
Abstract
Targeted protein degradation/downregulation (TPD/TPDR) is a disruptive paradigm for developing therapeutics. <2% of ~600 E3 ligases have been exploited for this modality, and efficacy for multi-subunit ion channels has not been demonstrated. NEDD4-2 E3 ligase regulates myriad ion channels, but its utility for TPD/TPDR is uncertain due to complex regulatory mechanisms. Here, we identify a nanobody that binds NEDD4-2 HECT domain without disrupting catalysis sites as revealed by cryo-electron microscopy and in vitro ubiquitination assays. Recruiting NEDD4-2 to diverse ion channels (CaV2.2; KCNQ1; and epithelial Na+ channel, ENaC, with a Liddle syndrome mutation) using divalent nanobodies (DiVas) strongly suppresses their surface density and function. Global proteomics indicates DiVa recruitment of endogenous NEDD4-2 to KCNQ1-YFP yields dramatically lower off-target effects compared to NEDD4-2 overexpression. The results establish utility of NEDD4-2 recruitment for TPD/TPDR, validate ion channels as susceptible to this modality, and introduce a general method to generate ion channel inhibitors.
Similar content being viewed by others

Introduction
Induced proximity of enzymes to control the stability or function of target proteins is a rapidly evolving and powerful approach to not only develop new therapeutics for a broad range of diseases but also to devise enabling research tools1. The premier example of this method recruits E3 ubiquitin ligases to foster targeted protein degradation (TPD) via the endogenous ubiquitin-proteasomal system (UPS)2,3. The most prevalent molecules for TPD fall into two classes—proteolysis targeting chimeras (PROTACs) are bivalent small molecules (or biologics, bioPROTACs) that simultaneously bind a target and an E3 ligase to form a ternary complex4; and molecular glues, exemplified by the immunomodulatory drugs (IMiDs), thalidomide and derivatives, which were found by serendipity to act as degraders by binding and reshaping the surface of an E3 ligase to recognize a new target protein5,6,7. TPD is either the underlying mechanism or has been successfully applied to conventionally undruggable targets including transcription factors IKZF1/IKZF35,6, androgen receptor8, the anti-apoptotic protein BCL2L19, and tau protein10, with several molecules employing this modality currently in clinical trials, primarily for oncology indications11. There are ~600 E3 ligases in the human genome, of which <2% have been exploited for TPD3. The most advanced PROTACs and molecular glues exploit the E3 ligases von Hippel–Lindau tumor suppressor (VHL) and cereblon, respectively, which are both members of the cullin-RING family of E3 ligases3,12. The current reliance of TPD approaches on only a small number of E3 ligases is a recognized limitation as it may limit efficacy due to development of resistance or lack of expression of the specific E3 ligase in relevant tissues13,14. Therefore, expanding the universe of E3 ligases suitable for TPD is a critical endeavor in this nascent field3.
To date, there has been scant application of TPD (or TPDR, a term reflecting the possibility of functional downregulation without frank degradation) to cell surface ion channels, a multi-family class of membrane proteins that are necessary for diverse biological functions, including generating the heartbeat, neuronal firing, hormonal secretion, gastric acidification, fluid homeostasis, and sensory perception15,16. Ion channel blockers are essential therapeutics for a wide variety of diseases such as cardiac arrhythmias, pain, epilepsy, multiple sclerosis, and hypertension17. Developing selective inhibitors for ion channels remains an incomplete and daunting task, typically approached by laborious high throughput screens for small molecule or toxin blockers of individual ion channels. TPD/TPDR offers a potentially modular and general method to generate selective inhibitors for a broad spectrum of surface ion channels, but this approach is largely unexplored. Adaptations of the classical bioPROTACs design have been developed specifically for targeting integral membrane proteins2. These include lysosomal targeting chimeras (LYTACs)18, antibody-based PROTACs (AbTACs)19, proteolysis targeting antibodies (PROTABs)20, and transferrin receptor targeting chimeras (TransTACs)21. LyTACs and TransTACs employ antibody conjugates that simultaneously bind to an extracellular portion of the target protein and, respectively, to either the cation-independent mannose-6-phosphate receptor (CI-M6PR), a cell-surface to lysosome shuttling receptor, or to the rapidly recycling transferrin receptor. AbTACs and PROTABs also utilize extracellular bivalent antibodies that concurrently bind substrate protein and single-pass RING E3 ligases RNF43 and ZNRF3, respectively19,20. However, none of these approaches have been applied to ion channels, which typically are macromolecular complexes with multiple membrane-spanning segments and distinctive subunits that may make them challenging for TPD/TPDR. Moreover, many ion channels have a large portion of their surface area in the cytosol with a comparatively small extracellular exposure, potentially making it challenging to develop extracellular antibodies for them22.
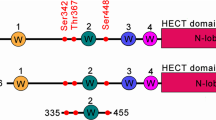
Conceptually, it could be advantageous to exploit E3 ligases which are known physiological ion channel regulators for designing TPD/TPDR approaches to this class of proteins. In this regard, the homologous to E6AP C-terminus (HECT) E3 ligase, NEDD4-2 (neural precursor cell expressed developmentally down-regulated 4-like) has been demonstrated to regulate the functional expression of numerous ion channels, including (but not limited to): voltage-gated Na+, Ca2+, and K+ channels; the epithelial sodium channel (ENaC); chloride channels (CFTR and ClC5); and water channels (AQP2)23. Nevertheless, there are several obstacles to potentially exploiting NEDD4-2 for TPD/TPDR of ion channels. NEDD4-2 (and NEDD4-1) are prototypes of the 9-member NEDD4 family of HECT E3 ligases which have a domain organization consisting sequentially of a C2 domain, 2-4 substrate-binding WW domains, and a catalytic HECT domain24. To date, there has been limited exploitation of the HECT E3 ligase family for TPD/TPDR25, and their mechanism of action, which involves direct transfer of ubiquitin to a catalytic cysteine on the HECT domain before delivery to substrate, is qualitatively different from the cullin-RING E3 ligases. Further, there are no available suitable ligands of NEDD4-2 that can be leveraged for TPD/TPDR. Finally, NEDD4-2 enzymatic activity is regulated by multi-layered mechanisms including phosphorylation26,27, protein-protein interactions28, and auto-inhibitory intramolecular interactions29, making it ambiguous whether its induced recruitment to ion channels would be effective in inhibiting their functional expression.
Another prominent approach for TPD/TPDR is the use of engineered E3 ligases that exert their actions intracellularly. The general principle involves the generation of chimeric proteins in which substrate-binding motifs of distinct E3 ligases are replaced by targeting moieties such as peptides, nanobodies or other antibody mimetics that direct the E3 catalytic domain to a chosen target30,31,32,33,34,35. This method has shown efficacy in degrading intracellular proteins and also in down-regulating abundance and/or function of membrane proteins, including ion channels34,35,36. A limitation of this chimeric approach is the potential for off-target effects due to over-expression of the E3 catalytic domain present in the engineered E3 ligases. This could be potentially circumvented by using engineered proteins to recruit endogenous E3 ligases to targeted proteins.
In this work, we isolate a nanobody that binds selectively to a unique site on NEDD4-2 HECT domain in a manner revealed by cryo-electron microscopy (cryoEM) to not interfere with sites on the protein necessary for catalysis (the E2 binding site, catalytic cysteine, and the ubiquitin exosite)37. Recruitment of endogenous NEDD4-2 to diverse ion channels—CaV2.2, KCNQ1, ENaC—and G-protein coupled receptors using a divalent (DiVa) nanobody format, in which the NEDD4-2 nanobody is coupled to a substrate-targeting nanobody, results in decreases in their surface density and function to a similar extent as seen with simple NEDD4-2 over-expression. Global proteome analyses indicated DiVa-mediated recruitment of endogenous NEDD4-2 to a test ion channel, KCNQ1-YFP, had minimal impact on cellular proteostasis, contrasting with changes in expression of hundreds of proteins observed with NEDD4-2 over-expression.
Results
Targeted recruitment of YFP-tagged NEDD4-2 HECT domain to reconstituted CaV2.2 inhibits channel surface density and ionic currents
We used a heterologous expression system to test whether recruitment of NEDD4-2 HECT domain could down-regulate functional expression of a plasma membrane ion channel, CaV2.2 (Fig. 1). The strategy was inspired by our previous finding that a construct consisting of a nanobody targeting the auxiliary β subunit of high-voltage activated calcium channels (nb.F3) fused directly to the NEDD4-2 HECT domain (CaV-aβlator) potently inhibits CaV1/CaV2 channels35. Nb.F3 by itself is functionally silent on CaV1/CaV2 channels expressed in heterologous cells35. CaV2.2 channels engineered with a double bungarotoxin binding site (BBS) in the extracellular domain IV S5-S6 loop (BBS-α1B) showed robust surface density by flow cytometry when co-expressed with CaVβ2a, YFP and separate nanobodies targeting these two proteins (nb.F3 and nb.YFP, respectively) (Fig. 1a, b; and Supplementary Fig. 1). Accordingly, cells co-expressing α1B + β2a + YFP +nb.F3 + nb.YFP yielded robust whole-cell currents (Fig. 1c, d). Fusing nb.F3 and nb.YFP together with a flexible linker (GGGGSGGGGS) yielded a divalent (DiVa) nanobody, DiVa nb[F3-YFP], which had no impact on either reconstituted CaV2.2 surface density (Fig. 1e, f; and Supplementary Fig. 1) or whole-cell currents (Fig. 1g, h). Co-expressing reconstituted CaV2.2, nb.F3 and nb.YFP with YFP-tagged NEDD4-2 HECT domain (YFP-HECT4-2) yielded small decreases in CaV2.2 surface density (Fig. 1i, j; and Supplementary Fig. 1) and whole-cell currents (Fig. 1k, l; and Supplementary Fig. 1), reflecting the potential for off-target effects of engineered E3 ligases comprised of enzymatic moieties directly fused to targeting modules. Co-expressing DiVa nb[F3-YFP] with reconstituted CaV2.2 and YFP-HECT resulted in robust decreases in channel surface density (Fig. 1m, n; and Supplementary Fig. 1) and whole-cell currents (Fig. 1o, p; Ipeak,0mV = -196.8 ± 44.8, n = 8 for α1B + β2a + α2δ-1 + nb.β + nb.YFP; and Ipeak,0mV = -59.8 ± 18.5, n = 8 for α1B + β2a + α2δ-1 + DiVa nb[F3-YFP] + YFP-HECT4-2, P < 0.05, one-way ANOVA and Tukey’s multiple comparisons test). Overall, these results establish the proof-of-concept that recruiting NEDD4-2 HECT domain to an ion channel using DiVa nanobodies can effectively inhibit ionic currents by down-regulating channel surface density, providing motivation to recruit endogenous NEDD4-2 for targeted downregulation of ion channels.

a Schematic of BBS-α1B + β + α2δ-1 + YFP + nb.F3 + nb.YFP. Cell surface α1B channels are selectively detected using Alexa Fluor 647-conjugated bungarotoxin (BTX-647) attaching to an extracellular bungarotoxin binding site (BBS) engineered into α1B. Created in BioRender. Colecraft, H. (https://BioRender.com/crkk1ct). b Exemplar flow cytometry contour and cumulative density frequency (CDF) plots. c Exemplar whole-cell currents for cells expressing α1B + β + α2δ-1 + YFP + nb.F3 + nb.YFP. d Population I-V curve for cells expressing α1B + β + α2δ-1 + YFP + nb.F3 + nb.YFP. Data points are means ± SEM; n = 8 for each point from three separate transfections. e Schematic; created in BioRender. Colecraft, H. (https://BioRender.com/crkk1ct). f Flow cytometry data for cells expressing BBS-α1B + β + α2δ-1 + YFP + DiVa nb[F3-YFP]. g, h Exemplar currents and population I-V curve for cells expressing α1B + β + α2δ-1 + YFP + DiVa nb[F3-YFP], n = 9 from three independent transfections (red traces, symbols and curves). Black trace in (h) is I-V curve reproduced from (d) to facilitate visual comparison. i Schematic; created in BioRender. Colecraft, H. (https://BioRender.com/crkk1ct). j Flow cytometry data for cells expressing BBS-α1B + β + α2δ-1 + YFP-HECT4-2 + nb.F3 + nb.YFP. (k, l) Exemplar currents and population I-V curve for cells expressing α1B + β + α2δ-1 + YFP-HECT4-2 + nb.F3 + nb.YFP. n = 8 from three independent transfections (blue traces, symbols and curve). Black trace in (l) is I-V curve reproduced from (d) to facilitate visual comparison. m Schematic; created in BioRender. Colecraft, H. (https://BioRender.com/crkk1ct). n Flow cytometry data for cells expressing BBS-α1B + β + α2δ-1 + YFP-HECT4-2 + DiVa nb[F3-YFP]. o, p Exemplar currents and population I-V curves for cells expressing α1B + β + α2δ-1 + YFP-HECT4-2 + DiVa nb[F3-YFP], n = 9 from three independent transfections (magenta traces, symbols and curve). Black trace in (p) is I-V curve reproduced from (d) to facilitate visual comparison. *p = 0.0398 by one-way ANOVA and Tukey’s multiple comparisons test.
Identification and characterization of a nanobody that binds NEDD4-2 HECT domain
Given the lack of appropriate binders of NEDD4-2 that can be used for targeted recruitment of this E3 ligase, we sought to identify a nanobody that binds NEDD4-2 HECT domain (HECT4-2) but does not interfere with the catalytic activity of the enzyme. We expressed HECT4-2 bearing His- and FLAG-tags on the N- and C-terminus, respectively, in E. coli, and purified the protein from bacterial lysates using a nickel column (Fig. 2a, b). Purified HECT4-2 was used as bait to isolate nanobody binders from a yeast nanobody display library containing 1 × 108 unique synthetic nanobodies presented on the extracellular surface of yeast (Fig. 2c; Supplementary Fig. 2)38. Following two rounds of magnetic-activated cell sorting using 1 μM HECT4-2 as bait, individual clones were sorted into a 96-well plate, and their binding to HECT4-2 determined using flow cytometry (Supplementary Fig. 2). We obtained 15 unique nanobodies from our initial 96-well on-yeast staining assay, and herein focus on one positive clone, termed nb.C11. We cloned nb.C11 into a mammalian expression vector in frame with Cerulean (Cer) cDNA sequence. We co-expressed Cer-nb.C11 with Venus-tagged HECT4-2 (HECT4-2-Ven) in HEK293 cells and used flow cytometry fluorescence coupled with fluorescence resonance energy transfer (flow-FRET)39 to probe binding between the two proteins in live cells (Fig. 2d). Cells expressing Cer.nb.C11 and HECT4-2-Ven displayed robust FRET that was well fit by a binding curve, confirming interaction between the two proteins in live mammalian cells (Fig. 2d, g). By contrast, Cer-nb.C11 did not bind HECT4−1-Ven, despite the high homology between NEDD4−1 and NEDD4-2 HECT domains (83% sequence identity) (Fig. 2d, g).

