
Abstract
GPR99/OXGR1 is a G protein-coupled receptor (GPCR) with two endogenous agonists, the tricarboxylic acid cycle derivative 2-oxoglutarate (α-ketoglutarate) and the inflammatory mediator cysteinyl leukotriene E4 (LTE4), hence also termed CysLT3 receptor. How GPR99/OXGR1 recognizes two distinct ligands is a biologically important question. Here we present cryo-EM structures of GPR99/OXGR1-Gq complexed with oxoglutarate and LTE4, respectively. The oxoglutarate-bound structure shows a binding pocket surrounded by the transmembrane domains (TM), with a primary site and an accessory site for simultaneous binding of two oxoglutarate molecules for full activation of the receptor. The TM binding pocket, however, is too small to accommodate the cysteinyl leukotriene LTE4. Alanine substitution of key residues for oxoglutarate binding had little impact on LTE4-induced signaling. A distinct site in between TM3/4/5 just above intracellular loop 2 was identified in the solved structure with LTE4, but the densities were less well-defined. Alanine substitution of amino acids potentially involved in LTE4 interaction at this site abrogated LTE4-induced receptor activation without affecting oxoglutarate-induced signaling. Both ligands activated GPR99/OXGR1 primarily through the Gq pathway, but LTE4 also induced Gi signaling. These findings illustrate the structural basis for GPR99/OXGR1 to interact with structurally distict oxoglutarate and LTE4.
Similar content being viewed by others


Introduction
GPR99 was initially identified as a homologue of P2Y1, a purinergic GPCR that binds nucleotide ligands1. Further studies found that GPR99 does not bind nucleotide ligands2 but recognize endogenous dicarboxylates such as oxoglutarate3. Oxoglutarate is mainly derived from the tricarboxylic acid (TCA) cycle and serves as an essential myometabokine for autocrine and paracrine signaling, as well as an immunometabolite4. GPR99 recognizes oxoglutarate with an EC50 of approximately 200 μM and was subsequently named OXGR15. GPR99/OXGR1 is expressed in various human tissues including the kidney, respiratory epithelium, placenta, muscle, fetal brain and immune cells1,4,6,7. As a receptor of oxoglutarate, GPR99/OXGR1 is characteristic with its low affinity and low potency for this ligand, which is present in various tissues at concentrations in the micromolar range. In renal tubules, high concentrations of oxoglutarate promote Gq-mediated IP1 accumulation that contributes to the regulation of acid-base homeostasis4. Variations in renal pH may alter the polarity of oxoglutarate, thereby influencing Gq signaling through the activation of GPR99/OXGR14,8. These context-dependent features highlight how physiological environment may shape GPR99/OXGR1 signaling.
In addition to oxoglutarate, leukotriene E4 (LTE4) was identified as a ligand of GPR99/OXGR19. LTE4 is a cysteinyl leukotriene with a branched structure and established functions in allergy and asthma10,11,12. Elevated concentrations of LTE4 are observed in severe asthma, acute exacerbation of asthma, and aspirin-exacerbated respiratory disease (AERD)13,14,15, where it mediates sustained tracheobronchial constriction. An enrichment of GPR99/OXGR1 expression on the respiratory epithelial cells further exacerbates the inflammatory condition. Compared to LTC4 and LTD4 that are cysteinyl leukotrienes binding to different G protein-coupled receptors, LTE4 is more stable12. This feature of LTE4 contributes to asthma development due to the chronic effect of receptor/ligand-related hyperresponsiveness16. Other experimental evidence also supports the presence of the third cysteinyl leukotriene GPCR with high affinity for LTE417, whereas the LTE4 concentrations required to activate CysLT1R and CysLT2R are much higher than its physiological levels18. Thus, GPR99/OXGR1 is recognized as the physiologically relevant LTE4 receptor (CysLT3R), in addition to CysLT1R and CysLT2R that primarily bind to LTC4 and LTD45,9,18. CysLT3R (GPR99/OXGR1) is markedly different from CysLT1R and CysLT2R in primary sequence with only 36% of amino acid identity9, and the binding affinity of LTE4 to GPR99/OXGR1 (Kd ~ 2.5 nM)9 is higher than that of LTC4 and LTD4 to CysLT1R and CysLT2R, respectively7,9. Extensive studies have associated LTE4-CysLT3R(GPR99/OXGR1) signaling with potent pro-inflammatory effects, such as enhanced bronchoconstriction3,19 and eosinophil influx into the airway6,7,15. The high affinity of LTE4 for GPR99/OXGR1 is in sharp contrast to the low affinity of oxoglutarate (Kd in hundreds of micromolar) for the same receptor, suggesting that the two structurally different ligands may interact with GPR99/OXGR1 in different manners. However, the structural basis for GPR99/OXGR1 to bind oxoglutarate and LTE4 is currently lacking despite progress in structural characterization of CysLT1R and CysLT2R20,21,22.
Here we report a cryo-EM structure of GPR99/OXGR1 bound to oxoglutarate. Structural and functional analyses identify a primary and an accessory binding site for oxoglutarate, featuring simultaneous binding of two oxoglutarate molecules for full activation of the receptor. Furthermore, we found that the transmembrane binding pocket in GPR99/OXGR1 is too small to accommodate the branched LTE4, and further cryo-EM analysis of a GPR99/OXGR1-Gq complex formed in the presence of LTE4 identified a potential binding site outside the transmembrane binding pocket. Through functional analysis, we confirmed the alternative binding mode of LTE4 to GPR99/OXGR1, which likely serves as a promising target for future drug candidates. We found that LTE4 primarily activates the Gq-signaling pathways and the Gi pathway to a lesser extent. Although the poor EM density at this site prevented us to definitively locate the LTE4 binding site, our study provides a rational framework of the structural basis for and mechanistic insights into ligand recognition by GPR99/OXGR1.
Results
Overall structure of the oxoglutarate-GPR99/OXGR1-Gq signaling complex
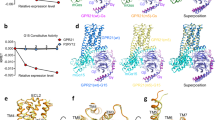
The human GPR99/OXGR1 and Gq proteins were co-expressed in Sf9 insect cells and purified. The endogenous ligand oxoglutarate was introduced to facilitate the formation of the GPR99/OXGR1-Gq signaling complex, which was purified and subjected to cryo-EM analysis (Supplementary Fig. 1a, b). The structure of the signaling complex was then determined at a global resolution of 3.16 Å (Supplementary Fig. 1c–f, Table 1). The structure models were confidently built, and most residues were assigned based on the well-defined density map (Fig. 1a and Supplementary Fig. 2). Two separate ligand densities were observed in the orthosteric binding pocket, which fit well with two ligand molecules of oxoglutarate (Fig. 1b). GPR99/OXGR1 holds the canonical seven-transmembrane architecture and G protein coupling manner of GPCR (Fig. 1c). There are two disulfide bridges in the extracellular domains: C24N-term-C2747.25, and C1063.25-C183ECL2 (using GPCR Ballesteros-Weinstein numbering as superscripts23). The disulfide bonds are conserved among most Class A GPCRs and contribute to stabilization of the extracellular domains (Supplementary Fig. 3). The GPR99/OXGR1 in our structure adopts a conformation in the active state, with an intracellular pocket accommodating the α5 helix in Gq (Fig. 1b).

a Cryo-EM density map of the oxoglutarate-GPR99/OXGR1-Gq complex. GPR99/OXGR1 is shown in cyan, Gαq in light purple, Gβ in light pink, Gγ in wheat yellow, and scFv16 in grey. b 3-D structure of oxoglutarate-GPR99/OXGR1-Gq complex (side view). The structure and EM density of the two oxoglutarate molecules (OG1, OG2) are highlighted in the inset. c, An enlarged view of OG1 and OG2 in the receptor, shown as yellow sticks. Molecular interactions between OG1 and OG2 and GPR99/OXGR1, in side view (d) and extracellular view (e). Polar interactions with key residues of the receptor are shown as red dashed lines.
