
Abstract
Loss-of-function variants in PALB2 give rise to defects in DNA damage repair by homologous recombination (HR), increasing the risk of breast cancer in female carriers. However, genetic testing frequently reveals missense variants of uncertain significance (VUS) for which the impact on protein function and cancer risk are unclear. Here we assay 84% of all possible missense variants in 11 out of 13 PALB2 exons using site-saturation functional screens with PARP inhibitor sensitivity as a readout for HR. These exons encode the coiled-coil and WD40 domains, which we identify as the minimal regions required for HR. Furthermore, we reveal the functional impact of 6718 missense variants, classifying 3904 variants as functional (58%), 2422 as intermediate (36%), and 392 as damaging (6%). A burden-type analysis shows that damaging missense variants in PALB2 are associated with a significantly increased risk of breast cancer, similar to that observed for truncating variants. These results will be valuable for the classification of PALB2 missense VUS and clinical management of carriers.
Similar content being viewed by others


Introduction
Germline protein-truncating variants (PTV) in PALB2 are associated with high breast and pancreatic cancer risk and moderate ovarian cancer risk1. However, genetic testing has identified numerous PALB2 missense variants for which the cancer risk is unclear. To improve the clinical interpretation of these variants of uncertain significance (VUS), complementary approaches that assess the molecular effects of such variants are needed. The American College of Medical Genetics and Genomics (ACMG) and Association for Molecular Pathology (AMP) have proposed variant interpretation guidelines that incorporate different types of evidence at various levels of strength. These guidelines provide rules for combining the different types of evidence for clinical classification of variants as benign, likely benign, uncertain significance, likely pathogenic, or pathogenic2,3. The reliable classification of variants depends heavily on genetic and clinical data which is lacking for the majority of the rare missense variants in PALB2. This indicates the urgent need for additional means to classify VUS in PALB2.
Functional analysis has been shown to provide accurate information on the impact of VUS on PALB2 protein function. Given that PALB2 plays a pivotal role in DNA double-strand break repair by homologous recombination (HR), a process that is key to tumor suppression, most assays measure the functional effect of PALB2 variants on HR4,5,6. These assays are mostly based on the use of well-established HR reporters such as DR-GFP, or measure cellular sensitivity to treatment with poly(ADP-ribosyl) polymerase (PARP) inhibitor. Generally, PALB2-deficient cells that ectopically express PALB2 cDNA with a particular variant were used in these assays. This way >150 different PALB2 variants, comprising >120 missense VUS, were functionally characterized7,8,9. VUS with an impact on protein function (>50% reduced HR activity compared to WT) were all located in the coiled-coil (CC) or WD40 domain of PALB2, highlighting the importance of these domains for PALB2’s role in HR. Indeed, PALB2 interacts with BRCA1 and BRCA2 through these domains, respectively. Both BRCA1 and BRCA2, similar to PALB2, function as tumour suppressors by playing essential roles in HR. Thus, the functional analysis of missense variants in PALB2 has the potential to identify damaging variants that may confer increased cancer risk, thereby aiding in the clinical classification of such variants.
The functional assays that have been used thus far for the analysis of missense variants in PALB2 are often time and resource intensive7,8,9, and the results may not be publicly available for years after the variant is encountered in carriers. These assays will therefore not be able to cope with the pace and scale at which genetic testing reveals single nucleotide variants (SNVs) in PALB2 that are classified as VUS (as of June 30, 2025, >2,900 SNVs in PALB2 were listed as VUS in ClinVar). Moreover, given that PALB2 variants are very rare, variant frequencies from extremely large breast cancer patient cohorts and functional data for large sets of variants are needed to link the functional impact of variants with cancer risk.
Here, we develop and apply a cDNA-based site-saturation screen in Palb2-deficient mouse embryonic stem (mES) cells to functionally characterize all possible missense variants in the CC and WD40 domains of human PALB2, which appear to be the sole domains required for its function in HR. Using this approach, more than 7,000 variants in PALB2 are functionally interrogated by determining their impact on cellular PARPi sensitivity, identifying 392 damaging missense variants. By combining these results with case-control association studies involving more than 485,000 breast cancer cases and nearly 600,000 non-cancer female controls, the functional impact of PALB2 missense variants can be linked to breast cancer risk. Importantly, damaging missense variants in PALB2 are associated with a similar high risk for breast cancer as observed for protein truncating variants10,11. Thus, the site-saturation functional screens presented here are a valuable source for the clinical classification of variants in PALB2, thereby improving clinical management of carriers.
Results
The region between the CC and WD40 domains of PALB2 is dispensable for HR
No missense variants outside of the N-terminal CC and C-terminal WD40 domains of PALB2 have been identified as damaging in functional assays that use HR as a read-out7,8,9. However, whether the region between the CC and WD40 domains, which includes the Chromatin Association Motif (ChAM) and MRG15-binding motif, is required for HR is not fully understood, with previous work implicating ChAM in promoting loading of the core-HR factor RAD51 onto damaged chromatin and in conferring resistance to DNA inter-strand crosslinks12. We therefore set out to examine the importance of this region for HR. We generated five PALB2 cDNA deletion constructs lacking either the ChAM (ΔChAM), MRG-binding motif (ΔMRG15), exon 4 (ΔExon4) exon 5 (ΔExon5), or exon 6 (ΔExon6) (Fig. 1a). All exon deletions were generated in-frame. We did not examine exon 3 as deletion largely disrupts the CC domain. The constructs were introduced site-specifically by recombination-mediated cassette exchange (RMCE) at the Rosa26 locus in Palb2KO mES cells carrying the DR-GFP reporter, which allows flow cytometry-based detection of HR after I-SceI endonuclease-induced DNA double-strand break formation7. The stable expression of these PALB2 deletion constructs rescued the HR defect of Palb2KO cells to levels observed in cells expressing wild-type PALB2 (Fig. 1b, c). Consistently, the expression of these deletion constructs also rescued the sensitivity of Palb2KO cells to treatment with PARPi (Fig. 1d). These data suggest that these regions are dispensable for the PALB2 function in HR, reducing the likelihood that missense variants therein are capable of impacting HR. We therefore focused on classifying all possible missense variants in the CC and WD40 domains of PALB2 by assessing the impact on HR en masse.

a Schematic representation of the PALB2 protein in which amino acid numbers are shown to specify the evolutionarily conserved functional domains of PALB2 (top). PALB2 exon numbers 1-13 are specified by cDNA numbers (bottom). In-frame exons, white; out-of-frame exons, grey. b Western blot showing the expression of five PALB2 deletion variants in Trp53KO/Palb2KO mES cells, as compared to wild-type (WT) PALB2 and the empty vector (Ev) control. Tubulin is used as a loading control. c DR-GFP HR assay in Trp53KO/Palb2KO mES cells expressing the indicated PALB2 deletion variants from (b). HR efficiencies were normalized to the WT PALB2 condition which was set to 100%. Mean ± s.e.m. are shown, n = 3 biological replicates, dots represent individual data points, one-way ANOVA with two-sided Dunnet’s multiple comparison test. ***P < 0.001, NS = not significant. d As in c, except for PARPi sensitivity assays. Values indicate the relative resistance to 0.5 μM PARPi treatment with the WT PALB2 condition set to 100%. ****P < 0.0001. Source data are provided as a Source Data file.
Site-saturation functional screens of missense variants in the CC and WD40 domains of PALB2
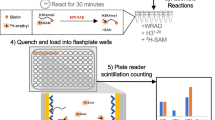
To comprehensively evaluate the functional impact of PALB2 variants, we conducted site-saturation screens, analysing all possible missense variants in the PALB2 CC and WD40 domains. In these screens, PARP inhibitor sensitivity was used as a readout for HR efficiency (Fig. 2a). We employed 6 libraries based on human PALB2 cDNA: one for the CC domain (amino acid 9-43) and 5 libraries that together cover the entire WD40 domain (amino acid 856-1,186). The libraries, which together contained >95% of all possible missense and nonsense variants, as well as 49 synonymous variants (Supplementary Fig. 1), were introduced in the Palb2KO mES cells by RMCE (6 and 3 biological replicates for the CC and WD40 domain, respectively). Clones expressing the PALB2 variants were selected, pooled and expanded. PARPi sensitivity assays were performed on these cells using non-treated cells as control conditions (3 technical replicates for the libraries of both the CC and WD40 domain). Following DNA isolation, the PALB2 cDNA region that encodes the CC domain and the 5 regions spanning the WD40 domain were amplified by PCR and sequenced. For each variant, depletion scores and standard errors (SE) were calculated using Enrich2 software (Fig. 2a)13,14. The depletion scores are based on the ratio of variant frequencies before and after PARPi treatment. SE were based on replicate measurements and used to filter out variants that could not be confidently scored (SE > 0.5). Finally, depletion scores were normalized to that of wild-typePALB2 (present in each library), which was set to ‘0’, and to the average score of all nonsense variants (of each library), which was set to ‘-1’. Biological replicates generally showed a moderate but significant correlation between depletion scores of variants in both the CC (Supplementary Fig. 2) and WD40 domain (Supplementary Fig. 3) based on Pearson correlation analysis (see Methods). Using these depletion scores, functional maps for variants in the CC and WD40 domain were generated (Figs. 2b and 3). Altogether, we obtained depletion scores for 6,718 missense variants, which comprises 97% of all possible missense variants in the CC and WD40 domains of PALB2 (609 for the CC domain, which comprises 92% of all possible variants, and 6,109 for the WD40 domain, which comprises 97% of all possible variants) (Supplementary Data 1). The median depletion score for the nonsense variants was -1.011 (n = 339, standard deviation = 0.413), while for the synonymous variants it was -0.096 (n = 49, standard deviation = 0.306). Importantly, depletion scores of nonsense and synonymous variants were bimodally distributed (Fig. 4a and Supplementary Fig. 4a, b), allowing the classification of missense variants as ‘damaging’, ‘intermediate’ or ‘functional’ by fitting a two-component mixture model. Using this mixture model analysis (Fig. 4b), thresholds were established to achieve 98% sensitivity and specificity, such that 2% of nonsense variants scored as functional and 2% of synonymous variants as damaging. Applying these thresholds (Fig. 4b), we classified 270 out of 339 nonsense variants (80%) as damaging and 18 out of 42 unique synonymous variants (43%) as functional. Among the missense variants, 392 (6%) were categorized as damaging, 2422 (36%) as intermediate, and 3904 (58%) as functional (Fig. 4c and Supplementary Data 1).