a Schematic of tagged NEDD4-2 HECT domain (HECT4-2) construct cloned into bacterial expression plasmid. b Coomassie-stained gel of purified HECT4-2. Inset: Structure of HECT4-2 domain (PDB ID: 3JW0) showing distinct N- and C-lobes in magenta and orange, respectively. Lys - cell lysate; Sup - supernatant; FT - flowthrough; E1-E3 - eluted fractions 1-3. Gel is representative of 3 independent experiments with similar results. c Schematic of yeast display selection method used to isolate nanobody binders to HECT4-2. d Top, FRET pair schematic, Cer-nb.C11 + HECT-Ven. Bottom, flow cytometry FRET (flow-FRET) scatter plots and binding curve fits for HECT4-2-Ven and HECT4−1-Ven, respectively. e Top, FRET pair schematic, Cer-nb.C11 + NEDD4-2-Ven. Bottom, flow-FRET scatter plot and binding curve fit. f Top, FRET pair schematic, Cer-nb.C11 + either Ven-N-lobe or Ven-C-lobe. Bottom, flow-FRET scatter plot and binding curve fits for Ven-N-lobe and Ven-C-lobe, respectively. g Relative dissociation constants (Kd) obtained from binding curve fits to flow-FRET scatter plots for binding interactions between nb.C11 and indicated FRET binding partners; N = 3 independent experiments for HECT4-1, N-lobe and C-lobe groups, N = 4 for NEDD4-2, and N = 5 for HECT4-2. Data are means ± SEM. *p < 0.0001 compared to HECT4-1 and C-lobe using one-way ANOVA and Tukey’s multiple comparisons test. h Bio-layer interferometry (BLI) sensorgrams for binding of HECT4-2 (analyte) to nb.C11 (captured ligand). Trace colors denote analyte concentrations of 7.5 nM (black), 15 nM (red), 30 nM (blue), 60 nM (green), and 120 nM (purple). i Binding curve of nb.C11 association with WW4-HECT4-2. j Kinetic parameters (kon, koff, Kd) obtained from fitting data in (h) to a global 1:1 binding model.
Full-length NEDD4-2 contains a Ca2+-dependent membrane-targeting C2 domain and four substrate-binding WW domains upstream of the catalytic HECT domain. NEDD4 family E3 ligases are known to adopt intramolecular autoinhibitory interactions involving association of the HECT domain with WW and C2 domains40. Thus, it was uncertain whether the epitope on HECT4-2 recognized by nb.C11 would be accessible in full-length NEDD4-2. Reassuringly, Cer-nb.C11 bound full-length NEDD4-2-Ven with a similar affinity as the interaction with HECT4-2-Ven (Fig. 2e, g). HECT4-2 has distinct N- (binds E2 enzymes and contains ubiquitin exosite) and C-lobes (contains the catalytic cysteine for ubiquitin transfer). Cer-nb.C11 preferentially bound HECT4-2 N-lobe (Fig. 2f, g). Thus, nb.C11 binds NEDD4-2, but not NEDD4-1, inside cells using an epitope on the HECT4-2 N-lobe that is accessible in the full-length NEDD4-2 protein.
We further used biolayer interferometry to directly measure binding affinity of nb.C11 to purified NEDD4-2 HECT domain. HIS-tagged nb.C11 was immobilized on Octet HIS1K biosensors and binding kinetics measured for application of different concentrations of purified WW4-HECT4-2 (Fig. 2h). Fitting the data to a 1:1 binding model indicated a high-affinity interaction with Kd = 2 nM (Fig. 2i, j).
Structural basis of nb.C11 interaction with NEDD4-2 HECT domain
We used cryogenic electron microscopy (cryoEM) to elucidate the structural basis for interaction of nb-C11 with HECT4-2. Pilot experiments indicated that inclusion of the WW4 domain stabilized the NEDD4-2 HECT domain and provided additional ordered mass to facilitate structural studies (Supplementary Fig. 3). We assembled a complex of WW4-HECT4-2 and nb.C11 and confirmed nanobody binding by mass photometry and coelution on size exclusion chromatography (Supplementary Fig. 3). CryoEM analysis (Supplementary Figs. 4–7) resulted in a reconstruction with a global resolution of 3.1 Å (Fig. 3a), with minimal directional anisotropy (Supplementary Fig. 4d). The average resolution in the nanobody was 3.1 Å, and 3.16 Å in the HECT-WW4 domain (Supplementary Fig. 6). Consistent with flow-FRET data (Fig. 2f), nb.C11 was bound to the N-lobe of the NEDD4-2 HECT domain (Fig. 3). Density quality was sufficient to support generation and refinement of a near-complete atomic model, including confident assignment of the nanobody-HECT domain interface and the WW4 domain (Fig. 3). The WW4 domain forms a well-ordered fold with three anti-parallel β strands (β1-β3)41,42 interacting with a non-canonical PY motif (589PAVPY593) on the proximal loop connecting WW4 to the α1’ helix at the start of the HECT4-2 domain (Fig. 3; Supplementary Figs. 8,9). Thus, this 589PAVPY593 motif diverges from the canonical L/PPxY recognized by most WW domains of NEDD4-143,44 and NEDD4-241.

a Final cryo-EM map of the WW4-HECT4-2:nb.C11 complex at 3.1 Å resolution (EMD-73821). Sharpened density map is contoured at 0.124. b Atomic model of WW4-HECT4-2:nb.C11 complex (PDB 9Z5Q) including a close-up view showing NEDD4-2 N-lobe small subdomain (NLSSD) interactions with CDR1-3 loops on nb.C11. c Superposition of previously published E2 enzyme (UBCH5B) (PDB: 3JW0; red), donor ubiquitin (UbD) (PDB: 3JW0; dark brown) and acceptor ubiquitin (UbA) (PDB: 4BBN; light brown) binding sites on the HECT4-2:nb.C11 complex (HECT4-2, light gray; nb.C11, dark gray).
The HECT domain N-lobe is organized into two subdomains, referred to as the N-lobe large subdomain (NLLSD; includes α1’-α5 and α9-α10 helices; β1-β4 strands) and N-lobe small subdomain (NLSSD; comprises α6-α8 helices and β5-β6 strands), respectively45,46 (Fig. 3; and Supplementary Fig. 9). Nb.C11 binds solely to the HECT4-2 NLSSD (Fig. 3; and Supplementary Fig. 9). Specifically, nb.C11 exclusively interacts with residues from the β5-β6 hairpin loop, the loop connecting β6 to α8, and α8 itself (Fig. 3; Supplementary Fig. 9). The binding interface is tightly packed and involves residues from all three nb.C11 complementarity-determining regions (CDR1-CDR3) and the scaffold (Fig. 3b). The interface buries surface area of 851.6 Å2 and contains a core region comprised of two asymmetric hydrophobic clamps. The first smaller clamp constitutes a hydrophobic boundary between HECT4-2 β5-β6 hairpin loop and nb.C11 CDR3, and involves face-to-face π-stacking between Y786 (HECT4-2) and Y105 (CDR3) and a CH/π face-to-face interaction47 between Phe782 (HECT4-2) and P106 (CDR3). The Y786 sidechain also forms a hydrogen bond with the backbone carbonyl group of P106. The second larger clamp comprises W32 and Y33 (CDR1) forming π-stacking and hydrogen bonding interactions, respectively, with W814 and E806 from α8, while Y58 (CDR2) contacts D809 (α8) via a side-chain mediated hydrogen bond. W32 also forms a hydrogen bond with the backbone carbonyl of L790. In addition, the backbone carbonyl group from P792 (β6-α8 loop) forms a hydrogen bond and a CH/π interaction with Y37 (nb.C11 backbone). Finally, Q108 (nb.C11 backbone) forms a hydrogen bond with the D789 backbone carbonyl (Fig. 3b).
Crystal structures of E6AP and NEDD4-2 HECT domains in complex with UbcH7 and UbcH5B+ubiquitin, respectively, localized binding of the E2 conjugating enzymes to a hydrophobic groove on the NLSSD, primarily involving interactions with residues on the α7 helix46,48. The nb.C11 binding site is on the opposite face of the NEDD4-2 NLSSD when compared to the E2 binding sites (Fig. 3c; Supplementary Fig. 9). This is a unique binding interface that has not been previously targeted by any ligand or observed as a site of protein-protein interactions, including with ubiquitin variants that bind NEDD4-2 HECT domain at either the E2 binding site or ubiquitin exosite37. A cryoEM structure of full-length NEDD4-2 found the C2 domain binds the E2 binding site on the HECT domain49, which is the opposite face of the nb.C11 binding site (Supplementary Fig. 8). The nb.C11/WW4-HECT4-2 structure also provided insights into the basis for selectivity of nb.C11 for NEDD4-2 over NEDD4-1, despite the high sequence conservation in the NLSSD between the two isoforms (Supplementary Fig. 9). Of the eight residues on HECT4-2 involved in binding nb.C11, four are not conserved in NEDD4-1 [Y786H, D789E, P792N, and D809Y] (Fig. 3b; and Supplementary Fig. 9); mutating the three non-conservative substitutions in NEDD4-1 to the corresponding residues in NEDD4-2 [H711Y, N717P, and Y734D] reconstituted binding to nb.C11 (Supplementary Fig. 10), confirming that variation at these positions underlies the selectivity of nb.C11 for NEDD4-2.
Impact of nb.C11 on NEDD4-2 HECT domain catalytic activity
We assembled an in vitro ubiquitination assay to directly determine the impact of nb.C11 on NEDD4-2 HECT catalytic activity. First, we established a time course for the enzymatic reaction by mixing E1, E2, WW4-HECT2, and Ub for increasing time periods. The reaction was stopped at various times, separated on an SDS-PAGE gel and probed with anti-Ub antibody to detect formation of polyubiquitin chains as a measure of NEDD4-2 HECT enzymatic activity (Fig. 4a). We observed a time-dependent assembly of ubiquitin chains of varying stoichiometries, ranging from di-ubiquitin to higher order polymers (Fig. 4a). With increasing reaction times, the intensity of ubiquitin polymers increased while the amount of single ubiquitin declined concomitantly (Fig. 4a). Under the assay conditions, the half-maximal time of the reaction was 10.9 ± 1.2 min (Fig. 4b). We obtained essentially identical results regarding the time course of polyubiquitination when we substituted full-length NEDD4-2 for WW4-HECT4-2, suggesting minimal autoinhibition by NEDD4-2 C2 domain under the assay conditions (Fig. 4b). In addition to forming independent polyubiquitin chains, immunoblotting using an anti-WW4-HECT4-2 antibody indicated that the enzyme itself underwent autoubiquitination, as previously reported50,51 (Fig. 4c).

a Time course of WW4-NEDD4-2 HECT catalytic activity assessed by monitoring formation of polyubiquitin chains in an in vitro ubiquitination assay probed with anti-ubiquitin antibody. Blot is representative of 3 independent experiments with similar results. b Time course of HECT domain in vitro catalytic activity for either purified WW4-HECT4-2 (black circles, n = 3) or full-length NEDD4-2 (red circles, n = 2). c Time course of WW4-HECT4-2 in vitro autoubiquitination visualized using anti-WW4-HECT4-2 antibody. Blot is representative of 3 independent experiments with similar results. d Impact of purified nb.C11 on capacity of WW4-HECT4-2 to catalyze formation of polyubiquitin chains in vitro. e Population data showing impact of nb.C11 on WW4-HECT4-2 in vitro catalytic activity (n = 6). *p < 0.0001 compared to WW4-HECT4-2 control by one-way ANOVA and Tukey’s multiple comparisons test. f Impact of purified nb.C11 on capacity of full-length NEDD4-2 to catalyze formation of polyubiquitin chains in vitro. g Population data showing impact of nb.C11 on full-length NEDD4-2 in vitro catalytic activity (n = 4). *p < 0.0001 compared to full-length NEDD4-2 control by one-way ANOVA and Tukey’s multiple comparisons test.
With the in vitro ubiquitination assay established, we assessed the impact of nb.C11 on WW4-HECT4-2 enzymatic activity. We assembled the enzymatic reaction (ATP, E1, E2, WW4-HECT4-2 and ubiquitin) ± 50 μM nb.C11 (Fig. 4d). Anti-ubiquitin staining showed the expected formation of polyubiquitin chains in the control (no nb.C11) lane. The polyubiquitin staining was even darker in the test lane, indicating that nb.C11 does not inhibit WW4-HECT4-2 enzymatic activity, but in fact, rather enhances it (Fig. 4d, e). As a negative control, we observed no polyubiquitin chains using a catalytically dead WW4-HECT4-2[C/S] mutant Fig. 4d, e). We obtained similar results with full length NEDD4-2, indicating that the presence of C2 and WW1-WW3 domains did not interfere with nb.C11 binding and enhancement of NEDD4-2 HECT catalytic activity (Fig. 4f, g).
Targeted recruitment of endogenous NEDD4-2 inhibits function of heterologously expressed and native CaV2.2 channels
We next determined the potential of using nb.C11 as a tool to recruit endogenous NEDD4-2 to ion channels as a method for inhibiting their function by reducing their cell surface density. We first focused on multi-subunit (membrane α1B, cytoplasmic CaVβ, and extracellular α2δ-1) CaV2.2 transiently expressed in HEK293 cells. We compared the impact of CaVβ-targeted nb.F3, NEDD4-2 over-expression, and targeted recruitment of endogenous NEDD4-2 with DiVa nb[C11-F3] on CaV2.2 surface density and whole-cell currents (Fig. 5a–c; and Supplementary Fig. 11). Nb.F3 had minimal impact on CaV2.2 surface density measured by flow cytometry of BTX-647 bound to surface BBS-α1B (Fig. 5b), in accord with previous findings that this nanobody is functionally inert on CaV1/CaV2 channels35. By contrast, NEDD4-2 co-expression resulted in a 50% decrease in CaV2.2 channel surface density compared to control (Fig. 5b). In agreement with this finding, co-expressing CaV2.2 with NEDD4-2 led to an ~50% decrease in whole-cell current amplitude compared to control cells (Fig. 5c; and Supplementary Fig. 11). Notably, co-expressing CaV2.2 subunits with DiVa nb[C11-F3] resulted in a virtually identical decrease in channel surface density and whole-cell current amplitude as observed with NEDD4-2 over-expression (Fig. 5b, c; and Supplementary Fig. 11), demonstrating the efficacy of targeted recruitment of endogenous NEDD4-2 to inhibit CaV2.2 currents. As a negative control, DiVa nb[C11-F3] had no impact on the surface density of a BBS-tagged potassium ion channel, BBS-KCNQ1-YFP (Supplementary Fig. 12), indicating substrate specificity of targeted recruitment of endogenous NEDD4-2 to inhibit ion channels.

a Schematic of experimental design. Created in BioRender. Colecraft, H. (https://BioRender.com/crkk1ct). Recombinant CaV2.2 (α1B + β2a + α2δ-1) was expressed with nb.F3, NEDD4-2 or DiVa nb[C11-F3]. b Impact of CFP (control), nb.F3, NEDD4-2 or DiVa nb[C11-F3] co-expression on BBS-tagged CaV2.2 surface density assessed by BTX-647 staining and flow cytometry; N = 3 independent experiments for nb.F3 and NEDD4-2 groups, and N = 4 for CFP and DiVa nb[C11-F3] groups. *p < 0.0001, one-way ANOVA and Tukey’s multiple comparisons test. c Population I-V curves evoked from HEK293 cells expressing CaV2.2 and nb.F3 (black squares, n = 32), NEDD4-2 (red circles, n = 28) or DiVa nb[C11-F3] (blue triangles, n = 9). Data are means ± SEM from at least three independent transfections. *p = 0.0009 and p = 0.0314 for NEDD4-2 and DiVa nb[C11-F3], respectively, compared to nb.F3 by one-way ANOVA and Dunnett’s multiple comparisons test. d–f Pulldown of CaV2.2 under different co-expression conditions probed with anti-α1B (d), anti-ubiquitin (e), and anti-CaVβ2 antibodies (f). g Population data of normalized ubiquitin to pulled down CaV2.2 ratio. h Confocal images of DRG neurons infected with adenovirus expressing tdTomato-P2a-DiVa nb[C11-F3]. Images are representative of results obtained from two independent experiments. i Exemplar families of whole-cell Ba2+ currents evoked by step depolarization protocols applied to DRG neurons infected with adenoviruses expressing mCherry (left) or tdTomato-P2a-DiVa nb[F3-C11] (right). j Population I-V curves from DRG neurons expressing mCherry (black squares, n = 8 cells from three independent experiments) or tdTomato-P2a-DiVa nb[F3-C11] (blue triangles, n = 11 cells from three independent experiments). *p = 0.0045 compared to control by two-tailed unpaired t test with Welch’s correction.
