
Abstract
Toll-like receptor 3 (TLR3), an innate immune sensor for double-stranded RNA (dsRNA), traffics from the endoplasmic reticulum (ER) after synthesis to endolysosomes for proteolytic cleavage and activation. However, the molecular mechanisms governing TLR3 trafficking remain largely unclear. Here, we identify the ER-resident E3 ligase HMG-CoA reductase degradation protein 1 (HRD1), a core component of ER-associated degradation (ERAD), as a key regulator that promotes TLR3 trafficking and downstream signaling. HRD1 deficiency in macrophages significantly impairs poly(I:C)-induced TLR3 signaling and inflammatory responses in vitro and in vivo, caused by a marked reduction in TLR3 transport into endolysosomes and subsequent proteolytic processing. Mechanistically, HRD1 mediates ubiquitination of ER-localized TLR3 at lysine 813, which is required for its recognition and sorting by the endosomal sorting complex required for transport (ESCRT) machinery. This HRD1 function is decoupled from its canonical ERAD activity and the ER stress sensor inositol-requiring enzyme 1 alpha (IRE1α). Hence, our study identifies a previously unrecognized mechanism controlling TLR3 signaling and links HRD1-mediated ubiquitination to immune sensor trafficking during innate immune responses.
Similar content being viewed by others


Introduction
Toll-like receptors (TLRs) are an evolutionarily ancient family of proteins operating as germline-encoded pattern recognition receptors (PRRs) to sense structurally conserved microbial components termed pathogen-associated molecular patterns (PAMPs)1. TLR3 detects dsRNA from genomes of dsRNA viruses or intermediates formed during replication of single-stranded RNA (ssRNA) viruses, to induce the transcription of type I interferons (IFNs) and proinflammatory cytokines and initiate antiviral immune response2. The full-length TLR3 is synthesized in the endoplasmic reticulum (ER) and transported through the Golgi apparatus to endosomes, where it undergoes cleavage by cathepsins to generate the activable form3,4,5. This strategically restricted activation of TLR3 in endolysosomes allow the receptor to mediate immune response against the internalized viral dsRNA but sequestering it away from self-nucleic acids6. Despite the vital role of TLR3 trafficking in initiating immune response, the molecular mechanisms governing this process remain largely unclear.
Proteins targeted to expression on membranes, including many immune sensors, are initially synthesized and folded in the ER7. The regulation of synthesis, modification and stability of membrane immune receptors within the ER can significantly impact the immune response. For example, folding and function of TLRs are dependent on the activity of ER-resident chaperones gp96 and Unc-93 homolog B1 (UNC93B1)8,9. Stimulator of interferon genes (STING) is an ER adaptor protein that mounts the body’s inflammatory response against cytosolic double-stranded DNA10, Toll interacting protein (TOLLIP), stromal interaction molecule 1 (STIM1) and STING ER exit protein (STEEP) control the stability, retention and exit of ER-resident STING to regulate its activation and signaling11,12,13,14. Thus, ER emerges as a central hub of immune regulation by monitoring the synthesis, maturation, degradation and trafficking of immune receptors. While TLR3 is synthesized in its precursor form and primarily resident in the ER under basal stage3,4, whether any ER factors can regulate TLR3 protein modification and thus its trafficking remains unknown.
Protein synthesis in the ER is coupled with stringent quality control to ensure that only correctly folded proteins exit the ER for further maturation and trafficking. ERAD is a fundamental quality control mechanism responsible for the recognition and removal of misfolded and aberrant proteins in the ER for cytosolic proteasomal degradation15,16,17. HRD1, an ancient and highly conserved ER membrane-embedded E3 ubiquitin ligase, forms a complex with adaptor protein SEL1L to nucleate the ERAD machinery17,18,19. Recent studies have collectively demonstrated the pivotal functions of HRD1 ERAD in critical physiological processes, such as food digestion, nutrient storage, glucose homeostasis, food intake, water balance and thermogenic regulation20,21,22,23,24,25,26,27. In immune cells, we and others have showed HRD1-mediated ERAD activity downregulates the protein level of ER-resident pre-B cell receptor complex (pre-BCR) and STING, negatively regulates pre-BCR- and STING-mediated signaling, and exerts critical roles in controlling early B cell development and innate immune response28,29,30. While TLR3 is mostly resident in the ER of resting cells, whether HRD1 modifies TLR3 and regulates its signaling has not thoroughly examined.
In this article, we report that ubiquitination of TLR3 by HRD1 within the ER is a prerequisite for its trafficking into endolysosomes and subsequent processing. Hrd1 deficiency or its inhibition reduces the levels of cleaved and activable TLR3, significantly dampening TLR3-mediated signaling and inflammatory response after ligand stimulation. Interestingly, HRD1 ubiquitinates TLR3 to promote its trafficking, decoupled from its degradation by ERAD. Hence, HRD1 moonlights as a positive regulator for TLR3 innate immune signaling by directly modifying the innate immune sensor.
Results
Identification of the HRD1-TLR3 interaction and its enhancement upon TLR3 stimulation
Our previous study has demonstrated that ER-resident E3 ligase HRD1 can ubiquitinate and negatively regulate the level of ER-resident STING protein, and Hrd1 deficiency in macrophages amplifies production of pro-inflammatory cytokines and type I interferon (IFN) upon STING stimulation30. To further elucidate the role of HRD1 in macrophage function, we performed immunoprecipitation on protein lysates from the wild-type (WT) RAW 264.7 macrophage cell line and Hrd1−/− cells, generated via the CRISPR/Cas9 system (Fig. S1a), using an anti-HRD1 antibody, followed by mass spectrometry (CoIP-MS) to identify proteome-wide endogenous HRD1 substrates. MS analyses of endogenous HRD1 interactome identified 812 hits specially in WT macrophage cells but not in Hrd1-deficient cells (Fig. 1a). Among the top specific hits were HRD1 itself and known components of the endogenous ERAD complex, including SEL1L and OS9 proteins (Fig. 1a). Intriguingly, the ER-synthesized innate immune sensor TLR3 was identified as a top-ranking hit (Fig. 1a). Given that TLR3 primarily resides in the ER membrane, we speculated whether TLR3 and its signaling is regulated by HRD1. Protein level of HRD1, but not the ER chaperon Binding immunoglobulin protein (BiP) or the unfolded protein response (UPR) sensors IRE1α and PKR-like endoplasmic reticulum kinase (PERK), was increased in primary peritoneal macrophage upon treatment with the dsRNA analog poly(I:C) (Fig. 1b), further suggesting a possible role of HRD1 in TLR3 signaling pathway.

a Comparative analysis of anti-HRD1 IP-MS in WT and HRD1−/− RAW 264.7 cells to identify HRD1-interacting candidates, and the top ten specific HRD1-interacting ER proteins shown. b Immunoblot analysis of indicated proteins in primary macrophages treated with 50 μg/ml poly(I:C) for the indicated times, representative of three biologically independent repeats. The quantitation of protein levels (normalized to the loading control) is shown below the blot. c Immunoblot analysis of indicated proteins following immunoprecipitation of Flag in HEK293T cells transfected with TLR3-Flag and HRD1-Myc plasmids for 24 h, and subsequently treated with 50 μg/ml poly(I:C) for 3 h. The quantitation of protein levels (normalized to the no poly(I:C) treatment) is shown below the blot. IP, immunoprecipitation. d Immunoblot analysis of indicated proteins following immunoprecipitation of endogenous HRD1 in RAW 264.7 macrophages at various time points following treatment with 50 μg/ml poly(I:C). The quantitation of protein levels (normalized to the 0 h) is shown below the blot. IgG, immunoglobulin G. e–h Diagrams of full-length HRD1 protein domains and various HRD1 truncate mutants (e) and TLR3 protein domains and various TLR3 truncate mutants (g), along with mapping of TLR3 and HRD1 interacting domains (f, h). WT, wild type; TM, transmembrane. LRR, leucine-rich repeat; TIR, cytosolic Toll/interleukin-1 receptor domain. Results showing immunoblot analysis following Myc immunoprecipitation (f) or Flag immunoprecipitation (h) in HEK293T cells transfected with various plasmids encoding WT and truncated HRD1-Myc or TLR3-Flag proteins, as indicated. The blot data are representative of three biologically independent repeats (b–d, f, h). Source data are provided as a Source Data file.
We then confirmed the interaction of HRD1 with TLR3. In HEK293T cells, immunoprecipitation of transfected Flag-tagged TLR3 effectively pulled down transfected Myc-tagged HRD1, and treatment with poly(I:C) led to a higher yield of HRD1 pulldown (Fig. 1c). Under basal condition, immunoprecipitation of endogenous TLR3 or HRD1 effectively pulled down endogenous HRD1 or TLR3, respectively in macrophages (Figs. 1d and S1b). The physical interaction of endogenous TLR3 and HRD1 was increased shortly after poly(I:C) treatment (Figs. 1d and S1b). We then tested how HRD1 interacts with TLR3. HRD1 is a multi-pass transmembrane protein with a large cytosolic domain. In HEK293T cells, immunoprecipitation of truncated HRD1 proteins revealed that both the transmembrane and cytosolic RING domains were required for its interaction with TLR3 (Figs. 1e, f and S1c). The TLR3 protein is composed of a N-terminal leucine-rich repeat (LRR) ectodomain, a transmembrane (TM) domain, and a cytosolic Toll/interleukin-1 receptor (TIR) domain (Fig. 1g)31. Immunoprecipitation of truncated TLR3 proteins showed that TM domain of TLR3 was required for the effective interaction between TLR3 and HRD1 (Figs. 1h and S1d). Taken together, HRD1 interacts with TLR3, and both the protein level of HRD1 and their interactions increase upon dsRNA stimulation.
HRD1 positively regulates TLR3-mediated immune signaling
To directly test the role of HRD1 in the regulation of TLR3-mediated immune signaling, we compared the immune response of WT and Hrd1−/− cells to TLR3 agonist stimulation. While protein levels of TLR3 downstream effectors TBK1, IRF3 and P65 were comparable, these proteins, as well as IκBα, were hypo-phosphorylated in Hrd1−/− RAW 264.7 macrophages compared to WT cells upon treatment with poly(I:C) (Fig. 2a). We then tested whether HRD1’s regulation of TLR3 signaling is applicable to human cells or more physiologically relevant primary macrophages. The overexpression of HRD1 in TLR3-stable-expression HEK293T-TLR3 cells increased the phosphorylation of TBK1, P65, and IκBα upon stimulation in a dose-dependent manner, while HRD1 deficiency in HEK293T-TLR3 cells decreased their phosphorylation (Figs. 2b and S2a). Knocking down Hrd1 in primary macrophages using siRNAs (Fig. S2b, c) or treating primary macrophages with LS-102 (Fig. 2c)—a specific small-molecule HRD1 inhibitor that suppresses its E3 ubiquitin ligase catalytic activity32,33,34—significantly reduced the phosphorylation levels of TLR3 downstream effectors TBK1, IRF3, P65 and IκB in response to poly(I:C) treatment.