a Schematic flow of the site-saturation functional screens performed in this study. NGS next-generation sequencing, AA = amino acid. b Amino acid function map of the CC domain of PALB2 spanning 35 amino acid residues from p.L9 to p.K43 (top). The map shows depletion scores for 638 PALB2 missense and nonsense variants as generated by Enrich2, n = 6 biological replicates. Amino acid characteristics for all variants are indicated (left). Dark red squares represent variants that were depleted in PARPi-treated conditions versus untreated conditions, n = 3 technical replicates. Blue squares represent variants that were enriched. Grey squares represent variants that were either missing in the library or filtered out during the analysis. Grey dots represent the original (wild-type) amino acids. Data for all variants were normalized to WT PALB2, which was set to ‘0’, and to the average of the nonsense variants, which was set to ‘−1’. Source data are provided as a Source Data file.

Amino acid function maps that comprise the entire WD40 domain of PALB2 spanning 331 amino acid residues from p.Q856 to p.S1186 (top of each map). Each map corresponds to a distinct WD40 variant library (left and Supplementary Fig. 1). The maps show depletion scores for 6419 PALB2 missense and nonsense variants as generated by Enrich2, n = 3 biological replicates. Amino acid characteristics for all variants are indicated (left of each map). Dark red squares represent variants that were depleted in PARPi-treated conditions versus untreated conditions, n = 3 technical replicates. Blue squares represent variants that were enriched. Grey squares represent variants that were either missing in the libraries or filtered out during the analysis. Grey dots represent the original (wild-type) amino acids. Data for all variants were normalized to WT PALB2, which was set to ‘0’, and to the average of the nonsense variants per library, which was set to ‘−1’. Source data are provided as a Source Data file.

a Histogram showing the distribution of 7106 depletion scores of nonsense, synonymous and missense PALB2 variants from Figs. 2b and 3. Dashed lines indicate functional classification thresholds determined by mixture modelling. Dashed line on the right separates functional (right) from intermediate (middle) variants at a depletion score of −0.0759. Dashed line on the left separates intermediate (middle) from damaging (left) variants at a depletion score of −0.7037. b ROC curve showing 98% sensitivity and specificity for classification of variants from ‘a’ was generated by fitting a two-component mixture model to depletion scores of 339 nonsense and 49 synonymous variants. c Histogram showing the distribution of 6,718 depletion scores of PALB2 CC and WD40 missense variants. Dashed lines indicate functional classification thresholds as in b. Number and fraction (%) of missense variants classified as functional (green), intermediate (orange) or damaging (red) are indicated. d Correlation analysis between outcomes of PARPi sensitivity assays and depletion scores from site-saturation functional screens of missense variants in PALB2. n = 140 PALB2 variants (43 CC and 97 WD40 variants). PARPi assays were performed twice with similar results (Supplementary Data 1 and Source Data Fig. 4d). Depletion scores are from Figs. 2b and 3. Dashed lines indicate functional thresholds as in (a). r = Pearson correlation coefficient. Two-sided P < 0.0001. e As in d, except for DR-GFP assays. f SGE was used to introduce SNVs across a region of exon 10 of PALB2 encoding p.G1000 to p.I1037 of the WD40 domain. A gRNA/Cas9 construct was transfected with a plasmid library containing SNVs within ~100 bp of genomic sequence, homology arms, and synonymous variants within the CRISPR target site to prevent re-cutting. Cells were collected 9 days after transfection and targeted sequencing was performed to quantify SNV abundances and calculate SGE scores. g Correlation analysis between SGE scores and depletion scores from site-saturation functional screens of 179 PALB2 variants in the WD40 region spanning p.G1000 to p.I1037 (Supplementary Data 1). SGE experiments were performed twice with similar results (Supplementary Data 1 and Source Data). Depletion scores are from Fig. 3. Dashed lines indicate the functional classification thresholds as in (b). r = Pearson correlation coefficient. Two-sided P < 0.0001. Source data are provided as a Source Data file.
Validation of site-saturation functional screens of missense variants in the CC and WD40 domains of PALB2
To validate our site-saturation functional screens, we measured the relative PARPi resistance levels for a panel of missense variants (n = 140). Of these variants, 96 were newly tested, while 44 had been previously tested7,15. When comparing PARPi resistance levels to the depletion scores from the screens (Supplementary Data 1), we observed a statistically significant correlation of good to very good strength for variants in the CC domain. (n = 43, r = 0.8055, p < 0.0001), WD40 (n = 97, r = 0.6500, p < 0.0001) and CC + WD40 domains (n = 140, r = 0.6494, p < 0.0001) (Fig. 4d and Supplementary Fig. 4c, d). Consistently, we also observed a significant correlation, ranging from good to very good, between the outcome of the site-saturation screens and those from DR-GFP reporter-based assays for the same variants in the CC (n = 43, r = 0.8715, p < 0.0001), WD40 (n = 97, r = 0.7131, p < 0.0001) and CC + WD40 domains (n = 140, r = 0.7086, p < 0.0001) (Fig. 4e and Supplementary Fig. 4e, f). Moreover, our site-saturation screens correctly categorized previously reported missense variants with no functional impact (e.g., p.E19V, p.E42K, p.G998E), a moderate impact (e.g., p.Y28C, p.R37H, p.L1070P), or a detrimental impact (comparable to that of truncating variants, e.g., p.L35P, p.W912G, p.I944N) as functional, intermediate or damaging, respectively (Supplementary Data 1)7,15.
Next we aimed at validating the outcome of our site-saturation screens in human cells using saturation genome editing (SGE)16. To this end, we performed a deep mutational scan of SNVs in exon 10 encoding p.G1000 to p.I1037 of the WD40 domain of PALB2 using a cell survival assay in haploid human HAP1 cells (Fig. 4f). Since PALB2 is essential in HAP1 cells17, damaging variants in this gene reduce cell survival. CRISPR/Cas9-based saturation genome editing was used in combination with a variant library to introduce all unique SNVs in the PALB2 exon 10 region. Cells were cultured to allow genome-editing and harvested 5 and 20 days later. DNA was extracted and targeted sequencing of the exon 10 region was performed. Sequencing data were processed to quantify the abundance of each variant and calculate a SGE score using the frequency difference of reads between day 20 and 5. Functional variants are abundant and will have high SGE scores, whereas damaging variants show low abundance due to depletion from the cell culture and will have low SGE scores (Fig. 4f). We obtained SGE scores for 276 SNVs, leading to 9 nonsense and 68 synonymous changes, as well as 199 amino acid substitutions. The nonsense and synonymous variants combined, as well as the missense variants showed an asymmetric distribution that is skewed towards low SGE scores (Supplementary Fig. 4g, h). Among these variants, we identified 218 unique nonsense (n = 9), synonymous (n = 31) and missense (n = 178) changes (see Fig. 4g in Source Data file). Importantly, we obtained depletion scores for 179 of these variants, comprising 170 missense and 9 nonsense variants, in the PARPi sensitivity-based site-saturation screens (Fig. 4g and Supplementary Data 1), allowing comparison of depletion scores and SGE scores. We observed a good and significant correlation between these scores (n = 179, r = 0.6439, p < 0.0001; Fig. 4g), indicating concordance between the outcomes of our functional analysis of PALB2 variants in mES and human HAP1 cells.
Finally, we examined the relationship between the functional categorization of variants and frequencies in large-scale variant databases, including gnomAD, FLOSSIES and BRAVO. GnomAD contains whole-genome and whole-exome sequencing data from more than 140,000 individuals18, including 186 missense, 9 nonsense and 8 synonymous PALB2 variants that were functionally assessed in the PARPi sensitivity-based site-saturation screens. Functional missense and synonymous variants (i.e. those with high depletion scores) tend to exhibit higher allele frequencies than damaging missense and nonsense variants (Supplementary Fig. 5a; Supplementary Data 2). A similar trend was observed in the BRAVO dataset (Supplementary Fig. 5b; Supplementary Data 2), which includes genome sequencing data from over 145,000 non-cancer individuals. The FLOSSIES dataset, composed of genome data form ~10,000 elderly, healthy women, lacks damaging missense and nonsense variants. Nevertheless, functional missense variants in FLOSSIES tend to show higher allele frequencies compared to intermediate variants (Supplementary Fig. 5c; Supplementary Data 2). Collectively, the concordance with PARPi-sensitivity and DR-GFP assays in mES cells, SGE in human HAP1 cells, and large-scale human variant databases supports the validity of the site-saturation functional screens.
Variants in the CC domain disrupt PALB2’s interaction with BRCA1 and its recruitment to DNA damage sites
We and others have previously shown that damaging missense variants in the PALB2 CC-domain have an impact on the interaction with BRCA1, as well as on the BRCA1-dependent recruitment of PALB2 to sites of DNA damage (e.g., p.L24S, p.L35P or p.R37H)7,9,19. To further validate findings from the site-saturation functional screens, we selected variants that scored as damaging (p.L21S, p.A22P, p.Y28D, p.T31P, p.A33P, p.R34P, p.Q36P), intermediate (p.S10T, p.C11W, p.E13A, p.L17S, p.K25E, p.E27G, p.Y28S, p.S29G, p.K30E) or functional (p.E19V, p.R26K) (Supplementary Data 1). The previously reported damaging p.L35P variant was included as a control. To examine the impact on the PALB2-BRCA1 interaction, we transiently expressed YFP-tagged PALB2 carrying these individual variants in U2OS cells and performed pull-downs using GFP Trap beads. The two functional variants p.E19V and p.R26K, as well as the three intermediate variants p.S10T, p.C11W and p.E13A, efficiently precipitated endogenous BRCA1 to similar levels as observed for wild-type PALB2 (Fig. 5a), whereas the other intermediate variants p.L17S, p.K25E, p.E27G, p.Y28S, p.S29G and p.K30E partially affected the co-precipitation of BRCA1 (Fig. 5a). In contrast, the damaging variants p.L21S, p.A22P, p.Y28D, p.T31P, p.A33P, p.R34P and p.Q36P, similar to p.L35P, failed to co-precipitate any endogenous BRCA1 (Fig. 5a).

