
Abstract
Atmospheric multiphase buffering capacity is a crucial physicochemical property of aerosol that significantly influences the Earth’s radiative budget, climate, air quality, and public health. However, its implications for air pollution remain largely unexplored. Here, we present four years of continuous observation and laboratory experiments as evidence that reactive nitrogen (Nr)-driven atmospheric multiphase buffering can induce unintended Concomitant Pollution. Specifically, gaseous pollutants undergo phase partitioning into aerosol aqueous phases during the process of pH change under buffering constraints, subsequently transforming into concomitant particulate pollution. Furthermore, from a global perspective, we reveal that NH4+/NH3-induced Concomitant Pollution exhibits greater intensity in South Asia during winter, while in summer, it intensifies in North America, Europe, and East Asia. And anthropogenic emissions, particularly buffering agents like NH3, exert a greater impact on Concomitant Pollution across continents than meteorological factors. Thus, the relentless rise in global NH3 emissions has positioned Concomitant Pollution as an urgent challenge.
Similar content being viewed by others
Introduction
Aerosol acidity (defined as aerosol pH), a critical regulator of terrestrial ecosystem dynamics, operates at the nexus of the Earth’s radiative budget, atmospheric chemistry, biogeochemical cycling, and climate feedbacks and public health1,2,3,4,5,6. While anthropogenic emissions of sulfur, nitrogen, and alkaline species (e.g., ammonia (NH3), dust) drive large-scale perturbations in aerosol pH, emerging evidence reveals a counterintuitive planetary feedback: aerosols possess the capacity to autonomously resist external acid-base disturbances through self-regulating proton (H+) exchange at gas-aqueous-particle interfaces7, known as atmospheric multiphase buffering capacity. This multiphase buffering capacity, enabled by dynamic buffering agents (conjugate acid-base pairs, including NH4+/NH3, HNO3/NO3−, and HSO4−/SO42−) that shuttle protons across phases7,8 (Fig. 1a), acts as an atmospheric chemical thermostat, stabilizing aerosol acidity against anthropogenic emission disturbance while recalibrating gas-to-particle partitioning of and climate-altering organics. Notably, NH4+, NH3, HNO3, and NO3− are key reactive nitrogen (Nr) species9, with multiphase buffering thereby driving phase-selective reconfiguration of Nr concentrations across gas-particle interfaces and affecting nitrogen deposition10. Deciphering the multiphase buffering deeply is thus pivotal for predicting cascading failures in air quality governance, carbon-climate feedbacks, and the acidification tipping points of vulnerable biomes.

a Schematic of the buffering mechanism. b Temporal variation trend of aerosol pH and Acid-Base buffering capacity at different PM2.5 concentration levels. c Decadal evolution of dominant buffering pairs. d The buffering capacity β curve for the average during the entire observation period.
In recent years, the scientific community has started providing important knowledge on the multiphase buffering capacity of aerosols. Studies commonly focus on the mechanistic interactions between multiphase buffering capacity and aerosol pH11,12. For example, there is literature revealing mechanisms behind the mismatch between changed NH3, nitrogen oxides (NOx), and sulfur dioxide (SO2) emissions and stable aerosol acidity in the southeastern United States and Canada based on multiple buffering capacity11, which represents only a part of the extensive impacts. Furthermore, subsequent research has expanded to examine the role of multiple buffering capacity in chemical reaction kinetics, emphasizing its significance in maintaining stable aerosol pH and increasing overall sulfate production across diverse regions, including the southeastern United States, North China Plain, northern India, western Europe, and other regions worldwide13. Notwithstanding the findings from earlier studies, however, a comprehensive understanding of the effects of emerging multiphase buffering capacity in the atmosphere has remained enigmatic. This study seeks to establish the critical significance of multiphase buffering capacity in regulating air pollution dynamics.
Since the industrial revolution, atmospheric pollution has gradually become a prominent issue, but its causative factors have evolved incrementally and vary distinctly across contexts. The study of the Los Angeles smog in 1943 led people to realize that some components of the atmosphere, through chemical reactions involving sunlight14, can be converted into harmful pollutants that further impact the environment. Another classic event is the London Smog in 1952, where they have since found that it’s not the SO2 directly emitted by coal burning, but the secondary sulfates formed by SO2 chemical conversion that caused the air pollution15. A more recent case is the severe photochemical haze pollution16,17, which is affected by the emissions of primary pollutants and the formation of secondary particulate matter (PM2.5) from gaseous and multi-phase heterogeneous reactions and hygroscopic growth of aerosols15,18,19. Notably, the rapid episodic enhancement of secondary inorganic aerosols (SIAs)-dominated by ammonium nitrate bursts-has emerged as a persistent enigma, which may be related to the interconversion between Nr species20. Our previous research has also shown that the chemical reactions of Nr in the atmospheric nitrogen cycle can regulate the formation of PM2.5-O3 co-pollution21. Here, inspired by the theory of multiple buffers, we propose a viewpoint that unintended concomitant pollution (defined as an increase in PM2.5 mass concentration resulting from the gas-to-particle conversion of buffering agents during aerosol acidity stabilization) always emerges during atmospheric multiphase buffering processes driven by key Nr species (Fig. 1a), through combining long-time field observations and global model analyses. Consequently, we have sufficient reason to suspect that unintended concomitant pollution caused by the multiphase buffering process could emerge as a critical driver behind the anomalously rapid SIAs accumulation observed in recent winter haze episodes.
To address this issue, we conduct exploration and discover a trade-off effect between buffering capacity and PM2.5 concentration, as well as a response mechanism between the two issues. Surprisingly, we find that concomitant pollution can be triggered by negative response mechanisms between buffering capacity and air pollution during the pH buffering process, thereby inadvertently exacerbating the pollution. Transmission electron microscopy experiments also provide solid evidence to confirm this phenomenon. Machine learning emphasizes the importance of relative humidity (RH), NH3, and temperature (T) in concomitant pollution. In addition, we further investigate the global spatiotemporal patterns of concomitant pollution. We provide conclusive quantitative evidence demonstrating that concomitant pollution caused by NH4+/NH3 is stronger than that caused by HNO3/NO3−. The concomitant pollution in summer is stronger than in winter, especially prevalent in North America, Europe, South Africa, and East Asia. The theoretical mechanism analysis disentangled the effects of complex and diverse factors on concomitant pollution on a global scale, highlighting that the anthropogenic factor (AF), buffering agent NH3, plays a predominant role in concomitant pollution. Agricultural sources are the main source of NH3, the 2024 Statistical Yearbook released by the Food and Agriculture Organization of the United Nations reports that inorganic fertilizers used in agriculture reached 185 million tonnes of nutrients in 2022, with 58% of this amount being nitrogen, which represents an increase of 37% compared with 200022. The NH4+/NH3 buffering-induced concomitant pollution highlights a self-reinforcing cycle: agricultural NH3 emissions sustain buffering capacity while simultaneously fueling ammonium nitrate-driven PM2.5 growth, creating policy dilemmas for simultaneous air quality and food security goals. Therefore, we propose that prior prevention and control of NH3 represents a crucial strategy for alleviating global secondary pollution and achieving the World Health Organization’s PM2.5 global air quality guidelines by 2050. This study not only advances our fundamental understanding of atmospheric chemistry but also provides a scientific basis for developing more effective air pollution control strategies in urban environments.