We assessed whether NEDD4-2 and DiVa nb[C11-F3] yielded overt signatures on the amount of ubiquitin on CaV2.2. We co-expressed CaV2.2 (α1B-mCherry/YFP-β2a/α2δ-1) separately with various proteins (CFP, nb.F3, nb.C11, NEDD4-2, and DiVa nb[C11-F3]) in HEK293 cells, pulled down α1B-mCherry and detected the protein by Western blotting (Fig. 5d). Under all transiently transfected cell conditions, we detected a robust band consistent with the expected ~280 kDa molecular weight of α1B-mCherry, that was absent in untransfected cells (Fig. 5d). We also detected fainter bands at higher molecular weights that could indicate a propensity for the formation of dimers and higher order oligomers (Fig. 5d). We stripped the blot and successively probed for ubiquitin (Fig. 5e) and co-immunoprecipitated YFP-β2a (Fig. 5f). In control cells expressing CaV2.2 + CFP, we observed a substantial ubiquitin signal on pulled down α1B-mCherry, reflecting the basal activity of endogenous E3 ligases on the channel (Fig. 5e). Unexpectedly, co-expressing NEDD4-2 led to a decrease in the biochemically detected ubiquitination of α1B-mCherry (Fig. 5e, g). By contrast, DiVa nb[C11-F3] did not result in a decrease in CaV2.2 ubiquitination, but rather showed a small increase that was not statistically significant (Fig. 5e, g). Neither nb.F3 or nb.C11 co-expression affected CaV2.2 basal ubiquitination (Fig. 5e, g). The amount of β2a-YFP co-immunoprecipitated with α1B-mCherry was comparable among the different conditions (Fig. 5f), suggesting that the decrease in CaV2.2 whole-cell current observed with NEDD4-2 and DiVa nb[C11-F3] is not due to either CaVβ or α1B degradation.
Finally, we tested the efficacy of recruitment of endogenous NEDD4-2 to inhibit native CaV1/CaV2 channels in their physiological context. We incorporated DiVa nb[F3-C11] (pilot experiments indicated similar effects as DiVa nb[C11-F3] in HEK293 cells) into an adenovirus vector with mCherry as a reporter, enabling expression in primary cultured murine dorsal root ganglion (DRG) neurons (Fig. 5h), a tissue that has been previously shown to express NEDD4-252. Whole-cell patch clamp recordings of control cultured DRG neurons expressing just tdTomato yielded large high-voltage activated (HVA) CaV1/CaV2 channel currents, reflecting the aggregate activity of all CaV1/CaV2 type channels present in these neurons (Fig. 5i, j). By contrast, DRG neurons expressing DiVa nb[F3-C11] expressed significantly reduced HVA Ca2+ currents (Fig. 5i, j), confirming the efficacy of recruiting endogenous NEDD4-2 to inhibit CaV1/CaV2 channels in their native environment.
Targeted recruitment of endogenous NEDD4-2 inhibits KCNQ1 channels
KCNQ1 pore-forming subunits tetramerize to form ion channels that are important for cardiac action potential repolarization and for salt and water transport in epithelial cells in organs including the lungs, cochlea, intestines, and kidneys53. KCNQ1 is known to be down-regulated by NEDD4-2 which binds a PY-motif located on KCNQ1 C-terminus54. As an initial test of the generality of the efficacy of DiVa-mediated recruitment of endogenous NEDD4-2 to generate ion channel inhibitors, we determined the effectiveness of the method on heterologously expressed KCNQ1 (Fig. 6a). Control HEK293 cells transiently transfected with KCNQ1-YFP and nb.YFP displayed large outward whole-cell currents in response to a family of test pulse potentials (from -60 mV to +100 mV in 20-mV increments) (Fig. 6a–c). Co-expressing NEDD4-2 with KCNQ1-YFP yielded substantially reduced whole-cell currents (Fig. 6a–c) mediated by a decrease in channel surface density (Fig. 6d, e). NEDD4-2 co-expression also increased ubiquitination of pulled down KCNQ1 (Fig. 6f–h) and reduced total KCNQ1 protein expression resolved by Western blot (Fig. 6i), in accord with previous reports54. We generated DiVa nb[YFP-C11] by fusing an anti-GFP/YFP nanobody55 to nb.C11. to enable recruitment of endogenous NEDD4-2 to GFP/YFP-tagged proteins. Reassuringly, DiVa nb[YFP-C11] co-expression yielded a similar suppression of KCNQ1-YFP whole-cell current amplitude (Fig. 6a–c) and channel surface density (Fig. 6d, e) as was achieved with NEDD4-2 over-expression. The biochemical effects of DiVa recruitment of endogenous NEDD4-2 on KCNQ1 ubiquitination and stability in HEK293 cells were relatively modest but became more evident when NEDD4-2 was co-expressed (Fig. 6f–i). Surprisingly, KCNQ1-YFP currents obtained from transient transfection of a different cell line, Chinese hamster ovary (CHO) cells, were unaffected by co-expressed DiVa nb[YFP-C11] (Fig. 6j), in sharp contrast to the observations in HEK293 cells (Fig. 6c). We hypothesized that this discrepancy was due to a difference in expression of endogenous NEDD4-2 between the two cell lines. Indeed, Western blotting showed that HEK293 cells express endogenous NEDD4-2 and the expression level is significantly up-regulated by transient transfection of exogenous NEDD4-2 (Fig. 6k, l). By contrast, we could not detect expression of endogenous NEDD4-2 in CHO cells (Fig. 6k, l). The sequences of the peptide immunogen used to generate the NEDD4-2 antibody used in Western blots, and the region containing nb.C11 binding site, are conserved between human and Chinese hamster NEDD4-2 sequences (Supplementary Fig. 13), ruling out the potential trivial explanation that species variations in NEDD4-2 sequence underlies the ineffectiveness of DiVa nb[YFP-C11] in CHO cells. Our finding of a lack of NEDD4-2 expression in CHO cells is serendipitous, as it provides a critical control, showing that endogenous expression of NEDD4-2 is required for the functional effect of DiVa nb[YFP-C11] on KCNQ1-YFP functional expression to be evident.

a Cartoon of KCNQ1-YFP (Q1-YFP). b Exemplar families of currents evoked from HEK293 cells co-transfected with Q1-YFP and nb.YFP (left); NEDD4-2 (middle) or DiVa nb[YFP-C11] (right). c Population I-V curves for Q1-YFP expressed with nb.YFP (black squares, n = 11), NEDD4-2 (red circles, n = 8), and or DiVa nb[YFP-C11] (blue triangles, n = 10). Data are means ± SEM from three independent transfections. d Exemplar flow cytometry CDF plots showing the effects of over-expression of nb.YFP (black); NEDD4-2 (red), or DiVa nb[YFP-C11] (blue) on co-transfected BBS-KCNQ1-YFP in HEK293 cells. e Impact of nb.YFP (control), NEDD4-2, or DiVa nb[YFP-C11] co-expression on BBS-KCNQ1-YFP surface density assessed by BTX-647 staining and flow cytometry; N = 4 independent experiments for each condition. *p = 0.0042 and p = 0.0003 for NEDD4-2 and DiVa nb[YFP-C11], respectively, compared to nb.YFP control by one-way ANOVA and Tukey’s multiple comparisons test. f, g Exemplar immunoblots of immunoprecipitated KCNQ1-YFP showing the effects of the indicated conditions on KCNQ1 steady-state ubiquitination. h Aggregate data quantification of changes in KCNQ1 ubiquitination; N = 3 independent experiments for each condition. *p = 0.0119, **p = 0.0007 by one-way ANOVA and Tukey’s multiple comparisons test. i Exemplar Western blot (top) and population data (bottom) showing impact of NEDD4-2 and DiVa nb[YFP-C11] co-expression on KCNQ1-YFP steady-state expression levels in transfected HEK293 cells; N = 5 independent experiments for each condition. #p = 0.0167, *p = 0.0017 by one-way ANOVA and Tukey’s multiple comparisons test; ns, not significant. Data are means ± SEM. j Population I-V curves evoked from CHO cells co-transfected with KCNQ1-YFP + nb.YFP (black) or KCNQ1-YFP + DiVa nb[YFP-C11] (blue); n = 10 cells per condition. Data are means ± SEM. k Representative immunoblot of endogenous and expressed NEDD4-2 in HEK293 and CHO cells, respectively. l Quantification of endogenous and expressed NEDD4-2 expression in HEK293 and CHO cells, respectively; N = 3 independent experiments for each condition. *p = 0.0368, #p = 0.068 compared to untransfected controls, two-tailed unpaired t test with Welch’s correction. Data are means ± SEM.
Breadth of targets down-regulated by targeted recruitment of endogenous NEDD4-2
Another ion channel known to be regulated by NEDD4-2 is the amiloride-sensitive epithelial Na+ channel (ENaC), a constitutively active channel on the apical surface of high-resistance epithelial cells that mediates absorption of Na+ ions from the lumen in the distal nephron, distal colon, and the airways56. ENaC-mediated Na+ absorption in the distal nephron regulates extracellular fluid (ECF) Na+ concentration, ECF volume and blood pressure, while ENaC in the lungs modulates airway surface liquid clearance56. Structurally, ENaC is a heterotrimeric channel, comprised of membrane-spanning α, β, and γ subunits arranged in a counterclockwise orientation57 (Fig. 7a). Physiologically, ENaC functional expression is regulated by NEDD4-2 which utilizes WW domains to bind PY motifs located on the carboxyl terminus of the β- and γ-subunits of the channel29 (Fig. 7a, b). Mutations (missense, frameshift, or premature stops) that eliminate ENaC PY motifs lead to channel over-expression and result in Liddle syndrome, an autosomal dominant inherited form of severe and early-onset hypertension attendant with hypokalemia and metabolic alkalosis58,59,60. HEK293 cells transiently transfected with ENaC α, YFP-tagged β, and γ subunits expressed constitutively-active amiloride-sensitive currents elicited by ramp and step protocols (Fig. 7c, d; and Supplementary Fig. 14). Cells co-expressing WT α and γ with a Liddle syndrome mutant β-subunit, YFP-β[P617L]61, displayed five-fold larger amiloride sensitive ENaC currents, consistent with the expected diminished restraint of channel surface density by endogenous NEDD4-2 (Fig. 7b–d; Supplementary Fig. 14). Recruitment of endogenous NEDD4-2 to Liddle syndrome mutant ENaC (α, YFP-β[P617L], γ) using DiVa nb[YFP-C11] resulted in a marked decrease in amiloride-sensitive currents to levels that were not significantly different from control WT channels (Fig. 7c, d), demonstrating a reassertion of NEDD4-2 control of channel expression.

a Cartoon of the epithelial sodium channel (ENaC) assembled with α, YFP-tagged β, and γ subunits. b Sequence alignment of conserved β-ENaC PY motif and showing position of a Liddle syndrome mutation, P617L. c Top, voltage ramp protocol. Bottom, exemplar ENaC currents from cells expressing WT ENaC + nb.YFP (left), ENaC[P617L] + nb.YFP (middle), or ENaC[P617L] + DiVa nb[YFP-C11] (right). d Population ENaC I-V curves evoked from HEK293 cells expressing WT ENaC + nb.YFP (black squares, n = 20), ENaC[P617L] + nb.YFP (red circles, n = 15), or ENaC[P617L] + DiVa nb[YFP-C11] (blue triangles, n = 9). *p = 0.0004, one-way ANOVA and Tukey’s multiple comparisons test. e Cartoon of BBS- and YFP-tagged G-protein coupled receptor representing LGR5, LPAR1 or SuccR. Created in BioRender. Colecraft, H. (https://BioRender.com/u49y109). f Population data showing impact of DiVa nb[YFP-C11] on surface density of BBS- and YFP-tagged LGR5, LPAR1 and SuccR; N = 5 independent experiments for each group. *p = 0.0321, **p = 0.0043, ***p = 0.0004, one-way ANOVA and Tukey’s test. Data are means ± SEM. g Impact of NEDD4-2, DiVa nb[YFP-C11], or DiVa nb[C11-F3] on CaVβ1b-YFP expression evaluated with relative YFP fluorescence; N = 3 independent experiments for each condition). *p = 0.0107, **p = 0.003, #p = 0.0042 compared to CFP control using one-way ANOVA and Tukey’s multiple comparisons test. Data are means ± SEM. (h) Impact of NEDD4-2, DiVa nb[YFP-C11] or DiVa nb[C11-F3] on CaVβ1b-YFP expression evaluated with relative YFP fluorescence. Data are means ± SEM.
We next examined the capacity of targeted recruitment of NEDD4-2 to down-regulate functional expression of other classes of proteins beyond ion channels, including G-protein coupled receptors (GPCRs) and cytosolic proteins. We engineered three GPCRs — leucine-rich repeat-containing G-protein coupled receptor 5 (LGR5), lysophosphatidic acid receptor 1 (LPAR1), and succinate receptor 1 (SuccR) — with extracellular BBS- and intracellular YFP-tags on the N- and C-termini, respectively (Fig. 7e). In comparison to control cells expressing tagged GPCRs and nb.YFP, cells co-expressing the GPCRs and DiVa nb[YFP-C11] displayed significantly decreased surface density of LGR5, LPAR1, and SuccR (Fig. 7f). Finally, we probed whether recruitment of NEDD4-2 would be effective for TPD of cytosolic proteins. As exemplar cytosolic proteins we used YFP-tagged voltage-gated Ca2+ channel auxiliary β1b subunit (CaVβ1b-YFP) and YFP, respectively. CaVβ1b-YFP expression was reduced by NEDD4-2 co-expression, and this effect was mimicked by both Diva nb[YFP-C11] and Diva nb[F3-C11] recruiting endogenous NEDD4-2 (Fig. 7g). By contrast, Diva nb[YFP-C11] was less effective in degrading YFP (Fig. 7h). Thus, the efficacy of targeted recruitment of NEDD4-2 to degrade cytosolic proteins may be substrate-dependent.
Comparative impact of NEDD4-2 over-expression and DiVa recruitment of endogenous NEDD4-2 on the global proteome
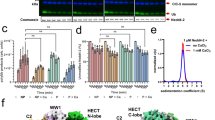
We next determined whether, and to what extent, recruiting endogenous NEDD4-2 to a target substrate impacted global cellular proteostasis. To address this question, we utilized tandem mass tag (TMT) multiplexed mass spectrometry to conduct comparative total proteome analysis in three experimental groups comprised of HEK293 cells transiently transfected with KCNQ1-YFP + CFP-P2A-nb.YFP (Group 1, control), KCNQ1-YFP + CFP-P2A-NEDD4-2- (Group 2) and KCNQ1-YFP + CFP-P2A-DiVa nb[YFP-C11] (Group 3) (Fig. 8a). Three independent transfections were done for Group 1 and four for each of Groups 2 and 3, respectively. We used FACS to select CFP- and YFP- double-positive cells for processing to preclude trivial differences arising from variations in transient transfection efficiency (Fig. 8a). 50 μg of protein from each sample was precipitated and digested with Lys-C (1:50 overnight at room temperature) and trypsin (1:50 for 6 hrs at 37 °C), followed by labeling with TMT 11plex reagents. The 11 TMT-labeled samples were pooled using amounts computed from a ratio check of each sample. Peptides were fractionated by reverse phase HPLC, desalted, and analyzed on an Orbitrap Lumos mass spectrometer. Principal component analysis of the dataset suggested that the second replicate from the different groups were clustered together and separated from other samples in their respective groups. Therefore, the second replicate from the three experimental groups was not included for data analysis.