a YPF/GFP pulldowns of the indicated PALB2 CC variant proteins following transient expression in U2OS cells. PALB2 CC variants are indicated in three colours reflecting their functional outcome in the site-saturation screens in ‘Fig. 2b’; green is functional, orange is intermediate, red is damaging. GFP-NLS and YFP-PALB2-L35P served as negative controls. Western blot analysis was performed using antibodies against GFP and BRCA1. Representative blots of two independent experiments with similar results are shown. b Western blot analysis of the expression of human PALB2 WD40 variants in Trp53KO/Palb2KO mES cells using an antibody directed against PALB2. The empty vector (Ev) served as a negative control on each blot. Tubulin was used as a loading control. *indicates a non-specific band. PALB2 variants are indicated in colour as in (a). Expression levels indicated below the blots are normalized to Tubulin and relative to the normalized WT PALB2 expression level, which was set to 1. Blots of one experiment are shown. c Fluorescence microscopy analysis (left) and quantification of the cellular distribution (right) of human YFP-PALB2 WD40 variants following transient expression in U2OS cells. Coloring of variants is as indicated in (a). Data represent the mean ±s.e.m. n = 3 biological replicates, dots represent individual data points, two-way ANOVA with two-sided Dunnet’s multiple comparison test. *P < 0.05, **P < 0.01, ***P < 0.001, NS = not significant. Source data are provided as a Source Data file.
Next, we examined for some of these variants whether they have an impact on the BRCA1-dependent localization of PALB2 to sites of DNA damage. To this end, YFP-tagged PALB2 carrying these variants was transiently expressed in U2OS cells and examined for localization at DNA damage-containing tracks generated by UV-A laser micro-irradiation. The localization of DNA damage sensor protein NBS1, which was tagged with mCherry and co-expressed in these cells, was used to control for equal DNA damage levels. We found that the intermediate variants p.S10T and p.E13A, similar to the functional variants p.E19V and p.R26K, did not impact the accumulation of PALB2 at sites of DNA damage, whereas p.L17S and p.E27G had a moderate impact when compared to that of wild-type PALB2 (Supplementary Fig. 6a, b). The damaging variants p.L21S and p.L35P had the strongest impact on the accumulation of PALB2 at sites of DNA damage. The accumulation of mCherry-NBS1 was unaffected in these cells (Supplementary Fig. 6a, b). The effect of these variants on the PALB2-BRCA1 interaction and on PALB2 localization at sites of DNA damage, despite the variability and non-significant differences, was consistent with the observed HR defect in DR-GFP assays, PARPi sensitivity assays and site-saturation functional screens (Figs. 2, 4d, e and Supplementary Data 1). Taken together, these analyses identified p.L17S, p.L21S, p.A22P, p.K25E, p.E27G, p.Y28D, p.Y28S, p.S29G, p.K30E, p.T31P, p.A33P, p.R34P and p.Q36P as new variants that impair PALB2 function in HR by impacting the interaction with BRCA1, which in turn affects PALB2 recruitment to DNA damage sites.
Variants in the PALB2 WD40, but not CC domain, affect protein stability
Prior studies, including ours, have demonstrated that damaging missense variants in the PALB2 WD40 domain impact the expression of PALB27,9,19. Reverse transcription-quantitative (RT-q)PCR analysis indicated that these variants did not affect expression at the mRNA level, but rather affected protein folding and/or stability, as shown in inhibition assays of translation and protein degradation using cycloheximide and proteasome inhibitor, respectively7,15. To further validate findings from the site-saturation functional screens, we selected 61 variants in the WD40 domain that were scored as damaging (25), intermediate (18) or functional (18), and examined effects on PALB2 expression by western blot analysis (Fig. 5b and Supplementary Data 1). We also included 33 variants in the CC domain that were scored as damaging (10), intermediate (21) or functional (2) (Supplementary Fig. 6c and Supplementary Data 1). For all functional variants in the WD40 domain, PALB2 expression was comparable to that of wild type. However, damaging variants generally showed very low levels of expression (e.g. 0.22 for p.W912S; Fig. 5b), whereas intermediate variants often partially reduced PALB2 expression (e.g. 0.70 for p.R976G; Fig. 5b). In contrast, variants in the CC domain generally showed near wild-type levels of PALB2 expression regardless of their functional classification, except for p.K20I, which showed reduced expression levels (0.31; Supplementary Fig. 6c).
WD40 variants showing reduced expression have been shown to mislocalise, likely as a result of protein instability and subsequent proteasomal degradation in the cytoplasm8,15. Consistent with these findings we found that 5 WD40 variants, which showed reduced expression (p.I887S, p.V895F, p.C933R, p.G971D, p.F1016C; Fig. 5b), were mislocalised when introduced in YFP-tagged PALB2 and transiently expressed in U2OS cells (Fig. 5c), an effect that was similar to that of the previously reported mislocalised p.L1027R variant15. In contrast, the WD40 variant p.E1083K, which was functional and expressed to WT levels (Fig. 5b and Supplementary Data 1), and several variants in the CC domain (p.S10T, p. E13A, p.L17S, p.Y28S, p.A22P, and p.Y28D), all of which showed near wild-type expression levels (Supplementary Fig. 6c), did not grossly mislocalise in the cytoplasm (Fig. 5c and Supplementary Fig. 6d).
Given the correlation between protein instability and loss of functionality for missense variants in the WD40 domain, we asked whether predicting the effect of WD40 missense variants on protein stability may constitute a step toward understanding the functional impact of these variants. Several methods have been developed to predict changes in the Gibbs free energy of unfolding (∆∆G) between wild-type and variant proteins using sequence and structure information. We used the FoldX BuildModel command to determine the ∆∆G values for all possible missense variants in the structured regions of the WD40 domain20. ∆∆G values were obtained for 5,596 missense variants in this domain. While highly elevated ∆∆G values were observed for several missense variants, a vast majority of variants showed moderately increased ∆∆G values, indicative of a mild protein destabilisation (Supplementary Fig. 7a). This suggests that only substitutions of a subset of residues may lead to severe protein destabilisation and loss of functionality. In agreement, we found that ∆∆G values only correlated weakly with depletion scores obtained for missense variants in the WD40 domain (Supplementary Fig. 7a; n = 5,596, r = -0.2137, p < 0.0001) (see Methods). A similar trend was observed when ∆∆G values were obtained using the deep-learning based method RaSP (Supplementary Fig. 7b; n = 6,071, r = -0.2460, p < 0.0001)21. Taken together, our results suggest that missense variants in the WD40 domain, but not in the CC domain, can lead to protein instability and subsequent loss of function, the latter of which is only poorly predicted by ∆∆G-based in silico models.
In silico tools show limited specificity in predicting the functional impact of PALB2 variants
We previously showed that the outcomes of in silico prediction tools such as PolyPhen22, SIFT23, Align-GVGD24 and REVEL25 generally show little to no correlation with those from PARPi sensitivity and DR-GFP assays7. We sought to examine this further by comparing the predictive performance of four more recently developed tools, Helix, AlphaMissense, EVE, and BayesDel, against the outcome of the PARPi sensitivity-based site-saturation screens for 6718 missense variants in the CC and WD40 domains26,27,28,29. All tools generated predictions for 1347 variants, showing moderate correlations with depletion scores for Helix (r = -0.4384), AlphaMissense (r = -0.4484), and BayesDel (r = -0.3541), while EVE showed a weaker correlation (r = -0.2154), all with p < 0.0001 (Supplementary Fig. 8). Notably, although all tools tended to overestimate the damaging impact of variants with high depletion scores, potentially leading to false positives, they were more accurate for variants with low depletion scores. Extending the analysis to all 6718 variants, Helix and AlphaMissense showed strong correlation for variants in the CC domain (Helix: r = -0.6509; AlphaMissense: r = -0.3408; n = 609), moderate to weak correlation for those in the WD40 domain (Helix: r = -0.2873; AlphaMissense: r = -0.2266; n = 6,109), and moderate correlation across the combined domains (Helix: r = -0.2882; AlphaMissense: r = -0.2527; n = 6,718), all with p < 0.0001 (Supplementary Fig. 9a–f). Again, both tools overestimated the damaging effects of variants with high depletion scores.
To further assess the performance of the in silico prediction tools, receiver operating characteristic (ROC) analysis was performed using missense variants classified as damaging (HR < 30%; n = 41) or functional (HR > 70%; n = 105) based on DR-GFP assays from both the current study (Fig. 4e) and previous research (Supplementary Fig. 9g)7. The analysis revealed that Helix, BayesDel and EVE performed moderately well (AUC values 0.846, 0.781 and 0.645, respectively). In contrast, AlphaMissense showed relatively better performance (AUC value 0.946), with good sensitivity (1) but limited specificity (0.8) (Supplementary Fig. 9g–i). This aligns with the Pearson correlation data, showing that AlphaMissense frequently overestimate the deleteriousness of variants with functional impact in our site-saturation screens (Supplementary Figs. 8 and 9a–f). In conclusion, while Helix and AlphaMissense may offer reliable predictions for missense variants within the CC domain, overall Helix, AlphaMissense, EVE, and BayesDel, like other in silico models previously evaluated7, generally lack sufficient accuracy in predicting the functional impact of PALB2 variants. Notably, these models tend to lack specificity, often overestimating the damaging impact of variants with high depletion scores.
