RNA G4s, cells were first treated with 50 µg/ml of RNase A for 10 min at 25 °C. After washing three times with PBS, K562 cells were blocked with 10% normal donkey serum in PBS for 1 h. The primary antibodies SP1 (Active Motif, 39058), YY1 (ABclonal, A19569), and ZNF507 (Bethyl, A303-274A-T) were diluted in PBST buffer and incubated with cells overnight at 4 °C. On the next day, the cells were washed with PBST for three times and then incubated with donkey anti-mouse 488 (life technology, A21202) and donkey anti-rabbit 594 (life technology, A21207) for 3 h at RT and counterstained with DAPI for 10 min. After washing three times with 0.1% Tween-20 in PBS, the cells were spread on cover slides and imaged with SIM on a DeltaVisionOMXV4 system (GE Healthcare) equipped with a 603/1.42 NA Plan Apo oil-immersion objective (Olympus) and three laser beams (405, 488 and 568 nm).
Purification of His-tagged SP1
The full-length SP1 with an N-terminal 6 × His tag was cloned into the pFastBac1 vector using BamHI and XhoI restriction sites, with construct identity confirmed by Sanger sequencing. The recombinant plasmid was transformed into DH10bac competent cells (AngYuBio, G6006-10) for bacmid generation through blue-white selection. Recombinant bacmid DNA was then transfected into Sf21 insect cells cultured in SF-900 II medium (Invitrogen, 10902088) at 27 °C using Cellfectin® II reagent (Invitrogen, 10362100) to produce P1 baculovirus stock. After 5 days transfection, P1 virus was harvested and used to infect fresh Sf21 cells for P2 virus amplification. The P2 virus was collected 4 days post infection and similarly used to generate P3 virus stock, which was aliquoted and stored at −80 °C for long-term preservation.
Log-phase Sf21 cells were infected with P3 baculovirus at a 5% inoculum density for recombinant SP1 expression. Cells were harvested 72 h post infection and resuspended in 50 ml of lysis buffer (20 mM Tris-HCl, pH 8.0, 500 mM NaCl, 20 mM imidazole, 10% glycerol). After complete resuspension, cells were lysed by sonication and centrifuged at 16,000 rpm for 30 min. The clarified supernatant was filtered (0.45 μm) and loaded at 1 ml/min (4 °C) onto a 5 ml Ni-NTA column (GE, 17524801) pre-equilibrated with buffer A using an ÄKTA FPLC system. Bound proteins were eluted with a linear imidazole gradient (20–500 mM) at 2 ml/min. SP1-containing fractions were pooled, concentrated, and further purified by size-exclusion chromatography using a Superdex 200 column (GE, 28990944) equilibrated with gel filtration buffer (20 mM Tris-HCl, pH 8.0, 200 mM NaCl, 1 mM DTT, 10% glycerol). Finally, high-purity SP1 fractions were pooled, aliquoted, and stored at −80 °C.
Circular dichroism spectroscopy
For intermolecular G4 formation, 500 μM biotinylated Sμ oligonucleotides were annealed in G4-forming buffer (25 mM Tris-HCl, pH 7.5, 1 M KCl, 1 mM DTT) by heating to 95 °C for 5 min followed by gradual cooling (−0.1 °C/s) to RT. The resulting structures were purified by size-exclusion chromatography using a Superdex 75 column (GE, 29148721) and buffer-exchanged into low-salt conditions (25 mM Tris-HCl, pH 7.5, 100 mM KCl, 1 mM DTT). Intramolecular G4 structures were prepared by heating 10 μM oligonucleotides in low-salt buffer to 95 °C for 5 min followed by immediate cooling on ice. To obtain dsDNA and ssDNA, LiCl buffer (25 mM Tris-HCl, pH 7.5, 100 mM LiCl, 1 mM DTT) was used to eliminate G4 structure. Briefly, mixed equal molar concentration of Sμ fragment and its complement strand performed the same annealing procedure, but merely KCl was replaced by LiCl. For linear DNA, Sμ fragment was incubated at 95 °C for 5 min and flash cooled on ice. All DNA samples were characterized by circular dichroism spectroscopy using a Chirascan Plus spectrometer (200–300 nm wavelength range, 1 nm/s scan rate) at RT.