a Schematic of experimental flow chart for tandem mass tag (TMT) mass spectrometry analyses of HEK293 cells proteome-wide changes. Partially created in BioRender. Colecraft, H. (https://BioRender.com/jd6uc7h) (b–d) Volcano plots comparing proteomes of HEK293 cells expressing (b) KCNQ1-YFP + nb.YFP vs KCNQ1-YFP + NEDD4-2, c KCNQ1-YFP + nb.YFP vs KCNQ1-YFP + DiVa nb[YFP-C11], and (d) KCNQ1-YFP + DiVa nb[YFP-C11] vs KCNQ1-YFP + NEDD4-2. Dotted lines demarcate fold-change (FC) cut-off = 1.5 and p-value = 0.05 using Welch’s two-sided unpaired t-test. Down-regulated genes fitting the cut-off criteria are colored in red and up-regulated genes in green. e, f Top terms from GO analyses of down-regulated and up-regulated genes, respectively. Adjusted p-values obtained from Welch’s two-sided unpaired t-test followed by BH-FDR adjustment.
The proteomic analysis profiled expression of 9142 proteins (Supplementary Data 1). Reassuringly, NEDD4-2 was highly over-expressed in the proteome of cells from group 2 (transfected with NEDD4-2) relative to either group 1 (nb.YFP) or group 3 (DiVa nb[YFP-C11]), providing an internal positive control of the assay (Fig. 8b, d). Comparing the proteomes of group 1 to group 2 indicated NEDD4-2 over-expression resulted in 189 down-regulated proteins and 606 up-regulated proteins compared to control cells expressing nb.YFP (p = 0.05 and fold change (FC) = 1.5 cutoff) (Fig. 8b). By contrast, expression of DiVa nb[YFP-C11] resulted in a dramatically lower change in the cellular proteome (1 down-regulated and 17 up-regulated proteins) compared to control (Fig. 8c). Comparing groups 2 and 3 indicated NEDD4-2 over-expression resulted in 120 down-regulated proteins (Supplementary Data 2) and 240 up-regulated proteins (Supplementary Data 3), respectively, compared to cells expressing DiVa nb[YFP-C11] (adj. p = 0.05 and FC = 1.5 cutoff) (Fig. 8d). Gene ontology (GO) analysis showed that genes down-regulated by NEDD4-2 over-expression were enriched in those involved in RNA binding or associated with ribosomes, with the top three biological processes potentially affected being protein translation, ubiquitin-dependent catabolism, and regulation of mRNA stability (Fig. 8e). Conversely, proteins that were up-regulated in NEDD4-2-over-expressing HEK293 cells were enriched for proteins in the extracellular space, the endoplasmic reticulum and Golgi lumen, the basement membrane, and the lysosomal lumen; the top three biological processes potentially affected include protein ubiquitination, cell adhesion, and cell migration (Fig. 8f). Altogether, the data indicate that DiVa-mediated recruitment of endogenous NEDD4-2 to KCNQ1-YFP (and presumably to other potential substrates) achieves the same functional effect as NEDD4-2 over-expression (Fig. 6), but without the overt impact on global proteostasis observed with NEDD4-2 over-expression (Fig. 8).
Discussion
Ion channels represent the second largest class of drug targets after GPCRs, and selective posttranslational ion channel blockers are highly sought after therapeutics for numerous diseases62. Nevertheless, <10% of ion channels are targeted by currently approved medicines, and very few drugs in this category have been approved over the last three decades63. A significant bottleneck is the identification of ligands that act as selective inhibitors of distinct ion channels. Typically, these are identified empirically by either high throughput screening and medicinal chemistry of small molecule libraries or isolated from complex mixtures of toxins that are present in venomous animals64. These approaches are laborious, relatively slow, and often end up with ion channel blockers that are not selective for a particular channel isoform. Targeted protein degradation or down-regulation (TPD/TPDR) offers a promising general approach to create new selective ion channel blockers. While there has been a widespread adoption of this technology to selectively eliminate proteins for either investigative or therapeutic purposes, the efficacy of the approach for composite ion channel protein complexes has not been previously demonstrated3. This work validates the general utility of creating ion channel inhibitors by targeted recruitment of endogenous NEDD4-2 to a chosen ion channel. Importantly, for some substrates, such as CaV2.2, we demonstrate current inhibition is mediated by increased channel intracellular retention without frank protein degradation, and thus, more consistent with TPDR rather than TPD nomenclature.
The minimal requirements to successfully engage TPD/TPDR boils down to identifying suitable binders for an endogenous E3 ligase and the target protein. An acknowledged deficiency in the TPD field is that < 2% of the ~600 E3 ligases present in the human genome have been exploited for the method3. We show here not only the utility of recruiting endogenous NEDD4-2 to diverse ion channels as being efficacious for their functional inhibition, but also the feasibility of using nanobodies for this purpose. The demonstration that an endogenous, full-length HECT family E3 ligase is suitable for TPD/TPDR is notable because they have a unique mechanism of enzymatic action relative to other E3 ligase families12,65. Moreover, their enzymatic activity is regulated by intricate intramolecular and posttranslational mechanisms that made it uncertain whether they would be suitable for TPD/TPDR66. NEDD4-2 was our strategic choice for this study because it has been demonstrated to physiologically regulate functional expression of many surface ion channels23,67. Beyond ion channels, NEDD4-2 also physiologically regulates the abundance, subcellular localization, and function of diverse transporters and G-protein coupled receptors raising the prospect that it could be generally useful in TPD/TPDR regulation of all classes of integral surface membrane proteins23.
A proteome-wide screen to discover viable protein degradation and stabilization effectors did not identify any HECT E3 ligase family members as being suitable for TPD68. The invisibility of HECT E3 ligases from this screen could be due to two main factors. First, multi-subunit ion channel complexes may be underrepresented in the choice of targets screened in this proteome-wide assay. Second, the sole focus on protein expression levels as a readout for efficacy may not be sufficient to identify effectors that are effective for down-regulation of ion channel function. Ion channels can be strongly inhibited by interventions that redistribute them from the plasma membrane to intracellular sites without their degradation, as shown here and previously reported34,35. The HECT domains of different NEDD4 family E3 ligases specify distinct polyubiquitin linkage types that can exert disparate functional outcomes on membrane protein degradation and trafficking69,70. The yeast homolog of NEDD4, Rsp5, preferentially catalyzes K63 polyubiquitin chains which are predominantly associated with non-degradative outcomes, including decreasing the surface density of target membrane proteins by promoting their net retention in intracellular sites23,71,72,73. A pertinent question is how effective recruitment of endogenous NEDD4-2 would be in degrading cytosolic proteins which represent the largest class of proteins targeted by PROTACs molecules to date. While global proteomic analysis indicated NEDD4-2 over-expression results in decreased expression of many cytosolic and nuclear proteins, it is unclear which of these are due to a direct interaction with NEDD4-2, or are secondary to changes in proteins involved in mRNA stability, protein translation, or ubiquitin-dependent catabolism as identified by our GO analysis. Indeed, of 43 proteins identified to bind and/or be ubiquitinated by NEDD4-2 in an in vitro assay that were present in our proteome analysis74, none were represented in the list of 120 down-regulated genes (NEDD4-2 vs. Diva nb[YFP-C11]), while one of these proteins (SENP2) was up-regulated. This could reflect either a lack of interaction between these proteins and NEDD4-2 in cells in contrast to observations in vitro, or that the ubiquitination catalyzed by NEDD4-2 yields predominantly non-degradative outcomes. Yet another possibility is that another signaling event is required for over-expressed NEDD4-2 to catalyze degradation of particular protein substrates. Nevertheless, our finding that Diva nb[F3-C11] decreased expression of CaVβ1b suggests targeted recruitment of NEDD4-2 could be effective for TPD of some cytosolic proteins.
The results emphasize the versatility of nanobodies as a platform for developing selective binders for both E3 ligases and target proteins. In addition to their relatively small size (12-15 kDa) and ability to recognize antigens in the cytoplasm when expressed intracellularly, the ability of nb.C11 to distinguish between highly conserved NEDD4-1 and NEDD4-2 HECT domains highlights the exquisite selectivity of these reagents, which may not be as easily attainable using small molecules. Methods for identifying nanobodies specific for target proteins using immunization of alpacas or screening of synthetic nanobody display libraries are now readily available38,75. The advent of in silico de novo nanobody/antibody or mini-protein binder design and optimization algorithms76,77 also lower the barrier for developing these useful reagents, potentially allowing for generation of nanobodies/binders against targets which are challenging to express and purify. The DiVa nanobody approach only requires identifying inert binders of the E3 ligase and the target protein. On the target protein side, this is a significantly lower bar than conventional approaches for identifying ion channel inhibitors. Overall, we envision that this work will enable a pipeline for high-throughput development of DiVa nanobody inhibitors for the entire surface ion channelome, with significant implications for both basic research and therapeutic development. Anticipated advantages of such DiVa nanobody inhibitors include the possibility of engineering tissue specificity by incorporating tissue-specific promoters and the potential to engineer chemogenetic or optogenetic variants that enable reversible control of channel inhibition by small molecules and light, respectively78. Other induced proximity approaches specialized for eliminating surface membrane proteins have been developed and include LYTACS, AbTACS, PROTABs, and TransTACs18,19,20,21. As part of their modes of action, all these approaches require generation of an extracellular antibody to the target membrane protein. The DiVa nanobody approach described here is fundamentally different from these prior methods in that both the target and E3 ligase effector are engaged intracellularly. The DiVa nanobodies are relatively small ( < 30 kDa) and fit comfortably within the packaging capacity of commonly used adeno-associated viral vectors. It may also be possible to pair a substrate-targeting nanobody with a ubiquitin variant that binds NEDD4-2 (or other HECT domain E3 ligase) without blocking catalysis to achieve targeted down regulation of protein expression37. Altogether, the DiVa nanobody method offers the unique advantages of being well-suited for membrane proteins with large intracellular and small extracellular footprints for which it may be challenging to raise outward facing antibodies, being amenable for gene therapy development, and minimizing off-target effects as compared to direct fusion of HECT domains to targeting moieties.
Methods
Molecular biology and plasmid construction
A full list of DNA constructs and related information for all DNA constructs used in this study is provided in the Excel document (Supplementary Data 4). In the following paragraphs, we describe cloning methods for constructs that were used for the first time in this study. For NEDD4-2 HECT domain (HECT4-2) purification, the HECT domain consisting of residues 615-994 of full-length NEDD4-2 (a gift from Joan Massague, Addgene plasmid # 27000)79 was cloned via Gibson assembly into a NYCOMPS expression vector (a generous gift from Dr. Filippo Mancia)80, with an N-terminal FLAG-10xHis-TEV cassette and a C-terminal FLAG tag.
Candidate HECT4-2 nanobodies were cloned into the PiggyBac CMV mammalian expression vector, and downstream of a cerulean marker using AgeI/NotI cloning sites. CFP-P2a-nb.C11 was generated by cloning nb.C11 into a previously described customized vector CFP-P2a-XX36 using BamHI and NotI sites. HECT4-2 and HECT4-1 were both cloned into PCDNA3.1 and upstream of a Venus (Ven) marker using EcoRI/HindIII cloning sites. Full-length NEDD4-2 was cloned into pCDNA3.1- HECT4-2-Venus using BamHI/NheI to create NEDD4-2-Venus. To generate Venus-tagged HECT4-2 N- and C-lobes, the N- and C-lobes of NEDD4-2 HECT domain were cloned separately into a customized mammalian expression vector and downstream of mVenus using NotI and XbaI sites.
Human epithelial sodium channel (ENaC) α, β, and γ subunits were gifts from Christie Thomas (Addgene plasmid # 83430, 83429, and 83428, respectively)81. To make Venus-β-ENaC, mVenus was cloned via BamHI/KpnI cloning sites into the human β-ENaC construct. Ven-β-ENaC P617L was created using PCR mutagenesis.
We used Gibson assembly to clone the mVenus fluorescence protein into the C-terminal position of the human leucine-rich repeat-containing G-protein coupled receptor 5 (LGR5; a gift from Toshiro Sato; Addgene plasmid # 129094)82. The Succinate Receptor 1 plasmid construct (SUCCR1) was a gift from Bryan Roth (Addgene plasmid # 66507)83. We generated SUCCR1-mVenus by cloning SUCCR1 into a customised Venus plasmid vector using AgeI and XhoI restriction sites. The Lysophosphatidic Acid Receptor 1 (LPAR1) plasmid was a gift from Bryan Roth (Addgene plasmid # 66418)83. We used Gibson assembly to generate LPAR1-mVenus. For each of LGR5-Venus, SUUCR1-mVenus, and LPAR1-mVenus, the 13-amino acid residue bungarotoxin binding site (BBS; TGGCGGTACTACGAGAGCAGCCTGGAGCCCTACCCCGAC) was introduced at the N-terminus using Gibson assembly.
Cell culture and transfection
Low passage HEK293 cells were cultured in DMEM supplemented with 8% fetal bovine serum (FBS) and 100 mg/mL of penicillin–streptomycin and maintained in humidified incubators at 37 °C and 5% CO2. HEK293 cell transfection was accomplished using the calcium phosphate precipitation method. Briefly, for transfections performed in 12-well plates, 1 µg of each plasmid to be transfected was mixed with 7.75 μL of 2 M CaCl2 and sterile deionized water (to a final volume of 62 μL). The mixture was then added dropwise, with constant tapping, to 62 μL of 2x HEPES buffered saline containing (in mM): HEPES 50, NaCl 280, Na2HPO4 1.5, pH 7.09. The resulting DNA–calcium phosphate mixture was incubated for 20 min at room temperature (RT) and then added dropwise to HEK293 cells (60–80% confluent). Cells were washed with Ca2+-free phosphate buffered saline after 4-6 h and maintained in supplemented DMEM. For other applications, the volume and amounts of transfection reagents were appropriately scaled based on the area of the transfection culture dish.
Chinese hamster ovary (CHO) cells were obtained from ATCC (Manassas, VA), and cultured at 37°C in Kaighn’s Modified Ham’s F-12K (ATCC) supplemented with 8% FBS and 100 mg/mL of penicillin–streptomycin. CHO cells were transfected using the Lipofectamine™ 3000 Transfection Reagent, following the manufacturer’s instructions.
