
Abstract
Subsurface regions critically govern surface events, such as the interactions with reactants in heterogeneous catalysis, thereby significantly modulating catalytic performance. However, precise control of subsurface atomic arrangement remains challenging due to complex metal-adsorbate interactions and limited structural accessibility. Here we achieve precise control of subsurface atomic layer in platinum-based intermetallic compounds through targeted positioning of heterometallic atoms to subsurface via in-situ constructed atomic diffusion pathways. This site-specific placement and subsequent thermodynamic-induced atomic rearrangement are governed by surface energy minimization and adsorbate-induced segregation. Through atomic-precision subsurface engineering, we successfully synthesize a series of L10 (face-centered tetragonal, fct)-PtFe@PtMsub, where Msub represents heteroatoms (Ru, Rh, Pd, Ag) incorporated into subsurface layers. As demonstrated, the as-synthesized L10-PtFe@PtPdsub simultaneously stabilizes ligand and strain effects, thereby breaking the trade-off in L10-PtM with Pt skin, where Pt skin typically quenches ligand effects while introducing strain effects. Consequently, L10-PtFe@PtPdsub/C catalyst demonstrates practical proton exchange membrane fuel cells performance, simultaneously delivering high activity and durability. This work provides a rational strategy for catalyst design that promotes the understanding of subsurface active sites in heterogeneous catalysis.
Similar content being viewed by others

Introduction
The catalytic performance of a heterogeneous catalyst is predominantly determined by its surface and near-surface atomic architecture, which dictates both geometric configurations and electronic environments1,2. Surface modifications, including surface doping3,4, small molecule functionalization5,6,7, and surface encapsulation8,9,10, are widely utilized to enhance catalytic performance in various industrial reactions3,5,7,8. However, surface structures directly involved in catalytic reactions frequently undergo irreversible reconstruction, leading to severe degradation of precisely engineered surface architectures11,12,13,14. Taking these into account, subsurface engineering has emerged as a robust alternative by leveraging electronically active yet structurally protected subsurface sites2,15,16,17.
Subsurface engineering targets the manipulation of 2–3 atomic layers beneath a material’s surface, which have been demonstrated to play a critical role in surface events—particularly in heterogeneous catalysis—through strain and ligand effects18. Motivated by this promise, various strategies have been explored to regulate subsurface composition, including: (i) bottom-up construction in bulk model catalysts, such as chemical vapor deposition19, electrodeposition20, and molecular beam epitaxy (MBE)15,21, (ii) in-situ incorporation of non-metallic species on catalysts under reaction conditions22, exemplified by subsurface metal carburization18. Nevertheless, directly extending these bottom-up construction protocols, primarily for bulk materials, to nanocatalysts remains a challenge. Self-limited synthesis techniques, represented by atomic layer deposition (ALD), exhibit promising capabilities for the atomic-precision construction of subsurface in nanocatalysts23,24. However, the ALD synthesis technique suffers from limitations such as the lack of general applicability and challenges in achieving scalable batch fabrication25. Given these constraints, developing conventional wet-chemical synthesis methods—advantageous for their simplicity and scalability—to achieve the precise subsurface tailoring is urgently needed, while still hindered by embedded atomic sites and complex diffusion dynamics.
In this work, Pt-based intermetallic compounds (IMCs) featuring Pt skins integrate well-defined atomic structures and superior catalytic functionalities, providing an ideal platform for atomic-scale subsurface engineering and corresponding structure-activity relationship studies. We achieved atomic-precision subsurface engineering of L10-PtFe@Pt by targeted placement of heterometallic atoms via a simple liquid phase synthesis approach. This process involves in-situ construction of atomic diffusion pathways, followed by energy-minimized atomic rearrangement governed by surface thermodynamics and metal-adsorbate interaction. Based on this, we successfully synthesized L10-PtFe@PtMsub, where Msub represents in-situ incorporated subsurface heterometallic atoms, including Ru, Rh, Pd, and Ag. Specifically, these heteroatoms occupy the Fe vacancies generated via galvanic replacement and acid leaching26,27, with subsequent atomic rearrangement stabilizing these subsurface sites. The as-synthesized L10-PtFe@PtPdsub/C demonstrates that subsurface engineering stabilized ligand and strain effects, breaking ligand-strain effects trade-off—as the Pt-skin typically quenches spatially dependent ligand effect while generating strain effect. As a result, the L10-PtFe@PtPdsub/C catalyst exhibits practical catalytic potential in proton exchange membrane fuel cells (PEMFCs), overcoming activity-durability trade-off in oxygen reduction reaction (ORR) through Pdsub atoms modulating electronic environments of active sites on the surface and stabilizing metals against leaching. In a membrane electrode assembly (MEA), the L10-PtFe@PtPdsub/C catalyst delivers a high initial mass activity (MA) of 1.17 A mgPt−1 at 0.9 V and a rated power density of 0.88 W cm−2, surpassing that of commercial Pt/C. Moreover, it exhibits a MA retention of 88.1%, and a potential loss of only 15 mV at 0.8 A cm−2 and a rated power loss of 8% after 30,000 cycles of accelerated stress testing, exceeding the U.S. DOE 2025 technical targets.
Results and discussion
Subsurface atomic site modulation engineering
Highly ordered IMCs, characterized by a fully ordered heterogeneous atomic arrangement, have emerged as promising catalysts. Pt-based IMCs with Pt skin, possessing structural regularity, enable the implementation of a precise subsurface atomic regulation strategy. In this study, carbon-supported highly ordered L10-PtFe nanoparticles served as the initial templates. Subsurface lattice vacancies generated by the cooperative effects of galvanic replacement and acid leaching facilitate the in-situ incorporation of foreign metals26,27. Subsequent thermal-driven atomic diffusion and rearrangement enable the targeted incorporation of heteroatoms into the subsurface, ultimately leading to a stabilized subsurface structure (Fig. 1a).

a Synthesis scheme for in-situ subsurface atomic site modulation strategy by conventional solution-phase synthesis. b Left: the redox potential-driven doping mechanism leads to selective doping, including self-nucleation, (sub)surface-modification, and no surface modification. Right: the lattice site occupancy of heterometallic atoms governed by key factors, including surface energy and metal-adsorbate interactions. The calculated surface energies of surface and subsurface sites were compared. Source data for the calculated surface energies are provided as a Source Data file.
To elucidate the underlying chemical mechanisms of subsurface engineering, we systematically investigated the incorporation behavior of various metals (Fig. 1b). The results reveal that the feasibility of incorporating foreign metals into the L10-PtFe nanoparticles is governed by the relative magnitudes of their reduction potentials \(({E}_{{{{{\rm{M}}}}}^{{{{\rm{n}}}}+}}/{{{\rm{M}}}})\) compared to that of Fe \(({E}_{{{{{\rm{Fe}}}}}^{3+}}/{{{\rm{Fe}}}})\)28,29. X-ray photoelectron spectroscopy (XPS) analysis confirms that the leached Fe species are primarily as Fe3+, due to the weakly oxidizing acidic reaction conditions (Supplementary Fig. 1). For instance, 3d transition metals such as Zn, Co, and Ni, which exhibit lower redox potentials than Fe (EM < EFe), are not incorporated into the nanoparticles due to their inability to replace Fe (lower-left in Fig. 1b). In contrast, metals with significantly higher redox potentials, such as Au, exhibit spontaneous nucleation and growth adjacent to the PtFe nanoparticles, rather than being integrated into the host structure (upper-left in Fig. 1b). To further investigate this behavior, we examined the elemental distribution of PtFe nanoparticles followed by Au introduction. The high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) image and energy-dispersive X-ray spectroscopy (EDS) spectra reveal the formation of discrete, large nanoparticles with brighter contrast, which are predominantly composed of Au (Supplementary Fig. 2a, b). In contrast, the smaller PtFe nanoparticles show negligible Au content (Supplementary Fig. 2c). Furthermore, we systematically altered key reaction parameters—including lowering Au precursor concentration, reducing acid reaction temperature, and extending annealing—and found that Au invariably undergoes spontaneous nucleation and growth in all resulting samples (Supplementary Fig. 3). These observations confirm that Au tends to undergo spontaneous nucleation rather than incorporate into the PtFe lattice.