Site-saturation functional screens aid in the clinical reclassification of missense VUS in PALB2
Genetic testing has led to a tremendous increase in the number of reported VUS in PALB2. As of June 30, 2025, 3014 SNVs in PALB2 were listed in ClinVar, 2925 of which encode 2800 unique missense VUS (Fig. 6a and Supplementary Data 3). We next investigated whether the results of our site-saturation functional screens could assist in the classification of VUS in the CC and WD40 domain. The screens produced depletion scores for 872 VUS in these domains, representing 31.1% of those reported in ClinVar, which we categorized as functional (n = 518), intermediate (n = 304), or damaging (n = 50). For only 41 of the VUS (1.5%) in the CC and WD40 domains no depletion scores were available (Fig. 6a). Of note, for 21 ClinVar-reported missense VUS located between the start and CC domain of PALB2 (pre-CC), no depletion scores were available as this region was not subjected to functional analysis (Fig. 6a and Supplementary Data 3). Thus, our site-saturation functional screens can aid in the categorization of 872 (31.1%) of the VUS in the CC and WD40 domains listed in ClinVar.

a Radial bar chart of PALB2 SNVs reported in ClinVar (as of June, 2025) (left), pie chart showing the distribution PALB2 missense VUS from ClinVar which were examined in site-saturation functional screens and are located in the CC and WD40 domains. Variants in the pre-CC (start codon to first codon of the CC domain) and middle regions were not examined in these screens. Histogram showing the frequency distribution of depletion scores of PALB2 CC and WD40 VUS from ClinVar (right). Dashed lines indicate thresholds from ‘Fig. 4a’, which were used to classify PALB2 CC and WD40 VUS from ClinVar as either functional (green), intermediate (orange) or damaging (red). b Schematic representation of the PALB2 protein in which amino acid numbers are shown to specify the evolutionarily conserved functional domains of PALB2 (bottom). PALB2 variants are color-coded as indicated. c DR-GFP HR assay in Trp53KO/Palb2KO mES cells expressing the indicated PALB2 variants from (b). HR efficiencies were normalized to the WT PALB2 condition which was set to 100%. Ev is the empty vector control. d PARPi sensitivity assay using Trp53KO/Palb2KO mES cells expressing the indicated PALB2 variants variants from (b) and (c). Values indicate the relative resistance to 0.5 μM PARPi treatment with the WT PALB2 condition set to 100%. In c and d, mean is shown, n = 2 biological replicates, dots represent individual data points. Source data are provided as a Source Data file.
We previously showed that the region between the CC and WD40 domain (encoding p.I44 to p.L855) is dispensable for PALB2 function in HR, suggesting it is unlikely that damaging missense variants will be identified therein (Fig. 1). ClinVar listed 1866 missense VUS in this region (Fig. 6a). To provide additional support that these variants may be functional rather than damaging, we randomly chose 26 variants, along with 5 synthetic missense variants in this region (Fig. 6b), and expressed them in the Palb2KO mES cells to determine the impact on HR (Supplementary Fig. 10). Expression of all 26 missense VUS rescued HR in the DR-GFP reporter to levels comparable to that in cells expressing wild-typePALB2 (Fig. 6c). This result was validated in PARPi assays, which demonstrated for 12 of these variants that expression rendered Palb2KO cells resistant to PARPi treatment (Fig. 6d). Corroborating these findings, 16 of the missense VUS exhibited near wild-type expression, whereas a synthetic variant causing a frameshift (p.S201fs) markedly reduced PALB2 expression, as expected (Supplementary Fig. 10). These findings further indicate that the 1,866 missense VUS located in region p.I44 to p.L855 are likely functional. Therefore, our functional data may also contribute to the reclassification of missense VUS located in the middle region of PALB2, including those reported in ClinVar.
Damaging missense variants in PALB2 are associated with increased breast cancer risk
Having categorized missense variants in PALB2 as functional, intermediate or damaging (Fig. 4c and Supplementary Data 1), we next asked if their functional impact correlated with increased breast cancer risk. The risks of breast cancer for individuals carrying PALB2 functional (depletion scores >-0.0759) and damaging (depletion scores <-0.7037) missense variants were estimated in case-control association analyses. Burden analysis of damaging missense variants in the BRIDGES, CARRIERS and CZECANCA case-control studies combined yielded a strong association with breast cancer (odds ratio (OR) = 4.67; 95%CI: 1.36 – 18.82; p = 0.009) (Table 1), consistent with previously reported results for protein truncating variants11,30. In contrast, functional variants were not associated with increased risk (OR = 1.07; 95%CI: 0.93 – 1.23; p = 0.333). Likewise, intermediate variants with functional scores between -0.7037 and -0.0759 were not associated with increased risk of disease (OR = 1.28; 95%CI: 1.00 – 1.64; p = 0.050) (Table 1).
To validate these findings, association studies for functionally characterized missense variants were conducted using breast cancer cases and controls from UK Biobank (see Methods and Table 1). Damaging missense variants were once more associated with an increased risk of breast cancer (OR = 4.94; 95%CI: 1.07 – 20.68; p = 0.043) whereas functional variants (OR = 0.99; 95%CI: 0.75 – 1.30; p = 1.000) and intermediate variants (OR = 1.63; 95%CI: 1.12 – 2.34; p = 0.013) were not. These findings were further confirmed for damaging (OR = 3.25; 95%CI: 1.58 – 7.51; p = 7.83 ×10-4), intermediate (OR = 1.00; 95%CI: 0.86 – 1.16; p = 1.000), and functional variants (OR = 1.10; 95%CI: 0.94 – 1.27; p = 0.228) using a clinical cohort and non-cancer female gnomAD controls, as well as for damaging (OR = 2.73; 95%CI: 1.16 –6.53; p = 0.013), intermediate (OR = 0.87; 95%CI: 0.73 – 1.03; p = 0.096), and functional variants (OR = 1.09; 95%CI: 0.93 – 1.28; p = 0.285) from the same clinical cohort using controls from the All of Us research program (see Methods and Table 1)18. Importantly all associations for damaging variants were significantly different from functional and intermediate variants, indicating that the damaging missense variants identified in the site-saturation functional screens represent a group of variants that are likely associated with a high risk of developing breast cancer.
Discussion
Loss-of-function variants in the PALB2 gene give rise to defects in DNA damage repair by HR, thereby increasing the risk of breast, pancreatic and ovarian cancer in carriers1. However, genetic testing frequently reveals missense VUS in PALB2 for which the impact on protein function and cancer risk are unclear (as of June 30, 2025, 3014 SNVs in PALB2 were listed in ClinVar, 2925 of which encode 2800 unique missense VUS (Fig. 6a and Supplementary Data 3). Functional assays can be used to determine the functional impact of VUS in PALB2, but the available assays are time-consuming and low throughput. Moreover, PALB2 VUS are very rare, complicating cancer risk analysis. To overcome these challenges, we functionally assessed 84% of all possible missense variants across 11 of 13 PALB2 exons using site-saturation screens, with PARP inhibitor sensitivity serving as a readout for PALB2 function in HR. These exons encode the coiled-coil and WD40 domains, which we identified as the minimal regions required for HR. The functional impact of 6718 missense variants was determined, categorizing 3904 variants as functional (58%), 2422 as intermediate (36%), and 392 as damaging (6%). The screen data correlated well with those from PARPi sensitivity and DR-GFP assays in mES cells and SGE in human HAP1 cells, as well as those from large-scale variant databases of human populations, including gnomAD, FLOSSIES and BRAVO. Moreover, the screen data could be validated in molecular follow-up studies assessing the PALB2 interaction with BRCA1, recruitment to DNA damage sites and stability/localisation, validating a high-quality functional dataset. Importantly, our data are expected to aid in the reclassification of VUS reported in ClinVar by showing that VUS located in region p.I44 to p.L855 are likely functional, while 872 VUS in the CC and WD40 domains could be categorized as functional, intermediate or damaging. Finally, burden case-control analysis revealed that damaging missense variants in PALB2 are associated with a significantly increased risk of breast cancer. These results will be essential for the clinical interpretation of PALB2 missense VUS and clinical management of carriers.
Advances in high-throughput screening have led to a sweeping expansion of functional data for missense variants in disease genes, including BRCA1 and BRCA231,32,33,34,35,36. Here we extend these screens by proving functional data for almost 7000 missense variants in PALB2. Although these screens provide unprecedented insight into how the functional impact of such variants may be linked to disease, they also have their limitations. For instance, high-throughput approaches can be inherently more noisy compared to single-variant functional analysis. In case of our site-saturation functional screens of PALB2 missense variants, this may complicate the interpretation of measured functional impacts, particularly for intermediate variants or for damaging variants with depletion scores close to the threshold of qualifying as intermediate or damaging. The latter makes it challenging to properly classify such variants and define the optimal clinical management. Validating the impact in single variant test and orthogonal assays may be necessary for such variants. Another limitation of our approach is that it cannot measure functional effects of missense variants caused by altered PALB2 mRNA splicing as it is a cDNA-based complementation assay. To this end, saturation genome editing of PALB2 or a bacterial artificial chromosome (BAC)-based assay (in which full-length PALB2 is expressed in Palb2KO cells) in combination with PALB2 mRNA expression analysis would be required. However, in silico analysis using SpliceAI (threshold >0.5) indicates that only 22 out of the 2078 missense variants (1.06%) assessed in our site-saturation functional screens are encoded by SNVs predicted to impact PALB2 mRNA splicing (see Supplementary Data 4)37. This suggest that these variants mostly impair PALB2 functionality at the protein level. Of note, SpliceAI (threshold > 0.5) predicts that only 20 of the 4758 missense variants (0.42%) located between the coiled-coil and WD40 domains may impact mRNA splicing (Supplementary Data 4). The SNVs encoding these missense variants could be pathogenic due to altered splicing. However, outside of these few cases, our data suggest that missense variants in this region rarely impair PALB2 protein function (Figs. 1 and 6b–d). Supporting this notion, ClinVar-reported missense variants within this region were not associated with an increased risk of breast cancer (Supplementary Data 5). Finally, PALB2 is a multi-functional protein that, in addition to its role in HR, plays roles in checkpoint maintenance and the recovery of stalled replication forks38. This questions whether functional screens of PALB2 variants should be extended to multiple read-outs. We previously showed that its role in HR and DNA damage-induced G2/M checkpoint maintenance may be linked, since all variants that impacted HR also showed an altered checkpoint response7. This suggests that using checkpoint responses as a read-out may provide limited new insights, if at all, into the functional impact of PALB2 variants. In contrast, defects in the recovery of stalled replication forks have been shown to be linked to cancer development and chemosensitivity39. Thus, determining the impact of PALB2 variants in this process may not only provide new mechanistic insight into the PALB2 mode of action during this process, but may also shed further light on the link between functional impact of PALB2 variants and breast cancer risk. A major challenge, however, may be the implementation of readouts for DNA replication (e.g. based on DNA fiber/combing assays), particularly in high-throughput screens.