DRG neuron isolation and culture
The isolation of murine dorsal root ganglion (DRG) neurons was performed following the Columbia University Institutional Animal Care and Use Committee guidelines. Adult WT C57BL mice were used in this study. Following decapitation of mice, DRGs were harvested and transferred to Ca2+-free, Mg2+-free Hanks’ balanced salt solution (HBSS; Thermo Fisher Scientific #14170161). Ganglia were treated with collagenase P (1.5 mg/mL; Sigma-Aldrich, #11249002001) in HBSS for 20 min at 37 °C (with gentle shaking), followed by a 2-min incubation at 37 °C (with gentle rotation) in 1 mL warm TrypLE (Thermo Fisher Scientific, #12604013). The majority of TrypLE was removed by pipetting after incubation. Leftover TrypLE was neutralized with culture medium (modified Eagle’s medium [MEM]; Thermo Fisher Scientific, #11095-080) supplemented with 10% horse serum (heat-inactivated; Thermo Fisher Scientific #26050070), 100 U/mL penicillin, 100 μg/mL streptomycin (Thermo Fisher Scientific, #15140122), 1% MEM vitamin solution (Thermo Fisher Scientific, #11120052), and 2% B-27 supplement (Thermo Fisher Scientific, #17504044). The DRGs were left in the tubes vertically for 5 min after which the serum-containing medium was decanted. DRGs were then resuspended with serum-free MEM containing the supplements listed above and triturated using a fire-polished Pasteur pipette. DRG neurons were plated on laminin-treated (0.05 mg/mL) glass coverslips previously rinsed with 70% ethanol and sterilized by UV light exposure for at least 15 min. DRG cultures were then incubated at 37 °C in 5% CO2 for ~3–6 hrs, after which adenoviral vectors (mCherry control; tdTomato-P2a-DiVa nb[F3-C11]; Vector Biolabs (Malvern, PA) were added separately to respective DRG culture wells. The DRG cultures were then maintained at 37 °C in a 5% CO2 humidified incubator and used for electrophysiological experiments at ~48 hrs post-adenoviral infection.
Flow cytometry-based cell surface labeling assay
Cell surface ion channel pools were assayed by flow cytometry in live, transfected HEK293 cells as previously described35. 48 h post-transfection, cells were gently washed with ice-cold phosphate-buffered saline (PBS) containing Ca2+ and Mg2+ (in mM: 0.9 CaCl2, 0.49 MgCl2, pH 7.4), and then incubated for 30 min in blocking medium (DMEM with 3% BSA) at 4 °C. Afterward, the HEK293 cells were incubated with 1 μM Alexa Fluor-647 conjugated α-bungarotoxin (BTX-647; Life Technologies) at 4 °C for 1 hr. Subsequently, the HEK293 cells were washed three times with PBS containing Ca2+ and Mg2+. HEK293 cells were then gently harvested in Ca2+-free PBS, and assayed by flow cytometry using either a BD Fortessa or BD LRSII Cell Analyzer (BD Biosciences, San Jose, CA, USA). CFP, YFP, and mCherry-tagged proteins were excited at 405 nm, 488 nm, and 561 nm, respectively, while Alexa Fluor-647 was excited at 633 nm (on the LRSII) or 640 nm (on the Fortessa).
Electrophysiology
For voltage-gated calcium channel and epithelial sodium channel (ENaC) electrophysiology experiments, whole-cell recordings from HEK293 cells and DRG neurons were conducted at room temperature 48 hrs after transfection using EPC-8 and EPC-10 patch-clamp amplifiers (HEKA Electronics) controlled by pulse software (HEKA). Micropipettes were prepared from 1.5 mm thin-walled glass (World Precision Instruments) using a P97 microelectrode puller (Sutter Instruments). For calcium channel recordings in both HEK293 cells and DRG neurons, the internal solution contained (in mM): 135 cesium methansulfonate (CsMeSO3), 5 CsCl, 5 EGTA, 1 MgCl2, 10 HEPES, and 2 mg/mL MgATP (pH 7.3). Series resistance was typically between 1-2 MΩ. There was no electronic resistance compensation. The external solution contained (in mM): 140 tetraethylammonium-MeSO3, 5 BaCl2, and 10 HEPES (pH 7.4). Whole-cell I-V curves were generated from a family of 20 ms-step depolarizations (-40 mV to +120 mV from an initial 10 ms hold at -90 mV). There was a final repolarization step to -90 mV for 20 ms. Currents were sampled at 20 kHz and filtered at 5 kHz. Traces were acquired at a repetition interval of 10 s. Leak and capacitive transients were subtracted using a P/4 protocol.
Whole-cell measurements of amiloride-sensitive currents were performed in HEK293 cells transfected with the alpha, YFP-tagged beta, and gamma subunits of the epithelial sodium channel (ENaC). The pipette solution was composed of (in mM): 150 KCl, 2 MgCl2, 5 EGTA, and 10 HEPES. The external solution contained (mM): 150 NaCl, 2 MgCl2, 2 CaCl2, 10 HEPES, and 0.01 amiloride. The initial test external solution did not contain amiloride. The current due to ENaC was defined as the amiloride-sensitive current, which is the difference between currents obtained in the absence and presence of 0.01 mM amiloride. Series resistance was typically between 2-3 MΩ. There was no electronic resistance compensation. To define the amiloride-sensitive ENaC currents, a voltage-ramp protocol was first applied to transfected HEK293 cells with the cells initially in an external solution without amiloride. For this voltage-ramp protocol, transfected HEK293 cells were held at +40 mV for 20 ms; followed by a step to +60 mV for 5 ms; then a 500 ms voltage-ramp to -100 mV; and a return to +40 mV holding potential for 20 ms. Currents were sampled at an interval of 1 ms and filtered at 500 Hz with a 2.5 s interval between successive sweeps. After currents became stable, a voltage-step protocol was applied with the cells in the external solution without amiloride. For the voltage-step protocol, the cells were held at +40 mV for 20 ms followed by a family of 500 ms steps from -120 mV to +80 mV in 20-mV increments, and a return to +40 mV holding potential for 20 ms. Currents were sampled at 1 kHz and filtered at 333 Hz with a 2.5 s sweep interval. After the initial recordings in the external solution without amiloride, the voltage-ramp and voltage-step protocols were repeated in the named order for the same cell a second time while perfusing an external solution containing 0.01 mM amiloride. A second voltage step protocol was then conducted in the presence of amiloride.
For electrophysiology recordings of KCNQ1 currents, transfected HEK293 and CHO cells were plated on 3.5 cm culture dishes on the stage of an inverted microscope (OLYMPUS BH2-HLSH, Precision Micro Inc, Massapequa, NY, USA). The external solution contained the following: 132 mM NaCl, 4.8 mM KCl, 2 mM CaCl2, 1.2 mM MgCl2, 10 mM HEPES, and 5 mM glucose (pH was adjusted to 7.4 with NaOH). The internal solution contained the following: 110 mM KCl, 5 mM ATP-K2, 11 mM EGTA, 10 mM HEPES, 1 mM CaCl2, and 1 mM MgCl2 (pH was adjusted to 7.3 with KOH). Pipette series resistance was typically 1.5–3 MΩ when filled with the internal solution. Whole-cell currents were recorded at room temperature using an Axopatch 200B amplifier (Axon Instruments, Foster City, CA, USA). Current-voltage relationship curves were generated using 2-sec depolarizing steps ranging from -60 mV to +100 mV (in 20 mV increments). The currents were triggered every 10 sec from an initial 500 ms hold at -70 mV, and a final repolarizing step to -40 mV for 5 sec. Currents were sampled at 10 kHz and filtered at 5 kHz.
Flow cytometry FRET (Flow-FRET) experiments
The experimental and analysis steps used for the flow cytometry-based fluorescence resonance energy transfer (Flow-FRET) assay have been previously described36,39,84. Briefly, HEK293 cells were transfected using the calcium phosphate transfection method or the polyethylenimine (PEI) transfection method. 1 μg each of cerulean (Cer)- and Venus (Ven)-tagged cDNA pair was mixed in 100 μL of serum-free DMEM media. Afterward, 5 μL of PEI was added to each experimental condition. The DNA-PEI mixture was allowed to sit at room temperature for 15–25 min following which the mixture was added to the cells. FRET experiments were performed ~48 hrs post-transfection. 100 μM cycloheximide was added to cells ≥2 hrs before experimentation to halt the synthesis of new fluorophores and to allow existing fluorophores to fully mature. Cells were gently washed with ice-cold PBS (containing Ca2+ and Mg2+), harvested in Ca2+-free PBS, and assayed by flow cytometry using a BD LSR II Cell Analyzer (BD Biosciences). Cerulean, Venus, and FRET signals were analyzed using the following laser and filter set configurations: BV421 (excitation, 405 nm; emission, 450/50 nm), FITC (excitation, 488 nm; emission, 525/50 nm) and BV520 (excitation, 405 nm; emission, 525/50 nm). Several controls were prepared for each experiment, including untransfected blanks for background subtraction, single-colour Venus and Cerulean for spectral unmixing, Cerulean and Venus co-expressed together for concentration-dependent spurious FRET estimation, as well as a series of Cerulean–Venus dimers for FRET calibration. Custom MATLAB software was used to analyze FRET donor and acceptor efficiency and generate FRET binding curves as a function of the concentrations of free acceptor and donor.
Protein purification for nanobody generation
For NEDD4-2 HECT domain purification, the NEDD4-2 HECT domain plasmid was transformed into Rosetta DE3 E. coli (Millipore Sigma), following manufacturers’ instructions. Cells were grown at 37 °C in 1 L 2xTY media supplemented with 50 μg/mL carbenicillin and 35 μg/mL chloramphenicol and shaken at 225 rpm. Protein expression was induced with 0.2 mM IPTG when the cells reached an OD of 0.6-0.8. The cells were then grown overnight at 22 °C. Cells were lysed using an Avestin® Emulsiflex-C3 homogenizer in a buffer containing 50 mM Tris, 150 mM KCl, 10% sucrose, 1 mM PMSF (phenylmethylsulfonyl fluoride), and EDTA-free cOmplete protease inhibitor cocktail (Roche), pH 7.4. The lysate was spun down at 35,000 g for 1 hr. NEDD4-2 HECT domain was subsequently isolated from the supernatant with anti-FLAG antibody (M2) affinity chromatography and eluted with 100 μg/mL FLAG peptide (Sigma Millipore) in 50 mM TrisHCl, 150 mM KCl, pH 7.4. The protein was then applied to an ion exchange column (MonoQ, GE) and eluted with a linear KCl gradient of 50 mM to 1 M. Peak fractions were collected and subjected to size exclusion chromatography (Superdex 200, GE) in a buffer containing 20 mM Tris, 150 mM KCl, pH 7.4. Proteins were brought to 20% glycerol, flash frozen, and stored at -80 °C.
HECT4-2 nanobody (nb.C11) isolation from yeast nanobody display
We used a synthetic yeast nanobody display library (kind gift from Dr. Andrew Kruse, Harvard University) to isolate nanobody binders to purified HECT4-2 using a previously described protocol38. The naive yeast library was initially grown in 250 mL sterilized ‘Trp/Gal’ media, composed of the following: 0.48 g Trp drop-out media supplement (Sigma Aldrich), 1.675 yeast nitrogen base, 2.6 g sodium citrate, 1.85 g citric acid monohydrate, 2.5 mL Pen/Strep (10,000 units/mL stock), and 5 g galactose, pH 4.5. Cells were incubated for 48 h at 25 °C, 220 rpm to induce nanobody expression. Induced cells were washed and incubated in the following buffer for selection: PBS, 0.1% BSA, and 5 mM maltose. For the first selection, 5 × 109 cells were first subject to a preclear step, to remove nanobodies that bind selection reagents. Cells were incubated with 250 μL anti-FITC microbeads (Miltenyi) and 75 μL FITC-conjugated anti-FLAG M2 antibody (Sigma Aldrich) at 4 °C for 30 min, and then passed through an LD column. These pre-cleared cells were then subjected to a positive selection by incubating the cells with 1 μM HECT4-2 and the same amount of microbeads and anti-FLAG FITC, at 4 °C for 1 hr and then passed through an LS column. The enriched population of yeast was quantified (to approximate the library size) and then grown for 48 h at 30 °C, 220 rpm. Subsequent selections used 10x the number of cells obtained after the previous selection, and 1 μM HECT4-2. After two rounds of MACS selection, the yeast were stained with 1 μM HECT4-2 and sorted into single wells of a 96-well plate using fluorescence-activated cell sorting (FACS). Individual clones were grown and used for binding validation and subsequent plasmid isolation.
Expression and purification of WW4-HECT4-2 domain and full-length NEDD4-2
The WW4-HECT4-2 domain (residues 560-994 of Addgene #2700), corresponding to residues 541-975 in the canonical full-length human NEDD4-2 (cFL-NEDD4-2) sequence (Uniprot id: Q96PU5-1) was cloned into NYCOMPS expression plasmid80,85 via the Gibson assembly method86 with N-terminal Flag-His10 tags followed by a TEV cleavage site (ENLYFQSY) and a C-terminal Flag tag. The plasmid containing WW4-HECT4-2 was transformed into E. coli BL21 (DE3) pLysS (Millipore Sigma). Cells were grown at 37°C, shaken at 280 rpm in 1 L Terrific broth supplemented with 50 μg/ml Carbenicillin, 35 μg/ml chloramphenicol and 0.4% glycerol until OD600 0.5, whereupon expression was induced by the addition of 1 mM IPTG, and cells were grown at 18 °C for a further 18-20 hours. The cells were harvested by centrifugation at 12,300 g for 10 mins at 4°C using Beckman Coulter JLA-10.500 Fixed angle rotor, resuspended in 1x Phosphate Buffered Saline to remove excess media, centrifuged again and stored at -80°C prior until use. Thawed cell pellets were resuspended in His Equilibration buffer (HE) containing 20 mM Tris HCl pH 7.5, 300 mM NaCl, 10% glycerol supplemented with 0.5 mM PMSF, 1 mM TCEP, 1 mM EDTA, 10 μg/mL DNase I and EDTA-free cOmpleteTM protease inhibitor cocktail (Roche) and lysed using Avestin® Emulsiflex-C3 homogenizer. Lysates were clarified by ultracentrifugation at 98,000 g for 45 min at 4 °C in a Type 45.1 Ti rotor (Beckman-Coulter). The supernatants were then passed through a 0.22 μm syringe sterile filter (Thermo-Fisher) before being applied to 10 mL of Ni-NTA XPure Agarose resin (UBPBio) pre-equilibrated with HE buffer without EDTA and with 20 mM imidazole. After the resin was extensively washed with 10 column volumes (CV) of wash buffer (HE + 70 mM imidazole, pH 7.5), the WW4-HECT4-2 protein was eluted with 2–4 column volumes of elution buffer (HE + 350 mM imidazole, pH 7.5). The eluate buffer was exchanged into a buffer containing 20 mM HEPES pH 7.5, 200 mM NaCl, and 10% glycerol using a desalting column to remove imidazole. WW4-HECT4-2 protein was treated with recombinant TEV protease (molar ratio 25:1) for 3 h at room temperature to cleave the N-terminal Flag and His tags. Uncleaved protein and cleaved tag peptide was captured by passing over a 1 mL Ni-NTA XPure Agarose Resin (UBPBio). The cleaved WW4-HECT4-2 protein was concentrated (Amicon Ultra-15 centrifugal filter, 10 kDa cutoff; EMD Millipore), filtered, and further purified by size-exclusion chromatography on a Superdex 200 Increase 10/300 GL column (Cytiva) in 20 mM HEPES pH 7.5, 150 mM NaCl, 1 mM TCEP and 1 mM EDTA. The peak fractions containing WW4-HECT4-2 domain were evaluated for purity by SDS-PAGE, pooled and concentrated to yield 25 mg purified protein per liter of cell culture as evaluated by absorbance at 280 nm, and flash frozen for future use.