a Immunoblot analysis of indicated proteins in wild-type (scramble) versus Hrd1−/− RAW 264.7 cells treated with 50 μg/ml poly(I:C) for the indicated times, representative of three independent repeats. gRNA, guide RNA. b Immunoblot analysis of indicated proteins in Hrd1+/+ and Hrd1−/− HEK293T cells with stable expression of TLR3, following treatment with 50 μg/ml poly(I:C) for the indicated times. The data are representative of three independent repeats. c Immunoblot analysis of indicated proteins in primary macrophages treated with vehicle or LS-102 (5 μM) for 24 h, and subsequently treated with 50 μg/ml poly(I:C) for the indicated times, representative of three independent repeats. d qPCR analysis of Ifnb1, Il1b, Tnfa, Il6, Ccl5, and Bip in WT versus Hrd1−/− RAW 264.7 cells treated with vehicle or 50 μg/ml poly(I:C) for the indicated times. n = 4 each, representative of at least three independent repeats. mRNA, messenger RNA. e ELISA analysis of TNFα, IL6 and IFNβ in the culture supernatants of vehicle- or LS-102 (5 μM for 24 h)-treated primary macrophages, followed by poly(I:C) (50 μg/ml) treatment for 6 h. n = 4 each, representative of three independent repeats. f Representative microscopy images showing replication of GFP-tagged HSV-1 in Vero cells under various treatments, with quantitation shown in Supplementary Fig. S2i). Vero cells were exposed to secretome obtained from poly(I:C)-stimulated WT and Hrd1−/− RAW 264.7 cells and incubated with 0.5 or 1.0 MOI HSV-1 for 24 h. GFP-HSV, GFP-tagged HSV-1. Quantitation of the ratio of phosphorylated to total protein (p/t) is shown below each blot. Values represent mean ± SEM, by unpaired, two-tailed, Student’s t-test (d, e). Source data are provided as a Source Data file.
The stimulation of TLR3 consistently resulted in a notable reduction in the expression of various inflammatory cytokine genes (Ifnb1, Il1b, Tnfa, Il6, and Ccl5) in Hrd1−/− macrophages compared to WT cells, using poly(I:C) (Fig. 2d). As expected, Hrd1−/− macrophages showed a significant upregulation of the UPR gene Bip (Fig. 2d). However, the reduced inflammatory gene expression is not due to UPR activation, as shown later. Treatment with LS-102 significantly reduced the transcription of inflammatory genes (Fig. S2d) and the secretion of cytokines, including TNFα, IL6 and IFNβ (Fig. 2e), in poly(I:C)-stimulated primary macrophages. LS-102 treatment did not reduce poly(I:C)-induced transcription of inflammatory genes in Hrd1−/− macrophages, excluding an off-target effect (Fig. S2e). Consistently, Hrd1 knockdown in primary macrophages also led to reduced transcription of inflammatory genes and decreased secretion of TNFα, IL6 and IFNβ upon poly(I:C)-stimulation (Fig. S2f, g). Poly(I:C)-induced NF-κB promoter activity and interferon-stimulated response element (ISRE) activity was also reduced in Hrd1−/− HEK293T-TLR3 cells compared to WT cells (Fig. S2h). Therefore, the HRD1-TLR3 signaling regulatory axis represents a general mechanism across different cells. We next evaluated the potency of secretomes from poly(I:C)-stimulated WT and Hrd1−/− macrophages in inhibiting HSV-1 replication. Replication of the HSV-1 was significantly higher in Vero cells treated with secretome from Hrd1−/− cells when compared to the control group (Figs. 2f and S2i). Taken together, we conclude that HRD1 enhances TLR3-mediated innate immune signaling.
HRD1 and TLR3 signaling form a positive feedback loop between each other
HRD1 is a positive regulator of TLR3 signaling (Fig. 2a–f), and its protein levels and transcription are increased upon poly(I:C) stimulation (Figs. 1b, and 3a), indicating that TLR3 signaling upregulates HRD1 to form a positive feedback loop. Consistent with previous study showing HRD1 expression can be induced via extra-cellular signal-regulated kinase1/2 (ERK1/2) activation and transcriptional activity of ETS135, poly(I:C) stimulation elevated ERK activation, as indicated by the hyper-phosphorylation of ERK1/2 (Fig. 3b), and increased the transcription of Ets1 and Hrd1 (Fig. 3c). Knocking down Ets1 in primary macrophages using siRNAs inhibited poly(I:C)-induced increase in Hrd1 transcription (Fig. S3a, b). Furthermore, both PD98059, an inhibitor of the ERK1/2 upstream mitogen-activated protein kinase (MAPK) kinase 1/2 (MEK1/2), and LS-102 inhibited the elevation of ERK phosphorylation and significantly reduced the transcription of Ets1 and Hrd1 following poly(I:C) stimulation (Fig. 3b, c). The suppressive effects of LS-102 on ERK phosphorylation and the transcription of Ets1 and Hrd1 were rescued by the addition of various TLR3 downstream cytokines (Fig. 3d, e, f).

a qPCR analysis of Hrd1 mRNA levels in primary macrophages pre- and post-poly(I:C) (50 μg/ml for 3 h) treatment. n = 4 each. b–c Immunoblot analysis of ERK1/2 phosphorylation levels (b) and qPCR analysis of Ets1 and Hrd1 mRNA levels (c) in macrophage cells pre-treated with LS-102 (5 μM) or ERK1/2 inhibitor PD98059 for 24 h, and then with poly(I:C) (50 μg/ml) for 3 h. n = 4 each in (c). d–e Immunoblot analysis of ERK1/2 phosphorylation levels (d) and qPCR analysis of Ets1 and Hrd1 mRNA levels (e) in macrophage cells pre-treated with LS-102 (5 μM for 24 h), and stimulate with poly(I:C) (50 μg/ml) and indicated cytokines for 3 h. n = 4 each in (e). f Immunoblot analysis of indicated proteins in macrophages treated with poly(I:C), TNFα, IL1β, IL6 or IFNβ for 12 h. g qPCR analysis of Tlr3 mRNA levels in macrophages pre- and post-poly(I:C) (50 μg/ml for 3 h) treatment. n = 4 each. h Immunoblot analysis of indicated proteins in macrophages pre-treated with vehicle or LS-102 for 24 h, followed by poly(I:C) (50 μg/ml) treatment for the indicated times. i–j Immunoblot analysis of STAT1 phosphorylation levels (i) and qPCR analysis of Irf1, Irf2 and Tlr3 mRNA levels (j) in macrophages pre-treated with LS-102 (5 μM) or STAT1 inhibitor Fludarabine for 24 h, and then stimulated with poly(I:C) (50 μg/ml) for 3 h. n = 4 for (j). k–l Immunoblot analysis of STAT1 phosphorylation levels (k) and qPCR analysis of Irf1, Irf2 and Tlr3 mRNA levels ( l) in macrophages pre-treated with LS-102 (5 μM) for 24 h, followed by stimulation with poly(I:C) (50 μg/ml) and IFNβ for 3 h. n = 4 for (l). All experiments were repeated for at least three times. Quantitation of the ratio of phosphorylated to total protein (p/t) and indicated protein is shown below each blot. Values represent mean ± SEM, NS, not significant, by unpaired, two-tailed, Student’s t-test (a, c, e, g, j, l). Source data are provided as a Source Data file.
Tlr3 transcription and protein levels were also increased in macrophage upon poly(I:C) stimulation (Fig. 3g, h), which was dependent on HRD1 and signal transducer and activator of transcription 1 (STAT1) activity, as both LS-102 and the STAT1 inhibitor Fludarabine suppressed poly(I:C)-induced Tlr3 upregulation (Fig. 3h–j). Previous studies showed that IFNβ and interferon regulatory factors (IRFs) promote TLR3 expression36. Adding back IFNβ directly induced STAT1 activation in the presence of LS-102 (Fig. 3k) and rescued the induction of Tlr3 transcription (Fig. 3l). Additionally, Irf1 and Irf2 induction was observed upon poly(I:C) stimulation (Fig. 3j, l), which was suppressed by LS-102 and Fludarabine (Fig. 3j, l) but rescued by IFNβ (Fig. 3l). Taken together, TLR3 signaling induces HRD1 expression through ERK1/2-ETS1 activity. In turn, HRD1 activity contributes to the increased transcription of Tlr3 in a STAT1-dependent manner, thereby amplifying the inflammatory response.
The effect of HRD1 on TLR3 signaling is uncoupled from ERAD and UPR
The classical function of HRD1 is to ubiquitinate misfolded or misfolding-prone substrate proteins and promote their proteasomal degradation17,18. For example, HRD1 deficiency leads to the accumulation and impaired function of pro-arginine vasopressin (proAVP) and pro-opiomelanocortin (POMC)23,24. Considering that TLR3 is synthesized and mostly resides in the ER of resting cells3,4, we then tested whether HRD1 promotes TLR3 signaling by regulating the abundance of TLR3 protein in the ER. Surprisingly, there was no difference in the abundance of the full-length TLR3 protein between WT and Hrd1−/− macrophages (Fig. 4a). Consistent with this, the translation shut-off assay using cycloheximide (CHX) revealed that the full-length TLR3 protein levels decreased by a comparable ratio within 9 h of treatment in both WT and Hrd1−/− macrophages (Fig. 4b). The overexpression of HRD1 in HEK293T-TLR3 cells did not change the protein levels of TLR3 at varying doses (Fig. 4c). These findings indicate that HRD1 does not directly regulate the degradation and abundance of the TLR3 protein in the ER. We also used Eeyarestatin I that targets Valosin-containing protein (VCP) to inhibit ERAD37,38, and ERAD inhibitor pretreatment induced expression of UPR genes, including Xbp1s and Bip, but did not reduce poly(I:C)-induced expression of pro-inflammatory genes in macrophages (Fig. 4d). Additionally, irrespective of Eeyarestatin I pretreatment, poly(I:C)-induced inflammatory gene expression remained consistently impaired in HRD1-deficient macrophages compared to WT macrophages (Fig. 4d).

a Immunoblot and quantitation analysis of TLR3 in WT and Hrd1−/− RAW 264.7 cells. n = 3. b Immunoblot analysis in WT and Hrd1−/− RAW 264.7 cells treated with CHX (50 µg/ml) for the indicated times, with quantitation from five independent experiments shown. c, Immunoblot analysis of indicated proteins in HEK293T-TLR3 cells transfected with increasing amounts of plasmid expressing HRD1 protein. d qPCR analysis of Xbp1s, Bip, Tnfa, Ifnb1, Il6 and Il1b in WT and Hrd1−/− RAW 264.7 cells treated with Eeyarestatin I (5 uM) for 8 h and/or poly(I:C) (50 µg/ml) for 6 h. n = 4 each. e Immunoblot analysis of indicated proteins in WT and Hrd1−/− RAW 264.7 cells treated with thapsigargin (300 nM) for 1.5 h and/or poly(I:C) (50 µg/ml) for 1 h. Tg, thapsigargin. p-, phosphorylated PERK; n-, non-phosphorylated PERK. f qPCR analysis of Bip, Xbp1s, Il6, Ccl5, Chop, Tnfa, Ifnb1 and Il1b in WT and Hrd1−/− RAW 264.7 cells treated with thapsigargin (300 nM) and/or poly(I:C) (50 µg/ml) for 6 h. n = 4 each. g qPCR analysis of Xbp1u, Xbp1s, Tnfa, Il1b, Ccl5, Il6 and Ifnb1 in WT and Hrd1−/− RAW 264.7 cells treated with 4μ8c (0.1 mM) for 30 h and/or poly(I:C) (50 µg/ml) for 6 h. n = 4 each. Quantitation of the ratio of phosphorylated to total protein (p/t) and indicated protein is shown below each blot (c, e). All data were representative of at least three independent repeats. Values represent mean ± SEM NS, not significant, by unpaired, two-tailed, Student’s t-test (a, b, d, f, g). Source data are provided as a Source Data file.