Molecular basis for oxoglutarate recognition by GPR99/OXGR1
In the oxoglutarate-GPR99/OXGR1-Gq complex, the transmembrane domains TM1-3, TM6-7, extracellular loop (ECL) 1 and ECL2 of GPR99/OXGR1 encircle a hydrophilic and positively charged orthosteric pocket suited for the binding of negatively charged oxoglutarate molecule (Supplementary Fig. 4). The oxoglutarates adopt a horizontal orientation (Supplementary Fig. 4). One oxoglutarate molecule (OG1 in Fig. 1b) is embedded at the bottom of the pocket with the carboxyl group pointing downwards, and the other oxoglutarate molecule (OG2) fits well in the solvent-accessible subpocket at the top of the TM pocket with the carboxyl group pointing upwards (Fig. 1b and Supplementary Fig. 4). OG1 is directly clamped by salt bridges with four positively charged amino acids: R1103.29 and R2887.39 for one carboxyl, and H1143.33 and R2616.55 for the other (Fig. 1d, e). There are sufficient interactions between OG1 and these amino acid residues that form a primary binding site for the ligand. In comparison, OG2 is salt-bridged with K321.31 and H2817.32 at one carboxyl, and a hydrogen bond with Y922.63 through the other carboxyl (Fig. 1d, e). The surrounding amino acids form an accessory site near ECL2, which is well exposed to the solvent environment and therefore less stable for OG2 binding. Notably, two gating arginine residues (R1103.29 and R2887.39), together with Y932.64 and D185ECL2, spatially separate OG1 from OG2 (Supplementary Fig. 5a). The interaction network of arginines3 stabilizes the extracellular domain of GPR99/OXGR1 and separates OG1 from the extracellular solvent environment (Supplementary Fig. 5b).
Structural basis of selectivity for GPCR binding of dicarboxylates
Our findings reveal a distinctive binding mode for dicarboxylates in GPR99/OXGR1 compared to GPR91, suggesting a molecular mechanism underlying dicarboxylate selectivity in these GPCRs. The essential residues K321.31, H2817.32 and Y922.63 for OG2 binding in GPR99/OXGR1 are not found in GPR91 (E22, N274 and S82 in GPR91; Fig. 2 and Supplementary Fig. 6). Positively charged arginine residues are critical for the recognition and binding of negatively charged carboxyl groups, which are present in oxoglutarate (from oxoglutaric acid) and succinate (from succinic acid) in physiological pH. Most arginine residues in the orthosteric pocket are conserved between GPR91 and GPR99/OXGR1, namely R1103.29, R2616.55, R2646.58, R268ECL3 and R2887.39, but R177ECL2 and R180ECL2 in GPR99/OXGR1 are absent in GPR91 (Fig. 2 and Supplementary Fig. 6). In GPR91, only R993.29 and R2817.39 are known to participate in direct interactions with identified agonists and antagonists, including succinate, epoxysuccinate, maleic acid, compound 31, and NF-56-EJ4024,25.

Side view (a) and top view (b) of the transmembrane binding pockets of GPR99/OXGR1 (cyan) bound with oxoglutarate (yellow) and GPR91/SUCNR1 (pink) bound with succinate (green). Ligands and key residues for ligand binding are shown as sticks.
We found that, in GPR99/OXGR1, R1103.29, R2616.55 and R2887.39 contributed to the binding of OG1 and were involved in receptor activation. The other four arginine residues, R177ECL2, R180ECL2, R2646.58 and R268ECL3, were positioned above the OG1 and at the top of the orthosteric pocket, with their positively charged guanidinium groups oriented down towards the orthosteric pocket (Supplementary Fig. 5b). This cluster of arginines likely acts as a gatekeeper of the orthosteric pocket, facilitating the recruitment of oxoglutarate. In particular, the flexibility of R177ECL2 and R180ECL2 at the tip of ECL2 in GPR99/OXGR1 may be crucial for capturing negatively charged molecules, allowing only appropriate ligands like oxoglutarate (here OG2) to engage with R2646.58 and R268ECL3 (Supplementary Fig. 5b). Interestingly, the positively charged R177ECL2 and R180ECL2 in GPR99/OXGR1 are replaced by the negatively charged D167ECL2 and N168ECL2 in GPR91. Additionally, the tip of ECL2 in GPR91 is positioned farther from the orthosteric pocket compared to GPR99/OXGR1. These observations suggest differences in ligand recognition and recruitment between GPR99/OXGR1 and GPR91, thus contributing to ligand selectivity in these two dicarboxylate receptors. In this structure, the closely positioned arginine residues at the orifice and surface of the orthosteric binding pocket form an “arginine network” that mediates the recruitment and binding of oxoglutarate to the receptor.
Structural insights into LTE4 binding to GPR99/OXGR1
Unlike oxoglutarate, LTE4 has a relatively large, hydrophobic tail and two charged carboxyl groups at the head (Fig. 3a). Like other CysLTs, the “Y” shaped carboxyl terminus of LTE4 differs from the straight chain of hydroxyacids found in LTB410,26. To learn how LTE4 binds GPR99/OXGR1, we compared our solved structure with available structures of CysLT1R and CysLT2R20,21, bound to Pranlukast (Fig. 3b) and ONO-2570366 (Fig. 3c), respectively. Amino acid residues known to interact with these ligands were identified with the superimposed receptor structures shown in the background. The binding pockets showed little resemblance at these critical residues, an observation consistent with the grouping of GPR99/OXGR1 in the P2Y nucleotide receptor subfamily and the two CysLT receptors in a different subfamily of GPCRs1,27. We also compared the receptor structures sliced at the TM binding pocket in the same plane. As shown in Fig. 3d–f, the binding pockets of CysLT1R and CysLT2R are similar in shape and volume (Fig. 3d, e), snugly holding their respective ligands. In comparison, the binding pocket of GPR99/OXGR1 differs drastically from these two receptors in shape and volume, suitable only for the much smaller oxoglutarate molecules. Other notable features include the absence of positively charged arginine network, which is important for binding of the bicarboxylic ligands such as oxoglutarate and succinate3,25,28, in the CysLT receptors. Moreover, the large access cleft from the lipid membrane between TM4 and TM5 in the CysLT receptors22,27, which allow lateral entry of the bulky tails of the lipid ligands, is missing in GPR99/OXGR1. While revising the manuscript, we compared the structure of GPR99/OXGR1 with the LTD4-bound CysLT2R structure that became available only recently22, and found little resemblance between the two at the orthosteric binding pockets (Supplementary Fig. 7). These results strongly suggest that LTE4 uses a different binding site in GPR99/OXGR1.

a Chemical structure of the LTE4 molecule, the hydrophilic head and the hydrophobic tail of LTE4 are highlighted in boxes. b Comparison of TM binding pocket residues in superimposed structures of CysLT1R (light pink, PDB ID: 6RZ4) and GPR99/OXGR1 (light cyan, this work). c Comparison of TM binding pocket residues in superimposed structures of CysLT2R (light green, PDB ID: 6RZ6) and GPR99/OXGR1 (light cyan, this work). d–f Superimposed receptor structures of CysLT1R-Pranlukast, CysLT2R-ONO-2570366 and GPR99/OXGR1-oxoglutarate, sliced at the TM binding pocket in the same plane. GPR99/OXGR1-oxoglutarate structure is aligned as a reference model for structural comparison.
To illustrate how LTE4 binds to GPR99/OXGR1, an LTE4-GPR99/OXGR1-Gq complex was obtained with an excessive amount of LTE4 ( ~ 1.00 μM), and then subjected to cryo-EM analysis (Supplementary Fig. 8a, b). The resulting structure of the complex was resolved at an overall resolution of 2.84 Å (Fig. 4a, Supplementary Fig. 8c–g and 9, Table 1). An analysis of the structure found no extra EM density for LTE4 in the orthosteric pocket. Instead, a binding site was identified above the intracellular loop 2 (ICL2) and surrounded by TM3-5. Although the EM density could not definitively indicate binding of LTE4 at this site, the shape fits well with an LTE4 molecule (Fig. 4b, c).

a Cryo-EM density map of the LTE4-GPR99/OXGR1-Gq complex. GPR99/OXGR1 is shown in wheat, Gαq in light purple, Gβ in light pink, Gγ in wheat yellow, and scFv16 in grey. b 3-D structure of the LTE4-GPR99/OXGR1-Gq complex (side view). The structure and EM density (inset) of the bound LTE4 are highlighted on the right.c An enlarged view of the LTE4 bound to the receptor. Molecular interactions between LTE4 (green) and GPR99/OXGR1, in side view (d) and extracellular view (e). Polar interactions with key residues of the receptor are shown as red dashed lines.
Several approaches were taken to further explore the potential site for LTE4. A structural complex model was built based on the EM density, and potential interactions between LTE4 and GPR99/OXGR1 were identified. Specifically, L1223.41, T1253.44, I1293.48, F1303.49, V1524.44, A1534.45, V1574.49, I1604.52, T2055.45, L2095.49 could form extensive hydrophobic interactions with the aliphatic chain of LTE4 (Supplementary Fig. 10a). The negatively charged head of LTE4 could be attached to a positively charged subpocket formed by ICL2 and TM3-4 (Fig. 4d and Supplementary Fig. 10b). In detail, R1484.40 could form a salt bridge with one carboxyl group, C141ICL2 could possibly form a hydrogen bond with the other carboxyl group, and C1494.41 could form a hydrogen bond with the hydroxyl group as well as another hydrogen bond with a carboxyl group (Fig. 4d, e). The presence of these interactions was validated with alanine substitutions and the effects on receptor signaling by LTE4 as well as oxoglutarate were compared (see below).
