
Abstract
In the realm of molecular construction, the skeletal editing techniques of heterocyclic compounds demonstrate unique efficiency, particularly in synthesizing molecular structures that are challenging to obtain through traditional synthetic methods. Compared to the ring-contraction reaction of saturated nitrogen heterocycles and aryl rings, the site selectivity and stereoselective skeletal editing of pyrimidine fused heterocycles remain relatively underdeveloped. Here we report a chiral hypervalent iodine(III)-catalyzed skeletal editing of pyrimidine moieties within polynitrogen heterocycles, which efficiently produces optically pure multi-substituted imidazoline rings. The reaction demonstrates exceptional functional group tolerance, as shown by the ring contraction of diverse polynitrogen heterocycles and the late-stage functionalization of M1G-dR and its analogues, including nucleosides, nucleotides, and oligonucleotides. Density functional theory calculations explore the details of the mechanism and the factors that determine the reaction’s stereoselectivity.
Similar content being viewed by others
Introduction
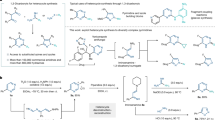
Fused nitrogen heterocyclic compounds have received increasing attention in recent decades due to their significant value in the field of medicine1,2. The physicochemical properties of these compounds are significantly influenced by the type and size of the ring structure, as well as the substituent groups on the core skeleton3. Particularly, quinazolinones and purines, as key structural motifs in many natural products and bioactive compounds4,5, often exhibit novel physicochemical or biological properties when added with an additional ring6. Studies have shown that 2D tricyclic derivatives exhibit enhanced fluorescence properties compared to their 2D bicyclic parent compounds, but with reduced bioactivity (Fig. 1a)7,8,9. In contrast, their 3D tricyclic derivatives exhibit superior pharmacokinetic properties and pharmacological activity, in line with the “escape from flatland” strategy (Fig. 1a)10,11,12,13. Furthermore, structure-activity relationship studies have confirmed that optically pure compounds possess significantly enhanced bioactivity compared to their racemic counterparts14,15. Consequently, the synthesis of chiral semisaturated aza-heterocycles has remained a hot topic in organic synthesis. Traditional synthetic methods rely on cyclization and annulation reactions10,16,17, which often involve multi-step reaction sequences, pre-functionalized starting materials, and/or harsh reaction conditions. Direct dearomatization of heteroaromatics is an efficient strategy for constructing semisaturated aza-heterocycles, but the reported methods primarily involve electron-rich indoles18. Methods for directly converting electron-deficient aza-fused heterocycles into chiral semisaturated aza-heterocycles are rarely reported19,20.

a Advantages of semi-saturated tricyclic nucleosides in pharmacokinetic properties or biological properties. b Ring contraction reactions: from 3D aliphatic six-membered ring to 3D aliphatic five-membered ring; from 2D aromatic six-membered ring to 2D aromatic five-membered ring. c Design and challenges of stereoselective ring contraction from a 2D six-membered ring to a 3D five-membered ring. d This work: stereoselective skeletal editing of fused polyazapolycycles.
The skeletal editing strategy is designed to generate reactive intermediates by disrupting the inherent molecular framework, followed by reconfiguration, thereby facilitating swift and facile alterations to the core structure21,22,23,24,25,26. This approach serves as a potent instrument for the rapid and efficient development of drug candidates within the field of medicinal chemistry27,28,29,30,31,32,33,34. Among these, the transformation of a three-dimensional (3D) aliphatic six-membered ring into a 3D aliphatic five-membered ring is particularly well-advanced (Fig. 1b)35,36,37,38. Notably, the photoinduced ring-rearrangement of saturated heterocycles, as recently reported by Sarpong’s group, has marked a significant advancement in this area39,40. The strategies for skeletal editing of two-dimensional (2D) aromatic six-membered rings to 2D aromatic five-membered rings have also been rapidly evolving (Fig. 1b)21,35. Harran41,42, Zheng43, and Fu44 have reported innovative methods for the transformation of pyridine structures into pyrrole and pyrazole frameworks, respectively. Levin’s group also introduced a novel approach to prepare indole compounds by removing carbon from quinoline skeleton through photochemical reaction45,46. Furthermore, McNally47, Sarpong48 and Fu49 have independently reported effective strategies for the synthesis of pyrazole and oxazole scaffolds through the deconstruction-reconstruction of pyrimidine skeletons. Despite these studies’ significant contributions to scientific progress in the field, asymmetric skeletal editing methods for aza-heterocycles are limited, with only You50 and Bi’s51 groups reporting asymmetric annulation reactions of indoles. Selective editing of pyrimidine-fused aza-heterocycles, particularly the stereoselective ring contraction rearrangement of 2D aza-heterocycles to chiral 3D aza-heterocycles, remains an underexplored area.