For other metals such as Mo, W, Ru, Rh, Pd, and Ag, which possess moderate redox potentials slightly higher than that of Fe, successful incorporation into the PtFe nanoparticles was achieved (center-left in Fig. 1b). The spatial distribution of these heteroatoms within the nanoparticles is subsequently governed by thermodynamic factors during the annealing process, particularly surface energy considerations and the metals’ affinity for adsorbates (right in Fig. 1b)30. Among these, Mo and W exhibit the difficulty in reducing to metallic states and their strong affinities for oxygen31,32, leading to the formation of Mo- and W-rich oxide clusters on the nanoparticle surface. As evidenced by HAADF-STEM images and EDS mappings (Supplementary Fig. 4), the intensity profiles of the mixed elemental mapping images show that Mo and W signals initiate further outward or overlap with Pt, confirming the surface localization of these oxide clusters. To understand the surface energy factor, we calculated the surface energy (Esurf) for various heterometals capable of incorporating into the host PtFe@Pt lattice. The Esurf for surface-alloyed (PtMsur) and subsurface-alloyed (PtMsub) configurations was compared to determine the preferential site occupancy of each element (right in Fig. 1b). The Esurf of pure Pt was determined to be 2.32 J m−2. Upon alloying with Ru, Rh, and Pd, the Esurf of the corresponding PtMsur structures were calculated to be 2.61, 2.45, and 2.31 J m−2, respectively. Notably, these values are consistently higher than those of their subsurface-alloyed structures (2.32, 2.30, and 2.28 J m−2 for PtRusub, PtRhsub, and PtPdsub, respectively). These results strongly indicate that, from a thermodynamic perspective, the subsurface incorporation of Ru, Rh, and Pd confers greater structural stability33. Although Ag exhibits a lower Esurf in the surface-alloyed configuration (2.34 J m−2) than that of PtAgsub configuration (2.37 J m−2), its preferential migration to subsurface layers is ultimately driven by its weak interactions with hydrogen adsorbates under reductive annealing conditions. We summarize the redox potentials, calculated surface energies, and resulting doping behaviors of all heterometals in Table S2.
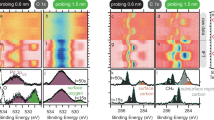
To experimentally confirm the distribution of M in the subsurface layer, we performed EDS elemental mapping and depth-resolved XPS to probe the surface and subsurface structures of PtFe@PtMsub nanostructures. First, the nanoscale distribution of M was confirmed by HAADF-STEM and EDS elemental mapping, with the intensity profile of mixed elemental mapping revealing the presence of M peaks located between those of Pt and Fe (Fig. 2a–d). Moreover, the depth-resolved XPS was performed using photons-energy-dependent excitation (Fig. 2e–g). The Pt 4f and M 3d core-level signals were acquired under identical kinetic energy conditions, thereby maintaining a constant information depth. At a kinetic energy of 140 eV, corresponding to a surface-sensitive probing regime, distinct Pt signals were observed, while no discernible signals from the M elements (M = Ag, Pd, Rh) were detected. As the kinetic energy increased to 170, 270, 370, and 470 eV, resulting in a greater probing depth, the intensities of M 3d peaks increase progressively from the background level (Fig. 2e–g). Furthermore, the M atomic concentration quantified by XPS was normalized to the value obtained at a kinetic energy of 470 eV (set as unity) to analyze its depth-dependent variation. As shown in Fig. 2h, the resulting profile rises rapidly from zero and then remains constant, indicating that M is located exclusively in subsurface sites, with no distribution on the surface or in the core regions. For Rusub, due to significant overlap between the Ru 3d5/2 (280.2 eV) and C 1s (284.6 eV) peaks, and further compounded by the high C content and low Ru concentration in the L10-PtFe@PtRu/C, the strong C 1s signal obscured the Ru 3d region, precluding reliable quantification. Moreover, the Ru 3p signal was too weak to be detected. To address this limitation and further support our findings, we supplemented more STEM-EDS results on L10-PtFe@PtRusub nanoparticles, thereby strengthening the overall robustness of the dataset (Supplementary Fig. 5). These results suggested that the M elements are predominantly located in the subsurface region.

HAADF-STEM image and corresponding EDS mapping of individual L10-PtFe@PtRu NP (a), L10-PtFe@PtRh NP (b), L10-PtFe@PtPd NP (c), L10-PtFe@PtAg NP (d). The corresponding intensity profile of mixed-element mapping is shown to the right of the elemental mapping image. Scale bar, 2 nm. e–g XPS spectra of M 3d, Pt 4 f in L10-PtFe@PtRhsub/C, L10-PtFe@PtRdsub/C, and L10-PtFe@PtAgsub/C excited by photons with different energies. h Depth profile of M concentration in L10-PtFe@PtMsub/C, with the concentration normalized to the value at a kinetic energy of 470 eV. Source data for the XPS are provided as a Source Data file.
The role of Fe vacancies in facilitating the subsurface migration of M elements was further investigated, with L10-PtFe@PtPdsub serving as a model system. Through controlling the acid etching time to modulate Fe vacancy density, we conducted extensive statistical analysis on a large ensemble of nanoparticles to investigate the heteroatom Pd content and its lattice site occupancy. The Fe vacancy density is quantitatively defined as the deviation from the ideal 1:1 stoichiometry of the L10-PtFe. Specifically, it is calculated as 50 at% minus the experimentally measured atomic percentage of Fe (at%) by STEM-EDS. Firstly, the EDS results reveal that Pd doping was unachievable in the absence of Fe vacancies (Supplementary Fig. 6a), confirming that the presence of Fe vacancies is an essential prerequisite for the incorporation of Pd atoms into the nanoparticles. As the Fe vacancy density increased, a positive correlation with the Pd incorporation efficiency was observed (Supplementary Fig. 6e). When Fe vacancies were confined exclusively to the nanoparticle surface, the insufficient vacancy concentration significantly reduced the kinetic driving force for inward diffusion, resulting in the preferential localization of Pd atoms within the outermost atomic layer (Supplementary Fig. 6b). Only when the Fe vacancy density was increased within approximately the first three surface layers, Pd could incorporate into subsurface sites (Supplementary Fig. 6c). In addition, it was observed that even with sufficient Fe vacancies, excessive Pd content could lead to the surface segregation of Pd atoms (Supplementary Fig. 6d). These results collectively demonstrate that sufficient Fe vacancy density is critical for enabling effective diffusion and subsequent stabilization of heterogeneous Pd atoms within subsurface layers.
In summary, we employed redox potential, surface energy, and metal-adsorbate interaction as key descriptors to systematically investigate the behavior of various heterometallic atoms during subsurface engineering. Guided by this understanding, we successfully synthesized L10-PtFe@PtMsub, where Msub represents the incorporation of foreign metals, including Ru, Rh, Pd, and Ag. We further extended this strategy to L12-Pt3Fe nanoparticles, demonstrating its general applicability across different ordered phases by using representative heterometallic atoms such as spontaneously-nucleated Au, subsurface-incorporated Pd and Ag, and surface-decorated WOx (Supplementary Fig. 7). The successful implementation of this strategy in L10-PtFe and L12-Pt3Fe demonstrates its general applicability and potential for expanding the range of metal systems, offering an efficient and versatile platform for precise modulation of the electronic environment at surface active sites.
Effects of subsurface engineering on structure
As a proof of concept, we demonstrated that subsurface engineering influenced the geometric and electronic structure of surface active sites and the practical catalytic potential of the L10-PtFe@PtPdsub/C catalyst for the ORR in PEMFCs (Fig. 3a). As elaborated above, we constructed thermodynamically favorable subsurface L10-PtFe@PtPdsub configuration by incorporating Pd atoms into the in-situ generated Fe vacancies followed by thermal annealing. Furthermore, DFT calculations reveal a low kinetic barrier of 1.12 eV for the migration of surface Pd atoms into subsurface sites, which can be readily surmounted by our synthetic annealing conditions (Fig. 3b). The result indicates that the subsurface incorporation of Pd is kinetically accessible.

a Schematic illustration of the subsurface atomic modulation strategy for L10-PtFe@PtPdsub nanocatalysts. b Calculated energy analysis of surface Pd atoms migrating into the subsurface layer. c XRD patterns of L10-PtFe/C, L10-PtFe@Pt/C, and L10-PtFe@PtPdsub/C catalysts. The standard peaks of L10-PtFe are also shown. d Aberration-corrected HAADF-STEM image of a L10-PtFe@PtPdsub NP and the line profile of the marked region in the HAADF-STEM image. Scale bar, 1.0 nm. e HAADF-STEM image and corresponding atomic EDS elemental mapping of L10-PtFe@PtPdsub NP. Scale bar, 0.5 nm. f Enlarged and rotated view of (d) for clear visualization of the atomic strain analysis. scale bar, 0.5 nm. The colored squares correspond to different strain values. g Pt 4 f XPS spectra of L10-PtFe/C, L10-PtFe@Pt/C, and L10-PtFe@PtPdsub/C. h Pt L3-edge XANES spectra of L10-PtFe@Pt/C, L10-PtFe@PtPdsub/C, and Pt foil. i Fourier transformed X-ray absorption fine structure k3-weighted χ(R) function spectra of L10-PtFe@Pt/C, L10-PtFe@PtPdsub/C, and Pt foil. The dotted lines indicate the fitting results. Source data are provided as a Source Data file.