Computational approaches were taken to further evaluate possible binding of LTE4 to this alternative site. Molecular docking was applied to dock LTE4 to the TM pocket and the alternative site, respectively. The corresponding binding energies were calculated, showing an energetic preference for LTE4 at the alternative binding site (Supplementary Fig. 11a). Furthermore, three independent 1-µs MD simulations captured a stable binding pose of LTE4 at this alternative site (Supplementary Fig. 11b, c).
Validation of GPR99/OXGR1-ligand interactions by site-directed mutagenesis
To further elucidate ligand interaction with GPR99/OXGR1, site-directed mutagenesis and functional assays were performed. Amino acid residues exhibiting polar interactions with oxoglutarate and LTE4 were substituted by alanine, and all mutants were positive for cell surface expression (Supplementary Fig. 12a). The accumulation of IP1, downstream of the Gq-phospholipase C (PLC) pathway, was measured for both WT and mutants of GPR99/OXGR1 (Fig. 5). Alanine substitution of residues interacting with OG1 (R110A, H114A, R261A, R288A) resulted in a significant reduction in both the potency and efficacy of IP1 accumulation following oxoglutarate stimulation (Fig. 5a). In comparison, mutations in OG2-interacting residues (K32A and Y92A) led to a more than 100-fold increase in EC50, but the maximum response (Emax) remained unchanged (Fig. 5a). These findings suggest that, while OG2 is essential for maintaining ligand occupation, it plays a less significant role in receptor activation compared to OG1.

Transfected cells (HeLa) expressing either WT or mutant receptors were stimulated with oxoglutarate or LTE4, the induced responses were measured to reflect G protein activation (cAMP reduction for Gαi, IP1 accumulation for Gαq). a 6 mutants with alanine substitutions of key residues for oxoglutarate binding were tested against WT in IP1 accumulation. Four of the 6 mutants abrogated GPR99/OXGR1 signaling for Gαq activation (IP1 accumulation), and the other 2 mutants reduced the response. b the same mutants had no or little effect on LTE4-induced IP1 accumulation. 3 alanine-substituted mutants of residues suspected to interact with LTE4 at the projected site were tested against WT in IP1 accumulation assay. All 3 mutants abrogated the LTE4-induced IP1 accumulation (c) while having little or much smaller effect on oxoglutarate-induced response (d). In e, LTE4, but not oxoglutarate, stimulated cells expressing the WT GPR99/OXGR1 with reduction of cAMP, indicating Gαi activation. In f, pertussis toxin (PTX), a bacterial toxin that blocks Gαi activation, abrogated LTE4-induced cAMP reduction, supporting coupling of GPR99/OXGR1 with Gi proteins in addition to Gq. In g, PTX had no effect on LTE4-induced IP1 accumulation that does not exclusively rely on Gi in the presence of Gq proteins. Data are shown as mean ± SEM from at least three independent experiments.
Consistent with the predicted structure for LTE4 binding, alanine substitution of the OG1 and OG2 sites had little effect on LTE4-induced receptor signaling (Fig. 5b). Substitution of the projected LTE4-binding residues (C141ICL2, R1484.40 and C1494.41) markedly reduced LTE4-induced IP1 accumulation (Fig. 5c), but had little effect on oxoglutarate-induced receptor activation (Fig. 5d). This finding suggests that LTE4 binding to GPR99/OXGR1 depends on hydrogen bonding with key residues C141ICL2, R1484.40 and C1494.41 in the binding site, but this site was not utilized by oxoglutarate. Altogether, results obtained from mutagenesis experiments provide functional clues that support the binding of LTE4 to a site distinct from the TM site for oxoglutarate. We have also noticed that some mutants (C141ICL2A, C1494.41A) exhibited a reduced Emax albeit having comparable cell surface expression with the WT GPR99/OXGR1. As C141ICL2 and C1494.41 are proximal to the ICL2 region where the receptor extensively interacts with G proteins, we suspect that mutations at these sites may alter the overall stability of ICL2 and consequently the recruitment and activation of G proteins, thus affecting the efficiency of Gq response. These functional data distinguish the TM binding mode of oxoglutarate from the distinct binding mode of LTE4 at GPR99/OXGR1.
Ligand-induced signaling through GPR99/OXGR1
Given the structural difference between oxoglutarate binding and LTE4 binding at GPR99/OXGR1, it is suspected that their downstream signaling properties might vary. We therefore examined the signaling pathways triggered by oxoglutarate and LTE4. Notably, oxoglutarate preferentially activated the PLC pathway downstream of Gq signaling, leading to increased IP1 accumulation; however, oxoglutarate failed to stimulate cAMP reduction downstream of Gi coupling (Fig. 5e). In contrast, LTE4 treatment caused reduction in cAMP accumulation (Fig. 5e), suggesting activation of Gαi. This interesting finding was further verified by treatment of the cells with pertussis toxin (PTX), which ADP-ribosylates Gαi and breaks the Gi activation cycle29. PTX abrogated the cAMP response elicited by LTE4 (Fig. 5f) without affecting IP1 accumulation (Fig. 5g). These findings indicate that LTE4 binding to GPR99/OXGR1 can engage Gi-mediated signaling in addition to its Gq-coupled activity.
To investigate the basal activity of GPR99/OXGR1, we used a NanoBiT-based G protein dissociation assay, with SmBiT fused to Gγ and LgBiT fused to Gα30. Gα dissociation from Gβγ, that accompanies G protein activation, reduces the NanoBiT signal. HEK293T cells were co-transfected with equal amounts of plasmids encoding tagged G proteins and varying amounts of plasmids encoding GPR99/OXGR1 or the constitutively active KSHV-GPCR as a control31. Basal G protein dissociation of GPR99/OXGR1 was compared to that of KSHV-GPCR. Increasing KSHV-GPCR plasmid levels resulted in a dose-dependent decrease in NanoBiT signal, indicating agonist-independent G protein dissociation, whereas GPR99/OXGR1 showed no signal change, suggesting negligible basal activity of GPR99/OXGR1 (Supplementary Fig. 12b). These results indicate that GPR99/OXGR1 is not a constitutively active GPCR that can complex with G proteins without agonist binding.
The G protein coupling interface of GPR99/OXGR1
The G protein interfaces are highly similar between oxoglutarate-bound and LTE4-bound GPR99/OXGR1 signaling complex. However, subtle differences are observed for sidechain orientations of the α5 helix of Gq protein (Fig. 6a). As a result, the G protein interaction profiles are slightly different between oxoglutarate-bound and LTE4-bound GPR99/OXGR1 structures. In the oxoglutarate-GPR99/OXGR1-Gq complex, more extensive polar interactions between Gq and GPR99/OXGR1 were identified (Fig. 6b). Specifically, Y351 of the α5 helix of Gq protein forms polar contacts with R1313.50 and S682.39 of GPR99/OXGR1. E350 forms a hydrogen bond with H145ICL2, and K349 interacts with D3078.48. Q345 forms polar interactions with Q230ICL3, and D341 has a polar contact with K2366.30 (Fig. 6b). In the LTE4-GPR99/OXGR1-Gq complex, four pairs of polar contacts are found between GPR99/OXGR1 and the Gq protein: R1313.50-Y351, D3078.48-K349, V1343.53-N347 and K2366.30-D341 (Fig. 6c). These subtle structural differences may help to explain the distinct biological outcomes induced by oxoglutarate versus LTE4.

a Superimposed structures of the Gq protein in the oxoglutarate-bound and LTE4-bound GPR99/OXGR1 signaling complex. Gq is shown in cyan for the oxoglutarate-bound structure and in champaign for the LTE4-bound structure. b Molecular interactions between Gq and GPR99/OXGR1 in the oxoglutarate-bound complex. Oxoglutarate is shown in cyan, with polar interactions highlighted by red dashed lines. c Molecular interactions between Gq and GPR99/OXGR1 in the LTE4-bound complex. LTE4 is shown in Champaign, with polar interactions marked by red dashed lines.