ERAD and UPR, two key quality-control machineries, are closely interconnected to maintain ER homeostasis16, we next examined whether HRD1 effect on TLR3 is mediated by UPR. Hrd1−/− cells exhibited mild ER stress compared to WT cells under basal conditions, as indicated by Eif2α phosphorylation levels (Fig. 4e) and expression of UPR genes Bip, Xbp1s, and Chop (Fig. 4f). Treatment with thapsigargin (Tg) induced robust ER stress in both WT and Hrd1−/− cells (Fig. 4e, f). However, ER stress induction did not suppress poly(I:C)-induced TLR3 signaling in WT cells, and regardless of Tg treatment, poly(I:C)-induced TLR3 signaling and inflammatory gene expression remained consistently reduced in Hrd1−/− macrophages compared to WT macrophages (Fig. 4e, f). Taken together, compromised TLR3 signaling in Hrd1-deficient cells is not due to the inhibition of ERAD activity or the induction of ER stress.
IRE1α-XBP1s branch of the UPR is reported to potentiate the production of pro-inflammatory cytokines upon TLR2 and TLR4 stimulation in macrophage39 and IRE1α is significantly increased in Hrd1−/− macrophages (Fig. 4e)30, we next asked whether IRE1α mediates HRD1 effect on TLR3 signaling. We used an IRE1α-specific inhibitor 4 μ8c40, which expectedly suppressed Xbp1 mRNA splicing in both WT and Hrd1−/− macrophages (Fig. 4g). 4μ8c pretreatment reduced the expression of pro-inflammatory genes in both genotypes but did not reverse the reduction in pro-inflammatory gene expression in Hrd1−/− RAW 264.7 cells upon poly(I:C) stimulation (Fig. 4g). Taken together, these data demonstrate that the effect of HRD1 on TLR3 pathway is ERAD-, UPR- and IRE1α- independent.
HRD1 modulates TLR3 signaling through ubiquitinating TLR3
Ubiquitination not only guides protein degradation by the proteasome but also exerts regulatory roles through diverse mechanisms41. We then tested whether HRD1 ubiquitinates TLR3 to regulate its signaling. In addition to the observed physical interaction between HRD1 and TLR3 (Fig. 1c–h), HRD1 indeed robustly ubiquitinated both exogenously overexpressed (Fig. 5a–b) and endogenous TLR3 proteins (Fig. 5c). Intriguingly, the ubiquitination of TLR3 by HRD1 was observed to increase significantly following poly(I:C) treatment, highlighting the physiological relevance of this modification (Fig. 5d). Moreover, HRD1 efficiently ubiquitinated ER-resident TLR3 when TLR3 transport from ER to Golgi was blocked by Brefeldin A (BFA) (Figs. 5e, and S3c). Similar as previously reported substrates42, the catalytic RING domain of HRD1 is critically needed to mediate the ubiquitination of TLR3, as demonstrated by the reduction of poly-ubiquitination of TLR3 in cells expressing HRD1 RING ligase-dead C291A/C294A (C2A) mutant (lanes 2 vs. 3, Figs. 5f, and S3d) or ΔRING mutant (lanes 2 vs. 4, Figs. 5f, and S3d). TM domain of TLR3 was required for the effective interaction between TLR3 and HRD1 (Fig. 1h), TM domain of HRD1 also plays a crucial role in facilitating the ubiquitination of TLR3 as ΔTM HRD1 mutant with intact RING domain failed to ubiquitinate TLR3 despite of its high expression level (lanes 2 vs. 5, Figs. 5f, and S3d). The overexpression of WT HRD1 significantly enhanced the poly(I:C)-triggered NF-κB promoter and ISRE activity, when compared to the overexpression of C2A, ΔRING or ΔTM mutants, further demonstrating the significance of HRD1 E3 ligase activity in TLR3 signaling (Fig. S3e). Consistent with the inhibition of TLR3 signaling by LS-102 (Figs. 2c, e, S2d), LS-102 also suppresses the ubiquitination of TLR3 by HRD1 (Figs. 5g, and S3f). Taken together, HRD1 directly ubiquitinates ER-located TLR3 via its RING domain, and the ubiquitin-ligation function of HRD1 is essential for its regulation of TLR3 signaling.

a Immunoblot analysis of TLR3 polyubiquitination following TLR3-Flag immunoprecipitation in HEK293T cells transfected with the specified plasmids, including tagged Ub, HRD1 and TLR3. b Immunoblot analysis of TLR3 polyubiquitination in vitro. Arrows indicated HRD1 and HRD1ΔTM proteins. c Immunoblot analysis of polyubiquitination following immunoprecipitation of endogenous TLR3 in WT and Hrd1−/− RAW 264.7 cells. Ub, ubiquitin. d Immunoblot analysis of polyubiquitination following immunoprecipitation of endogenous TLR3 in RAW 264.7 cells treated with 50 μg/ml poly(I:C) for the indicated times. e Immunoblot analysis of TLR3 polyubiquitination following TLR3-Flag immunoprecipitation in HEK293T cells that were transfected with the indicated plasmids for 18 h and treated with vehicle or 10 μg/ml Brefeldin A for 8 h. BFA, Brefeldin A. Immunoblot data of the input are shown in Supplementary Fig. 3c. f Immunoblot analysis of TLR3 polyubiquitination following TLR3-Flag immunoprecipitation in HEK293T cells transfected with the indicated plasmids. C2A, HRD1-dead variant; ΔRING, RING domain-deleted truncations; ΔTM, transmembrane domain-deleted truncation. Immunoblot data of the input are shown in Supplementary Fig. 3d. g Immunoblot analysis of TLR3 polyubiquitination following TLR3-Flag immunoprecipitation in HEK293T cells transfected with the indicated plasmids for 18 h, and sequentially treated with or without LS-102 (5 μM) for 8 h. Immunoblot data of the input are shown in Supplementary Fig. 3f. h Immunoblot analysis following immunoprecipitation of TLR3-Flag in the lysates of HEK293T cells transfected with TLR3-Flag, HRD1-Myc, and HA-Ub (WT and K-loss mutants). K-R, Lys mutated to Arg. i Immunoblot analysis of the indicated proteins following immunoprecipitation of TLR3-Flag or VCP-V5 in the lysates of HEK293T cells transfected with TLR3-Flag, HRD1-Myc, VCP-V5 and HA-Ub. Immunoblot data of the input are shown in Supplementary Fig. 3h. j Immunoblot analysis of the indicated proteins following immunoprecipitation of TLR3-Flag in the lysates of Hrd1+/+ and Hrd1−/− HEK293T cells transfected with TLR3-Flag. All blot data were representative of at least three independent repeats. Source data are provided as a Source Data file.
The type of ubiquitin linkage formed could be critical for the fate of the target protein, we next defined the polyubiquitin chain topology involved in TLR3 ubiquitination. Mutations of ubiquitin at K48 and K63 (K48R and K63R mutants) led to a significant reduction in the level of TLR3 polyubiquitination (Fig. 5h). In consistent, the K48- and K63-linked ubiquitination of TLR3 increased significantly following poly(I:C) treatment in macrophage in a HRD1-dependent way (Fig. S3g). VCP engages with ERAD substrates and is required for retrotranslocation and proteasomal degradation of misfolded proteins43. We further tested the interaction between VCP and TLR3 and found that ubiquitinated TLR3 interacted with HRD1 but not with VCP, instead reducing the interaction between HRD1 and VCP (Figs. 5i, and S3h). Taken together, HRD1-mediated ubiquitination of TLR3 primarily occurs through the K48 and K63 residues of ubiquitin, in contrast to K 27-linked ubiquitination observed in other HRD1 substrates, such as STING and CREBH30,44. This conformation of ubiquitin chain, together with other unrecognized features of TLR3 might allow it escape VCP engagement and proteasomal degradation after ubiquitination by HRD1.
In addition, no interaction was observed between TLR3 with SEL1L, BIP, or OS9, which facilitate the recruitment of ERAD substrates, both in WT and Hrd1−/− cells (Fig. 5j). While the adaptor protein SEL1L normally forms a complex with HRD1 and is required for HRD1 stability20, previous studies suggest that they may have independent functions21,45.
To further test the role of SEL1L, we utilized siRNA to reduce SEL1L protein levels without bringing down HRD1 in primary macrophages (Fig. S4a, b), and this approach demonstrated that reducing SEL1L alone did not decrease the expression of various inflammatory cytokine genes upon poly(I:C) stimulation (Fig. S4c). Furthermore, SEL1L deficiency did not reduce exogenously expressed HRD1-mediated TLR3 ubiquitination levels in HEK293T cells (Fig. S4d). Thus, HRD1 could modify TLR3 and regulate its signaling independent of ERAD activities.
HRD1 promotes TLR3 Trafficking into endolysosome for cleavage
Ubiquitination can facilitate the efficient sorting and the accurate delivery of membrane proteins to their destination organelles46. TLR3 primarily resides in the ER, but it undergoes trafficking through Golgi to endolysosomes upon poly(I:C) stimulation for proteolytic cleavage and activation3,4. Next, we investigated whether intracellular trafficking of TLR3 is impaired due to deficient HRD1 ubiquitination activity in Hrd1−/− cells. We performed confocal microscopy to examine TLR3 location at different organelles in WT and Hrd1−/− macrophages treated with or without poly(I:C) for the indicated times, including the ER (KDEL), trans-Golgi network (TGN38), and lysosomes (LAMP1) (Figs. 6a-c, and S5a–c). Our confocal microscopy studies revealed that in resting-state WT and Hrd1−/− cells, TLR3 was primarily located in the ER (Figs. 6a, d and S5a). Upon treatment with poly(I:C), the localization levels of TLR3 in the Golgi did not change in Hrd1−/− cells compared to WT cells (Figs. 6b, e, and S5b). In WT cells, poly(I:C) significantly increased TLR3 localization in the endolysosome, whereas TLR3 transportation to the endolysosome was substantially decreased in Hrd1−/− cells compared to WT cells (Figs. 6c, f, and S5c). Similarly, by using TLR3-Flag expressing WT and Hrd1−/− HEK293T cells, we observed that the location of TLR3 in the ER (Calnexin) and its transportation to the Golgi (GM130) did not change between WT and Hrd1−/− cells (Figs. 6g, h, and S5d-e, h-i).

a–f Confocal microscopic images of TLR3 co-stained with DAPI and ER marker KDEL (a), trans-Golgi network marker TGN38 (b) and lysosome marker LAMP1 (c) in WT and Hrd1−/− RAW 264.7 cells with or without poly(I:C) (50 µg/ml) treatment. Quantitation of the fraction of TLR3 in the ER (d, from left to right, n = 60, 58, 60, 60, 60, 60), trans-Golgi network (e, n = 60 for all groups), and lysosomes (f, n = 60 for all groups) in these macrophages were shown. Mander’s overlap coefficient is used for measurement of colocalization. g–j Confocal microscopic images of TLR3-Flag co-stained with DAPI and ER marker Calnexin (g), Golgi marker GM130 (h), early endosome marker Rab5 (i) and late endosome marker Rab7 (j) in WT and Hrd1−/− HEK293T cells transfected with TLR3-Flag plasmid and with or without poly(I:C) treatment. k–m Immunoblot analysis of full-length (FL) and cleaved TLR3 (CL) in WT and Hrd1−/− HEK293T cells transfected with TLR3-Flag plasmid and treated with 50 μg/ml poly(I:C) for 12 h (k), LS-102 (5 μM) for 8 h (l), and with Endo H or PNGase F (m). Arrow, Endo H-sensitive; Red box, Endo H-resistant. n Immunoblot analysis of indicated proteins following the isolation of the ER, Golgi and endosomes from WT and Hrd1−/− HEK293T cells transfected with TLR3-Flag for 24 h, followed by 50 µg/ml poly(I:C) stimulation for 1 h. Relative quantitation of indicated TLR3 bands shown below the blot. o–q Immunoblot analysis of indicated proteins following immunoprecipitation of TLR3-Flag in the lysates of HEK293T cells transfected with TLR3-Flag, HRD1-myc (WT, C2A, and ΔRING), and HRS-V5 (o), or TGS101-V5 (p), or VPS36-V5 (q). C2A, HRD1-dead variant; ΔRING, RING domain-deleted truncate. Quantitation of the protein level is shown below the blot. All confocal image and blot data were representative of at least three independent repeats (a–q). Values represent mean ± SEM, by unpaired, two-tailed, Student’s t-test (d–f). Source data are provided as a Source Data file.