Results and discussion
Declining buffering capacity precipitates concomitant pollution
To advance our understanding of the complex interactions governing buffering capacity and particulate pollution, we monitored buffering capacity (β) and aerosol pH dynamics across PM2.5 gradients, applying the multiphase buffer theory framework established by Zheng et al.7 (see Methods). Utilizing four years long-term observation data (2018–2021), we quantified the total buffering capacity β values and individual buffering capacity β of three buffer agents NH4+/NH3 or HNO3/NO3−, HSO4−/SO42− (Supplementary Fig. 1). Aerosol pH displayed an initial marginal increase, but then stabilized as the level of pollution worsened (Fig. 1b). Conversely, β manifested a pronounced decreasing pattern, undergoing a marked 49% decline in response to rising PM2.5 levels reaching approximately 200 μg m−3. This period was considered the pollution accumulation period. Interestingly, β sustained substantially depressed levels across the severe pollution period. We hypothesized that there exists an inverse systemic interaction between β and PM2.5 concentration. However, throughout the entire observation period, aerosol acidity maintained remarkable consistency. This phenomenon was consistently observed across almost all of the four years in this study (Supplementary Fig. 2), and was systematically evidenced by the diurnal variations of β and PM2.5 concentration (Supplementary Fig. 3).
Through systematic quantitative assessment of different buffering agents’ regulatory capacity, we found NH4+/NH3 dominated buffering capacity β, accounting for 80% of the total β, followed by HNO3/NO3−, with almost negligible contribution from HSO4−/SO42− (Fig. 1c). This can be attributed to anthropogenic activities have significantly augmented Nr, predominantly NOx and NH323,24, since pre-industrial times. NOx originates primarily from fossil fuel combustion in energy systems25, NH3 emissions are dominated by agricultural sources, including volatilization from livestock excreta and synthetic nitrogen fertilizers25,26,27, collectively constituting >80% of anthropogenic NH3 releases28. The centrality of NH4+/NH3 as the dominant buffering agent can also be confirmed on pH–β curves (Fig. 1d and Supplementary Fig. 4). Therefore, these findings establish NH4+/NH3 as the principal regulator of aerosol acidity and a critical amplifier of pollution episodes in this work, which was consistent with Gu et al.29. Above quantitative analysis reveals that atmospheric multiphase buffering can induce concomitant air pollution.
Mechanisms of pH buffering-induced concomitant pollution
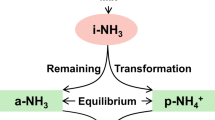
We systematically investigated the mechanistic basis of pH buffering-induced concomitant pollution. Based on nine pollution events during the four years of long-term observation, we found that the trade-off effect between buffering capacity and PM2.5 concentration is universal in the ambient atmosphere (Supplementary Figs. 5–7 and Text 2), which regulates aerosol acidity and air pollution. Therefore, we proposed a novel perspective that the dynamic exchange between gas-phase and particle-phase buffering agents plays a pivotal role in both exacerbating and alleviating air pollution during the acid-base buffering process. To better uncover this response mechanism, we first unveiled the essence of buffering for NH4+/NH3. Wherein, the chemical mechanism of acid buffering is that H+ can be depleted by bonding with NH3 to form NH4+ when acid (such as SO42− from other sources) is added into the liquid phase of the aerosol30,31, thereby maintaining the stability of the aerosol acidity. In this process, since NH3 enters the aerosol phase to form NH4+, it amplifies the load of inorganic PM2.5. This process was termed the “acid-base buffering-air pollution” negative response mechanism, characterized by triggering of concomitant pollution (Fig. 2a). Likewise, for base buffering, the OH- brought by base (such as calcium ions (Ca2+) from other sources) will be neutralized by to generate H2O and release NH3 to gas phase, which maintains the stability of the aerosol acidity. In this process, since NH4+ escapes into the gas phase as NH3, it suppresses inorganic PM2.5 formation. This process was termed the “acid-base buffering-air pollution” positive response mechanism, connected with pollution diminishing. As a result, pH values can be stabilized within a specific range without large fluctuations despite the addition of abundant acids or bases, while PM2.5 loading can be either enhanced or reduced (Supplementary Text 3 and Supplementary Figs. 8–10). We have incorporated high-time-resolution online monitoring experiments to conduct an in-depth investigation of the negative response mechanism (Supplementary Text 4 and Supplementary Figs. 11–13). Our experimental results demonstrate the validity of this response mechanism.

a Conceptual schematic of the mechanism of concomitant pollution in the negative response mechanism, based on the common trends observed across the nine studied events. This schematic diagram illustrates the trade-off relationship between pollution levels and buffering capacity. As pollution intensifies, the buffering capacity (yellow curve) exhibits a declining trend. The total pollution burden consists of secondary pollution (pink curve) and concomitant pollution (blue curve). b, c Field emission transmission electron microscopy experiment. The samples during the cleaning period were collected at a fine particulate matter (PM2.5) of 31 μg m−3 on October 30, 2023, while the samples during the pollution period were collected at a PM2.5 of 185 μg m−3 on the same day. All other sampling conditions were the same. d Probability density distribution (Bin width = 10) of the concomitant pollution induced by a slight change in pH (ΔpH = 0.5) under the NH4+/NH3-dominated buffering regime during the clean period. The blue bars represent the distribution of this concomitant pollution. The x-axis indicates the magnitude of the concomitant pollution triggered by the pH perturbation, while the y-axis represents the corresponding probability density.
Long-term observational data (2018–2021) provide quantitative evidence for substantiating the emergence of concomitant pollution. The ε(NH4+) (fraction of total ammonia (NH4T) that partitions to the particle phase) and PM2.5 concentration showed synchronous variation patterns (Supplementary Fig. 14). From the clean stage to the pollution stage, the aerosol pH value regulated by the buffering agent NH4+/ NH3 remained relatively stable (Supplementary Fig. 15), fluctuating within a narrow range of approximately 2 units (Supplementary Fig. 16) (see Supplementary Text 5 and “Materials and Methods” for specific calculations), which sustained consistently high nitrate partitioning efficiency (ε(NO3−) ≈ 0.9) (Supplementary Fig. 17). Crucially, ε(NH4+) showed a remarkable increasing trend (approximately from 0.5 to 0.7) with the increment of PM2.5 concentration (Supplementary Fig. 14), reflecting that a large amount of NH3 was converted into NH4+ to neutralize the acidic components formed in the atmosphere, that is, to form the concomitant nitrate particulate pollution. We also utilize single-particle transmission electron microscopy to provide technical verification of these findings: samples from the clean period mostly exhibit a circular shape and have a slightly elemental mass, while samples from the polluted period predominantly show irregular shapes and heavier elemental mass (Fig. 2b, c and Supplementary Fig. 18). Therefore, we have ample reason to believe that long-term observation and laboratory analysis results are strong evidence that buffering capacity can bring unexpected concomitant pollution while maintaining aerosol pH stability. Notably, through theoretical calculations, we found that the concomitant pollution caused by buffering agents varied within the range of approximately 0–200 mol kg−1 (about 0–13 μg m−3 after conversion) when causing 0.5 pH changes, with an average of 23.2 mol kg−1, during clean period (PM2.5 ≤ 75 μg m−3) (Fig. 2d). Therefore, quantitative theoretical analysis mechanistically demonstrated that atmospheric multiphase buffering can induce serve concomitant pollution.