Chiral hypervalent iodine reagents are pivotal tools in organic synthesis, increasingly playing a significant role in the field of asymmetric synthesis52. Recent research on chiral hypervalent iodine reagents has made remarkable progress, particularly in applications such as asymmetric dearomatization, α-functionalization of carbonyls, difunctionalization of alkenes, alkene oxidation rearrangements, and oxidative coupling reactions52,53. However, the electron-deficient nature, high polarity, poor solubility of pyrimidine-fused heterocycles, and the presence of multiple reactive sites54,55,56 pose significant challenges to the high chemo-, regio-, and enantioselective ring contraction rearrangement of pyrimidine catalyzed by chiral hypervalent iodine. Building on our previous work with hypervalent iodine mediated dialkoxylation of electron-rich indole compounds57, we hypothesized that the introduction of a methoxy group into the electron-deficient pyrimidine ring, followed by rearrangement with hypervalent iodine, could be a viable approach (Fig. 1c). The in situ introduction of the methoxy group not only alters the electronic properties of the compound but also enhances its solubility. Moreover, the methoxy group is an important pharmacophore group that plays a significant role in ligand-target interactions, the physicochemical properties of drugs, and ADME (absorption, distribution, metabolism, and excretion) characteristics58. We herein report a chiral hypervalent iodine(III)-catalyzed ring contraction rearrangement of pyrimidine moieties to form multi-substituted imidazolidine rings, successfully achieving a highly chemo-, regio- and stereo-selective transformation from 2D nitrogen heterocycles to fused 2D/3D nitrogen heterocycles (Fig. 1d). Compared with existing methods for imidazoline synthesis59,60,61, this approach exhibits superior atom economy, step economy, and substrate versatility.
Results and discussion
Reaction design and optimization
To initiate this study, tricyclic quinazolone 1a was selected as a model substrate for investigating the asymmetric ring-contraction reaction catalyzed by chiral hypervalent iodine. The utilization of chiral iodoarene 3a as a precatalyst and MeOH as both reagent and solvent did not lead desired result (Fig. 2, entry 1). Consequently, alternative chiral iodoarene precursors with distinct backbones were explored. Encouragingly, when chiral iodoarene 4a was employed, the desired compound 2a was obtained with excellent stereoselectivity, albeit a poor enantioselectivity, yielding 66% (Fig. 2, entry 3). To enhance the reaction outcome, the effect of acid additives was investigated (Fig. 2, entries 5-8). Employing TfOH resulted in an increased yield of compound 2a, reaching 84%. Notably, replacing MeOH with DCM as the solvent further enhanced the enantioselectivity to 56% ee (Fig. 2, entry 9). Subsequently, additional investigations focused on exploring the effectiveness of different chiral iodoarenes. Using the (S)-proline-derived chiral organoiodine 5c, the desired compound 2a was obtained with excellent stereoselectivity, exhibiting a yield of 89% and an impressive enantioselectivity of 97% ee (Fig. 2, entry 13). Regrettably, the expansion to other nucleophiles did not yield the expected results. The addition of benzylamine triggered the decomposition of starting material 1a, while nucleophiles such as acetic acid, thiophenol, and methyl acetoacetate failed to induce rearrangement, with most of 1a being recovered (see ESI, Table S5).

Reaction conditions: 1 (0.1 mmol, 1.0 equiv), MeOH (0.4 mL, 99.0 equiv), chiral ArI (20 mol%), selectfluor (2.0 equiv), acid (3.0 equiv), solvent (4.0 mL) at 30 °C of x h. Yield of isolated product. The enantiomeric excess (ee) values were determined by HPLC. The diastereomeric ratio (d.r.) was determined by 1H NMR spectroscopy of the crude reaction mixture.