Following the initial experimental investigations, we conducted a detailed characterization of the microstructures of the L10-PtFe/C, L10-PtFe@Pt/C, and L10-PtFe@PtPdsub/C catalysts. X-ray diffraction (XRD) patterns in Fig. 3c showed that the characteristic (001) and (110) peaks of the L10-PtFe/C, L10-PtFe@Pt/C, and L10-PtFe@PtPdsub/C matched well with the ordered L10-PtFe intermetallic phase. The ordering degree of fresh L10-PtFe/C samples was calculated to be approximately 71.85%, determined by the ratio of the peak intensity of the (110) superlattice peak to the sum of the (111) and (200) peaks in XRD pattern34, and neither acid-etching nor Pdsub-doping treatments significantly reduced the ordering degree (Supplementary Fig. 8). Additionally, the as-synthesized L10-PtFe/C samples exhibited a uniform NP size distribution, as shown in the HAADF-STEM images (Supplementary Fig. 9). L10-PtFe@Pt and L10-PtFe@PtPdsub nanoparticles were uniformly dispersed on the carbon supports with an average particle size of approximately 3 nm, which is similar to that of the fresh L10-PtFe/C samples (Supplementary Figs. 10 and 11). In addition, atomic HAADF imaging revealed that the Pt and Fe atomic columns in the L10-PtFe are arranged in a periodic order (Supplementary Fig. 12). In the case of L10-PtFe@Pt, approximately three Pt layers covering the ordered intermetallic core can be observed (Supplementary Fig. 13). The HAADF-STEM image of the individual ordered PtFe@PtPdsub NP is viewed along the [100] zone axis and the corresponding fast Fourier transform pattern shows the presence of the (001) superlattice spots (Fig. 3c and Supplementary Fig. 14). Intensity profiles extracted from the shell layer reveal the presence of weaker intensity columns within the otherwise uniform intensity atom columns, suggesting that Pd atoms may occupy some Pt sites (Fig. 3d). Furthermore, EDS elemental mappings were conducted to analyze the elemental distribution of the as-synthesized catalysts (Supplementary Fig. 15–17). The composition of L10-PtFe/C, L10-PtFe@Pt/C and L10-PtFe@PtPdsub/C was quantitatively analyzed by EDS, and the results were consistent with those obtained by inductively coupled plasma-mass spectrometry (ICP-MS), where the atomic ratio of Pt/Fe/Pd components was about 23:12.6:1 (Table S3).
To investigate in detail the distribution of Pd atoms in the surface layer, we performed atomic elemental mapping. It is more intuitively apparent in the atomic-level EDS mappings that Pd is mainly located in the subsurface layer, which is also confirmed by EDS spectra acquired from the Pt shell and PtFe core regions (Fig. 3e and Supplementary Fig. 18). To further confirm the presence of Pd in the subsurface layer, we conducted CO-adsorption diffuse reflectance infrared Fourier transform spectroscopy (CO-DRIFTS) measurements. Since the carbon supports are black in color and exhibit strong light absorption, thus negatively impacting the signal-to-noise ratio and the accuracy of the results35. To address this issue, we replaced the carbon supports with light-colored irreducible supports, SBA-15, and confirmed the consistent structure of the L10-PtFe@Pt and L10-PtFe@PtPdsub nanoparticles on both supports by XRD and STEM measurements (Supplementary Figs. 19–22). There was one peak located at 2063 cm−1 on L10-PtFe@Pt/SBA-15 assigned to linear CO on Pt, which exhibited a slight red shift to 2062 cm−1 after incorporation of Pd, suggesting a modification in the electronic structure of Pt36,37. Notably, no new adsorption peaks emerged following Pd doping, indicating that the atomic structures of the surfaces of the two samples are comparable (Supplementary Fig. 23). In summary, Pd is predominantly located in the subsurface layer of the nanoparticles, which can be supported by experimental evidences.
Effects of subsurface engineering on electronic structure
Since the atomic radius of Pd is only marginally smaller than that of Pt (Pt: 139 pm, Pd: 137 pm), the strain alteration induced by the substitution of Pd for Pt sites should be minimal, which is proved by atomic-level strain analysis (Fig. 3f and Supplementary Fig. 24). The coordinate positions of each atomic column were determined using StatSTEM software38, followed by measuring the spacing between the (110) planes. By comparing this spacing with the theoretical value for pure Pt, we derived the strain mapping image. Furthermore, XPS was conducted to analyze the electronic structure of the samples. As shown in Fig. 3g, the Pt 4f5/2 and Pt 4f7/2 spectra of L10-PtFe/C, L10-PtFe@Pt/C and L10-PtFe@PtPdsub/C were deconstructed into two peaks assigned to Pt0 and Pt2+, with Pt0 peak exhibiting larger peak area, indicating that Pt is predominantly in the metallic state. The binding energy of the Pt 4f7/2 peak in L10-PtFe/C (71.89 eV) is positively shifted by 0.10 eV compared to that of commercial Pt/C (71.79 eV), suggesting that the alloying between Pt and Fe has altered the electronic structure of Pt (Supplementary Fig. 25)39. Acid etching to form the Pt shell layer results in a negative shift of 0.09 eV in the binding energy of Pt 4f7/2 compared to L10-PtFe/C. Additionally, the binding energy of the Pt 4f7/2 peak in L10-PtFe@PtPdsub/C (71.75 eV) exhibits a slight negative shift of 0.05 eV relative to that of L10-PtFe@Pt/C, proving that Pdsub effectively transfers electrons to Pt, thereby regulating the electronic environment of Pt. In X-ray absorption near-edge structure (XANES) spectra, the oxidation state of Pt can be determined by analyzing the white-line peak intensity at the Pt L3-edge as it relates to the probability of electronic transition from the 2p orbital to the unoccupied 5 d orbitals40. As shown in Fig. 3h, the white-line peak intensity of L10-PtFe@PtPdsub/C is lower than that of L10-PtFe@Pt/C, indicating that the lower valence state of Pt is due to the extra electron transfer from Pd to Pt, in accordance with the XPS results. This indicates that the presence of Pdsub can alter the electronic structure of Pt, thereby exerting a ligand effect.
We further revealed the bonding information of the samples by analyzing the Fourier transforms extended X-ray absorption fine structure (FT-EXAFS)41. The FT-EXAFS spectra in R-space of Pt L3-edge (Fig. 3i) show that the Pt-M coordination peaks of the samples are within the range of 2–3 Å. Compared to Pt foil, the Pt-Pt bond lengths of the studied catalysts were shortened by Pt-M (M = Fe, Pd) alloying, which could potentially enhance the ORR activity. Furthermore, the EXAFS spectra were subjected to a fit in order to infer local atomic coordination structural information (Fig. 3i and Supplementary Fig. 26). The Pt–Pt bond with an average coordination number of 5.7 in L10-PtFe@PtPdsub/C exhibits a slight reduction in coordination compared to that of L10-PtFe@Pt/C (6.3), which confirms the successful doping of Pdsub (Table S4). The L10-PtFe@Pt/C and L10-PtFe@PtPdsub/C exhibited similar peak positions corresponding to the Pt-M bonds, suggesting that the effect of Pdsub on the strain effect is negligible. The wavelet transforms of Pt L-edge EXAFS results in Supplementary Fig. 27 show that the k-space maxima of L10-PtFe@Pt/C and L10-PtFe@PtPdsub/C are located at 7.0 Å−1 and 7.2 Å−1, respectively, which are lower than that of the Pt foil (9.7 Å−1), suggesting shortened Pt–M bonds in the studied catalysts. In addition, analysis of the Fe K-edge XAS data shows that the valence state and bond length of Fe remained unchanged (Supplementary Fig. 28). This indicates that the presence of Pdsub exerted minimal effect on the Fe within the core structure. These results suggest that the incorporation of subsurface Pd successfully modulates the electronic environment of Pt without affecting the original compressive strain.