Ligand-induced activation mechanism of GPR99/OXGR1
To elucidate the activation mechanism induced by orthosteric coupling of oxoglutarate, the recently reported structures of GPR91 in the active state25 (PDB: 8WOG) and inactive state24 (PDB: 6RNK) were used as references for comparison. The outward movement of TM5-6 and inward movement of TM7, which accommodated the α5 helix of Gq, illustrated a typical conformational change associated with receptor activation (Fig. 7a,b). Additionally, the structural motifs responsible for GPCR activation, specifically DRY, CWXP, NPXXY and PIF (corresponding to FRY, CFLP, NLLLY and PIF in GPR99), were compared between GPR99/OXGR1 and GPR91 (PDB: 8WOG). The oxoglutarate-GPR99/OXGR1-Gq complex represented a molecular mechanism for receptor activation triggered from the orthosteric pocket. Specifically, the salt bridge between OG1 and R2616.55 gave rise to an inward turning of TM6, which was transduced through the CFLP motif (Fig. 7c). The turning of TM6, along with the bridge between OG1 and H1143.33, caused a rearrangement of the hydrophobic core of the PIF motif (Fig. 7d). Besides, the salt bridge between OG1 and H1143.33, and the hydrogen bond between OG1 and R2887.39, triggered a conformational change that propagates through TM7. The salt bridge between oxoglutarate and R1103.29 initiated conformational changes that were transduced through TM3. Furthermore, OG2 played an accessory role, alongside OG1, in initiating receptor signaling. In a synergistic manner, the extracellular signal induced a rearrangement of the NLLLY (NPXXY) motif (Fig. 7e) and the FRY(DRY) motif (Fig. 7f) in the intracellular domain, opening the intracellular pocket to accommodate α5 helix of the Gq protein (Fig. 7e, f).

a Superimposed structures of GPR99/OXGR1 (this study, oxoglutarate-bound) and GPR91/SUCNR1 in the active state (PDB ID: 8WOG). b Superimposed structures of GPR99/OXGR1 (this study) and GPR91/SUCNR1 in the inactive state (PDB ID: 6RNK). Conformational changes at key activation motifs. Shown are side chain conformation at the CWxP (CFxP in GPR99/OXGR1 and GPR91/SUCNR1) motif (c), conformational changes at the PIF motif (d), conformational changes at the NPxxY (NLxxY in GPR99/OXGR1) motif (e), and rotameric conformation changes at the DRY (FRY in GPR99/OXGR1) motif (f). GPR99/OXGR1 in the active state is shown in cyan, GPR91/SUCNR1 in the active state is shown in pink, and GPR91/SUCNR1 in the inactive state is shown in orange.
The reported inactive state structures of CysLT1R and CysLT2R were used as references to illustrate the activation mechanism of GPR99/OXGR1 induced by LTE420,21 (Fig. 8). In this structure, polar interactions caused the ICL2 of GPR99/OXGR1 to move outward when LTE4 is present (Fig. 8a), thus inducing the opening of the G protein-coupling pocket (Fig. 8b). The hydrophobic interaction driven by the aliphatic chain of LTE4 directly triggered the rearrangement of the NLxxY motif (Fig. 8c) and the FRY motif (Fig. 8d). Consequently, the CFxP and PIF motifs rearranged synergistically, with TM5-6 moving outward and TM7 moving inward (Fig. 8e, f). Additionally, a salt bridge formed between R318 and D312, further stabilizing the activated conformation and resulting in an ordered helix8, which was absent in the inactive structure due to its flexibility.

a Superimposed structures of GPR99/OXGR1 (this study, LTE4-bound), CysLT1R in the inactive state (PDB ID: 6RZ4) and CysLT2R in the inactive state (PDB ID: 6RZ6). LTE4 is shown as yellow sticks. b Superimposed structures of GPR99/OXGR1, inactive CysLT1R and inactive CysLT2R. Conformational changes are marked with red arrows. Conformational changes at key activation motifs. Shown are conformational changes at the NPxxY (NLxxY in GPR99/OXGR1, DPxxY in CysLT1R) motif (c), rotameric conformation changes at the DRY (FRY in GPR99/OXGR1, FRC in CysLT1R, VRY in CysLT2R) motif (d), conformational changes at the CWxP (CFxP in GPR99/OXGR1 and CysLT2R, SFxP in CysLT1R) motif (e), and conformation changes at the PIF motif (f). GPR99/OXGR1 (CysLT3R) in the active state is shown in cyan, CysLT1R in the inactive state is shown in green, and CysLT2R in the inactive state is shown in salmon pink.
Discussion
The structural mechanisms by which both dicarboxylates and CysLTs bind their native receptors remain underexplored. Persistent questions revolve around how GPCRs recognize small, charged dicarboxylates with high specificity but low efficacy. There have been great computational efforts, but the issues remain due to the lack of effective structural references32,33. For example, an exhaustive metadynamics analysis led to the proposal that there are two low-energy succinate binding sites in the orthosteric pocket of GPR9132, although subsequent structural studies demonstrated the presence of only a single succinate binding site in this pocket25,28. Furthermore, in the absence of a resolved structure for the oxoglutarate receptor GPR99/OXGR1, it remains challenging to address how dicarboxylates are selectively recognized by their respective receptors. This understanding is crucial for developing targeted drugs with high specificity.
In this study, we present a high-resolution cryo-EM structure of GPR99/OXGR1 bound to the dicarboxylate oxoglutarate. Our structural and functional analyses reveal the molecular basis for ligand selectivity and G protein coupling preferences. Dicarboxylates are critical components in physiological fluids, often present at micromolar concentrations. As a result, their native GPCRs may have evolved orthosteric adaptations that attenuate or desensitize activation to prevent unnecessary receptor activation under normal conditions. Notably, GPR99/OXGR1 and GPR91/SUCNR1 have shared features including the arginine network at their binding pockets, but also distinct features in GPR99/OXGR1 allowing simultaneous binding of two oxoglutarate molecules. In SUCNR1, however, the positively charged R177ECL2 and R180ECL2 in GPR99/OXGR1 are absent (replaced by the negatively charged D167ECL2 and N168ECL2), and the tip of ECL2 in GPR91 is positioned farther from the orthosteric pocket compared to GPR99/OXGR1. These diffences may explain the capture of only one succinate molecule but not two by GPR91 in experimentally resolved SUCR1 structures24,25, although metadynamics analysis indicated two low-energy succinate binding sites in the orthosteric pocket of GPR9132. As proposed in the work by Shenol, Schwartz and coworkers32, an advantage of binding two dicarboxylates simultaneously is to ensure full activation of the receptor when ligand concentrations reach a necessary threshold, which is shown experimentally in the present work of GPR99/OXGR1.
In contrast, LTE4, a lipid mediator that preferentially remains associated with the membrane lipids, can easily access the distinct site and efficiently activate the receptor, thereby mediating inflammatory responses. This alternative activation occurs with high affinity and efficiency, contributing to the overactivation of the receptor and leading to diseases such as asthma and allergies7,15,18,21. Moreover, the DRY-to-FRY mutation in GPR99/OXGR1 may have evolved to accommodate activation signals from LTE4 at its distinct binding pocket. As the third cysteinyl leukotriene receptor (CysLT3R), GPR99/OXGR1 is highly expressed in kidney cells1, fetal brain cells, fibroblast34, as well as immune-related cells including mast cells, eosinophils, and respiratory epithelial mucosa cells6,7,9,35. GPR99/OXGR1 is associated with normal kidney pH-maintaining functions, central nervous system development, fibrosis pathogenesis, allergies and inflammation. Under different physiological contexts, ligand availability and expression levels of GPR99/OXGR1 and G proteins vary, affecting the activation and signaling of the receptor. For instance, in inflammatory microenvironments rich in leukotrienes, the sustained presence of LTE4 likely stabilizes its binding to the distinct pocket of GPR99/OXGR1, amplifying Gαi signaling to exacerbate inflammation through leukocyte recruitment and activation7,11,36. Moreover, an enrichment of GPR99/OXGR1 expression on the respiratory epithelial cells further exacerbates the inflammatory condition. An early study on LTE4-induced bronchoconstriction suggested that the high stability of LTE4, compared to LTC4 and LTD4, promotes asthma development due to the chronic effect of receptor/ligand-related hyperresponsiveness16. Conversely, in renal tubules, high oxoglutarate concentrations promote Gαq-mediated IP1 accumulation, regulating acid-base homeostasis in kidney4. Acidic renal pH may enhance the polarity of oxoglutarate, thereby favoring Gq signaling through the activation of GPR99/OXGR14,8. These context-dependent activation features highlight how patho-physiological environments may shape GPR99/OXGR1 signaling.