Deciphering concomitant pollution drivers with machine learning
Disentangling the drivers behind this concomitant pollution change requires understanding the role of influencing factors. To this end, we assessed the importance of factors, including meteorological conditions (T, RH), non-volatile ions (SO42−, Ca2+, etc., emitted from pollution sources), volatile compound (HNO3/NO3−), and concentration of the buffering agents (NH4T), using a random forest (RF) algorithm with partial dependence plot (PDP) analysis. Factor importance rankings revealed that RH contributed the most to concomitant pollution, followed by NH4T and T, while Ca2+, SO42−, and NO3T exerted minimal influence (Supplementary Fig. 20), indicating that the variations in T, RH, and NH4T significantly influence the concomitant pollution. We further applied the random forest-partial dependence plot (RF-PDP) method to describe the response of concomitant pollution to drivers (Fig. 3). The ten-fold cross-validation was applied to ensure the generalization ability and reliability of the model, with average R2 values for the training set and test set being 0.97 and 0.80 (Supplementary Fig. 21), respectively. These data-driven methods provided basic information for the change of concomitant pollution. RF-PDP simulations demonstrated nonlinear concomitant pollution responses: the increase of RH, NO3T, and SO42− dampened concomitant pollution increased, while the effects of T, NH4T, and Ca2+ were opposite (Fig. 3). We found that when RH increased from 20% to 40%, concomitant pollution decreased significantly by 72.1 mol kg−1, while the changes in concomitant pollution caused by variations in other factors were basically below 15 mol kg−1. Additionally, to gain a deeper understanding of the response of concomitant pollution to driver change, from a theoretical mechanism perspective, we constructed a thermodynamic model (see “Methods”) and quantified the effects of various factors on concomitant pollution through sensitivity simulation experiments (Supplementary Text 6 and Supplementary Figs. 22–27). The results indicate that changes in these factors primarily influence concomitant pollution by affecting pH and aerosol water content (AWC). For instance, increases in RH, SO42−, and NO3T compounds enhance aerosol acidity and AWC, leading to a reduction in the molar concentration of buffering agents and consequently a decrease in concomitant pollution. In contrast, the effects of NH4T and Ca2+ are the opposite. The sensitivity experiments for T show some discrepancies with the machine learning results, which may be because the RF-PDP simulations reflect the average concomitant pollution outcomes across multiple data points, whereas the sensitivity experiments demonstrate the actual variations in concomitant pollution under specific scenarios. The results for sensitivity simulation experiments mirrored those obtained for machine learning, corroborating the findings. Altogether, this dual-method convergence establishes RH, NH4T, and T as pivotal levers in concomitant pollution modulation, a finding critical for targeted pollution mitigation in anthropogenically perturbed atmospheres.

a Relative humidity (RH), b Temperature (T), c NH4T, d NO3T, e SO42−, and f Ca2+. The solid line represents the mean prediction, and the shaded region indicates ±1 standard deviation of the predictions across all Bootstrap iterations, reflecting model uncertainty due to data variability.
It should be noted that the above analysis was based on our long-term observations, and this situation mainly applies to regions with abundant ammonia emissions, which are dominated by NH4+/NH3 buffering. However, the dominant buffering agents and their drivers exhibit spatiotemporal heterogeneity globally. Despite this diversity, the underlying mechanisms are fundamentally the same: concomitant pollution is governed by a negative response mechanism, in which gas-phase buffering agents (NH3 or HNO3) are exchanged for particulate-phase counterparts (NH4+ or NO3−) to preserve aerosol acidity stability. Building on this framework, we further extend our investigation to global scales, evaluating region-specific buffering regimes, concomitant pollution, and their environmental implications in various regions worldwide.
Global spatiotemporal pattern and driving of concomitant pollution
We further combined response mechanisms with global simulation to explore the spatiotemporal pattern of concomitant pollution and its driving factors from a global perspective. Disparate dominated regimes of buffering capacity prevailed in different regions and seasons, depending on meteorological conditions, aerosol properties, and buffer agent concentrations. This raises critical questions regarding the spatiotemporal patterns of concomitant pollution across global regions. Supplementary Fig. 28 and Text S7 showed the global distribution of the buffering capacity in January, April, July, and October 2019. The analysis revealed that NH4+/NH3 constitutes the primary source of buffering capacity, followed sequentially by HNO3/NO3−, whereas HSO4−/SO42− makes a substantially reduced contribution globally (Supplementary Figs. 29–31). Additionally, buffering capacity levels are relatively low in January and high in July, with April and October showing similar conditions (Supplementary Figs. 28–31).
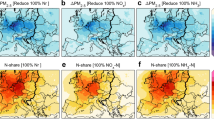
We presented the global distribution of NH4+/NH3 buffering-induced concomitant pollution (Co-P-NH4+/NH3) and HNO3/NO3− buffering-induced concomitant pollution (Co-P-HNO3/NO3−) in winter (January) and summer (July). The result indicated that the Co-P-NH4+/NH3 was stronger than Co-P-HNO3/NO3−, and concomitant pollution in summer was higher than that in winter (Fig. 4). Co-P-NH4+/NH3 Hotspots occurred across the globe in areas characterized by dense populations and intensive agricultural activities32,33, most notably in South Asia but also United States, Mexico, and Brazil in winter, which aligned spatially with buffering capacity distribution. Meanwhile, the Co-P-NH4+/NH3 at 40°–60°N in winter remained weak, but remained at a high level in summer due to the seasonal NH3 emission cycles in these areas (Supplementary Fig. 32). Overall, the Co-P-HNO3/NO3− was weaker than Co-P-NH4+/NH3, which is consistent with their respective contributions to buffering capacity. Furthermore, the Co-P-HNO3/NO3− was weaker in winter than in summer, which was similar to the seasonal variation in Co-P-NH4+/NH3. The weaker Co-P in winter is associated with the typical meteorological conditions of this season. Such an atmospheric environment greatly promotes the conversion process of gaseous components to the particulate phase. This rapid and efficient conversion mechanism effectively consumes the Co-P in the atmosphere, resulting in a relatively low level of Co-P. In winter, the Co-P-HNO3/NO3− was stronger in Central Africa and South Asia. In summer, strong Co-P-HNO3/NO3− appeared in South Africa, Europe, and Central North China, which mirrors HNO3 source regions (Supplementary Fig. 33). Another interesting phenomenon was that during summer, areas with strong concomitant pollution gradually expand towards the north latitude, which may also be related to the increase in temperature. Therefore, on a global scale, concomitant pollution was relatively weak in winter and strong in summer. And Co-P-NH4+/NH3 was stronger than Co-P-HNO3/NO3−. Meanwhile, we found that the Co-P-NH4+/NH3 can reach up to about 200 mol kg−1 when causing 0.5 pH changes in winter, while Co-P-HNO3/NO3− can only reach up to 100 mol kg−1. When subjected to equivalent pH changes, Co-P-NH4+/NH3 and Co-P-HNO3/NO3− concentrations during summer periods remain predominantly below the 50 mol kg−1 threshold (Supplementary Fig. 34). This hierarchy reflects the thermodynamic dominance of NH4+/NH3 buffering in aerosol acidity regulation and air pollution formation, consistent with their relative β contributions.

a Concomitant pollution caused by NH4+/NH3 in January. b In July. c Concomitant pollution caused by HNO3/NO3− in January. d In July.
We further discovered how driving factors affect the global distribution and seasonal variations of concomitant pollution. To address these distinctions, we decomposed the driving factors into meteorological factor (MF, consisting of T and AWC) and anthropogenic factor (AF, consisting of gas-phase buffering agents and ionic composition). A normalized influence metric, MF/AF (details in Methods), was developed to quantify their relative contributions. When MF/AF is greater than 1, MF have a greater influence; otherwise, AF play a more significant role.