For expression and purification of full-length NEDD4-2, 6xHis-TEV-cFL-NEDD4-2 obtained as a gift from Lalit Deshmukh (Addgene plasmid # 186806)87 was transformed into BL21 T7 express (New England Biolabs), and a single clone from the transformant was inoculated into Terrific Broth (TB) with 0.4% glycerol for overnight growth. 5.5 mL of overnight culture was inoculated into 500 mL TB in a 2 L Erlenmeyer flask supplemented with 50 μg/ml carbenicillin and 0.8% glycerol, grown at 37°C with shaking at 280 rpm to OD600 ~ 0.5. The culture was precooled gradually to 18°C before cells were induced with 0.2% arabinose for 20–22 h for cFL-NEDD4-2 protein expression. Cells were harvested and stored at -80°C prior to use as described above for WW4-HECT4-2. The cells were lysed in 20 mM Tris pH 8.0, 250 mM NaCl, 0.5 mM PMSF, 20 μg/ml DNase I, 1 mg/mL Lysozyme, and EDTA-free cOmpleteTM protease inhibitor cocktail (Roche) with Avestin® Emulsiflex-C3 and clarified with ultracentrifugation at 98,000 g for 45 min at 4 °C in a Type 45.1 Ti rotor (Beckman-Coulter). The supernatant was loaded to Ni-NTA XPure Agarose Resin (UBPBio), and bound cFL-NEDD4-2 was washed with 10 CV of lysis buffer without lysozyme, followed by 10 CV of wash buffer (20 mM Tris HCl pH 8.0, 250 mM NaCl + 15 mM imidazole) and eluted in same buffer with 500 mM imidazole. The eluted cFL-NEDD4-2 was concentrated (Amicon Ultra-15 centrifugal filter, 10 kDa cutoff; EMD Millipore), and loaded onto Superdex 200 Increase 10/300 GL column (Cytiva) pre-equilibrated with 50 mM Tris pH 8.0, 150 mM NaCl, 1 mM EDTA, and 1 mM DTT. The peak fractions containing cFL-NEDD4-2 were evaluated for purity by SDS-PAGE and pooled fractions were used in the ubiquitination assays.
nb.C11 expression and purification
Nb.C11 was cloned into a periplasmic expression vector (NYCOMPS) containing an N-terminal pelB leader sequence and a C-terminal hexahistidine tag and expressed in 2 L cultures of E. coli BL21 (DE3) pLysS strain (Millipore Sigma). Expressed nb.C11 was nickel-affinity purified using a previously described protocol38. The nanobody eluted from 5 mL Ni Sepharose 6 Fast Flow resin (Cytiva) in 20 mM HEPES pH7.5, 100 mM NaCl supplemented with 400 mM Imidazole was concentrated to 400 μl and further purified by size-exclusion chromatography on a Superdex 200 Increase 10/300 GL column (Cytiva) in 20 mM HEPES pH 7.5, 150 mM NaCl, 5% glycerol. The peak fractions of the nb.C11 were pooled, concentrated and flash frozen for future use.
Biolayer interferometry
We used biolayer interferometry to measure the binding affinity of nb.C11 (ligand) to purified WW4-HECT(NEDD4-2) (analyte). The binding kinetics of nb.C11 were assessed using the 4-Channel System: Octet® R4 (Sartorius). After an initial equilibration step of 60 sec, HIS-tagged nb.C11 at 50 nmol/L in buffer (20 mM HEPES and 150 mM NaCl), was loaded on HIS1K biosensors (Sartorius) to a final immobilization level of 0.4 nm. The immobilization time for the ligand binding was 100 sec. Binding kinetics were measured against the analyte WW4-HECT-4-2 at concentrations of 7.5, 15, 30, 60, and 120 nmol/L obtained by serial dilutions in buffer (20 mM HEPES, 150 mM NaCl, 1% BSA, and 0.005% Tween-20). The association and dissociation periods are 600 and 900 sec, respectively, at a temperature of 25 °C with the plate under continuous shaking (1000 rpm). Curves were aligned to a baseline immediately before association and reference-subtracted using a non-loaded control sensor run in parallel. A blank (buffer) injection subtraction (“double referencing”) was used to exclude bulk/refractive effects. No smoothing or detrending beyond the software’s default inter-step correction was applied. Data were analyzed using global fitting of a 1:1 Langmuir binding model with Octet Data Analysis software, version 12.0.2.59. Rmax was shared (global) across a sensor set to account for minor loading differences.
WW4-HECT4-2:nb.C11 complex formation and cryo-EM specimen preparation
Size-exclusion chromatography fractions of purified WW4-HECT4-2 protein were mixed with nanobody at a molar ratio of 1:1.5 and incubated for 30 mins on ice. The mixture was subjected to size-exclusion chromatography on a Superdex 200 Increase 10/300 GL column in a buffer containing 20 mM HEPES pH 7.5, 150 mM NaCl, 1 mM TCEP, and 1 mM EDTA. The fractions corresponding to WW4-HECT4-2:nb.C11 complex were validated for purity and the presence of both components by SDS-PAGE. The peak fractions of the complex were then concentrated to ~11 mg/mL (157 μM). The complex was further subjected to mass photometry (Refeyn OneMP) analysis which showed a sharp, 69-kDa molecular weight peak corresponding to the expected molecular weight of the WW4-HECT4-2:nb.C11 complex with a 17 kDa shift from measurement of the WW4-HECT4-2 alone. Right before plunge cooling, 1 μL of 0.3% CHAPS was added to a 6 μL sample of WW4-HECT4-2:nb.C11 complex and homogenously mixed by pipetting so that the final protein and CHAPS concentrations were at 9 mg/mL and 0.043% respectively. 3 μL of the WW4-HECT4-2:nb.C11 complex was applied to the foil side of glow-discharged UltraAuFoil 0.6/1 µm holey gold grids (SPT Labtech), and blotted using 595 filter paper (Ted Pella, Inc) for 9 s with a blot force of 4 and a wait time of 30 sec using a Vitrobot Mark IV (Thermo Fisher Scientific) operated at 4°C and 100% humidity. Each grid was vitrified by plunging into liquid ethane precooled in liquid N2.
EM data acquisition
Cryo-EM data for the WW4-HECT4-2:nb.C11 complex were recorded on a Titan Krios electron microscope (Thermo Fischer Scientific) operated at 300 kV, equipped with Gatan K3 Bio-Quantum with a slit width of 20 eV and a Gatan K3 direct electron detector. The energy filter slit was aligned automatically every hour using Leginon. All the cryo-EM movies were recorded in counting mode and with a nominal magnification of 165,000x which corresponds to a pixel size of 0.5135 Å, calibrated by crystal structure correlation using an apoferritin dataset collected under the same conditions. The data collection was performed using a dose of ~109.2 e-/Å2 88,89,90 across 100 frames at a dose rate of ~16.1 e-/pix/s, corresponding to dose per frame of 1.092 e-/Å2 and using a set defocus range of -1 to -2.5 μm. A 100 µm objective aperture was used. In total, 7,790 micrographs were recorded over a two-day collection.
EM data processing
CryoSPARC v4.4 was used for all processing steps unless otherwise indicated. 7790 movies were imported into CryoSPARC, and dose-weighted and motion corrected with Patch Motion. Patch-based CTF estimation was performed using Patch CTF on the aligned, non-dose-weighted averages followed by manual curation of micrographs, leaving 5742 micrographs for further processing. Blob picker was used to pick particles with 100 Å and 200 Å minimum and maximum particle diameter respectively. 1,631,714 particles were extracted and downsampled from 512 to 200-pixel box size. The particles were subjected to two rounds of 2D classification with 80 online-EM iterations and 5 full iterations. 2D classes with well-defined high-resolution features were selected, leaving 116,543 particles. Ab initio reconstruction was performed using 4 classes, with default settings. This resulted in one unique class with the nb.C11 bound to the HECT domain in the T-state conformation, and three less well-defined classes apparently corresponding to the HECT domain alone. Particles from the four ab initio reconstructions were used as input for heterogeneous refinement, with default settings. The nanobody-bound class from heterogeneous refinement was subjected to non-uniform refinement, yielding a final resolution of 3.95 Å (23,078 particles). Particles were extracted with recentering while downsampling from 512 to 200-pixel box size as before, followed by non-uniform refinement, including on-the-fly refinement of til/trefoil and global refinement of beam tilt and trefoil aberrations yielding an initial consensus refinement resolution of 3.74 Å (22, 904 particles). A Topaz model was trained91 using these 22,904 particles, with a downsampling factor of 8, and an estimated number of particles per micrograph of 50. The resulting model was used to pick particles using Topaz Extract. Heterogenous refinement was performed on the 742,103 particles picked with Topaz using the four ab initio models and four random density decoys, resulting in identification of one good nanobody-bound class of 152,408 particles, which was refined to 3.16 Å using non-uniform refinement. Refinement after splitting into image shift groups further improved resolution to 3.11 Å. We performed 3D classification without alignments on the 152,408 particles using a focused mask around the nanobody, allowing identification of a class with improved density quality for the nanobody. Non-uniform refinement of this class resulted in the final 3.06 Å reconstruction from 90,979 particles. FSC validation was calculated with a mask around the nanobody and another mask around the WW4-HECT4-2 domain with a 12 pix soft edge which allowed for calculating the overall resolution in the nanobody and the HECT domain to be 3.10 Å and 3.16 Å respectively. We then ran orientation diagnostics analysis in CryoSPARC 4.4 on the final reconstruction to analyze the orientation distribution and directional anisotropy of the data, which showed minimal anisotropy92,93. Detailed data collection, refinement and validation statistics are provided in Supplementary Table 1.
Atomic model building and refinement
We used the NEDD4-1 HECT domain crystal structure (PDB: 4BE8) as an initial model and placed it in the corresponding non-uniform refined and sharpened map and fit as rigid body in UCSF Chimera94. For the nanobody, we used a ColabFold predicted structure of the nanobody C1195 as a model and fit as rigid body in UCSF chimera. For the WW4 domain, a previously published NMR structure of NEDD4-2 WW3 domain (PDB: 2MPT)41 was used as an initial model, rigid-body fit in UCSF Chimera, and after combining the three parts of the atomic model, the complete assembly was completed and adjusted by real-space fitting in Coot96,97. Because the EM map did not exhibit any density for the C-terminus tail of HECT domain, and the residues 581-587 of the WW4 domain, these parts were truncated for the final maps of the WW4-HECT4-2:nb.C11 complex. Finally, iterative rounds of real space-refinement of the whole complex were performed in PHENIX v1.21-5207. The atomic model was validated using Molprobity98. A complete summary of data collection and refinement statistics are provided in Supplementary Table 1.
Immunoprecipitation and immunoblotting
Transfected HEK293 cells cultured in 60 mm dishes were harvested in PBS and then centrifuged at 2,000 g (4 °C) for 5 min. The cell pellets were resuspended in RIPA lysis buffer containing (mM): 150 NaCl, 50 Tris HCl, 1 EDTA, 0.1% (w/v) SDS, 1% Triton X-100, 0.5% sodium deoxycholate, and supplemented with protease inhibitor mixture (10 μL/mL, Sigma Aldrich), 1 PMSF, 2 N-ethylmaleimide, 0.05 PR-619 deubiquitinase inhibitor (LifeSensors). Cells were lysed on ice for 1 hr with intermittent vortexing and then centrifuged at 10,000 x g for 10 min at 4 °C. The cell lysate was collected following which protein concentration was determined with the bicinchoninic acid (BCA) protein estimation kit (Pierce Technologies).
For KCNQ1 pulldowns, cell lysates were precleared with 10 μL of protein A/G Sepharose beads (Rockland) for 1 hr at 4 °C and then incubated with 1 μg anti-KCNQ1 antibody (Alomone Labs, Israel) for 1 hr at 4 °C. Equivalent amounts of protein were then added to spin columns with 25 μL equilibrated protein A/G Sepharose beads and rotated overnight at 4 °C. Immunoprecipitates were washed five times with RIPA buffer and then eluted with 40 μL elution buffer (50 mM Tris, 10% (vol/vol) glycerol, 2% SDS, 100 mM DTT, and 0.2 mg/mL bromophenol blue) at 55 °C for 15 min. Proteins were resolved on a 4-12% Bis-Tris gradient precast gel (Life Technologies) in MOPS-SDS running buffer (Life Technologies) at 200 V constant for ~1 h. Protein bands were transferred by tank transfer onto a polyvinylidene difluoride (PVDF, EMD Millipore) membrane in transfer buffer (25 mM Tris pH 8.3, 192 mM glycine, 15% (vol/vol) methanol, and 0.1% SDS). The membranes were blocked with a solution of 5% non-fat dry milk (BioRad) in tris-buffered saline-Tween20 (TBS-T) (25 mM Tris pH 7.4, 150 mM NaCl, and 0.1% Tween-20) for 1 hr at RT and then incubated overnight at 4 °C with primary anti-KCNQ1 (Alomone Labs) antibody in blocking solution. The blots were washed with TBS-T three times for 10 min per wash and then incubated with a goat anti-rabbit secondary horseradish peroxidase-conjugated antibody (Thermo Fisher Scientific) for 1 hr at RT. After washing in TBS-T, the blots were developed with a chemiluminescent detection kit (Pierce Technologies) and then visualized on a gel imager. Membranes were then stripped with harsh stripping buffer (2% SDS, 62 mM Tris pH 6.8, 0.8% ß-mercaptoethanol) at 50 °C for 30 min, rinsed under running water for 2 min, and washed with TBST (3 times, 10 min per wash). Membranes were pre-treated with 0.5% glutaraldehyde and re-blotted with anti-ubiquitin (VU1, LifeSensors) according to the manufacturers’ instructions.
For immuno-detection of total protein fractions, equal amounts of proteins from each treatment group were loaded. Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), gel transfer, and immunoblotting were performed as described above, but with appropriate antibodies for the detection of KCNQ1 and actin.
For CaV2.2 co-immunoprecipitation studies, HEK293 cells cultured in 10 cm plates were co-transfected with mCherry-tagged α1B (10 µg), YFP-β2a (10 µg), α2δ1 (10 µg) and either CFP (10 µg), CFP-P2A-nb.F3 (10 µg), CFP-P2A-nb.C11 (10 µg), CFP-P2A-DiVa nb[C11-F3], or CFP-P2A-NEDD4-2 (10 µg). ~48 h post-transfection, the RFP-Trap agarose kit (ChromoTek) was used to pulldown α1B-mCherry in complex with the auxiliary subunits using the manufacturer’s protocol. SDS-PAGE and Western blotting were performed using a similar protocol as described above for KCNQ1.
In vitro ubiquitination assay
To monitor the time course for in vitro ubiquitination/autoubiquitination of WW4-HECT4-2 or cFL-NEDD4-2, 500 nM of purified human WW4-HECT4-2 cleaved of its N- and C-terminal tags, or 500 nM of purified cFL-NEDD4-2 in a base buffer (20 mM HEPES pH 7.5, 150 mM NaCl) was mixed with 5 mM MgCl2, 5 mM ATP, 80 nM UBE1 (E1 enzyme, Boston Biochem), 500 nM UBCH5C (E2 enzyme, Boston Biochem) on ice. Lastly, 6 μM ubiquitin (Boston Biochem) was added to initiate autoubiquitination reaction at room temperature and a 17 μL aliquot was immediately taken from the master mix for each of the different time intervals (0, 5, 10, 15, 20, 25, 30, 45, 60, 80,100, 120 mins). The reaction was quenched with 2x SDS sample buffer containing 715 mM β-mercaptoethanol at the indicate times and the reaction mixtures were separated by SDS/PAGE and analyzed by immunoblotting with pan-ubiquitin antibody (Biorad, P4D1) to detect ubiquitin chains, or with either anti-HECT4-2 or anti-WW4-HECT4-2 to detect ubiquitinated and non-ubiquitinated WW4-HECT4-2 and cFL-NEDD4-2 forms.