LTE4 is a high-affinity agonist of GPR99/OXGR1, based on pharmacological characterization of the receptor in transfected cell lines9. As a result, GPR99 is also named CysLT3R5,9. How LTE4 binds to a GPCR with low sequence similarity to the other two CysLT receptors remained unclear as we began this study. In previous structural studies of antagonist-bound CysLT1R and CysLT2R, the endogenous agonists CysLTs LTC4 and LTD4 were proposed to bind to the orthosteric pocket just as the antagonists, which was validated by functional assays20,21. A recent work of the LTD4-bound CysLT2R structure, published while this study was under review, provides a solid proof of the binding mode of LTD4 to its cognate CysLT2R receptor22. LTD4 binds within the transmembrane pocket of CysLT2R, with its polar head reaching to the polar top of the TM pocket and nonpolar interactions spanning TM helices TM4 and TM5, thus stabilizing its bulky tail (Supplementary Fig. 7a–c). The long bulky tail of LTD4 can therefore extend through an orifice opening formed between TM4 and TM5 of CysLT2R, similar to the previously proposed binding mode for CysLT1R and CysLT2R27. In contrast, the GPR99/OXGR1 TM pocket is much smaller, more polar and more hydrophilic. It has no cleft space between TM4 and TM5 for the accommodation of the long bulky tail of LTE4, hence poorly suited for LTE4 binding (Supplementary Fig. 7d–f). While the superimposed active-state structures of GPR99/OXGR1 and CysLT2R are generally similar, the TM3 of CysLT2R slightly shifts outwards (Supplementary Fig. 7g), and their binding pockets are very different. By examining the geometry of residues surrounding the TM binding pocket of CysLT2R, an opening is observed around the hydrophobic tail of LTD4. For the superimposed GPR99/OXGR1 structure at this site, larger amino acid side chains are found to block such an opening (Supplementary Fig. 7h). Despite the ability of being activated by LTE4, the sequence of GPR99/OXGR1 is quite different from CysLT1R and CysLT2R receptors with about 30% sequence identity (Supplementary Fig. 7i)3,5,9. Besides, as CysLT1R and CysLT2R can both selectively recognize LTC4 and LTD4 with high affinity but LTE4 with only low affinity, it is plausible that the binding mode of LTE4 is distinct from that of LTC4 and LTD4 to the respective cysteinyl leukotriene receptors37,38. Differing from Gq coupling of GPR99/OXGR1 when stimulated with oxoglutarate, the receptor is found to activate both Gq and Gi signaling pathways when stimulated with LTE4, thus contributing to its proinflammatory and allergy-inducing properties including induction of leukocyte chemotaxis7,18. These results highlight the common and distinct features in the signaling and activation of cysteinyl leukotriene receptors from structural and functional perspectives39. While our studies provide a solid structural foundation, further physiological and pharmacological research is required to advance the development of new cysteinyl leukotriene receptor-targeting drugs.
GPCR drug discovery now marches beyond the canonical transmembrane pocket, with increasing efforts focusing on allosteric ligands that bind to alternative binding sites and modulate receptor functions with signaling bias40,41,42,43,44,45. For some GPCRs such as S1PR3, GPR88, GPR35, GPR174, PTHR and CB2, alternative allosteric sites and mechanisms determine their downstream signaling pathways. For instance, the sphingosine-1-phosphate (S1P) receptor S1PR3 can be allosterically modulated by ligands binding to its extracellular vestibule (S1P, and its analogs) or deep within the transmembrane pocket (CYM-5541, CBP-307, BAF-312, FTY720-P, and VPC23019), fine-tuning its response and influencing G protein coupling bias towards Gi, Gq and G1346,47,48. As an important GPCR regulating neurological functions, GPR88 has its synthetic agonist 2-PCCA binding to a cavity at the cytosolic ends of TM5 and TM6 close to ICL3, allowing direct interaction with the α5 helix of Gi and further stabilization of the receptor-Gi complex49. In this case, the binding site is”orthosteric” to the particular ligand but non-canonical when compared with most other GPCRs that use TM binding pocket for ligand interaction. The LTE4-GPR99/OXGR1 interaction falls within this category, and the non-canonical binding site may offer a strategy to achieve biased agonism and antagonism through stabilization of conformations that favor either Gi or Gq coupling in different physiological environments.
In summary, the present study provides structural insights into oxoglutarate binding to GPR99/OXGR1. The characteristic binding of two oxoglutarate molecules simultaneously may help to address how a GPCR recognizes the small and charged dicarboxylates with high specificity but low efficacy. Despite our effort in resolving the LTE4-bound GPR99/OXGR1 structure, no EM density was found in the orthosteric binding pocket as originally expected. Results from a multitude of experiments and comparative analysis indicate that the TM pocket in GPR99/OXGR1 is too small to accommodate the bulky cysteinyl leukotriene. It is likely that LTE4 uses a distinct binding site for activation of GPR99/OXGR1, which has been clearly identified as CysLT3R through rigorous pharmacological experiments. The present work could not conclusively identify the LTE4 binding site due to poor EM density, but the collected information supports the presence of an LTE4 binding site that differs from the TM binding pocket for oxoglutarate. Our results may help further exploration of the LTE4 binding mechanism and unlock its therapeutic potential.
Method
Design of constructs
The cDNA encoding human wild-type GPR99 was synthesized by General Biol (Chuzhou, China). The constructs were created by cloning the full-length coding sequence into a pFastbac1 vector. To facilitate protein expression and purification, a hemagglutinin (HA) signal peptide, a FLAG tag, a human rhinovirus 3 C (HRV 3 C) protease cleavage site (LEVLFQGP) and a thermostabilized apocytochrome b562RIL (BRIL) were added to the N terminus, following our previously reported method25. The Gq protein was designed based on the Gi backbone by inserting the C terminus of Gq (residues 332 to 359) to DNGαi1 (dominant negative Gαi1 with G203A and A326S mutation) backbone (residues 1 to 326), which provided an additional site for the single-chain antibody variable fragment scFv16 to stabilize the complex. This strategy and the resulting chimeric Gq were widely used, as demonstrated in the reported structure determination of GPCR-Gq complexes50,51,52. Human Gβ1 and Gγ2 cDNAs with N-terminal 6×His tag were respectively cloned into a pFastBac-Dual vector. scFv16 was fused with a GP67 signal peptide at the N-terminal and an 8× His tag at the C-terminal. For the functional assay, the human GPR99 coding sequence was cloned into the pcDNA3.1 vector. Point mutations were introduced using PCR-mediated site-directed mutagenesis.
Purification of scFv16
Secreted scFv16 was purified from expression media of baculovirus-infected Sf9 insect cell (Invitrogen, Cat #: 11496015) culture using Ni-NTA. The supernatant from 2 L of culture was collected and loaded onto a gravity column packed with Ni-NTA resin. The resin was washed with 20 mM HEPES pH 7.5, 500 mM NaCl, and the protein was eluted with 20 mM HEPES pH 7.5, 150 mM NaCl and 250 mM imidazole. Eluted protein was concentrated and loaded onto Superdex 200 increase 10/300 size exclusion column (GE Healthcare Life Sciences, Sweden). The peak fractions were collected and concentrated, rapidly frozen in liquid nitrogen, and stored at −80 °C.
Expression and purification of the signaling complex
The bac-to-bac baculovirus expression system and Sf9 insect cells were used to express protein for structure determination. The baculoviruses were prepared according to the manual (Thermo Fisher Scientific). For protein expression, baculoviruses of GPR99, Gαq, and Gβ1γ2 were co-transfected into Sf9 cells when they reached a density of 2 × 106 cells/mL. The ratio of baculoviruses for transfection was 1:1:1, and then the proteins were expressed over a 48-hour period. The cell culture was harvested by centrifugation at 2000 × g for 15 min and kept frozen at −80 °C.
To purify the complex, the ligand (oxoglutarate or LTE4) was added to induce GPR99-Gq complex formation. Cell pellets were resuspended in buffer containing 25 mM HEPES pH 7.4, 50 mM of NaCl, 5 mM of KCl, 5 mM of CaCl2, 5 mM of MgCl2, 5% glycerol, 25 mU/mL apyrase, 2.5 μg/ml leupeptin, 0.16 mg/ml benzamidine, 100 μM of oxoglutarate or 1 μM of LTE4. Cell membranes were collected by centrifugation after 30 minutes of incubation. The cell membranes were then solubilized in 20 mM HEPES pH 7.4, 100 mM NaCl, 0.8% LMNG, 0.1% CHS, 10% glycerol, 2.5 μg/ml leupeptin, 0.16 mg/ml benzamidine, 50 μM of oxoglutarate or 1 μM of LTE4. After 2 h incubation at 4 °C, the supernatant was cleared by centrifugation at 20,000 × g for 30 min. A gravity-flow column with anti-FLAG affinity resin (GenScript Biotech) was used to capture the complex, followed by washing with 10 column volumes of wash buffer containing 20 mM of HEPES (pH 7.4), 100 mM of NaCl, 5% glycerol, 2 mM of CaCl2, 2 mM of MgCl2, 0.05% (w/v) LMNG, 0.05% (w/v) GDN (Anatrace), 0.003% (w/v) CHS, 20 μM of oxoglutarate or 0.4 μM of LTE4. The protein complex was eluted with buffer containing 20 mM of HEPES (pH 7.4), 100 mM of NaCl, 2 mM of CaCl2, 2 mM of MgCl2, 0.01% (w/v) LMNG, 0.001% (w/v) GDN (Anatrace), 0.001% (w/v) CHS, 0.2 mg/ml FLAG peptide (Sigma-Aldrich), 20 μM of oxoglutarate or 0.4 μM of LTE4. The eluate was concentrated by an Amicon Ultra-15 Centrifugal Filter Unit (Millipore) and separated on a Superose 6 10/300 size-exclusion chromatography column (Cytiva) pre-equilibrated with 20 mM HEPES (pH 7.4), 100 mM NaCl, 0.0015% LMNG, and 0.0005% GDN, 0.0003% CHS, 20 μM of oxoglutarate or 0.4 μM of LTE4. The relevant peak fractions of the protein complex were collected, analyzed on SDS-PAGE, analyzed on western-blotting, concentrated to approximately 10 mg/mL, and stored at -80 °C.