Substrate scope of pyrimidine fused heterocycles and alcohols
With the optimized reaction conditions established, the substrate scopes and limitations of this protocol were explored (Fig. 3). The quinazolinone bearing different groups on the phenyl ring afforded the polysubstituted fused-ring guanidines (2a-2l) with moderate to good yields (35-89%), excellent enantioselectivities (91-99% ee) and diastereoselectivities (>20:1 d.r.). 2a was synthesised in 73% yield and 98% ee in a 1 mmol scale reaction. The absolute configuration of products 2a and 2 f was unambiguously confirmed by single-crystal X-ray diffraction. The polyfluoro-substituted and fused tetracyclic substrates also reacted smoothly, and the ee values of the products 2j-2l were all above 90%. Subsequently, using primary alcohols (MeOH-d4, ethanol and cyclopropylmethanol) as nucleophiles, the desired products 6a-6c were obtained in 52-80% yields with up to 97% ee. In addition, the versatility of this synthetic approach for nitrogen-containing bicyclic substrates was investigated. Under standard reaction conditions, two pyrimidine-fused pyrimidines 7a and 7b were successfully converted to pyrimidine-fused imidazoline products 8a and 8b. The methodology was extended to the synthesis of 5,5-fused bicyclic systems, in particular using triazole-fused pyrimidine 7 d to produce triazole-fused imidazoline 8 d in moderate yields and excellent enantioselectivity (93% ee).

Reaction conditions: 1 or 7 (0.1 mmol, 1.0 equiv), 5c (20 mol%), selectfluor (2.0 equiv), TfOH (3.0 equiv), co-solvent DCM:ROH (v/v 10:1, 4.4 mL) at 30 °C. Yield of isolated product. The ee values were determined by HPLC. The diastereomeric ratio (d.r.) was determined by 1H NMR spectroscopy of the crude reaction mixture. aDCM:MeOH (v/v 3:2, 4.4 mL) was used as solvent. bDCM:ROH (v/v 1:1, 4.4 mL) was used as solvent.
Application in late-stage functionalization
Encouraged by the aforementioned experimental results, we intend to apply the developed method to the late-stage functionalization of endogenous nucleoside M1G-dR and its derivatives (Fig. 4). Initially, alkyl-substituted tricyclic nucleosides (ethyl, n-butyl and benzyl) were used as substrates, giving the target products 10a-10c in 63-65% yields and 93-95% ee values. Acyclovir, an antiviral drug against herpes simplex virus, and its series of amino acid and peptide-linked derivatives react with TMOP to form M1G-like acyclic nucleosides62. These substrates also underwent selective skeletal editing of the pyrimidine ring under standard conditions, yielding the annulated products 10d-10i with good yields, high diastereoselectivity and excellent enantioselectivity (>90% ee). The method developed can be directly applied to the late-stage functionalization of unprotected M1G-dR 9j and M1G-R 9 l, albeit with less than ideal yields. We have tried changing different chiral columns and mobile phases, as well as derivatising the product, but failed to separate the diastereoisomers by HPLC. Under the standard reaction conditions, the Obz-protected derivatives of M1G-dR 9k and M1G-R 9 m exhibited significantly enhanced reactivity, yielding the desired products 10k and 10 m with yields of 28% and 67%, respectively. The tricyclic guanosine derivative with a 5’-position azide substitution also reacted to form the corresponding target product 10n, with the azide group serving as a reactive site for further structural modification. In nature, nucleosides require activation by kinases to form nucleotides, and modified nucleosides are challenging to phosphorylate in vivo, thus making the late-stage functionalization of nucleotides highly significant. Using the tricyclic nucleotides 9o and 9p as substrates, the target products 10o and 10p were synthesized under standard conditions. Notably, when the substrates were M1G-containing oligonucleotides, the desired products 10q and 10r were obtained with yields of 18% and 57%, respectively. Compared with their acyclic counterparts, nucleoside, nucleotide, and oligonucleotide substrates performed poorly under identical conditions. We attribute this outcome to the lability of the glycosidic bond, which leads to partial decomposition of the starting material. Attempts to optimize the reaction parameters provided no improvement.