We demonstrated that damaging missense variants are associated with a clinically relevant increased risk of breast cancer. Dorling et al., however, reported limited evidence of an association between missense variants in PALB2 and cancer risk30. This divergence may be attributed to the less stringent cut-off for HR deficiency, and the reliance on in silico prediction tools to categorize missense variants as damaging. Combined with the relatively small proportion of risk-associated PALB2 missense variants, this may reflect limited statistical power to detect an association. In contrast, our study showed that damaging missense variants in PALB2 are associated with an increased risk of breast cancer, similar to that observed for PTVs (Table 1). As most variants are rare, it is unlikely that we will be able to estimate variant specific risks, although it might be possible to perform domain-specific analyses for such variants.
To facilitate clinical classification of genetic variants, the American College of Medical Genetics and Genomics (ACMG) and Association for Molecular Pathology (AMP) have proposed variant interpretation guidelines that incorporate different types of evidence (including functional assessment) at various levels of strength. These guidelines also provide rules for combining the different types of evidence to result in a final classification (benign, likely benign, uncertain significance, likely pathogenic, pathogenic), each with defined clinical significance2,3. While PTVs are generally readily classified, missense variants require more comprehensive information to attain a robust classification. However, the availability of clinical evidence, encompassing phenotypic and segregation data, is often limited. As a result, functional data will become crucial for the clinical classification of the majority of genetic missense variants (both currently known and those yet to be discovered). Notably, the Clinical Genome Resource (ClinGen) Sequence Variant Interpretation (SVI) Working Group recently refined the PS3/BS3 criteria, which define the impact of a variant on protein function as determined by a well-established functional assay (Brnich et al., 2020). In the case of PALB2, the absence of pathogenic missense variants poses challenges for validating and calibrating the assay up to the ACMG/AMP standards. Yet, the potential use of PTVs as pathogenic controls could be deemed acceptable under the condition that both PTVs and missense variants exhibit similar behaviour in functional assays, and evidence shows that both types of variants confer a comparable disease risk. The findings presented in this study confirm that our assay meets the necessary prerequisites, thereby facilitating the use of our functional data for ACMG/AMP-based variant classification.
Methods
Cell culture and generation of Trp53 KO/Palb2 KO mES cells with DR-GFP and RMCE
Trp53KO/Palb2KO mES cells carrying the DR-GFP reporter and RMCE system at the Pim1 and Rosa26 locus, respectively, were generated previously7 and cultured as previously described40. HAP1 cells (Horizon Discovery) were maintained in IMDM with 10% fetal bovine serum and 1% Penicillin/Streptomycin. U2OS cells were cultured in 5% CO2 at 37 °C in DMEM (Dulbecco’s modified Eagle’s medium) supplemented with 10% fetal calf serum and 1% Penicillin/Streptomycin. Human HAP1 and U2OS cells were authenticated using Short Tandem Repeat (STR) analysis by ATCC services (100% match). Cells were used only when confirmed free of mycoplasma contamination using the MycoAlert™ detection kit (LONZA).
Introducing variants and deletions into human PALB2 cDNA
The RMCE vector (pRNA-251-MCS-RMCE) (TaconicArtemis GmbH) containing human PALB2 cDNA driven by an Ef1α promotor was generated previously7. PALB2 variants, as well as ChAM (c.1183 - c.1338) and MRG15 (c.1831 - c.2292) deletions, were introduced by site-directed mutagenesis using the Quick-Change Lightning protocol (Agilent Technologies). Exon deletions (exon 4, c.212 - c.1684; exon 5, c.1684 - c.2514, includes the last nucleotide of exon 4 to generate an in-frame deletion; exon 6, c.2515 – c.2586) were generated using primer pairs that flank the exons. Primer phosphorylation by T4 PNK (NEB) was followed by PCR with Phusion Polymerase (NEB), DpnI (NEB) digestion and ligation with T4 DNA ligase (NEB). All constructs were verified by Sanger sequencing and used for downstream mES cell-based assays.
PARPi sensitivity assays
Functional analysis of single PALB2 variants using proliferation-based PARPi (Olaparib; Selleckchem, S1060) sensitivity assays was performed for selected PALB2 missense variants as previously described7. Briefly, cells were exposed to various concentrations of PARPi for two days. Thereafter, cells were incubated for one more day in drug free media, after which viability was measured using flow cytometry (using only forward scatter and side scatter).
HR reporter assays
HR assays using 2×106Trp53KO/Palb2KO mES cells carrying the DR-GFP reporter and RMCE system were performed as previously described7. Briefly, cells that were complemented with human PALB2 cDNA with or without a variant (or an empty vector) and were treated with neomycin to select for cells with integrated PALB2 variant cDNA. Two days after transfection of an I-Scel and mCherry co-expression vector41, GFP expression was measured using flow cytometry.
Generation of site-saturation variant libraries for CC and WD40 domains
Using the RMCE vector (pRNA-251-MCS-RMCE) (TaconicArtemis GmbH) containing human PALB2 cDNA7, a variant library specific for the CC region of PALB2 was generated by Ranomics Incorporated in Toronto, Canada, by using a series of in-house computational tools and multiplex PCR assembly reactions. The library was validated by the Leiden Genome Technology Center (LGTC) of the Leiden University Medical center (LUMC), and was shown to contain ~95% of all possible missense and nonsense variants in the CC region of (p.L9-p.K43) PALB2 (Supplementary Fig. 1).
Five WD40 variant libraries were generated by TWIST Bioscience using the RMCE vector (pRNA-251-MCS-RMCE) (TaconicArtemis GmbH) containing wild-typePALB2 cDNA. Altogether, the five variant libraries cover the entire WD40 domain of PALB2; WD40 library 1 (p.Q856-p.Q921), WD40 library 2 (p.I922-p.Q987), WD40 library 3 (p.Q988-p.D1053), WD40 library 4 (p.S1054-p.L1119), WD40 library 5 (p.E1120-p.S1186). For each variant library, TWIST supplied a double stranded linear fragment containing the entire WD40 region with the same added flanking sequences resulting in a final product of 1,071 bp for each library. For cloning, the RMCE vector containing wild-typePALB2 cDNA was used to amplify the entire vector lacking only the WD40 region of PALB2, using the site-directed mutagenesis kit and Quick-Change Lightning protocol (Agilent Technologies) and the following primers; Fw: 5’- CTTAATTAAGGCCAGGGATCTTCAAGC-3’, Rv: 5’-ATGCTATCAGAAGCAGGAAGCTCTG-3’. The reaction mix was subjected to DpnI digestion for 1 hour at 37 °C and the amplified vector was gel extracted. Each WD40 library was then cloned (in a separate reaction) into the amplified vector by Gibson Assembly (NEB) for 40 min at 50 °C using 50 ng amplified vector (RMCE vector backbone containing the entire PALB2 cDNA except the WD40 region) and 29 ng of WD40 library. The entire ligation mix was transformed into DH5α and grown o/n in 50 ml lysogeny broth in the presence of ampicillin. After midiprep of each WD40 library, all five libraries were validated by the Leiden Genome Technology Center (LGTC) of the Leiden University Medical center (LUMC), and were shown to contain >90% of all possible missense and nonsense variants in the WD40 region of PALB2 (Supplementary Fig. 1).
Integration of site-saturation variant libraries for CC and WD40 domains
The PALB2 variant libraries were integrated in 100 × 106Trp53KO/Palb2KO mES cells (six times the CC library and three times each WD40 library, representing biological replicates). Cells were divided in fractions of 10 × 106 cells for which each fraction was subjected to co-transfection of 1 μg FlpO expression vector (pCAGGs-FlpO-IRES-puro)42 with 1 μg RMCE exchange vector containing the variant libraries, as previously described7. Transfected cells were divided over twenty 10 cm tissue-culture plates and treated one day later with 50 μg/ml neomycin/G418 sulphate (ThermoFisher, 10131035) for 6-7 days. Resistant colonies expressing PALB2 variant cDNAs were pooled (50-100 × 103 colonies per variant library integration), mixed well and plated over three 10 cm tissue-culture plates containing neomycin. Two plates were trypsinized and stored at -80 °C as backup and one plate was used for the PARPi sensitivity assay.
High-throughput PARPi sensitivity assays
PARPi sensitivity after PALB2 variant library integration was assessed in triplo (representing technical replicates) by using 0.57×106 cells seeded on 6 cm tissue-culture plates. One day after seeding, cells were treated with 0.5 µM PARPi (Olaparib; Selleckchem, S1060) for two days, after which the medium was refreshed with drug-free medium and cells were cultured for one more day. A non-treated plate was taken along as a control at the start of seeding. DNA was eventually isolated from the surviving cells and subjected to next-generation sequencing.