However, poly(I:C) treatment significantly increased TLR3 transport to the early endosome (Rab5) and endolysosome (Rab7) in WT cells, whereas this transport was inhibited in poly(I:C)-treated Hrd1−/− cells (Figs. 6i, j, and S5f-g, j–k).
We then used Western blot analysis to directly examine the levels of cleaved TLR3 in HEK293T cells. Remarkably, we observed a significant reduction in the level of cleaved, activable TLR3 in poly(I:C)-treated Hrd1−/− cell compared to WT cells (Fig. 6k, with quantitation shown in Fig. S6a), and in LS102-treated cells compared to control cells (Fig. 6l, with quantitation shown in Fig. S6b). The uncleaved TLR3 were primarily high-mannose endoglycosidase (Endo) H-sensitive in both WT and Hrd1−/− cells, suggesting uncleaved TLR3 molecules were largely retained within the ER in both cell types (Fig. 6m, lanes 1–2 vs. 3-4, highlighted with arrow). A fraction of uncleaved TLR3 was endoH-resistant and peptide-N-glycosidase F (PNGase F)-sensitive (Fig. 6m, lanes 3-4 vs. 5-6, highlighted in red box), indicating that this specific fraction of TLR3 is located in post-ER compartments. And the relative protein levels of Endo H-resistant TLR3 in WT cells were higher compared to Hrd1−/− cells (Fig. 6m, lanes 3 vs. 4, with quantitation shown in Fig. S6c). In line with earlier reports suggesting that TLR3 underwent trafficking through the Golgi network before reaching endosomes for cleavage47, the cleaved form of TLR3 exhibited partial resistance to Endo H treatment (Fig. 6m, lanes 3-4 vs. 5-6), and the level of cleaved TLR3 is decreased in Hrd1−/− cells compared to WT cells (Fig. 6m, lanes 1 vs. 2, lanes 3 vs. 4, lanes 5 vs. 6). We further examined the distribution of TLR3 within the ER, Golgi and endosome following the isolation of these organelles. We observed a significant reduction in both un-cleaved and cleaved TLR3 within the fraction of endosomes in Hrd1−/− cells as compared to WT cells after poly(I:C) treatment (Fig. 6n). The regulation of TLR3 cleavage and intracellular trafficking by HRD1 was also examined in mouse embryonic fibroblasts (MEFs). HRD1 deficiency and LS-102 treatment resulted in a marked decrease of cleaved TLR3 levels (Fig. S6d-e). Furthermore, the proportion of post-ER un-cleaved TLR3 was significantly lower in Hrd1−/− MEFs compared to WT cells (Fig S6f). Following poly(I:C) stimulation, both uncleaved and cleaved TLR3 were notably reduced in the endosomal fraction of Hrd1−/− MEFs versus WT cells (Fig. S6g). Together, these results strongly indicate that HRD1 is essential for proper endosomal localization of TLR3.
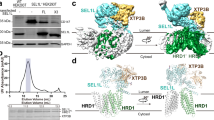
The ESCRT (endosomal sorting complexes required for transport) machinery play a critical role in the sorting of ubiquitinated membrane proteins to endolysosome48. Hepatocyte growth factor-regulated tyrosine kinase substrate (HRS), Tumor susceptibility gene 101(TSG101), and Vacuolar protein-sorting-associated protein 36 (VPS36), the key components of the ESCRT-0, ESCRT-I, and ESCRT-II complexes respectively, were previously reported to recognize, bind and collectively orchestra the sorting of polyubiquitinated TLR3 to endolysosome for cleavage3. Overexpression of WT HRD1, but not the HRD1 C2A mutant, significantly enhanced the binding of TLR3 with HRS, TSG101 and VPS36 (Fig. 6o–q). Furthermore, the interaction between endogenous TLR3 with HRS, TSG101 and VPS36 was significantly augmented in macrophage upon poly(I:C) treatment in a HRD1-dependent way (Fig. S7a).
E3 ligase TRIM3, localized in the Golgi apparatus and endosomes, mediates the ubiquitination of TLR3 at K831 to enhance its trafficking3. To delineate the relationship between TRIM3 and HRD1 in TLR3 trafficking, we utilized siRNA to knock down Trim3 in Hrd1−/− RAW 264.7 cells, and found that the reduction of Trim3 further decreased the expression of inflammatory cytokine genes upon poly(I:C) stimulation in cells with Hrd1 deficiency (Fig. S7b). Additionally, the expression of HRD1 did not affect TRIM3-mediated ubiquitination of TLR3 (Fig. S7c). These findings collectively suggest that HRD1’s E3 ubiquitination activity is a prerequisite for ESCRT-mediated recognition and sorting of TLR3 to endosomes.
Ubiquitination of TLR3 at K813 by HRD1 is indispensable for TLR3 trafficking
The cytosolic domain of TLR3, rather than the N-terminal ectodomain truncate, underwent polyubiquitination by HRD1 (Figs. 7a, and S8a), suggesting that the ubiquitination of TLR3 primarily takes place within its cytosolic domain. To further map the exact ubiquitination site(s), we next replaced each of the 11 cytosolic K residues to Arg (R) individually. TLR3 K813R mutant showed mostly reduced level of ubiquitination by HRD1 (Fig. S8b, c), and polyubiquitin chains composed of WT, K48 and K63 ubiquitin moieties were consistently observed to be conjugated on TLR3 via K813 residue (Fig. 7b). Moreover, Brefeldin A treatment led to the stuck of TLR3 K813R in the ER but did not induce its ubiquitination by HRD1, indicating HRD1 ubiquitinates ER-located TLR3 at K813 (Fig. S8d).

a Immunoblot analysis of TLR3 polyubiquitination following TLR3-Flag immunoprecipitation in HEK293T cells transfected with HA-Ub, HRD1-myc, and TLR3-Flag (WT and truncations). FL, full length; LRR, leucine-rich repeat truncation; TM + TIR, transmembrane domain and cytosolic Toll/interleukin-1 receptor domain truncation. b Immunoblot analysis of TLR3 polyubiquitination following immunoprecipitation of TLR3-Flag in the lysates of HEK293T cells transfected with HRD1-myc, HA-Ub (WT and K48-, K63-only mutants), and TLR3-Flag (WT and K813-loss mutant). K813R, K813-loss. c Immunoblot analysis of FL and cleaved TLR3 in MEF cells stably expressing WT TLR3-Flag or K813R mutant TLR3-Flag and treated with 50 μg/ml poly(I:C) for the indicated times. d Immunoblot analysis of TLR3-Flag in MEF cell lysates treated with or without Endo H or PNGase F. The cells stably expressed WT TLR3-Flag or K813R mutant TLR3-Flag and treated with 50 μg/ml poly(I:C) for 12 h before preparing the cell lysates. Arrow, Endo H-sensitive; Red box, Endo H-resistant. e Immunoblot analysis of the indicated proteins following the isolation of the ER, Golgi and endosomes from MEF cells stably expressing WT TLR3-Flag or K813R mutant TLR3-Flag and treated with 50 μg/ml poly(I:C) for 60 minutes. Relative quantitation of TLR3 bands in indicated organelles shown below the gel. f Immunoblot analysis of indicated proteins in HEK293T cells stably expressing WT TLR3 or K813R TLR3 and treated with 50 μg/ml poly(I:C) for the indicated times. Quantitation of the ratio of phosphorylated to total protein (p/t) is shown below each blot. All data were representative of at least three independent repeats (a–f). Source data are provided as a Source Data file.
Ubiquitination-defective TLR3 K813R mutant exhibited reduced levels of cleaved TLR3 at basal stage and upon poly(I:C) treatment in MEF cells (Fig. 7c, and quantification shown in Fig. S8e), further supporting a crucial role of HRD1-mediated TLR3 ubiquitination in promoting its cleavage. By using Endo H and PNGase F assays, we further observed that the level of uncleaved TLR3 K813R in the post-ER fraction was lower compared to wild-type TLR3, indicating a traffic defect of this mutant (Fig. 7d, lanes 3 vs. 4, highlighted in red box, and quantification shown in Fig. S8f). Following the isolation of cellular organelles, we observed a significant reduction in both uncleaved and cleaved TLR3 K813R within the fraction of endosomes as compared to WT TLR3 after poly(I:C) treatment (Fig. 7e). Furthermore, K813R mutation also resulted in a substantial impairment of signaling transduction following poly(I:C) treatment, as indicated by a notable decrease in the phosphorylation levels of TBK1 and P65, along with an elevated stabilization of IκBα (Fig. 7f). Thus, in contrast to HRD1-mediated ubiquitination of STING and non-secreted immunoglobulin light chain, which occurs at multiple types of amino acids to promote the degradation of these target proteins30,49, HRD1 specially mediates the ubiquitination of TLR3 at residue K813 to positively regulate its trafficking and signaling.
HRD1 promotes TLR3-mediated inflammation in vivo
To assess the physiological relevance of TLR3 regulation by HRD1, we examined whether targeting HRD1 could mitigate the systemic inflammation and tissue damage in mice following poly(I:C)-injection. Consistent with its in vitro effects (Fig. 2c, e), LS-102 treatment in vivo significantly reduced serum cytokines levels of TNFα, IL6 and IFNβ (Fig. 8a), as well as lethality rates (Fig. 8b), in C57BL/6 mice following poly(I:C)-injection. Histological analysis revealed that poly(I:C)-induced leukocyte infiltration, alveolar wall thickening, and edema in the lung were markedly alleviated in LS-102-treated mice (Fig. 8c). Additionally, hepatocyte cell death, indicated by TUNEL staining, was also significantly reduced (Fig. 8c).

a ELISA analysis of serum cytokines TNFα, IL6 and IFNβ in mice from vehicle- and LS-102- pretreated groups at 2 h after poly(I:C) (0.7 mg/kg body weight) injection. LS-102 was injected at 10 mg/kg body weight per day for three days (n = 8 or 10 mice for vehicle and LS-102-treated group). b Survivorship of C57BL/6 mice treated with vehicle or LS-102 followed by poly(I:C) plus D-GalN (0.5 g/kg body weight) injection (n = 6 mice each). c Representative images showing lung inflammation and hepatocyte cell death in the indicated mice. Sex- and age-matched C57BL/6 mice treated with vehicle or LS-102 for three days, followed by poly(I:C) plus D-GalN injection for 4 h, then lung sections were used for HE staining (n = 17 each), and liver sections were used for TUNEL analysis (n = 15 for mock group, n = 17 for LS-102 group). d qPCR analysis of indicated genes in BMDMs isolated from WT or TLR3K813R KI mice and treated with poly(I:C) for 3 h (n = 4 each). e Immunoblot analysis of the indicated proteins in BMDMs isolated from WT or TLR3K813R KI mice and treated poly(I:C). Ratio of phosphorylated to total protein (p/t) is shown below each blot. f ELISA analysis of serum cytokines TNFα, IL6 and IFNβ from sex- and age-matched WT or TLR3K813R KI mice after poly(I:C) injection for 2 h. n = 9 for each group. g Survivorship of WT and TLR3K813R KI mice after i.p. poly(I:C) plus D-GalN injection. n = 9 for each group. h Representative images showing lung inflammation and hepatocyte cell death in the indicated mice. WT and TLR3K813R KI mice were injected i.p. with poly(I:C) plus D-GalN for 4 h, then lung sections were used for HE staining (n = 12 for WT, n = 15 for TLR3K813R KI), and liver sections were used for TUNEL analysis (n = 12 for WT, n = 15 for TLR3K813R KI). All above data are from at least three independent repeats. Values represent mean ± SEM, by unpaired, two-tailed, Student’s t-test (a, d, f), and log-rank (Mantel–Cox) test (b, g). Source data are provided as a Source Data file.