Cryo-grid preparation and EM data collection
Negative stain electron microscopy was performed on all the samples to confirm homogeneity and complex formation. For cryo-grid preparation, aliquots of 3 µL of purified protein complex were applied onto an Ultrafoil 300 mesh R1.2/1.3 holy gold (Au) grid, which had already been glow-discharged by Tergeo-EM plasma cleaner. Afterward, the grids were blotted for 3.5 s with a blot force of 1 in 100% humidity at 4 °C to remove excess samples, and then quickly plunged into liquid ethane using a Vitrobot Mark IV (Thermo Fisher Scientific). The prepared grids were stored in liquid nitrogen.
The grid sample screening and data collection were performed using SerialEM software (Mastronarde, 2005). The final data sets were collected by a Gatan K3 direct electron detector installed on a 300 kV Titan Krios G3 microscope. During data collection, a GIF Quantum energy filter (Gatan, USA) was used to exclude inelastically scattered electrons, with the energy slit width set to 20 eV. Movie stacks were acquired at a nominal magnification of 105,000, resulting in a calibrated pixel size of 0.85 Å. The total exposure time was 2.5 s, fractionated into 50 frames at a dose rate of 20.8 e/pixel/s. The defocus range for dataset collection was set from -1.2 to -2.5 μm.
Image processing and 3D reconstructions
The cryoSPARC version v4.2.153 and Relion version 4.054 were used to process the cryo-EM datasets. The pipeline was similar to that reported previously25. Motion correction and dose-weighting were applied to align the movie stacks. After contrast transfer function (CTF) estimation, micrographs were manually inspected, and obviously bad micrographs were discarded. Representative particles were then manually picked to generate initial two-dimensional (2D) templates for auto-picking.
For the oxoglutarate-GPR99-Gq dataset, a total of 2,676,089 particles were template-based picked and subjected to 2D classification. After three rounds of 2D classification, particles were selected from the 2D averages with clear secondary features. Ab initio reconstruction was performed by cryoSPARC to generate initial three-dimensional (3D) templates, followed by rounds of 3D classification. Finally, a dataset containing 167,941 particles was used for homogeneous refinement, non-uniform refinement, and local refinement. The global resolution was 3.16 Å, estimated by the ‘gold standard’ criterion (FSC = 0.143).
For the LTE4-GPR99-Gq dataset, template-based particle picking resulted in a dataset of 2,043,778 particles, which was subjected to three rounds of 2D classification. Good particles with clear secondary features in the 2D averages were selected. Then, after multiple rounds of 3D classification, a final dataset containing 100,083 particles was obtained, resulting in a final map with an estimated global resolution of 2.84 Å.
Model building and refinement
As no experimental GPR99 structures had been reported previously, we used the predicted structure from the AlphaFold Protein Structure Database (AF-Q96P68-F1) as an initial template for model building. The model building of the G protein heterotrimer and scFv16 was facilitated by our previously reported structure (PDB: 8WOG) as the starting template. The model was docked into the electron microscopy density by UCSF chimera55 and manually rebuilt with Coot56, as well as iterative refinement with Phenix57. Final model validation and statistical analysis were performed using Molprobity58. The graphic structural figures were prepared using UCSF Chimera, ChimeraX and PyMOL.
IP-one accumulation assay
Human GPR99, both in its wild-type form and in mutants, were expressed in HeLa cells (ATCC, Cat #: CCL-2) 24 hours before harvesting (Invitrogen, L3000001). Gq downstream signaling, which leads to the accumulation of IP1, was measured using the IP-One Gq HTRF kit (Cisbio). Cells were resuspended in the stimulation buffer (Cisbio) and incubated with varying concentrations of oxoglutarate (A610290, Sangon Biotech, Shanghai, China) or leukotriene E4 (HY-113465, MedChemExpress, Monmouth Junction, NJ) diluted in the stimulation buffer for 30 minutes at 37 °C. IP1 accumulation was then assessed according to the manufacturer’s instructions. Fluorescence intensities were recorded using an Envision 2105 multimode plate reader (PerkinElmer). Intracellular IP1 levels were determined based on the fluorescence signals from the samples and IP1 standards.
cAMP inhibition assay
Human GPR99 (WT/mutants) were expressed in HeLa cells for 24 hrs (Invitrogen, L3000001), and pretreated with 250 ng/mL PTX or vehicle for 12 h at 37 °C. Gi downstream signaling, which results in the inhibition of cAMP accumulation was measured. Cells were harvested in HBSS supplemented with 5 mM HEPES, 0.1% (w/v) BSA, and 0.5 mM 3-isobutyl-1-methylxanthine. The plated cells were subsequently stimulated with varying concentrations of chemerin alongside 2.5 μM forskolin for 30 minutes. Intracellular cAMP levels were quantified using the LANCE Ultra cAMP kit (TRF0263, PerkinElmer) according to the manufacturer’s protocol, with readings taken on an EnVision 2105 multimode plate reader (PerkinElmer).
NanoBiT-based GPR99-G protein dissociation and Gα-Gβγ protein dissociation assays
HEK293T cells (ATCC, Cat #: CRL-3216) were seeded in 24-well plates and incubated for 24 hours. For GPR99-G protein dissociation assay, cells were co-transfected with plasmids encoding GPR99-SmBiT (333 ng/well) and Gαi-LgBiT (167 ng/well). For Gα-Gβγ protein dissociation, cells were co-transfected with plasmids encoding GPR99 (92 ng/well), Gαi-LgBiT (46 ng/well), Gβ (230 ng/well), and SmBiT-Gγ (230 ng/well). Following a 24-hour incubation at 37 °C, the transfected cells were harvested and seeded into white 384-well plates. For the luminescence assay, coelenterazine H (Yeasen Biotechnology, Shanghai, China) was added to a final concentration of 10 μM for 1-hour incubation at room temperature, and baseline luminescence signals were measured for 10 minutes using an Envision 2105 multimode plate reader (PerkinElmer). Ligands were then added, and luminescence detection continued for 1 hour.
Flow cytometry analysis
HEK293T cells were transfected with expression plasmids encoding FLAG-tagged wild-type (WT) or mutant GPR99 for 24 hours at 37 °C. The cells were then collected and washed with HBSS containing 0.5% BSA. After washing, the cells were incubated with a FITC-labeled anti-FLAG antibody (Sigma, Cat #F4049; diluted 1:50 in HBSS buffer) for 30 minutes on ice and washed again with HBSS. Flow cytometry (CytoFLEX, Beckman Coulter) was used to quantify the FITC fluorescence signals on cell surfaces. The fluorescence signals were then analyzed to determine the relative expression levels of GPR99 mutants.
Molecular docking analysis
Flexible docking followed by Binding Free Energy calculations were performed using the BIOVIA Discovery Studio (v2019) to evaluate the differential binding affinity of the ligand LTE4 at the transmembrane pocket and the distinct site. Flexible docking in DS is an induced fit protocol to simulate both protein and ligand flexibility. During flexible docking, there are three distinct phases: (i) protein side-chain conformation generation through CHARMM-based ChiFlex method, (ii) generation of ligand conformations and rigid ligand placement into each protein conformation through LibDock, and (iii) induced fit protein flexibility through ChiRotor-based side-chain reconstruction and final pose refinement through CDOCKER. Protein flexibility is introduced in the first and third phases.