PALB2 cDNA amplification and next-generation sequencing
The variant containing region of the integrated human PALB2 cDNA was amplified from 100 ng genomic DNA. Reactions contained 2* Kapa HiFi MasterMix polymerase (KR0370) and primers specific for either the CC region (Fw: 5’-GATGTGTATAAGAGACAGCGAGCTCGGATCCACTAGTAACG-3’; Rv:’-CGTGTGCTCTTCCGATCTCTGAGTGTTTTAGCTGCGGTGAG-3’), WD40 region 1 (Fw: 5’- GATGTGTATAAGAGACAGCGAACAGACTGAAACAGCAGAGC-3’; Rv 5’- CGTGTGCTCTTCCGATCTCAAAGCTACACACACGAGATTATACAC-3’), WD40 region 2 (Fw: 5’- GATGTGTATAAGAGACAGCTTTATACCTGGCACTTCGCAGAG-3’; Rv 5’- CGTGTGCTCTTCCGATCTGGTTTTCTTTGCCTCCTCCATCTTC-3’), WD40 region 3 (Fw: 5’- GATGTGTATAAGAGACAGGCCTGACAAAGAGGAGGCTAGTTAG -3’; Rv 5’- CGTGTGCTCTTCCGATCTGGATGACTCAGGACAATAAAGAGAAGCC-3’),WD40 region 4 (Fw: 5’- GATGTGTATAAGAGACAGGCTCTGCTTGGTACTACTATTATGAACAAC-3’; Rv 5’- CGTGTGCTCTTCCGATCTGGCAATTGTTCCAGAAGTCAAGATTGC-3’), or WD40 region 5 (Fw: 5’- GATGTGTATAAGAGACAGGGTGTGATGCTGTACTGTCTTCCTC-3’; Rv 5’- CGTGTGCTCTTCCGATCTCTGTAGGTCTGCTTGAAGATCCCTG-3’). PCR was performed under the following conditions; 98 °C for 1 minute; 18 cycles of 98 °C for 20 seconds, 65 °C for 30 seconds, and 72 °C for 30 seconds; and 72 °C for 2 minutes. The reactions produced amplicons specific for each of the integrated human PALB2 variant libraries. After clean up with Ampure XP beads (Beckman Coulter) the PCR product was checked on a Agilent Bio-Analyzer 2100 HS chip. A second PCR with Illumina index primers was performed under the following conditions; 98 °C for 1 minute; 10 cycles of 98 °C for 20 seconds, 60 °C for 30 seconds, and 72 °C for 30 seconds; and 72 °C for 2 minutes. The resulting PCR products were equimolarly pooled. All samples were sequenced on an Illumina MiSeq.
Variant scoring and analysis
FASTQ files from each sample were analysed using the Enrich2 software package13. Enrich2 grouped and counted identical amino acid changes even if they arose from different nucleotide changes. Reads containing insertions, deletions or multiple amino acid substitutions were removed from the analysis. The number of sequencing reads supporting each amino acid variant was then calculated. Variants that showed noisy or unreliable results across replicates, based on a standard error (SE) greater than 0.5, were filtered out. Depletion scores were calculated for each remaining variant. These scores reflect the ratio of a variant’s frequency in PARPi-treated samples compared to untreated samples, normalized to the abundance of wild-type PALB2 (set to ‘0’). Only variants that passed the SE filter and were detected in all replicate experiments (six for the CC library and three for each WD40 library) were included in the final analysis. Each library included a varying number of nonsense variants. An average depletion score was calculated for the nonsense variants in each integration experiment. All variant scores were normalized by setting the average nonsense score to ‘-1’, using the following Eq. (1):
A final depletion score for each variant was calculated as the mean of its normalized depletion scores across all replicate library integration experiments (six for the CC library and three for each WD40 library). The SE for each variant was determined by dividing the standard deviation of its normalized scores by the square root of the number of replicates. Final depletion scores were visualized in a heatmap using the matrix analysis software MORPHEUS (https://software.broadinstitute.org/morpheus; see Supplementary Data 1).
Mixture modelling
We fitted a two-component mixture model to Enrich2 scores of the combined data. Since the data were heavier-tailed than normal, each of the components was assumed to have a scaled and shifted t-distribution. The scores of synonymous variants were all assumed to come from mixture component 1, those from nonsense variants from component 2, and the missense were assumed to be a true mixture. The resulting seven-parameter model was fitted by maximum likelihood. Mixture modelling was applied to estimate the probability that each variant is damaging (Pd), based on its average depletion score across replicates. Classification thresholds were defined as follows: variants with Pd <0.02 were classified as ‘functional’, those with Pd > 0.98 as ‘damaging’, and variants with 0.02 <Pd <0.98 were assigned to the ‘intermediate’ category. ROC curves and AUC were calculated for the raw data and for the model. For the data-based AUC a confidence interval was calculated using a previously reported method43.
Generation of site-saturation mutagenesis libraries and Cas9/sgRNA plasmids for SGE
PALB2 exon 10 and adjacent upstream and downstream 10 nucleotide introns flanking the exon (GRCh38 chr.16: 23,621,352-23,621,488 (137nucleotides)) were selected for SGE. A single guideRNA (sgRNA) (protospacer sequence CCAATTTTTGATGCCCCCTG) was designed by Benchling design tool. sgRNA annealed oligos were ligated into pSpCas9(BB)-2A-Puro (PX459 v2.0) (Addgene; 62988) following BbsI (New England Biolabs, R0539L) digestion to create Cas9-sgRNA co-expression construct for SGE. For the SGE, 600-1000 bp homologous arms upstream and downstream of the target region were amplified from WT HAP1 genomic DNA and cloned into BamHI-HF digested pUC19 vector using the NEBuilder HiFi DNA assembly Cloning Kit. Cloned plasmid backbones were subject to site-saturation mutagenesis by inverse PCR using mutagenized codon “NNN” primers for all possible nucleotide changes at each amino acid position. A Protospacer Protection Edit (PPE) encoding a synonymous mutation (c.3030 G > A; p.Glu1010Glu) was introduced by site-directed mutagenesis into the protospacer adjacent motif (PAM) site of the target region to prevent re-cutting by the Cas9/sgRNA after successful editing. This variant received a maximum delta score of zero across SpliceAI predictions for donor and acceptor gain and loss, indicating no predicted impact on RNA splicing. Furthermore, a 3-nucleotide mutation was introduced into the intronic region of the homologous arm to strengthen reamplification of the DNA within that target region.
Saturation genome editing
In the SGE experiment, 5 million haploid-sorted HAP1 cells were co-transfected with 4 μg target-specific variant library and 16 μg Cas9/sgRNA targeting construct using Turbofectin 8.0 (Origene). Cells were selected in puromycin (1 μg/ml) for 3 days. Cells were harvested at day 5 (24 hrs after puromycin selection) and day 20 post-transfection and genomic DNA (gDNA) was extracted using Monarch Genomic DNA Purification Kit (New England Biolabs, T3010L). The target region was amplified by PCR to add barcodes for multiplexing. All PCR reactions were performed in 50 μL reactions using Q5 High-Fidelity 2X Master Mix (New England Biolabs, M0492L). Primers for genomic DNA amplification are included in Supplementary Data 6). All reactions were cleaned and concentrated using Ampure XP beads prior to sequencing for 150 cycles on an Illumina MiSeq (approximately 5 million reads per run).
Sequence analysis after SGE
FASTQ files of sequenced samples from Illumina MiSeq were trimmed for adapter sequences using cutadapt (v3.5). SeqPrep (v1.2) converted the paired-end reads into single reads. The single reads were aligned to the human reference genome (GRCh38) utilizing bwa-mem (v0.7.17). Following alignment, a custom-developed tool ‘CountReads‘ was used for mutation identification and characterization. ‘CountReads‘ included the preparation of reference amino acid and DNA sequences, validation of sequencing data integrity, and precise trimming of reads to relevant regions. The method also differentiated between various variant types and confirmed the presence of specific variants and aggregated and reported variant data. ‘CountReads‘ produced a VCF (Variant Call Format) file which was annotated with CAVA. The log2 ratio between the frequency of day 20 and day 5 read counts was used to measure the depletion/enrichment effect for each variant. Variants with under-represented read counts (< 10) in the library and day 5 were excluded from further analysis. Log2 ratios of variants were linearly scaled relative to median values for synonymous and nonsense SNVs using within exon normalization. SGE was performed in duplicate. Replicates of individual SNVs with Log2 ratios >1 relative to mean replicate values were excluded. Mean SGE scores were calculated if the difference between replicate SGE scores was between -1.5 and 1.5 (Supplementary Data 1).
Pulldown assays
Pulldown assays were performed as previously described7. Briefly, 20 μg of pGFP-NLS or pYFP-PALB2 plasmid12 was transfected into ~5 × 106 U2OS cells on a 15 cm dish using Lipofectamine 2000. The next day cells were trypsinized, and lysed in 1 ml EBC buffer (50 mM Tris pH 7.3, 150 mM NaCl, 0.5% NP-40, 2.5 mM MgCl2) containing 1 tablet protease inhibitor (Roche) per 10 ml buffer. Lysates were incubated with benzonase and centrifuged. The supernatant was then added to 25 μl of pre-washed GFP-trap beads (ChromoTek) and incubated for 1.5 hours at 4 °C on a rotating wheel. The beads were washed 5–6 times with EBC buffer and eventually resuspended in 25 μl Laemmli buffer after which about half of each sample was analysed by western blot analysis using an antibodies against human BRCA1 (1:1,000, MS110, Merck, cat. nr. OP92) and GFP (1:10,000, Abcam, cat nr. ab290).