To further establish the physiological relevance of HRD1-mediatd K813 ubiquitination of TLR3 in vivo, we generated knock-in (KI) mice carrying the K813R mutation on the C57BL/6 J genetic background using the CRISPR/Cas9-based methods (Fig. S9a). Three genotype-confirmed F0 mice were used to generate F1 mice independently by crossing each of them with C57BL/6 J WT mice. F1 heterozygous KI mice were inter-crossed to generate two independent homozygous KI mice lines, with genotype confirmed by PCR and sequencing (Fig. S9b). Each KI line was characterized independently, and similar results were obtained from each line. Bone marrow-derived macrophages (BMDMs) from TLR3K813R KI mice displayed a notably decreased response to poly(I:C) stimulation compared to those from WT littermates, including lower transcriptional levels of various inflammatory cytokines (Figs. 8d, and S9c), as well as reduced phosphorylation levels of TBK1, P65, and IκBα (Fig. 8e). Consistently, serum cytokines levels of TNFα, IL6 and IFNβ (Fig. 8f), as well as lethality rates (Fig. 8g), were significantly lower in TLR3K813R mice compared to their WT littermates after poly(I:C) treatment. Histological analysis confirmed that poly(I:C)-induced lung inflammation and hepatocyte cell death were markedly reduced in TLR3K813R cohorts (Fig. 8h). Taken together, HRD1-mediated regulation and K813 ubiquitination of TLR3 are essential for TLR3 agonist-driven inflammation in vivo.
Discussion
TLR3 plays a critical role as a membrane receptor in recognizing viral dsRNA and initiating antiviral immune responses2, and TLR3 stimulation can potentially enhance anti-tumor immunity50. Initially synthesized in the ER, TLR3 activation is strategically constricted in endolysosome compartment, the location where external pathogens are internalized and degraded1,6,51. Despite the clear significance of TLR3 trafficking, the specific molecular mechanism that directly regulates this process remains in its infancy. Interestingly, certain regulatory mechanisms known to be involved in other Toll-like receptors (TLRs) do not apply to TLR3. For instance, the ER-resident protein gp96 acts as a master chaperone for multiple TLRs, and PRAT4A is required for multiple TLR responses, but both proteins are dispensable for TLR38,52,53. Small calcium-binding protein S100A9 was reported to interact with TLR3 and required for delivery of TLR3 into late endosome, but the mechanism is still not well understood54. In a more recent study, it was shown that TRIM3, localized in the Golgi apparatus and endosomes, mediates the ubiquitination of TLR3 at K831 to enhance its trafficking3. Taken together, it remains unclear whether there is any ER protein(s) that can modify TLR3 to regulate its transport. In this study, we have uncovered that HRD1, an ER-resident E3 ligase, is a critical regulator of TLR3 trafficking. Through direct ubiquitination of TLR3 at K813, HRD1 positively modulates the engagement of TLR3 with ESCRT components HRS, TSG101, and VPS36, thereby enhancing its transport to endolysosomes for cleavage and ligand recognition (Fig. S9d). Interestingly, HRD1 ubiquitinates TLR3 within the ER (Fig. 5e); thus, this modification serves as a feed-forward control for TLR3 trafficking from Golgi to Endosome. The blockage of HRD1-mediated TLR3 ubiquitination significantly compromises the activity of its downstream signaling effectors and the transcription of target inflammatory genes. Therefore, HRD1 ubiquitination activity is a crucial determinant in the overall TLR3 immune response.
UNC93B1 is an ER membrane protein that is indispensable for the functions of endosomal TLRs, including TLR355. The mechanisms by which UNC93B1 mediates these regulatory effects include its association with TLR from the ER through post-Golgi steps, guiding the trafficking of TLR to the endosome56,57. Alternatively, it has been recently discovered that UNC93B1 deficiency resulted in near-complete loss of TLR3 and UNC93B1 works as a chaperone to maintain the stability of TLR3 independently of endosomal transport9. In contrast to UNC93B1, our data demonstrated that HRD1 is not involved in TLR3 abundance and stability, and it is unlikely HRD1 remains bound to TLR3 during its trafficking. Instead, as an ER-resident E3 ligase, HRD1 directly ubiquitinates TLR3 to facilitate its trafficking process. UNC93B1 and HRD1 thus represent two different mechanisms for regulating TLR3 signaling. Understanding how these two ER proteins coordinate their activities deserves further investigation in the future.
HRD1 forms a complex with adaptor protein SEL1L; this interaction is essential for stabilizing HRD1 and forming the core of the ERAD machinery15,16,17. The primary function of SEL1L-HRD1 complex is to recruit, ubiquitinate and target ER-resident substrate proteins for cytosolic proteasomal degradation19,58,59. In a context-dependent manner, HRD1 ERAD functions either as a triage mechanism for misfolding-prone substrates or as a regulator of substrate protein abundance. For example, misfolded proAVP and POMC are triaged and degraded by SEL1L-HRD1 ERAD to ensure ER homeostasis and proper protein maturation23,24. On the other hand, SEL1L-HRD1 ERAD serves as a quantity control mechanism by regulating the abundance of pre-BCR, IRE1α, cAMP responsive element-binding protein H (CREBH), Sigma non-opioid intracellular receptor 1 (SigmaR1), transforming growth factor-beta receptor 1 (TGF-β receptor 1), and STING, respectively, and therefore controlling the amplitudes of downstream signaling15,22,28,30,60,61,62. Contrary to the fate of previously identified HRD1 substrates, the stability of TLR3 and its abundance in the ER were not affected in Hrd1−/− macrophages. Additionally, our study shows, through both pharmacological and genetic approaches, that the inhibition of VCP/ERAD activity or the induction of ER stress does not compromise TLR3 signaling, and inhibition of IRE1α activity in Hrd1−/− cells does not rescue TLR3 signaling. Thus, the ubiquitination of TLR3 by HRD1 is uncoupled from proteasomal degradation, and HRD1 regulates TLR3 signaling independent of ERAD activity or ER stress. Instead, this HRD1-mediated protein ubiquitination serves as a regulatory mechanism to ensure TLR3 is transported into the endolysosome. Of note, in contrast to previously reported ubiquitination pattern that leads to degradation of HRD1 substrates30,44, HRD1-mediated ubiquitination of TLR3 primarily occurs at the K813 residue through the K48- and K63-linked ubiquitin chains. In addition, TLR3 does not interact with SEL1L, BIP, or OS9, which are key facilitators in recruiting substrates for ERAD (Fig. 5j). This unique ubiquitin linkage and biochemical characteristic may play a crucial role in determining the destiny of TLR3. Therefore, through its ubiquitination activity, HRD1 moonlights as a controller of TLR3 trafficking and regulates its activation. Future studies are needed to further understand how HRD1 distinguishes between its substrates and directs them either toward ERAD-mediated degradation or endo-lysosomal trafficking.
Our recent study indicates that SEL1L-HRD1 ERAD controls the abundance of ER-resident STING, an innate immune effector against ds-DNA viral infections, at the basal stage to limit the potential of STING signaling30. Here, we have further identified HRD1-mediated regulation of TLR3 trafficking significantly influences innate immunity. By modulating the sorting and cleavage of TLR3, HRD1 acts as a crucial determinant in the overall immune response against dsRNA viral infections. The dual role of HRD1 in STING and TLR3 sheds light on the complex interplay between ER pathways and innate immune signaling. Since TLR3 and STING agonists are both being investigated as vaccine adjuvants50, while HRD1 inhibitors could either boost STING-mediated or damp TLR3-mediated innate immune responses, our findings have relevant implications for the develop of new vaccines.
Methods
Mice
TLR3K813R KI mice were generated at GemPharmatech Co., Ltd using the CRISPR-Cas9 technology. The single guide RNA (sgRNA) was designed to target mouse genome TLR3 exon 6 where the mutation is located: 5’-TGATAACGAAAATGATTTTT-3’. The sgRNA was transcribed in vitro, and the homologous recombination vector (donor vector) carrying Tlr3 target mutation sequence was constructed and verified through sequencing. The CRISPR/Cas9 system, donor vector, and sgRNA were mixed and microinjected into fertilized mouse eggs of C57BL/6 JGpt background. The surviving injected fertilized eggs were then transferred into pseudopregnant females. Tail and toe samples were collected from 1-week-old pups born from the recipient mice to extract genomic DNA. A pair of primers was designed for PCR and sequencing: F (5’-GTTTCAGTGCATCGGATTCTTGG-3’) and R (5’-CTCAATAGCTTGCTGAACTGCGTG-3’). Genotype-positive mice carrying the desired mutations were established as F0 founders. Three independent founders were then bred separately to WT C57BL6/J mice to obtain F1 heterozygous TLR3K813R KI mice. F1 heterozygous mice were inter-crossed to generate homozygous TLR3K813R KI mice, WT and heterozygous littermates. C57BL/6J mice were purchased from the Model Animal Research Center of Nanjing University (China). All mice were housed in 20 °C, 40–60% humidity room with 12 h light cycle and fed a normal-chow diet (13% fat, 57% carbohydrate, and 30% protein). All animal procedures were approved by and done in accordance with the Ethics Committee of the First Affiliated Hospital, School of Medicine, Zhejiang University (2021-0135).
Power analysis of the animal size
Based on sample size formula of the power analysis, N = 8(CV)2[1 + (1-PC)2]/(PC)2, to reach the error = 0.05, Power = 0.80, percentage change in means (PC) = 20%, co-efficient of variation (CV) = 10–15% (varies between the experiments), 4–6 mice per group are the minimal number of mice to obtain statistical significance and to ensure adequate power. Mice in each group were randomly chosen based on the age, genotype and gender.
Cell culture and drug treatment
Peritoneal macrophages were obtained 4 d after 2 ml aged 4% brewed thioglycollate broth (VWR 90000-294) was intraperitoneal injected. Mice were euthanized and macrophages were collected by injection of 1 x PBS into the peritoneal cavity. The peritoneal exudate cells were centrifuged at ~1000 rpm for 10 min, followed with red blood cell lysis buffer treatment to remove red blood cells and resuspended in culture medium in six-well plates for further analyses as previously described. HEK293T (Cat. #CRL-3216), RAW 264.7 (Cat. #TIB-71), and MEF (Cat. #CRL-2991) cell lines were originally obtained from ATCC. Cells were maintained in DMEM (Cytiva) supplemented with 10% FBS (Braserum) and 1% penicillin/streptomycin at 37 °C under 5% CO2. 5 μM Eeyarestatin I (MedChemExpress), and 0.1 mM 4μ8c (MedChemExpress) were used in cell culture. TLR3 ligand poly(I:C) (High molecular weight, InvivoGen, tlrl-pic) was dissolved in sterile endotoxin-free physiological water and directly added into the culture medium at 50 µg/ml. LS-102 (MedChemExpress) was dissolved to 10 mM in DMSO and used at 5 µM. Thapsigargin (EMD Calbiochem) was dissolved to 0.6 mM in DMSO and used at 300 nM; Cycloheximide (CHX, Sigma) was dissolved in DMSO and used at 50 µg/ml; MG132 (MedChemExpress) was dissolved in DMSO and used at 25 μM; BFA (Sellck) was dissolved in DMSO and used at 10 µg/ml. The recombinant cytokines (MedChemExpress) were dissolved in complete medium and used at a final concentration 25 ng/ml in cell culture.