ORR performance studies
The electrocatalytic performance of L10-PtFe@PtPdsub/C catalyst for ORR was investigated by using rotating disk electrode (RDE) and MEA techniques. For comparison, undoped L10-PtFe@Pt/C and commercial Pt/C (20 wt%, TKK) were also tested under the same conditions for comparison. Figure 4a displays the RDE polarization curves of L10-PtFe@PtPdsub/C, L10-PtFe@Pt/C and commercial Pt/C catalysts in O2-saturated 0.1 M HClO4 solution. Among these catalysts, L10-PtFe@PtPdsub/C exhibited the most positive half-wave potential (E1/2) of 0.94 V versus reversible hydrogen electrode (RHE). The MA of L10-PtFe@PtPdsub/C at 0.9 V versus RHE was calculated to be 4.04 ± 0.17 A mgPt−1, which is higher than 1.51 ± 0.04 A mgPt−1 for L10-PtFe@Pt/C, and 0.30 ± 0.06 A mgPt−1 for commercial Pt/C (Fig. 4b). The electrochemical surface areas (ECSAs) of the studied catalysts were quantified by the area of underpotential deposition of the hydrogen region (HUPD). The specific activity (SA) of L10-PtFe@PtPdsub/C, L10-PtFe@Pt/C, and commercial Pt/C catalysts was accordingly calculated to be 4.87 ± 0.24, 1.98 ± 0.05, and 0.48 ± 0.10 mA cmPt−2, respectively (Fig. 4b). Among current research, there is a consensus that the performance of PtM@Pt is superior to that of Pt, which is attributed to the strain regulation42. Furthermore, the presence of Pdsub has nearly quadrupled the activity of L10-PtFe@PtPdsub/C compared to L10-PtFe@Pt/C, strongly confirming that the ligand effect introduced by Pdsub significantly enhances ORR activity. Accelerated durability tests (ADTs) were conducted in O2-saturated 0.1 M HClO4 solution between 0.6 and 1.0 V versus RHE at a sweep rate of 100 mV s−1 to evaluate the durability of the catalysts. After 30,000 ADT cycles, L10-PtFe@Pt/C exhibited a significant loss of activity, with decreases of 21.85% in MA and 8.22% in ECSA (Supplementary Fig. 29). In contrast, L10-PtFe@PtPdsub/C demonstrated markedly enhanced stability, retaining 95.79% of its MA and 99.21% of its ECSA after 30,000 ADT cycles. Remarkably, L10-PtFe@PtPdsub/C maintained 88.86% of its initial MA and 96.11% of its initial ECSA even after 70,000 ADT cycles, highlighting its exceptional durability (Fig. 4c, d and Supplementary Fig. 30). Additionally, we conducted CO poisoning resistance tests on both samples, as this is a key metric of practical acidic ORR performance. The peak potential of CO stripping on L10-PtFe@PtPdsub/C (852 mV) was smaller than that of L10-PtFe@Pt/C (881 mV) catalyst, verifying that L10-PtFe@PtPdsub/C exhibited robust CO tolerance exceeding that of L10-PtFe@Pt/C (Supplementary Fig. 31). The introduction of subsurface Pd atoms on the L10-PtFe@Pt/C significantly enhances the ORR performance, which is attributed to the synergy of strain and ligand effects. Moreover, to confirm the observed enhancements indeed originate from the subsurface-specific effects rather than general Pd alloying effects, we synthesized a reference L10-PtFe@PtPdsur/C catalyst with marginally increased Pd content, thereby allowing Pd to occupy not only subsurface but also surface sites (Supplementary Fig. 32). After 30,000 ADT cycles, this L10-PtFe@PtPdsur catalyst exhibited more pronounced performance degradation compared to the L10-PtFe@PtPdsub configuration (Supplementary Fig. 33). These experimental results strongly support the conclusion that the subsurface positioning of Pd plays a critical and distinct role in enhancing catalytic durability. Beyond this, we also evaluated the electrochemical performance of other L10-PtFe@PtMsub/C catalysts (Supplementary Figs. 34–37). This finding provides a novel strategy for significantly enhancing the ORR activity and stability of Pt-based catalysts by regulating the subsurface atomic structure.

a ORR polarization curve of L10-PtFe@Pt/C, L10-PtFe@PtPdsub/C, and commercial Pt/C in O2-saturated 0.1 M HClO4 (pH = 1.01 ± 0.03; electrode surface area, 0.196 cm2; rotating speed, 1600 rpm; scan rate, 10 mV s−1). The iR was determined to be 19.7 ± 3.51 Ω. b MA and SA of the as-prepared catalysts at 0.9 V versus RHE. The error bars correspond to the standard deviation derived from five independent measurements. c ORR polarization curves of L10-PtFe@PtPdsub/C in O2-saturated 0.1 M HClO4 (pH = 1.01 ± 0.03; electrode surface area, 0.196 cm2; rotating speed, 1600 rpm; scan rate, 10 mV s−1) before and after ADT. The iR was determined to be 19.7 ± 3.51 Ω. d The MA and ECSA of L10-PtFe@PtPdsub/C before and after ADT. e H2-air single-cell polarization curves and power density plots of the L10-PtFe/PtPdsub cathodes before (BOL) and after (EOL) AST test. Test conditions: cathode loading of 0.1 mgPt cm−2, 80 °C, 100% relative humidity, 150 kPaabs. f MA at 0.9 V in H2–O2 fuel cells and the current density at 0.7 V in H2-air fuel cells before and after AST test. Source data for the electrocatalytic performance data are provided as a Source Data file.
The L10-PtFe@PtPdsub/C catalyst was used as a cathode catalyst layer for MEA and commercial Pt/C as the anode catalyst to evaluate the practical performance of as-prepared catalysts in PEMFCs. The cathodic and anodic catalytic layers of MEA were prepared by ultrasonic spraying with a Pt loading of 0.10 mgPt cm−2 for the cathode and 0.05 mgPt cm−2 for the anode. A 30 wt% Pt/C (TKK) commercial cathode catalyst was tested under the same conditions for comparison. The intrinsic activity of the as-prepared catalysts was evaluated under H2–O2 conditions, where it was virtually unaffected by mass transport issues. Notably, the L10-PtFe@PtPdsub/C catalyst achieved an initial MA value of 1.17 A mgPt−1 at 0.9 V, which is considerably higher than commercial Pt/C (0.3 A mgPt−1) and the U.S. Department of Energy (DOE) 2025 target (0.44 A mgPt−1) (Fig. 4f)43. The PEMFC performance of MEAs under H2/air conditions was also investigated for practical applications. The peak power density of L10-PtFe@PtPdsub/C catalyst reached 1.22 W cm−2 at a Pt loading of 0.10 mgPt cm−2, which was significantly higher than that of the Pt/C catalyst (0.85 W cm−2) (Fig. 4e and Supplementary Fig. 38). In addition, the current density of the L10-PtFe@PtPdsub/C catalyst is considerably higher than that of commercial Pt/C catalyst within the typical operating voltage range (>0.6 V), in particular, a high current density of 1.05 A cm−2 can be achieved at 0.7 V. At the critical heat rejection limit, L10-PtFe@PtPdsub/C cathode exhibited a rated power density of 1.26 W cm−2 at 0.67 V, performed at 94 °C and 250 kPaabs (Supplementary Fig. 39). After being normalized to the total cathode Pt loading, the MEA made with L10-PtFe@PtPdsub/C cathode showed an unprecedented Pt utilization efficiency of 0.079 gPt kW−1, which surpassed the DOE 2025 target (<0.1 gPGM kW−1).
The durability of the as-prepared catalysts was evaluated by the accelerated stress test (AST) at 0.6 to 0.95 V under H2/N2 conditions. After 30,000 voltage cycles, the L10-PtFe@PtPdsub/C catalyst retained 88.1% of the initial MA value, which is much higher than the 47% observed in commercial Pt/C catalyst (Fig. 4f). Notably, the end-of-life (EOL) MA (1.03 A mgPt−1) was 3.96 times that of the DOE target value (0.26 A mgPt−1) and 7.36 times the commercial Pt/C catalyst (0.14 A mgPt−1). Furthermore, L10-PtFe@PtPdsub/C also maintained a high EOL peak power of 1.14 W cm−2, significantly outperforming most of the previously reported catalysts. Notably, the current density loss of L10-PtFe@PtPdsub/C at 0.67 V is only 8.0%, and the voltage loss at 0.8 A cm−2 is about 15 mV, which is superior to Pt/C (72 mV) and also exceeds the DOE 2025 target of <30 mV. The L10-PtFe@PtPdsub/C catalyst is one of the superior catalysts reported to date in terms of performance on many fuel cell metrics (Table S6). The above results confirm the high activity and structural stability of core-shell L10-PtFe@PtPdsub/C catalyst under the conditions of practical PEMFC operation, demonstrating its prospects for practical application.
Identical location STEM characterization during ORR process
Structural reconstructions triggered by catalytic reactions are worth being investigated, as it provides valuable insights into the active sites of catalysts and guides the development of novel catalysts44,45,46. Identical location STEM (IL-STEM), as a quasi-in-situ technique, delivers high operational convenience and compatibility without any sacrifice in resolution47,48. In this study, we employed IL-STEM method to study the structural evolution of the L10-PtFe@PtPdsub/C catalyst under ORR conditions. We first customized a working electrode capable of holding the TEM grid. After dropping the catalyst onto the TEM grid, it was then fixed onto the customized working electrode. At the desired reaction stage, the reaction is terminated and the grid can be removed from the electrode and observed using STEM, allowing for the examination of catalyst structural alterations in the same region, as shown in Supplementary Fig. 40. These operations can be repeated multiple times.