Enzyme-linked immunosorbent assay
BeaverNano™ streptavidin coated 96-well plate (Beaver, 22351) was washed twice with wash buffer (25 mM Tris-HCl, pH 7.5, 10.5 mM NaCl, 110 mM KCl, 1 mM MgCl2 and 0.1% Tween-20), followed by incubation with 100 μl of 5 nM biotinylated oligonucleotides at RT for 30 min. After washing twice with wash buffer, the plate was blocked with blocking buffer (5% BSA in wash buffer) at RT for 1 h. The blocked plate was incubated with the gradient concentration of full-length SP1 protein in binding buffer (25 mM Tris-HCl, pH 7.5, 10.5 mM NaCl, 110 mM KCl, 1 mM MgCl2, 3% BSA, 20 μM ZnCl2, and 3% glycerol) at RT for 1 h. After washing three times to remove unbound proteins, the plate was incubated with anti-SP1 antibody (Active Motif, 39058; 1:2000) diluted in binding buffer at RT for 1 h. The plate was subsequently washed three times with wash buffer, and incubated with goat anti-rabbit-HRP antibody (Invitrogen, A-11012; 1:5000) at RT for 1 h, followed by washing three times. Signal development was performed using TMB substrate (Biolegend, 421101) for 15 min at RT before stopping the reaction with an equal volume of 0.18 M sulfuric acid. Absorbance at 450 nm was measured using a microplate reader, and dissociation constants (Kd) were determined by analyzing saturation binding curves in GraphPad Prism.
HiChIP
HiChIP was performed as previously described69. Briefly, ~30 million K562 cells were cross-linked with 1% formaldehyde for 10 min at RT, quenched with 0.125 M glycine, and washed three times with PBS. Cell pellets were resuspended in 500 μl ice-cold Hi-C lysis buffer (10 mM Tris-HCl, pH 7.5, 10 mM NaCl, 0.2% NP-40, 1× protease inhibitors), rotated at 4 °C for 30 min, centrifuged (2500 × g, 5 min), and washed once with 500 μl Hi-C lysis buffer. The resulting nuclei were resuspended in 100 μl of 0.5% SDS and incubated at 62 °C for 10 min, followed by addition of 50 μl 10% Triton X-100 (final 1.5%) to quench SDS. After 15-min incubation at 37 °C, chromatin was digested with 50 U of MboI restriction enzyme (NEB, R0147L) in 500 μl NEB buffer 2 at 37 °C for 2 h, then heat-inactivated at 65 °C for 20 min.
The digested nuclei were pellet down by centrifugation at 2500 × g for 5 min at 4 °C. After discarding the supernatant, the nuclei were resuspended in 100 μl of fill-in master mix, which contained 0.6 mM biotin-14-dATP (Invitrogen, 19524016), 0.6 mM dTTP/dCTP/dGTP, and 100 U of DNA Polymerase I (Large Klenow fragment; NEB, M0210L), and incubated at 37 °C for 2 h with rotation. After fill-in reaction, 900 μl of ligation master mix (0.83% Triton X-100, 1 mg/ml BSA, 8000 U of T4 DNA ligase (NEB, M0202L) in 1× NEB T4 DNA ligase buffer) was added to the pelleted nuclei and ligate the DNA ends in proximity. The reaction tube was continually rotated at RT for 4 h. The nuclei were pelleted down at 2500 × g for 5 min and the supernatant was discarded.
The nuclei were resuspended with 350 μl of nuclear lysis buffer (10 mM Tris–HCl, pH 8.0, 100 mM NaCl, 1 mM EDTA, 0.5 mM EGTA, 0.1% sodium deoxycholate, 0.5% N-lauroylsarcosine, and 1× protease inhibitor), and transferred to a new 1.5 ml Bioruptor Microtubes with Caps. The chromatin was sheared using the following setting: 4 cycles, 30 s on/off. The sample was centrifuged at 12,000 × g for 10 min, then 1/10 volume of 10% triton X-100 was added, the supernatant was transferred to a new 1.5 ml DNA LoBind tube. Immunoprecipitation was performed by adding 8 μg SP1 antibody (Active mofit, 39058) to the lysate and rotating at 4 °C overnight. At the same time, 50 μl of Protein A/G beads were washed twice with nuclear lysis buffer, and resuspended with 1.5 ml of blocking buffer (Nuclear lysis buffer containing 10 mg/mL BSA), incubated at 4 °C overnight with rotation. On the next day, the blocked protein A/G beads were added into the above sample, rotated at 4 °C for an extra 4 h. Beads were washed three times with nuclear lysis buffer, twice with LiCl buffer (10 mM Tris–HCl, pH 7.5, 250 mM LiCl, 1 mM EDTA, 1% NP-40, 1% Sodium deoxycholate), once with 1 ml of low-salt buffer (10 mM Tris-HCl, pH 8.0, 100 mM NaCl, 1 mM EDTA). The thoroughly washed beads were resuspended with 100 μl of elution buffer (low salt buffer containing 0.2 mg/mL proteinase K), incubated at 37 °C for 2 h, followed by 65 °C overnight with shaking to reverse cross-linking. The eluted DNA was finally purified using Zymo Oligo clean & concentrator (Zymo, D4061) and quantified by Qubit dsDNA HS Assay Kit (Invitrogen, Q32854).