Reaction conditions: 9 (0.1 mmol, 1.0 equiv), 5c (20 mol%), selectfluor (2.0 equiv), TfOH (3.0 equiv), co-solvent DCM:MeOH (v/v 1:1, 4.4 mL) at 30 °C. Yield of isolated product. The ee values were determined by HPLC. The diastereomeric ratio (d.r.) was determined by 1H NMR spectroscopy of the crude reaction mixture. aKOH (3.0 equiv) was used as an additive. bThe ee values were determined by HPLC from the hydrolysed product. See Supplementary Information for details.
To showcase the synthetic utility of the current methodology, the recycling of chiral catalysts and derivatization reactions of the tricyclic nucleosides were performed (Fig. 5). The chiral catalyst re-5c was reused and recycled three times with minimal change in the yield and enantiomeric excess of the target product 2a (Fig. 5a). Compounds 10k and 10 m were deprotected to give 10j and 10 l in excellent yields. The azide-substituted product 10n underwent a click reaction with 6-bromo-9-(prop-2-yn-1-yl)−9H-purine to afford the triazole-linked oligonucleotide derivative 11 in a 92% yield (Fig. 5b).

a The catalyst recovery and recycling experiments. The scale-up synthesis. b The derivatization reaction of tricyclic nucleosides.
Mechanistic considerations
Subsequently, we conducted a preliminary mechanistic investigation of the rearrangement reaction (Fig. 6). Under the standard conditions, pyrido[1,2-a]quinoxaline 12 failed to react, underscoring the pivotal role of the N-1 atom in 1a in initiating the reaction (Fig. 6a, eq 1). When m-CPBA or PhI(OAc)₂ were employed as oxidants in place of Selectfluor, m-CPBA afforded the product 2a in 46% yield with 98% ee, whereas PhI(OAc)₂ delivered the product 2a in excellent yield but as a racemate (91% yield, 0% ee). These results suggest that Selectfluor first oxidizes the chiral aryl iodide 5c to a hypervalent iodine species, and this newly formed catalyst then drives the rearrangement reaction (Fig. 6a, eq 2 and 3). When PhI(OMe)₂ was employed directly as the catalyst, rac-2a could be obtained in 55% yield, indicating that the in-situ-generated Ar*I(OMe)₂ is the active catalytic species (Fig. 6a, eq 4).

a Control experiments. b Calculated potential energy profile for Ar*I(OMe)2-catalyzed stereoselective skeletal editing of 1a. See Supplementary Data 1 for the cartesian coordinates of the optimized structures. c Calculated geometries of transition structures and intermediates.
Guided by literature precedents63,64 and the above experimental findings, we elucidated the mechanism by means of DFT calculations (Fig. 6b). First, substrate 1a adds to the in-situ-generated chiral hypervalent iodine (Ar*I(OMe)2) through TS1 to give Int1. The transition state TS1 is 9.2 kcal/mol lower in energy than the TS1’, indicating that the S-configured Int1 is favored (Fig. 6c). Ligand exchange with TfOH then converts Int1 into Int2. In Int2, the methoxy group attached to the hydrogenated pyrimidine moiety significantly stabilizes the iodine(III) cation, with an energy 5.5 kcal/mol lower than that of Int2’. A syn-addition (TS2) of the MeO- and the hypervalent iodine (Ar*IOMe) across Int2 furnishes Int3, consistent with Ariafard’s observation of facile syn-additions of phenols with MeOH and hypervalent iodine reagents65. The low barrier of 5.0 kcal/mol for this step dictates the high chemo-, regio-, and stereoselectivity observed. Int3 subsequently releases MeO- to afford Int4. The hydropyrimidine unit in Int4 has three cis-oriented substituents that cause severe steric hindrance, resulting in a highly distorted conformation. Upon rearrangement (TS3), this motif transforms into an imidazoline ring. C-2 migrates to an exocyclic position, resulting in the loss of stereochemistry. Meanwhile, C-3 undergoes an intramolecular nucleophilic substitution that inverts its configuration, delivering the trans-configured Int5 and releasing Ar*I (4c). Finally, Int5 reacts with MeO- to deliver the product 2a.