CRISPR-mediated gene KO cells
Generation of CRISPR-based Hrd1−/− RAW 264.7 cells, Hrd1−/− HEK293T cells, Sel1l−/− HEK293T cells, and Hrd1−/− MEF cells was performed using the lentiCRISPRv2 vector from the Zhang laboratory at MIT. The Hrd1 and Sel1l sgRNAs were designed, synthesized and cloned into the lentiCRISPRv2 vector, which expresses the sgRNA, Cas9 protein, and pruomycin resistance gene. The mouse sgRNA sequence for Hrd1 was GAUCCAUGCGGCAUGUCGGG. The human sgRNA sequence for Hrd1 was CUUGGUCAGGUACACCACAG. The sgRNA sequences for Sel1l were sgRNA-1: GCUGACGCUGCUGCUGUGUG and sgRNA-2: GGAGGGAAGAUGGCAGACUG.
Generation of BMDMs
Bone marrow cells were isolated from tibiae and femur of mice. After filtration and centrifugation, the bone marrow cells were cultured in DMEM medium containing 10% FBS and 50 ng/ml M-CSF (PeproTech) for 3-5 days to generate BMDMs.
Western blot and image quantitation
Cells were lysed in RIPA buffer (50 mM Tris-HCl pH 7.4, 150 mM NaCl, 1 % NP-40, 0.5 % sodium deoxycholate, 1 mM EDTA, 1 mM PMSF, 1× Protease inhibitor cocktail). Phosphatase Inhibitor (LC laboratories) was added to inhibit phosphatase activity in lysates. Protein concentration was determined by BCA assay (Beyotime). Samples were subjected to SDS-PAGE, transferred to PVDF and incubated with primary antibodies (4 °C, overnight) and HRP-conjugated secondary antibodies (RT, 1 h). Signals were developed with ECL. For Endo H treatment, the lysates were treated with Endo H or PNGase F (New England BioLab) per manufacturer’s instruction. Antibodies used in this study were: Flag (Sigma F1804; 1:2000); HA (Proteintech 51064-2-AP; 1:4000), HRD1 (Proteintech 13473-1-AP; 1:1000), SEL1L (Abclonal; A12073, 1:2000), IRE1α (Cell Signaling 3294; 1:3000), PERK (Proteintech 20582-1-AP; 1:3000), Eif2α(Cell Signaling 5324; 1:3000), p-Eif2α(Cell Signaling 5324; 1:2000), β-tubulin (Proteintech 10068-1-AP; 1:3000), HSP90 (Proteintech 13171-1-AP; 1:5000), Myc (ABclonal AE010; 1:2000), V5 (ABclonal AE017; 1:2000), phospho-IκBα (ABclonal AP0707; 1:1000), HRS (ABclonal A1790; 1:3000), TSG101 (ABclonal A1692; 1:1000), VPS36 (ABclonal A15157; 1:1000), TLR3 (NOVUS NBP2-24875; 2 μg/ml),, p-TBK1 (Ser172) (Cell Signaling D52C2; 1:1000), TBK1 (Cell Signaling E9H5S; 1:2000), p-IRF3 (Ser396) (Cell Signaling D601M; 1:2000), IRF3 (Cell Signaling D83B9; 1:2000), IκBα (Cell Signaling 9242; 1:2000), β-actin (Cell Signaling 13E5; 1:5000), p65 (Cell Signaling D14E12; 1:2000), phospho-p65 (Cell Signaling 93H1; 1:1000), Ubiquitin (linkage-specific K48) (Cell Signaling D9D5; 1:2000), Ubiquitin (linkage-specific K63) (Cell Signaling D7A11; 1:2000), GOLPH2 (ABclonal A26409PM; 1:1000), EEA1 (ABclonal A5057; 1:2000), Ubiquitin (Santa Cruz P4D1; 1:200), TGN38 (Santa Cruz sc-166594; 1:200), and mouse IgG (Santa Cruz sc-2025), Rabbit IgG (Abclonal; AC005)ERK1/2 (Cell Signaling 4695; 1:2000), p-ERK1/2 (Cell Signaling 9101; 1:2000), STAT1 (Cell Signaling 14994; 1:2000), p-STAT1 (Cell Signaling 9167;1:2000). Western blot band density was quantitated by the use of Image Lab software 4.1 on the ChemiDOC XRS+ system (Bio-Rad). Protein levels were normalized to HSP90, β-tubulin or β-actin. Phosphorylated forms of proteins were normalized to levels of total proteins. Data are presented as mean ± SEM unless otherwise specified.
Coimmunoprecipitation
Cells were incubated with 20 mM N-ethylmaleimide (NEM) in PBS for 10 min on ice, snap frozen and lysed in lysis buffer (100 mM NaCl, 1 mM EDTA, 20 mM Tris-HCl pH7.5, protease inhibitor cocktail (Sigma), 0.5% Nonidet P-40 (NP40), 10 mM NEM) on ice for 15 min. Cells were centrifuged at 12,000 g at 4 °C for 10 min. Supernatants were collected and protein concentration was measured using the BCA assay (Beyotime). For each immunoprecipitation, the supernatant was incubated with 0.5 μg of the indicated antibody or control IgG at 4 °C for overnight, followed by incubation with 20 μl of Protein A/G PLUS-Agarose (Santa Cruz) at 4 °C for another 3 h. The agarose beads were washed for five times with lysis buffer before immunoblot analysis. For ubiquitination analysis, the transfected cells were lysed in lysis buffer containing 1% SDS and denatured by boiling for 5 min. The supernatant was diluted tenfold with lysis buffer before immunoprecipitation using the indicated antibodies. Immunocomplexes were eluted by boiling for 5 min in SDS sample buffer followed by SDS-PAGE and Western blot analysis.
IP-MASS
The WT and Hrd1−/− RAW 264.7 cells were lysed and immunoprecipitated using anti-HRD1 antibody and Protein A/G PLUS-Agarose (Santa Cruz). The binding proteins were eluted and denaturized from the beads after washing, and subjected to SDS-PAGE. Protein samples in gel were digested into peptides by trypsin, and then analyzed by liquid chromatography-tandem mass spectrometry using an UltiMate 3000 nanoLC system and Orbitrap Exploris 480 (Thermo Fisher Scientific). Peptides were separated on a home-made reversed-phase analytical column (30 cm, 100 μm i.d.) packed with 1.9 μm Reprosil-Pur C18 beads (Dr. Maisch, Ammerbuch, Germany) at a constant flow rate of 450 nl/min on an UltiMate 3000 nanoLC system. Solvent A was 0.1% formic acid. The gradient was comprised of an increase from 3% to 10% solvent B (0.1% formic acid in 98% acetonitrile) over 3 min, 10% to 24% in 37 min, 24% to 38% in 12 min and climbing to 80% in 4 min, then holding at 80% for the last 4 min. For data acquisition, the peptides were subjected to NSI source followed by tandem mass spectrometry (MS/MS) in Orbitrap Exploris 480 (Thermo Fisher Scientific) coupled online to the UPLC. The electrospray voltage applied was 2.0 kV. The m/z scan range was 400 to 1200 for full scan, and intact peptides were detected in the Orbitrap at a resolution of 60,000. Peptides were then selected for MS/MS using NCE setting as 27 and the fragments were detected in the Orbitrap at a resolution of 15,000. Each precursor ion was fragmented with HCD. The collision energy of the HCD was set to 27. A data-dependent procedure that alternated between one MS scan followed by 20 MS/MS scans with 30 s dynamic exclusion. Automatic gain control (AGC) was set at 5E4. The resulting MS/MS data were processed using Proteome Discoverer software v2.5 (Thermo Fisher). Tandem mass spectra were searched against SwissProt Mouse database using the SEQUEST algorithm. Trypsin (full) was specified as cleavage enzyme allowing up to 2 missing cleavages. The minimum peptide length was 6 amino acids with a maximum of 5 modifications per peptide. The mass tolerance for precursor ions was set as 10 ppm, and the mass tolerance for fragment ions was set as 0.02 Da. Proteins and PSMs were filtered with a maximum FDR of 1%, which constituted the full list of proteins identified from the respective Hrd1+/+ and Hrd1−/− immunoprecipitates. To identify HRD1-specific binding proteins, we excluded those proteins found in both Hrd1+/+ and Hrd1−/− immunoprecipitates. Given that HRD1 localizes to the ER, we focused on selecting ER-resident proteins that interact with HRD1. Fig 1a shows the top ten specific HRD1-interacting ER proteins, ranked by sum PEP score. The sum PEP score is calculated based on the posterior error probability (PEP) values of the peptide spectrum matches (PSM), with PEP indicating the likelihood that an observed PSM is a random event. The sum PEP score is the negative logarithm of the PEP values of the connected PSMs.
Plasmid construction
Flag-tagged TLR3 and V5-tagged HRS, TSG101, VPS36 and VCP were inserted into pEGFP-N2 plasmid, with the EGFP deletion. Myc-tagged HRD1 was generated as previously reported30. Plasmids containing HA-tagged Ub, its mutant and truncation were constructed by standard molecular biology techniques. For stable expression, WT and K813R TLR3 with or without Flag tag were individually subcloned into pCDH-CMV-MCS-EF1-copGFP plasmid. All of the plasmids were confirmed by DNA sequencing.
RNA extraction, reverse transcription (RT) and quantitative PCR (Q-PCR)
Total RNA was isolated from cells using RNAiso plus kit (Takara). cDNA was generated using PrimeScript™ RT reagent Kit with gDNA Eraser (Perfect Real Time, Takara), and amplified using TB Green Premix Ex Taq (Tli RNaseH Plus, Takara) per the manufacturer’s instructions. Q-PCR data were collected using QuantStudio™ 5 (Applied Biosystems) and gene expression was normalized to ribosomal L32 gene for each sample.
The sequences of the primers used are listed below:
Tnfa (TCAGCCGATTTGCTATCTCATA; AGTACTTGGGCAGATTGACCTC),
Il6 (AGACAAAGCCAGAGTCCTTCAG; TGCCGAGTAGATCTCAAAGTGA),
Il1b (CCAAGCAACGACAAAATACC; GTTGAAGACAAACCGTTTTTCC),
Ifnb1 (AGATCAACCTCACCTACAGG; TCAGAAACACTGTCTGCTGG),
Ccl5 (GCTGCTTTGCCTACCTCTCC; TCGAGTGACAAACACGACTGC),
Bip (TGTGGTACCCACCAAGAAGTC; TTCAGCTGTCACTCGGAGAAT),
Chop (TATCTCATCCCCAGGAAACG; GGGCACTGACCACTCTGTTT),
Xbp1u (ACTATGTGCACCTCTGCAGC, GTCCAGAATGCCCAAAAGG),
Xbp1s (GAGTCCGCAGCAGGTG; GTGTCAGAGTCCATGGGA),
L32 (GAGCAACAAGAAAACCAAGCA; TGCACACAAGCCATCTACTCA),
Ets1 (TGGGTGATGTGGGCTGTG; GGCTCCATTAACCTGATATGGT),
Hrd1 (AGCTACTTCAGTGAACCCCACT; CTCCTCTACAATGCCCACTGAC),
Irf1 (CGAGGGGACATTGGGATAG; CCTGAATAGAGGGCGGCA),
Irf2 (GAAAGAAACCAAAGACAGAAAAAG; TATAGCTGAAGTCAGGACCGC),
Tlr3 (AGTGTCACAGGACCCCAGA; GAACTACAGAAGCCAAAAAGC),
Sel1l (TGGGTTTTCTCTCTCTCCTCTG; CCTTTGTTCCGGTTACTTCTTG),
Trim3 (CGTCTCAGCCCTACAAAACAA; ACAGGCCTCACAGTAAAACTCC).