To assess the impact of nb.C11 on either WW4-HECT4-2 or cFL-NEDD4-2 catalytic activity, 500 nM of purified human WW4-HECT4-2 or cFL-NEDD4-2 was incubated with or without 50 μM purified nb.C11 in a base buffer (20 mM HEPES pH 7.5, 150 mM NaCl) on ice for 1 h. After this incubation period, 5 mM MgCl2, 5 mM ATP, 80 nM E1, 500 nM E2 and 6 μM ubiquitin was added to the mixture (30 μL total) to initiate the ubiquitination reaction. The reaction was allowed to proceed for 30 mins at room temperature and quenched by addition of 2x SDS sample buffer containing 715 mM β-mercaptoethanol. For mock control, purified human WW4-HECT4-2 or cFL-NEDD4-2 was kept in the base buffer without addition of E1, E2, and ubiquitin. Reactions were analyzed by immunoblotting and probing with anti-ubiquitin (Biorad, P4D1), to detect ubiquitin chains or with anti-WW4-HECT4-2 to detect ubiquitinated and non-ubiquitinated forms of WW4-HECT4-2 and cFL-NEDD4-2, respectively.
Confocal microscopy
For confocal microscopy, freshly isolated murine DRG neurons were cultured on 12 mm diameter glass coverslips. The glass coverslips were placed within 35 mm tissue culture plates containing DRG culture media (described under the DRG culture section). Five days post-adenovirus infection, cultured DRG neurons were washed once in PBS and then fixed with 4% paraformaldehyde at room temperature (RT) for 15 min. The fixed DRG neurons were washed three times with 0.1 M glycine in PBS following which the glass coverslips containing the fixed DRG neurons were placed on glass microscope slides and mounted in 4′,6-diamidino-2-phenylindole (DAPI) stain. Images were captured with a Nikon Eclipse Ti2 inverted microscope equipped with 405 nm, 488 nm, 561 nm, 638 nm lasers and run by NIS-Elements AR 5.02.00 64-bit software. Images were detected using the Nikon PLAN APO λD 20X/0.80 objective and analyzed using Fiji software.
Florescence-activated cell sorting (FACS)
For fluorescence-activated cell sorting (FACS), low passage HEK293 cells were cultured on 10 cm tissue culture plates and grown in DMEM supplemented with 8% fetal bovine serum (FBS) and 100 mg/mL of penicillin–streptomycin. The cells were maintained in humidified incubators at 37 °C and 5% CO2. HEK293 cells were transiently co-transfected with YFP-tagged KCNQ1 (10 µg) and one of the following: CFP-P2a-nb.YFP (10 µg); or CFP-P2a-NEDD4-2 (10 µg); or CFP-P2a-DiVa[nb.YFP-nb.C11] (10 µg). FACS was done ~48 hrs post-transfection. In preparation for FACS, adherent HEK293 cells were dissociated from the respective cell culture plates using TrypLE; re-suspended in 2% heat-inactivated FBS in phosphate-buffered saline (PBS); passed through separate 40 μm filters; and kept on ice before sorting. FACS was carried out on either a BD FACSAria II or BD Influx cell sorter each equipped with 405 nm, 488 nm, 561 nm, and 638 nm excitation lasers. For cell sorting, untransfected, YFP-only expressing, and CFP-only expressing HEK293 cells were used to define the region of interest. HEK293 cells expressing both YFP (positive for KCNQ1-YFP) and CFP (positive for anti-YFP-nanobody, NEDD4-2 or DiVa) were collected after sorting. ~1 million cells were sorted for each group with three to four independent transfections. Immediately following sorting, the sorted cells were re-suspended in 10% FBS in PBS and kept on ice. Afterward, the cells were washed twice in PBS by centrifugation to remove residual FBS. The cell pellets were stored at -80oC before and during transport for mass spectrometry studies.
Sample preparation for mass spectrometry
Mass spectrometry sample preparation and experiments were performed at the Thermo Fisher Center for Multiplexed Proteomics at Harvard Medical School (HMS-TCMP). Samples for protein analysis were prepared essentially as previously described99,100. Proteomes were extracted using a buffer containing 200 mM EPPS pH 8.5, 8 M urea, 0.1% SDS and protease inhibitors. Following lysis, each sample was reduced with 5 mM TCEP. Cysteine residues were alkylated using 10 mM iodoacetamide for 20 min at RT in the dark. Excess iodoacetimide was quenched with 10 mM DTT. A buffer exchange was carried out using a modified SP3 protocol101. Briefly, ~250 µg of Cytiva SpeedBead Magnetic Carboxylate Modified Particles (65152105050250 and 4515210505250), mixed at a 1:1 ratio, were added to each sample. 100% ethanol was added to each sample to achieve a final ethanol concentration of at least 50%. Samples were incubated with gentle shaking for 15 mins. Samples were washed three times with 80% ethanol. Protein was eluted from SP3 beads using 200 mM EPPS pH 8.5 containing trypsin (ThermoFisher Scientific, 90305R20) and Lys-C (Wako, 129-02541). Samples were digested overnight at 37 °C with vigorous shaking. Acetonitrile was added to each sample to achieve a final concentration of ~33%. Each sample was labeled, in the presence of SP3 beads, with ~60 µg of TMT 11plex reagents (ThermoFisher Scientific). Following confirmation of satisfactory labeling ( > 97%), excess TMT was quenched by the addition of hydroxylamine to a final concentration of 0.3%. The full volume from each sample was pooled and acetonitrile was removed by vacuum centrifugation for 1 h. The pooled sample was acidified using formic acid and peptides were de-salted using a Sep-Pak 50 mg tC18 cartridge (Waters). Peptides were eluted in 70% acetonitrile, 1% formic acid and dried by vacuum centrifugation.
Basic pH reversed-phase separation (BPRP)
TMT labeled peptides were solubilized in 5% ACN/10 mM ammonium bicarbonate, pH 8.0 and ~300 µg of TMT labeled peptides were separated by an Agilent 300 Extend C18 column (3.5 μm particles, 4.6 mm ID and 250 mm in length). An Agilent 1260 binary pump coupled with a photodiode array (PDA) detector (Thermo Scientific) was used to separate the peptides. A 45-min linear gradient from 10% to 40% acetonitrile in 10 mM ammonium bicarbonate pH 8.0 (flow rate of 0.6 mL/min) separated the peptide mixtures into a total of 96 fractions (36 sec). A total of 96 Fractions were consolidated into 24 samples in a checkerboard fashion and vacuum-dried to completion. Each sample was desalted via Stage Tips and re-dissolved in 5% FA/ 5% ACN for LC-MS3 analysis.
Liquid chromatography separation and tandem mass spectrometry (LC-MS3)
Proteome data were collected on an Orbitrap Fusion Lumos mass spectrometer (ThermoFisher Scientific) coupled to a Proxeon EASY-nLC 1000 LC pump (ThermoFisher Scientific). Fractionated peptides were separated using a 120 min gradient at 550 nL/min on a 35 cm column (i.d. 100 μm, Accucore, 2.6 μm, 150 Å) packed in-house. MS1 data were collected in the Orbitrap (60,000 resolution; maximum injection time 50 ms; AGC 4 × 105). Charge states between 2 and 6 were required for MS2 analysis, and a 120 s dynamic exclusion window was used. Top 10 MS2 scans were performed in the ion trap with CID fragmentation (isolation window 0.5 Da; Rapid; NCE 35%; maximum injection time 50 ms; AGC 2 × 104). An on-line real-time search algorithm (Orbiter) was used to trigger MS3 scans for quantification102. MS3 scans were collected in the Orbitrap using a resolution of 50,000, NCE of 55%, maximum injection time of 200 ms, and AGC of 3.0 × 105. The close out was set at two peptides per protein per fraction.
Data analysis
Raw files were converted to mzXML, and monoisotopic peaks were re-assigned using Monocle103. Searches were performed using the Comet search algorithm against a human database downloaded from Uniprot in May 2021. A 50 ppm precursor ion tolerance, 1.0005 fragment ion tolerance, and 0.4 fragment bin offset for MS2 scans were collected in the ion trap. TMT on lysine residues and peptide N-termini ( + 229.1629 Da) and carbamidomethylation of cysteine residues ( + 57.0215 Da) were set as static modifications, while oxidation of methionine residues ( + 15.9949 Da) was set as a variable modification. Each run was filtered separately to a 1% False Discovery Rate (FDR) on the peptide-spectrum match (PSM) level. Then proteins were filtered to the target 1% FDR level across the entire combined data set. For reporter ion quantification, a 0.003 Da window around the theoretical m/z of each reporter ion was scanned, and the most intense m/z was used. Reporter ion intensities were adjusted to correct for isotopic impurities of the different TMTpro reagents according to manufacturer specifications. Peptides were filtered to include only those with a summed signal-to-noise (SN) ≥ 110 across all TMT channels. The signal-to-noise (S/N) measurements of peptides assigned to each protein were summed (for a given protein). These values were normalized so that the sum of the signal for all proteins in each channel was equivalent thereby accounting for equal protein loading.
Data and statistical analyses
Data were analyzed off-line using HEKA FITMaster (v2x73.5), FlowJo (vl0.6.0), Microsoft Excel (v16.27), MATLAB, and GraphPad Prism 7 software. Statistical analyses were performed in GraphPad Prism and Microsoft Excel using built-in functions. Statistically significant differences between means (set at P < 0.05) were evaluated using specified statistical tests. Data are presented as means ± SEM.
Ethical statement
This study uses adult dorsal root ganglia neurons isolated from mice performed in accordance with the guidelines and approval of the Institutional Animal Care and Use Committee at Columbia University (Protocol # AC-AABU2659).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Plasmid and viral constructs for non-commercial purposes can be obtained by request from the corresponding author. The authors declare that all data supporting the results of this study can be found in the article, supplementary, and source data files. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD069051. The cryoEM density maps and structural models for nb.C11 in complex with NEDD4-2 HECT domain have been deposited in the Electron Microscopy Data Bank (EMDB) and the Protein Data Bank (PDB) respectively, with accession codes EMD-73821 and 9Z5Q. Source data are provided with this paper.
References
Stanton, B. Z., Chory, E. J. & Crabtree, G. R. Chemically induced proximity in biology and medicine. Science https://doi.org/10.1126/science.aao5902 (2018).
Zhao, L., Zhao, J., Zhong, K., Tong, A. & Jia, D. Targeted protein degradation: mechanisms, strategies and application. Signal Transduct. Target Ther. 7, 113 (2022).
Bekes, M., Langley, D. R. & Crews, C. M. PROTAC targeted protein degraders: the past is prologue. Nat. Rev. Drug Discov. 21, 181–200 (2022).
Sakamoto, K. M. et al. Protacs: chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation. Proc. Natl Acad. Sci. USA 98, 8554–8559 (2001).
Lu, G. et al. The myeloma drug lenalidomide promotes the cereblon-dependent destruction of Ikaros proteins. Science 343, 305–309 (2014).
Kronke, J. et al. Lenalidomide causes selective degradation of IKZF1 and IKZF3 in multiple myeloma cells. Science 343, 301–305 (2014).
Fischer, E. S. et al. Structure of the DDB1-CRBN E3 ubiquitin ligase in complex with thalidomide. Nature 512, 49–53 (2014).
Salami, J. et al. Androgen receptor degradation by the proteolysis-targeting chimera ARCC-4 outperforms enzalutamide in cellular models of prostate cancer drug resistance. Commun. Biol. 1, 100 (2018).
He, Y. et al. DT2216-a Bcl-xL-specific degrader is highly active against Bcl-xL-dependent T cell lymphomas. J. Hematol. Oncol. 13, 95 (2020).
Chu, T. T. et al. Specific knockdown of endogenous tau protein by peptide-directed ubiquitin-proteasome degradation. Cell Chem. Biol. 23, 453–461 (2016).
Mullard, A. Targeted protein degraders crowd into the clinic. Nat. Rev. Drug Discov. 20, 247–250 (2021).
Zheng, N. & Shabek, N. Ubiquitin ligases: structure, function, and regulation. Annu Rev. Biochem 86, 129–157 (2017).
Zhang, L., Riley-Gillis, B., Vijay, P. & Shen, Y. Acquired resistance to BET-PROTACs (Proteolysis-Targeting Chimeras) caused by genomic alterations in core components of E3 ligase complexes. Mol. Cancer Ther. 18, 1302–1311 (2019).
Shirasaki, R. et al. Functional genomics identify distinct and overlapping genes mediating resistance to different classes of heterobifunctional degraders of oncoproteins. Cell Rep. 34, 108532 (2021).
Ackerman, M. J. & Clapham, D. E. Ion channels-basic science and clinical disease. N. Engl. J. Med. 336, 1575–1586 (1997).
Subramanyam, P. & Colecraft, H. M. Ion channel engineering: perspectives and strategies. J. Mol. Biol. 427, 190–204 (2015).
Waszkielewicz, A. M. et al. Ion channels as drug targets in central nervous system disorders. Curr. Med Chem. 20, 1241–1285 (2013).
Banik, S. M. et al. Lysosome-targeting chimaeras for degradation of extracellular proteins. Nature 584, 291–297 (2020).
Cotton, A. D., Nguyen, D. P., Gramespacher, J. A., Seiple, I. B. & Wells, J. A. Development of antibody-based PROTACs for the degradation of the cell-surface immune checkpoint protein PD-L1. J. Am. Chem. Soc. 143, 593–598 (2021).
Marei, H. et al. Antibody targeting of E3 ubiquitin ligases for receptor degradation. Nature 610, 182–189 (2022).
Zhang, D. et al. Transferrin receptor targeting chimeras for membrane protein degradation. Nature, https://doi.org/10.1038/s41586-024-07947-3 (2024).
Colecraft, H. M. & Trimmer, J. S. Controlling ion channel function with renewable recombinant antibodies. J. Physiol. 600, 2023–2036 (2022).
Foot, N., Henshall, T. & Kumar, S. Ubiquitination and the regulation of membrane proteins. Physiol. Rev. 97, 253–281 (2017).
Ingham, R. J., Gish, G. & Pawson, T. The Nedd4 family of E3 ubiquitin ligases: functional diversity within a common modular architecture. Oncogene 23, 1972–1984 (2004).
He, X. et al. Covalent recruitment of NEDD4 for targeted protein degradation: rational design of small molecular degraders. J. Am. Chem. Soc. 147, 21512–21525 (2025).
Debonneville, C. et al. Phosphorylation of Nedd4-2 by Sgk1 regulates epithelial Na(+) channel cell surface expression. EMBO J. 20, 7052–7059 (2001).
Snyder, P. M., Olson, D. R., Kabra, R., Zhou, R. & Steines, J. C. cAMP and serum and glucocorticoid-inducible kinase (SGK) regulate the epithelial Na(+) channel through convergent phosphorylation of Nedd4-2. J. Biol. Chem. 279, 45753–45758 (2004).
Pohl, P., Joshi, R., Petrvalska, O., Obsil, T. & Obsilova, V. 14-3-3-protein regulates Nedd4-2 by modulating interactions between HECT and WW domains. Commun. Biol. 4, 899 (2021).
Rotin, D. & Staub, O. Nedd4-2 and the regulation of epithelial sodium transport. Front Physiol. 3, 212 (2012).
Ma, Y. et al. Targeted degradation of KRAS by an engineered ubiquitin ligase suppresses pancreatic cancer cell growth in vitro and in vivo. Mol. Cancer Ther. 12, 286–294 (2013).
Zhou, P., Bogacki, R., McReynolds, L. & Howley, P. M. Harnessing the ubiquitination machinery to target the degradation of specific cellular proteins. Mol. Cell 6, 751–756 (2000).