In conclusion, we have developed a highly chem-regio- and stereoselectivity method for the construction of polysubstituted 3D fused-ring guanidines via a chiral hypervalent iodine(III)-catalyzed ring-contraction rearrangement reaction of pyrimidine rings within polynitrogen heterocycles. The application of this method to the late-stage skeletal modification of endogenous nucleoside M1G-dR and its analogues involving serial polyatom changes is demonstrated. To demonstrate its versatility, the scaled-up synthesis, recycling of chiral catalysts and derivatization reactions of the tricyclic nucleoside derivatives were successfully carried out.
Methods
General procedure for the synthesis of chiral products
General procedure A
Nitrogen-containing heterocycle (0.1 mmol, 1.0 equiv), 5c (0.02 mmol, 20 mol%), selectfluor (0.2 mmol, 2.0 equiv) and DCM:ROH (v/v 10:1, 4.4 mL) were added to a screw-capped vial with a magnetic stirrer. TfOH (0.3 mmol, 3.0 equiv) was then rapidly added to the reaction mixture. The vial was tightly sealed and stirred at 30 °C for 3-24 h. The reaction was diluted with DCM and washed with a saturated aqueous solution of NaHCO3. Extract the aqueous phase with DCM. The combined organic phases were dried over anhydrous Na2SO4 and concentrated under reduced pressure. The residue was subjected to silica gel chromatography to give target compounds. The enantiomeric excess was determined by HPLC with a chiral column.
General procedure B
KOH (0.3 mmol, 3.0 equiv), 5c (0.02 mmol, 20 mol%), selectfluor (0.2 mmol, 2.0 equiv) and DCM:MeOH (v/v 1:1, 4.4 mL) were added to a screw-capped vial with a magnetic stirrer. TfOH (0.3 mmol, 3.0 equiv) was then rapidly added to the reaction mixture followed by tricyclic nucleoside (0.1 mmol, 1.0 equiv). The vial was tightly sealed and stirred at 30 °C for 12 h. The reaction was diluted with DCM and washed with a saturated aqueous solution of NaHCO3. Extract the aqueous phase with DCM. The combined organic phases were dried over anhydrous Na2SO4 and concentrated under reduced pressure. The residue was subjected to silica gel chromatography to give target compounds. The enantiomeric excess was determined by HPLC with a chiral column.
General procedure C
Tricyclic nucleoside (0.1 mmol, 1.0 equiv), 5c (0.02 mmol, 20 mol%), selectfluor (0.2 mmol, 2.0 equiv) and DCM:MeOH (v/v 1:1, 4.4 mL) were added to a screw-capped vial with a magnetic stirrer. TfOH (0.3 mmol, 3.0 equiv) was then rapidly added to the reaction mixture. The vial was tightly sealed and stirred at 30 °C for 12 h. The reaction was diluted with DCM and washed with a saturated aqueous solution of NaHCO3. Extract the aqueous phase with DCM. The combined organic phases were dried over anhydrous Na2SO4 and concentrated under reduced pressure. The residue was subjected to silica gel chromatography to give target compounds. The enantiomeric excess was determined by HPLC with a chiral column.
Data availability
Crystallographic data for compounds 2a (CCDC 2335284), 2 f (CCDC 2335049), and 9q (CCDC 2347563) are available free of charge from the Cambridge Crystallographic Date Centre. Additional optimization, experimental procedures, characterization of new compounds, and all other data supporting the findings are available in the Supplementary Information and Supplementary Data 1. Data supporting the findings of this manuscript are also available from the corresponding author upon request.
References
Kumar, A. et al. Nitrogen containing heterocycles as anticancer agents: a medicinal chemistry perspective. Pharmaceuticals 16, 299 (2023).
Marshall, C. M., Federice, J. G., Bell, C. N., Cox, P. B. & Njardarson, J. T. An update on the nitrogen heterocycle compositions and properties of U.S. FDA-approved pharmaceuticals (2013–2023). J. Med. Chem. 67, 11622–11655 (2024).
Vitaku, E., Smith, D. T. & Njardarson, J. T. Analysis of the structural diversity, substitution patterns, and frequency of nitrogen heterocycles among U.S. FDA approved pharmaceuticals. J. Med. Chem. 57, 10257–10274 (2014).
Li, H., Fu, G. & Zhong, W. Natural quinazolinones: from a treasure house to promising anticancer leads. Eur. J. Med. Chem. 245, 114915 (2023).