The Q-PCR conditions were: 94 °C for 5 min, 40 cycles of (94 °C for 15 s, 58 °C for 15 sec and 72 °C for 30 sec), followed by dissociation curve analysis.
Dual-luciferase reporter assay
Cells were transfected with the NF-κB or ISRE luciferase reporter with TK Renilla and the indicated plasmids using lipo8000 (Beyotime). 24 hours after transfection, the cells were either left untreated or subjected to treatment with poly(I:C) for the indicated durations. Subsequently, whole-cell lysates were collected, and luciferase activity was measured using a dual-luciferase reporter assay system (Promega) per the manufacturer’s instructions.
Stable cell line establishment
HEK293T cells were cotransfected with the plasmids of TLR3-containing pCDH-CMV-MCS-EF1-copGFP, psPAX2, and pMD2G. The culture medium was collected 48–72 h after transfection and filtered. WT and Hrd1−/− cells were infected with the virus-containing medium in the presence of polybrene (8 μg/ml, Beyotime). The positive cells were selected by sorting GFP-positive cells.
In vitro ubiquitination assays
The full-length and ΔTM mutant of HRD1 were constructed into the pMBP vector to generate MBP-fusion proteins. Recombinant proteins were expressed in the E. coli BL21 (DE3) Codon-Plus strain (TransGen Biotech) induced with 0.1 mM IPTG at 16 °C for 20 h, and purified using Ni-NTA affinity chromatography (Beyotime). The purified proteins were then processed by MBP tag removal (Beyotime) to obtain HRD1 and HRD1-ΔTM proteins, and further identified by western blotting. Flag-tagged TLR3 was immunoprecipitated from HEK293T cells ectopically expressing TLR3-Flag using an anti-Flag affinity gel (MCE).
For in vitro ubiquitination assays, the immunoprecipitated Flag-tagged TLR3 was incubated with HRD1, HRD1-ΔTM proteins, ubiquitin, E1 (Abcam, ab139467) and E2 (UBE2G2 from MCE) in the ubiquitination buffer (Abcam, ab139467) containing MgCl2 and ATP for 3 h at 37 °C according to the instructions. The reactions were terminated by adding an equal volume of 2× non-reducing gel loading buffer and analyzed by immunoblotting.
Immunofluorescent staining
Cells were mounted on coverslips and left untreated or treated with poly(I:C) for the indicated times, then fixed with 4% paraformaldehyde for 15 min. After rinse with PBS, the cells were permeabilized and blocked in a buffer containing 0.1% saponin and 5% normal goat serum in PBS at room temperature for 1 hour. Subsequently, the cells were stained with the indicated primary antibodies at 4 °C overnight, followed by secondary antibodies at room temperature for 1 h. Following extensive washes with PBS containing 0.1% saponin, slides were covered with antifade mounting medium with DAPI (Beyotime). Primary antibodies for immunofluorescent staining included: TLR3 (Proteintech 17766-1-AP, 1: 200), Calnexin (Proteintech 10427-2-AP, 1: 200), GM130 (Proteintech 11308-1-AP, 1:1000), Rab5 (Cell Signaling C8B1; 1: 200), Rab7 (Cell Signaling D95F2; 1: 200), KDEL (Novus Biologicals NBP1-97469, 1:200), TGN38 (Santa Cruz sc-166594,1:200), LAMP1 (Proteintech 65050-1-Ig, 1:1000), Flag (Sigma F1804, 1: 200). Fluorescence images were captured by a Leica TCS SP8 microscope with a 100× objective at the Imaging Core Facility of the First Affiliated Hospital at Zhejiang University School of Medicine. Quantitative analysis of colocalization images was performed using Fiji (ImageJ) software.
HSV1 infection
WT or Hrd1−/− RAW 264.7 cells were cultured in six-well plates, and once reaching 100% confluency, they were stimulated by 100 μg/mL poly(I:C) for 18 h (1.5 mL per well). The supernatants were collected as secretomes and filtered to remove cell debris. Vero cells were seeded on 12-well plates with 600 μL of the indicated secretome per well, and infected with GFP-HSV1 virus at indicated concentrations. After 24 h of incubation, fluorescence images were captured using a Leica DMi8 Fluorescence microscope at the Imaging Core Facility of the First Affiliated Hospital at Zhejiang University School of Medicine. Quantitative analysis of the GFP-positive area was performed using Fiji (ImageJ) software.
Poly(I:C) challenge
Sex- and age-matched mice were injected i.p. with poly(I:C) (InvivoGen, tlrl-pic) at 0.7 mg/kg body weight plus D-galactosamine (Sigma-Aldrich, G1639) at 0.5 g/kg body weight and observed for survival every 1 h. Serum was collected at 2 h time point post injection for cytokine analysis. The tissues for HE and TUNEL were collected at 4 h post injection.
Tissues HE and TUNEL staining
The lung and liver tissues were fixed in 4% formaldehyde for 24 h immediately after dissection from the mice. Fixed tissues were then processed through a series of ethanol solutions for dehydration and cleared in xylene before being embedded in paraffin wax. Paraffin-embedded tissues are sectioned into 4 μm thick slices and mounted onto glass slides. The sections were deparaffinized in xylene and rehydrated through graded ethanol solutions. For HE staining, the lung sections staining begins with Mayer’s hematoxylin, which binds to nucleic acids, imparting a blue color to nuclei. After differentiation in running tap water, sections are counterstained with eosin Y, which stains cytoplasmic and extracellular matrix components pink. Following staining, sections are dehydrated in absolute alcohol, cleared in xylene, and mounted with a resinous medium. The final preparation is examined under a Leica DM2500 LED microscope for histological analysis. For liver TUNEL staining, the liver sections were stained using One Step TUNEL Apoptosis Assay Kit (Beyotime). Briefly, the liver sections were digested with protease K (20 μg/ml) at 37°C for 30 min and rinsed with PBS. The liver sections incubated in the TUNEL detection solution at 37 °C for 60 min in the dark. After rinsing with PBS, the slides were covered with antifade mounting medium with DAPI (Beyotime). Fluorescence images were captured by a Leica TCS SP8 microscope at the Imaging Core Facility of the First Affiliated Hospital at Zhejiang University School of Medicine.
ELISA analysis
Macrophages were cultured in 12 well plate (1 × 106 cells/well), stimulated with poly(I:C) for indicated time, and the supernatant was collected. The levels of mouse TNFα, IL6, and IFNβ in the cell culture supernatants and mouse serum were quantified using ELISA commercial kits (BioLegend). All ELISAs were performed per suppliers’ protocols.
Isolation of ER, Golgi and endosome
ER Enrichment kit (Invent Biotechnologies), Golgi Apparatus Enrichment Kit (Invent Biotechnologies) and Endosome Isolation and Cell Fractionation kit (Invent Biotechnologies) were used to isolate ER, Golgi and endosome according to the manufacturer’s instructions, respectively. Briefly, 3 × 107 cells were collected and washed in cold PBS. For ER enrichment, the cells were freezed and resuspended in Buffer A, and transferred to a filter cartridge for centrifugation twice. All the supernatants were transferred to a new fresh tube and centrifuged. The supernatant was mixed with buffer B and centrifuged. The pellet was resuspended with buffer A and vortexed vigorously. After mixing with buffer C, the sample was centrifuged. Buffer D was added into the supernatant for the final centrifugation. The pellet was dissolved in SDS loading buffer. For Golgi enrichment, the cells suspended in buffer A were transferred to a filter cartridge, followed by centrifugation. The supernatant mixed with buffer B and centrifuged. After centrifugation, the pellet was resuspended in cold buffer A and centrifuged. Cold buffer C was added into the supernatant with vigorous vortex. After centrifugation, the pellet was dissolved in SDS loading buffer. For endosome isolation, the cell suspension with buffer A was transferred to a filter cartridge and centrifuged. Then, the supernatant was mixed with buffer B and incubated at 4 °C for overnight. Following centrifugation, the pellet was dissolved in SDS loading buffer.
siRNA
The siRNAs used in this study were synthesized by Sangon Biotech. The sequences targeting Hrd1 were siHrd1-1: sense, GCAUGGCAGUCCUGUACAUTT and antisense, AUGUACAGGACUGCCAUGCTT; siHrd1-2: sense, GAGCCAUCCGCAACAUGAATT and antisense, UUCAUGUUGCGGAUGGCUCTT. The sequences targeting Ets1 were siEts1: sense, GCUUCAUCACAGAGUCCUATT and antisense, UAGGACUCUGUGAUGAAGCTT; siEts1-2: sense, AGGUGUUAACUCCAGACAATT and antisense, UUGUCUGGAGUUAACACCUTT. The sequences targeting Sel1l were siSel1l-1: sense, GGCUCUUGUAUAUUAUACUTT and antisense, AGUAUAAUAUACAAGAGCCTT; siSel1l-2: sense, GUAGUUGCUGGCCAGAUAUTT and antisense, AUAUCUGGCCAGCAACUACTT. The sense and antisense sequences of siTrim3 are CUGAGGUUUUGCUAGUAAGTT and CUUACUAGCAAAACCUCAGTT, respectively.
Statistical analysis
Results were expressed as mean ± SEM. Comparisons between groups were made using unpaired two-tailed Student’s t-test. Data distribution was assumed to be normal, but this was not formally tested. No sample size calculation was performed for in vitro experiments. No animals or samples were excluded from the analysis. All experiments were repeated with at least three independent biological replicates.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The coIP-MASS raw data generated in this study have been deposited in iProX (IPX0009703001) with a sharing URL (https://www.iprox.cn//page/SCV017.html?query=IPX0009703001). Source data are provided with this paper.
References
Fitzgerald, K. A. & Kagan, J. C. Toll-like receptors and the control of immunity. Cell 180, 1044–1066 (2020).
Alexopoulou, L., Holt, A. C., Medzhitov, R. & Flavell, R. A. Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature 413, 732–738 (2001).
Li, W. W. et al. Ubiquitination of TLR3 by TRIM3 signals its ESCRT-mediated trafficking to the endolysosomes for innate antiviral response. Proc. Natl. Acad. Sci. USA 117, 23707–23716 (2020).
Johnsen, I. B. et al. Toll-like receptor 3 associates with c-Src tyrosine kinase on endosomes to initiate antiviral signaling. EMBO J. 25, 3335–3346 (2006).
Garcia-Cattaneo, A. et al. Cleavage of Toll-like receptor 3 by cathepsins B and H is essential for signaling. Proc. Natl. Acad. Sci. USA 109, 9053–9058 (2012).
Lind, N. A., Rael, V. E., Pestal, K., Liu, B. & Barton, G. M. Regulation of the nucleic acid-sensing Toll-like receptors. Nat. Rev. Immunol. 22, 224–235 (2022).