Prior to docking, LTE4 was extracted from the cryo-EM structure and preprocessed using the “Prepare Ligands” and “Minimize Ligands” protocols in DS. This step ensured correction of valency errors, adjustment of protonation states, and geometry optimization under the CHARMM force field. The receptor extracted from the cryo-EM structure was prepared by adding hydrogen, defining the binding site, and specifying flexible side chains for induced fit modeling. Residues within 5 Å of the bound ligand in the cryo-EM structure were selected as flexible for ChiFlex/ChiRotor processing. Docking regions for the two binding sites were defined using a spherical selection with a radius of 20 Å centered on the geometric center of LTE4 and oxoglutarate. For each receptor, multiple low-energy conformations were generated by ChiFlex. Ligand conformations were generated using the BEST mode in CatConf. Rigid docking poses were generated using LibDock based on binding-site hotspots, which were clustered and filtered before a final refinement stage using CDOCKER.
After the flexible docking, poses were sorted by -CDOCKER_ENERGY, and the top-ranked poses were selected for binding free energy calculations. The “Calculate Binding Energies” section in Discovery Studio estimates the free energy of binding for a receptor-ligand complex by calculating the free energies of the complex, the receptor, and the ligand. Binding free energies were computed using CHARMM-based molecular mechanics and implicit solvation via the PBSA model. Ligand conformational entropy was included in the estimation by enabling the “Estimate Entropy” and “Ligand Conformational Entropy” options. The reported energy terms include van der Waals, electrostatic, ligand strain, receptor strain, and solvation contributions, providing an overall estimate of binding energy for each pose. The poses with the lowest binding energy were selected as the final poses for further analysis.
Molecular dynamics (MD) simulations and analysis
Molecular dynamics (MD) simulations started with the atomic coordinates of LTE4 and GPR99 extracted from the cryo-EM structure of LTE4-GPR99-Gq complex resolved in this study; the Gq protein heterotrimer complex was removed before the system setup. The protonation states of the ligand-receptor complex were assigned for a pH of 7.4 using the H + + web server. Next, the CHARMM-GUI membrane builder was used to embed the complex within a POPC (1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine) lipid bilayer59. The entire system was then solvated in a periodic water box (TIP3P model) containing 0.1 M NaCl. CHARMM36m force field was applied to model the system. Simulations were produced on GPUs using GROMACS (version 2024.2)60. The system was maintained at 310 K and 1.0 bar, and energy minimized in 10,000 steps. Then a 200-ns restrained MD simulations was performed to equilibrate and relax the system. After initial energy minimization and equilibration, three independent production simulations were performed, each lasting 1 μs. The production runs were conducted under an NPT ensemble, using the leap-frog algorithm. Bonds involving hydrogen atoms were constrained using the LINCS algorithm. Long-range electrostatic interactions were computed with the Particle Mesh Ewald (PME) method, while a 1.2 nm cutoff was used for short-range van der Waals and Coulombic interactions. The stability of the ligand-receptor complex was monitored throughout the simulations by calculating the root mean square deviation (RMSD). Trajectory frames were first aligned to the protein backbone Cα atoms of the initial structure, and the ligand’s RMSD was then computed relative to its starting pose. Analysis of these trajectories confirmed that LTE4 consistently adopted a stable conformation in the alternative pocket above ICL2 of GPR99. Initial coordinate and simulation input files and a coordinate file of the final output of molecular dynamics simulations are provided as Supplementary Data 1.
Statistical analysis
The data analysis was performed using Prism 9.5.0 (GraphPad, San Diego, CA). Dose-response curves for agonist analysis were generated using the log[agonist] vs. response equation (three parameters) within the software. For the IP1 and cAMP assay, data points were expressed as percentages (mean ± SEM) relative to the maximum response level for each sample, based on at least three independent experiments, as indicated in the figure legends. For NanoBiT assays, data points were expressed as fold changes (mean ± SEM) relative to negative control (cells treated without ligands) for each sample, based on at least three independent experiments, as indicated in the figure legends. EC50 values were derived from the dose-response curves. For cell surface expression, data points were shown as percentages (mean ± SEM) of the flow cytometry fluorescence signals of wild-type GPR99. Statistical comparisons were made using one-way analysis of variance (ANOVA). A p-value of 0.05 or less was considered statistically significant.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The atomic coordinates for the oxoglutarate-GPR99-Gq complex and the LTE4-GPR99-Gq complex have been deposited in the Protein Data Bank with accession codes 8YYW and 8YYX, respectively. The corresponding EM maps have been deposited in the Electron Microscopy Data Bank with accession codes EMD-39681 and EMD-39682, respectively. All data needed to evaluate the conclusions in the paper are present in the main text or the supplementary materials. Initial coordinate and simulation input files and a coordinate file of the final output of molecular dynamics simulations are provided as Supplementary Data 1. Source data are provided with this paper.
References
Wittenberger, T. et al. GPR99, a new G protein-coupled receptor with homology to a new subgroup of nucleotide receptors. BMC Genomics 3, 17 (2002).
Abbracchio, M. P. et al. The recently deorphanized GPR80 (GPR99) proposed to be the P2Y15 receptor is not a genuine P2Y receptor. Trends Pharm. Sci. 26, 8–9 (2005).
He, W. et al. Citric acid cycle intermediates as ligands for orphan G-protein-coupled receptors. Nature 429, 188–193 (2004).
Tokonami, N. et al. alpha-Ketoglutarate regulates acid-base balance through an intrarenal paracrine mechanism. J. Clin. Invest 123, 3166–3171 (2013).
Davenport, A. P. et al. International Union of Basic and Clinical Pharmacology. LXXXVIII. G protein-coupled receptor list: recommendations for new pairings with cognate ligands. Pharm. Rev. 65, 967–986 (2013).
Shirasaki, H., Kanaizumi, E. & Himi, T. Expression and localization of GPR99 in human nasal mucosa. Auris Nasus Larynx. 44, 162–167 (2017).
Kanaoka, Y. & Austen, K. F. Roles of cysteinyl leukotrienes and their receptors in immune cell-related functions. Adv. Immunol. 142, 65–84 (2019).
Guerrero, A. et al. alpha-Ketoglutarate Upregulates Collecting Duct (Pro)renin Receptor Expression, Tubular Angiotensin II Formation, and Na(+) Reabsorption During High Glucose Conditions. Front. Cardiovasc. Med. 8, 644797 (2021).
Kanaoka, Y., Maekawa, A. & Austen, K. F. Identification of GPR99 protein as a potential third cysteinyl leukotriene receptor with a preference for leukotriene E4 ligand. J. Biol. Chem. 288, 10967–10972 (2013).
Montuschi, P. Role of Leukotrienes and Leukotriene Modifiers in Asthma. Pharm. (Basel) 3, 1792–1811 (2010).
Lee, M., Boyce, J. A. & Barrett, N. A. Cysteinyl Leukotrienes in Allergic Inflammation. Annu Rev. Pathol. 20, 115–141 (2025).
Lee, T. H., Woszczek, G. & Farooque, S. P. Leukotriene E4: perspective on the forgotten mediator. J. Allergy Clin. Immunol. 124, 417–421 (2009).
Yuan, Y. et al. Exercise-induced alpha-ketoglutaric acid stimulates muscle hypertrophy and fat loss through OXGR1-dependent adrenal activation. EMBO J. 39, e103304 (2020).
Husted, A. S., Trauelsen, M., Rudenko, O., Hjorth, S. A. & Schwartz, T. W. GPCR-Mediated Signaling of Metabolites. Cell Metab. 25, 777–796 (2017).
Yokomizo, T., Nakamura, M. & Shimizu, T. Leukotriene receptors as potential therapeutic targets. J. Clin. Invest 128, 2691–2701 (2018).
Arm, J. P. et al. Asthmatic airways have a disproportionate hyperresponsiveness to LTE4, as compared with normal airways, but not to LTC4, LTD4, methacholine, and histamine. Am. Rev. Respir. Dis. 142, 1112–1118 (1990).
Maekawa, A., Kanaoka, Y., Xing, W. & Austen, K. F. Functional recognition of a distinct receptor preferential for leukotriene E4 in mice lacking the cysteinyl leukotriene 1 and 2 receptors. Proc. Natl. Acad. Sci. USA 105, 16695–16700 (2008).
Yamamoto, T., Miyata, J., Arita, M., Fukunaga, K. & Kawana, A. Current state and future prospect of the therapeutic strategy targeting cysteinyl leukotriene metabolism in asthma. Respir. Investig. 57, 534–543 (2019).
Varney, M. J. & Benovic, J. L. The Role of G Protein-Coupled Receptors and Receptor Kinases in Pancreatic beta-Cell Function and Diabetes. Pharm. Rev. 76, 267–299 (2024).