Motter, J. et al. Purine nucleoside antibiotics: recent synthetic advances harnessing chemistry and biology. Nat. Prod. Rep. 41, 873–884 (2024).
Lish, M. S. et al. Pharmacophore establishment and optimization of saturated 1,6-naphthyridine-fused quinazolinones that inhibit meningoencephalitis-causing naegleria fowleri. J. Med. Chem. 67, 18265–18289 (2024).
Jahnz-Wechmann, Z., Framski, G., Januszczyk, P. & Boryski, J. Bioactive fused heterocycles: Nucleoside analogs with an additional ring. Eur. J. Med. Chem. 97, 388–396 (2015).
Goslinski, T., Golankiewicz, B., De Clercq, E. & Balzarini, J. Synthesis and biological activity of strongly fluorescent tricyclic analogues of acyclovir and ganciclovir. J. Med. Chem. 45, 5052–5057 (2002).
Golankiewicz, B. et al. Fluorescent tricyclic analogues of acyclovir and ganciclovir. A structure− antiviral activity study. J. Med. Chem. 44, 4284–4287 (2001).
Luise, N. & Wyatt, P. G. Generation of polar semi-saturated bicyclic pyrazoles for fragment-based drug-discovery campaigns. Chem. Eur. J. 24, 10443–10451 (2018).
Lovering, F., Bikker, J. & Humblet, C. Escape from flatland: increasing saturation as an approach to improving clinical success. J. Med. Chem. 52, 6752–6756 (2009).
Wu, W.-L. et al. Discovery of novel tricyclic heterocycles as potent and selective DPP-4 inhibitors for the treatment of type 2 diabetes. ACS Med. Chem. Lett. 7, 498–501 (2016).
Karloff, D. B. et al. Glyoxal caging of nucleoside antivirals toward self-activating, extended-release prodrugs. J. Am. Chem. Soc. 146, 29402–29406 (2024).
Seri-Levy, A., West, S. & Richards, W. G. Molecular similarity, quantitative chirality, and QSAR for chiral drugs. J. Med. Chem. 37, 1727–1732 (1994).
Kahlon, D. K., Lansdell, T. A., Fisk, J. S. & Tepe, J. J. Structural-activity relationship study of highly-functionalized imidazolines as potent inhibitors of nuclear transcription factor-κB mediated IL-6 production. Bioorg. Med. Chem. 17, 3093–3103 (2009).
Long, A., Oswood, C. J., Kelly, C. B., Bryan, M. C. & MacMillan, D. W. C. Couple-close construction of polycyclic rings from diradicals. Nature 628, 326–332 (2024).
Twigg, D. G. et al. Partially saturated bicyclic heteroaromatics as an sp3 -enriched fragment collection. Angew. Chem. Int. Ed. 55, 12479–12483 (2016).
Xia, Z. L., Xu-Xu, Q. F., Zheng, C. & You, S. L. Chiral phosphoric acid-catalyzed asymmetric dearomatization reactions. Chem. Soc. Rev. 49, 286–300 (2020).
Escolano, M. et al. Recent strategies in the nucleophilic dearomatization of pyridines, quinolines, and isoquinolines. Chem. Rev. 124, 1122–1246 (2024).
Anjirwala, S. N. & Patel, S. K. Efficient synthetic strategies for fused pyrimidine and pyridine derivatives: a review. J. Heterocycl. Chem. 61, 1481–1516 (2024).
Jurczyk, J. et al. Single-atom logic for heterocycle editing. Nat. Synth. 1, 352–364 (2022).
Li, J., Tang, P., Fan, Y. & Lu, H. Skeletal editing of pyrrolidines by nitrogen-atom insertion. Science 389, 275–281 (2025).
Wu, F.-P., Tyler, J. L. & Glorius, F. Diversity-generating skeletal editing transformations. Acc. Chem. Res. 58, 893–906 (2025).
Wang, Z., Xu, P., Guo, S.-M., Daniliuc, C. G. & Studer, A. C-to-N atom swapping and skeletal editing in indoles and benzofurans. Nature 642, 92–98 (2025).
Puriņš, M., Nakahara, H. & Levin, M. D. Bridging the pyridine-pyridazine synthesis gap by skeletal editing. Science 389, 295–298 (2025).