Figure 5 delineates the global spatiotemporal patterns of indicator MF/AF, which characterizes the importance of factors on NH4+/NH3 buffering-induced concomitant pollution (Co-P-NH4+/NH3) and HNO3/NO3− buffering-induced concomitant pollution (Co-P-HNO3/NO3−) in winter (January) and summer (July). For Co-P-NH4+/NH3, in winter, the breakdown indicated that the areas with weak Co-P-NH4+/NH3 in January were mostly dominated by MFs (Fig. 5). In Russia and Canada, the cold air and increased water content hindered the rise of concomitant pollution. Conversely, the strong Co-P-NH4+/NH3 in South Asia stemmed from intensive agricultural activities emitting high NH3 concentrations34 (Supplementary Fig. 32). The contribution of MF to strong Co-P-NH4+/NH3 in North America was higher than that in South Asia, which exhibited mixed drivers. The combination of high temperature, low AWC, and appropriate aerosol acidity collectively contributes to the high levels of Co-P-NH4+/NH3 (Supplementary Fig. 35–37). The MF contributed more to Co-P-NH4+/NH3 in Brazil and South Africa, which may be due to the high T and low AWC environment (Supplementary Fig. 35–38). In summer, the contribution of MF to high Co-P-NH4+/NH3 was gradually increasing in North America and Europe, possibly because of increased T and diminished AWC (Supplementary Fig. 35–38). These meteorological conditions favor the partitioning of NH4+/NH3 as gaseous NH3 rather than aerosol-phase NH4+, elevating the NH4+/NH3 Concomitant Pollution Index. Additionally, high-temperature and low-humidity conditions enhance aerosol buffering capacity. Consequently, the elevated NH4+/NH3 Concomitant Pollution Index, combined with increased buffering capacity, synergistically intensifies concomitant pollution during summer. This dual mechanism explains the significantly higher concomitant pollution levels in regions like North America and Europe during summer compared to winter. Basically, most regions globally were dominated by AFs, which may be a result of the rising anthropogenic NH3 burdens worldwide (Supplementary Fig. 32). For Co-P-HNO3/NO3−, in winter, the contribution of AF to the strong Co-P-HNO3/NO3− gradually decreases poleward, mirroring Co-P-NH4+/NH3 gradients. The strong Co-P-HNO3/NO3− in Central Africa and South Asia was mainly dominated by AFs owing to the abundant concentration of HNO3 (Supplementary Fig. 33). Moreover, in the Sahara desert, the combination of a strong alkaline aerosol environment and low T results in relatively weak Co-P-HNO3/NO3−, and the contributions of MF and AF were comparable. In summer, the contribution of AF to Co-P-HNO3/NO3− gradually strengthened, possibly due to the increase in HNO3 concentration, such as in East Asia and North America. Increased HNO3 concentration was the result of enhanced photochemical reactions. The global distributions of oxidants OH radical indicated that the OH radical has undergone strong increases in summer compared to winter, especially in regions with latitudes between 0° and 60°N (Supplementary Fig. 39). The OH radical concentrations in East Asia, South Asia, Europe, and North America are significantly higher during summer than in winter (Supplementary Fig. 40). The elevated nitrate production rate for the photochemical reactions pathway further confirmed the above results (Supplementary Figs. 40 and 41).

a MF/AF to concomitant pollution caused by NH4+/NH3 in January. b In July. c MF/AF to concomitant pollution caused by HNO3/NO3− in January. d In July. In Figs (a–d), MF/AF serves as an indicator characterizing the relative contributions of meteorological factors (MF) and anthropogenic factors (AF), defined as the ratio of meteorological to anthropogenic factors. When MF/AF exceeds 1 (depicted in green in the figures), meteorological factors dominate; conversely (depicted in red), anthropogenic factors exert a greater influence. e The season variation of concomitant pollution, HNO3 concentration, and NH3 concentration in East Asia, South Asia, Europe, South Africa, and North America.
Implications of concomitant pollution
Our work reveals that pH buffering-induced concomitant pollution is indeed a critical driver behind the anomalously rapid SIAs accumulation observed in recent winter haze episodes. We find that the concomitant pollution can be triggered by the acid-base buffering-air pollution negative response mechanism, which paradoxically amplifies PM2.5 initiation and evolution. This phenomenon is mainly achieved by maintaining pH stability through the gas-liquid exchange of buffering agents composed of important reactive nitrogen species (NH4+/NH3 or HNO3/NO3−). Theoretical analysis and field emission transmission electron microscopy experiments have both confirmed our finding. Machine learning attribution analysis identifies RH as a key leverage point: curbing ammonia emissions during periods of low RH (<40%) could prove significantly more effective, which particularly emphasizes the timing of ammonia emissions controls in northern China. Sensitivity experiments and theoretical analysis also confirm this result (Supplementary Text S8; Supplementary Figs. 42 and 43). Strategic emission curtailment during meteorologically favorable windows (e.g., RH < 40%) could disrupt the self-reinforcing pollution-buffering cycle while maintaining ecosystem nitrogen thresholds. Therefore, this finding underscores the importance of preemptive action against gaseous buffering agents (e.g., NH3, NOx) before a rise in pollution levels.
Global spatiotemporal analysis reveals pronounced seasonality in concomitant pollution. Overall, the concomitant pollution in summer is higher than that in winter. Thus, NH4+/NH3-induced concomitant pollution in North America, Europe, South Africa, and East Asia requires more attention. In contrast, South Asia exhibits inverse seasonality, where winter NH3 emissions from intensive agriculture and biomass burning elevated concomitant pollution compared to monsoon seasons. Furthermore, the importance of anthropogenic emissions is emphasized globally. Global NH3 emissions have surged from 20.6 Tg N yr−1 in 1860 to 58.2 Tg N yr−1 in 1993, with current trajectories projecting a doubling to 118 Tg N yr−1 by 205035. Therefore, this threefold anthropogenic acceleration of buffering agent NH3-from preindustrial baselines to Anthropocene extremes—portends a commensurate amplification of PM pollution burdens. We estimated that the concomitant pollution caused by NH3 could reach up to about 24 μg m−3 by 2050 (based on the maximum concomitant pollution that can be caused by buffering 0.5 pH), which will be 1.8 times higher than in 2019. These findings corroborate the status of reactive nitrogen as a pressing global environmental challenge. As a response measure, the United Nations Environment Assembly has established an unprecedented ambition to halve anthropogenic nitrogen waste by 2030 in its Sustainable Nitrogen Management Resolution (UNEP/EA. 4/L16)36 and the 2019 Colombo Declaration37. Therefore, our analyses underscore the significance of persistent NH3 control to achieve the WHO Global Air Quality Guidelines targets for PM2.5 by 2050, and they have been proven to be cost-effective29,38. This strategy balances ecological nitrogen thresholds with air quality imperatives in the Anthropocene.