For library preparation, 5 μl of Dynabeads MyOne Streptavidin C1 beads (Invitrogen, 65001) were washed with Tween wash buffer and resuspended in 10 μl of 2× biotin binding buffer (10 mM Tris-HCl, pH 7.5, 2 M NaCl, 1 mM EDTA) and added to each sample. After incubation at RT for 20 min with rotation, beads were washed with twice with Tween wash buffer, once with 200 μl TE buffer (10 mM Tris-HCl, pH 7.5, 2 M NaCl, 1 mM EDTA), and incubated with 0.5 μl Tn5 transposase mix (TruePrep DNA Library Prep Kit V2, Vazyme, TD501). After incubation at 55 °C for 10 min, 2 μl of 5× TS buffer was added to strip Tn5 enzyme. Beads were subsequently washed twice with 200 μl of Tween wash buffer (5 mM Tris-HCl pH 7.5, 1 M NaCl, 0.5 mM EDTA, 0.05% Tween-20) at 55 °C for 2 min with shaking, once with TE buffer, then resuspended with PCR mix (25 μl of 2× KAPA HiFi HotStart ReadyMix, 2.5 μl of 10 mM N5XX forward primer, 2.5 μl of 10 mM N7XX reverse primer, 20 μl of ddH2O). The PCR program was set as follows:72 °C 3 min; 95 °C 3 min; 14–15 cycles of 98 °C 15 s, 60 °C 20 s, 72 °C 3 min; 72 °C 5 min. HiChIP libraries were size selected to 300–700 bp by using 0.5×/0.3× Ampure XP beads, and eluted in 15 μl of ddH2O.
HiChIP data analysis
HiChIP raw data was first aligned to hg19 reference genome with HiC-Pro70 pipeline, within default parameters for bowtie271 global and local alignment. Paired reads with two different MboI restriction cutting sites were retained. FitHiChIP72 was used to call significant interactions from the mapped reads. FitHiChIP applies fixed-size 5 kb binning and defines a peak-bin if that bin overlaps with HiChIP peaks called by MACS2 with restriction of q = 0.01. Otherwise, that bin is defined as non-peak-bin. Peak-to-Peak (interactions involving peak bins at both ends) mode was used to estimate the contact probability with genomic distance between 5 kb and 2 Mb. Interactions with q-value < 0.05 were regarded as significant loops and used for downstream analysis. Significant loop with its chromosome name and middle nucleotide region of each bin can be visualized in IGV as arcs by adding ‘track graphType=arc’ to the head of BED files.
Dual-luciferase assay
CNDP2, DMTF1, SIRT6, and TUSC2 promoters were amplified from the human genomic DNA and cloned into pGL3-Basic vectors between BmtⅠ and HindⅢ restriction sites (Supplementary Data 4). The mutant (MT) reporters were generated through site-directed mutagenesis protocol, as we previously described73. 200 ng of the WT or MT reporters were co-transfected with 200 ng of pRL-TK Renilla luciferase plasmids into 293T cells at the 24-well plate using GeneTwin™ reagent (Biomed, TG101-02). On the next day, the cells were washed three times with PBS and resuspended in 100 μl of 1× cell lysis buffer (Promega, E1910), followed by incubation at RT for 10 min. Half of the lysate was centrifuged at 12,000 rpm for 2 min at RT, and the supernatant was transferred to a new 1.5 ml EP tube for quantification using the Dual-Luciferase Reporter Assay System (Promega, E1910) on a GloMax® 96 Microplate Luminometer (Promega, E6531). The other half of the lysate was used to extract total RNA using TRIzol LS (Thermo Fisher Scientific, 10296028). RT-qPCR was performed on the StepOnePlus real-time PCR machine (Applied Biosystems, 4376592) with Hieff qPCR SYBR Green Master Mix (YEASEN, 11203ES08).
Knock-in of risk variants at CNDP2 and TUSC2 promoters
The relevant gRNAs were designed using CRISPRdirect74 (http://crispr.dbcls.jp /) and inserted into the px330-mCherry plasmid. The donor template containing the corresponding mutations was cloned into the pEGFP-N1 plasmid using NotI restriction site. The gRNA and donor plasmids were co-transfected into K562 cells using GeneTwin™ reagent (Biomed, TG101-02). After 2 days of transfection, EGFP and mCherry double-positive cells were sorted into 96-well plates for further culture of at least 1 week. The genomic DNA was extracted from single colony derived cells for genotyping by Sanger sequencing. Successfully knock-in cells were expanded and used for subsequent experiments.
Cell growth assay
The CNDP2 and TUSC2 mutant cells were diluted to a concentration of 10,000 cells/ml in RPMI 1640 medium, and seeded 100 μl per well into a 96-well plate. All cells were cultured in a 37 °C humidified incubator with 5% CO₂. 20 μl of MTS (CellTiter96® AQueous One Solution Reagent, Promega, G3508) was added to each well of the cells at different time points and incubated at 37 °C for 1 h. The absorbance at 490 nm was measured using Multiskan™ FC (Thermofisher, 1410101).