Liu, L.-J. et al. Indole-quinoline transmutation enabled by a formal rhodium carbynoid. Angew. Chem. Int. Ed. 64, e202501966 (2025).
Zhang, P., Hua, L., Takahashi, T., Jin, S. & Wang, Q. Recent advances in the dearomative skeletal editing of mono-azaarenes. Synthesis 56, 55–70 (2023).
Roque, J. B., Kuroda, Y., Göttemann, L. T. & Sarpong, R. Deconstructive diversification of cyclic amines. Nature 564, 244–248 (2018).
Reisenbauer, J. C., Green, O., Franchino, A., Finkelstein, P. & Morandi, B. Late-stage diversification of indole skeletons through nitrogen atom insertion. Science 377, 1104–1109 (2022).
Kennedy, S. H., Dherange, B. D., Berger, K. J. & Levin, M. D. Skeletal editing through direct nitrogen deletion of secondary amines. Nature 593, 223–227 (2021).
Conboy, A. & Greaney, M. F. Synthesis of benzenes from pyridines via N to C switch. Chem 10, 1940–1949 (2024).
Woo, J., Stein, C., Christian, A. H. & Levin, M. D. Carbon-to-nitrogen single-atom transmutation of azaarenes. Nature 623, 77–82 (2023).
Ma, C., Lindsley, C. W., Chang, J. & Yu, B. Rational molecular editing: a new paradigm in drug discovery. J. Med. Chem. 67, 11459–11466 (2024).
Li, E.-Q., Lindsley, C. W., Chang, J. & Yu, B. Molecular skeleton editing for new drug discovery. J. Med. Chem. 67, 13509–13511 (2024).
Joynson, B. W. & Ball, L. T. Skeletal editing: Interconversion of arenes and heteroarenes. Helv. Chim. Acta 106, e202200182 (2023).
Xu, Y.-A., Xiang, S.-H., Che, J.-T., Wang, Y.-B. & Tan, B. Skeletal editing of cyclic molecules using nitrenes. Chin. J. Chem. 42, 2656–2667 (2024).
Hui, C., Craggs, L. & Antonchick, A. P. Ring contraction in synthesis of functionalized carbocycles. Chem. Soc. Rev. 51, 8652–8675 (2022).
Tanifuji, R. Skeletal editing: Recent progress on ring-contraction. J. Syn. Org. Chem. Jpn. 80, 778–779 (2022).
Jurczyk, J. et al. Photomediated ring contraction of saturated heterocycles. Science 373, 1004–1012 (2021).
Kim, S. F. et al. Mechanistic investigation, wavelength-dependent reactivity, and expanded reactivity of N-aryl azacycle photomediated ring contractions. J. Am. Chem. Soc. 146, 5580–5596 (2024).
Hurlow, E. E. et al. Photorearrangement of [8]−2,6-pyridinophane N-oxide. J. Am. Chem. Soc. 142, 20717–20724 (2020).
Feng, Z., Allred, T. K., Hurlow, E. E. & Harran, P. G. Anomalous chromophore disruption enables an eight-step synthesis and stereochemical reassignment of (+)-marineosin a. J. Am. Chem. Soc. 141, 2274–2278 (2019).
Luo, J., Zhou, Q., Xu, Z., Houk, K. N. & Zheng, K. Photochemical skeletal editing of pyridines to bicyclic pyrazolines and pyrazoles. J. Am. Chem. Soc. 146, 21389–21400 (2024).
Xu, K. et al. Synthesis of 2-formylpyrroles from pyridinium iodide salts. Org. Lett. 22, 6107–6111 (2020).
Woo, J. et al. Scaffold hopping by net photochemical carbon deletion of azaarenes. Science 376, 527–532 (2022).
Woo, J., Zeqiri, T., Christian, A. H., Ryan, M. C. & Levin, M. D. Carbon-atom scavengers enable divergent, selective carbon deletion of azaarenes. J. Am. Chem. Soc. 147, 20120–20131 (2025).
Uhlenbruck, B. J. H., Josephitis, C. M., de Lescure, L., Paton, R. S. & McNally, A. A deconstruction-reconstruction strategy for pyrimidine diversification. Nature 631, 87–93 (2024).