Western blot analysis
Expression of all PALB2 variants was examined by Western blot analysis as previously described7. Two different primary rabbit polyclonal antibodies directed against the N-terminus of human PALB2 (1:1,000, kindly provided by Cell Signalling Technology prior to commercialization) were used. Wild-type human PALB2 and empty vector (Ev) were used as controls on the blot, while Tubulin (1:10,000, Sigma, T6199 clone DM1A) was used as loading control. For protein stability and degradation assays, cells were treated with 100 μg/ml cycloheximide (Sigma, C7698-1G) for up to 3 hours, or 0.5 or 3 μM MG-132 (Selleckchem, S2619) for 24 hours, after which western blot samples were collected and analysed.
Laser micro-irradiation and PALB2 recruitment
U2OS cells were grown on 18-mm coverslips and sensitized with 10 µM 5′-bromo-2-deoxyuridine (BrdU) for 24 h before micro-irradiation. Cells were co-transfected with 1 µg YFP-PALB2, with or without a variant, and 0.5 µg mCherry-NBS1 expression vector using lipofectamine 2000 (Invitrogen). For micro-irradiation, cells were placed in a live-cell imaging chamber set to 37 °C in CO2-independent Leibovitz’s L15 medium supplemented with 10% FCS and penicillin–streptomycin (Invitrogen). Micro-irradiation experiments were carried out with a Zeiss Axio Observer microscope driven by ZEN software using a 63x/1.4 oil immersion objective coupled to a 355 nm pulsed DPSS UV-laser (Rapp OptoElectronic). To monitor the recruitment of YFP-PALB2 to laser-induced DNA damage sites, cells were imaged before and 360 s after laser irradiation. The fluorescence intensity of YFP-PALB2 and mCherry-NBS1 at DNA damage sites relative to that in an unirradiated region of the nucleus was quantified and plotted over time. Kinetic curves were obtained by averaging the relative fluorescence intensity of cells displaying positive recruitment (n > 30 cells per condition).
Cellular localization assay
Quantification of YFP-PALB2 subcellular localization was based on transient expression in U2OS cells that were fixed using 4% formaldehyde and permeabilized using Triton X-100. Cells were immunostained with anti-GFP and DAPI prior to immunofluorescence analysis and quantification (based on ~100 cells per condition per replicate). Images were acquired on a Zeiss AxioImager M2 wide-field fluorescence microscope with 63x PLAN APO (1.4 NA) oil-immersion objectives, running ZEN 2012 blue edition v1.1.0.0 (Zeiss). Assays were conducted in triplicate and average values and SEM were calculated to generate the respective plots.
Destabilization energy analysis for variants in the WD40 domain
Structural analysis of WD40 domain variants was performed using a previously reported X-ray crystal structure from the Protein Data Bank (https://www.rcsb.org/: 2W18)44. The structure was prepared using the FoldX RepairPDB command to identify and repair clashes and bad torsion angles in the side-chains, keeping the backbone of the structure fixed. Destabilization energies (kcal/mol) for variants identified with deep mutational scanning were computed using the FoldX BuildModel command, taking the average of triplicate runs20. Alternatively, a list of destabilization energies for variants the WD40 domain, which was prepared using a structure from the Alpha-fold Protein Structure Database (https://alphafold.ebi.ac.uk/: Q86YC2) in combination with the deep learning-based RaSP method21, was obtained from the Electronic Data Research Archive (https://sid.erda.dk/public/).
Cancer risk analysis
Associations between pooled PALB2 variants and breast cancer risk were assessed using two-sided Fisher’s Exact tests, based on data from breast cancer cases and age-matched female controls without cancer from the population-based BRIDGES, CARRIERS, and CZECANCA studies11,30,45. Association testing by logistic regression for pooled variants was also conducted using female breast cancer cases compared to non-cancer female controls from UK Biobank (adjusted for age and ancestry); for female breast cancer cases receiving cancer genetic testing by Ambry Genetics compared to the gnomAD v.4.1. non-UK Biobank female controls (adjusted for ancestry), and All of Us research program non-cancer female controls (adjusted for ancestry and for age of diagnosis of cases and age at enrolment for controls). Analyses were conducted for pooled functional, intermediate and damaging variants and also for variants in the middle region of PALB2 using Firth logistic regression, adjusting for age and ancestry and deriving P-values from the likelihood ratio of the Firth logistic regression. All variants with minor allele frequencies >0.001 were excluded to avoid dilution of associations by recurrent variants (Supplementary Data 1).
Data analysis
Missense, nonsense, and synonymous variants were selected by filtering prior to download. Variants were aligned with our functional dataset following the conversion of the variantId (based on genomic location) into HGVS-compliant variant descriptions using the Variant Validator tool available at: https://variantvalidator.org/service/validate. Throughout this study, NCBI transcript NM_024675.4 and NP_078951.2 were used to indicate nucleotide and amino acid positions in PALB2, respectively. Frequency data derived from gnomAD18, BRAVO, FLOSSIES and classification data from the ClinVar database were harmonized with the 7106 PALB2 variants for which depletion scores were available (Supplementary Data 2 and 3). For 57 codons, multiple nucleotide variants resulted in the same amino acid alteration (Supplementary Data 1). As data analysis and correlation with functional data were performed at the amino acid level, we merged the available information for distinct nucleotide variants, yielding a unified entry for a specific protein variant.
Pearson’s correlation coefficients (r) were used to assess the strength of linear relationships between variables. Positive correlations were classified as weak (r < 0.25), moderate (0.25 ≤ r < 0.50), good (0.50 ≤ r < 0.75) and very good (r ≥ 0.75), while negative correlations were classified as weak (r > -0.25), moderate (-0.25 ≥ r > -0.50), good (-0.50 ≥ r > -0.75) and very good (r ≤ -0.75). Statistical significance was determined using corresponding P-values.
ROC analysis of in silico prediction outcomes was performed using a set of missense variants that were individually tested via the DR-GFP assay in both the current study and previous work7. Variants exhibiting <30% HR activity were used as proxies for damaging variants, while those with >70% HR activity were considered functional. Although these thresholds are arbitrary, they provide a practical framework for comparative analysis in the absence of definitive clinical classifications for PALB2 missense variants. Based on these analysis, key performance metrics, including sensitivity, specificity, positive predictive value (PPV), negative predictive value (NPV), and overall accuracy, were calculated to evaluate the predictive power. Finally, 2 × 2 contingency tables were generated to compare the predictions of each in silico tool with the experimentally determined functional impact.
Regarding frequency data, we computed the mean frequency for nucleotide variants giving rise to identical protein variants. For ClinVar submissions, we joined the clinical significance classifications for variants and subsequently performed a majority voting approach to attribute the protein variant to a singular category (akin to handling variants with conflicting interpretations, outlined below). ClinVar employs the term “conflicting interpretations” to denote variants for which varying clinical significance classifications have been submitted. The presence of these “conflicting” variants added complexity to the breakdown of variant classes, as they would be tallied threefold (i.e., counted in VUS, (L)B, and the “conflicting” category). To resolve this, a majority voting strategy was applied, assigning each variant to either VUS, (L)B or (L)P, based on the most frequent classification (see Fig. 6a in Source Data file). Finally, splice site prediction was performed using SpliceAI version 1.3.1. (https://github.com/Illumina/SpliceAI)37. SpliceAI was run locally with maximum distance of 10k nucleotides (4999 nucleotides on either site from the variant of interest) with masked scores. SpliceAI cutpoints to predict spliceogenicity were based on the maximum delta score of donor/acceptor loss and donor/acceptor gain >0.5 for predicted splicing impact (Supplementary Data 4). All statistical analysis was performed using Graphpad Prism (version 10.2.3).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
In silico prediction scores for PALB2 missense variants were obtained for Helix27, AlphaMissense29, EVE26, and BayesDel (no allele frequencies (noAF))28. Variant frequency data were obtained from GnomAD v.2.1.1. (non-cancer; https://gnomad.broadinstitute.org/gene/ENSG00000083093?dataset=gnomad_r2_1_non_cancer)18, UK Biobank (access@ukbiobank.ac.uk), BRAVO (August 2, 2023; freeze 10; https://bravo.sph.umich.edu/gene.html?id=PALB2), FLOSSIES (June 8, 2023; https://whi.color.com/gene/ENSG00000083093), ClinVar (June 30, 2025; https://www.ncbi.nlm.nih.gov/clinvar/), BRIDGES30, CARRIERS11, and CZECANCA45. UK Biobank is a large-scale biomedical database and research resource containing genetic, lifestyle and health information from half a million UK participants. UK Biobank’s database, which includes blood samples, heart and brain scans and genetic data of the 500,000 volunteer participants, is globally accessible to approved researchers who are undertaking health-related research that’s in the public interest. UK Biobank recruited 500,000 people aged between 40–69 years in 2006–2010 from across the UK. With their consent, they provided detailed information about their lifestyle, physical measures and had blood, urine and saliva samples collected and stored for future analysis. Re-used with the permission of the NHS England and/or UK Biobank (application 65898 to F.J.C.). This research used data assets made available by National Safe Haven as part of the Data and Connectivity National Core Study, led by Health Data Research UK in partnership with the Office for National Statistics and funded by UK Research and Innovation (research which commenced between (March 31, 2023). Data used in this study were obtained from participants enrolled in the All of Us Research Program. Participant-provided information was accessed through the National Institutes of Health’s All of Us Research Program. All analyses were conducted in accordance with the program’s data use policies and ethical guidelines. The raw NGS data from the site-saturation functional screens and SGE have been deposited in the Sequence Read Archive (SRA, https://www.ncbi.nlm.nih.gov/sra/, accession nr. PRJNA1074372) and in the Gene Expression Omnibus (GEO, https://www.ncbi.nlm.nih.gov/geo/, accession nr. GSE255117), respectively. The remaining data are available within the Article, Supplementary Information, Supplementary Data or Source Data file. Source data are provided with this paper.
References
Yang, X. et al. Cancer risks associated with germline PALB2 pathogenic variants: an international study of 524 families. J. Clin. Oncol. 38, 674–685 (2020).