After 50,000 cycles of the ADT, the carbon support structure morphology and the pore structure changed slightly (Supplementary Figs. 41, 42). Additionally, the migration of NPs was noted, some of which were marked with blue circles (Supplementary Fig. 42). Furthermore, no disappearance of whole NPs due to dissolution was observed, and the formation of a limited number of clusters was observed (illustrated by orange circles), which should originate from the reassembly of single atoms dissolved from other particles. In addition, we investigated the atomic structure changes of the catalyst at the atomic scale (Fig. 5a). After 30,000 electrochemical potential cycles, the overall structure of the nanoparticles and the number of atomic layers remained almost unchanged, and the outermost layers of unsaturated coordination atoms were still present. The outermost layers exhibited only weak atomic dissolution and rearrangement, with very few single atoms observed at the periphery of the particle. After 50,000 potential cycles, the dissolution and rearrangement of the severely unsaturated coordinated atomic layers on the exposed (001) and (011) planes occurred, with preferential dissolution at the steps. This resulted in a reduction of two atomic layers on the (001) plane of the nanoparticle, while the number of atomic layers on the (011) plane remained unchanged. Additionally, clusters consisting of dispersed single atoms were observed in the vicinity. Some nanoparticles are somewhat amorphous on the Pt surface due to dissolution and rearrangement of atoms on the surface (Supplementary Fig. 43). However, the particle sizes remain unchanged, indicating that the nanoparticles have not undergone structural collapse. Additionally, the grain boundaries in nanoparticles remain unaffected following the reaction (Supplementary Fig. 43).

a IL-STEM images of L10-PtFe@PtPdsub/C catalysts of initial, after 30,000 potential cycles, and after 50,000 potential cycles. Scale bar, 2 nm. b IL-EDS quantification of elemental content in catalysts at different reaction stages. c Charge density difference of L10-PtFe@PtPdsub/C. The blue and yellow isosurfaces indicate charge accumulation and depletion, respectively. Gray spheres represent Pt atoms, and red spheres represent Pd atoms. d d-band PDOS (surface Pt layer) for L10-PtFe@Pt. e d-band PDOS (surface Pt layer and Pd 4d) for L10-PtFe@PtPdsub. The Fermi level is set at zero energy (gray dashed line). f Calculated free energy diagram for L10-PtFe@PtPdsub (111), L10-PtFe@Pt (111), and Pt (111) catalysts under an electrode potential of 0 V. g Vacancy formation energy (Evacancy) of surface Pt atoms for L10-PtFe@Pt, L10-PtFe@PtPdsur, and L10-PtFe@PtPdsub. Source data are provided as a Source Data file.
The changes in the elemental composition of L10-PtFe@PtPdsub/C in the same region at different reaction cycles were quantitatively analyzed by EDS (Fig. 5b and Supplementary Fig. 41). We found that the changes in the composition of the Pt, Fe, and Pd on the L10-PtFe@PtPdsub/C were insignificant, especially for Fe, which was barely dissolved. It is well established that the dissolution of non-precious metals in alloy catalysts is highly likely to occur, thereby accelerating the deactivation of the catalyst. For the L10-PtFe@Pt/C catalyst after 50,000 cycles, a similar phenomenon was observed, with the Fe content decreasing from 32.62 to 8.72 at.% (Supplementary Fig. 44 and Table S7). In contrast, for the L10-PtFe@PtPdsub/C catalyst, the mitigation of Fe atom dissolution is proposed to arise from the presence of Pdsub. The Pdsub atoms greatly attenuate the structural collapse caused by surface Pt dissolution, thus preventing the formation of dissolution channels for Fe atoms. In addition, for the L10-PtFe@PtPdsur/C catalyst with Pd on the surface, STEM-EDS mapping results of post-ADT catalysts revealed severe Pd leaching in multiple nanoparticles—contrast to the minimal Pd loss observed in the L10-PtFe@PtPdsub/C catalyst (Supplementary Fig. 45). These results confirm that the enhanced stability originates specifically from the unique electronic and structural effects induced by the subsurface occupancy of Pd, which effectively mitigates metal dissolution and preserves the structural integrity.
Mechanistic investigation
DFT calculations were carried out to elucidate the effects of subsurface atomic modulation strategy on the electronic structures of surface active sites and the resulting ORR kinetics. Based on the information about the structural characterization of the catalysts, we constructed atomistic models of L10-PtFe@Pt, L10-PtFe@PtPdsub, and Pt slab, and the optimized structures are shown in Supplementary Fig. 46. The charge density difference of L10-PtFe@PtPdsub catalyst in Fig. 5c reveals that electrons are transferred from Pd to Pt, where the blue and yellow isosurfaces indicate charge accumulation and depletion, respectively. In addition, Bader charge analysis showed that Pd lost 0.51 electrons, which further proved the effect of Pd on the surface Pt electronic structure, in alignment with the XAS results. This charge redistribution confirms that the incorporation of Pd effectively introduces ligand effects to the surface active sites. Furthermore, a comparison of the projected density of states (PDOS) between L10-PtFe@Pt (Fig. 5d) and L10-PtFe@PtPdsub reveals that the incorporation of Pd induces strong d-orbital hybridization between subsurface Pd and surface Pt atoms. This electronic interaction results in a downshift of the d-band center, from −2.14 eV in L10-PtFe@Pt to −2.23 eV in L10-PtFe@PtPdsub, indicating a modulation of the surface electronic structure and weakened adsorption of oxygenated intermediates.
Given that the ORR reaction kinetics are closely related to the adsorption strength of the reaction intermediates on the Pt surface, we constructed models for the adsorption of different intermediate states in the four-electron associative ORR process (Supplementary Fig. 47). The computational hydrogen electrode (CHE) method was implemented to predict the free energy evolution along the four-electron ORR pathway on these catalysts when no external potential was applied (U = 0 V). The desorption kinetics of *OH intermediates is widely recognized as a key descriptor for catalytic performance in the ORR. The free energy diagrams in Fig. 5f indicate that the rate-determining step is the desorption of *OH for L10-PtFe@Pt and L10-PtFe@PtPdsub, and the introduction of Pd can weaken the interaction between the *OH intermediate and catalysts, thereby accelerating the reaction. Notably, the compressive strain from the PtFe core alone weakens the *OH adsorption energy on the surface Pt sites by 0.06 eV compared to pure Pt. While this confirms a significant improvement over pure Pt, the incorporation of subsurface Pd in the PtFe@PtPdsub model further weakens the *OH adsorption energy by 0.08 eV. This comparison allows us to quantitatively decouple the strain effects from PtFe core and ligand effects from Pd atoms. The baseline strain effect counts for approximately 42.86% of the total weakening of *OH binding. The additional 57.14% improvement is directly attributable to the electronic effect induced by subsurface Pd, which provides a distinct benefit beyond pure lattice compression. This additional weakening of *OH binding by subsurface Pd is critical for optimizing the ORR activity. Moreover, given that Pt dissolution represents a major degradation pathway in electrocatalysts, the vacancy formation energy of surface Pt (Esur) was calculated to evaluate the catalyst’s stability. The Esur values for L10-PtFe@Pt and L10-PtFe@PtPdsub were 3.05 and 3.30 eV, respectively, demonstrating that subsurface Pd atoms can inhibit Pt dissolution (Fig. 5g). In contrast, surface Pd lowers the Esur, which may due to the higher dissolution tendency of Pd compared to Pt in acidic electrolytes. These results highlight the critical role of subsurface sites in stabilizing surface Pt. The theoretical insights support experimental findings, thereby providing additional evidence that the subsurface engineering strategy employed in L10-PtFe@Pt can effectively enhance ORR performance through combining ligand effects and compressive strain.
In summary, we developed a precise subsurface atomic modulation strategy through in-situ introducing foreign metal into the subsurface atomic layer, providing direct tuning of coordination and electronic structures. Guided by the principles of surface energy minimization and metal-adsorbate interactions, we successfully synthesized stabilized L10-PtFe@PtMsub structures, Msub represents in-situ incorporated heterometallic atoms, including Ru, Rh, Pd, and Ag. As a proof of concept, the as-synthesized L10-PtFe@PtPdsub simultaneously stabilizes ligand effects and maintains strain effects, thereby breaking the trade-off between ligand and strain effects in Pt-based IMCs with Pt skin. And the L10-PtFe@PtPdsub catalyst breaks the activity-durability trade-off in ORR, delivering practical fuel cell performance with both high activity and enhanced durability. The subsurface Pd atoms optimized the surface-intermediates interaction strength by modulating the surface electronic environments, while simultaneously suppressing the dissolution of metal atoms. This work presents an atomic-precision subsurface modulation strategy on tuning surface electronic structures of heterogeneous catalysts and opens new avenues for the development of novel catalysts.