Bartholomew, G. L., Carpaneto, F. & Sarpong, R. Skeletal editing of pyrimidines to pyrazoles by formal carbon deletion. J. Am. Chem. Soc. 144, 22309–22315 (2022).
Li, S. et al. Skeletal editing of 4-arylpyrimidines into diverse nitrogen heteroaromatics via four-atom synthons. Nat. Commun. 16, 7112 (2025).
Huang, X.-Y., Xie, P.-P., Zou, L.-M., Zheng, C. & You, S.-L. Asymmetric dearomatization of indoles with azodicarboxylates via cascade electrophilic amination/aza-prins cyclization/phenonium-like rearrangement. J. Am. Chem. Soc. 145, 11745–11753 (2023).
Zhang, X. et al. Asymmetric dearomative single-atom skeletal editing of indoles and pyrroles. Nat. Chem. 17, 215–225 (2024).
Yoshimura, A. & Zhdankin, V. V. Advances in synthetic applications of hypervalent iodine compounds. Chem. Rev. 116, 3328–3435 (2016).
Parra, A. Chiral hypervalent iodines: active players in asymmetric synthesis. Chem. Rev. 119, 12033–12088 (2019).
Keder, R., Dvorakova, H. & Dvorak, D. New approach to the synthesis of N7-arylguanines and N7-aryladenines. Eur. J. Org. Chem. 2009, 1522–1531 (2009).
Deetz, M. J., Malerich, J. P., Beatty, A. M. & Smith, B. D. One-step synthesis of 4(3H)-quinazolinones. Tetrahedron Lett. 42, 1851–1854 (2001).
Merino, P. Chemical synthesis of nucleoside analogues. (John Wiley & Sons, 2013).
Chen, N. et al. Hypervalent iodine(III)-mediated umpolung dialkoxylation of N-substituted indoles. J. Org. Chem. 87, 12759–12771 (2022).
Chiodi, D. & Ishihara, Y. The role of the methoxy group in approved drugs. Eur. J. Med. Chem. 273, 116364 (2024).
Liu, H. & Du, D.-M. Recent advances in the synthesis of 2-imidazolines and their applications in homogeneous catalysis. Adv. Synth. Catal. 351, 489–519 (2009).
Mehedi, M. S. A. & Tepe, J. J. Recent advances in the synthesis of imidazolines (2009–2020). Adv. Synth. Catal. 362, 4189–4225 (2020).
Li, J., Yu, B. & Lu, Z. Chiral imidazoline ligands and their applications in metal-catalyzed asymmetric synthesis. Chin. J. Chem. 39, 488–514 (2021).
Deng, T.-T. et al. Synthesis of nucleoside and nucleotide analogues by cyclization of the guanine base with 1,1,3,3-tetramethoxypropane. Org. Lett. 24, 7834–7838 (2022).
Zhu, W. et al. Catalytic asymmetric nucleophilic fluorination using BF3.Et2O as fluorine source and activating reagent. Nat. Commun. 12, 3957 (2021).
Zhou, B., Haj, M. K., Jacobsen, E. N., Houk, K. N. & Xue, X. S. Mechanism and origins of chemo- and stereoselectivities of aryl iodide-catalyzed asymmetric difluorinations of beta-substituted styrenes. J. Am. Chem. Soc. 140, 15206–15218 (2018).
Ganji, B. & Ariafard, A. DFT mechanistic investigation into phenol dearomatization mediated by an iodine(III) reagent. Org. Biomol. Chem. 17, 3521–3528 (2019).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (21772236 grant to B.W.) and the Fundamental Research Funds for the Central Universities, South-Central Minzu University (CZQ23024 grant to W.-W.S.).
Author information
Authors and Affiliations
Contributions
W.-W.S. and B.W. conceived the concept, directed the project; W.-W.S. and Y.-B.X. carried out experimental work. W.-W.S. wrote the paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Yan Xiong, and the other, anonymous, reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Sun, WW., Xie, YB. & Wu, B. Chiral hypervalent iodine catalyzed stereoselective skeletal editing of pyrimidine fused heterocycles. Nat Commun 17, 284 (2026). https://doi.org/10.1038/s41467-025-67000-3
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-67000-3