Co-IP and western blotting
Co-IP was performed as previously described73. Briefly, 293T cells in 15 cm dishes were co-transfected with 15 μg of pCDNA3.0-Flag-SP1 and pCDNA3.0-HA-SP1 using GeneTwin™ transfection reagent (Biomed, TG101). After 48 h, cells were washed with ice-cold PBS and lysed in co-IP buffer (10 mM Tris-Cl pH 7.4, 100 mM NaCl, 2.5 mM MgCl₂, 0.5% NP-40, 1× protease inhibitors) with rotation at 4 °C for 30 min. Lysates were sonicated (Branson SLPe, 40% amplitude, 10 s on/30 s off, 2 cycles) and supernatants were aliquoted for incubation with 6 μg of IgG (Santa Cruz, sc-2025), anti-HA (Dia-An, 2063), or anti-FLAG (Sigma, F1804) antibodies overnight at 4 °C, followed by addition of pre-blocked Protein A/G beads (Thermo, 26162; 30 μl/sample) and rotated at 4 °C for 4 h. After thoroughly washing with co-IP buffer, beads were further treated with DNase I/RNase A (Thermo, AM2238) or control buffer at RT for 10 min. The proteins were eluted from beads with 1× LDS buffer at 70 °C for 15 min and resolved by 10% SDS-PAGE for western blotting analysis. The following antibodies were used: anti-FLAG (Sigma, F1804; 1:1000), anti-HA (Dia-An, 2063; 1:1000), and HRP-conjugated goat anti-mouse IgG (Invitrogen, 31430; 1:10,000).
ssG4-seq mapping and visualization
Sequencing adapters and polyG at the end of raw ssG4-seq reads were removed using the Trimmomatic75 (v0.36) and Cutadapt (v2.10), respectively. After filtering, clean reads were mapped to the reference genome using the mem command in the BWA software76 (v0.7.17). Of note, the version of genome assembly is hg19 for human, mm10 for mouse, chlsab2 for green monkey, and dm6 for Drosophila. PCR duplicates were discarded using the Picard MarkDuplicates function (http://broadinstitute.github.io/picard/). Paired-end reads that aligned to the forward or reverse strand were separated by the SAMtools77 package (v0.1.19). Mapping results were stored in BAM format files which could be converted into BigWig format files by the bedGraphToBigWig78 program and then visualized by the IGV43 visualization tool (v2.3.92).
Analysis of G4 peaks, motifs, and TF binding
The ssG4-seq peaks were identified using the MACS279 software (v2.1.1) with input DNA as a control, under the following parameters: macs2 callpeak -t ssG4.bam -c Input.bam -f BAMPE -n name --nomodel --keep-dup all -p 0.00001. Peaks that were identified in all three replicates were considered as high confidence peaks for K562 cells. Consensus motifs for ssG4-seq peaks were identified using the genomic sequence in the high confidence peaks by the MEME80 package (v5.0.4) with the following parameters: meme -revcomp -dna -nmotifs 10 -evt 0.01 -minw 3 -maxw 12.
The association between G4 peaks and TF binding was quantified by intersecting Class I and Class II G4 peak coordinates with TF binding sites using bedtools intersect (with the -u option). For each class, the binding percentage was derived by dividing the count of overlapping peaks by the total number of peaks in that class.
Enhancer and promoter definition
Enhancers and promoters were defined as previously described81, with minor modifications, using H3K4me1, H3K4me3, and H3K27ac ChIP-seq data generated in each cell line. Promoters were defined as H3K4me3-enriched peaks within ±2.5 kb of TSSs. Enhancers were defined by stitching together H3K27ac peaks located within 12.5 kb of each other and classifying the resulting clusters as enhancers. Super-enhancers were screened out using the ROSE algorithm82 based on the coverage of H3K27ac ChIP-seq signals in enhancer regions.
G4 conservation
To calculate the correlation between ssG4-seq datasets among different species, the human genome (hg38 version) was selected as a standard reference. First, the genomic coordinates of alignments for all ssG4-seq datasets were converted to the hg38 version using the CrossMap83.py script. Next, non-overlapped 1-kb windows in the hg38 genome that are syntenic to the defined genomic region (e.g., enhances or promoters) for both species were selected using the LiftOver84 tool. Finally, the number of ssG4-seq reads in these 1-kb windows was calculated and used to calculate the Pearson correlation coefficient. G4 sites in human chromosome 14 that are syntenic to G4 sites in mouse or green monkey chromosomes were identified using the LiftOver tool and illustrated by the Circos85 software (v0.69-5).
TF binding sites identification
ChIP-seq datasets for 344 transcription factors (TF) were downloaded from the ENCODE database (https://www.encodeproject.org), including data for SP1 (ENCSR991ELG) and ZNF507 (ENCSR598TIR). Binding peaks for each TF were identified by the MACS2 software with default parameters. Notably, YY1 binding peaks identified by Li et al.23 were directly used. The YY1 ChIP-seq data were downloaded from the GEO repository with accession number GSE128106. Consensus motifs and corresponding logos for all TFs were downloaded from the JASPAR86 database.