Brnich, S. E. et al. Recommendations for application of the functional evidence PS3/BS3 criterion using the ACMG/AMP sequence variant interpretation framework. Genome Med. 12, 3 (2019).
Richards, S. et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–424 (2015).
Nepomuceno, T. C. et al. The role of PALB2 in the DNA damage response and cancer predisposition. Int. J. Mol. Sci. 18, 1886 (2017).
Boonen, R., Vreeswijk, M. P. G. & van Attikum, H. Functional characterization of PALB2 variants of uncertain significance: toward cancer risk and therapy response prediction. Front. Mol. Biosci. 7, 169 (2020).
Nepomuceno, T. C. et al. PALB2 variants: protein domains and cancer susceptibility. Trends Cancer 7, 188–197 (2021).
Boonen, R. et al. Functional analysis of genetic variants in the high-risk breast cancer susceptibility gene PALB2. Nat. Commun. 10, 5296 (2019).
Rodrigue, A. et al. A global functional analysis of missense mutations reveals two major hotspots in the PALB2 tumor suppressor. Nucleic Acids Res. 47, 10662–10677 (2019).
Wiltshire, T. et al. Functional characterization of 84 PALB2 variants of uncertain significance. Genet. Med. 22, 622–632 (2020).
Breast Cancer Association, C. et al. Breast cancer risk genes—association analysis in more than 113,000 women. N. Engl. J. Med. 384, 428–439 (2021).
Hu, C. et al. A population-based study of genes previously implicated in breast cancer. N. Engl. J. Med. 384, 440–451 (2021).
Bleuyard, J. Y., Buisson, R., Masson, J. Y. & Esashi, F. ChAM, a novel motif that mediates PALB2 intrinsic chromatin binding and facilitates DNA repair. EMBO Rep. 13, 135–141 (2012).
Rubin, A. F. et al. A statistical framework for analyzing deep mutational scanning data. Genome Biol. 18, 150 (2017).
Rubin, A. F. et al. Correction to: A statistical framework for analyzing deep mutational scanning data. Genome Biol. 19, 17 (2018).
Ng, P. S. et al. Characterisation of protein-truncating and missense variants in PALB2 in 15 768 women from Malaysia and Singapore. J. Med. Genet. 59, 481–491 (2021).
Starita, L. M. et al. Variant interpretation: functional assays to the rescue. Am. J. Hum. Genet. 101, 315–325 (2017).
Blomen, V. A. et al. Gene essentiality and synthetic lethality in haploid human cells. Science 350, 1092–1096 (2015).
Karczewski, K. J. et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581, 434–443 (2020).
Foo, T. K. et al. Compromised BRCA1-PALB2 interaction is associated with breast cancer risk. Oncogene 36, 4161–4170 (2017).
Schymkowitz, J. et al. The FoldX web server: an online force field. Nucleic Acids Res. 33, W382–W388 (2005).
Blaabjerg, L. M. et al. Rapid protein stability prediction using deep learning representations. Elife 12, e82593 (2023).
Adzhubei, I. A. et al. A method and server for predicting damaging missense mutations. Nat. Methods 7, 248–249 (2010).
Sim, N. L. et al. SIFT web server: predicting effects of amino acid substitutions on proteins. Nucleic Acids Res. 40, W452–W457 (2012).
Tavtigian, S. V., Samollow, P. B., de Silva, D. & Thomas, A. An analysis of unclassified missense substitutions in human BRCA1. Fam. Cancer 5, 77–88 (2006).
Ioannidis, N. M. et al. REVEL: an ensemble method for predicting the pathogenicity of rare missense variants. Am. J. Hum. Genet. 99, 877–885 (2016).
Frazer, J. et al. Disease variant prediction with deep generative models of evolutionary data. Nature 599, 91–95 (2021).
Vroling, B. & Heijl, S. White paper: The Helix Pathogenicity Prediction Platform. https://arxiv.org/abs/2104.01033 (2021).
Feng, B. J. PERCH: a unified framework for disease gene prioritization. Hum. Mutat. 38, 243–251 (2017).
Cheng, J. et al. Accurate proteome-wide missense variant effect prediction with AlphaMissense. Science 381, eadg7492 (2023).
Dorling, L. et al. Breast cancer risks associated with missense variants in breast cancer susceptibility genes. Genome Med. 14, 51 (2022).
Findlay, G. M. et al. Accurate classification of BRCA1 variants with saturation genome editing. Nature 562, 217–222 (2018).
Starita, L. M. et al. A multiplex homology-directed DNA repair assay reveals the impact of more than 1,000 BRCA1 missense substitution variants on protein function. Am. J. Hum. Genet. 103, 498–508 (2018).
Ikegami, M. et al. High-throughput functional evaluation of BRCA2 variants of unknown significance. Nat. Commun. 11, 2573 (2020).
Li, H. et al. Functional annotation of variants of the BRCA2 gene via locally haploid human pluripotent stem cells. Nat. Biomed. Eng. 8, 165–176 (2024).
Huang, H. et al. Functional evaluation and clinical classification of BRCA2 variants. Nature 638, 528–537 (2025).
Sahu, S. et al. Saturation genome editing-based clinical classification of BRCA2 variants. Nature 638, 538–545 (2025).
Jaganathan, K. et al. Predicting splicing from primary sequence with deep learning. Cell 176, 535–548.e524 (2019).
Ducy, M. et al. The tumor suppressor PALB2: inside out. Trends Biochem. Sci. 44, 226–240 (2019).
Sidorova, J. A game of substrates: replication fork remodeling and its roles in genome stability and chemo-resistance. Cell Stress 1, 115–133 (2017).
Boonen, R. et al. Functional analysis identifies damaging CHEK2 missense variants associated with increased cancer risk. Cancer Res. 82, 615–631 (2022).
Bouwman, P. et al. A high-throughput functional complementation assay for classification of BRCA1 missense variants. Cancer Discov. 3, 1142–1155 (2013).
Kranz, A. et al. An improved Flp deleter mouse in C57Bl/6 based on Flpo recombinase. Genesis 48, 512–520 (2010).
DeLong, E. R., DeLong, D. M. & Clarke-Pearson, D. L. Comparing the areas under two or more correlated receiver operating characteristic curves: a nonparametric approach. Biometrics 44, 837–845 (1988).
Oliver, A. W., Swift, S., Lord, C. J., Ashworth, A. & Pearl, L. H. Structural basis for recruitment of BRCA2 by PALB2. EMBO Rep. 10, 990–996 (2009).
Soukupova, J. et al. Validation of CZECANCA (CZEch CAncer paNel for Clinical Application) for targeted NGS-based analysis of hereditary cancer syndromes. PLoS ONE 13, e0195761 (2018).
Acknowledgements
The authors thank Jos Jonkers and Peter Bouwman for providing the pTT5-Puro (RMCE acceptor cassette), pRNA-251-MCS-RMCE (RMCE exchange vector) and pCMV-Red-I-SceI constructs, Maria Jasin and Francis Stewart for sharing the DR-GFP reporter and FlpO constructs, Cell Signaling for providing antibodies directed against PALB2 prior to commercialization, and Noel de Miranda, Robin van Schendel, Diana van den Heuvel, Michael Parsons and Thom Hartog for help with data analysis. This research has been conducted using the UK Biobank Resource under application number 65898 with help from Nicholas Boddicker who initiated breast cancer case control analyses performed by F.J.C. and C.H. The CZECANCA consortium was financially supported by grants from the Ministry of Health of the Czech Republic (DRO-VFN-64165 and LM2023067), the Charles University (COOPERATIO, SVV260631, UNCE/24/MED/022), and the Ministry of Education Youth and Sports of the Czech Republic (EXCELES program, project LX22NPO5102, funded by the European Union—Next Generation EU). This work was furthermore financially supported by the P30 CA008748 grant (MSKCC), the Niehaus Center for Inherited Cancer Genomics (to J.V.), the Breast Cancer Research Foundation (to J.V. and F.J.C.), NIH grants (R35 CA253197 and R01 CA225662 to F.J.C.), the European Union’s Horizon 2020 Research and Innovation Programme (BRIDGES, grant number 634935, M.P.G.V. and H.v.A.), and grants from the Dutch Cancer Society (grant number 7473 to H.v.A., and grant number 12754 to M.P.G.V. and H.v.A.).
Author information
Authors and Affiliations
Consortia
Contributions
R.A.C.M.B. introduced PALB2 variants and variant libraries in the RMCE exchange construct, and performed the site-saturation functional screens, PARPi survival assays, DR-GFP reporter assays and western blot analysis in mES cells. S.C.K. introduced PALB2 variants, and performed the site-saturation functional screens, PARPi survival assays, DR-GFP reporter assays, co-immunoprecipitation experiments, cellular localisation assays and western blot analysis in mES and U2OS cells. R.M. analysed all NGS data and generated functional maps for the site-saturation functional screens with help from D.R. and S.L.K. M.E.B. and M.P.G.V. performed population database-related analysis. M.B.R. studied YFP-PALB2 recruitment to laser-induced DNA damage. P.K., M.J., C.H., V.J., M.C., M.E.R. and F.J.C. performed the cancer risk association analysis with F.J.C. and C.H. using data obtained under UKBB application 65898. M.E.R. provided the clinical cohort data. S.H. and B.V. provided in silico predictions of functional variant effects. J.G. performed mixture modelling analysis. M.V. provided in silico predictions of protein destabilization effects with help from M.E.R. H.H. and F.J.C. performed SGE. H.v.A. conceived and supervised the project. H.v.A. wrote the paper, prepared figures and performed data analysis with help from R.A.C.M.B. and M.P.G.V.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Sara Gutierrez-Enriquez, who co-reviewed with Joanna Domènech-Vivó, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Boonen, R.A., Knaup, S.C., Menafra, R. et al. Site-saturation functional screens identify PALB2 missense variants associated with increased breast cancer risk. Nat Commun 17, 775 (2026). https://doi.org/10.1038/s41467-025-67252-z
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-67252-z