The main potential for NH3 abatement lies in agricultural activities, as they are the primary source of NH3 emissions39. This study emphasizes that the environmental benefits of emission reduction measures (such as reducing PM2.5) are highly dependent on the region and season in which they are implemented. To maximize cost-effectiveness, emission reduction measures should be prioritized during meteorological windows most sensitive to NH3 emissions (e.g., periods of low RH). This measure mainly includes two aspects: optimizing nitrogen fertilizer and improving manure management40. For optimizing nitrogen fertilizer, deep placement of N fertilizer is a good option, as it mainly reduces NH3 volatilization by applying the fertilizer deeper into the soil rather than surface broadcasting41. The use of efficient nitrogen fertilizers containing urease or nitrification inhibitors can also suppress the release of NH3 into the atmosphere42. It is particularly important that the timing of these measures is crucial. For instance, in regions like Northern China and South Asia, emphasis should be placed on implementing these measures during the pre-monsoon dry season (spring), as the low RH conditions during this period cause a significant portion of gaseous NH3 to remain in the gas phase without converting into particulate matter. For North America and Europe, focus should be placed on summer months-these periods coincide with peak fertilizer application, and are accompanied by low RH and high solar radiation, which facilitate the rapid formation of secondary particulate matter. At this stage, emission reduction proves most effective in lowering PM2.5 concentrations. And optimized management practices such as the “4 R Nutrient Stewardship” approach promoted by the International Fertilizer Association43,44 are the best management practices, which help reduce agricultural NH3 emissions without causing crop yield loss. This approach mainly includes the right fertilizer type, the right amount, the right placement, and the right time. Therefore, we propose to refine the “Right Time” principle in the “4 R Nutrient Stewardship” framework into concrete practice: minimizing fertilizer application during adverse weather conditions characterized by low RH (<40%) and limited precipitation. This approach leverages targeted best management practices to achieve optimal emission reduction outcomes. For livestock production systems, NH3 abatement measures include manipulation of feed rations and manure management practices, such as improvement of storage facilities and covering the surface to reduce gas diffusion45,46. Given that manure application similarly constitutes a significant source of NH3, the aforementioned recommendation regarding optimized fertilization timing remains equally applicable to the field application of manure. Strategic timing of fertilizer and manure application to coincide with beneficial meteorological windows, alongside the synergistic adoption of high-efficiency fertilizers and improved manure management, can dramatically cut agricultural NH3 emissions without sacrificing crop yields47. Additionally, mitigating combustion-related NH3 emissions is another option for reducing NH3 emissions, which can alleviate pressure on agricultural NH3 mitigation efforts48,49,50.
Additionally, buffering agents NH3 and NOx, recognized as short-lived climate forcers51, exert complex climate impacts through dual radiative pathways. Their gas-to-particle conversion generates secondary inorganic aerosols, which induce net negative radiative forcing via light scattering and cloud interactions52. This aerosol-driven cooling temporarily offsets anthropogenic CO2 warming, creating a precarious climate penalty. Nevertheless, future improvements in air quality could paradoxically intensify global warming. Therefore, it’s crucial to integrate considerations among “acid-base buffering-concomitant pollution-climate”, to better develop an effective emission reduction strategy that is beneficial to both air quality and climate change, which is a critical frontier in Anthropocene sustainability science.
Methods
Field observations
The 1-h resolution observations were collected at the air quality research supersite located in Nankai University from January 1, 2018, to November 31, 2021. The PM2.5 mass concentrations were measured using the beta-ray particulate matter automatic monitor. Water-soluble ions (NH4+, NO3−, SO42−, Cl−, Na+, K+, Mg2+, and Ca2+) in PM2.5 and semi-volatile gas components (NH3, HNO3, and HCl) were measured using an ambient ion monitor (AIM URG-9000D, URG Corporation). The concentrations of SO2, NO2, and O3 were measured using corresponding gas analyzers (Teledyne API T101, T201, and T400). The temperature (T) and RH were recorded using a miniature weather station (WS600-UMB, Lufft). Details of the observation stations were described in our previous work21,53.
Global simulation
To conduct global distribution of concomitant pollution, we used monthly average meteorological data T and RH, gaseous component data HNO3, NH3, HCl, and ion data Na+, Mg2+, K+, Ca2+, NO3−, NH4+. Cl−, SO42− from the three-dimensional chemical transport model GEOS-Chem (version 14.5.3) in January, April, July, and October 2019 (https://doi.org/10.1038/s41467-024-48793-1)54. The relevant datasets for reference and validation can be referred to https://doi.org/10.6084/m9.figshare.28532636. GEOS-Chem is driven by the Modern-Era Retrospective Analysis for Research and Applications, version 2 (MERRA-2) reanalysis meteorological fields with 2°(latitude) × 2.5°(longitude) horizontal resolution within the regional domains. The specific model settings can be found in our previous research54.
For data calibration, the simulation results of PM2.5 in this work are verified by comparing with the reanalysis datasets or observation-constrained datasets. The global reanalysis dataset “Satellite-derived PM2.5” from the Atmospheric Composition Analysis Group at Washington University in St. Louis (https://sites.wustl.edu/acag/datasets/surface-pm2-5/) is utilized (Supplementary Fig. S44). In addition, we thoroughly discuss the spatiotemporal resolution of the NH3 emission inventory and evaluate how emission uncertainties impact the results (Supplementary Text 9 and Figs. 45–48).
pH estimation
Aerosol pH and AWC were estimated using the thermodynamic equilibrium model ISORROPIA-II (http://isorropia.eas.gatech.edu), assuming that the aerosol system is in equilibrium55. This model solves the equilibrium state of K+-Ca2+-Mg2+-NH4+ Na+-SO42−-NO3−-Cl−-H2O aerosol system, and it has been widely used in many other studies56,57,58. The forward mode and metastable state were selected in this study. The aerosol pH was calculated by the following equation:
Where \({{{{\rm{c}}}}}_{{{{{\rm{H}}}}}^{+}}\) is the concentration of H+ per volume of air (μg m−3), \({{{{\rm{\gamma }}}}}_{{{{{\rm{H}}}}}^{+}}\) is the activity coefficient of H+ (assumed to be 1.0)55, and AWC is the concentration of AWC (μg m−3).
Buffering capacity β
The buffering capacity β, also known as differential buffer capacity, is defined as the amount of strong base (db (mol)) that needs to be added to a 1 L solution to increase its pH by dpH units; Or conversely, to reduce the pH of a 1 L solution by dpH units, the amount of strong acid that needs to be added to the solution (da (mol))59. The mathematical expression is as follows:
Based on this formula, the specific mathematical expression of buffer value β for conjugate acid-base equivalent buffer systems is derived7,59, which is the sum of the self-buffering capacity of water and the buffering capacity of the buffering agent:
Where [H+] and [OH−] are the concentrations of H+ and OH− in the solution, respectively. The c represents the total equivalent molality of the buffer agent. And the \({K}_{{{{\rm{a}}}},{{{\rm{i}}}}}\) is the effective dissociation constant of the buffer agent, which can be expressed by:
for volatile base BOH and volatile acid HA that dissociate in the form
where \({K}_{{{{\rm{a}}}},{{\mathrm{BOH}}}}\) and \({K}_{{{{\rm{a}}}},{{\mathrm{HA}}}}\) are acid dissociation constants, \({\rho }_{{{{\rm{w}}}}}\) is the density of water (1012 μg m−3), \({H}_{{{{\rm{i}}}}}\) is Henry’s law constant, R is the ideal gas constant (0.0825 atm L mol−1 kg−1), T is the temperature in K and AWC is aerosol water content in μg m−3.
Therefore, for NH4+/NH3 buffering agent:
Where \([{{{{\rm{NH}}}}}_{4}^{{{{\rm{T}}}}}]\) is total equivalent molality of NH3 and NH4+ in mol kg−1; \({{K}_{{{{\rm{a}}}},{{{\mathrm{NH}}}}_{3}}}^{ * }\) is the effective ionization equilibrium constant of the conjugate acid of NH3 in mol kg−1; \({\gamma }_{{{{\mathrm{NH}}}}_{4}^{+}}\) is the activity coefficient of NH4+; \({H}_{{{{\mathrm{NH}}}}_{3}}\) is the Henry’s law constant of NH3 in mol kg−1 atm−1; \({K}_{{{{\rm{b}}}},{{{\mathrm{NH}}}}_{3}}\) is the base dissociation constant of NH3 in mol kg−1; \({K}_{{{{\rm{W}}}}}\) is the water dissociation constant in mol2 kg−2.