RNA-seq analysis
Briefly, adapters in RNA-seq reads were removed using the Trimmomatic75 software (v0.36). After filtering, clean reads were mapped to the reference genome (assembly version: hg19) by the HISAT2 program (v2.1.0). The abundance of each gene or isoform was calculated by the StringTie87 software (v2.0.4).
3C-qPCR
3C-qPCR was performed as previously described88. Briefly, ~10 million cells per condition were crosslinked with 1% formaldehyde for 10 min at RT, and the reaction was quenched with 125 mM glycine. Nuclei were isolated and lysed in ice-cold lysis buffer. Chromatin was digested overnight with 400 units of MboI (NEB, R0147M) per 10 million cells. After restriction enzyme inactivation, chromatin fragments were ligated using T4 DNA Ligase (NEB) in a large-volume reaction for 4 h at 16 °C. Crosslinks were reversed by overnight incubation with proteinase K at 65 °C, followed by DNA purification via phenol-chloroform extraction and ethanol precipitation. The resulting 3C libraries were quantified using a Nanodrop spectrophotometer. qPCR was performed using Hieff SYBR Green Master Mix (YEASEN, 11203ES08) on the StepOnePlus real-time PCR machine (Applied Biosystems, 4376592). Primers were designed to amplify ligation junctions corresponding to specific chromatin interactions. The interaction frequency for each target locus was normalized to that of a control primer (GAPDH) set amplifying a nearby, constitutively interacting genomic region. Data are presented as the mean relative interaction frequency from at least three independent biological replicates.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
We obtained the following datasets from the GEO repository for K562 cells: BG4 ChIP-seq (GSE107690), Chem-map (GSE209713), G4access (GSE187007), and G4 CUT&Tag-Hui (GSE181373). Genomic coordinates for PQS34 were obtained from the GEO repository (GSE133379) and used directly in our analysis. Histone ChIP-seq data were obtained from public repositories: U2OS (GEO: GSE139190), Vero (GEO: GSE154761), S2 (GEO: GSE245077, GSE50369, GSE50366), and Ramos (GEO: GSE62063) from the Gene Expression Omnibus (GEO); and CH12F3, K562, HeLa, and HepG2 from the ENCODE database (http://hgdownload.cse.ucsc.edu/goldenPath/mm9/encodeDCC/wgEncodeLicrHistone for CH12F3 cells, and http://hgdownload.cse.ucsc.edu/goldenPath/hg19/encodeDCC/wgEncodeBroadHistone for other three cell lines). G4-CUT&Tag-Li peaks for K562 cells were kindly shared by Prof. Kaiwei Liang at Wuhan University. All the sequencing data generated in this paper, including ssG4-seq, RNA-seq and SP1 HiChIP, have been deposited in the Genome Sequence Archive (GSA) under accession number CRA025444 and the Genome Sequence Archive for Human (GSA-Human) under accession number HRA011386. Source data are provided with this paper.
Code availability
Homemade scripts for all the analysis has been deposited in https://github.com/ZhangJinyueibp/ssG489 at GitHub repository.
References
Bochman, M. L., Paeschke, K. & Zakian, V. A. DNA secondary structures: stability and function of G-quadruplex structures. Nat. Rev. Genet. 13, 770–780 (2012).
Varshney, D., Spiegel, J., Zyner, K., Tannahill, D. & Balasubramanian, S. The regulation and functions of DNA and RNA G-quadruplexes. Nat. Rev. Mol. Cell Biol. 21, 459–474 (2020).
Hsu, S. T. et al. A G-rich sequence within the c-kit oncogene promoter forms a parallel G-quadruplex having asymmetric G-tetrad dynamics. J. Am. Chem. Soc. 131, 13399–13409 (2009).
Esain-Garcia, I. et al. G-quadruplex DNA structure is a positive regulator of MYC transcription. Proc. Natl. Acad. Sci. USA 121, e2320240121 (2024).
Du, Z., Zhao, Y. & Li, N. Genome-wide analysis reveals regulatory role of G4 DNA in gene transcription. Genome Res. 18, 233–241 (2008).
Siddiqui-Jain, A., Grand, C. L., Bearss, D. J. & Hurley, L. H. Direct evidence for a G-quadruplex in a promoter region and its targeting with a small molecule to repress c-MYC transcription. Proc. Natl. Acad. Sci. USA 99, 11593–11598 (2002).
Chen, Y. et al. An upstream G-quadruplex DNA structure can stimulate gene transcription. ACS Chem. Biol. 19, 736–742 (2024).
Foulk, M. S., Urban, J. M., Casella, C. & Gerbi, S. A. Characterizing and controlling intrinsic biases of lambda exonuclease in nascent strand sequencing reveals phasing between nucleosomes and G-quadruplex motifs around a subset of human replication origins. Genome Res. 25, 725–735 (2015).
Besnard, E. et al. Unraveling cell type-specific and reprogrammable human replication origin signatures associated with G-quadruplex consensus motifs. Nat. Struct. Mol. Biol. 19, 837–844 (2012).