Sun, S., Tang, X., Guo, Y. & Hu, J. Endoplasmic reticulum composition and form: Proteins in and out. Curr. Opin. Cell Biol. 71, 1–6 (2021).
Yang, Y. et al. Heat shock protein gp96 is a master chaperone for toll-like receptors and is important in the innate function of macrophages. Immunity 26, 215–226 (2007).
Pelka, K. et al. The Chaperone UNC93B1 regulates toll-like receptor stability independently of endosomal TLR transport. Immunity 48, 911–922.e917 (2018).
Barber, G. N. STING: infection, inflammation and cancer. Nat. Rev. Immunol. 15, 760–770 (2015).
Pokatayev, V. et al. Homeostatic regulation of STING protein at the resting state by stabilizer TOLLIP. Nat. Immunol. 21, 158–167 (2020).
Zhang, B. C. et al. STEEP mediates STING ER exit and activation of signaling. Nat. Immunol. 21, 868–879 (2020).
Srikanth, S. et al. The Ca2+ sensor STIM1 regulates the type I interferon response by retaining the signaling adaptor STING at the endoplasmic reticulum. Nat. Immunol. 20, 152–162 (2019).
Luo, Y., Chang, L., Ji, Y. & Liang, T. ER: a critical hub for STING signaling regulation. Trends Cell Biol. 34, 865-881 (2024).
Bhattacharya, A., Qi, L. ER-associated degradation in health and disease—from substrate to organism. J. Cell Sci. 132, jcs232850 (2019).
Hwang, J. & Qi, L. Quality control in the endoplasmic reticulum: crosstalk between ERAD and UPR pathways. Trends Biochem. Sci. 43, 593–605 (2018).
Guerriero, C. J. & Brodsky, J. L. The delicate balance between secreted protein folding and endoplasmic reticulum-associated degradation in human physiology. Physiol. Rev. 92, 537–576 (2012).
Vasic, V. et al. Hrd1 forms the retrotranslocation pore regulated by auto-ubiquitination and binding of misfolded proteins. Nat. Cell Biol. 22, 274–281 (2020).
Mueller, B., Klemm, E. J., Spooner, E., Claessen, J. H. & Ploegh, H. L. SEL1L nucleates a protein complex required for dislocation of misfolded glycoproteins. Proc. Natl. Acad. Sci. USA 105, 12325–12330 (2008).
Sun, S. et al. Sel1L is indispensable for mammalian endoplasmic reticulum-associated degradation, endoplasmic reticulum homeostasis, and survival. Proc. Natl. Acad. Sci. USA 111, E582–E591 (2014).
Sha, H. et al. The ER-associated degradation adaptor protein Sel1L regulates LPL secretion and lipid metabolism. Cell Metab. 20, 458–470 (2014).
Sun, S. et al. IRE1α is an endogenous substrate of endoplasmic-reticulum-associated degradation. Nat. Cell Biol. 17, 1546–1555 (2015).
Shi, G. et al. ER-associated degradation is required for vasopressin prohormone processing and systemic water homeostasis. J. Clin. Investig. 127, 3897–3912 (2017).
Kim, G. H. et al. Hypothalamic ER-associated degradation regulates POMC maturation, feeding, and age-associated obesity. J. Clin. Investig. 128, 1125–1140 (2018).
Bhattacharya, A. et al., Hepatic Sel1L-Hrd1 ER-associated degradation (ERAD) manages FGF21 levels and systemic metabolism via CREBH. EMBO J. 37, e99277 (2018).
Zhou, Z. et al., Endoplasmic reticulum-associated degradation regulates mitochondrial dynamics in brown adipocytes. Science (2020).
Shrestha, N. et al. Sel1L-Hrd1 ER-associated degradation maintains β-cell identity via TGFβ signaling. J. Clin. Investig. (2020).
Ji, Y. et al. The Sel1L-Hrd1 endoplasmic reticulum-associated degradation complex manages a key checkpoint in B cell development. Cell Rep. 16, 2630–2640 (2016).
Yang, Y. et al. The endoplasmic reticulum-resident E3 ubiquitin ligase Hrd1 controls a critical checkpoint in B cell development in mice. J. Biol. Chem. 293, 12934–12944 (2018).
Ji, Y. et al. SEL1L-HRD1 endoplasmic reticulum-associated degradation controls STING-mediated innate immunity by limiting the size of the activable STING pool. Nat. Cell Biol. 25, 726–739 (2023).
Liu, L. et al. Structural basis of toll-like receptor 3 signaling with double-stranded RNA. Science 320, 379–381 (2008).
Wu, T. et al. Hrd1 suppresses Nrf2-mediated cellular protection during liver cirrhosis. Genes Dev. 28, 708–722 (2014).
Xu, Y. et al. The ER membrane-anchored ubiquitin ligase Hrd1 is a positive regulator of T-cell immunity. Nat. Commun. 7, 12073 (2016).
Yagishita, N. et al. RING-finger type E3 ubiquitin ligase inhibitors as novel candidates for the treatment of rheumatoid arthritis. Int. J. Mol. Med. 30, 1281–1286 (2012).
Gao, B., Calhoun, K. & Fang, D. The proinflammatory cytokines IL-1beta and TNF-alpha induce the expression of Synoviolin, an E3 ubiquitin ligase, in mouse synovial fibroblasts via the Erk1/2-ETS1 pathway. Arthritis Res. Ther. 8, R172 (2006).
Heinz, S. et al. Species-specific regulation of Toll-like receptor 3 genes in men and mice. J. Biol. Chem. 278, 21502–21509 (2003).
Wang, Q., Li, L. & Ye, Y. Inhibition of p97-dependent protein degradation by Eeyarestatin I. J. Biol. Chem. 283, 7445–7454 (2008).
Shibuya, K. et al. AAA-ATPase valosin-containing protein binds the transcription factor SREBP1 and promotes its proteolytic activation by rhomboid protease RHBDL4. J. Biol. Chem. 298, 101936 (2022).
Martinon, F., Chen, X., Lee, A. H. & Glimcher, L. H. TLR activation of the transcription factor XBP1 regulates innate immune responses in macrophages. Nat. Immunol. 11, 411–418 (2010).
Cross, B. C. et al. The molecular basis for selective inhibition of unconventional mRNA splicing by an IRE1-binding small molecule. Proc. Natl. Acad. Sci. USA 109, E869–E878 (2012).
Swatek, K. N. & Komander, D. Ubiquitin modifications. Cell Res. 26, 399–422 (2016).
Kikkert, M. et al. Human HRD1 is an E3 ubiquitin ligase involved in degradation of proteins from the endoplasmic reticulum. J. Biol. Chem. 279, 3525–3534 (2004).
DeLaBarre, B., Christianson, J. C., Kopito, R. R. & Brunger, A. T. Central pore residues mediate the p97/VCP activity required for ERAD. Mol. Cell 22, 451–462 (2006).
Wei, J. et al. HRD1-ERAD controls production of the hepatokine FGF21 through CREBH polyubiquitination. EMBO J. 37, e98942 (2018).
Lu, Y. et al. ER-localized Hrd1 ubiquitinates and inactivates Usp15 to promote TLR4-induced inflammation during bacterial infection. Nat. Microbiol. 4, 2331–2346 (2019).
Foot, N., Henshall, T. & Kumar, S. Ubiquitination and the regulation of membrane proteins. Physiol. Rev. 97, 253–281 (2017).
Toscano, F. et al. Cleaved/associated TLR3 represents the primary form of the signaling receptor. J. Immunol. 190, 764–773 (2013).
Vietri, M., Radulovic, M. & Stenmark, H. The many functions of ESCRTs. Nat. Rev. Mol. Cell Biol. 21, 25–42 (2020).
Shimizu, Y., Okuda-Shimizu, Y. & Hendershot, L. M. Ubiquitylation of an ERAD substrate occurs on multiple types of amino acids. Mol. Cell 40, 917–926 (2010).
Pulendran, B., Arunachalam, P. S. & O’Hagan, D. T. Emerging concepts in the science of vaccine adjuvants. Nat. Rev. Drug Discov. 20, 454–475 (2021).
Majer, O., Liu, B. & Barton, G. M. Nucleic acid-sensing TLRs: trafficking and regulation. Curr. Opin. Immunol. 44, 26–33 (2017).
Liu, B. et al. Folding of Toll-like receptors by the HSP90 paralogue gp96 requires a substrate-specific cochaperone. Nat. Commun. 1, 79 (2010).
Takahashi, K. et al. A protein associated with Toll-like receptor (TLR) 4 (PRAT4A) is required for TLR-dependent immune responses. J. Exp. Med. 204, 2963–2976 (2007).
Tsai, S. Y. et al. Regulation of TLR3 Activation by S100A9. J. Immunol. 195, 4426–4437 (2015).
Tabeta, K. et al. The Unc93b1 mutation 3d disrupts exogenous antigen presentation and signaling via Toll-like receptors 3, 7 and 9. Nat. Immunol. 7, 156–164 (2006).
Lee, B. L. et al. UNC93B1 mediates differential trafficking of endosomal TLRs. Elife 2, e00291 (2013).
Kim, Y. M., Brinkmann, M. M., Paquet, M. E. & Ploegh, H. L. UNC93B1 delivers nucleotide-sensing toll-like receptors to endolysosomes. Nature 452, 234–238 (2008).
Hampton, R. Y., Gardner, R. G. & Rine, J. Role of 26S proteasome and HRD genes in the degradation of 3-hydroxy-3-methylglutaryl-CoA reductase, an integral endoplasmic reticulum membrane protein. Mol. Biol. Cell 7, 2029–2044 (1996).
Plemper, R. K. et al. Genetic interactions of Hrd3p and Der3p/Hrd1p with Sec61p suggest a retro-translocation complex mediating protein transport for ER degradation. J. Cell Sci. 112, 4123–4134 (1999).
Bhattacharya, A. et al. Hepatic Sel1L-Hrd1 ER-associated degradation (ERAD) manages FGF21 levels and systemic metabolism via CREBH. EMBO J. 37, e99277 (2018).
Shrestha, N. et al. Sel1L-Hrd1 ER-associated degradation maintains β cell identity via TGF-β signaling. J. Clin. Investig. 130, 3499–3510 (2020).
Zhou, Z. et al. Endoplasmic reticulum-associated degradation regulates mitochondrial dynamics in brown adipocytes. Science 368, 54–60 (2020).
Acknowledgements
We thank Dr. Pinglong Xu (Life Sciences Institute, Zhejiang University) for HSV-1 virus. This work was supported by National Natural Science Foundation of China projects (82171731, 82371737 to Y.J.; 82303596 to Y.L.; 82188102 to T.L.); and Investigator Start-up Funds of First Affiliated Hospital of Zhejiang University (B2021010-12 to Y.L.; B20735 to Y.J.).
Author information
Authors and Affiliations
Contributions
L.Z. performed the most of the experiments; Z.H. performed HSV1 infection and some Western blots; S.Y. performed confocal microscopy; Y.L., M.S., and Z.L. assisted with some experiments; L.Q. and Z.Z. provided some reagents and discussions; L.Z., Y.L., and Y.J. designed experiments and analyzed the data; Y.J. wrote the manuscript; L.Z. wrote the methods; Y.J., T.L., and Y.L. supervised the experiment; everybody edited and approved the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Dandan Lin, Gerardo Lederkremer and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhao, L., He, Z., Sun, Y. et al. Ubiquitination by HRD1 is essential for TLR3 trafficking and its innate immune signaling. Nat Commun 17, 525 (2026). https://doi.org/10.1038/s41467-025-67219-0
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-67219-0