For HNO3/NO3− buffering agent:
Where \([{{{{\rm{NO}}}}}_{3}^{{{{\rm{T}}}}}]\) is total equivalent molality of HNO3 and NO3− in mol kg−1; \({{K}_{{{{\rm{a}}}},{{{\mathrm{HNO}}}}_{3}}}^{ * }\) is the effective acid dissociation constants of the conjugate acid of HNO3 in mol kg−1; \({\gamma }_{{{{\mathrm{NO}}}}_{3}^{-}}\) is the activity coefficient of NO3−; \({H}_{{{{\mathrm{HNO}}}}_{3}}\) is the Henry’s law constant of HNO3 in mol kg−1 atm−1.
For HSO4−/SO42− buffering agent:
Where \([{{{{\rm{SO}}}}}_{4}^{{{{\rm{T}}}}}]\) is total equivalent molality of HSO4− and SO42− in mol kg−1; \({{K}_{{{{\rm{a}}}},{{{\mathrm{HSO}}}}_{4}^{-}}}^{ * }\) is the effective acid dissociation constants of the conjugate acid of HSO4− in mol kg−1; \({\gamma }_{{{{\mathrm{HSO}}}}_{4}^{-}}\) and \({\gamma }_{{{{\mathrm{SO}}}}_{4}^{2-}}\) is the activity coefficient of HSO4− and SO42−.
And total buffering capacity:
The meaning of all parameters is the same as above. The calculation of the acid dissociation constant and Henry’s coefficient is shown in Supplementary Table 1.
The calculation of pH change by integral buffer capacity α
In the actual world environment, the buffering process is usually a continuous process, and the buffering capacity β described above describes the instantaneous buffering capacity of aerosols. To describe the entire continuous buffering process, we introduce the integral buffering capacity αproposed by Dai et al.60. The corresponding mathematical expression (Eq. (17)) is defined as the amount of strong acid or base required to change the pH of 1 L of solution within the specified pH range.
Among them, C is the required acidity and alkalinity, pH1 is the initial pH value before the buffering process occurs, and pH2 is the pH value after the buffering process occurs. The specific derivation process of integral buffering capacity α is as follows.
According to the Brønsted–Lowry theory of acids and bases, a buffer solution is usually a balanced system of weak acids and their conjugate acids in water. For a solution consisting of a weak acid HA and a conjugate acid A−, a dissociation equilibrium is defined as follows:
Therefore, the pH of the buffer solution is obtained:
Where, \({K}_{a}\) is the acid dissociation equilibrium constant, C is the total concentration of weak acid HA and conjugated acid A−.
Based on the equation of differential buffer capacity with [H+] and total concentration C:
Combining Eq. 19, we obtain:
Integrate both sides of Eq. 22 after separating variables, take the difference of n units between the upper and lower limits of the pH value, that is \({{{\mathrm{pH}}}}_{2}={{{\mathrm{pH}}}}_{1}+n\). [A−]1 is the concentration of A− corresponding to pH1, and [A−]2 is the concentration of A− corresponding to pH2.
Thus, we have:
We then obtain the expression of the integral buffer capacity when base is added:
Similarly, when adding acid, we set pH2 = pH1 − n to obtain the corresponding integral buffer capacity as follows:
Where n is the change of pH, and the remaining variables are the same as in β.
Therefore, when adding acidity or alkalinity, the above equation can also be transformed into Eqs. 28–29 to calculate the change in pH value.
Field emission transmission electron microscopy experiment
We collected single particle samples during the clean and polluted periods using the DKL-2 single particle sampler on the second floor of the School of Environmental Science and Engineering at Nankai University on October 30, 2023. Aerosol particles are deposited on a copper TEM grid coated with carbon film using a single-stage cascade impactor with a nozzle diameter of 0.5 mm and an air flow rate of 1.0 l min−1. The sampling time range is 30 to 120 s, depending on visibility. After sampling, seal the TEM grid in dry plastic capsules to prevent contamination. Afterwards, the sample was analyzed using a field emission transmission electron microscope (TEM). TEM images are used to determine particle size, morphology, and mixing state. Use energy dispersive X-ray spectroscopy (EDX) to determine elemental composition.
The calculation of concomitant pollution
The essence of the buffering process is neutralization of acid and base, which is accompanied by the gas-particle partitioning process. Therefore, the concentration and ratio of gas-phase and particle-phase buffering agents in the ambient air are crucial for the final generation of particle-phase pollutants, ultimately leading to the degree of pollution. For this purpose, the ratio of gas-phase buffering agent NH3 concentration to total buffering agent (NH3 + NH4+) concentration is defined as “Concomitant Pollution Index” (Co-P index, Eq. 30). Moreover, the calculation of “concomitant pollution” (Co-P) has been further defined, with the following equations:
where, [NH3] and [NH4+] are the concentration of NH3 and NH4+ in ambient environment, respectively. \({C}_{{{{\mathrm{NH}}}}_{4}^{{{{\rm{T}}}}}}\) is the total concentration of NH3 and NH4+ in μg m−3, \({M}_{{{{\mathrm{NH}}}}_{3}}\) is the relative molecular mass of NH3.
Similarly, HNO3/NO3− induced concomitant pollution is as follows:
where, [HNO3] and [NO3−] are the concentration of HNO3 and NO3− in ambient environment, respectively. \({C}_{{{{\mathrm{NO}}}}_{3}^{{{{\rm{T}}}}}}\) is the total concentration of HNO3 and NO3− in μg m−3, \({M}_{{{{\mathrm{HNO}}}}_{3}}\) is the relative molecular mass of HNO3.
Machine learning
Machine learning methods, including RF, Xgboost, and Neural Networks, can effectively utilize observation datasets, construct models from the perspective of measured data, and evaluate the effects of pollutant influencing factors. Among them, RF performs well in classification and importance assessment in the environmental field61,62,63, which was applied to evaluate the importance of multiple factors (SO42−, Ca2+, T, RH, NH4T, and NO3T) to concomitant pollution. Here, randomly extract 70% of the samples from all datasets, forming a new training dataset for model training. The remaining 30% of the samples will be used as the test dataset to evaluate the performance of the model. As shown in Supplementary Fig. 49, the coefficient of determination (R2) is 0.88, indicating good model performance.
Random forest-partial dependence plot (RF-PDP)
PDP is a visualization tool used to understand how a model’s prediction depends on one or more features. It provides a visual understanding of the internal working mechanism of the model by showing how the average value of the model’s prediction changes as the feature value varies. The calculation principle of RF-PDP is as follows:
Where \({{{\rm{f}}}}\left({{{{\rm{X}}}}}_{{{{\rm{S}}}}}\right)\) represents the predicted value of the RF model when the input is \({X}_{{{{\rm{S}}}}}\); RF stands for the trained RF model; \({X}_{{{{\rm{S}}}}}\) is the feature selected for calculating the partial dependence function in the RF model; \({X}_{{{{\rm{C}}}}}^{({{{\rm{i}}}})}\) represents the features that are not selected in the RF model, and these features are input into the model as fixed values; n is the number of samples.
The hyperparameter settings and validation methods of the machine learning model was set as follows:
Hyperparameter settings: in the RF regressor, we set the hyperparameter n_estimators = 500, indicating that the model will train 500 decision trees. Another hyperparameter random_state = 0 to ensure consistent results in each run. The main hyperparameters in pdp.pdp_isolate include (1) percentile_range = (1, 100), this hyperparameter sets the range of features for the PDP graph from the 1st percentile to the 100th percentile, which determines the range of feature data used to generate the PDP; (2) num_grid_points = 20, this hyperparameter indicates the number of discrete points of the PDP graph, meaning that the feature values will be divided into 20 discrete points.