Lerner, L. K. & Sale, J. E. Replication of G quadruplex DNA. Genes 10, 95 (2019).
Lemmens, B., van Schendel, R. & Tijsterman, M. Mutagenic consequences of a single G-quadruplex demonstrate mitotic inheritance of DNA replication fork barriers. Nat. Commun. 6, 8909 (2015).
Williams, S. L. et al. Replication-induced DNA secondary structures drive fork uncoupling and breakage. EMBO J. 42, e114334 (2023).
Liu, Z., Lee, A. & Gilbert, W. Gene disruption of a G4-DNA-dependent nuclease in yeast leads to cellular senescence and telomere shortening. Proc. Natl. Acad. Sci. USA 92, 6002–6006 (1995).
Vannier, J. B., Pavicic-Kaltenbrunner, V., Petalcorin, M. I., Ding, H. & Boulton, S. J. RTEL1 dismantles T loops and counteracts telomeric G4-DNA to maintain telomere integrity. Cell 149, 795–806 (2012).
Jansson, L. I. et al. Telomere DNA G-quadruplex folding within actively extending human telomerase. Proc. Natl. Acad. Sci. USA 116, 9350–9359 (2019).
Wang, G. & Vasquez, K. M. Dynamic alternative DNA structures in biology and disease. Nat. Rev. Genet. 24, 211–234 (2023).
Hansel-Hertsch, R. et al. Landscape of G-quadruplex DNA structural regions in breast cancer. Nat. Genet. 52, 878–883 (2020).
Li, P. et al. Targeting G-quadruplex by TMPyP4 for inhibition of colorectal cancer through cell cycle arrest and boosting anti-tumor immunity. Cell Death Dis. 15, 816 (2024).
Yuan, L. et al. Existence of G-quadruplex structures in promoter region of oncogenes confirmed by G-quadruplex DNA cross-linking strategy. Sci. Rep. 3, 1811 (2013).
Simone, R., Fratta, P., Neidle, S., Parkinson, G. N. & Isaacs, A. M. G-quadruplexes: emerging roles in neurodegenerative diseases and the non-coding transcriptome. FEBS Lett. 589, 1653–1668 (2015).
Lago, S. et al. Promoter G-quadruplexes and transcription factors cooperate to shape the cell type-specific transcriptome. Nat. Commun. 12, 3885 (2021).
Spiegel, J. et al. G-quadruplexes are transcription factor binding hubs in human chromatin. Genome Biol. 22, 117 (2021).
Li, L. et al. YY1 interacts with guanine quadruplexes to regulate DNA looping and gene expression. Nat. Chem. Biol. 17, 161–168 (2021).
Yangyuoru, P. M., Bradburn, D. A., Liu, Z., Xiao, T. S. & Russell, R. The G-quadruplex (G4) resolvase DHX36 efficiently and specifically disrupts DNA G4s via a translocation-based helicase mechanism. J. Biol. Chem. 293, 1924–1932 (2018).
Gomez-Del Arco, P. et al. The G4 resolvase Dhx36 modulates cardiomyocyte differentiation and ventricular conduction system development. Nat. Commun. 15, 8602 (2024).
Huber, M. D., Lee, D. C. & Maizels, N. G4 DNA unwinding by BLM and Sgs1p: substrate specificity and substrate-specific inhibition. Nucleic Acids Res. 30, 3954–3961 (2002).
Huber, M. D., Duquette, M. L., Shiels, J. C. & Maizels, N. A conserved G4 DNA binding domain in RecQ family helicases. J. Mol. Biol. 358, 1071–1080 (2006).
Tang, W. et al. The Werner syndrome RECQ helicase targets G4 DNA in human cells to modulate transcription. Hum. Mol. Genet. 25, 2060–2069 (2016).
Ketkar, A., Voehler, M., Mukiza, T. & Eoff, R. L. Residues in the RecQ C-terminal domain of the human werner syndrome helicase are involved in unwinding G-quadruplex DNA. J. Biol. Chem. 292, 3154–3163 (2017).
Schaffitzel, C. et al. In vitro generated antibodies specific for telomeric guanine-quadruplex DNA react with Stylonychia lemnae macronuclei. Proc. Natl. Acad. Sci. USA 98, 8572–8577 (2001).
Henderson, A. et al. Detection of G-quadruplex DNA in mammalian cells. Nucleic Acids Res. 42, 860–869 (2014).
Liu, H. Y. et al. Conformation selective antibody enables genome profiling and leads to discovery of parallel G-quadruplex in human telomeres. Cell Chem. Biol. 23, 1261–1270 (2016).
Biffi, G., Tannahill, D., McCafferty, J. & Balasubramanian, S. Quantitative visualization of DNA G-quadruplex structures in human cells. Nat. Chem. 5, 182–186 (2013).
Zheng, K. W. et al. Detection of genomic G-quadruplexes in living cells using a small artificial protein. Nucleic Acids Res. 48, 11706–11720 (2020).