Methods
Chemicals
Hexachloroplatinic hexahydrate (H2PtCl6·6H2O, AR), Iron(II) sulfate heptahydrate (FeSO4·7H2O, AR, 99.0~101.0%), palladium(II) chloride (PdCl2, AR), ruthenium(III) chloride hydrate (RuCl3·xH2O, GR), rhodium(III) chloride hydrate (RhCl3·xH2O, 98%), silver nitrate (AgNO3, AR), tetrachloroauric(III) acid trihydrate (HAuCl4·4H2O, AR, ≥47.8%), ammonium molybdate tetrahydrate (Na2MoO4·2H2O, AR, ≥99.0%), sodium tungsten oxide dihydrate (Na2WO4·2H2O, AR, ≥99.5%), and acetone (AR, ≥99.5%) were purchased from Sinopharm Chemical Reagent Co., Ltd (Shanghai, China). Perchloric acid (GR, 70.0–72.0% HClO4) was purchased from Sigma-Aldrich. Ketjenblack EC-600J carbon black was purchased from Cabot. All chemicals were used as received without further purification. Deionized (DI) water from Milli-Q System (Millipore, Billerica, MA) was used in all our experiments.
Synthesis of the L10-PtFe/C and L10-PtFe@Pt/C catalysts
Carbon-supported L10-PtFe IMCs catalysts were prepared by a wet-impregnation method. Specifically, 0.03 mmol H2PtCl6·6H2O and 0.03 mmol FeSO4·6H2O were first dissolved in 20 mL of acetone, followed by the addition of 36 mg of Ketjenblack EC-600J. The mixture was stirred for 12 h to obtain a homogeneous solution, and then dried naturally at 25 °C. Finally, the dried powder was annealed in a tube furnace to 900 °C at 5 °C/min in an H2/Ar (5:95) atmosphere and maintained for 2 h and then cooled to 25 °C to obtain L10-PtFe/C. To synthesize L10-PtFe@Pt/C, 10 mg L10-PtFe/C powder was dispersed in 15 mL of a 0.1 M HClO4 solution, stirred and heated to 60 °C for 12 h. Then the solution was centrifuged, washed three times with DI water, and dried to obtain the powder, and the powder was annealed in a tube furnace to 400 °C at 5 °C/min in an H2/Ar (5:95) atmosphere and maintained for 2 h.
Synthesis of the L10-PtFe@PtMsub/C catalysts
The synthesis process of L10-PtFe@PtMsub/C was demonstrated using L10-PtFe@PtPdsub/C as an example. Specifically, 10 mg L10-PtFe/C powder was dispersed in 15 mL of a 0.1 M HClO4 solution and subjected to ultrasonic treatment to form a homogeneous suspension. Subsequently, an aqueous solution of PdCl2 (0.038 mg) was added, and the mixture was stirred and heated at 60 °C for 12 h. After the reaction, the solution was centrifuged, washed three times with DI water, and dried to obtain the final powder. Finally, the powder was annealed in a tube furnace to 400 °C at 5 °C/min in an H2/Ar (5:95) atmosphere and maintained for 2 h. The synthesis of L10-PtFe@PtMsub/C followed the same procedure as L10-PtFe@PtPdsub/C, except that the PdCl2 precursor was substituted with an equimolar amount of the M precursor, including RuCl3·xH2O, RhCl3·xH2O, AgNO3, HAuCl4·4H2O, Na2MoO4·2H2O, and Na2WO4·2H2O.
Structural characterizations
XRD patterns of as-prepared catalysts were examined using Philips X’Pert Pro Super diffractometer with Cu Kα radiation (λ = 1.54178 Å). Low magnification high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) images and EDS mapping were carried out on the Thermo Fisher Talos F200X with the Super X-EDS system. Aberration-corrected HAADF-STEM images and atomic EDS mapping were obtained on the Thermo Fisher Themis Z. XPS experiments were conducted in the Thermo ESCALAB MK II X-ray photoelectron spectrometer with Al Kα as the excitation source. Quantitative analysis of material element composition was conducted by inductively coupled plasma-mass spectrometry (ICP-MS, Thermo Fisher, iCAP RQ). Samples were ground with graphite for XAFS analysis on the BL14W1 and BL20U beamline of the Shanghai Synchrotron Radiation Facility.
XPS depth characterizations
XPS depth profiling analysis was achieved by using synchrotron radiation X-ray as an excitation source and implemented at BL10B photoemission end stations in the National Synchrotron Radiation Laboratory. Probing depth was achieved by regulating the kinetic energy of the emitted photoelectrons of specific core electron energy level to the relative predefined value. The binding energies were calibrated by C 1 s (284.6 eV) and the collecting energy step was 0.1 eV.
Stain mapping
Van Aert et al. proposed an efficacious model-based estimation algorithm to quantify the positions and intensities of atomic columns from atomic resolution (S)TEM images and developed a statSTEM software package using the MATLAB programming language. The atomic positions in the HAADF-STEM image of a L10-PtFe@PtPdsub NP were identified using statSTEM software. Based on these atomic position coordinates, we derived the spacing of the neighboring atomic columns and calculated the strain values compared to the theoretical values.
CO-adsorption diffuse reflectance infrared Fourier transform spectroscopy
CO-DRIFTS characterization was performed on a Nicolet iS10 spectrometer. After mixing and grinding the samples with KBr and loading them into an in-situ reaction cell (Praying Mantis Harrick), the samples were first treated in 10% O2/Ar at 150 °C for 30 min and then reduced in 10% H2/Ar at 150 °C for 30 min. Subsequently, the samples were continuously vented with Ar at a flow rate of 20 mL/min, cooled to 25 °C and a background spectrum was obtained until a stable baseline was achieved. Subsequently, a switch to 10% CO was made until the sample was saturated with adsorption. Next, the sample was switched back to Ar for purging and DRIFT spectra were collected until the CO gas-phase peak disappeared. Spectral acquisition parameters were as follows: 256 scans with a resolution of 4 cm−1.
Electrochemical measurements (rotating disk electrode test)
Electrochemical ORR testing was conducted using a CHI700E electrochemical station and a Pine ASR rotator. The Pine ASR instrument was equipped with a glassy carbon electrode as the working electrode (0.196 cm2), Pt sheet as the counter electrode, Ag/AgCl (saturated KCl) electrode as the reference electrode, and the electrolyte was 0.1 M HClO4 solution (pH = 1.01 ± 0.03, determined from five independent measurements). 0.1 M HClO4 electrolyte was prepared by diluting 2.16 mL high-purity concentrated HClO4 (70–72%) with DI water in a 250 mL volumetric flask. The electrolyte was prepared immediately before use. The Ag/AgCl electrode was calibrated for a RHE with a Pt sheet in a high-purity H2-saturated electrolyte. All catalyst inks were prepared by dispersing 1 mg of the as-prepared catalysts in a mixture containing 0.985 mL isopropanol and 15 µL Nafion® and ultrasonicating for 30–60 min to form a 1 mg/mL ink. 10–15 µL of ink was cast on the glassy carbon and dried under ambient conditions. The final loadings were about 10 µg cm−2 for L10-PtFe@Pt/C and L10-PtFe@PtMsub/C electrocatalysts according to ICP-MS.
For electrochemical tests, CV curves were scanned in N2-saturated 0.1 M HClO4 from 0.05 to 1.2 V vs. RHE at a scan rate of 50 mV s−1 to obtain. Only when the CV curves reached a steady state could they be used for background subtraction and ECSA determination. And the ORR polarization curves, corrected for iR drop (E-iR), were carried out in O2-saturated 0.1 M HClO4 at 10 mV s−1. ADT tests were performed by scanning between 0.6 and 1.0 V vs. RHE in O2-saturated 0.1 M HClO4 at 100 mV s−1. The solution resistance was determined to be 19.7 ± 3.51 Ω by the CHI700E electrochemical station.
CO stripping measurement involved three processes: high-purity CO was first bubbled into a 0.1 M HClO4 solution holding the potential at 0.05 V vs. RHE for 10 min, then pure N2 was bubbled into the electrolyte for 10 min, and finally, the voltammetry curve was scanned from +0.05 to +1.15 V vs. RHE at a scanning rate of 50 mV s−1.
All measured potentials were converted to the RHE using the Nernst equation:
Calculation methods for MA, SA, and ECSA
Firstly, the CV curve was obtained by scanning at a rate of 50 mV s−1 within the potential range of 0.0–1.2 V in a N2-saturated 0.1 M HClO4 solution. Subsequently, the ECSA was calculated by integrating the hydrogen adsorption peaks (approximately 0.05–0.4 V) on the CV curve to obtain the charge quantity (QH), with the assumption that the value for a monolayer of adsorbed hydrogen is 210 μC/cm². The specific equation is as follows:
Where MPt (μgPt) represents the Pt loading on the working electrode.