Validation methods: In terms of data partitioning, the data is randomly partitioned into a training set and a test set. We set train_test_split (X, Y, test_size = 0.3, random_state = 8), where 30% of the data is used for testing and 70% of the data is used for training. random_state = 8 ensures the reproducibility of the data division. We split the data into a training set and a test set, fitted the model using the training set and tested its performance. In addition, the ten-flod cross-validation was applied to ensure the model’s generalizability. The results show that the average R2 values for the training set and test set are 0.97 and 0.80 (Supplementary Fig. 21), respectively, indicating that the model used in this study has strong predictive capabilities and can effectively capture the main trends and relationships in the data.
The calculation of dominated influence factors
For concomitant pollution caused by NH4+/NH3, based on Eq. 30, we take the logarithm ln on both sides of the formula to obtain formula 37:
Where \({{\mathrm{ln}}}\left(\frac{2.303\cdot {10}^{3}}{{M}_{{{{\mathrm{NH}}}}_{3}}}\right)\) is a constant term, \({C}_{{{{\mathrm{NH}}}}_{4}^{{{{\rm{T}}}}}}\) and H+ are both influenced by AF, where \({{K}_{{{{\rm{a}}}},{{NH}}_{3}}}^{ * }\) and AWC are both influenced by MF.
Moving the constant term to the left of the equation, Eq. 34 is transformed into:
Therefore, for Co-P caused by NH4+/NH3, the contributions of anthropogenic and MF to Co-P can be roughly calculated using the following equation:
Similarly, for HNO3/NO3− induced Co-P, the contributions of anthropogenic and MF to Co-P can be roughly calculated using the following equation:
It is important to note that the MF/AF ratio employed in this study is a conceptual simplification. In reality, meteorological and AF are highly interdependent and exist on a continuum rather than as discrete categories. For instance, AWC, a key parameter influencing heterogeneous chemistry, is co-determined by meteorological conditions (e.g., RH) and anthropogenic aerosol composition. The discrete categorization, while enabling a clear and practical diagnostic framework for global analysis, may introduce uncertainties in precisely quantifying the absolute contributions of each factor and might underestimate their synergistic effects. Nevertheless, the MF/AF indicator serves as a valuable diagnostic tool for identifying the dominant drivers of concomitant pollution across different regions and seasons.
Data availability
The authors declare that the main data supporting the findings of this study are available within the article, its Supplementary Information file, and the Source data. Source data can be accessed online via the Figshare https://doi.org/10.6084/m9.figshare.28532636.
Code availability
The ISORROPIA-II model is available at http://isorropia.epfl.ch.
References
Pye, H. O. T. et al. The acidity of atmospheric particles and clouds. Atmos. Chem. Phys. 20, 4809–4888 (2020).
Freedman, M. A., Ott, E. J. E. & Marak, K. E. Role of pH in aerosol processes and measurement challenges. J. Phys. Chem. A 123, 1275–1284 (2019).
Liu, Y. C. et al. Dust emission reduction enhanced gas-to-particle conversion of ammonia in the North China plain. Nat. Commun. 13, 6887 (2022).
Shi, X. R. et al. High-resolution data sets unravel the effects of sources and meteorological conditions on nitrate and its gas-particle partitioning. Environ. Sci. Technol. 53, 3048–3057 (2019).
Weber, R. J., Guo, H. Y., Russell, A. G. & Nenes, A. High aerosol acidity despite declining atmospheric sulfate concentrations over the past 15 years. Nat. Geosci. 9, 282–285 (2016).
Faherty, T., Raymond, J. E., Mcfiggans, G. & Pope, F. D. Acute particulate matter exposure diminishes executive cognitive functioning after four hours regardless of inhalation pathway. Nat. Commun. 16, 1339 (2025).
Zheng, G. J. et al. Multiphase buffer theory explains contrasts in atmospheric aerosol acidity. Science 369, 1374–1377 (2020).
Pan, D. et al. Regime shift in secondary inorganic aerosol formation and nitrogen deposition in the rural United States. Nat. Geosci. 17, 617–623 (2024).
Liu, Z. H. et al. Optimal reactive nitrogen control pathways identified for cost-effective PM2.5 mitigation in Europe. Nat. Commun. 14, 4246 (2023).
Zhu, J. X. et al. Changing patterns of global nitrogen deposition driven by socio-economic development. Nat. Commun. 16, 46 (2025).
Zheng, G., Su, H. & Cheng, Y. Revisiting the key driving processes of the decadal trend of aerosol acidity in the U.S. ACS Environ. Au 2, 346–353 (2022).
Zheng, G., Su, H., Wan, R., Duan, X. & Cheng, Y. Rising alkali-to-acid ratios in the atmosphere may correspond to increased aerosol acidity. Environ. Sci. Technol. 58, 16517–16524 (2024).
Zheng, G. J., Su, H., Andreae, M. O., Pöschl, U. & Cheng, Y. F. Multiphase buffering by ammonia sustains sulfate production in atmospheric aerosols. AGU Adv. 5, e2024AV001238 (2024).
Kwon, H. J. et al. Long-lifetime water-washable ceramic catalyst filter for air purification. Nat. Commun. 14, 520 (2023).
He, H. et al. Mineral dust and NOx promote the conversion of SO2 to sulfate in heavy pollution days. Sci. Rep. 4, 4172 (2014).
The Asian Brown Cloud (ABC) - UNEP and C-4 (2002) The Asian brown cloud: climate and other environmental impacts. UNEP, Nairobi—Executive summary. Environ. Sci. Pollut. Res. 9, 289–295 (2002).
Solomon, S., Qin, D., Manning, M., Chen, Z. & Miller, H. L. Contribution of Working Group I to the Fourth Assessment Report of the Intergovernmental Panel on Climate Change (Cambridge University Press, 2007).
Ma, J. Z., Xu, X. B., Zhao, C. S. & Yan, P. A review of atmospheric chemistry research in China: Photochemical smog, haze pollution, and gas-aerosol interactions. Adv. Atmos. Sci. 29, 1006–1026 (2012).
Zhao, X. J. et al. Analysis of a winter regional haze event and its formation mechanism in the North China Plain. Atmos. Chem. Phys. 13, 5685–5696 (2013).
Zhao, Y. et al. Rising importance of agricultural nitrogen oxide emissions in China’s future PM2.5 pollution mitigation. npj Clim. Atmos. Sci. 8, 93 (2025).
Wang, F. et al. Drivers of PM2.5-O3 co-pollution: from the perspective of reactive nitrogen conversion pathways in atmospheric nitrogen cycling. Sci. Bull. 67, 1833–1836 (2022).
Food and Agriculture Organization. World food and agriculture: Statistical Yearbook 2024 (FAO, 2024).
Liu, X. J. et al. Enhanced nitrogen deposition over China. Nature 494, 459–462 (2013).
Galloway, J. N. et al. Transformation of the nitrogen cycle: recent trends, questions, and potential solutions. Science 320, 889–892 (2008).
Canfield, D. E., Glazer, A. N. & Falkowski, P. G. The evolution and future of Earth’s nitrogen cycle. Science 330, 192–196 (2010).
Sutton, M. A. et al. Too much of a good thing. Nature 472, 159–161 (2011).
Stokstad, E. Ammonia pollution from farming may exact hefty health costs. Science 343, 238–238 (2014).