Hänsel-Hertsch, R., Spiegel, J., Marsico, G., Tannahill, D. & Balasubramanian, S. Genome-wide mapping of endogenous G-quadruplex DNA structures by chromatin immunoprecipitation and high-throughput sequencing. Nat. Protoc. 13, 551–564 (2018).
Li, C. et al. Ligand-induced native G-quadruplex stabilization impairs transcription initiation. Genome Res. 31, 1546–1560 (2021).
Hui, W. W. I., Simeone, A., Zyner, K. G., Tannahill, D. & Balasubramanian, S. Single-cell mapping of DNA G-quadruplex structures in human cancer cells. Sci. Rep. 11, 23641 (2021).
Yu, Z. et al. Chem-map profiles drug binding to chromatin in cells. Nat. Biotechnol. 41, 1265–1271 (2023).
Esnault, C. et al. G4access identifies G-quadruplexes and their associations with open chromatin and imprinting control regions. Nat. Genet. 55, 1359–1369 (2023).
Xu, W. et al. The R-loop is a common chromatin feature of the Arabidopsis genome. Nat. Plants 3, 704–714 (2017).
Zhang, Y. et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137 (2008).
Hänsel-Hertsch, R. et al. G-quadruplex structures mark human regulatory chromatin. Nat. Genet. 48, 1267–1272 (2016).
Robinson, J. T. et al. Integrative genomics viewer. Nat. Biotechnol. 29, 24–26 (2011).
Landt, S. G. et al. ChIP-seq guidelines and practices of the ENCODE and modENCODE consortia. Genome Res. 22, 1813–1831 (2012).
Sen, D. & Gilbert, W. Formation of parallel four-stranded complexes by guanine-rich motifs in DNA and its implications for meiosis. Nature 334, 364–366 (1988).
Machanick, P. & Bailey, T. L. MEME-ChIP: motif analysis of large DNA datasets. Bioinformatics 27, 1696–1697 (2011).
McHaourab, Z. F., Perreault, A. A. & Venters, B. J. ChIP-seq and ChIP-exo profiling of Pol II, H2A.Z, and H3K4me3 in human K562 cells. Sci. Data 5, 180030 (2018).
Consortium, E. P. An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74 (2012).
Gerstein, M. B. et al. Architecture of the human regulatory network derived from ENCODE data. Nature 489, 91–100 (2012).
Raiber, E. A., Kranaster, R., Lam, E., Nikan, M. & Balasubramanian, S. A non-canonical DNA structure is a binding motif for the transcription factor SP1 in vitro. Nucleic Acids Res. 40, 1499–1508 (2012).
Cawley, S. et al. Unbiased mapping of transcription factor binding sites along human chromosomes 21 and 22 points to widespread regulation of noncoding RNAs. Cell 116, 499–509 (2004).
Song, J. et al. Two consecutive zinc fingers in Sp1 and in MAZ are essential for interactions with cis-elements. J. Biol. Chem. 276, 30429–30434 (2001).
Hasegawa, Y. & Struhl, K. Different SP1 binding dynamics at individual genomic loci in human cells. Proc. Natl. Acad. Sci. USA 118, e2113579118 (2021).
Maizels, N. Dynamic roles for G4 DNA in the biology of eukaryotic cells. Nat. Struct. Mol. Biol. 13, 1055–1059 (2006).
Mumbach, M. R. et al. Enhancer connectome in primary human cells identifies target genes of disease-associated DNA elements. Nat. Genet. 49, 1602–1612 (2017).
Weintraub, A. S. et al. YY1 is a structural regulator of enhancer-promoter loops. Cell 171, 1573–1588.e28 (2017).
Yusufzai, T. M., Tagami, H., Nakatani, Y. & Felsenfeld, G. CTCF tethers an insulator to subnuclear sites, suggesting shared insulator mechanisms across species. Mol. Cell 13, 291–298 (2004).
Pascal, E. & Tjian, R. Different activation domains of Sp1 govern formation of multimers and mediate transcriptional synergism. Genes Dev. 5, 1646–1656 (1991).
Mastrangelo, I. A., Courey, A. J., Wall, J. S., Jackson, S. P. & Hough, P. V. DNA looping and Sp1 multimer links: a mechanism for transcriptional synergism and enhancement. Proc. Natl. Acad. Sci. USA 88, 5670–5674 (1991).
Courey, A. J., Holtzman, D. A., Jackson, S. P. & Tjian, R. Synergistic activation by the glutamine-rich domains of human transcription factor Sp1. Cell 59, 827–836 (1989).
Zhang, J. et al. The International Cancer Genome Consortium Data Portal. Nat. Biotechnol. 37, 367–369 (2019).
Huber, C. D., Kim, B. Y. & Lohmueller, K. E. Population genetic models of GERP scores suggest pervasive turnover of constrained sites across mammalian evolution. PLoS Genet. 16, e1008827 (2020).