Gusach, A. et al. Structural basis of ligand selectivity and disease mutations in cysteinyl leukotriene receptors. Nat. Commun. 10, 5573 (2019).
Luginina, A. et al. Structure-based mechanism of cysteinyl leukotriene receptor inhibition by antiasthmatic drugs. Sci. Adv. 5, eaax2518 (2019).
Jiang, M. et al. Structural basis of the cysteinyl leukotriene receptor type 2 activation by LTD4. Proc. Natl. Acad. Sci. USA 122, e2417148122 (2025).
Ballesteros, J. A. & Weinstein, H. in Methods in Neurosciences Vol. 25 (ed Stuart C. Sealfon) 366-428 (Academic Press, 1995).
Haffke, M. et al. Structural basis of species-selective antagonist binding to the succinate receptor. Nature 574, 581–585 (2019).
Liu, A., Liu, Y., Zhang, W. & Ye, R. D. Structural insights into ligand recognition and activation of the succinate receptor SUCNR1. Cell Rep. 43, 114381 (2024).
Yokomizo, T., Izumi, T., Chang, K., Takuwa, Y. & Shimizu, T. A G-protein-coupled receptor for leukotriene B4 that mediates chemotaxis. Nature 387, 620–624 (1997).
Luginina, A. et al. Structural diversity of leukotriene G-protein coupled receptors. J. Biol. Chem. 299, 105247 (2023).
Li, C. et al. Molecular basis of ligand recognition and activation of the human succinate receptor SUCR1. Cell Res 34, 594–596 (2024).
Bokoch, G. M. & Gilman, A. G. Inhibition of receptor-mediated release of arachidonic acid by pertussis toxin. Cell 39, 301–308 (1984).
Inoue, A. et al. Illuminating G-Protein-Coupling Selectivity of GPCRs. Cell 177, 1933–1947 e1925 (2019).
Liu, A. et al. Structural insights into KSHV-GPCR constitutive activation and CXCL1 chemokine recognition. Proc. Natl. Acad. Sci. USA 121, e2403217121 (2024).
Shenol, A. et al. Molecular dynamics-based identification of binding pathways and two distinct high-affinity sites for succinate in succinate receptor 1/GPR91. Mol. Cell 84, 955–966 e954 (2024).
Trauelsen, M. et al. Receptor structure-based discovery of non-metabolite agonists for the succinate receptor GPR91. Mol. Metab. 6, 1585–1596 (2017).
Pu, S. et al. Montelukast prevents mice against carbon tetrachloride- and methionine-choline deficient diet-induced liver fibrosis: Reducing hepatic stellate cell activation and inflammation. Life Sci. 325, 121772 (2023).
Bankova, L. G. et al. Leukotriene E4 elicits respiratory epithelial cell mucin release through the G-protein-coupled receptor, GPR99. Proc. Natl. Acad. Sci. USA 113, 6242–6247 (2016).
Sasaki, F. & Yokomizo, T. The leukotriene receptors as therapeutic targets of inflammatory diseases. Int Immunol. 31, 607–615 (2019).
Lynch, K. R. et al. Characterization of the human cysteinyl leukotriene CysLT1 receptor. Nature 399, 789–793 (1999).
Heise, C. E. et al. Characterization of the human cysteinyl leukotriene 2 receptor. J. Biol. Chem. 275, 30531–30536 (2000).
Qiao, A. et al. Structural basis of G(s) and G(i) recognition by the human glucagon receptor. Science 367, 1346–1352 (2020).
Slosky, L. M., Caron, M. G. & Barak, L. S. Biased Allosteric Modulators: New Frontiers in GPCR Drug Discovery. Trends Pharmacol. Sci. 42, 283–299 (2021).
Aranda-Garcia, D. et al. Large scale investigation of GPCR molecular dynamics data uncovers allosteric sites and lateral gateways. Nat. Commun. 16, 2020 (2025).
Conflitti, P. et al. Functional dynamics of G protein-coupled receptors reveal new routes for drug discovery. Nat. Rev. Drug Discov. https://doi.org/10.1038/s41573-024-01083-3 (2025).
Kugawa, M. et al. Structural insights into lipid chain-length selectivity and allosteric regulation of FFA2. Nat. Commun. 16, https://doi.org/10.1038/s41467-025-57983-4 (2025).
Zheng, Y. et al. Development of an allosteric adhesion GPCR nanobody with therapeutic potential. Nat. Chem. Biol. https://doi.org/10.1038/s41589-025-01896-2 (2025).
Lopez-Balastegui, M. et al. Relevance of G protein-coupled receptor (GPCR) dynamics for receptor activation, signalling bias and allosteric modulation. Br. J. Pharm. https://doi.org/10.1111/bph.16495 (2024).
Maeda, S. et al. Endogenous agonist-bound S1PR3 structure reveals determinants of G protein-subtype bias. Sci Adv 7, https://doi.org/10.1126/sciadv.abf5325 (2021).
Ti, R. et al. Fine-tuning activation specificity of G-protein-coupled receptors via automated path searching. Proc. Natl. Acad. Sci. USA 121, e2317893121 (2024).
Zhao, C. et al. Structural insights into sphingosine-1-phosphate recognition and ligand selectivity of S1PR3-Gi signaling complexes. Cell Res 32, 218–221 (2022).
Chen, G. et al. Activation and allosteric regulation of the orphan GPR88-Gi1 signaling complex. Nat. Commun. 13, 2375 (2022).
Mao, C. et al. Unsaturated bond recognition leads to biased signal in a fatty acid receptor. Science 380, eadd6220 (2023).
Cao, C. et al. Structure, function and pharmacology of human itch GPCRs. Nature 600, 170–175 (2021).
Kim, K. et al. Structure of a Hallucinogen-Activated Gq-Coupled 5-HT(2A) Serotonin Receptor. Cell 182, 1574–1588 e1519 (2020).
Punjani, A., Rubinstein, J. L., Fleet, D. J. & Brubaker, M. A. cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296 (2017).
Kimanius, D., Dong, L., Sharov, G., Nakane, T. & Scheres, S. H. W. New tools for automated cryo-EM single-particle analysis in RELION-4.0. Biochem J. 478, 4169–4185 (2021).
Pettersen, E. F. et al. UCSF Chimera-a visualization system for exploratory research and analysis. J. Comput Chem. 25, 1605–1612 (2004).
Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. Features and development of Coot. Acta Crystallogr. D. Biol. Crystallogr. 66, 486–501 (2010).
Liebschner, D. et al. Macromolecular structure determination using X-rays, neutrons and electrons: recent developments in Phenix. Acta Crystallogr D. Struct. Biol. 75, 861–877 (2019).
Chen, V. B. et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D. Biol. Crystallogr. 66, 12–21 (2010).
Park, S., Choi, Y. K., Kim, S., Lee, J. & Im, W. CHARMM-GUI Membrane Builder for Lipid Nanoparticles with Ionizable Cationic Lipids and PEGylated Lipids. J. Chem. Inf. Model 61, 5192–5202 (2021).
Abraham, M. J. et al. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1-2, 19–25 (2015).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (grant number: 82402140), The Medical Scientific Research Foundation of Guangdong Province, China (grant number: B2025178), Special Project for Clinical and Basic Sci & Tech Innovation of Guangdong Medical University (grant number: GDMULCJC2024121, GDMULCJC2024156 and GDMULCJC2025140), Shenzhen Science and Technology Program Grant No. RCBS20221008093330067 (to A.L.). This work was also supported by Shenzhen Medical Research Fund SMRF-D2403009 (to R.D.Y.), Yunnan Key Research and Development Program Grant 202402AA310032 (to R.D.Y.), Ganghong Young Scholar Development Fund (R.D.Y.), Kobilka Institute of Innovative Drug Discovery at The Chinese University of Hong Kong, Shenzhen (R.D.Y.), fund from Shenzhen-Hong Kong Cooperation Zone for Technology and Innovation (HZQB-KCZYB-2020056), and China Postdoctoral Science Foundation 2022M713049 (to A.L.) and start-up funds from Guangdong Medical University (to A.L.).
Author information
Authors and Affiliations
Contributions
A.L. and R.D.Y. conceived, initiated, and designed the project. A.L. and R.D.Y. supervised the study. A.L., Y. Liu and Y. Long performed the experiments, analysis the data and prepare the figures. A.L., Y. Liu and R.D.Y. wrote the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declares no competing interests.
Peer review
Peer review information
Nature Communications thanks Slawomir Filipek, Anastasiia Gusach and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Liu, A., Liu, Y., Long, Y. et al. Molecular insights into ligand recognition and signaling of OXGR1. Nat Commun 16, 11205 (2025). https://doi.org/10.1038/s41467-025-67101-z
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-67101-z