Liu, L. et al. Exploring global changes in agricultural ammonia emissions and their contribution to nitrogen deposition since 1980. Proc. Natl. Acad. Sci. USA 119, e2121998119 (2022).
Gu, B. J. et al. Abating ammonia is more cost-effective than nitrogen oxides for mitigating PM2.5 air pollution. Science 374, 758–762 (2021).
Chen, Z., Liu, P., Liu, Y. & Zhang, Y. H. Strong acids or bases displaced by weak acids or bases in aerosols: reactions driven by the continuous partitioning of volatile products into the gas phase. Acc. Chem. Res. 54, 3667–3678 (2021).
Chen, Z., Liu, P., Su, H. & Zhang, Y. H. Displacement of strong acids or bases by weak acids or bases in aerosols: thermodynamics and kinetics. Environ. Sci. Technol. 56, 12937–12944 (2022).
Zhou, M., Xie, Y., Wang, C., Shen, L. & Mauzerall, D. L. Impacts of current and climate induced changes in atmospheric stagnation on Indian surface PM2.5 pollution. Nat. Commun. 15, 7448 (2024).
Qin, Y. et al. Amplified positive effects on air quality, health, and renewable energy under China’s carbon neutral target. Nat. Geosci. 17, 411–418 (2024).
Ge, Y., Vieno, M., Stevenson, D. S., Wind, P. & Heal, M. R. Global sensitivities of reactive N and S gas and particle concentrations and deposition to precursor emissions reductions. Atmos. Chem. Phys. 23, 6083–6112 (2023).
Galloway, J. N. et al. Nitrogen cycles: past, present, and future. Biogeochemistry 70, 153–226 (2004).
Assembly UNE. Sustainable Nitrogen Management Resolution (UNEP/EA.4/L.16) https://leap.unep.org/en/content/unea-resolution/sustainable-nitrogen-management (2019).
Management UNGCoSN. Colombo Declaration on Sustainable Nitrogen Management. United Nations (2019).
Guo, Y. X. et al. Aspirational nitrogen interventions accelerate air pollution abatement and ecosystem protection. Sci. Adv. 10, eado0112 (2024).
Zhan, X. Y. et al. Improved estimates of ammonia emissions from global croplands. Environ. Sci. Technol. 55, 1329–1338 (2021).
Gu, B. J. et al. Cost-effective mitigation of nitrogen pollution from global croplands. Nature 613, 77–84 (2023).
Ti, C. P., Xia, L. L., Chang, S. X. & Yan, X. Y. Potential for mitigating global agricultural ammonia emission: a meta-analysis. Environ. Pollut. 245, 141–148 (2019).
Zhang, X. M. et al. Societal benefits of halving agricultural ammonia emissions in China far exceed the abatement costs. Nat. Commun. 11, 4357 (2020).
Bruulsema, T. Managing nutrients to mitigate soil pollution. Environ. Pollut. 243, 1602–1605 (2018).
United Nations Economic Commission for Europe. “Guidance document on integrated sustainable nitrogen management” (UNECE, 2021).
Liu, M. X. et al. Ammonia emission control in China would mitigate haze pollution and nitrogen deposition, but worsen acid rain. Proc. Natl. Acad. Sci. USA 116, 7760–7765 (2019).
Xu, P. et al. Fertilizer management for global ammonia emission reduction. Nature 626, 792–798 (2024).
Zhang, W. F. et al. Closing yield gaps in China by empowering smallholder farmers. Nature 537, 671–674 (2016).
Liang, X. Y. et al. Air quality and health benefits from fleet electrification in China. Nat. Sustain. 2, 962–971 (2019).
Suarez-Bertoa, R., Pechout, M., Vojtísek, M. & Astorga, C. Regulated and non-regulated emissions from Euro 6 diesel, gasoline and CNG vehicles under real-world driving conditions. Atmosphere 11, 204 (2020).
Wang, Y. J. et al. Vehicular ammonia emissions significantly contribute to urban PM2.5 Pollution in two Chinese megacities. Environ. Sci. Technol. 57, 2698–2705 (2023).
Fu, B. et al. The contributions of individual countries and regions to the global radiative forcing. Proc. Natl. Acad. Sci. USA 118, e2018211118 (2021).
Chen, Y. et al. Substantial cooling effect from aerosol-induced increase in tropical marine cloud cover. Nat. Geosci. 17, 404–410 (2024).
Gao, J. et al. Targeting atmospheric oxidants can better reduce sulfate aerosol in China: H2O2 aqueous oxidation pathway dominates sulfate formation in haze. Environ. Sci. Technol. 56, 10608–10618 (2022).
Gao, J. et al. Hydrogen peroxide serves as pivotal fountainhead for aerosol aqueous sulfate formation from a global perspective. Nat. Commun. 15, 4625 (2024).
Fountoukis, C. & Nenes, A. ISORROPIA II: a computationally efficient thermodynamic equilibrium model for K+-Ca2+-Mg2+-NH4+-Na+-SO42−-NO3−-Cl−-H2O aerosols. Atmos. Chem. Phys. 7, 4639–4659 (2007).
Andersen, S. T. et al. Extensive field evidence for the release of HONO from the photolysis of nitrate aerosols. Sci. Adv. 9, eadd6266 (2023).
Xu, Y. et al. Aerosol liquid water promotes the formation of water-soluble organic nitrogen in submicrometer aerosols in a suburban forest. Environ. Sci. Technol. 54, 1406–1414 (2020).
Holt, J., Selin, N. E. & Solomon, S. Changes in inorganic fine particulate matter sensitivities to precursors due to large-scale US emissions reductions. Environ. Sci. Technol. 49, 4834–4841 (2015).
Van Slyke, D. D. On the measurement of buffer values and on the relationship of buffer value to the dissociation constant of the buffer and the concentration and reaction of the buffer solution. J. Biol. Chem. 52, 525–570 (1922).
Dai, M. Integral capacity of buffer. Microchim. Acta 82, 159–169 (1984).
Vu, T. V. et al. Assessing the impact of clean air action on air quality trends in Beijing using a machine learning technique. Atmos. Chem. Phys. 19, 11303–11314 (2019).
Zhang, Y. M. et al. Significant changes in chemistry of fine particles in wintertime Beijing from 2007 to 2017: impact of clean air actions. Environ. Sci. Technol. 54, 1344–1352 (2020).
Shi, Z. B. et al. Abrupt but smaller than expected changes in surface air quality attributable to COVID-19 lockdowns. Sci. Adv. 7, eabd6696 (2021).
Acknowledgements
This work is supported by the National Key R&D Program of China (2023YFC3709500 to S.S. and 2023YFC3709502 to S.S.), the National Natural Science Foundation of China (42477105 to S.S.), the Blue Sky Foundation, and Tianjin Science and Technology Plan Project (PTZWHZ00120 to Y.F.). We extend our gratitude to Professor Liu Zirui and Professor Pan Yuepeng at the Institute of Atmospheric Physics, Chinese Academy of Sciences, as well as Dr. Sun Qian at the Institute for Carbon-Neutral Technology, Shenzhen Polytechnic University, for their invaluable support in providing the ACSM data.
Author information
Authors and Affiliations
Contributions
G.S. designed the research. Y.W. and J.G. performed research. H.W. and H.X. helped with model simulations. Y.W. analyzed and interpreted model results. Y.W. and G.S. drafted the manuscript. S.S., Y.F., and A.R. provided comments on the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wei, Y., Gao, J., Wang, H. et al. Reactive nitrogen-driven atmospheric multiphase buffering induces unintended concomitant pollution. Nat Commun 17, 472 (2026). https://doi.org/10.1038/s41467-025-67167-9
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-67167-9