Jolma, A. et al. Multiplexed massively parallel SELEX for characterization of human transcription factor binding specificities. Genome Res. 20, 861–873 (2010).
Consortium, G. T. et al. Genetic effects on gene expression across human tissues. Nature 550, 204–213 (2017).
Albert, F. W. & Kruglyak, L. The role of regulatory variation in complex traits and disease. Nat. Rev. Genet. 16, 197–212 (2015).
Hagège, H. et al. Quantitative analysis of chromosome conformation capture assays (3C-qPCR). Nat. Protoc. 2, 1722–1733 (2007).
Lyu, J., Shao, R., Kwong Yung, P. Y. & Elsasser, S. J. Genome-wide mapping of G-quadruplex structures with CUT&Tag. Nucleic Acids Res. 50, e13 (2022).
Li, L., He, S., Sun, J. M. & Davie, J. R. Gene regulation by Sp1 and Sp3. Biochem. Cell Biol. 82, 460–471 (2004).
Mumbach, M. R. et al. HiChIP: efficient and sensitive analysis of protein-directed genome architecture. Nat. Methods 13, 919–922 (2016).
Servant, N. et al. HiC-Pro: an optimized and flexible pipeline for Hi-C data processing. Genome Biol. 16, 259 (2015).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–U54 (2012).
Bhattacharyya, S., Chandra, V., Vijayanand, P. & Ay, F. Identification of significant chromatin contacts from HiChIP data by FitHiChIP. Nat. Commun. 10, 4221 (2019).
Chen, J. et al. The RNA-binding protein ROD1/PTBP3 cotranscriptionally defines AID-loading sites to mediate antibody class switch in mammalian genomes. Cell Res. 28, 981–995 (2018).
Naito, Y., Hino, K., Bono, H. & Ui-Tei, K. CRISPRdirect: software for designing CRISPR/Cas guide RNA with reduced off-target sites. Bioinformatics 31, 1120–1123 (2015).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Li, H. et al. The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Kent, W. J., Zweig, A. S., Barber, G., Hinrichs, A. S. & Karolchik, D. BigWig and BigBed: enabling browsing of large distributed datasets. Bioinformatics 26, 2204–2207 (2010).
Feng, J. X., Liu, T., Qin, B., Zhang, Y. & Liu, X. S. Identifying ChIP-seq enrichment using MACS. Nat. Protoc. 7, 1728–1740 (2012).
Bailey, T. L. et al. MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res. 37, W202–W208 (2009).
Cai, Z. K. et al. RIC-seq for global in situ profiling of RNA-RNA spatial interactions. Nature 582, 432–437 (2020).
Whyte, W. A. et al. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell 153, 307–319 (2013).
Zhao, H. et al. CrossMap: a versatile tool for coordinate conversion between genome assemblies. Bioinformatics 30, 1006–1007 (2014).
Hinrichs, A. S. et al. The UCSC Genome Browser Database: update 2006. Nucleic Acids Res. 34, D590–D598 (2006).
Krzywinski, M. et al. Circos: an information aesthetic for comparative genomics. Genome Res. 19, 1639–1645 (2009).
Castro-Mondragon, J. A. et al. JASPAR 2022: the 9th release of the open-access database of transcription factor binding profiles. Nucleic Acids Res. 50, D165–D173 (2022).
Pertea, M., Kim, D., Pertea, G. M., Leek, J. T. & Salzberg, S. L. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 11, 1650–1667 (2016).
Rebouissou, C., Sallis, S. & Forne, T. Quantitative chromosome conformation capture (3C-qPCR). Methods Mol. Biol. 2532, 3–13 (2022).
Li, S. et al. ssG4-seq for global profiling of strand-specific G-quadruplex structures in mammalian genomes. ssG4. https://doi.org/10.5281/zenodo.17519174 (2025).
Acknowledgements
This work was supported by the National Key Research and Development Program of China (2022YFA1303300), the National Natural Science Foundation of China (32025008 and 32130064), and the Beijing Municipal Natural Science Foundation (Z220011) to Y.X.
Author information
Authors and Affiliations
Contributions
Y.X. conceived and supervised the project. S.L. developed the ssG4-seq method with the help from R.W. S.L. constructed HiChIP libraries, performed ELISA assay, purified SP1 proteins, and constructed shRNA plasmids against SP1. J.C. performed G4 staining and SIM analysis. R.W. created knock-in mutations and performed relevant analysis. Z.C. performed 3C-qPCR analysis with the help from R.Y. W.L., C.L., and R.L. critically revised the manuscript for scientific accuracy. J.Z. conducted the bioinformatics analysis with the help of C.C. and J.B. Y.X. wrote the manuscript with help from S.L., R.W., and J.Z.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Cyril Esnault and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Li, S., Wang, R., Zhang, J. et al. ssG4-seq for global profiling of strand-specific G-quadruplex structures in mammalian genomes. Nat Commun 17, 195 (2026). https://doi.org/10.1038/s41467-025-66895-2
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-66895-2