Caussinus, E., Kanca, O. & Affolter, M. Fluorescent fusion protein knockout mediated by anti-GFP nanobody. Nat. Struct. Mol. Biol. 19, 117–121 (2011).
Hatakeyama, S., Watanabe, M., Fujii, Y. & Nakayama, K. I. Targeted destruction of c-Myc by an engineered ubiquitin ligase suppresses cell transformation and tumor formation. Cancer Res. 65, 7874–7879 (2005).
Kanner, S. A., Morgenstern, T. & Colecraft, H. M. Sculpting ion channel functional expression with engineered ubiquitin ligases. Elife https://doi.org/10.7554/eLife.29744 (2017).
Morgenstern, T. J., Park, J., Fan, Q. R. & Colecraft, H. M. A potent voltage-gated calcium channel inhibitor engineered from a nanobody targeted to auxiliary Ca(V)beta subunits. Elife https://doi.org/10.7554/eLife.49253 (2019).
Morgenstern, T. J. et al. Selective posttranslational inhibition of Ca(V)beta(1)-associated voltage-dependent calcium channels with a functionalized nanobody. Nat. Commun. 13, 7556 (2022).
Zhang, W. et al. System-wide modulation of HECT E3 ligases with selective ubiquitin variant probes. Mol. Cell 62, 121–136 (2016).
McMahon, C. et al. Yeast surface display platform for rapid discovery of conformationally selective nanobodies. Nat. Struct. Mol. Biol. 25, 289–296 (2018).
Lee, S. R., Sang, L. & Yue, D. T. Uncovering aberrant mutant PKA function with flow cytometric FRET. Cell Rep. 14, 3019–3029 (2016).
Mari, S. et al. Structural and functional framework for the autoinhibition of Nedd4-family ubiquitin ligases. Structure 22, 1639–1649 (2014).
Escobedo, A. et al. Structural basis of the activation and degradation mechanisms of the E3 ubiquitin ligase Nedd4L. Structure 22, 1446–1457 (2014).
Wang, Z. et al. A multi-lock inhibitory mechanism for fine-tuning enzyme activities of the HECT family E3 ligases. Nat. Commun. 10, 3162 (2019).
Kanelis, V., Rotin, D. & Forman-Kay, J. D. Solution structure of a Nedd4 WW domain-ENaC peptide complex. Nat. Struct. Biol. 8, 407–412 (2001).
Bobby, R. et al. Structure and dynamics of human Nedd4-1 WW3 in complex with the alphaENaC PY motif. Biochim Biophys. Acta 1834, 1632–1641 (2013).
Maspero, E. et al. Structure of the HECT:ubiquitin complex and its role in ubiquitin chain elongation. EMBO Rep. 12, 342–349 (2011).
Kamadurai, H. B. et al. Insights into ubiquitin transfer cascades from a structure of a UbcH5B approximately ubiquitin-HECT(NEDD4L) complex. Mol. Cell 36, 1095–1102 (2009).
Zondlo, N. J. Aromatic-proline interactions: electronically tunable CH/pi interactions. Acc. Chem. Res. 46, 1039–1049 (2013).
Huang, L. et al. Structure of an E6AP-UbcH7 complex: insights into ubiquitination by the E2-E3 enzyme cascade. Science 286, 1321–1326 (1999).
Janosev, M. et al. Structural basis of ubiquitin ligase Nedd4-2 autoinhibition and regulation by calcium and 14-3-3 proteins. Nat. Commun. 16, 4875 (2025).
Wright, K. M. et al. NEDD4L intramolecular interactions regulate its auto and substrate Na(V)1.5 ubiquitination. J. Biol. Chem. 300, 105715 (2024).
Bruce, M. C. et al. Regulation of Nedd4-2 self-ubiquitination and stability by a PY motif located within its HECT-domain. Biochem J. 415, 155–163 (2008).
Laedermann, C. J. et al. Dysregulation of voltage-gated sodium channels by ubiquitin ligase NEDD4-2 in neuropathic pain. J. Clin. Invest 123, 3002–3013 (2013).
Jespersen, T., Grunnet, M. & Olesen, S. P. The KCNQ1 potassium channel: from gene to physiological function. Physiol. (Bethesda) 20, 408–416 (2005).
Jespersen, T. et al. The KCNQ1 potassium channel is down-regulated by ubiquitylating enzymes of the Nedd4/Nedd4-like family. Cardiovasc Res. 74, 64–74 (2007).
Kubala, M. H., Kovtun, O., Alexandrov, K. & Collins, B. M. Structural and thermodynamic analysis of the GFP:GFP-nanobody complex. Protein Sci. 19, 2389–2401 (2010).
Hanukoglu, I. & Hanukoglu, A. Epithelial sodium channel (ENaC) family: phylogeny, structure-function, tissue distribution, and associated inherited diseases. Gene 579, 95–132 (2016).
Noreng, S., Bharadwaj, A., Posert, R., Yoshioka, C. & Baconguis, I. Structure of the human epithelial sodium channel by cryo-electron microscopy. Elife https://doi.org/10.7554/eLife.39340 (2018).
Abriel, H. et al. Defective regulation of the epithelial Na+ channel by Nedd4 in Liddle’s syndrome. J. Clin. Invest 103, 667–673 (1999).
Hansson, J. H. et al. Hypertension caused by a truncated epithelial sodium channel gamma subunit: genetic heterogeneity of Liddle syndrome. Nat. Genet 11, 76–82 (1995).
Shimkets, R. A. et al. Liddle’s syndrome: heritable human hypertension caused by mutations in the beta subunit of the epithelial sodium channel. Cell 79, 407–414 (1994).
Rossi, E. et al. Liddle’s syndrome caused by a novel missense mutation (P617L) of the epithelial sodium channel beta subunit. J. Hypertens. 26, 921–927 (2008).
Santos, R. et al. A comprehensive map of molecular drug targets. Nat. Rev. Drug Discov. 16, 19–34 (2017).
Hutchings, C. J., Colussi, P. & Clark, T. G. Ion channels as therapeutic antibody targets. MAbs 11, 265–296 (2019).
Kaczorowski, G. J., McManus, O. B., Priest, B. T. & Garcia, M. L. Ion channels as drug targets: the next GPCRs. J. Gen. Physiol. 131, 399–405 (2008).
Yang, Q., Zhao, J., Chen, D. & Wang, Y. E3 ubiquitin ligases: styles, structures and functions. Mol. Biomed. 2, 23 (2021).
Sluimer, J. & Distel, B. Regulating the human HECT E3 ligases. Cell Mol. Life Sci. 75, 3121–3141 (2018).
Bongiorno, D., Schuetz, F., Poronnik, P. & Adams, D. J. Regulation of voltage-gated ion channels in excitable cells by the ubiquitin ligases Nedd4 and Nedd4-2. Channels (Austin) 5, 79–88 (2011).
Poirson, J. et al. Proteome-scale discovery of protein degradation and stabilization effectors. Nature, https://doi.org/10.1038/s41586-024-07224-3 (2024).
Scheffner, M. & Kumar, S. Mammalian HECT ubiquitin-protein ligases: biological and pathophysiological aspects. Biochim Biophys. Acta 1843, 61–74 (2014).
Hehl, L. A. et al. Structural snapshots along K48-linked ubiquitin chain formation by the HECT E3 UBR5. Nat. Chem. Biol. 20, 190–200 (2024).
Kim, H. C. & Huibregtse, J. M. Polyubiquitination by HECT E3s and the determinants of chain type specificity. Mol. Cell Biol. 29, 3307–3318 (2009).
MacGurn, J. A., Hsu, P. C. & Emr, S. D. Ubiquitin and membrane protein turnover: from cradle to grave. Annu Rev. Biochem 81, 231–259 (2012).
Shanmugam, S. K. et al. Decoding polyubiquitin regulation of K(V)7. 1 (KCNQ1) surface expression with engineered linkage-selective deubiquitinases. Nat. Commun. 16, 5805 (2025).
Persaud, A. et al. Comparison of substrate specificity of the ubiquitin ligases Nedd4 and Nedd4-2 using proteome arrays. Mol. Syst. Biol. 5, 333 (2009).
Pardon, E. et al. A general protocol for the generation of Nanobodies for structural biology. Nat. Protoc. 9, 674–693 (2014).
Hie, B. L. et al. Efficient evolution of human antibodies from general protein language models. Nat. Biotechnol. 42, 275–283 (2024).
Li, L. et al. Machine learning optimization of candidate antibody yields highly diverse sub-nanomolar affinity antibody libraries. Nat. Commun. 14, 3454 (2023).
Colecraft, H. M. Designer genetically encoded voltage-dependent calcium channel inhibitors inspired by RGK GTPases. J. Physiol. 598, 1683–1693 (2020).
Gao, S. et al. Ubiquitin ligase Nedd4L targets activated Smad2/3 to limit TGF-beta signaling. Mol. Cell 36, 457–468 (2009).
Mancia, F. & Love, J. High throughput platforms for structural genomics of integral membrane proteins. Curr. Opin. Struct. Biol. 21, 517–522 (2011).
Raikwar, N. S. & Thomas, C. P. Nedd4-2 isoforms ubiquitinate individual epithelial sodium channel subunits and reduce surface expression and function of the epithelial sodium channel. Am. J. Physiol. Ren. Physiol. 294, F1157–F1165 (2008).
Shimokawa, M. et al. Visualization and targeting of LGR5(+) human colon cancer stem cells. Nature 545, 187–192 (2017).
Kroeze, W. K. et al. PRESTO-Tango as an open-source resource for interrogation of the druggable human GPCRome. Nat. Struct. Mol. Biol. 22, 362–369 (2015).
Kanner, S. A., Shuja, Z., Choudhury, P., Jain, A. & Colecraft, H. M. Targeted deubiquitination rescues distinct trafficking-deficient ion channelopathies. Nat. Methods 17, 1245–1253 (2020).
Bruni, R. & Kloss, B. High-throughput cloning and expression of integral membrane proteins in Escherichia coli. Curr. Protoc. Protein Sci. 74, 29 26 21–29 26 34 (2013).
Gibson, D. G. et al. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 6, 343–345 (2009).
Nelson, S. L., Li, Y., Chen, Y. & Deshmukh, L. Avidity-based method for the efficient generation of monoubiquitinated recombinant proteins. J. Am. Chem. Soc. 145, 7748–7752 (2023).
Grant, T. & Grigorieff, N. Measuring the optimal exposure for single particle cryo-EM using a 2.6 A reconstruction of rotavirus VP6. Elife 4, e06980 (2015).
Greber, B. J. et al. High-resolution cryo-EM of a small protein complex: The structure of the human CDK-activating kinase. Structure, https://doi.org/10.1016/j.str.2024.03.003 (2024).
Sun, M. et al. Practical considerations for using K3 cameras in CDS mode for high-resolution and high-throughput single particle cryo-EM. J. Struct. Biol. 213, 107745 (2021).
Bepler, T. et al. Positive-unlabeled convolutional neural networks for particle picking in cryo-electron micrographs. Nat. Methods 16, 1153–1160 (2019).
Baldwin, P. R. & Lyumkis, D. Non-uniformity of projection distributions attenuates resolution in Cryo-EM. Prog. Biophys. Mol. Biol. 150, 160–183 (2020).
Tan, Y. Z. et al. Addressing preferred specimen orientation in single-particle cryo-EM through tilting. Nat. Methods 14, 793–796 (2017).
Pettersen, E. F. et al. UCSF Chimera-a visualization system for exploratory research and analysis. J. Comput Chem. 25, 1605–1612 (2004).
Mirdita, M. et al. ColabFold: making protein folding accessible to all. Nat. Methods 19, 679–682 (2022).
Casanal, A., Lohkamp, B. & Emsley, P. Current developments in Coot for macromolecular model building of Electron Cryo-microscopy and Crystallographic Data. Protein Sci. 29, 1069–1078 (2020).
Emsley, P. & Cowtan, K. Coot: model-building tools for molecular graphics. Acta Crystallogr D. Biol. Crystallogr 60, 2126–2132 (2004).
Williams, C. J. et al. MolProbity: more and better reference data for improved all-atom structure validation. Protein Sci. 27, 293–315 (2018).
Li, J. et al. TMTpro-18plex: the expanded and complete set of TMTpro reagents for sample multiplexing. J. Proteome Res 20, 2964–2972 (2021).
Navarrete-Perea, J., Yu, Q., Gygi, S. P. & Paulo, J. A. Streamlined tandem mass tag (SL-TMT) protocol: an efficient strategy for quantitative (Phospho)proteome profiling using tandem mass tag-synchronous precursor selection-MS3. J. Proteome Res. 17, 2226–2236 (2018).
Hughes, C. S. et al. Single-pot, solid-phase-enhanced sample preparation for proteomics experiments. Nat. Protoc. 14, 68–85 (2019).
Schweppe, D. K. et al. Full-featured, real-time database searching platform enables fast and accurate multiplexed quantitative proteomics. J. Proteome Res. 19, 2026–2034 (2020).
Rad, R. et al. Improved monoisotopic mass estimation for deeper proteome coverage. J. Proteome Res. 20, 591–598 (2021).
Acknowledgements
We thank Ming Chen for technical support; Dr. Chi-Kun Tong for help with DRG neuron isolations; Kookjoo Kim for technical support and assistance with figure preparation; and Mary Smith (Barnard College) for helping with in vitro ubiquitination experiments. This work was supported by grants R21-NS130409, RO1-HL121253, RO1-HL142111, R01-NS126850 and P01-HL164319 from the NIH (to HMC); F31 DK118866 (to T.J.M); AHA predoctoral fellowship 20PRE35210815 (to ADB); and AHA postdoctoral fellowship POST1019343 (to SKS). Flow cytometry experiments were performed in the CCTI Flow Cytometry Core, supported in part by the NIH (S10RR027050). Confocal images were collected in the HICCC Confocal and Specialized Microscopy Shared Resource, supported by NIH (P30 CA013696). Cryo-EM data were collected at the Columbia University Cryo-Electron Microscopy Center, with technical support from Robert A. Grassucci and Zhening Zhang.
Author information
Authors and Affiliations
Contributions
A.D.B., E.A., T.J.M., O.B.C., H.M.C. designed experiments; A.D.B., E.A., T.J.M., S.K.S., X.Z., Y.D.L., P.C., M.D., F.V. performed experiments; A.D.B., E.A., T.J.M., S.K.S., X.Z., M.D., F.V., O.B.C., H.M.C. analyzed data; A.D.B., E.A., T.J.M., O.B.C., H.M.C. wrote the paper; T.J.M., A.D.B., E.A., R.S.K., F.V., O.B.C., H.M.C. edited the paper; R.S.K., O.B.C., H.M.C. supervised the work; A.D.B., T.J.M., R.S.K., O.B.C., H.M.C. obtained funding.
Corresponding authors
Ethics declarations
Competing interests
T.J.M. and H.M.C. have filed a patent application through Columbia University based on this work. Travis J. Morgenstern and Henry M. Colecraft, 2019. Composition and methods for genetically-encoded high voltage-activated calcium channel blockers using engineered ubiquitin ligases (U.S. Application Serial No. 62/830,142). H.M.C. is a scientific co-founder and on the SAB of two startups, Stablix, Inc. and Flux Therapeutics, pursuing targeted protein stabilization therapeutics. The remaining authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Alejandro Rojas Fernandez and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Darko-Boateng, A., Afriyie, E., Morgenstern, T.J. et al. Ion channel inhibition by targeted recruitment of NEDD4-2 with divalent nanobodies. Nat Commun 17, 378 (2026). https://doi.org/10.1038/s41467-025-67068-x
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-67068-x