The kinetic currents (Ik) can be calculated using the Koutecký-Levich equation:
where I is the measured current and Id is the diffusion-limited current. Id can be determined using the Levich equation:
where n is the number of electrons transferred, F is the Faraday constant (96485 C/mol), A is the geometric area of the working electrode (0.196 cm2), D is the diffusion coefficient of oxygen in 0.1 M HClO4 (1.93 × 10−5 cm2/s), ν is the kinematic viscosity of the 0.1 M HClO4 (1.01 × 10−2 cm2/s), ω is the rotation rate, and CO2 is the concentration of oxygen molecules in 0.1 M HClO4 (1.26 × 10−6 mol/cm3).
The MA of the catalyst is calculated by normalizing the kinetic current at 0.9 V vs. RHE to the Pt loading (μgPt) on the working electrode.
The SA of the catalyst is obtained by dividing the MA by the ECSA.
Fuel-cell measurements
The 30 mg L10-PtFe@PtPdsub/C catalysts, and commercial 30 wt% Pt/ (TEC10E30E) were mixed with 10 mL of water/isopropanol with a 1:1 ratio in a serum bottle. Nafion solution (Aquivion® D79-25BS Perfluorosulfonic Acid, 25%) was adapted as proton conductor and cross-linking agent. The mixture was ultrasonicated for 120 min to obtain a homogeneous ink. The concentration of all inks was controlled to be 3 mgcat. mL−1. The optimized I/C ratio of ink was controlled to 0.6. The catalyst-coated-membrane (CCM) was prepared on GORE Nafion membrane (active area of 5 cm2) with an ultrasonic spray equipment (SONO·TEK). The carbon paper (22BB SGL Carbon) with a 215 μm-thick microporous layer was used as a gas diffusion layer (GDL). The MEA contained five parts with two GDLs, two gaskets (140 μm), and the prepared CCM. The Pt loadings of all MEAs at cathode and anode were controlled to be 0.1 and 0.05 mgPt cm−2, respectively.
Then the MEA was packaged in a single cell and tested in the 850e fuel cell test system (Scribner Associates, Inc.), with seven channel serpentine flow field. The cell pre-activation was performed before data collection as followed. As the cell and humidifiers to 80 °C, the cell voltage was swept from open circuit to 0.3 V under H2–O2 conditions at 150 kPaabs and 100% relative humidity (RH) with several cycles until the current density was stable. The MA was obtained from H2–O2 conditions (0.2/0.5 L min−1) at 80 °C, 150 kPaabs and 100% RH. The fuel cell was operated at 0.9 ViR‑correct for 15 min, and the MA was obtained from the average current density at the last 1 min. The H2-air polarization curve was collected under 80 °C, 150 kPaabs and 100% RH (or 94 °C, 250 kPa and 90% RH), with the H2/air flow of 0.5/2 L min−1. The accelerated stress test (AST) was adopted 30,000 cycles of square wave holding 3 s at voltage 0.6 V and then 3 s at voltage 0.95 V under 80 °C, 100 kPaabs and 100% RH with H2/N2 flow of 0.2/0.075 L min−1, according to DOE standards.
Quasi-in-situ identical location-(S)TEM (IL-STEM) measurements
The IL-STEM experimental procedure is described in four sequential steps:
-
1.
Structural characterization of as-prepared catalysts at identical locations
Firstly, the catalyst ink was deposited onto Au TEM grids selected for the chemical inertness of Au. Initial STEM characterization focused on nanoparticles in the central region of the Au grid—an area with distinct morphological features that enabled precise relocation for comparative post-reaction analysis. Following atomic-resolution HAADF-STEM imaging of target nanoparticles, a series of multi-magnification images were acquired to establish precise positional references.
-
2.
Fabrication of the working electrode assembly for IL-STEM characterization
Firstly, we modified the conventional working electrode by affixing a helical cap on the glassy carbon electrode side, the cap featuring a central aperture with a diameter slightly smaller than that of the Au grid, thereby ensuring effective electrode-electrolyte interfacial contact. Subsequently, the Au grid dropped with catalysts was positioned at the center of the glassy carbon electrode with the catalyst-coated side facing outward, and securely immobilized by tightening the helical cap.
-
3.
Electrochemical cycling tests
The IL-STEM electrochemical cycling protocol mirrored standard RDE methodologies. After the working electrode integrated the Au grid was installed in a Pine Research RDE holder, ADTs were performed. Notably, despite the ultralow catalyst loading on the IL-STEM working electrode, the system maintained characteristic CV profiles.
-
4.
Structural characterization of catalysts at identical locations in the desired reaction stage
Upon reaching the desired reaction stage, the Au grid is removed from the reaction system and transferred to the TEM. After the central region of the Au grid was found under the low-magnification TEM imaging, precisely relocating the target catalyst nanoparticles by using pre-acquired multi-scale reference images, and performing the HAADF-STEM imaging.
The above process can be repeated at different reaction stages in order to study the structural evolution of the catalyst under electrocatalytic reaction conditions.
Theoretical calculations
All the density functional theory calculations were conducted using the Vienna ab initio Simulation Package with the projector-augmented wave pseudopotentials. The generalized gradient approximation with the Perdew–Burke–Ernzerhof function was used to describe the exchange-correlation energy. The plane-wave energy cutoff is 500 eV, and the convergence criterion of the electronic self-consistent iteration was set to be 10−6 eV. The structures were relaxed, and the convergence criterion of the force on each atom is 0.01 eV Å−1. The Monkhorst-Pack 3 × 3 × 1 k-point grid was used to sample the Brillouin zone.
The slab model of L10-PtFe@Pt(Pdsub) was constructed based on the Pt (111) surface model in a 4 × 4 in-plane supercell. The vacuum layer was set to be 15 Å to ensure the slabs were separated.
The elementary steps in the four-electron ORR occurs are believed to involve surface-adsorbed OOH*, O*, and OH* species, as follows:
The Gibbs free energy change (ΔG) for each adsorbate can be calculated by employing the CHE model developed by Nørskov et al.:
where ΔE is the adsorption energy obtained from DFT calculation, ΔZPE is the zero-point energy correction, T is the temperature (298.15 K), ΔS is the entropy change. U is the implemented potential, ΔGpH represents the free energy correction of the pH, kB is the Boltzmann constant.
The surface energies (Esurf) of all the relevant surfaces were calculated by the following formula:
where Eslab and Ebulk are the total energies of the slab and a bulk unit, respectively; n represents the number of atoms in the slab, and A is the exposed surface area of one side of the slab.
The formation energy (Ef) was defined as follows:
where Evacancy and Epristine are the total energies of structures with and without a vacancy, respectively, and μPd is the chemical potential of Pd.
Data availability
The experiment data that support the findings of this study are provided in the Source Data file. The atomic coordinates of the optimized L10-PtFe@PtPdsub and L10-PtFe@Pt models are provided as Supplementary Data 1 and 2, respectively. Source data and Supplementary Data are provided with this paper. Source data are provided with this paper.
References
Tao, F. & Salmeron, M. Surface restructuring and predictive design of heterogeneous catalysts. Science 386, eadq0102 (2024).
Ledentu, V. et al. Heterogeneous catalysis through subsurface sites. J. Am. Chem. Soc. 122, 1796–1801 (2000).
Li, P. et al. Manipulation of oxygen species on an antimony-modified copper surface to tune the product selectivity in CO2 electroreduction. J. Am. Chem. Soc. 146, 26525–26533 (2024).
Jalil, A. et al. Nickel promotes selective ethylene epoxidation on silver. Science 387, 869–873 (2025).
Nam, D.-H. et al. Molecular enhancement of heterogeneous CO2 reduction. Nat. Mater. 19, 266–276 (2020).
Chen, S. et al. Selective CO2 reduction to ethylene mediated by adaptive small-molecule engineering of copper-based electrocatalysts. Angew. Chem. Int. Ed. 62, e202315621 (2023).
Zhao, K. et al. A molecular design strategy to enhance hydrogen evolution on platinum electrocatalysts. Nat. Energy 10, 725–736 (2025).
Zhao, Z. et al. Graphene-nanopocket-encaged PtCo nanocatalysts for highly durable fuel cell operation under demanding ultralow-Pt-loading conditions. Nat. Nanotech. 17, 968–975 (2022).
Ma, X. et al. Carbonate shell regulates CuO surface reconstruction for enhanced CO2 electroreduction. Nat. Synth. 4, 53–66 (2025).
Cao, X. et al. Sandwiching intermetallic Pt3Fe and ionomer with porous N-doped carbon layers for oxygen reduction reaction. Nat. Commun. 16, 2851 (2025).
Li, Y. et al. Accelerated selective electrooxidation of ethylene glycol and inhibition of C-C dissociation facilitated by surficial oxidation on hollowed PtAg nanostructures via in situ dynamic evolution. JACS Au 5, 714–726 (2025).
Peng, B. et al. Embedded oxide clusters stabilize sub-2 nm Pt nanoparticles for highly durable fuel cells. Nat. Catal. 7, 818–828 (2024).
Tao, F. et al. Reaction-driven restructuring of Rh-Pd and Pt-Pd core-shell nanoparticles. Science 322, 932–934 (2008).
Song, Y. et al. Bimetallic synergy from a reaction-driven metal oxide-metal interface of Pt-Co bimetallic nanoparticles. ACS Catal. 13, 13777–13785 (2023).
Mu, R. et al. Synergetic effect of surface and subsurface Ni Species at Pt-Ni bimetallic catalysts for CO Oxidation. J. Am. Chem. Soc. 133, 1978–1986 (2011).
Lin, Y. et al. Optimizing surface active sites via burying single atom into subsurface lattice for boosted methanol electrooxidation. Nat. Commun. 16, 286 (2025).
Vitale-Sullivan, M. & Stoerzinger, K. A. Interplay of surface and subsurface contributions in electrocatalysis. Curr. Opin. Electrochem. 39, 101252 (2023).
Teschner, D. et al. The roles of subsurface carbon and hydrogen in palladium-catalyzed alkyne hydrogenation. Science 320, 86–89 (2008).
Jin, G. et al. Heteroepitaxial van der Waals semiconductor superlattices. Nat. Nanotech. 16, 1092–1098 (2021).
Stephens, I. E. L. et al. Tuning the activity of Pt(111) for oxygen electroreduction by subsurface alloying. J. Am. Chem. Soc. 133, 5485–5491 (2011).
Zhao, X. et al. Engineering covalently bonded 2D layered materials by self-intercalation. Nature 581, 171–177 (2020).
Derouin, J. et al. Thermally selective formation of subsurface oxygen in Ag(111) and consequent surface structure. ACS Catal. 6, 4640–4646 (2016).
Lu, J. Atomic lego catalysts synthesized by atomic layer deposition. Acc. Mater. Res. 3, 358–368 (2022).
Guan, Q. et al. Bimetallic monolayer catalyst breaks the activity-selectivity trade-off on metal particle size for efficient chemoselective hydrogenations. Nat. Catal. 4, 840–849 (2021).
Jin, S. et al. Atom-by-atom design of Cu/ZrOx clusters on MgO for CO2 hydrogenation using liquid-phase atomic layer deposition. Nat. Catal. 7, 1199–1212 (2024).
Yang, L. et al. Cation exchange in colloidal transition metal nitride nanocrystals. J. Am. Chem. Soc. 146, 12556–12564 (2024).
Wang, Z. et al. Tailored lattice compressive strain of Pt-Skins by the L12-Pt3M intermetallic core for highly efficient oxygen reduction. Adv. Mater. 35, 2301310 (2023).
Shi, Y. et al. Noble-metal nanocrystals with controlled shapes for catalytic and electrocatalytic applications. Chem. Rev. 121, 649–735 (2021).
Zhao, M. et al. Hollow metal nanocrystals with ultrathin, porous walls and well-controlled surface structures. Adv. Mater. 30, 1801956 (2018).
Tran, R. et al. Surface energies of elemental crystals. Sci. Data 3, 160080 (2016).
Huang, X. et al. High-performance transition metal-doped Pt3Ni octahedra for oxygen reduction reaction. Science 348, 1230–1234 (2015).
Luo, H. et al. Amorphous MoO with high oxophilicity interfaced with PtMo alloy nanoparticles boosts anti-CO hydrogen electrocatalysis. Adv. Mater. 35, 2211854 (2023).
Ramirez Caballero, G. E. & Balbuena, P. B. Surface segregation phenomena in Pt-Pd nanoparticles: dependence on nanocluster size. Mol. Simul. 32, 297–303 (2006).
Shao, R. Y. et al. Promoting ordering degree of intermetallic fuel cell catalysts by low-melting-point metal doping. Nat. Commun. 14, 5896 (2023).
Hu, J. et al. Unique applications of carbon materials in infrared stealth: a review. Chem. Eng. J. 452, 139147 (2023).
Aleksandrov, H. A. et al. Can the state of platinum species be unambiguously determined by the stretching frequency of an adsorbed CO probe molecule? Phys. Chem. Chem. Phys. 18, 22108–22121 (2016).
Bazin, P. et al. FT-IR study of CO adsorption on Pt/CeO2: characterisation and structural rearrangement of small Pt particles. Phys. Chem. Chem. Phys. 7, 187–194 (2005).
De Backer, A. et al. StatSTEM: an efficient approach for accurate and precise model-based quantification of atomic resolution electron microscopy images. Ultramicroscopy 171, 104–116 (2016).
Xie, M. et al. Pt-Co@Pt octahedral nanocrystals: enhancing their activity and durability toward oxygen reduction with an intermetallic core and an ultrathin shell. J. Am. Chem. Soc. 143, 8509–8518 (2021).
Liu, X. et al. Introducing electron buffers into intermetallic Pt alloys against surface polarization for high-performing fuel cells J. Am. Chem. Soc. 146, 2033–2042 (2024).
Yang, L. et al. Rare earth evoked subsurface oxygen species in platinum alloy catalysts enable durable fuel cells. Angew. Chem. Int. Ed. 63, e202315119 (2024).
Wang, Z. et al. Strain engineering of Pt-based electrocatalysts for oxygen reaction reduction. Front. Energy 18, 241–262 (2024).
Office of Energy Efficiency & Renewable Energy. Energy. gov. 2016. https://www.energy.gov/eere/.
Beermann, V. et al. Real-time imaging of activation and degradation of carbon supported octahedral Pt-Ni alloy fuel cell catalysts at the nanoscale using in situ electrochemical liquid cell STEM. Energy Environ. Sci. 12, 2476–2485 (2019).
Huang, H. & Russell, A. E. Approaches to achieve surface sensitivity in the in situ XAS of electrocatalysts. Curr. Opin. Electrochem. 27, 100681 (2021).
Ortiz Peña, N. et al. Morphological and structural evolution of Co3O4 nanoparticles revealed by in situ electrochemical transmission electron microscopy during electrocatalytic water oxidation. ACS Nano 13, 11372–11381 (2019).
Hrnjic, A. et al. Observing, tracking and analysing electrochemically induced atomic-scale structural changes of an individual Pt-Co nanoparticle as a fuel cell electrocatalyst by combining modified floating electrode and identical location electron microscopy. Electrochim. Acta 388, 138513 (2021).
Zhi-Peng, W. et al. Applications of in situ electron microscopy in oxygen electrocatalysis. Microstructures 2, 2022002 (2022).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (52122212, Y.L.; 12274391. Y.L.; 22321001, C.W. and Y.L.; 12404225, X.H.; Grant nos. 22525013, C.W.), the Youth Innovation Promotion Association of CAS (2020458, Y.L.), the Key Research Program of Frontier Sciences, CAS (ZDBS-LY-SLH003, Y.L.), CAS Project for Young Scientists in Basic Research (YSBR-070, C.W.), the National Key Research and Development Program of China (2022YFB3807602, Y.L.; 2022YFA1203600, C.W.), China Postdoctoral Science Foundation (2023M743348 and 2024T170888, X.H.), Postdoctoral Fellowship Program of CPSF (GZC20232511, X.H.) and the University Synergy Innovation Program of Anhui Province (GXXT-2022-007, H.D.). The calculations were performed on the Supercomputing Center of the University of Science and Technology of China. We thank the photoemission end stations MCD-A, MCD-B, BL10B, BL01B in the National Synchrotron Radiation Laboratory (NSRL) and BL14W1, BL20U in the Shanghai Synchrotron Radiation Facility (SSRF) for help in characterizations.
Author information
Authors and Affiliations
Contributions
Y.L. and C.W. conceived the idea and co-wrote the paper. X.T., S.X., and X.H. carried out the sample synthesis, characterization, and electrochemical performance measurement. Y.Z. calculated the DFT calculation. L.X. analyzed the XAS results. Y.X. drew the schematic diagram. H.D., R.G., and M.M. discussed with results and helped with the modification of the paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Qing Li and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Tai, X., Zhou, Y., Xu, S. et al. Atomic diffusion pathway mediated subsurface engineering. Nat Commun 17, 593 (2026). https://doi.org/10.1038/s41467-025-67296-1
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-67296-1
